
Abstract
Specialized secretory cells, including keratinocytes in the last viable layers of mammalian epidermis, utilize lysosome-related organelles (LROs) to exocytose distinct cargoes vital for tissue function. Here, we demonstrate that the Flower isoform, hFWE4, a putative Ca2+ channel that permits endocytic retrieval of presynaptic vesicles and lytic granules, also resides on epidermal lamellar bodies (LBs), an LRO that extrudes a proteinaceous lipid-rich matrix to finalize the epidermal barrier. In differentiated keratinocyte cultures, we show that hFWE4-positive LB-like vesicles associate with a distinct ensemble of LRO trafficking mediators and demonstrate that hFWE4 liberates Ca2+ from intracellular stores to enable the surface presentation of cargo contained within these vesicles. Finally, supporting a critical role for hFWE4-dependent trafficking in establishing the epidermal barrier, we demonstrate that this process is dysregulated in genetic diseases of cornification that are driven by impairments in keratinocyte Ca2+ handling. Our results provide new insight into the biogenesis and trafficking of epidermal LBs and more broadly suggest that hFWE4 may serve as a core component of LRO trafficking machinery that endows Ca2+ dependency to distinct stages of the transport process depending on the cell of origin.
Similar content being viewed by others


Introduction
The epidermal barrier is continuously regenerated by keratinocytes that execute a Ca2+-dependent terminal differentiation program as they move apically towards the body surface. This process culminates in the production of ‘corneocytes’, anucleate cellular corpses in the stratum corneum (SC) with a covalently crosslinked ‘cornified envelope’ underlying the plasma membrane1,2. Corneocytes are embedded within a lipid-rich extracellular matrix that is secreted apically from lamellar bodies (LBs) in the underlying, Ca2+-rich keratinocyte layers of the stratum granulosum (SG). LBs are heterogeneous lysosome-related organelles (LROs) (reviewed in ref. 3) that contain unique lipid contents and carry lipid-modifying enzymes4, antimicrobial peptides5,6, proteases7,8, and adhesion molecules8 required for proper SC function. In addition to directing apical secretion of LBs, keratinocytes in the second layer of the SG (SG2) establish a functional tight-junction (TJ) barrier at apico-lateral plasma membranes, restricting water and solute permeability across the tissue9,10,11,12. It is well appreciated that both LB secretion and TJ formation are sensitive to perturbations in extracellular and cytosolic Ca2+ levels13,14, however, the molecular events that establish elevated Ca2+ levels in the SG, and the mechanisms by which this is translated into the apically polarized LRO transport required for building the epidermal barrier are unknown.
Evidence has emerged in a number of tissues to suggest that cell-type-specific LROs serve as acidic intracellular Ca2+ stores and that release of these stores can facilitate their polarized secretion15. However, this phenomenon has not been investigated in the apical secretion of epidermal LBs. While typically attributed to Ca2+ flux through lysosome associated Ca2+ permeable channels such as TRPML16, TPCs17, P2X418,19 or RyRs20, work in mouse and Drosophila models has recently suggested that proteins encoded by the understudied Flower (Fwe or Cacfd1) gene regulate Ca2+-dependent polarized transport of LROs in neurons21,22,23 and facilitate endocytic retrieval of LRO-derived cargo in cytotoxic T-lymphocytes (CTLs)24. We previously demonstrated that the canonical protein isoform encoded by the human hFWE gene, hFWE4, is a small four-transmembrane pass protein that resides in the membrane of cytosolic vesicles and is trafficked for transient integration into the plasma membrane of HEK293 cells25. While the composition of hFWE4-positive vesicles in human epithelial cells is unknown, hFWE4 exhibits robust structural conservation to its orthologues in mouse and Drosophila, which are known to regulate endocytic retrieval of lytic granules and presynaptic vesicles, respectively. While one previous study has suggested that FWE-deficient mice are modestly resistant to DMBA/TPA-induced skin carcinogenesis26, the molecular function of FWE in skin is unknown. We reasoned that, given the function of its orthologues in neurons and CTLs, hFWE4 may similarly regulate epidermal LBs, which are LROs that are dysfunctional in disorders of cornification exhibiting impairments in the skin barrier.
Here, we present the first investigation into the expression and function of FWE in human epidermis, revealing localization of hFWE4 to epidermal LBs in the SG layers where it elevates cytosolic Ca2+ to facilitate surface trafficking of LB cargo toward the apical keratinocyte membrane. We demonstrate the importance of this process to the proper formation of the epidermal barrier and provide evidence that FWE-positive LBs fail to be properly secreted in epidermal pathologies where premature cornification is driven by impaired Ca2+ handling.
Results
Flower (FWE) is up-regulated in terminally differentiating epidermal keratinocytes and localizes to apically polarized vesicles
In human epidermis, confocal microscopy revealed that FWE was largely restricted to keratinocytes in the upper stratum spinosum (SS) and throughout the three layers of the stratum granulosum (SG), termed SG3, SG2, and SG1 from inner to outer, respectively. Cells in the uppermost SG1 layer frequently displayed the most intense, apically polarized signal for FWE (Fig. 1a). Double immunostaining for the desmosomal cadherin desmoglein-1 (DSG-1) and FWE demonstrated that in upper SS and lower SG3 layers, FWE puncta accumulated just underneath the apical plasma membrane and appeared to fuse with this membrane in SG2 and SG1 cells (Fig. 1a). Airyscan2 super-resolution microscopy further revealed FWE-positive puncta of 140-250 nm in diameter aligning between DSG-1 positive puncta (marking desmosomes between each cell layer) along the apical membrane of SG1, and to a lesser extent SG2 cells (Fig. 1b). To validate the specificity of the FWE antibody used in these immunofluorescence and subsequent immunoblotting assays, FWE knockout (KO) N/TERT-2G keratinocytes were generated using Cas9:sgRNA RNP delivery to target exon 1 which is shared amongst all FWE transcript variants, and monoclonal knockout cell lines were subsequently derived (Supplementary Fig. 1A, B). Immunoblotting of differentiated monolayers and immunofluorescence in epidermal organoid culture revealed specificity for a single < 25 kDa product and apically polarized signal in the SG, respectively (Supplementary Fig. 1c, d). These observations suggested that FWE labels a subset of apically secreted vesicles in SG keratinocytes of the upper epidermis, where the extracellular lipid-rich matrix that fortifies the cutaneous barrier is actively assembled.

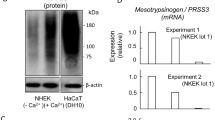
a Representative confocal microscopy of double immunofluorescence for desmoglein-1 (DSG-1) and Flower (FWE) in normal human epidermis (n = 3) with Hoechst-labeled nuclei (blue). b Airyscan2 super resolution confocal image of the inset stratum granulosum area in (A) showing stratum spinosum (SS) and SG layers 1-3. The line scan indicated by the yellow line in the image shows fluorescence intensity values for DSG-1 and FWE along the apical SG1 membrane. Scale bars (a) 10 µm, (b) 500 nm. c Representative (n = 3) immunoblots for FWE and differentiation markers in whole cell lysates collected from monolayer NHEK or N/TERT-2G keratinocytes in a subconfluent (SC) undifferentiated state or after differentiation in 1.4 mM CaCl2 containing media for 1, 3, or 5 days post confluence. d Representative FLAG immunoblot (n = 2) of monolayer N/TERT-2G keratinocytes harboring a 3XFLAG knock-in (3XFLAG-KI) preceding the stop codon in exon 6B of hFWE (see Supplementary Fig. 2A). Whole cell lysates from unedited cells (Mock) and KI cells were collected from cells as described in (c). e hFWE expression values retrieved from bulk RNA seq experiments (GSE127223 and GSE228631) performed on subconfluent or differentiated NHEK monolayers (n = 3 replicates per timepoint, * = DESeq2, p- or q-value < 0.05).
To determine whether this differentiation-associated expression pattern could be recapitulated in vitro, normal human epidermal keratinocytes (NHEKs) and TERT-immortalized NHEKs (N/TERT-2G) were differentiated for either one, three, or five days in high calcium (1.4 mM) medium. Immunoblotting revealed a progressive increase in total FWE protein during differentiation along with the differentiation markers keratin 10 (K10) and loricrin (LOR) (Fig. 1c). As additional validation of the results obtained with the FWE antibody, a CRISPR-Cas9 strategy was used to knock-in a 3 x FLAG tag immediately upstream of the stop codon in the shared exon 6b of hFWE3 and hFWE4 in N/TERT-2G cells and a heterozygous knock-in (KI) clone was isolated (Supplementary Fig. 2A–C). N/TERT-2G were subjected to a Ca2+ induced differentiation time course and immunoblotting revealed a progressive increase in 3 x FLAG-tagged FWE levels during differentiation that mirrored endogenous expression (Fig. 1d). Differentiation kinetics in KI cells were unaffected as evidenced by similar rates of K10 and filaggrin (FLG) increase relative to WT controls (Supplementary Fig. 2C) Importantly, immunoblotting for epitope-tagged protein yielded only a single specific band that runs at the expected molecular weight for hFWE4-3 x FLAG (Fig. 1d). The second most abundant isoform, hFWE3, is ~ 5 kDa smaller than the canonical hFWE4, but was not observed. Detection of only the canonical FWE isoform (hFWE4) is consistent with our recent analysis of the GTEX database, which suggests hFWE4 accounts for ~ 80% of all hFWE transcripts25, and is in line with protein-level data in mice, where only the hFWE4 orthologue (mFwe2) is detected24. Lastly, to assess whether FWE expression changes are evident at the transcript level during keratinocyte differentiation, we reanalyzed two publicly available bulk RNA-sequencing (RNA-seq) datasets from NHEK differentiation time courses conducted in refs. 27,28, which also revealed a progressive increase in hFWE expression during differentiation (Fig. 1e). Collectively, these data identify FWE as a novel marker of late-stage terminal differentiation localized to the upper epidermal layers and suggest that FWE is trafficked to the apical surface of SG keratinocytes as they begin to cornify.
FWE-deficient epidermal organoids exhibit impaired barrier function
Given the distribution of FWE in the differentiated layers of human epidermis and its elevated expression at both transcript and protein levels during keratinocyte differentiation in vitro, we hypothesized that FWE plays an active role in the epidermal differentiation program. As late terminal differentiation and cornification are not well replicated in two-dimensional monolayer cultures, we assessed the effect of FWE depletion on the maturation of epidermal organoid cultures grown at the air-liquid interface. This organotypic model of human epidermis undergoes cornification over the course of 10–14 days to produce histologically normal in vitro epidermis and is amenable to subsequent barrier function testing. During organoid maturation, FWE accumulation in SG keratinocytes peaked between 4–6 days of air-liquid interface culture and declined abruptly after 10 days (Fig. 2a), consistent with FWE accumulation preceding complete acquisition of epidermal barrier function, which occurs at day 10 as previously measured using electrical impedance spectroscopy (EIS) in our recent work29.

