
Abstract
Low back pain (LBP), primarily driven by intervertebral disc degeneration (IVDD), has become a core challenge in public health. DDX1, an RNA-binding protein, plays key roles in RNA metabolism but its function in IVDD remains unclear. We identify DDX1 as a substrate of methyltransferase EZH2, which methylates DDX1 at lysine 234 (K234), promoting IVDD in vitro and in vivo. EZH2 inhibition restores matrix homeostasis in nucleus pulposus (NP) cells and slows IVDD progression. Methylation at DDX1 K234 disrupts its interaction with splicing factors and RNA targets, promoting exon 14 skipping in MATR3. This truncated MATR3 disrupts nuclear architecture, increases chromatin accessibility, and activates signaling pathways such as Wnt, leading to NP cell senescence and apoptosis. Notably, delivery of MATR3-L-overexpressing mRNA via cationic lipid nanoparticles reduces NP cell degeneration and significantly alleviates IVDD, offering important insights into IVDD pathogenesis and potential therapeutic strategies.
Similar content being viewed by others


Introduction
Low back pain (LBP) is a significant health burden in the elderly and the second leading cause of medical consultations in industrialized countries1. As the most common and burdensome musculoskeletal condition, an estimated 40% of LBP cases are linked to intervertebral disc degeneration (IVDD), a widespread and progressive age-associated pathology2. Extensive research has explored the pathogenesis of IVDD, revealing a strong association between aging, apoptosis, and disc degeneration3. In addition, IVDD manifests as a pro-aging and apoptotic intervertebral disc (IVD) microenvironment, characterized by hypoxia, inflammation, and disturbances in metabolic and nutritional factors4. Notably, epigenomic alterations influencing chromatin architecture are key regulators of cell fate and aging. In nucleus pulposus (NP) cells, such changes may exert long-term effects across cell generations and uncover previously unrecognized aspects of the aging process5,6. However, the renewal and differentiation of NP cells in IVD tissues during degeneration are still unclear, and the related epigenomic regulatory mechanisms are still worth exploring.
Alternative splicing (AS) refers to the process of generating mRNA splice isoforms through different splicing modes from one mRNA precursor, adding complexity to gene regulation by influencing protein isoform diversity, mRNA stability, and translation efficiency7. When this event occurs abnormally, different proteomes will be produced through different splicing methods, which will further affect normal cell physiological functions and may lead to pathological changes8. Previous studies have shown that senescence relies on a defined, alternatively spliced transcriptome, but this has not been comprehensively defined on a transcriptome scale, and the molecular mechanisms underlying this program are poorly understood9. Increasing evidence suggests that splicing is co-transcriptional and is directly affected by the epigenetic landscape and dynamics of the underlying transcription machinery at these sites; on the other hand, different mRNA transcripts generated by AS may also play a regulatory role in chromatin reprogramming10. However, the molecular couplers linking chromatin reprogramming and AS during IVDD remain unknown.
RNA-binding proteins (RBP) stand out as pivotal post-transcriptional regulators due to their capacity to broadly influence the activity of multiple target RNA and their involvement in cellular senescence, where they orchestrate the expression of apoptotic genes, growth factors, and cell cycle proteins11,12. Even in minute quantities, RBP can elicit significant changes in gene expression, often attributed to aberrant patterns of post-translational modifications (PTM), among which lysine methylation has emerged as a key player13,14, impacting various biological functions such as protein-protein interactions, stability, subcellular localization, and transcription15. So far, large-scale screens have gained insight into the intricate network of interactions between RBP and their targets, laying the groundwork for a systematic exploration of post-transcriptional regulatory mechanisms, studies on RBP-mediated regulation of AS exemplify this endeavor16. Therefore, an analysis of NP cells phenotypes from the perspective of PTM-mediated RBP structure and status promises to unveil previously uncharacterized post-transcriptional regulatory pathways underlying IVDD and may identify candidate molecules bridging the epigenome and AS in this pathological process.
In this study, we demonstrated that EZH2-mediated DDX1 lysine methylation promotes MATR3 exon 14 skipping and facilitates NP cells senescence and apoptosis during the progression of IVDD. Functional gain and loss experiments suggest that EZH2 deficiency rescues the diminished synthesis metabolism and enhanced degradation metabolism in NP cells, thereby mitigating the IVDD progression. Mechanistically, the increased expression of EZH2 enhances the binding of DDX1, leading to the mono-methylation of lysine residue 234 on DDX1 (DDX1 K234me1). Methylated DDX1 weakens its binding to the exon-intron region near MATR3 splicing sites and reduces recruitment of splicing factors. Consequently, MATR3 exon 14 skipping results in the formation of MATR3 short isoforms (MATR3-S), which genetically induce whole-genome chromatin reprogramming and aberrant activation of senescence and apoptosis signaling pathways such as Wnt and MAPK in NP cells. Importantly, a strategy based on engineered cationic lipid nanoparticles (LNPs) for the delivery of MATR3-L overexpressed mRNA drug system can effectively alleviate reduced anabolism, enhanced catabolism, as well as senescence in NP cells, and significantly alleviate IVDD progression.
Results
K234 methylation of DDX1 is increased in NP cells during IVDD
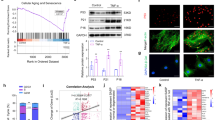
To identify key non-histone proteins regulated by lysine methylation17, cells from non-degenerative and degenerative NP tissues, obtained from individuals with lumbar fractures, idiopathic scoliosis, or IVDD, were lysed and incubated with a pan-lysine methylation-specific antibody. LC-MS/MS was used to analyze compounds. (Fig. 1A). NP tissue degeneration was assessed using the Pfirrmann grading based on medical imaging technology—MRI, which showed that the T2-weighted high-intensity white signal decreased and was heterogeneous, and eventually being replaced by a high-intensity black signal, suggesting NP tissue during IVDD progression18. Water content in the medium declined, while fibrosis increased. Safranin O-Fast Green further validated the degenerative phenotype of NP tissue, revealing pronounced matrix degradation, calcification, chondroid proliferation, and cell clustering, particularly evident in the severely degenerated group19 (grade IV) (Supplementary Fig. 1A). In addition, immunohistochemical (IHC) staining revealed a decline in collagen II-positive (anabolic) cells and a corresponding increase in MMP3-expressing (catabolic) cells within degenerated NP tissue (Supplementary Fig. 1B, E). Notably, DDX1-derived peptides were significantly identified in degenerative NP tissue—methylation modification at an evolutionarily conserved lysine residue (K234) in the SPRY (231-240aa) domain (Supplementary Fig. 1C). Considering the proposed roles for SPRY domain, methylation within this domain may be of significance. LC-MS showed that K234 was monomethylated (Fig. 1B). Additionally, lysine at position 234 is highly conserved across DDX1 proteins from multiple species (Supplementary Fig. 1D), indicating its potential functional significance. Notably, K234 methylation was detected in both NP cells and HEK-293T tool cells (Fig. 1C, D, Supplementary Fig. 1F). Given that DDX1 is an RBP, its function may be affected by lysine methylation, which in turn regulates intracellular signaling pathways and gene expression networks. We cultured NP cells with the oxidant tert-butyl peroxide (TBHP) to construct an in vitro degeneration model to study whether pharmacologically induced oxidative stress damage would lead to differential expression of DDX1 K234 methylation20. Following TBHP treatment, collagen II and aggrecan showed a downward trend whereas MMP3 and ADAMTS5 showed an opposite trend. (Supplementary Fig. 1H, I). Immunofluorescence (IF) staining demonstrated type II collagen-positive decreased and MMP3 increased in NP cells treated with TBHP (Fig. 1E, Supplementary Fig. 1G), indicating that TBHP-induced degeneration in cultured NP cells reflects the degenerative state observed in IVDD21,22. Interestingly, DDX1 K234 methylation was significantly induced in TBHP-treated NP and HEK-293T cells (Fig. 1F, Supplementary Fig. 1J). Then, lysine residue 234 in the SPRY domain was replaced with arginine to generate a DDX1 mutant23,24. Substituting lysine (K) with arginine (R) preserves the positive charge while preventing methylation, thus mimicking the unmethylated protein form. The DDX1 K234R mutant remained unmethylated in TBHP-treated HEK-293T cells (Fig. 1G). Collectively, these findings identify Lys234 as a DDX1-specific methylation site in SPRY, linked to NP cell degeneration.

A LC-MS/MS experimental workflow schematic. Cell lysates from degenerated and non-degenerated NP tissues were digested and applied to LC-MS/MS. B LC-MS/MS of the Lys234 residue of the peptide segment NQALFPACVLK, with a molecular weight of +14.016 Da. C, D Detection of endogenous DDX1 lysine methylation in NP and HEK-293T cells by CO-IP (n = 3). E IF analysis of collagen II and MMP3 expression (green: collagen II; red: MMP3; scale bar: 50μm). F Expression of endogenous DDX1 lysine methylation was detected by CO-IP in normal and TBHP-treated NP cells (n = 3). G CO-IP of the DDX1 K234 methylation (R2 was the R1 clone system, n = 3). H, J Schematic illustration showing the experiment design. Compared with the control group (n = 5), the rat IVDD model was established by needle puncture (model group, n = 20), LV-scrambled or LV-shDDX1 were used to knock down DDX1 in model group rats (scrambled group, n = 5 and shDDX1 group, n = 5). Subsequently, shRNA-resistant LV-DDX1 WT (DDX1 WT group, n = 5) and LV-DDX1 K234R (DDX1 K234R group, n = 5) were re-overexpressed in the shDDX1 group rat and injected weekly into the NP. I H&E and SO&FG of discs from rats. Scale bar: 1 mm. K–M X-ray K, CT L, MRI M of rat vertebrae (n = 5). N, O DHI N, Pfirrmann grades (O) of discs (n = 10). Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Data are represented as mean ± SEM. P value was quantified by two-way ANOVA (N, O). Panels (C–G) show the results of a representative similar result from one of three independent experiments. Panels (I, K–M) the results of a representative similar result from one of five independent experiments.
Since K234 was identified endogenously via unbiased mass spectrometry, we examined how its mono-methylation affects DDX1 function and contributes to IVDD. To explore the biological potential of methylation in the SPRY domain, wild-type DDX1 (WT) and the methylation-deficient mutant (K234R) were ectopically expressed in DDX1-knockdown NP cells. Compared to the control (siControl), DDX1 knockdown reversed TBHP-induced anabolic decline and catabolic increase in NP cells in vitro, whereas re-expression of DDX1, but not the K234R, significantly worsened TBHP-induced degeneration (Supplementary Fig. 1K, L). To avoid off-target effects, the knockdown efficiency of another siDDX1 in NP cells (Supplementary Fig. 2B, C) was detected by western blot. Meanwhile, DDX1 WT and K234R mutants were re-overexpressed as expected, which were almost twice as much as the control. To further explore the in vivo role of lysine methylation, a needle puncture-induced IVDD animal model was established in Sprague-Dawley (SD) rats25,26. Furthermore, we used lentiviruses (LVs) carrying short hairpin RNA targeting DDX1 (shDDX1) to knock down DDX1 in vivo and subsequently ectopically expressed shRNA-resistant DDX1 WT-LV and DDX1 K234R-LV, injected weekly into the NP area of the tailbone IVD of SD rats for a total of 4 weeks27 (Fig. 1H, J). Notably, X-ray and computed tomography (CT) analyzes showed that shDDX1-LV significantly alleviated the constricted disc and subchondral bone destruction in the coccyx, while re-expression of DDX1-LV but not DDX1 K234R-LV further resulted in loss of IVD height (Fig.1K, L). Magnetic resonance imaging (MRI), a more sensitive radiological diagnostic technique, showed an increase in T2-weighted signal in the coccyx IVDs28, indicating that the water content of the IVD was retained after DDX1 knockdown and that re-expression of normal DDX1, but not the arginine mutant, led to reduced water content in the IVDs (Fig. 1M). Histological analysis using HE and SO&FG staining confirmed that LV-mediated DDX1 deficiency alleviated the degenerative phenotype in discs, relieving the ambiguous interface between the disordered extracellular matrix (ECM), annulus fibrosus (AF), and NP in these IVDs and reducing the presence of inflammatory cells within AF (Fig. 1I, N, O, Supplementary Fig. 1M). By immunohistochemistry (IHC) staining, an increase in collagen II and a decrease in MMP3 were found in discs with sh-DDX1-LV, indicating that IVDD progression was restored (Supplementary Fig. 1N, O). Importantly, reinjection of DDX1 WT-LV but not DDX1 K234R-LV resulted in atrophy of the NP tissue, unclear boundary between the NP and AF, and disordered matrix structure, and promoted progressive degeneration of the coccyx IVD (Fig. 1I). In addition, re-ectopic expression of DDX1 instead of DDX1 K234R resulted in a decrease in collagen II-positive NP cells and an increase in MMP3-positive NP cells (Supplementary Fig. 1N, O). To avoid off-target effects, the knockdown efficiency of another shDDX1-LV in IVDs was tested by IHC (Supplementary Fig. 2H, I). DDX1 K234R-LV was overexpressed again as expected, almost doubling compared to the scrambled. Therefore, these findings confirm that DDX1 K234 methylation may lead to NP degeneration and promote IVDD progression both in vivo and in vitro.