a Representative FWE immunofluorescence on N/TERT-2G derived epidermal organoids grown at air-liquid interface for the indicated number of days (n = 2 organoids per timepoint, scale bars, 20 µm). Heatmap beneath images reports average percentage of maximal EISdiff AUC for each timepoint from ref. 29. b Full impedance spectra from control and hFWE KO N/TERT-2G after 10 d at air-liquid interface, as well as AUC calculations (c) for impedance values in EISdiff range are shown normalized to control (* = p < 0.05, *** = p < 0.001, **** = p < 0.0001, one-way ANOVA with Bonferroni’s multiple comparison test). CLDN1 KO N/TERT-2G previously developed in ref. 29 were included as a positive control for barrier deficit. (n = 6 organoids for Control, hFWE KO pool and hFWE KO clone8 over two independent experiments; n = 3 organoids for hFWE KO clone5 and clone11). d Representative (n = 3) H&E and differentiation marker (LOR, FLG) immunohistochemistry on epidermal organoids. Dashed lines on H&E indicate the border of keratohyalin granule-containing cells in the SG (scale bars, 50 µm). e Volcano plot of DESeq2 results from bulk RNA-seq comparing all hFWE KO organoids (n = 11, including pool and clonal organoids) to control (n = 3) organoids. f Dotplot (left) and emap (right) of enriched GO:BP terms from gene-set enrichment analysis on significantly (p-adj < 0.05) downregulated genes in KO organoids. Enriched nodes related to vesicle function and lipid metabolism are denoted with red and blue outlines on emap, respectively.
Further suggesting that FWE functionally contributes to epidermal barrier development, FWE KO organoids showed a significant reduction in electrical impedance within the range of frequencies that correlates with differentiation of viable keratinocytes (EISdiff)29 (Fig. 2b, c). This phenotype did not appear to result from a proliferation defect upon FWE KO, as in monolayer culture, there was a modest increase in the S-phase fraction of some KO clones, while in organoid cultures, there was no apparent change in the fraction of proliferating Ki67 + cells between conditions (Supplementary Fig 3A–D). These findings are consistent with the previous report of a small but significant increase in the S-phase fraction of breast cancer cells upon KO of hFWE30. On routine histology, we observed minimal differences in the morphology of KO organoids compared to WT, but did observe a modest reduction in the number of FLG and LOR positive granular layer cells detected by IHC (Fig. 2d). Collectively, these data indicated that FWE expression functionally contributes to the development of the epidermal barrier.
To identify other factors contributing to the significant barrier deficit, transcriptomic profiling using bulk RNA-seq was performed on WT and KO organoids, showing significant alterations in the expression of 733 genes (438 UP, 295 DOWN; p-adj < 0.05) (Fig. 2e and Supplementary Fig 4A–C). Consistent with the morphological analysis showing minor changes in the uppermost epidermal layers of KO cultures, no major differences in markers of various stages of keratinocyte differentiation were observed. Across the 57 genes comprising the epidermal differentiation complex31; only 7 (IVL, LCE3C-E, LCE5A, S100A11, S100A16, TCHH, and CRNN) showed significant reduction in expression in FWE KO organoids (Supplementary Fig. 4C, D). However, we were intrigued by gene set enrichment analysis (GSEA) showing that downregulated genes were enriched for biological functions related to vesicle function, including organelle acidification, lysosome maturation, autophagy, and cation homeostasis, along with those related to lipid metabolism, all of which are known drivers of the latest stages of epidermal differentiation32 (Fig. 2f and Supplementary Fig. 4B). As FWE orthologues in mouse and fly have previously been described to function as putative calcium-permeable channels22 and regulate the trafficking of lysosome-related organelles or their derivative cargo in other cell types (e.g., synaptic vesicles and lytic granules), we reasoned that FWE may be exerting calcium-dependent control over biogenesis and trafficking of epidermal lamellar bodies (LBs), LROs potentially derived from the trans Golgi network33 that are generated and secreted in the SG to drive cutaneous barrier formation.
FWE localizes to epidermal lamellar bodies (LBs) and facilitates their apically-directed secretion
Based on the apically polarized subcellular distribution of FWE in SG cells, we hypothesized that FWE may physically associate with epidermal LBs, which are secreted apically by cells in SG2 and SG1. Supporting this hypothesis, quantitative Airyscan2 super-resolution microscopy revealed partial colocalization of FWE with three distinct LB cargo molecules; corneodesmosin (CDSN) (Mander’s coefficient 0.532 ± 0.033), skin-derived antileukoprotease (SKALP) (0.410 ± 0.051), and kallikrein-5 (KLK5) (0.572 ± 0.037) in the SG of normal human skin (N = 3) (Fig. 3a, b). FWE + /CDSN+ structures were found in the cytosol and at the apical membrane of both SG2 and SG1 cells, while FWE + /KLK5 + and FWE + /SKALP + structures were found almost exclusively at the apical membrane of SG1 or the cytosol of SG2, respectively (Fig. 3a and Supplementary Fig. 5A). In contrast to their colocalization with LB cargo, FWE positive vesicles exhibited minimal colocalization with the early endosome marker EEA1, or the desmosomal cadherin DSG-1 (Fig. 3a and Supplementary Fig. 5A).

a Representative Airyscan2 imaging of double immunofluorescence for FWE and either desmoglein-1 (DSG-1), early endosome antigen 1 (EEA1), corneodesmosin (CDSN), kallikrein-5 (KLK5), or skin-derived antileukoprotease (SKALP) in the SG of normal human epidermis. Inset regions indicated by yellow dashed boxes. The white dashed line demarcates the border between SG1 and SC. b Mander’s coefficient (n = 12–15 micrographs across three biologically independent samples) for colocalization of FWE and the indicated marker in double-positive cell layers of Airyscan2 micrographs. Large dots indicate the average of n ≥ 4 distinct imaging fields (small dots) for each independent sample. c CDSN and SKALP IHC in epidermal organoids (n = 3) derived from control or hFWE KO N/TERT-2G cells. Inset regions indicated by dashed boxes. Scale bars, (a) 1 µm (full image), 1 µm (inset), (c) 20 µM, 10 µm (inset).
Based on these imaging data, we hypothesized that FWE may directly contribute to LB biogenesis and/or apical trafficking for secretion during cornification. Supporting this hypothesis, we found that CDSN-positive puncta begin accumulating in the SG of epidermal organoids on day 4 of stratification and reach maximum accumulation in the SC before subsequently declining after day 10, the timepoint after which FWE expression in viable keratinocyte layers declines substantially (Supplementary Fig 5B and Fig. 2a). We next directly tested this hypothesis by performing IHC for LB markers on WT and KO epidermal organoids. CDSN IHC on WT organoids revealed strong expression and apical polarization in SG keratinocytes along with SC signal, reflecting its localization to corneodesmosomes (Fig. 3c), while in contrast, FWE deficient organoids displayed diminished signal that had lost polarized distribution in SG and was absent from SC layers (Fig. 3c). Similarly, SKALP IHC revealed minimal SC signal and near absent apically polarized SG1 signal in in addition to fewer total SKALP-positive cell layers in KO organoids relative to WT controls (Fig. 3c). Collectively, these data provided evidence that proper delivery of epidermal LBs to the stratum corneum is impaired in the absence of FWE.
Proteomic profiling defines the composition of hFWE4-positive vesicles and their association with LB-specific trafficking machinery
Several well-studied trafficking regulators, including Rab11A34,35, Myo5b36, and Cav137 have been identified as mediators of LB transport. However, the broader network of machinery required for apical LB secretion in the SG is undefined38. It is also unclear whether LBs primarily traffic secretory cargo or whether they also carry membrane-associated proteins involved in distinct aspects of epidermal barrier formation that occur at the apical plasma membrane, such as tight junction (TJ) formation. Intriguingly, a previous study identified TJ components in proteomic analysis of the LB fraction isolated from human epidermis39, suggesting that these proteins could be cargoes of LBs, or alternatively, of a precursor vesicle that cofractionates with mature LBs. Such precursor vesicles have been hypothesized to arise from the trans-Golgi network (TGN)3,33. However, direct evidence for this remains limited, and purified LB fractions from human epidermis may be slightly contaminated by other membranous compartments, complicating interpretations. Having demonstrated that in human epidermis, FWE labels both LBs carrying canonical cargo and apically polarized vesicles without such cargo, we sought to comprehensively profile the proteome of intact FWE-positive vesicles in differentiated keratinocyte cultures to gain insight into their composition, cellular origin, and transport mechanisms.
To achieve this, N/TERT-2G cell lines stably expressing either a TetON 3 x NLS EGFP (TetON-GFP) or 3 x NLS EGFP P2A hFWE4-3 x FLAG (TetON-hFWE4) transgene were generated through lentiviral transduction (Supplementary Fig. 6A). In TetON-hFWE4 cells, hFWE4 expression was robustly induced in response to doxycycline, assumed a subcellular localization in line with our previous work25, and ran at the same molecular weight as the 3 x FLAG tagged endogenous hFWE4 (Supplementary Fig. 6B–D). To profile their composition, following 4 d of differentiation, hFWE4-positive vesicles were immuno-isolated by anti-FLAG IP under non-denaturing conditions and subject to label-free quantitative mass spectrometry (LFQ-MS) (Fig. 4a). Similar immuno-isolation strategies have recently been employed to profile the composition of endosomes40, lysosomes41, Golgi42, and other membrane-bound organelles43. A total of 180 proteins were differentially enriched (L2FC > 1, FDR < 0.05) in IPs from TetON-hFWE4 (Flower-IP) compared to TetON-GFP (Control-IP), including many membrane and vesicular proteins, suggesting an association with hFWE4-positive vesicles or membrane fragments (Fig. 4b). Application of consensus graph-based subcellular annotation from ref. 43 to all proteins identified in our LFQ-MS data revealed that proteins enriched in Flower-IPs predominantly localized to plasma membrane, trans-Golgi, and early and recycling endosomes (Fig. 4c). In addition, proteins associated with hFWE4-positive vesicles overlapped significantly with those found in the LB proteome of human epidermis39 (32.2% overlap, Fisher’s exact test of the hypergeometric distribution = 2.52e-32) (Fig. 4d).

a Schematic of non-denaturing immunoprecipitation strategy for isolation of intact vesicles harboring 3XFLAG-tagged hFWE4 from differentiated (D4) keratinocytes. Created in BioRender. Rudd, J. (2025) https://BioRender.com/zvwuxf0. b LFQ-MS volcano plot showing proteins enriched (pink) in Flower-IP relative to Control-IP (n = 5) using cutoffs of FDR < 0.05 and log2FC > 1. c Graph-based annotation (from ref. 43) of subcellular localization for all proteins identified in the LFQ-MS dataset. Proteins identified as significantly enriched in (b) are shown in pink. Box plots show median and 25th–75th percentile values with whiskers extending to minimum and maximum values. d Overlap of Flower-IP enriched proteins with those identified as part of the 984-component human LB proteome (from ref. 39). Fisher’s exact test of the hypergeometric distribution was calculated to determine the significance of the overlap. e Representation of Flower-IP enriched proteins (FDR < 0.05) organized by functional network or multi-subunit complex. f Immunoblot validation of Flower-IP enrichment for select interactions, including SNARE complex and plasma membrane cargo. Unrelated intervening lanes were removed from the TROP2 blot. g Representative Airyscan2 imaging of double immunofluorescence for CDSN and VAMP3 or STXBP3 showing partial colocalization CDSN positive LBs in the SG of human epidermis. Mander’s coefficient (n = 12–15 micrographs across three biologically independent samples) for colocalization of CDSN and the indicated marker in double-positive cell layers of Airyscan2 micrographs. Scale bars, 1 µm.
Amongst the proteins associated with hFWE4-positive vesicles, we identified a diverse set of plasma membrane cargos including the serine peptidase inhibitor SPINT1 and TJ components TROP2 and JAM-A (Fig. 4e, f). While many prototypical secretory LB cargos were not found to be enriched in hFWE4-positive vesicles, we hypothesized that in vitro, LB-like vesicles may exist in only a subset of highly differentiated cells and represent a small fraction of vesicles used as input for immunoprecipitation. To test this hypothesis and determine whether these LB-defining cargo associate with hFWE4-positive vesicles in vitro, we performed parallel experiments in which hFWE4-EGFP expressing N/TERT-2G were differentiated for four days in 1.4 mM Ca2+ containing media and subject to immunocytofluorescence against TGN46, KLK5, SKALP, and CDSN (Supplementary Fig. 7). Airyscan2 microscopy revealed that in basal keratinocytes hFWE4 occupies a perinuclear TGN46-positive trans-Golgi compartment, and that as cells reach the suprabasal layer, this vesicle population undergoes significant dispersion. We found that while expression of LB markers was restricted to a few select suprabasal cells, double-positive cells exhibited significant colocalization with hFWE4 (Supplementary Fig. 7). In line with our findings in human epidermis, colocalization with KLK5 was most robust (Mander’s coefficient, 0.796 ± 0.088), while SKALP (0.623 ± 0.053) and CDSN (0.615 ± 0.066) showed a mix of colocalized vesicles and more complex structures with partial colocalization potentially representing fusion events. To investigate the lipid composition of these vesicles, we next performed pulse chase assays with NBD-C6-ceramide, a fluorescently labeled lipid which labels Golgi-derived vesicles and serves as a precursor to glucosylceramides and sphingomyelin that populate LBs44. In suprabasal cells of differentiated cultures hFWE4-EGFP positive vesicles were loaded with NBD-C6-ceramide (Supplementary Fig. 8). Collectively, these data suggested that in vitro differentiation drives the generation of sparse, hFWE4-positive, LB-like vesicles that derive from the TGN.
In addition to cargo molecules, MS identified many proteins associated with hFWE4-positive vesicles that could be further organized according to functional relationships or integration into multi-subunit complexes. These included V-ATPase subunits that regulate organelle acidification, proteins with varied roles in lipid metabolism, signaling, and lipoprotein trafficking, and an extensive Rab and Rho GTPase network (Fig. 4e). hFWE4-positive vesicles also associated with many SNARE complex subunits, including v- and t-snares, regulatory Sec1/Munc18-like (SM) proteins, and SNAPs, which regulate both fusion and complex disassembly (Fig. 4e, f). Given that interaction between cognate v-snares, t-snares, and regulatory SM proteins provides specificity for targeting secretory vesicles to defined plasma membrane domains, we investigated whether these identified subunits are associated with CDSN-positive LBs in the SG of human epidermis. In Airyscan2 imaging of double immunofluorescence for STXBP3 or VAMP3 with CDSN, colocalization with CDSN-positive LBs were observed, suggesting that these SNARE complex components may serve as previously unappreciated contributors to polarized LB trafficking in human epidermis (Fig. 4g).
Ectopic FWE expression facilitates plasma membrane delivery of LB-associated cargo including the tight junction component, TROP2
To determine whether elevated expression of hFWE4 was sufficient to promote cell surface presentation of LB-related cargo contained in hFWE4-positive vesicles, cell surface proteins were biotinylated and affinity purified from TetON-GFP and hFWE4 cells (N = 5) after differentiating monolayer cultures for four days in 1.4 mM Ca2+. LFQ-MS profiling of cell surface proteomes from these two cell populations revealed significantly increased surface presentation (FDR < 0.1) of 471 proteins in TetON-hFWE4 cells, while only 25 showed a significant reduction in surface presentation (Fig. 5a). Surface enriched proteins shared significant overlap with those associated with hFWE4-positive vesicles (37/471, Fisher’s exact test of the hypergeometric distribution p = 1.30e-24) (Fig. 5b) and those found in the LB proteome (209/471, Fisher’s exact test of the hypergeometric distribution p = 4.43e-151) (Fig. 5c).

a LFQ-MS volcano plot showing enriched (pink) and depleted (green) cell surface proteins in TetON-hFWE4 N/TERT-2G cells relative to TetON-GFP control after four days of differentiation (n = 5, FDR < 0.1). Overlap of cell surface enriched proteins with the LB proteome (Raymond 2008) (b) and with Flower-IP enriched proteins (c). Fisher’s exact test of the hypergeometric distribution was calculated to determine the significance of the overlap. d Representative TROP2 immunoblot of 100 µg affinity-purified cell surface protein relative to 10 µg total protein from D7 differentiated TetON-GFP or TetON-hFWE4 N/TERT-2G in the presence (n = 6) or absence (n = 3) of doxycycline. Densitometric quantification of immunoblots across two independent experiments is presented (* = p < 0.05, one-way ANOVA with Dunnet’s multiple comparison test). e Representative (n = 4 independent experiments) spinning disc confocal imaging of live N/TERT-2G expressing a TetON-hFWE4-EGFP and SNAP-tagged TROP2. Inset (right) shows colocalization on hFWE4-EGFP positive vesicles. Scale bars, 10 µm (full image), 1 µm (inset).
Of the 25 candidates that met both of these criteria, the putative LB cargo TROP2 stood out as a multifunctional EPCAM homolog involved in both TJ stabilization and calcium signal transduction. While TROP2 is highly expressed in the upper epidermis and its contribution to epidermal barrier function has been evidenced in both animal45 and tissue culture46 models, little is known about molecular mechanisms regulating its surface presentation. To validate hFWE4-dependent surface trafficking of TROP2, biotinylated cell surface protein from TetON-GFP and TetON-hFWE4 cells treated with doxycycline or vehicle for seven days of differentiation was affinity-purified and used to compare surface abundance to total TROP2 levels by immunoblotting. In response to doxycycline, the ratio of cell surface to total TROP2 increased significantly (9.3x, p < 0.05) in TetON-hFWE4 but not in control TetON-GFP cells (Fig. 5d), while vehicle-treated controls exhibited no difference in this ratio. In a complementary approach, we generated N/TERT-2G cells expressing both SNAP-tagged TROP2 and an inducible hFWE4-EGFP transgene. Live confocal imaging revealed significant colocalization of JFX650-labeled TROP2 with hFWE4-EGFP positive vesicles, further suggesting that hFWE4-containing vesicles physically traffic TROP2 to the surface (Fig. 5e).
hFWE4 potentiates Ca2+ release from intracellular stores to facilitate trafficking of LB cargo in differentiated keratinocytes
Seminal work suggested that the Drosophila ortholog of hFWE4 multimerizes to form a Ca2+-permeable ion channel that mediates transport of neurotransmitter at the presynaptic terminal bouton22. While this direct Ca2+ channeling function has been debated47, work done in murine T-lymphocytes also demonstrated that endocytic defects caused by FWE KO can be rescued by extracellular Ca2+ supplementation, suggesting that FWE may regulate vesicular trafficking by influencing local Ca2+ handling24. Given the critical importance of Ca2+ in the final stages of keratinocyte differentiation32,48, we hypothesized that hFWE4-mediated trafficking during late-stage keratinocyte differentiation may be similarly reliant on alterations in cytosolic Ca2+. To determine whether hFWE4 expression altered cytosolic calcium levels in epidermal keratinocytes, bi- and tri-cistronic expression vectors allowing either inducible expression of a nuclear localized mCherry with cytosolic GCaMP6s alone or co-expressed with hFWE4 were stably introduced into N/TERT-2G via lentivirus (Fig. 6a). Following doxycycline induction, live imaging revealed that cells co-expressing GCaMP6s and hFWE4 exhibited greater elevation of GCaMP6s signal in response to thapsigargin treatment and subsequent extracellular calcium addition than cells expressing GCaMP6s alone (Fig. 6b–d). To further interrogate the source of Ca2+ release potentiated by hFWE4 expression, we pulsed cells with either thapsigargin or the lysosomotropic agent Glycyl-L-phenylalanine 2-naphthylamide (GPN) in nominal Ca2+ free media. In both of these experimental paradigms, hFWE4 expressing cells exhibited more robust Ca2+ responses, with GPN provoking a more robust response than thapsigargin (Supplementary Fig. 9–D). Non-selective release of intracellular stores by treatment with ionomycin and EGTA in nominal Ca2+ free media also produced a more significant Ca2+ response in hFWE4 expressing cells than in controls, and produced the largest response amongst treatments within the hFWE4 expressing population (Supplementary Fig. 9A–D).