DDX1 was methylated at lysine 234 through its interaction with EZH2
To identify physiologically relevant upstream methyltransferases responsible for this modification, co-immunoprecipitation (Co-IP) assays were conducted in HEK-293T cells to isolate DDX1-binding partners. Based on an abundance greater than ten and a P-value < 0.05, KMT5A, SMYD3, SETD7, EZH2, and G9a were identified as candidate lysine methyltransferases (Fig. 2A, B). Substantial lysine methylation was subsequently observed in anti-DDX1 immunoprecipitates from HEK-293T induced with the EZH2 plasmid (Fig. 2C). IP using Flag antibody, immunoblotting (IB) with DDX1, of lysates from HEK-293T cells induced with specific plasmids revealed that EZH2 exhibited the strongest interaction with DDX1 (Fig. 2D). EZH2 is aberrantly activated in both osteoarthritis and rheumatoid arthritis29,30, but its potential regulatory role in IVDD remains unclear. We found that in the in vitro degeneration model of NP cells cultured with TBHP, pharmacologically induced oxidative stress led to a marked upregulation of EZH2 expression (Fig. 2E, I, Supplementary Fig. 2F). IHC staining revealed an elevated gradient of EZH2-positive cells in degenerated NP tissue (Fig. 2G, H), suggesting that the degenerative state of NP cells during IVDD is related to the increased expression of EZH2. IF staining showed that DDX1 expressed by degenerative NP cells exposed to TBHP co-localized with EZH2 in the nucleus (Fig. 2F, Supplementary Fig. 2G). Furthermore, pull-down assays using His-tagged DDX1 and GST-EZH2 confirmed a direct physical interaction between the two proteins (Fig. 2L, Supplementary Fig. 2A). We observed strong physical interactions between both endogenous and exogenous DDX1 and EZH2 in NP and HEK-293T cells (Fig. 2J, K, N), indicating a direct association. This interaction suggests methylation regulation, which was confirmed by a reduction in DDX1 methylation in siEZH2-NP cells, as detected using anti-pan-methyl antibodies (Fig. 2O). In contrast, in EZH2-overexpressing NP cells, the lysine methylation levels of DDX1 were significantly increased (Fig. 2P). Considering that DDX1 was mono-methylated at lysine 234 located in the SPRY domain, we further predicted that the EZH2 methylation site was on K234. The DDX1 mutant was created by substituting lysine 234 with arginine. As shown in Fig. 2Q, Co-IP analysis using an anti-lysine methylation antibody confirmed that this substitution markedly diminished DDX1 methylation. Notably, the K234R and K234M mutants did not affect the interaction of EZH2 with DDX1 (Fig. 2M), this suggests that EZH2 serves as a potential DDX1 methyltransferase responsible for methylation of lysine 234 in DDX1.

A Chordal graph and B GO biological process enrichment showing differential abundance of DDX1-interacting lysine methyltransferase in HEK-293T cells transfected with DDX1 WT. C CO-IP of the lysine methylation of exogenous DDX1 in HEK-293T (n = 3). D Interaction analysis between exogenous DDX1 and lysine methyltransferase (n = 3). E Protein level analysis of EZH2, β-actin was used as the loading control (n = 3, P = 0.0055). F IF of EZH2 and DDX1 in NP cells. Scale bar: 10μm (n = 3). G IHC of the EZH2 in NP. Scale bar: 100μm (n = 3). H Association assessment between the EZH2 and Pfirrmann grades (n = 3, P < 0.0001). I mRNA analysis of EZH2 in NP cells by RT-PCR (n = 3, P = 0.0009, P = 0.0021). J, K Co-IP of endogenous EZH2 with anti DDX1 antibody J and endogenous DDX1 with anti EZH2 antibody (K) in NP cells (n = 3). L, M Affinity pulldown using 6×His-tagged DDX1 followed by IB with anti-His and anti-GST antibodies, as well as CO-IP, were performed in HEK-293T cells. The latter was used to assess the binding between exogenously expressed EZH2 and either WT or mutant (KR, KM) forms of DDX1 under overexpression conditions (n = 3). N CO-IP was carried out in HEK-293T cells transiently transfected with the indicated constructs to characterize the interaction profiles between DDX1 and EZH2 (n = 3). O, P To investigate the regulatory effect of EZH2 on the lysine methylation of DDX1 under oxidative stress, CO-IP were conducted in TBHP-treated NP cells. Comparisons were made between EZH2-knockdown and EZH2-overexpressing conditions to determine the functional relevance of EZH2-mediated PTM (n = 3). Q CO-IP of the methylation of WT and K234R DDX1 in HEK-293T transfected with Flag-EZH2 (n = 3). Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Data are represented as mean ± SEM. P value was quantified by Student’s t-test (B, E, I) and two-way ANOVA (H). Panels (C–G, J–Q) show the results of a representative similar result from one of three independent experiments.
EZH2-mediated K234 methylation of DDX1 promotes IVDD progression
To investigate the role of EZH2-dependent DDX1 methylation in IVDD regulation, we re-expressed DDX1 WT or DDX1 K234M mutant in NP/siDDX1 cells. Notably, the K to methionine (M) substitution mimics the methylation pattern of the protein13,14,23. Compared to controls, EZH2 knockdown reversed TBHP-induced anabolic decline and catabolic increase in NP cells in vitro, while re-expression of the DDX1 K234M mutant but not DDX1 WT could significantly exacerbate TBHP-driven NP cells degeneration (Fig. 3A–D). To further understand the impact of EZH2-dependent DDX1 methylation in IVDD, we used LVs carrying shEZH2 to knock down EZH2 in SD rats, followed by re-ectopic expressing DDX1 WT-LV and DDX1 K234M-LV, were injected into the NP area of the tailbone IVD of SD rats every week for 4 weeks (Fig. 3E, I). Further experiments showed that shEZH2-LVs significantly alleviated the loss of coccygeal disc height and subchondral bone destruction, while re-expression of DDX1 K234M-LV but not DDX1-LV could further cause the loss of IVD height (Fig. 3F, G, J). MRI results showed an increase in the T2-weighted signal of the coccyx IVD, indicating that the water content of the IVD was retained after EZH2 knockdown, and re-expression of the DDX1 K234M mutant but not DDX1 WT resulted in a loss of water content of the IVD (Fig. 3H). HE and SO&FG staining results showed that LV-mediated EZH2 deficiency rescued the acquisition of coccygeal IVD degeneration-like phenotypes, alleviating the disordered ECM between AF and NP with a blurry interface and reducing inflammatory cells within the AF (Fig. 3K–M). Consistent with the in vivo results, IVDs treated with shEZH2-LVs showed increased collagen II-positive and decreased MMP3-positive NP cells, indicating attenuation of IVDD progression (Fig. 3N, O). Importantly, reinjection of LVs carrying the DDX1 K234M mutant, but not wild-type DDX1, caused NP tissue atrophy, blurred NP-AF boundaries, disrupted ECM, and accelerated coccygeal IVD degeneration (Fig. 3K). In addition, re-ectopic expression of DDX1 K234M instead of DDX1 inhibited collagen II-positive but enhanced MMP3-positive NP cells (Fig. 3N, O). Therefore, these results confirm that EZH2-dependent DDX1 methylation may lead to NP degeneration and promote IVDD progression both in vivo and in vitro.

A, B The siControl and siEZH2 plasmids were transfected into TBHP-treated NP cells, and the Collagen II (P < 0.0001) and MMP3 (P = 0.0022) was measured (n = 3). C, D Cells derived from NP/siEZH2 cells overexpressed with indicated DDX1-WT or DDX1-K234M were used to detect Collagen II (P = 0.0194) and MMP3 (P = 0.0208) expression by western blot (n = 3). E, I Illustration presenting the experiment design. The LV-scrambled or LV-shEZH2 were used to knock down EZH2 in needle puncture model rats (scrambled group, n = 5 and shEZH2 group, n = 15). Subsequently, LV-DDX1 WT (DDX1 WT group, n = 5) and LV-DDX1 K234M (DDX1 K234R group, n = 5) were overexpressed in the shEZH2 group rats and injected weekly into the NP. The duration was 4 weeks. F–H X-ray F), CT (G), MRI results H) of rat vertebrae (n = 5). J, L-M) DHI (P < 0.0001, P = 0.0019), Pfirrmann grades (P = 0.0017, P = 0.0036) and histological points (P < 0.0001, P = 0.0001) of rat discs (n = 10). K HE and SO&FG of rat discs. Scale bar: 1 mm. N, O IHC of collagen II (P < 0.0001, P < 0.0001) and MMP3 (P < 0.0001, P = 0.0003) in rat discs. Scale bar: 150μm (n = 5). Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Data are represented as mean ± SEM. P value was evaluated by Student’s t-test (B, D) and two-way ANOVA (J, L, M, O). Panels (A, C) show the results of a representative similar result from one of three independent experiments. Panels (F–H, K, N) show the results of a representative similar result from one of five independent experiments.
DDX1 methylation promotes cellular senescence and apoptosis
To elucidate the functional significance of DDX1 methylation, we performed mRNA transcriptome sequencing (bulk RNA-seq) analysis to compare the gene collections of normal and TBHP-cultured NP cells, as well as NP/siDDX1 and HEK-293T/siDDX1 cells re-expressing siRNA-resistant DDX1 K234M or DDX1 K234R mutations31 (Fig. 4A) (Supplementary Data 1). Compared with the control, a total of 3205 down-regulated and 3618 up-regulated genes were identified in NP cells cultured by TBHP (Supplementary Fig. 3A). Compared with the DDX1 K234R, a total of 217 down-regulated and 172 up-regulated genes were determined in DDX1 K234M group HEK-293T cells (Supplementary Fig. 3B, D). Compared with the DDX1 K234R group, a total of 349 down-regulated genes (P < 0.05) and 486 up-regulated genes were identified in NP cells of DDX1 K234M group (P < 0.05) (Supplementary Fig. 3C). Comprehensive analysis showed that 66 up-regulated genes in normal NP cells overlapped in NP and HEK-293T cells overexpressing DDX1 K234R (Supplementary Fig. 3E), 71 up-regulated genes in TBHP-treated cells overlapped in NP and HEK-293T cells overexpressing DDX1 K234M (Supplementary Fig. 3F). Subsequently, GSEA revealed a clear correlation between cellular senescence and apoptosis pathways and the degenerative phenotype of TBHP-treated NP cells (Fig. 4B). Interestingly, the GSEA demonstrated that DDX1 K234M overexpression similarly regulated cellular senescence and apoptotic signaling pathway (Fig. 4C, Supplementary Fig. 4A, C). GO showed that DDX1 K234M mediated genes were enriched in mitotic cell cycle, apoptosis regulation, G2/M transition, cell proliferation, and division (Fig. 4D, Supplementary Fig. 4B, D). Previous studies identified hundreds to thousands of senescence- and apoptosis-related genes in various cell types, including NP cells32. Among them, 29 classical senescence- and apoptosis-related genes were elevated in TBHP-stimulated NP cells compared to controls (Fig. 4E). Notably, these genes (AIFM1, TNFRSF10A, RELA, BAX, BCL2L1) expression levels had similar trend in NP/siDDX1 cells and HEK-293T/siDDX1 cells re-expressing the siRNA-resistant DDX1 K234M compared to the DDX1 K234R (Fig. 4F, Supplementary Fig. 4E) (Supplementary Data 2). These results indicate that high levels of DDX1 K234 methylation may exert a pro-senescence and pro-apoptosis effect in NP cells.