a Schematic of bi- and tri-cistronic lentiviral vectors used to stably coexpress a nuclear-localized mCherry indicator with GCaMP6s alone or with GCaMP6s and hFWE4 in N/TERT-2G cells. Created in BioRender. Rudd, J. (2025) https://BioRender.com/gtefyvp. Immunoblot validation shows inducible expression of hFWE4-3XFLAG in N/TERT-2G cells. b Representative spinning disc confocal imaging of GCaMP6s in control (n = 120 cells across 24 independent experiments) or hFWE4 overexpressing (n = 193 cells across 18 independent experiments) cells following 1 µM thapsigargin treatment and 1.4 mM CaCl2 supplementation. Snapshots from representative movies show intensity profiles of GCaMP6s at indicated timepoints before and after treatments. Scale bars, 10 µm. Max fold change in GCaMP6s intensity in response to thapsigargin (c) and extracellular Ca2+ supplementation (d) are shown. Mean fold change is represented by dashed lines in (d) (**** = p < 0.0001, unpaired two-tailed t test). e Immunoblotting for TROP2 using 300 µg cell surface protein and 10 µg total protein from doxycycline-treated TetON-GFP and TetON-hFWE4 (n = 2) cell cultures that were loaded with either 40 µM BAPTA-AM or DMSO prior to switching to 1.4 mM CaCl2 for 1, 2, or 4 d differentiation. D4 differentiated cells were reloaded with BAPTA or DMSO after 48 h of differentiation. Densitometry quantification under each panel reflects intensity values relative to the DMSO-treated control for each genetic condition. For inputs, tubulin normalized intensities were used. Values are the average of duplicate samples for each experimental group.
To determine whether this effect on cytosolic Ca2+ was responsible for hFWE4-mediated transport of LB cargo such as TROP2 to the cell surface, TetON-GFP and TetON-hFWE4 cells were pretreated with the cell-permeant Ca2+ chelator, BAPTA-AM, or vehicle alone prior to differentiation. Immunoblotting for TROP2 in affinity-purified cell surface fractions after one, two, or four days of differentiation in doxycycline-containing media demonstrated that BAPTA treatment significantly reduced hFWE4-mediated TROP-2 surface presentation at all timepoints, while there was no equivalent effect of BAPTA in TetON-GFP cells. Intriguingly, total levels of TROP2 were significantly reduced in TetON-hFWE4 cells treated with BAPTA-AM (Fig. 6e), suggesting that impaired calcium- and hFWE4-dependent plasma membrane delivery of TROP2 may result in its routing into a degradative compartment.
In the course of performing experiments using TetON-GFP and TetON-hFWE4 expressing cells, a proliferation deficit was observed in TetON-hFWE4 when induced in undifferentiated conditions. To determine whether this phenotype was similarly dependent on elevation of cytosolic Ca2+, which can trigger terminal keratinocyte differentiation, in response to hFWE4 expression, cell cycle profiles were first assayed in response to 24 h of vehicle or doxycycline treatment in 0.4 mM Ca2+ conditions revealing a small, but significant G1-arrested population in uninduced TetON-hFWE4 cells (likely a result of leaky construct expression (Supplementary Fig. 6B, C)) that was significantly increased in response to doxycycline treatment (Supplementary Fig. 10A). To determine whether the G1-arrest was Ca2+ dependent, cells were preloaded with 40 µM BAPTA-AM or DMSO prior to 24 h of vehicle or doxycycline treatment. Following BAPTA treatment to chelate intracellular calcium, there was a complete attenuation of G1-arrest in uninduced TetON-hFWE4 cells and partial attenuation in induced cells. Importantly, BAPTA treatment had no effect on the cell cycle profile in TetON-GFP cells (Supplementary Fig. 10B). These data suggest that in addition to facilitating surface trafficking, the elevation of cytosolic Ca2+ induced by hFWE4 expression is sufficient to elicit G1-arrest. To determine whether this cell cycle effect was sufficient to elicit keratinocyte differentiation in a more physiologically relevant model, we generated epidermal organoids comprised of ten percent TetON-GFP or TetON-hFWE4 and induced expression throughout stratification. Imaging of wholemount organoids revealed that TetON-hFWE4 cells were predominantly found in suprabasal layers while TetON-GFP cells maintained a larger population in the basal progenitor layer (Supplementary Fig. 10C). Collectively, these data indicated that ectopic expression of hFWE4 promotes differentiation of epidermal keratinocytes.
FWE-positive LBs are mislocalized in cornification disorders driven by impaired cytosolic Ca2+ handling
Proper execution of end-stage terminal differentiation and cornification in the epidermis is largely governed by calcium-dependent signal transduction. The critical role of calcium homeostasis in the epidermis is well illustrated by disorders of cornification, such as Darier disease (DD), which is caused by a germline mutation in the ER calcium reuptake pump, SERCA2 (encoded by ATP2A2)49. Impaired cytosolic calcium handling in DD keratinocytes prevents normal desmosome trafficking, leading to loss of keratinocyte cohesion (acantholysis) in suprabasal layers and accelerates differentiation to produce prematurely cornified cells known as ‘corps ronds’ and ‘grains’ that are typically found in the upper epidermal layers. Recent work has also demonstrated that Grover disease (GD), which shares identical histological features with DD, can be driven by somatic mutations in ATP2A2 50. Given the data presented here that describe FWE as a new regulator of calcium-dependent polarized LB trafficking during cornification, we hypothesized that expression or subcellular localization of FWE might be dysregulated in these diseases.
Supporting this hypothesis, reanalysis of bulk RNA-seq data from recent work characterizing transcriptomic alterations in skin biopsies from patients with DD and GD51 revealed a significant increase in FWE expression in both lesion types relative to normal skin (Fig. 7a). FWE immunofluorescence performed on clinical samples from Grover’s and Darier patients (N = 3) as well as normal human skin (N = 5) demonstrated that, relative to normal skin, the apically polarized FWE signal was expanded downward into additional spinous layers in acanthotic regions of the epidermis (Fig. 7b). Interestingly, the most striking increase in FWE signal intensity was observed in dyskeratotic foci characterized by numerous ‘corps ronds’ and grains on H&E-stained tissue sections (Fig. 7b, arrows). Cells in these regions displayed a marked loss of the polarized distribution of FWE, which often encased the entire plasma membrane (Fig. 7b, arrows) or exhibited aberrant basal or lateral polarity (Fig. 7b, arrowheads). Double immunofluorescence for CDSN and FWE in DD disease suggested that these alterations in FWE signal likely reflect an accumulation and loss of polarity of FWE-associated LBs as there was markedly increased intensity of both proteins in lesional areas of epidermis (Fig. 7c). Airyscan2 super resolution imaging of dyskeratotic foci in DD samples showed co-occurrence of FWE and CDSN in ‘corps ronds’ with both proteins lacking apparent subcellular polarity, suggesting a failure of polarized delivery of LBs to the cell surface (Fig. 7d). Collectively, these findings suggest that dysregulation of FWE expression and aberrant localization of FWE-positive LBs occur in dyskeratotic regions of DD and GD lesions.

a hFWE expression retrieved from bulk RNA-seq of normal skin (n = 4) and Darier (n = 11), Grover (n = 10), or Hailey-Hailey disease (n = 7) from GSE233000 (* = DESeq2 p-adj < 0.05). b Representative FWE immunofluorescence performed on normal (n = 5), Grover (n = 3) and Darier (n = 3) patient samples. Following fluorescent imaging, H&Es were performed on the same section to allow co-registration of FWE signal with histopathology. Arrows show significant elevation of FWE signal in corps ronds and grains of dyskeratotic foci, arrowheads show examples of cells with improper FWE polarization. c Representative double immunofluorescence for FWE and CDSN on Darier skin biopsies (n = 3 biologically independent samples) showing significant colocalization in dyskeratotic foci. Arrows indicate representative corps ronds imaged using Airyscan2 super resolution confocal in (d) showing accumulation of double positive vesicles around the entire cell periphery. Scale bars, (b, c) 100 µm, (d) 10 µm (full image), 1 µm (inset).
Discussion
Lysosome-related organelles (LROs) are responsible for the exocytosis of diverse secretory cargo across many cell types. This includes keratinocytes found in the stratum granulosum (SG) of mammalian epidermis, where apical extrusion of lamellar bodies (LBs) is essential for establishing barrier function. As this region of the epidermis is not easily modeled in monolayer culture, the molecular events that facilitate the polarized secretion of these vesicles are poorly understood. However, their exquisite sensitivity to perturbations in extracellular and cytosolic Ca2+ is well appreciated13. Our results provide new insight into this process by demonstrating that hFWE4, a small integral membrane protein with no prior documented function in normal skin, localizes to epidermal LBs and, in differentiated keratinocytes, facilitates release of intracellular Ca2+ stores to facilitate delivery of LB cargo to the cell surface. Our proteomic and image-based analysis of hFWE4-positive, LB-like vesicles in terminally differentiated keratinocytes identifies cargoes specific to this specialized keratinocyte LRO, but also importantly highlights that there is significant redundancy between the trafficking machinery associated with LBs and with LROs found in a wide range of tissues. hFWE4 and its orthologues have been demonstrated to regulate Ca2+-dependent transport of LROs in presynaptic neurons22, endocytic retrieval of LRO-derived cargo in cytotoxic T-lymphocytes24, and, now, apically-directed secretion of an LRO in terminally differentiated epidermal keratinocytes. Thus, we hypothesize that hFWE4 is a core component of the LRO transport machinery and likely plays a functional role in a number of specialized secretory cell types. The data presented here provide a foundation for interrogating the biology of hFWE4-mediated LRO trafficking across tissues.
Supporting this point, we have identified VAMP3, STXBP3, STX4, and NAPA as SNARE components associated with hFWE4-positive vesicles and demonstrate that in human epidermis, both VAMP3 and STXBP3 are partially colocalized to CDSN containing LBs. From our data, it remains unclear whether these molecules directly interact during LB fusion events. Future work is required to determine whether these molecules are necessary for hFWE4-dependent LB trafficking and to dissect the mechanisms by which hFWE4 might regulate their behavior during this process. As interaction of STXBP3 and STX4 regulates fusion events for many specialized vesicles, including GLUT4 storage vesicles (GSVs) in adipocytes and skeletal muscle52 and zymogen granules in pancreatic acinar cells53, we hypothesize that this process is also relevant to LB biology and that FWE may perform an important regulatory function. These associations with SNARE components are particularly noteworthy in skin as mutations in other SNARE encoding genes, including SNAP29 54 and VPS33B55,56 cause rare neuroichthyotic syndromes (CEDNIK syndrome and ARC syndrome) that manifest with impaired LB secretion leading to improper cornification. Thus, hFWE4 and the LB-associated SNARE components reported here could be involved in ichthyosis pathogenesis, including genetic ichthyoses of unknown origin.