A Schematic workflow showing bulk RNA-seq of normal or TBHP-treated NP cells (n = 3), HEK-293T/siDDX1 cells infected with DDX1 K234M or DDX1 K234R (n = 3), and NP/siDDX1 cells infected with DDX1 K234M or DDX1 K234R (n = 3), which were used to identify DDX1 methylation mediated degeneration-associated phenotypes in NP cells. B GSEA of DEGs in normal and TBHP-treated NP cells. C GSEA of DEGs in HEK-293T/siDDX1 cells infected with DDX1 K234M or DDX1 K234R. D GO biological process enrichment analysis of transcriptome from HEK-293T/siDDX1 cells infected with DDX1 K234M or DDX1 K234R. E Heatmap representing senescence- and apoptosis-related genes determined by transcriptome in (Supplementary Data Fig. 2A). Twenty-nine genes were discovered to be significantly elevated in TBHP-treated cells were highlighted in orange (n = 3). F Heatmap representing senescence- and apoptosis-related genes determined by transcriptome in (Supplementary Data Fig. 2B). Twenty-five genes significantly elevated in K234M cells were highlighted in orange (n = 3). G–H Apoptotic cells were detected by FC in G TBHP treated NP cells (n = 3) and H TBHP treated HEK-293T (n = 3), including early and late period. I SA-β-gal staining of NP cells. Scale bar: 100μm (n = 3). J Quantitative analysis of flow cytometry showing the apoptotic cells percentage of NP (P = 0.0001, P = 0.0010) and HEK-293T cells (P = 0.0006, P = 0.0002) with different indicated treatments (n = 3). Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Data are represented as mean ± SEM. P value was evaluated by Student’s t-test (B–D) and two-way ANOVA (J). Panels (E, F, J) show the results of a representative similar result from one of three independent experiments.
Consistent with this, we next measured apoptosis in NP cells and HEK-293T cell mediated by DDX1 K234 methylation using Annexin V-FITC/Propidium Iodide-PE staining and flow cytometry (FC). As shown, siDDX1 significantly rescued NP cell apoptosis compared with the control in vitro. However, expression of siRNA-resistant normal DDX1 again, rather than K234R, markedly intensified TBHP-induced apoptosis in NP cells (Fig. 4G, J, Supplementary Fig. 4G). Similar findings were also appeared in HEK-293T cells (Fig. 4H, J). Furthermore, DDX1 knockdown suppressed senescence and apoptosis in TBHP-stimulated NP cells, while expression of DDX1 again, but not the K234R, elevated both processes, as shown by TUNEL and P53 staining (Supplementary Fig. 3H, K). Western blot analysis showed that DDX1 inactivation decreased CASP3 and Bax levels while increasing Bcl-2 expression. However, overexpression of DDX1 WT but not DDX1 K234R had opposite effects (Supplementary Fig. 3G, I). Additionally, DDX1 knockdown via siRNA reduced staining of SA-β-gal in TBHP-stimulated NP cells, whereas overexpression of DDX1 WT—but not the K234R mutant—increased SA-β-gal positivity, indicating that DDX1 methylation promotes lysosomal senescence activity (Fig. 4I, Supplementary Fig. 4F). Further analysis effectively confirmed increased senescence-related proteins in NP cells transfected with DDX1 WT compared to the mutant (Supplementary Fig. 3J, L). Collectively, these findings suggest that DDX1 lysine methylation enhances senescence and apoptosis in NP cells.
Methylation results in a transcriptome reduction of DDX1 binding sites on RNA
To construct a genome-wide maps of DDX1-RNA interactions mediated by lysine methylation, RNA immunoprecipitation with next generation sequencing (RIP-seq) were performed on normal and methylated DDX1 in normal and TBHP-treated NP cells33. The Control-RIP group yielded a total of 6,724,986 reads, while the TBHP-RIP group obtained 5,783,158 reads. Notably, 93.75% of the reads from the Ctrl-RIP and 95.86% from the TBHP-RIP were mapped to an annotated human genome. Due to RNA degradation, it may not be possible to obtain a uniform distribution of reads on transcripts, which shows that proteins are relatively enriched in their direct binding sites compared with non-binding sites, which also makes RIP-seq possible with peak calling. Peak distribution analysis showed that DDX1 was enriched in exons, 3’UTRs and 5’UTRs (Fig. 5A, B), with the highest enrichment located near the 5’UTRs end of the exon (Fig. 5C, G). In total, RIP-seq identified 22,477 and 10,517 DDX1 enriched peaks in control and degenerative NP cells transcripts (excluding errors caused by reduced spontaneous expression in transcripts) (Fig. 5D). Subsequently, gene expression was quantified using RPKM. Cluster analysis revealed that methylated DDX1 bound approximately 2000 fewer annotated human genes compared to unmethylated DDX1 (Fig. 5E). To account for nonspecific sequence binding34, analysis of protein-RNA interaction sites showed that, relative to the TBHP-RIP, the Ctrl-RIP exhibited stronger enrichment at shared binding regions (Fig. 5F, G). Correlation exploration of RIP- and RNA-seq data further indicated that DDX1 preferentially binds near exon-intron junctions, particularly around the 3’ and 5’ splice sites, with binding intensity modulated by lysine methylation (Fig. 5H).

A, B PEAK distribution of gene functional regions of DDX1-binding transcripts in (A) normal and (B) TBHP-treated NP cells. C Distribution of DDX1-binding regions on genomic elements. D PEAK number analysis of RIP-Seq results of normal and TBHP-treated NP cells. E Heatmaps and line plots show the location of normal or methylated DDX1-enriched functional regions and the number of annotated human genes as indicated by cluster analysis. F Scatter plot of Control-RIP and TBHP-RIP read counts versus DDX1 binding regions. G Distribution of Control-RIP and TBHP-RIP reads across genes. H The intensity of RIP reads distributed around alternative exons. High-intensity exons (Control-RIP, blue) are included, and low-intensity exons (TBHP-RIP, deep blue) are excluded after methylation of DDX1. Randomly selected exons are used as controls (yellow). I, J, M GO analysis of DDX1-binding genes in Control-RIP and TBHP-RIP. K De novo motif analysis results of DDX1 RIP fragments, showing the top four enriched DDX1 binding sequence features. L Visualization concerned with localization of motifs 1 to 4 in the DDX1 binding region. N Comparison of the distribution differences between RIP reads and input in different genomic functional regions. O Analysis of the enrichment of DDX1 binding motifs in exonic regions and near intron-exon boundaries near the 5’splice site. To exclude the influence of different exon lengths, all exon intervals were normalized to 300 bp. P Statistical ranking analysis of the top four motifs bound by DDX1 based on the P calculated by HOMER. Statistical legend: *P < 0.05, **P < 0.01, ***P < 0.001. All data are expressed as mean ± SEM. Significant differences were evaluated by Student’s t test (I, J, M, P).
To elucidate the functional significance of DDX1-enriched peaks, GO analysis revealed that methylation mediated DDX1 differential target genes located in the perinuclear region of cytoplasm and nucleolus were enriched in DNA damage and repair, cell division and cell cycle by binding multiple substrates. (Fig. 5I, J, M). To further elucidate DDX1-RNA interactions, the HOMER algorithm was used to identify DDX1-binding RNA motifs, revealing AGUGGAA 7-mer35 as the most enriched element (Fig. 5K, L, P). Notably, over 81.5% of these motifs were located within exonic and intronic regions (Fig. 5N). Among the top four highest-scoring motifs, motifs 1 and 4 were enriched near 5’ splice site intron-exon junctions, while motifs 2 and 3 were positioned within exons (Fig. 5O). Collectively, these findings indicate that lysine methylation diminishes DDX1’s direct association with pre-mRNA substrates.
Methylated DDX1 reduces splicing factor recruitment and promotes MATR3 exon 14 skipping
To explore DDX1-regulated AS, transcriptome from normal and TBHP-treated NP cells, along with HEK-293T/siDDX1 cells and NP/siDDX1 cells that re-expressing siRNA-resistant DDX1 K234M and K234R mutants, were analyzed. AS variation events include exon omission (SE), intron retention (RI), selective switching of 5’ and 3’ sites (A5SS and A3SS), and exon mutually exclusive (MEX) expression patterns36 (Fig. 6A). Skipped exons predominated in TBHP-treated NP cells (72%), decreasing to 68% in HEK-293T cells and 67% in NP cells overexpressing DDX1 K234M (Fig. 6B, Supplementary Fig. 5A). Coincidentally, the second-ranked AS events in both types of gene profiles are mutually exclusive exons with the same proportion (17%). Overall, similar proportions of various AS events were observed in NP and HEK-293T cells. To identify functional targets linked to IVDD, we integrated DDX1-bound genes with SE-associated genes applied transcriptome and RIP data. Comprehensive analysis showed that only six exon-skipping genes bound to DDX1 overlapped in TBHP-treated NP cells, as well as NP and HEK-293T cells overexpressing DDX1 K234M (Fig. 6C) (Supplementary Data 3). Given that RNA-seq analysis combined with functional assays revealed DDX1 methylation on lysine, activating senescence and apoptosis signaling pathways leading to NP cell degeneration. Meanwhile, GO analysis based on RIP-seq revealed DDX1 target genes located in the perinuclear region were enriched in BP, such as DNA damage and repair, cell division, and cycle (Fig. 5I, J). Therefore, of the DDX1-bound and alternatively spliced transcripts, MATR3 (Matrin3), a nuclear protein-coding gene, caught our attention. The Matrin3 protein plays various crucial roles in the nucleus, including maintaining nuclear matrix structure, regulating gene transcription, participating in mRNA metabolism, DNA repair, and regulating apoptosis. Abnormalities in these functions may exacerbate the occurrence and development of various diseases37,38. We analyzed RNA-seq and found that both TBHP treatment and overexpression of DDX1 K234M (NP and HEK-293T cells) promoted skipping of exon 14, producing a shorter MATR3 isoform (MATR3-S). Notably, RIP-seq data showed DDX1 binding near MATR3 exon 14, indicating MATR3 as a direct DDX1 target (Fig. 6D, E, Supplementary Fig. 5C, D). Sequence analysis revealed that exon 14 skipping causes a frameshift but does not introduce a premature stop codon, suggesting the resulting isoform affects function structurally rather than through expression changes (Fig. 6F). Using isoform-specific primers, we measured the MATR3-L/MATR3-S ratio and found that TBHP treatment significantly reduced this ratio in NP cells, with DDX1 K234M overexpression producing a similar effect (Fig. 6G, Supplementary Fig. 5B). Semi-quantitative RT-PCR further confirmed that TBHP treatment increased exon 14 skipping, generating more MATR3-S and fewer full-length isoforms, an effect mirrored by DDX1 K234M overexpression (Fig. 6H, Supplementary Fig. 5E). Together, these results strongly support that DDX1 promotes MATR3 alternative splicing via direct pre-mRNA binding.

A Schematic diagram showing different types of AS events. B Pie chart showing the composition ratio of each type of AS event in transcriptome of NP cells and HEK-293T cells. C Venn diagram showing the overlap of genes with SE events in the following four data sets: NP cells between Control and TBHP treatment groups (2542 genes), NP cells under K234R and K234M expression conditions (1266 genes), HEK-293T cells under K234R and K234M conditions (1077 genes), and 366 target genes directly bound by DDX1 identified in RIP-seq. D Combining RNA-seq and RIP-seq data, IGV was used to visualize the alternative splicing pattern of the MATR3 gene and the distribution of its DDX1 binding sites. E Sashimi plot concerned with SE of MATR3 exon 14 in TBHP-treated NP cells (blue), HEK-293T cells re-expressing K234M in the siDDX1 background (orange), and respective control cells (red or green). F Schematic diagram of the two transcript structures of MATR3: MATR3-L is the full-length form containing exon 14, and MATR3-S is the splicing variant lacking this exon. G Analysis of the changes in the expression ratio of MATR3-L/MATR3-S in NP cells (P = 0.0030) and HEK-293T cells (P = 0.0092), the results are based on three independent replicates (n = 3). H Semi-quantitative RT-PCR was used to verify the AS events of MATR3 in NP and HEK-293T cells under different treatments. I IP-MS was used to analyze the GO enrichment of associated proteins with DDX1 in HEK-293T cells. J Biological process enrichment in proteins from DDX1 proximal proteomes. K Correlation between the DDX1 enrichment ratios obtained via LC-MS from different HEK-293T cells transfected with DDX1 K234R or DDX1 K234M mutant. L, M Adjusted p values of (L) GO and (M) KEGG terms enriched in DDX1 proximal proteomes in HEK-293T cells with indicated treatment. Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Data are represented as mean ± SEM. A significant P value was demonstrated by Student’s t-test (G, I, L, M). Panels (H) show the results of a representative similar result from one of three independent experiments.
To uncover how DDX1 regulates AS, we performed immunoprecipitation using a DDX1 antibody combined with MS in siDDX1/HEK-293T cells overexpressing DDX1 K234R and K234M mutants. GO enrichment analysis revealed enrichment of proteins concerned with spliceosome-mediated mRNA splicing and RNA splicing regulation among DDX1 interactors (Fig. 6I). Compared to methylated DDX1, various RNA splicing factors were more abundant in normal DDX1, including HNRNPA2B1, RBM26 and SF3B2 (Fig. 6J, Supplementary Fig. 5F–H). Known DDX1 interactors such as HNRNPK2 and DHX36 were enriched under both conditions, validating the dataset (Fig. 6J). RNA helicase obtains energy by hydrolyzing ATP to unwind RNA double strands or secondary structures. By detecting the consumption rate of ATP in the reaction system, the helicase activity or the regulatory effect of RNA-binding proteins on the unwinding process can be indirectly evaluated. The K234R and K234M mutants did not affect the helicase activity of DDX1 towards nonspecifically bound RNA probes, and then DDX1 K234M weakened the interaction with MATR3 compared with DDX1 WT and K234R (Supplementary Fig. 5I). Intriguingly, based on GO and KEGG analysis, proteins proximal to DDX1 K234R exhibited higher enrichment in mRNA splicing (Fig. 6K–M), whereas rRNA processing was roughly the same between DDX1 K234R and K234M, suggesting that the interaction of DDX1 with splicing regulators is self-standing of its helicase activity. Taken together, these data indicate that methylation impedes DDX1 from recruiting proteins involved in mRNA processing to regulate AS.
MATR3-S triggers excessive chromatin accessibility involved in senescent and apoptotic regions
To assess the significance of MATR3 exon 14 skipping, we performed ATAC-seq to examine genome chromatin accessibility in NP cells overexpressing either MATR3-L (including exon 14) or MATR3-S (lacking exon 14). The ATAC-seq data sets of both types of NP cells show a similar length of about 200 base pairs. The existence of double peaks means different chromatin structures or specific DNA sequence regions (Fig. 7A). Consistent with transcriptional studies, the number of chromatin peak changes in NP cells overexpressing MATR3-S exceeded those in NP cells overexpressing MATR3-L, and the repeated elements have less ATAC peak signal in MATR3-L human NP cells suggesting a more compact chromatin structure. (Fig. 7B, Supplementary Fig. 6A). Principal component analysis (PCA) and correlation analysis based on accessible chromatin peaks separated these two types of NP cells (Supplementary Fig. 6B, C). As expected, chromatin peaks predominantly localized to introns, exons, promoters, and distal regions, which typically contain non-coding regulatory elements like enhancers (Fig. 7C). Moreover, in NP cells, the chromatin peaks altered by MATR3 isoform variation were almost entirely distal (Supplementary Fig. 6D), suggesting that non-coding regulatory elements are especially responsive to changes in MATR3 isoforms. Interestingly, MATR3 isoforms have similar effects on the dynamic chromatin accessibility in NP cells. Most dynamic chromatin peaks in NP cells are closed with the increase of MATR3-L, whereas they are opened with the increase of MATR3-S (Fig. 7D–F). These data suggest that during NP cells degeneration, MATR3-L leads to a more repressive chromatin state, while MATR3-S results in a more relaxed chromatin state.

A Histogram of all paired-end reads in the ATAC dataset of NP cells overexpressing MATR3-L or MATR3-S. B Bar graph showing differential ATAC-seq peak numbers in MATR3-L human NP cells versus MATR3-S human NP cells. C Visualization of the mean distribution of ChiPseeker annotations in MATR3-L human NP cells. D Heat map showing differential ATAC signal in MATR3-L human NP cells versus MATR3-S human NP cells. E Chromosome visualization of ATAC differentially accessible peaks for different MATR3 isoform changes. Each vertical line represents the corresponding dynamic ATAC-seq peak of different types of NP cells. F Volcano map shows the differentially accessible ATAC-seq peaks in MATR3-L human NP cells versus MATR3-S human NP cells. G Schematic diagram of the interaction network of Wnt signaling pathways based on GO annotations. H Heat map showing differential ATAC signal of CTNNB1 in MATR3-L human NP cells versus MATR3-S human NP cells. I GO analysis of up-regulated ATAC peak in MATR3-S human NP cells. J GO analysis of down-regulated ATAC peak in MATR3-S human NP cells. K Heatmaps showing the chromatin accessibility changes of differential peaks associated with the “Wnt signaling pathway” (left) and “MAPK signaling pathway” (right) in different MATR3 isoforms. Some representative genes associated with these differential peaks are annotated in the figure. L Violin plots quantify the ATAC-seq signal difference between MATR3-L and MATR3-S isoforms at the CTNNB1 locus in human nucleus pulposus cells (P = 0.032555). M, N Visualization of differential chromatin accessibility of the CTNNB1 gene region under different MATR3 isoform conditions from the chromosome level (M) and IGV browser view (N), respectively (FDR < 0.05). Data information: *P < 0.05, **P < 0.01, ***P < 0.001. All data are expressed as mean ± SEM, and statistical analysis was performed using Student’s t test (F, I, J, L).
Network analysis highlights the Wnt as a key regulator of various biological processes, including the cell cycle, apoptosis, and differentiation, with intricate crosstalk involving the MAPK pathway39,40 (Fig. 7G). Upon activation, β-catenin trans-locates and interacts with TFs to promote expression of cell cycle genes like Cyclin D1, facilitating progression into the S phase41. While generally associated with cell survival and proliferation, excessive activation of Wnt/β-catenin can, under certain conditions, trigger apoptosis42. Indeed, GO and KEGG analysis reveals that chromatin accessibility in regions involved in promoting cellular senescence and apoptosis (“stress-activated MAPK cascade”, “Ras protein signal transduction”, “Wnt signal pathway”, and “cell-cell signaling by Wnt”) is generally increased in degenerated NP cells overexpressing MATR3-S (Fig. 7I, Supplementary Fig. 6E). Conversely, chromatin accessibility in regions concerned with inhibiting cell cycle and growth development (“G/M transition of mitotic cell cycle”, “cell morphogenesis”, and “skeletal system development”) is generally decreased in normal NP cells overexpressing MATR3-L (Fig. 7J, Supplementary Fig. 6F). From NP cells overexpressing MATR3-S, it is shown that chromatin accessibility in regions involved in the Wnt signaling pathway (e.g. Wnt family members (WNT2, WNT3) (Fig. 7K), Catenin (CTNNB1) (Fig. 7H, L–N), glycogen synthase kinase (GSK3B) (Supplementary Fig. 6J)), MAPK signaling pathway (e.g. MAPK family members (MAPK1, MAPK3) (Fig. 7K)), and genes positively regulating apoptosis (e.g. RIPK1, CASPASE3) (Supplementary Fig. 6K, M) is increased, whereas chromatin accessibility in regions concerned with cell cycle (e.g. cyclin-dependent kinases, CDK1) (Supplementary Fig. 6L) is decreased. With overexpression of MATR3-L, NP cells exhibit opposite changes in chromatin accessibility in the aforementioned gene regulatory regions (Supplementary Fig. 6G–I). Thus, MATR3 isoforms exert contrasting effects on the chromatin landscape of NP cells, inducing opposite changes in accessibility at regulatory regions linked to senescence and apoptosis.
Cationic LNP-based MATR3-L mRNA cargo mitigates IVDD progression
Considering the large nucleotide sequence of MATR3, limited feasible packaging methods, and the excellent performance of LNPs in the lateral transfer of cargo between adjacent cells43, we construct cationic lipid LNPs overexpressing MATR3-L or MATR3-S based on microfluidic technology (Supplementary Fig. 7A). Microfluidic technology effectively controls size and generates various types of LNPs, characterized by its ability to accurately distribute nanoscale volumes, predominantly diffuse axial mixing, and low-volume continuous operation44. In this process, DOTAP can directly interact with cell surfaces through electrostatic interactions, cholesterol contributes to lipid stability, DOPE enhances nucleic acid delivery efficiency, and polyethylene glycol (PEG) limits plasma protein binding and nonspecific uptake, thereby increasing in vivo circulation half-life45. To construct functional mRNA transcripts, DNA vectors containing the MATR3 gene were built. The transcribed MATR3-L has a length of approximately 2544 nucleotides, while MATR3-S has a length of approximately 2385 nucleotides, consistent with the predicted lengths. Transmission electron microscopy (TEM) images revealed well-dispersed MATR3-L-LNPs, MATR3-S-LNPs, and vector-LNPs, all displaying irregular spherical or cup-shaped morphologies consistent with their size distribution (Fig. 8A). To assess the mRNA transcripts’ ability to overcome challenges such as nuclease sensitivity, antigen presentation difficulties, and effective carrier-mediated delivery in vivo, agarose gel electrophoresis was used to evaluate mRNA binding with cationic LNPs. Results confirmed complete encapsulation of MATR3-L and MATR3-S when the cationic liposome-to-mRNA ratio (w/w) exceeded 8:1 (Fig. 8B). Nanoparticle tracking analysis (NTA) indicated that the average particle size of vector-LNPs was 80.94 ± 12.28 nm, with a zeta potential of 59.56 ± 5.41 mV. The average particle size of MATR3-L-LNPs was 193.73 ± 10.30 nm, with a zeta potential of 27.56 ± 7.81 mV. For MATR3-S-LNPs, the average particle size was 168.19 ± 11.61 nm, with a zeta potential of 35.78 ± 6.11 mV (Fig. 8C, D).

A TEM of LNPs loading with MATR3-L, MATR3-S or vector, Scale bar: 100 nm. B Gel retardation assay to determine the optimal ratio of LNP to mRNA. C, D NTA shows the size distribution (C) and zeta potential D of cationic LNPs/mRNA complexes. E, G DHI of rat coccygeal discs (n = 10, P = 0.0001, P = 0.0005, P = 0.0171, P = 0.0040). F, H, I X-ray F, CT H, MRI results (I) of rat discs (n = 5). J HE and SO&FG of rat discs. Scale bar: 1 mm. K, L Pfirrmann degenerative grades (P = 0.0209, P = 0.0429, P = 0.0377, P = 0.0215) and histological score (P < 0.0001, P = 0.0002, P = 0.0101, P = 0.0011) of rat coccygeal discs (n = 10). M, N IF (Scale bar: 500μm) staining of collagen II (P < 0.0001, P < 0.0001, P = 0.0001, P < 0.0001) and P21 (P < 0.0001, P < 0.0001, P = 0.0010, P = 0.0004) in rat coccygeal discs (n = 5). Data information: *P < 0.05, **P < 0.01, ***P < 0.001. Data are represented as mean ± SEM. P value was demonstrated by two-way ANOVA (G, K, L, N). Panels (A, B) show the results of a representative similar result from one of three independent experiments. Panels (F, H-J, M) show the results of a representative similar result from one of five independent experiments.