While hFWE4 is involved in direct transport of LROs and endocytic retrieval of LRO-derived cargo, our work adds to the body of literature suggesting that its precise function in LRO biology differs across tissue types. Here we show that instead of the endocytic role described for its orthologues in presynaptic neurons and cytotoxic T-lymphocytes (CTLs), in the SG of epidermis, hFWE4 controls apically polarized exocytosis of epidermal LBs, a process vital for epidermal barrier formation. We hypothesize that this discrepancy in endo- versus exocytic function across cell types is in part dependent on tissue-specific differences in cargo contained within the lumen of hFWE4-positive vesicles. LBs released by SG1 and SG2 keratinocytes contain a lipid-rich proteinaceous matrix and fuse to the apical membrane, which eventually undergoes extensive cross-linking to form the rigid cornified envelope. Collectively, these factors may slow exocytic release of LBs, effectively preventing endocytic retrieval, leading to exclusion of hFWE4 from the early endosomal compartment in these cells. This kind of delayed exocytic release impeding subsequent endocytosis occurs during the secretion of LBs in alveolar type II epithelial cells18,57. However, it is not known whether such a mechanism is at play in epidermal LBs.
We documented an elevation in cytosolic Ca2+ upon mobilization of intracellular stores in hFWE4-expressing cells and Ca2+-dependent hFWE-mediated trafficking of TROP2 in keratinocytes. However, the precise molecular mechanisms by which hFWE4 confers Ca2+ dependency to vesicular transport in epidermal keratinocytes is unclear. One hypothesis compatible with our data is that hFWE4 itself acts as a Ca2+-permeable channel that releases Ca2+ from the lumen of hFWE4-positive vesicles into the surrounding cytosol. This would be consistent with our data demonstrating hFWE4-dependent cytosolic Ca2+ elevation in response to thapsigargin and GPN, as acidic organelles like LROs are sensitive to these agents58,59,60. While we find that these responses occur in nominal Ca2+ free media, suggesting that intracellular stores are those predominantly affected, it remains possible that a small pool of plasma-membrane associated hFWE4 facilitates entry of extracellular Ca2+ into the cytosol. Our data show that hFWE4-expressing cells exhibit larger cytosolic Ca2+ elevation in response to extracellular Ca2+ addition after ER depletion, which could be explained by plasma-membrane-associated hFWE4 facilitating store-operated calcium entry (SOCE) via an unknown mechanism.
While direct Ca2+ channel functionality was initially attributed to the Drosophila ortholog of hFWE422, this conclusion has been contested47 and direct evidence for channeling through hFWE4 orthologues remains limited, largely because the membranes of small intracellular vesicles are not easily amenable to patch-clamp methods. Importantly, our data are also compatible with a model in which hFWE4 indirectly facilitates Ca2+ release from vesicular stores by modulating the activity of unknown channel proteins. In either circumstance, hFWE4-positive vesicles may serve as a Ca2+ reservoir that when released, could recruit Ca2+-dependent adapter proteins to facilitate polarized vesicular transport and/or facilitate SNARE complex assembly at the target membrane by activating requisite Ca2+ sensors. Similar mechanisms have been demonstrated to regulate exocytosis of lysosomes and lysosome related organelles (LROs) in other tissues15,61. Regardless of the mechanism, one outstanding question posed by our work is whether hFWE4-positive LBs in human epidermis contribute to the global elevation of cytosolic Ca2+ levels in the SG that triggers the final stage of cornification, or whether these vesicles only transiently elevate local Ca2+ levels for purposes of vesicular transport regulation. In addition, given our findings that ectopic hFWE4 expression facilitates cell-cycle arrest and differentiation in basal keratinocytes, future work may seek to investigate whether hFWE4-dependent Ca2+ elevation contributes to differentiation commitment in this population. It is possible that such an effect might exert additional FWE-mediated influence on cutaneous barrier formation.
Proteomic and imaging analysis demonstrated that hFWE4-positive vesicles in differentiated keratinocytes carry many putative LB cargo, including lipid regulators linked to cornification and junctional proteins essential for Ca2+-dependent TJ formation. This reflects the heterogeneity of epidermal LBs, which are believed to originate from the trans-Golgi network (TGN)33 before subpopulations are loaded with spatially segregated cargo through an unknown mechanism prior to secretion7,8,62. Our data strongly support the notion that LBs arise from the TGN, revealing that hFWE4-positive LB-like vesicles are highly enriched for proteins that annotate to this compartment. As a prototypical cargo for mechanistic studies, we focused on TROP2, a cell surface glycoprotein and TJ component, that is trafficked to the cell surface in hFWE4-positive vesicles and had previously been identified in the LB fraction of human epidermis39. While LB secretion and TJ biogenesis/turnover are spatially and temporally linked in SG211,63,64, the relationship between these processes is not well understood. As we also identified proteases such as SPINT1 and ADAM family members that influence skin barrier function by regulating ectodomain shedding and intramembrane proteolysis of TJ components such as claudins and TROP246, it is possible that hFWE4-positive LBs assemble the requisite machinery for TJ assembly and turnover at the plasma membrane.
Finally, we present data suggesting disease relevance of hFWE4 function in skin, showing that mislocalized FWE-associated LBs accumulate in prematurely cornified keratinocytes that typify two epidermal barrier disorders previously linked to abnormal Ca2+ handling, Darier (DD) and Grover (GD) disease. While the Ca2+ handling abnormalities that drive these epidermal diseases are thought to result from dysregulation of ER stores downstream of SERCA2 mutation49,65, it is plausible that the abundance of FWE-positive LBs in these cells could act as additional Ca2+ stores whose release further accelerates inappropriate cornification. Accumulation of LBs in ‘corps ronds’, which were then called Odland bodies66, was noted over sixty years ago in EM studies of Darier disease performed by Caulfield and Wingram67 and later by Peck et al.68. However, there has been little follow-up on these findings. In light of the data presented here, future studies may investigate whether targeting FWE expression or function to normalize LB secretion and restore proper cornification is a valuable therapeutic strategy in DD, GD, or other skin barrier disorders in need of novel treatments.
Methods
Cell culture
N/TERT-2G69 a gift from James Rheinwald, Brigham and Women’s Hospital, Boston, MA, USA) and NHEK (Lonza 00192906) were maintained in EpiLife (Gibco MEPI500CA) medium supplemented with 1x HKGS (Gibco S0015), 1% pen-strep (Gibco 15070063), and brought to 0.4 mM CaCl2. HEK293FT cells (Invitrogen R70007) for lentiviral packaging were maintained in high-glucose DMEM supplemented with L-glutamine and sodium pyruvate (Gibco 1195065), 10% FBS (Sigma 0926), and 1% pen-strep (Gibco 14130122). All cells were grown in an air-jacketed humidified incubator at 37 oC, 5% CO2.
For differentiation in submerged culture, 100% confluent N/TERT-2G or NHEK were switched to fully supplemented EpiLife medium containing 1.4 mM CaCl2. Cells were refed daily with this medium post-confluence until the desired endpoint was reached.
CRISPR/Cas9 Editing and clonal cell line generation
CLDN1 KO N/TERT-2G keratinocytes were a kind gift from Matthew Brewer, Rochester University Medical Center, Rochester, NY, USA. For cell lines generated in house, CRISPR/Cas9 editing of N/TERT-2G cells was performed using transient electroporation of purified Cas9:sgRNA RNPs as described in detail70. Briefly, for KO of hFWE, 600 ng of a modified sgRNA (Synthego CRISPRevolution sgRNA EZ Kit) targeting exon 1 of hFWE (5’-GCAUGACGUGGUGGUACCGC-3’) was complexed with 3ug Alt-R Cas9 V3 (IDT 1081058) in vitro. Where indicated, a scrambled negative control sgRNA (Synthego, Negative Control, Scrambled sgRNA#1, mod-sgRNA) was complexed to Alt-R Cas9 V3. The assembled RNP complex was subsequently electroporated into 2 × 105 N/TERT-2G using the NEON transection system to deliver 1 pulse of 1700 V for 20 ms. Cells were then seeded in fully supplemented EpiLife medium.
For KI of a 3XFLAG tag into the endogenous hFWE locus, a similar strategy was used. Briefly, 300 ng of an Alt-R modified sgRNA (IDT) targeting the 3’ end of the exon 6b coding sequence (5’-AGGGGGAGCUGUGAAGGGCU-3’) was complexed with 3 µg Alt-R Cas9 V3 (IDT 1081058) in vitro. Prior to electroporation, 0.45 µg of a ~ 150 bp ssODN (IDT) encoding 5’ and 3’ homology arms flanking a 3XFLAG-AUG was added to the mixture. Cells were electroporated under conditions described above and seeded into fully supplemented EpiLife medium containing 1 µM Alt-R HDR Enhancer V2 (IDT 10007910). Cells were refed with enhancer-free EpiLife medium 16 h after electroporation.
Sanger sequencing (Genewiz) of target regions was performed on gDNA isolated from polycloncal cell pools to estimate editing efficiency prior to limiting dilution based clonal isolation. Briefly, the edited polyclonal cell pools were seeded at 1 cell/well in 96-well tissue culture plates and expanded for cryopreservation and gDNA isolation. PCR amplicons of genomic regions targeted during editing were Sanger sequenced (Genewiz), and editing relative to the wild-type locus was analyzed using interference of CRISPR edits (ICE, Synthego).
sgRNA and ssODN sequences used for editing can be found in Supplementary Table 1. Primer sequences used for genotyping of exon 1 and exon 6b can be found in Supplementary Table 2.
Lentiviral production and generation of stable cell lines
Lentiviral particles were packaged in HEK293FT by lipofection (Lipofectamine LTX Plus) of psPAX2 (Addgene 12260), pMD2.G (Addgene 12259), and transfer vector. Lentiviral supernatants were concentrated to 100x using Lenti-X concentrator (Takara cat), and N/TERT-2G were infected in suspension in 0.5 μg/ml polybrene. Infected cells were selected in 3 μg/ml blasticidin and maintained either as a stable polyclonal pool or subject to limiting dilution cloning as described for CRISPR/Cas9 engineered cell lines.