To assess the in vivo effects of MATR3-LNP, engineered nanoparticles were injected into the discs (Fig. 8E). Radiographic measurements revealed that compared to the vector-LNP, MATR3-S-LNP led to a decrease in disc height (Fig. 8F, G), subchondral bone destruction (Fig. 8H), and deficient of water content in the discs (Fig. 8I, K), while MATR3-L-LNP significantly reduced the aforementioned radiological pathological changes. Histological staining demonstrated that LNP-mediated overexpression of MATR3-S promoted the acquisition of degenerative-like phenotypes in the coccygeal IVDs, whereas overexpression of MATR3-L alleviated the disordered ECM, blurry interface between the AF and NP, and reduced inflammatory cells within the AF (Fig. 8J, L). As expected, MATR3-S-LNP significantly reduced NP cells expressing synthetic metabolism markers while markedly increasing those with aging-related markers. Notably, in the needle puncture rat model, MATR3-L-LNP treatment decreased senescence markers and boosted collagen II expression in NP cells (Fig. 8M, N, Supplementary Fig. 7B, C), demonstrating that lipid-mediated MATR3-L overexpression effectively alleviates NP cell aging and IVDD progression in coccygeal discs. Collectively, these findings reveal molecular mechanisms and offer potential therapeutic strategies for mitigating NP cell aging and improving IVDD outcomes via MATR3-L liposomal delivery.
Discussion
While chromatin reprogramming-driven cellular senescence is linked to the early onset of age-related diseases25, the mechanisms triggering abnormal transcriptome AS and heightened chromatin accessibility during aging and apoptosis-related degeneration remain largely unclear. This study elucidates the mechanism linking elevated EZH2-mediated DDX1 methylation to regulate AS of MATR3 and NP cells chromatin reprogramming during the progression of IVDD (Fig. 9). MS screening of proteome methylation modifications in volunteer samples and histological staining, coupled with extensive RNA-seq gene expression profiling of aging and apoptosis-related markers, and methylation-deficient variant treatment rescued rat IVDD progression, indicating that EZH2-mediated DDX1 methylation promotes NP cells senescence and IVDD progression. Functional gain and loss experiment further demonstrated that EZH2 deficiency rescued NP cells from reduced anabolism and enhanced catabolism, and led to reduced DDX1 methylation, which is the trigger for the acquisition of DDX1-MATR3 axis-dependent degenerative phenotypes in IVDs. Mechanistically, analysis of the DDX1 interactome combined with endogenous and exogenous immune interactions revealed that loss of the EZH2-DDX1 interaction resulted in reduced DDX1 lysine 234 methylation modification, which promoted DDX1 binding in non-coding regulatory regions of MATR3. Subsequently, DDX1 recruits splicing factors and enriches them at the splice site of MATR3, thereby driving exon 14 inclusion to form MATR3-L. Ultimately, MATR3-L genetically maintains whole-genome chromatin accessibility, preventing excessive activation of senescence and apoptosis signaling pathway in NP cells. Notably, mRNA-engineered LNPs carrying MATR3-L effectively alleviated DDX1 K234M-induced catabolism and senescence in NP cells and greatly alleviated IVDD.

In degenerated NP cells, upregulated EZH2 enhances the methylation of lysine 234 in DDX1. Methylated DDX1 interacts less with MATR3, leading to reduced enrichment of splicing factors at splicing sites and consequently promoting the generation of MATR3-S through exon 14 skipping. MATR3-S promotes excessive opening of chromatin accessibility, thereby aberrantly activating Wnt signaling pathways to promote NP cells aging and apoptosis. Additionally, a strategy based on cationic LNPs for delivering mRNA cargo overexpressing MATR3-L effectively alleviates the progression of IVDD.
IVDD is a common major cause of LBP worldwide, and recent years have seen numerous studies aimed at elucidating its pathogenesis. Although previous publications have elucidated the crucial role of epigenetic regulation in IVDD from various perspectives46, uncovering the epigenomic regulatory mechanisms and depicting the epigenomic landscape during IVDD remains challenging for the development of efficacious treatment methods for IVDD and associated back pain. Various etiological and risk factors contribute to IVDD progression, but age remains the sole well-established risk factor linked to its onset and development. Recent research highlights abnormal responses of resident cell populations to aging and cell-driven tissue degradation within the IVD, including age-associated phenotypic alterations and extracellular matrix remodeling47. Various stresses, including oxidative stress, abnormal mechanical loading, and inflammatory factors, disrupt the biomechanical homeostasis and repair responses mediated by the ECM microenvironment, leading to increased proliferation cycles and acquisition of aging phenotypes in NP cells48. Chromatin accessibility is essential for controlling cellular fate and aging49. However, epigenomic research on NP cells has so far focused mainly on gene variation at the pre- or post-transcriptional aspects driven by abnormal DNA methylation or histone modifications. Importantly, the transcriptional level structural dependency of PTM-mediated epigenomic changes in in vivo NP cells remains unknown, and such alterations in the chromatin landscape may exert more enduring effects on the microenvironment. Based on protein PTMs triggering intrinsic stress or exogenous interference-induced aging, as well as abnormal methylation modifications accumulated in terminally differentiated cells during organismal aging, our study further elucidates that RBP-dependent post-transcriptional regulatory defects during the progression of IVDD trigger nuclear transcriptome aberrant splicing and induce NP cells aging and apoptosis, enhancing our understanding of the driving factors of epigenetic changes during IVDD.
DDX1, a member of the DEAD-box family defined by the preserved D-E-A-D (aspartate-glutamate-alanine-aspartate) sequence, functions as an RNA-binding protein concerned with key RNA metabolic such as pre-mRNA splicing, mRNA transport, RNA editing and decay, and ribosome biogenesis50. It plays a vital role in maintaining genome integrity and regulating genetic dynamics across various cellular stages through multiple independent pathways51. DDX1 has been detected in cytoplasmic RNA granules—complexes of RNA and proteins essential for localized translation—within neuronal dendrites and axons, where its binding to RNA increases under oxidative stress52,53. Additionally, in highly proliferative cells, DDX1 acts as a critical DNA damage response factor, recruited to DNA double-strand break sites by the phosphorylated MRN complex, facilitating repair by clearing Ataxia Telangiectasia Mutated proteins at these loci. Constitutive DDX1 deficiency in adult mice results in chromatin condensation, loss of nuclear membrane integrity, disrupts tissue homeostasis, drives the onset of progeria phenotypes, and is accompanied by progenitor cell exhaustion, leading to organ dysfunction54. Considering that maintaining quiescent or ultimately stable differentiation states in the face of internal stress and external perturbations is essential for tissue renewal and homeostasis, DDX1 is indispensable in the developmental process, participating in regulating the cell cycle phases of NP cells and the regenerative capacity of IVD tissue.
Currently, new research increasingly emphasizes the mutually independent evolutionary potential of AS and transcription, both equally important for regulating gene function55. RBPs participate in the regulation of AS through multiple mechanisms and make an essential contribution in the diversity and modulation of gene expression. Under the regulation of PTMs, their functional abnormalities or mutations may lead to abnormal AS, which is closely associated to the evolution of various pathologies. The amino side chain (-NH₂) of DDX1 K234 is positively charged and can interact with adjacent negatively charged residues (such as E237 glutamic acid and N235 aspartic acid) through hydrogen bonds. When K234 is methylated, the chemical properties of its amino side chain change, and the positive charge is replaced by a methyl group (-CH₃), thereby weakening the charge interaction with other molecules or residues. It is worth noting that N235 after K234 is located in the center of the α helix, and the two ends form a β-strand structure56, thereby maintaining the stability of the local structure. Considering that the methylation occurs within the α helix, this modification may change the polarity and charge of the residue, consequently impacting its stability or causing helical bending. In addition, when methylation occurs in the β-strand or its adjacent region, the methylated lysine may cause conformational changes in the β-turn by changing its chemical properties (such as reducing hydrogen bond formation), thereby may affect the RNA binding region and the protein interaction platform. Therefore, a comprehensive understanding of the mechanism of action of DDX1 in AS and how it is regulated is of great significance for revealing gene regulatory networks, disease mechanisms, and developing related treatment strategies.
Chromatin bridging to the perinuclear region is cell-type specific during development and differentiation, and dynamically regulates gene accessibility, playing a key role in gene expression control57. Among nuclear core proteins, Matrin-3 (MATR3) interacts with various structural and regulatory factors to govern RNA processing, and mutations in MATR3 cause rare genetic disorders38. In pituitary cells, MATR3 has been implicated in protein folding regulation, while studies in neural stem cells suggest its expression maintains an undifferentiated state, though these findings remain largely morphological. The contribution of MATR3 to chromatin during growth remains largely unexplored58. Recently, the presence of MATR3 was detected in a protein associated with sex chromosomes, highlighting its chromatin-level regulatory ability. Over a dozen MATR3 point mutations are associated with hereditary distal myopathy59. Furthermore, mis-localization and altered expression of MATR3 were observed in motor neurons of ALS patients, suggesting a link between MATR3 dysregulation and neuromuscular disease60. It is necessary to further explore how MATR3, a widespread regulator of genomic stability, affects the development and progression of IVDD through mediating cellular senescence and apoptosis pathways61,62. This work explores MATR3’s critical function in chromatin structure and epigenetic regulation, elucidating its molecular mechanism as a key factor coupling epigenetic reprogramming to drive nucleus pulposus cell senescence and apoptosis via alternative splicing during IVDD.
In summary, we found that during the progression of IVDD, EZH2-mediated DDX1 methylation improving chromatin accessibility via the MATR3 splicing-dependent pathway, promoting synthetic and catabolism imbalance, and aging of NP cells. Mechanistically, enhanced interaction between EZH2 and DDX1 promotes methylation at the K234. The increased methylation of DDX1 reduces its binding to the non-coding regulatory region of MATR3 and recruitment of splicing factors, leading to the skipping of exon 14 of MATR3. Ultimately, MATR3-S leads to an excessive increase in chromatin accessibility, culminating in aberrant NP cell synthesis, dysregulation of catabolic processes, and the manifestation of senescence-associated phenotypes. Importantly, the MATR3-L overexpression mRNA cargo system delivered by LNPs effectively alleviates DDX1 K234M-induced NP cells degeneration and significantly mitigates the progression of IVDD.
Methods
Ethics approval
All procedures involving patient medical record review, NP collection and use, animal acquisition, surgical interventions, and tissue processing were executed according to the prescribed protocols supported by the Huazhong University of Science and Technology—Tongji Medical College’s Ethics Committee (Approval Nos. S341 and S2394).
Human NP samples
NP tissues were obtained without compromising health from 24 volunteers undergoing spinal surgery for thoracolumbar fractures, scoliosis, or IVDD. Degeneration grades were assessed using MRI data and clinical records based on the Pfirrmann classification18. All enrolled individuals provided written informed consent for the use of their NP tissues and the release of identifiable information. The collected samples were utilized for cell culture, LC-MS, and histological analyses. Comprehensive personal details were provided in Supplementary Table 1.
Cell cultivation and treatments
According to previously reported standard operating procedures, grade I NP (case 1–5, 7) was obtained from patients undergoing spinal surgery, including individuals with lumbar fractures or idiopathic scoliosis. The obtained tissue samples were cut into small pieces of approximately 1 mm³ under sterile conditions and rinsed twice with pre-cooled PBS to remove impurities. Subsequently, the blocks were placed in a digestion solution comprising 0.4% collagenase and digested for 4 h to fully release cells. The digested cell suspension was spun at 800 rpm for 5 min to recover the cells and washed twice with PBS to remove residual enzyme solution. The cells were seeded in a humidified incubator. When the cells reached an appropriate density, they were passaged. Subsequent experiments used second-generation NP cells. To simulate the oxidative stress environment in which cells degenerate, 100 μM tert-Butyl hydroperoxide (TBHP) was used to treat the second-generation NP cells and HEK-293T cells for 24 h before the start of relevant experiments to induce degenerative changes, thereby providing a reliable in vitro model for subsequent mechanism studies and intervention experiments.
Western blotting
Cells were lysed for 30 min with RIPA lysis buffer containing 1% PMSF to extract total protein. The sample was spun at 12,000 rpm for 15 min, after which the supernatant was carefully harvested for further examination. Protein levels were measured using the BCA assay kit (Beyotime, China). According to the experimental requirements, 20–50 μg of protein from each sample was separated by 6–12% SDS-PAGE gel, and the separated proteins were electrotransferred to PVDF membrane (Millipore, USA). After transfer, the membrane was blocked with 5% skim milk powder solution at room temperature for 1 h to block nonspecific binding. Subsequently, the membrane was incubated with the corresponding primary antibody at 4 °C overnight. On the following day, the membrane underwent three washes with TBS buffer plus 0.1% TBS-T, each lasting 10 min, followed by incubation with HRP-conjugated secondary antibody at room temperature for one hour. After washing, enhanced chemiluminescence substrate (Thermo Fisher, USA) adopted for color development and the signals were captured by ChemiDoc MP Imaging System (Bio-Rad, USA). Detailed information of all antibodies used is given in Supplementary Table 2.
Co-IP
Cells were resuspended in NP-40 containing 1% protease inhibitor cocktail and 1% PMSF and lysed on ice for 30 mins to extract total protein. The sample was subsequently spun at 12,000 rpm for 15 min, and the resulting supernatant was carefully harvested. The protein abundance was quantified using the BCA method (Beyotime, China) to ensure that the concentration of each sample was consistent, to ensure the reliability of the co-immunoprecipitation (Co-IP) experiment. A total of 500–1000 µg (final concentration 1–2 µg/µL) of protein samples for Co-IP were taken and incubated with specific primary antibodies pre-coupled to Protein A/G magnetic beads at 4 °C overnight, and continuously mixed under rotation to promote antigen-antibody binding. After incubation, the immune complexes attached to magnetic beads were rinsed multiple times to remove non-specific binding components and finally eluted with 1 × SDS loading buffer. The eluted samples were separated by SDS-PAGE and further subjected to immunoblotting (IB) analysis to detect the target protein and its interacting proteins.
LC-MS/MS
NP tissues (non-degenerative and degenerative) were obtained from a patient (n = 1, case 5 and 20) undergoing spinal surgery for thoracolumbar fractures, scoliosis, or IVDD. Samples were rinsed with PBS and homogenized on ice in NP-40 lysis buffer. After centrifugal separation at 12,000 rpm for 25 min at 4 °C, the supernatant was collected. Similarly, HEK293T cells transfected with EV, WT, or its K234R/K234M mutants were lysed 48 h post-transfection, and protein complexes were enriched by immunoprecipitation using anti-DDX1 or control IgG and Protein A/G agarose beads. All samples were resolved by SDS-PAGE and visualized with Coomassie Brilliant Blue. Protein bands of interest were excised and subjected to in-gel digestion. After decolorization, proteins were reduced with 15 mM DTT, alkylated with 60 mM iodoacetamide, and digested 12 h at 37 °C with sequencing-grade trypsin (Promega, USA). The resulting peptides were desalted using C18 columns or StageTips, vacuum-dried, and reconstituted in 0.2% formic acid. Peptides were analyzed using LC-MS/MS on either a Q Exactive Orbitrap coupled to an EASY-nLC 1200 setup. Chromatographic separation was performed on C18 columns (75 µm × 25 cm, particle size 1.9–2 µm), with linear elution gradients 120 min at flow rates of 200–300nL/min; for some runs, a pre-concentration step using a C18 trapping column (100 µm × 2 cm, 5 µm) was included, and column temperature was maintained at 50 °C. Mass spectrometric acquisition was performed in DDA mode. The Q Exactive system used a full scan resolution of 70,000 (m/z 200), while the Fusion Lumos used 120,000. The top 20 precursor ions (charge ≥2) were selected for MS/MS using HCD, with resolutions of 17,500–30,000, normalized collision energy of 28, and dynamic exclusion of 30 s. Data were processed using Proteome Discoverer (v1.4 or v2.4) and Mascot v2.6.1 against the UniProt human proteome database (20,417 entries, 2024 release). Search parameters included trypsin-specific digestion, allowing up to two missed cleavages, a precursor ion mass tolerance of ±10ppm, fragment ion tolerance of ±0.02 Da, minimum peptide length of seven amino acids, and Mascot ion score >20. The FDR for both peptide and protein identification was controlled below 1% using a target-decoy approach. PTM sites were localized using the ptmRS algorithm, retaining only those with confidence ≥0.75. For lysine methylation, a + 14.0157 Da mass shift was confirmed by manual inspection of b/y ion series in MS/MS spectra. DDX1 interactors were identified by subtractive comparison with IgG controls, and key spectra were manually validated to ensure accurate peptide assignments.
RT-qPCR
RNA from treated NP cells was separated using TRIzol reagent (Invitrogen, USA) in line with the manufacturer’s recommendations. The extracted RNA was tested for concentration and purity. After the quality was confirmed, HiScript III RT SuperMix (Vazyme, China) was used to perform reverse transcription to synthesize the first-strand cDNA. qPCR reactions were performed using AceQ SYBR Green Master Mix (Vazyme) on a CFX96 Touch Real-Time PCR System (Bio-Rad, USA). Technical replicates were set for each sample, and GAPDH served as the internal control gene to normalize expression levels. Primer sequences are detailed in Supplementary Table 3.
GST and His pulldown assay
The recombinant plasmids encoding GST-tagged EZH2 (pGEX-5X-1) and His-tagged DDX1 (pET-28a) were transformed into Escherichia coli BL21 competent cells. They were pre-cultured overnight at 37 °C in 5 mL of LB liquid medium containing appropriate antibiotics, and then inoculated into 500 mL of fresh LB medium for further amplification. When the bacterial liquid grew to the mid-logarithmic phase (OD₆₀₀ was approximately 0.8), IPTG was added at a final concentration of 0.25 mM to induce expression, and low-temperature induction was performed at 16 °C for 16 h. The induced bacteria were collected by centrifugation at 4 °C and 5000 rpm for 10 min, resuspended in pre-cooled lysis buffer, and ultrasonically disrupted in an ice bath. The lysate was clarified by centrifugation at 15,000 rpm and 4 °C for 15 min, and the supernatant was incubated with glutathione Sepharose 4B at 4 °C for 8 h with gentle rotation to bind the target protein. The bound magnetic beads were eluted according to different tags: GST fusion protein was eluted with 10 mM reduced glutathione solution; His-tagged protein was purified with 250 mM imidazole after washing with binding buffer. Each step was performed according to the instructions of the corresponding kit to ensure protein purity and activity.
RNA interference and plasmid transfection
DDX1 and EZH2 knockdown in NP cells (Supplementary Data 4) was conducted using small interfering RNA (siRNA). Upon confirming efficient gene silencing, NP cells were subjected to subsequent treatments. Lentiviral vectors encoding DDX1, EZH2, and a scrambled control (LV-shDDX1, LV-shEZH2, and LV-Scrambled) were produced using the VP003-CMV-MCS-EF1-mCherry-T2A-Puro system (OBIO, China). Detailed siRNA and shRNA sequences are listed in Supplementary Table 3. To generate mammalian expression plasmids, human wild-type DDX1 cDNA (NM_004939.3) and mutants (K234R, K234M) were cloned into pEn-CMV (HA-tag), human wild-type EZH2 cDNA (NM_004456.5), G9a cDNA (NM_006709.5), SETD7 cDNA (NM_030678.4), SMYD3 cDNA (NM_022743.3), and KMT5A cDNA (NM_020382.7) were cloned into pCDNA3.1-CMV (Flag-tag), human wild-type DDX1 cDNA (NM_004939.3) was cloned into pET-28a-CMV (His-tag); human EZH2 cDNA (NM_004456.5) was cloned into pGEX-5X-1-CMV-ZsGreen (GST-tag). For transient expression, NP and HEK293T cells were transfected with the respective eukaryotic expression vectors using Lipofectamine™ 2000 (Invitrogen, USA) and cultured for 72 h. Separately, BL21 cells were chemically transformed with prokaryotic expression plasmids and incubated for 1 h at 37 °C in LB medium before plating.
Animal model and lentivirus injection
Two-month-old female SD rats weighing 200 ± 20 g were maintained under a 12 h light/dark cycle at 21 °C. All procedures were supported by the institution’s animal ethics committee (Approval No. S2394). An IVDD model was induced in three-month-old rats by needle puncture of the tail IVDs using a 20-gauge needle (Co6-8 levels). Under anesthesia with 3% pentobarbital, rats were positioned prone, and the needle was inserted parallel to the endplates, advancing 5 mm into the NP center. The needle was then rotated 180° axially and held for 10 s. The adjacent discs (5–6 and 8–9) served as untreated internal controls. Following group assignment (n = 5), a 33-gauge Hamilton microsyringe was used to inject 2 µL of lentiviral solutions—LV-shDDX1, LV-DDX1 WT, LV-DDX1 K234R, LV-DDX1 K234M, LV-shEZH2, or LV-Scrambled—slowly into the NP region of the Co6-7 and Co7-8 discs. The injections were maintained over one month. After this period, animals were euthanized for radiological and histological evaluations.
Bulk-RNA sequencing
Human NP cells and HEK-293T cells were digested and separated using 0.4% collagenase II. After testing to meet quality requirements, 1 μg of RNA from each sample was used to construct libraries using the Illumina TruSeq RNA Sample Preparation Kit, and the library construction steps were improved in combination with the Ovation RNA-Seq System V2. The library quality was then evaluated on the Bioptic Qsep100 analyzer, and finally, next-generation sequencing was carried out on the Illumina HiSeq X Ten platform. When transcriptome data were obtained, GSEA v4.3.2 software was used to perform 1000 permutations under default parameters for gene set enrichment analysis. The annotation gene sets used were based on the C2 (curated) and C5 (ontology) sets in MSigDB v7.5.1.
IF analysis
NP cells were seeded on pre-prepared slides and treated as required by the experiment. Subsequently, the cells were rinsed using PBS and fixed in 4% paraformaldehyde for 20 mins, then treated with 0.5% Triton X-100 at 22 °C for 15 mins to permeabilize the membranes. To reduce nonspecific binding, normal goat serum was used for blocking for 30 min. Subsequently, the samples were maintained in the presence of antibodies targeting DDX1, EZH2, collagen II, MMP3, and P21 at 4 °C overnight. The next day, subsequent to rigorous washing with PBS incorporating 0.1% TBS-T, fluorescent secondary IgG conjugated to Alexa Fluor 488 or 568, matched to the corresponding species, were added for incubation at room temperature. Nuclear structures were observed following DAPI staining, and images were finally acquired using an Olympus BX53 fluorescence microscope for analysis.
FC and TUNEL assays
Apoptosis was recognized through Annexin V-FITC/PI detection kit (Vazyme, A213-01). First, cells were enzymatically dissociated using EDTA-free trypsin for five minutes, harvested, washed once with pre-cooled PBS, and resuspended in 100 µL binding buffer (approximately 5 × 10⁵ cells/mL). Subsequently, 5 µL of reaction solution were administered to the cell mixture in liquid and incubated for 30 min away from light. After staining, 400 µL of binding buffer was administered and measured within 20 min using CytoFLEX S flow cytometer. No less than 10,000 were harvested for each sample, and the data were examined using CytExpert v2.4. In addition, cell apoptosis was also evaluated by TUNEL method by using kit.