Epidermal organoid culture
Epidermal organoid cultures were performed as previously described70 with minor adjustments. In short, cell culture inserts in a 24 wells carrier plate (Nunc, Thermo Fisher Scientific, 141002) were coated using 100 µg/mL rat tail collagen (Sigma–Aldrich, C3867) for 1 h at 4 °C. After phosphate–buffered saline (PBS) washing the filters, 150,000 cells were seeded and submerged in CnT–prime medium (CELLnTEC, CnT–PR) at the lowest insert stand. After 72 h, the medium was switched to differentiation medium (40% Dulbecco’s modified Eagle’s Medium (Sigma–Aldrich, D6546) and 60% 3D barrier medium (CELLnTEC, CnT–PR–3D)) and the organoids were lifted to the highest stand, air–exposed. Medium was refreshed every other day until the end of the culture (10 days) organoids were harvested by taking punch biopsies (4 mm for RNA, 4 mm for immunohistochemistry (IHC).
In select experiments, organoids were seeded with starting populations composed of 10% TetON-GFP or –hFWE4 cells and 90% WT N/TERT-2G. In these experiments, the differentiation medium used at the air-liquid interface was supplemented with 200 ng/ml doxycycline to induce expression at the onset and for the duration of stratification. Punch biopsies retrieved from these cultures after 10 days of stratification were fixed and then subject to nuclear counterstaining with NucSpot 650 (Biotium 41035), before optical clearing in FUnGI71 clearing solution for wholemount imaging.
Electrical impedance spectroscopy
Locsense Artemis (Locsense, Enschede, the Netherlands), equipped with a custom–made incubator-compatible smart lid, was used to measure the electrical impedance spectrum as previously described29. First, a PBS-only control was measured for each electrode, including device self-calibration. Meanwhile, the organoids were acclimated to room temperature and lowered to the middle position in the transwell plate. 1600 μL of PBS was added below, and 500 μL PBS was added on top of the insert. Thereafter, the smart lid was placed on the wells, ensuring both electrodes being submerged. Impedance was measured over a frequency range from 10 Hz to 100,000 Hz in 30 logarithmic intervals. The PBS-only blank measurement was subtracted from the impedance output for each electrode individually. For EISdiff (127–2212 Hz), the area under the curve was calculated using the frequency range.
RNA-seq and data analysis
Total RNA was isolated from epidermal organoids after 10 days of air-liquid interface culture using the Favorprep total tissue RNA kit (Favorgen Biotech, Tung, Taiwan) according to the manufacturer’s instructions. Library preparation, sequencing, and data analysis were performed in the Innovative Genomics and Bioinformatics Core at Creighton University (RRID:SCR_024816).
To prepare sequencing libraries, a total RNA input of 500 ng was used for library preparation with the NEBNext Ultra II Directional RNA Library Prep kit for Illumina, along with NEBNext Poly(A) mRNA Magnetic Isolation Module (both New England BioLabs Inc.) according to the manufacturer’s protocol. The library quality and quantity of RNA were assessed using RNA ScreenTape Assay (Agilent) and Agilent 4200 TapeStation System following the manufacturer’s protocol. Libraries were pooled equimolar for sequencing to a final concentration of 3 nM. The pooled libraries were sequenced on the Illumina NextSeq 2000 system (Illumina) using NextSeq 1000/2000 P2 Reagent Cartridge 100 cycles and P2 Flow Cell (Illumina).
For analysis of bulk RNA-seq data, adapters were trimmed with Trim Galore v.0.6.5, and reads were aligned to hg38 as the reference genome (GRCh38 release 45 from Gencode). STAR v. 2.7.11b72 was used for read mapping, and Salmon v.1.10.173 for transcript quantification. Differential expression analysis was carried out using DESeq2 v. 1.41.1374. Sample A11 (a technical replicate of KO clone 8) was excluded because it was identified as an outlier based on PCA analysis and Euclidean distances of all samples after variance stabilizing transformation. Genes that were not expressed in any sample were excluded from the analysis. GO and KEGG enrichment was carried out in R (v.4.3.2) using clusterProfiler (v.4.10)75. For GSEA, genes were ranked by their values in the “stat” column of the DESeq2 results. This column represents Wald test values, which are calculated by dividing the log fold change by the standard error for each gene. The cutoff for significance of the enrichment scores was p-adj ≤ 0.05.
Plasmids and molecular cloning
pCW57-MCS1-P2A-MCS2-Blast (Addgene 80921) was used as a base vector to construct all lentiviral TetON constructs. Inserts were synthesized as double-stranded gBlocks (IDT) with In-Fusion-compatible overhangs or generated via standard PCR-based amplification using Q5 High-Fidelity mastermix (NEB M0492L). pLVX-puro-GCaMP6s (Addgene 164589) was used as a PCR template for the GCaMP6s coding sequence. Relevant primer sequences can be found in Supplementary Table 2. Inserts were cloned into EcoRI/BamHI or BsrgI digested vector using In-Fusion Snap Assembly mastermix (Takara Bio 638947). Final versions of all vectors were subject to whole-plasmid sequencing by Plasmidsaurus for sequence validation. Lenti CMV TROP2-V5-SNAP encoding a C-terminal V5-SNAP fusion to the TROP2 coding sequence was synthesized and whole-plasmid sequenced by VectorBuilder.
Organelle immunoprecipitation
To isolate intact hFWE4-positive vesicles from differentiated keratinocytes for proteomic profiling, TetON-GFP and TetON-hFWE4 N/TERT-2G were grown to 100% confluence in 15 cm dishes (N = 5) and switched into 1.4 mM CaCl2 EpiLife containing 200 ng/ml doxycycline. After four days of differentiation with daily refeeding, cell sheets were subject to non-denaturing lysis and rapid immuno-isolation using techniques similar to those described elsewhere40,41,42,43. Briefly, cell sheets were washed 1x with cold PBS, then scraped on ice and transferred into 1.2 ml cold KPBS buffer (25 mM KCL, 100 mM potassium phosphate, pH 7.2) with protease and phosphatase inhibitor (HALT, Pierce) on ice. Cells were mechanically homogenized with 25 strokes of a Tenbrock tissue grinder and subsequently homogenized with five passes through a 22 G needle. Lysed cells were centrifuged at 1000 × g for 5 m at 4 C to pellet nuclei, and a 100 μL aliquot of post-nuclear supernatant was frozen as input while 1.1 ml was added to 100 μL of prewashed anti-DYKDDDK magnetic agarose (Pierce A36797) and rotated for 50 m at 4 C for immunoprecipitation. Beads were washed three times by rotating in cold KPBS and subsequently eluted for 30 m with 1% NP-40 in cold KPBS with inhibitors. Eluates were aliquoted and frozen for label-free quantitative mass spectrometry (LFQ-MS) and immunoblotting. For immunoblotting, protein estimation was performed on input samples, and loading volumes of input and IPs were adjusted to ensure equal starting material.
Cell surface biotinylation
To isolate surface protein from differentiated keratinocytes for proteomic profiling, TetON-GFP and TetON-hFWE4 N/TERT-2G were grown to 100% confluence and switched into 1.4 mM CaCl2 EpiLife containing 200 ng/ml doxycycline. After the differentiation time point was reached, cell sheets were subject to surface biotinylation as described in ref. 25 with slight modifications. Briefly, cells were washed with cold PBS and then incubated on ice in 0.5 mg/ml EZ link sulfo NHS ss biotin (Thermo Fisher #21331) for 30 m. Unreacted biotin was quenched three times in cold 50 mM glycine, and cells were subsequently sonicated in cold lysis buffer (1% Triton-X 100, 150 mM NaCl, 1 mM EDTA) with protease and phosphatase inhibitor cocktail. Clarified lysates were kept at -80C until processing. For LFQ-MS experiments, 5 mg whole cell lysate per sample (N = 5) was incubated at 4 C overnight with 250 μl High Capacity NeutrAvidin resin (Pierce 29202). Resin was washed three times for 5 m each at 4 C in cold lysis buffer, then eluted twice in 6 M urea, 100 mM DTT at 65 C for 15 m. For smaller-scale immunoblotting experiments, 100 μg lysates were incubated at 4 C overnight with 30 μl NeutrAvidin resin (Pierce 29200). Resin was washed as described above, then eluted in 2X LAEMMLI buffer. For immunoblotting assays, protein quantities indicated in the figure legends for input and pulldowns were loaded on an SDS-PAGE gel to compare surface:total protein ratio. BAPTA loading was performed where indicated in surface biotinylation experiments by incubating cells in 0.08% Pluronic with 40 µM BAPTA-AM (AAT Bioquest 126150-97-8) or DMSO in PBS (Gibco 10010023) for 30 m at 37 C. Cells were washed twice in PBS and refed with 1.4 mM CaCl2 EpiLife.
LFQ-MS and proteomic data analysis
LFQ-MS proteomics experiments were performed at the UNMC proteomics core facility using standard protocols. The protein concentration was estimated in each sample using the BCA Protein Assay Kit (Pierce). 100 μg of proteins from each sample was diluted to 100 μL volume with 100 mM ammonium bicarbonate (ambic). Proteins were reduced with 5 μL of 200 mM tris(2-carboxyethyl) phosphine (TCEP) (1 h incubation, 55 ⁰C) and alkylated with 5 μL of 375 mM iodoacetamide (IAA) (30 min incubation in the dark, room temperature). The reduced and alkylated proteins were purified with acetone precipitation at − 20 ⁰C overnight. The next day, protein precipitates were collected by centrifugation at 8000 × g for 10 min at 4 ⁰C and pellets were briefly air-dried and resuspended in 100 μL of 50 mM ambic. The protein digestion was carried out using 2.5 μg of trypsin per sample (16 h incubation, 37 ⁰C). The next day, samples were dried out using a speed vacuum and then desalted with C18 spin columns (Pierce). Clean peptides were dried out again with speed vacuum and resuspended in 0.1% formic acid, and next analyzed using a high-resolution mass spectrometry nano-LC-MS/MS Tribrid system, Orbitrap Fusion™ Lumos™ coupled with an UltiMate 3000 HPLC system (Thermo Scientific).
1.5 μg of each sample was loaded onto trap column Acclaim PepMap 100 (75 μm × 2 cm C18 LC Columns, Thermo Fisher Scientific) at a flow rate of 4 μl/min, and next separated with a Thermo RSLC Ultimate 3000 (Thermo Fisher Scientific) on a Thermo Easy-Spray PepMap RSLC C18 column (75 μm × 50 cm C-18 2 μm, Thermo Fisher Scientific) at a flow rate 0.3 μl/min and 50 °C, with a step gradient of 9%–25% solvent B (0.1% FA in 80% acetonitrile) from 10–15 min and 25%–40% solvent B from 15–40 min, with a 70 min total run time. The MS scan was done using detector: Orbitrap resolution 120000; scan range 350–1800 m/z; RF lens 30%; AGC target 4.0 e5; maximum injection time 100 ms. The most intense ions with charge states 2–6 isolated in 3 s cycles were selected in the MS scan for further fragmentation. MS2 scan parameters set: activation HCD with 35% normalized collision energy, detected at a mass resolution of 30000. The AGC target for MS/MS was set at 5.0 e4, and ion filling time set to 60 ms.
Protein identification was performed by searching MS/MS data against the Uniprot database (selected for Human) in Proteome Discoverer (Thermo Fisher Sci, vs 3.0.), assuming the digestion enzyme trypsin. The parameters for Sequest HT were set as follows: Enzyme: trypsin, Max missed cleavage: 2, Precursor mass tolerance: 10 ppm, Peptide tolerance: ± 0.02 Da, Fixed modifications: carbamidomethyl (C); Dynamic modifications: oxidation (M), acetyl (N-term). The parameters for the Precursor ions quantifier were set as follows: peptides to use unique + razor, precursor abundance based on intensity; normalization mode: total peptide amount; scaling mode: on all average. Raw LFQ intensities were further processed and analyzed for differential protein abundance in the Perseus Computational Platform76. Briefly, LFQ intensities were log2 transformed and filtered to exclude proteins with > 50% missing values. Missing values were replaced from a normal distribution (width 0.3, down shift 1.8), and two-sided t tests were used to compare groups (permutation-based FDR < 0.05 for Organelle IP, FDR < 0.1 for surface proteomics).
For subcellular annotation of the Flower-IP dataset, consensus graph based annotation for subcellular compartments from ref. 43 was downloaded (https://organelles.czbiohub.org/Subcellular_UMAP) and used to classify Uniprot IDs. For analysis of gene set overlap between two independent datasets, one tailed Fisher’s exact test of the hypergeometric distribution was performed using the complete human proteome as the population size.
Antibodies
Primary antibodies used for immunofluorescence, immunohistochemistry, and immunoblotting assays can be found in Supplementary Table 3.
Immunofluorescence, Immunohistochemistry and Fluorescent Ceramide labeling
Human skin was collected from deidentified, discarded surgical specimens and FFPE processed using standard techniques. Epidermal organoids were excised as 4 mm biopsies and processed for FFPE using standard techniques. For immunofluorescence assays on human skin or epidermal organoids, standard protocols were used, including citrate-based antigen retrieval (VectorLabs) prior to blocking and antibody incubation in 1% BSA/5% goat serum/0.3% TritonX-100. Species-specific fluorophore-conjugated secondary antibodies were used for detection, and nuclei were counterstained with Hoechst 33342 before mounting coverslips with Vectashield Antifade mounting medium (VectorLabs). For immunocytofluorescence assays, cells were grown on collagen coated glass bottom 96 well plates (Cellvis P96-1.5H-N) under indicated conditions, fixed in 10%NBF, permeabilized with 0.3% TritonX-100, and subject to immunostaining against indicated targets using standard techniques. Nuclei were counterstained with Hoechst 33342 before imaging.
Standard LSM confocal and Airyscan2 superresolution confocal imaging were performed on immunostained or otherwise fluorescently labeled human skin, organoid specimen, or cultured cells using a Zeiss LSM980 with Airyscan2. For all experiments, 405, 488, 561, and 633 nm laser lines and a Plan-Apochromat 63x/1.4NA objective were used. All Airyscan2 data was deconvolved using Airyscan joint deconvolution (jDCV) in Zen Blue. Standard LSM confocal images were subject to median or Gaussian filtering in Zen Blue, applied identically to all images within a given experiment. Orthogonal projections of maximum intensity for Airyscan2 and LSM confocal micrographs are presented unless otherwise specified. Mander’s colocaliziation coefficients in Airyscan2 micrographs were quantified using the JaCOP Fijij PlugIn77 Analysis was restricted to cell layers expressing both markers of interest, and thresholds were generated using single color controls.
For immunohistochemistry of epidermal organoids, sections were blocked for 15 min with 5% serum in PBS and subsequently incubated with primary antibody against the protein of interest for 1 h at room temperature. Next, a 30 min incubation step with biotinylated secondary antibody (Vector Laboratories, Burlingame, CA) was performed, followed by a 30 min incubation with avidin-biotin complex (Vector Laboratories). The peroxidase activity of 3-amino-9-ethylcarbazole was used to visualize the protein expression, and the sections were mounted using glycerol gelatin (Sigma-Aldrich). For routine histology, deparaffinized sections were stained with hematoxylin (Klinipath, 4085.9005) and eosin (Klinipath, 4082.9002).
RT-qPCR
Total RNA was isolated from cultured cells using Qiagen RNeasy mini (Qiagen 74104), and cDNA was synthesized using SuperScript III First-Strand Synthesis System (Invitrogen 18080051) according to the manufacturers instructions. Actin and hFWE2/4 qPCR was subsequently performed on cDNAs using SsoAdvanced Universal SYBR Green Supermix (BioRad 1725270) and a BioRad CFX384 Touch Real-Time according to manufacturers instructions. Primer sequences used for RT-qPCR are listed in Supplementary Table 2.
Cell cycle analysis
For cell cycle assays, subconfluent N/TERT-2G were induced to express the indicated transgene with 200 ng/ml doxycycline for 24 h in 0.4 mM CaCl2 EpiLife media. Assays were performed using the Click-IT EdU Alex Fluor 647 Flow Cytometry Assay Kit (Life Technologies C10419). Briefly, cells were pulsed with 1 µM EdU for 1.5hm, then trypsinized for fixation, permeabilization, and detection following the manufacturer’s protocol. Total DNA content was labeled with FxCycle Violet Ready Flow Reagent (Invitrogen R37166) prior to analysis on a Yeti flow cytometer. More than 20,000 events were analyzed per condition in FlowJo. Representative gating strategy for cell cycle experiments is presented in Supplementary Fig. 3C.
Live cell imaging
Live cell imaging experiments were performed on a Nikon TiE equipped with a Yokagawa CSU-X1 spinning disc, Hamamatsu Orca Flash camera, and Nikon Perfect Focus System (PFS) using 488, 561, and 647 nm laser lines, and either a Plan Apo λ 100x/1.45NA or S Fluor 40x/1.3 oil objective were used for imaging. Some live cell imaging experiments were performed on a Zeiss LSM980 with Airyscan2 using 405, 488, 561, and 633 nm laser lines and a Plan-Apochromat 40x/1.3NA or 63x/1.4NA objective. Cells for live imaging were grown on either collagen-coated glass bottom 96-well plates (Cellvis P96-1.5H-N) or collagen-coated ibidi 8-well 1.5 glass bottom chamber slides (ibidi 80807) and were maintained in a stagetop incubator at 37 C, 5% CO2 during imaging.
For experiments involving live two-color imaging of TROP2-V5-SNAP and hFWE4-EGFP, stable N/TERT-2G cells were induced to express 3xNLS mCherry 2 A hFWE4-EGFP for 24 h, then treated with 5 µM JFX650 in 1.4 mM CaCl2 EpiLife medium for 30 m at 37 C, washed, and imaged in 1.4 mM CaCl2 EpiLife. Sequential acquisition of EGFP (488 nm, 200 ms exposure) and JFX650 (647 nm, 200 ms exposure) were performed as a timelapse with a frame rate of 1 s.
For experiments involving live three-color imaging of 3xNLS mCherry, hFWE4-EGFP, and C6-NBD-Ceramide, stable N/TERT-2G cells were induced to express 3xNLS mCherry 2 A hFWE4-EGFP for 24 h after 72 h of differentiation. Cells were labeled on ice with 5 µM 1:1 NBD-C6-Ceramide:BSA (Invitrogen N22651) in HBSS for 30 m, before washing and incubating in warmed 1.4 mM Ca2+ EpiLife medium for 30 m prior to imaging.
For experiments involving live imaging of GCaMP6s, stable N/TERT-2G cells were induced to express the indicated construct for 24 h in 0.06 mM CaCl2 EpiLife. Alternatively, for experiments in nominal Ca2+ free media, cells were induced to express the indicated construct for 48 h in 0.06 mM CaCl2 EpiLife, and were then switched to 0 mM Ca2+ EpiLife 20 min prior to imaging. Positive cells were identified on the basis of the nuclear mCherry indicator, then timelapse imaging of the GCaMP6s signal (488 nm) was performed at indicated time intervals. Where indicated, cells were either fed with either 1 µM thapsigargin (Cell Signaling Technologies 12758) in 0.06 mM CaCl2 containing EpiLife medium and then switched to thapsigargin-free EpiLife containing 1.4 mM CaCl2, or fed with 1 µM thapsigargin, 200 µM GPN (SantaCruz Biotechnology, sc-252858), or 5 µM ionomycin (Invitrogen I24222) + 0.25 mM EGTA and imaged as described above. Single-cell intensity traces were analyzed in NIS Elements software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Bulk RNA seq data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database (GSE269871) [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE269871]. Publicly available datasets reanalyzed in this study can be found in GEO under accessions GSE127223, GSE228631, and GSE233000. LFQ-MS proteomics data generated in this study have been deposited in the MassIVE repository (MSV000095056) [https://doi.org/10.25345/C5ZP3WB6C]. Source data are provided in this paper.
References
Candi, E., Schmidt, R. & Melino, G. The cornified envelope: a model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 6, 328–340 (2005).
Simpson, C. L., Patel, D. M. & Green, K. J. Deconstructing the skin: cytoarchitectural determinants of epidermal morphogenesis. Nat. Rev. Mol. Cell Biol. 12, 565–580 (2011).
Mahanty, S. & Setty, S. R. G. Epidermal Lamellar Body Biogenesis: Insight Into the Roles of Golgi and Lysosomes. Front. Cell Dev. Biol. 9, https://doi.org/10.3389/fcell.2021.701950 (2021).
Rassner, U., Feingold, K. R., Crumrine, D. A. & Elias, P. M. Coordinate assembly of lipids and enzyme proteins into epidermal lamellar bodies. Tissue Cell 31, 489–498 (1999).
Oren, A., Ganz, T., Liu, L. & Meerloo, T. In human epidermis, beta-defensin 2 is packaged in lamellar bodies. Exp. Mol. Pathol. 74, 180–182 (2003).
Braff, M. H., Nardo, A. D. & Gallo, R. L. Keratinocytes store the antimicrobial peptide Cathelicidin in Lamellar Bodies. J. Invest. Dermatol. 124, 394–400 (2005).
Ishida-Yamamoto, A. et al. LEKTI is localized in lamellar granules, separated from KLK5 and KLK7, and is secreted in the extracellular spaces of the superficial stratum granulosum. J. Invest. Dermatol 124, 360–366 (2005).