RIP sequencing analysis
To preserve the interaction between RNA and protein, NP cells were first disrupted in a lysis buffer supplemented with protease and RNase inhibitors. After centrifugation to eliminate cell fragments, the clarified solution was incubated with anti-DDX1. Subsequently, the supernatant was washed with low-salt and high-salt buffers to remove nonspecific binders as much as possible. The immune complex was incubated with proteinase K to degrade proteins and release RNA bound to DDX1. Subsequently, polyadenylated RNA was concentrated using the Dynabeads™ DIRECT™ RNA Purification Kit. The purified RNA was processed with the NEBNext Ultra™ Library Construction Kit to construct sequencing libraries, and the quality was assessed on the Bioptic Qsep100 analysis system. Next-generation sequencing was performed on the Illumina’s NovaSeq. The sequencing data were aligned with the human genome GRCh38/hg38 through Bowtie software, and the uniquely aligned reads were sorted using SAMtools. The exomePeak was used for peak identification and differential binding analysis, and the annotation information was based on the GRCh38 reference genome. Finally, combined with motif enrichment analysis, the RNA-binding preference characteristics of DDX1 were further clarified.
AS analysis
AS analysis was conducted using HISAT2 for read alignment, followed by identification of splicing events with rMATS (v4.1.0). The study included NP cells treated with TBHP and untreated controls, as well as HEK-293T/NP cells subjected to DDX1 knockdown and reconstitution with either DDX1 K234M or K234R mutants (n = 3). AS events were classified into five categories: ES, RI, ASS (5’ or 3’) and ME. Events were considered significant if they met the criteria and an absolute inclusion level difference >0.1. ASprofile was also used to catalog splicing patterns in each sample. Further validation was performed using MISO (v0.5.4), for which clean reads were realigned to the hg19 reference to match the tool’s index requirements. MATR3 exon 14 skipping was visualized using Sashimi plots. Additionally, a global AS landscape was generated using IsoformSwitchAnalyzeR.
ATAC sequencing analysis
Cells were pretreated with DNase to eliminate extracellular and dead cell-derived DNA. Following treatment, cells were spun at 500 rpm, and the resulting precipitate was resuspended in 50 µL of cold lysis buffer. After lysis, 1 mL of ATAC-seq RSB was added, and nuclei were collected by spinning at 500 rpm. The nuclear precipitate was then redispersed in 50 µL of transposition mix (comprising 10 µL of 5 × TD buffer, 5 µL of Tn5 transposase, and 35 µL of nuclease-free water). Transposition was carried out at 37 °C for 30 min in a thermomixer at 1000 rpm. The resulting DNA was amplified in a 25 µL reaction containing 2 × HiFi PCR mix, 1 µL of Nextera i5 (N5xx) and i7 (N7xx) indexing primers. Post-PCR, libraries were size-selected using EpiTM DNA Clean Beads. First, 35 µL of beads were added, the supernatant was pipetted into a fresh container, and an additional 10 µL of beads was used to enrich fragments between 250–350 bp. Library quality was assessed with the Bioptic Qsep100 Analyzer (Bioptic Inc.), and genome determination was carried out by NovaSeq 6000 using paired-end 150 bp reads.
Radiological analysis
Radiological assessments of rat coccygeal IVDs were performed using X-ray, CT, and MRI. After one month of surgery, rats were sedated with 5% SP and euthanized for tail collection. X-ray imaging was conducted using the DRX system with the following settings: 0.06 s exposure time, 100 cm source-to-image distance, 160 mA tube current, and 50 kV tube voltage. CT scans were acquired on a SkyScan1176 system (Bruker, Germany) with a resolution of 18 µm, a current of 100 µA, and a voltage of 60 kV. Three-dimensional reconstructions of coccygeal IVDs were generated using CT-Vox software (Bruker, Germany). For MRI, T2WI were obtained using a fast spin echo sequence on a 7.0 T animal-dedicated MRI scanner with repetition time of 3000 ms, echo time of 70 ms, slice thickness of 0.5 mm, and no inter-slice gap.
Histological analysis
Rat caudal IVDs were washed with PBS after X-ray examination and evaluation, immobilized in 4% PFA for 48 h, demineralized by means of 10% EDTA solution. The decalcified tissues were also dehydrated, embedded, and sectioned. The obtained sections were treated with HE and SO&FG to evaluate the alterations in the IVD tissue structure, and images were taken under an Olympus BX53 microscope. For IHC (case 1–24), formaldehyde-preserved, paraffin-infiltrated tissue sections or tissue microarrays were dewaxed in dimethylbenzene, rehydrated in ethanol gradients, and epitope exposure was performed using EDTA buffer by microwave heating. Nonspecific binding sites were then blocked with serum, and were maintained with first antibodies (against EZH2, collagen II, MMP3, and P21). Signals were detected with species-specific HRP-conjugated secondary antibodies and visualized by DAB color development. The intensity and distribution of positive areas of IHC staining were independently scored by two pathologists who were blinded to the experimental grouping to ensure the objectivity and consistency of the results.
In vitro ATP detection test
Prepare the reaction system (including purified DDX1, mutant (K234R/K234M), double-stranded RNA probe (non-differential binding sequence), MATR3, 1 mM ATP, reaction buffer, and diluent), and set up groups. Preheat the reaction system at 37 °C to start the reaction, take samples at 30 min, add EDTA-containing stop solution, and inactivate enzyme activity in an ice bath. Subsequently, use the luciferase method to detect ATP content: add the sample and standard (0–10 μM ATP gradient) to a 96-well plate, add an equal volume of luciferase working solution, and incubate in the dark for 10 min, read the relative light unit, and calculate the ATP through the standard curve.
Preparation of mRNA by in vitro transcription
The gene was inserted into a vector containing a T7 promoter. The plasmid was purified using a plasmid extraction kit, yielding a final concentration of 2 µg/ml. The in vitro transcription reaction system was prepared as follows: 5 µl of 10× RNA polymerase buffer, 4 µl of 10 mM NTPs, 2 µg of template DNA, 2 µl of T7 RNA polymerase, 0.5 µl of RNase inhibitor, to a final volume of 50 µl. These components were added to a 1.5 ml RNase-free reaction tube in the specified proportions and gently mixed. After transcription, residual template DNA and enzymes were removed using a commercial purification kit. DNase I was added to digest the remaining DNA. The reaction was terminated with the appropriate buffer. After mRNA was recovered by isopropanol precipitation, the precipitate was harvested by spinning and rinsed with 70% ethanol to remove impurities. The washed precipitate was naturally dried and reconstituted in double-distilled water. Subsequently, the RNA was calculated using a NanoDrop, and its concentration was assessed with A260/A280 ratio. The processed samples were stored to ensure their stability and reliability of downstream experiments.
Synthesis of mRNA loaded LNPs
LNPs loaded with mRNA are prepared using cholesterol, polyethylene glycol phospholipids (e.g., DSPE-MPEG2000), helper phospholipid (e.g., DOPE), and cationic ionizable lipids (e.g., DPTAP) as the main lipids. The required RNA concentration is calculated based on the nitrogen-to-phosphate ratio of 8 and the two-phase flow rate ratio (FRR) of 3. The mRNA mass that can be loaded per mM total lipid is calculated as 1 mmol × 0.5 × 106/ (8 × RNA phosphate content). A 100 mM solution of citric acid monohydrate and sodium citrate dihydrate is prepared separately by mixing 50 ml of each with DEPC-treated water, and then 33 ml of the citric acid solution and 17 ml of the sodium citrate solution are combined. After adding DEPC, the mixture is left to stand for 30 min, followed by autoclaving to remove DEPC. After sterilization, DEPC water is added to 100 ml. The mRNA concentration is determined, and then diluted with citrate buffer solution according to the lipid concentration. The lipid-ethanol mixture and citrate buffer solution are filtered through a 0.22μm filter membrane. The lipid-ethanol mixture is drawn into a 3 ml syringe (approximately 0.5 ml volume, flow rate of 3 ml/min), and the mRNA-citrate buffer solution is drawn into another 3 ml syringe (approximately 1.5 ml volume, flow rate of 9 ml/min), with air bubbles expelled from both syringes. The outlet of the syringes is connected to the sample inlet tube and fixed on the injection pump. After stabilizing the flow rate of the outlet tube, the first 100 μl of liquid is discarded, and then the sample is collected in a collection tube.
mRNA-LNPs identification
Load 20 µl of LNPs or mRNA-LNP complexes onto formvar/carbon-coated grids for 5 min, then air-dry on filter paper. Immediately afterward, negatively stain the grids with 3% phosphotungstic acid for 1 min and allow them to dry overnight. Imaging is conducted using a FEI Tecnai G20-TEM. For particle measurement, dilute 20 µl of LNPs or mRNA-LNP complexes 100–500-fold in pre-cooled PBS. Size and concentration are analyzed by NTA using a ZetaView multi-parameter particle tracker. The mRNA binding capacity of LNPs is evaluated by agarose gel retardation assay: 0.5 µg of specific mRNA is mixed with LNPs at various mass ratios, subjected to 1% agarose gel electrophoresis, stained with Golden View™, and imaged using the Bio-Rad ChemiDoc XRS system.
Intradiscal injection of LNPs
Groups of normal or model SD rats (n = 5) were anesthetized with 3% pentobarbital. Using a 33-gauge needle, 2 µl of PBS, vector-LNPs (100 µg/µl), MATR3-S-LNPs (100 µg/µl), or MATR3-L-LNPs (100 µg/µl) was slowly injected into the NP region (6–8 segments) once weekly. This procedure confirmed successful delivery of engineered LNPs into the NP. Rats were sacrificed for imaging and evaluation, while Co5-6 and Co8-9 discs served as internal negative controls.
Statistics and reproducibility
All data are presented as mean ± SD, and the results are derived from at least three independent replicates. The sample size was set with reference to previous research reports to ensure sufficient statistical power25. To reduce bias, the sample groups were processed blindly during the experiment and the result evaluation stage. Unpaired Student’s t test served to comparison between the dual groups, and two-way ANOVA served to comparison of several groups. All statistical analyses were performed in GraphPad Prism. Statistical significance is marked in the charts and legends, and the standards are as follows: *P < 0.05, **P < 0.01, ***P < 0.001. The specific number of repetitions has been clearly stated in the corresponding charts and legends. In addition, all representative images of experiments, such as IB, IHC, IF, semiquantitative RT-PCR, and FC are derived from at least three independent replicates to ensure the reproducibility and representativeness of the results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq, RIP-seq and ATAC-seq data generated in this study are available on the National Center for Biotechnology Information (NCBI) database under the BioProject number PRJNA1245287, PRJNA1245112 and PRJNA1245087 (https://www.ncbi.nlm.nih.gov/bioproject). The protein mass spectrometry data are available in the ProteomeXchange partner repository (PXD064432) (https://proteomecentral.proteomexchange.org/). The proteomic datasets are available in Source Data. Source data is available for Figs. 1b–d, f, g, n, o, 2b–e, h, j–q, 3c, d, j–l, o, 4g, 5a–c, 6g, h, j, 7f, k, l, 8c, g, l, m and Supplementary Data Figs. 1e–l, n, 2a–f, l 3i–l, 5b, e–i, 6g, i, 7b, d in the associated Source Data. Detailed individual information, PCR genotyping primer sequences, siRNA sequences for transfection, and antibody data are provided in Supplementary Tables 1–3 within the Supplementary Information File. Additionally, the Source Data supporting this study are included with the article. Requests for further supplementary information can be addressed to the corresponding author and will be fulfilled accordingly. Source data are provided with this paper.
References
Knezevic, N. N., Candido, K. D., Vlaeyen, J. W., Van Zundert, S. J. & Cohen, S. P. Low back pain. Lancet 398, 78–92 (2021).
Weifeng, Z. et al. Self-powered triboelectric-responsive microneedles with controllable release of optogenetically engineered extracellular vesicles for intervertebral disc degeneration repair. Nat. Commun. 15, 5736 (2024).
Tang, S., Stokasimov, E., Cui, Y. & Pellman, D. Breakage of cytoplasmic chromosomes by pathological DNA base excision repair. Nature 606, 930–936 (2022).
Silagi, E., Schipani, E., Shapiro, I. & Risbud, M. The role of HIF proteins in maintaining the metabolic health of the intervertebral disc. Nat. Rev. Rheumatol. 17, 426–439 (2021).
Zhu, J. et al. CRLF1/CLCF1 heterodimer involvement in intervertebral disc degeneration via exacerbation of extracellular matrix degradation and nucleus pulposus cell senescence. Osteoarthr. Cartil. 33, 574–589 (2025).
Maximilian, H. F. & Giacomo, C. Molecular mechanisms of transgenerational epigenetic inheritance. Nat. Rev. Genet. 23, 325–341 (2022).
Leclair, N. K. Poison exon splicing regulates a coordinated network of SR protein expression during differentiation and tumorigenesis. Mol. Cell 80, 648–665.e649 (2020).
Matthias, K. V. et al. Mechanism for the initiation of spliceosome disassembly. Nature 632, 443–450 (2024).
Eleftherios, Z., Rachel, Q., Alexis, C. & Simon, J. C. Mapping the transcriptome: Realizing the full potential of spatial data analysis. Cell 186, 5677–5689 (2023).
Wang, E. & Aifantis, I. RNA splicing and cancer. Trends Cancer 6, 631–644 (2020).
Charlotte, J. W., Christopher, W. J. S. & Chris, D. J. Alternative splicing as a source of phenotypic diversity. Nat. Rev. Genet. 23, 697–710 (2022).
Florence, B. M. & Luc, D. G. The Staufen1-dependent cell cycle regulon or how a misregulated RNA-binding protein leads to cancer. Biol. Rev. Camb. Philos. Soc. 96, 2192–2208 (2021).
Fang, Y. et al. Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic Acids Res 49, 5779–5797 (2021).
Jijun, H. et al. Regulation of postnatal cardiomyocyte maturation by an RNA splicing regulator RBFox1. Circulation 148, 1263–1266 (2023).
Cornett, E. M., Ferry, L., Defossez, P. A. & Rothbart, S. B. Lysine methylation regulators moonlighting outside the epigenome. Mol. Cell 75, 1092–1101 (2019.
Mohammad, N. et al. Alternative splicing of a chromatin modifier alters the transcriptional regulatory programs of stem cell maintenance and neuronal differentiation. Cell Stem Cell 31, 754–771 (2024).
Bo, Y. et al. KMT5A-methylated SNIP1 promotes triple-negative breast cancer metastasis by activating YAP signaling. Nat. Commun. 13, 2192 (2022).
Pfirrmann, C., Metzdorf, A., Zanetti, M., Hodler, J. & Boos, N. Magnetic resonance classification of lumbar intervertebral disc degeneration. Spine 26, 1873–1878 (2001).
Vera, F. et al. A new immunometabolic perspective of intervertebral disc degeneration. Nat. Rev. Rheumatol. 18, 47–60 (2022).
Sheng, L. et al. Restoration of lysosomal function attenuates autophagic flux impairment in nucleus pulposus cells and protects against mechanical overloading-induced intervertebral disc degeneration. Autophagy 21, 979–995 (2025).
Goupille, P., Jayson, M. I., Valat, J. P. & Freemont, A. J. Matrix metalloproteinases: the clue to intervertebral disc degeneration?. Spine (Philos. Pa 1976) 23, 1612–1626 (1998).
Trefilova, V. V. et al. The role of polymorphisms in collagen-encoding genes in intervertebral disc degeneration. Biomolecules 11, 1279 (2021).
Li, W. et al. A methylation phosphorylation switch determines Plk1 kinase activity and function in DNA damage repair. Sci. Adv. 5, eaau7566 (2019).
Liang, H. et al. Lysine methylation of PPP1CA by the methyltransferase SUV39H2 disrupts TFEB-dependent autophagy and promotes intervertebral disc degeneration. Cell Death Differ. 30, 2135–2150 (2023).
Li, G. et al. WTAP-mediated m (6)A modification of lncRNA NORAD promotes intervertebral disc degeneration. Nat. Commun. 13, 1469 (2022).
Cheng, X. et al. Circular RNA VMA21 protects against intervertebral disc degeneration through targeting miR-200c and X linked inhibitor-of-apoptosis protein. Ann. Rheum. Dis. 77, 770–779 (2018).
Zoe, K., Gadi, P., Dan, G. & Zulma, G. Stem cells and exosomes: New therapies for intervertebral disc degeneration. Cells 10, 2241 (2021).
Runge, V., Richter, J. & Heverhagen, J. Speed in clinical magnetic resonance. Invest. Radiol. 52, 1–17 (2017).
Trenkmann, M., Brock, M. & Gay, R. Expression and function of EZH2 in synovial fibroblasts: epigenetic repression of the Wnt inhibitor SFRP1 in rheumatoid arthritis. Ann. Rheum. Dis. 70, 1482–1488 (2011).
Zhe, W. et al. A targeted exosome therapeutic confers both CfDNA scavenging and macrophage polarization for ameliorating rheumatoid arthritis. Adv. Mater. 35, e2302503 (2023).
Wang, Z. et al. Splicing factor BUD31 promotes ovarian cancer progression through sustaining the expression of anti-apoptotic BCL2L12. Nat. Commun. 13, 6246 (2022).
Smith, J. & Pereira-Smith, O. Replicative senescence: implications for in vivo aging and tumor suppression. Science 273, 63–67 (1996).
Teruhiko, S. et al. DDX1 is required for non-spliceosomal splicing of tRNAs but not of XBP1 mRNA. Commun. Biol. 8, 92 (2025).
Ya-Ru, Z. et al. Whole exome sequencing analyses identified novel genes for Alzheimer’s disease and related dementia. Alzheimers Dement 20, 7062–7078 (2024).
Yena, C. & Yong, K. K. ROS-mediated cytoplasmic localization of CARM1 induces mitochondrial fission through DRP1 methylation. Redox Biol. 73, 103212 (2024).
Salima, B. et al. GPATCH8 modulates mutant SF3B1 mis-splicing and pathogenicity in hematologic malignancies. Mol. Cell 84, 1886–1903 (2024).
Lin, Z. et al. Cathepsin B S-nitrosylation promotes ADAR1-mediated editing of its own mRNA transcript via an ADD1/MATR3 regulatory axis. Cell Res 33, 546–561 (2023).
Malik, A. et al. Neuronal activity regulates Matrin 3 abundance and function in a calcium-dependent manner through calpain-mediated cleavage and calmodulin binding. Proc. Natl Acad. Sci. Usa. 120, e2206217120 (2023).
Nuria, O. et al. Wnt/β-catenin and NFκB signaling synergize to trigger growth factor-free regeneration of adult primary human hepatocytes. Hepatology 79, 1337–1351 (2024).
Rim, E., Clevers, H. & Nusse, R. The Wnt pathway: From signaling mechanisms to synthetic modulators. Annu. Rev. Biochem. 91, 571–598 (2022).
Burgess, A. et al. SnapShot: S-phase entry and exit. Cell 179, 802–802 (2019).
Ruscitto, A. et al. Lgr5-expressing secretory cells form a Wnt inhibitory niche in cartilage critical for chondrocyte identity. Cell Stem Cell 30, 1179–1198.e7 (2023).
Cheng, L. & Hill, A. Therapeutically harnessing extracellular vesicles. Nat. Rev. Drug Discov. 21, 379–99 (2022).
Naidu, G. et al. A combinatorial library of lipid nanoparticles for cell type-specific mRNA delivery. Adv. Sci. (Weinh.). 10, e2301929 (2023).
Seo-Hyeon, B. et al. A lipid nanoparticle platform incorporating trehalose glycolipid for exceptional mRNA vaccine safety. Bioact. Mater. 38, 486–498 (2024).
Jiang, C. et al. Inhibition of EZH2 ameliorates cartilage endplate degeneration and attenuates the progression of intervertebral disc degeneration via demethylation of Sox-9. EBioMedicine 48, 619–629 (2019).
Zhao, X. et al. Degenerated nucleus pulposus cells derived exosome carrying miR-27a-3p aggravates intervertebral disc degeneration by inducing M1 polarization of macrophages. J. Nanobiotechnology 21, 317 (2023).
Wu, J. et al. Self-amplifying loop of NF-κB and periostin initiated by PIEZO1 accelerates mechano-induced senescence of nucleus pulposus cells and intervertebral disc degeneration. Mol. Ther. 30, 3241–3256 (2022).
Shu-Hsien, S. et al. A serotonergic axon-cilium synapse drives nuclear signaling to alter chromatin accessibility. Cell 185, 3390–3407 (2022).
Anthony, F. T. M., Yepeth, B., Isaac, S. W. & Eda, K. Substrate Specificities of DDX1: A Human DEAD-Box Protein. ACS Omega 10, 2598–2607 (2025.
Robertson-Anderson, R. M. et al. Single-molecule studies reveal that DEAD box protein DDX1 promotes oligomerization of HIV-1 Rev on the Rev response element. J. Mol. Biol. 410, 959–971 (2011).
Miller, L. et al. Combinations of DEAD box proteins distinguish distinct types of RNA: protein complexes in neurons. Mol. Cell. Neurosci. 40, 485–495 (2009).
Jain, S. et al. ATPase- modulated stress granules contain a diverse proteome and substructure. Cell 164, 487–498 (2016).
Jain, S. et al. DDX1 vesicles control calcium-dependent mitochondrial activity in mouse embryos. Nat. Commun. 13, 3794 (2022).
Carlos, A. et al. Sites of transcription initiation drive mRNA isoform selection. Cell 186, 2438–2455 (2023).
Kellner, J. N. & Meinhart, A. Structure of the SPRY domain of the human RNA helicase DDX1, a putative interaction platform within a DEAD-box protein. Acta Crystallogr. F. Struct. Biol. Commun. 1, 1176–1188 (2015).
Barral, A. et al. SETDB1/NSD-dependent H3K9me3/H3K36me3 dual heterochromatin maintains gene expression profiles by bookmarking poised enhancers. Mol. Cell 82, 816–832 (2022).
Pandya-Jones, A. et al. A protein assembly mediates Xist localization and gene silencing. Nature 587, 145–151 (2020).
Caputo, M. et al. Duplication of exons 15 and 16 in matrin-3: a phenotype bridging amyotrophic lateral sclerosis and immune-mediated disorders. Neurol. Sci. 43, 1419–1421 (2022).
Skowronska-Krawczyk, D. et al. Required enhancer-matrin-3 network interactions for a homeodomain transcription program. Nature 514, 257–261 (2014).
Malik, A. & Barmada, S. Matrin 3 in neuromuscular disease: physiology and pathophysiology. Jci. Insight 6, e143948 (2021).
Ji, M. et al. Preclinical development of a microRNA-based therapy for intervertebral disc degeneration. Nat. Commun. 9, 5051 (2018).
Acknowledgements
This research was funded by the National Science Foundation of China (NSFC: 81772401, 82130072, 82072505), the Fundamental Research Funds for Central Universities (HUST: YCJJ20242118), and the Natural Science Foundation of Hubei Province (2023AFB532). Thanks for the technical support by the Huazhong University of Science & Technology Analytical & Testing center, Medical sub-center. We appreciate the technical assistance provided by the Wuhan Center for Magnetic Resonance at the Innovation Academy for Precision Measurement Science and Technology, Chinese Academy of Sciences. We also thank the members of the research groups and colleagues for their valuable feedback and suggestions. Finally, we thank the Home for Researchers (www.home-for-researchers.com) for providing an illustration in Figs. 1a, 3e, 4a, 9, and Supplementary Fig. 7a (WPYATA7C42).
Author information
Authors and Affiliations
Contributions
C.Y., D.Z., Y.S., H.L., and B.T. formulated the experiments. D.Z., H.L., B.T., and Z.D. conducted the majority of experiments and carried out data analysis. G.L., W.Z., D.W., X.Z., J.L., X.Z. assisted with the animal surgery. L.M., B.W., X.F., K.W., and L.T. gathered the clinical samples. D.Z., Y.S., and C.Y. drafted the manuscript. All authors reviewed and endorsed the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Richard Bennett and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, D., Liang, H., Tong, B. et al. DDX1 methylation mediated MATR3 splicing regulates intervertebral disc degeneration by initiating chromatin reprogramming. Nat Commun 16, 6153 (2025). https://doi.org/10.1038/s41467-025-61486-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61486-7