Ishida-Yamamoto, A. et al. Epidermal lamellar granules transport different cargoes as distinct aggregates. J. Invest. Dermatol. 122, 1137–1144 (2004).
Furuse, M. et al. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1–deficient mice. J. Cell Biol. 156, 1099–1111 (2002).
Yoshida, K. et al. Functional tight junction barrier localizes in the second layer of the stratum granulosum of human epidermis. J. Dermatol Sci. 71, 89–99 (2013).
Yokouchi, M. et al. Epidermal cell turnover across tight junctions based on Kelvin’s tetrakaidecahedron cell shape. ELife 5, e19593 (2016).
Rübsam, M. et al. E-cadherin integrates mechanotransduction and EGFR signaling to control junctional tissue polarization and tight junction positioning. Nat. Commun. 8, 1250 (2017).
Elias, P. M. et al. The secretory granular cell: the outermost granular cell as a specialized secretory cell. J. Investig. Dermatol. Symp. Proc. 3, 87–100 (1998).
Stuart, R. O. et al. Critical role for intracellular calcium in tight junction biogenesis. J. Cell Physiol. 159, 423–433 (1994).
Patel, S. & Cai, X. Evolution of acidic Ca2+ stores and their resident Ca2+-permeable channels. Cell Calcium 57, 222–230 (2015).
Faouzi, M. & Penner, R. TRPM2. in Mammalian Transient Receptor Potential (TRP) Cation Channels: Volume I (eds. Nilius, B. & Flockerzi, V.) 403–426 (Springer, Berlin, Heidelberg, 2014).
Calcraft, P. J. et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459, 596–600 (2009).
Miklavc, P. et al. Fusion-activated Ca2+ entry via vesicular P2X4 receptors promotes fusion pore opening and exocytotic content release in pneumocytes. Proc. Natl. Acad. Sci. USA 108, 14503–14508 (2011).
Qureshi, O. S., Paramasivam, A., Yu, J. C. H. & Murrell-Lagnado, R. D. Regulation of P2X4 receptors by lysosomal targeting, glycan protection and exocytosis. J. Cell Sci. 120, 3838–3849 (2007).
Miklavc, P., Frick, M., Wittekindt O. H., Haller, T. & Dietl, p. Fusion-activated Ca2+ entry: An “active zone” of elevated Ca2+ during the postfusionstage of lamellar body exocytosis in rat type II pneumocytes. PLoS ONE5, https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0010982 (2010).
Yao, C.-K., Liu, Y.-T., Lee, I.-C., Wang, Y.-T. & Wu, P.-Y. A Ca2+ channel differentially regulates Clathrin-mediated and activity-dependent bulk endocytosis. PLoS Biol. 15, e2000931 (2017).
Yao, C.-K. et al. A synaptic vesicle-associated Ca2+ channel promotes endocytosis and couples exocytosis to endocytosis. Cell 138, 947–960 (2009).
Li, T.-N. et al. A positive feedback loop between Flower and PI(4,5)P2 at periactive zones controls bulk endocytosis in Drosophila. Elife 9, e60125 (2020).
Chang, H.-F. et al. Cytotoxic granule endocytosis depends on the Flower protein. J. Cell Biol. 217, 667–683 (2018).
Rudd, J. C. et al. Membrane structure and internalization dynamics of human Flower isoforms hFWE3 and hFWE4 indicate a conserved endocytic role for hFWE4. J. Biol. Chem. 299, https://doi.org/10.1016/j.jbc.2023.104945 (2023).
Petrova, E., López-Gay, J. M., Rhiner, C. & Moreno, E. Flower-deficient mice have reduced susceptibility to skin papilloma formation. Dis. Models Mech. 5, 553–561 (2012).
Chen, X., Lloyd, S. M., Kweon, J., Gamalong, G. M. & Bao, X. Epidermal progenitors suppress GRHL3-mediated differentiation through intronic polyadenylation promoted by CPSF-HNRNPA3 collaboration. Nat. Commun. 12, 448 (2021).
Pedro, M. P. et al. GPCR Screening reveals that the metabolite receptor HCAR3 regulates epithelial proliferation, migration, and cellular respiration. J. Invest. Dermatol.144, 1311–1321 (2023).
van den Brink, N. J. M. et al. Electrical impedance spectroscopy quantifies skin barrier function in organotypic in vitro epidermis models. J. Invest. Dermatol.144, 2488–2500 (2024).
Madan, E. et al. Flower isoforms promote competitive growth in cancer. Nature 572, 260–264 (2019).
Kypriotou, M., Huber, M. & Hohl, D. The human epidermal differentiation complex: cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp. Dermatol. 21, 643–649 (2012).
Matsui, T. et al. A unique mode of keratinocyte death requires intracellular acidification. Proc. Natl. Acad. Sci. 118, e2020722118 (2021).
Yamanishi, H., Soma, T., Kishimoto, J., Hibino, T. & Ishida-Yamamoto, A. Marked Changes in Lamellar Granule and Trans-Golgi Network Structure Occur during Epidermal Keratinocyte Differentiation. J. Invest. Dermatol. 139, 352–359 (2019).
Ishida-Yamamoto, A., Kishibe, M., Takahashi, H. & Iizuka, H. Rab11 Is Associated with epidermal lamellar granules. J. Invest. Dermatol. 127, 2166–2170 (2007).
Reynier, M. et al. Rab11a is essential for lamellar body biogenesis in the human epidermis. J. Invest. Dermatol. 136, 1199–1209 (2016).
Reynier, M. et al. The Actin-Based Motor Myosin Vb Is Crucial to Maintain Epidermal Barrier Integrity. J. Invest. Dermatol. 139, 1430–1438 (2019).
Sando, G. N. et al. Caveolin expression and localization in human keratinocytes suggest a role in lamellar granule biogenesis. J. Invest. Dermatol. 120, 531–541 (2003).
Prado-Mantilla, A. & Lechler, T. Polarity in skin development and cancer. Curr. Top. Dev. Biol. 154, 317–336 (2023).
Raymond, A.-A. et al. Lamellar bodies of human epidermis: Proteomics characterization by high throughput mass spectrometry and possible involvement of CLIP-170 in their trafficking/secretion. Mol. Cell. Proteom. 7, 2151–2175 (2008).
Park, H. et al. Spatial snapshots of amyloid precursor protein intramembrane processing via early endosome proteomics. Nat. Commun. 13, 6112 (2022).
Abu-Remaileh, M. et al. Lysosomal metabolomics reveals V-ATPase and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813 (2017).
Fasimoye, R. et al. Golgi-IP, a tool for multimodal analysis of Golgi molecular content. Proc. Natl. Acad. Sci. USA 120, e2219953120 (2023).
Hein, M. Y. et al. Global organelle profiling reveals subcellular localization and remodeling at proteome scale. Cell 188, 1137–1155 (2025).
Madison, K. C. & Howard, E. J. Ceramides are transported through the Golgi apparatus in human keratinocytes in vitro. J. Invest. Dermatol. 106, 1030–1035 (1996).
Szabo, R., Ward, J. M., Artunc, F. & Bugge, T. H. EPCAM and TROP2 share a role in claudin stabilization and development of intestinal and extraintestinal epithelia in mice. Biol. Open 11, https://doi.org/10.1242/bio.059403 (2022).
Wu, C.-J., Lu, M., Feng, X., Nakato, G. & Udey, M. C. Matriptase cleaves EpCAM and TROP2 in keratinocytes, destabilizing both proteins and associated claudins. Cells 9, 1027 (2020).
Xue, L. et al. Voltage-dependent calcium channels at the plasma membrane, but not vesicular channels, couple exocytosis to endocytosis. Cell Rep. 1, 632–638 (2012).
Bikle, D. D., Xie, Z. & Tu, C.-L. Calcium regulation of keratinocyte differentiation. Expert Rev. Endocrinol. Metab. 7, 461–472 (2012).
Sakuntabhai, A. et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat. Genet. 21, 271–277 (1999).
Seli, D. et al. Association of Somatic ATP2A2 Damaging Variants With Grover Disease. JAMA Dermatol. 159, 745–749 (2023).
Roth-Carter, Q. R. et al. Transcriptional profiling of rare acantholytic disorders suggests common mechanisms of pathogenesis. JCI Insight 8, e168955 (2023).
D’Andrea-Merrins, M., Chang, L., Lam, A. D., Ernst, S. A. & Stuenkel, E. L. Munc18c Interaction with Syntaxin 4 Monomers and SNARE Complex Intermediates in GLUT4 Vesicle Trafficking*. J. Biol. Chem. 282, 16553–16566 (2007).
Dolai, S. et al. Depletion of the membrane-fusion regulator Munc18c attenuates caerulein hyperstimulation-induced pancreatitis. J. Biol. Chem. 293, 2510–2522 (2018).
Sprecher, E. et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am. J. Hum. Genet. 77, 242–251 (2005).
Rogerson, C. & Gissen, P. VPS33B and VIPAR are essential for epidermal lamellar body biogenesis and function. Biochim. Biophys. Acta 1864, 1609–1621 (2018).
Choi, H.-J., Lee, M.-W., Choi, J.-H., Moon, K.-C. & Koh, J.-K. Ichthyosis associated with ARC syndrome: ARC syndrome is one of the differential diagnoses of ichthyosis. Pediatr. Dermatol. 22, 539–542 (2005).
Haller, T. et al. Fusion pore expansion is a slow, discontinuous, and Ca2+-dependent process regulating secretion from alveolar type II cells. J. Cell Biol. 155, 279–289 (2001).
Haller, T., Völkl, H., Deetjen, P. & Dietl, P. The lysosomal Ca2+ pool in MDCK cells can be released by ins(1,4,5)P3-dependent hormones or thapsigargin but does not activate store-operated Ca2+ entry. Biochem. J. 319, 909–912 (1996).
Menteyne, A., Burdakov, A., Charpentier, G., Petersen, O. H. & Cancela, J.-M. Generation of specific Ca2+ signals from Ca2+ stores and endocytosis by differential coupling to messengers. Curr. Biol. 16, 1931–1937 (2006).
Yuan, Y. et al. The lysosomotrope GPN mobilises Ca2+ from acidic organelles. J. Cell Sci. 134, jcs256578 (2021).
Reddy, A., Caler, E. V. & Andrews, N. W. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 106, 157–169 (2001).
Zeeuwen, P. L. J. M. et al. Colocalization of cystatin M/E and cathepsin V in lamellar granules and corneodesmosomes suggests a functional role in epidermal differentiation. J. Invest. Dermatol. 127, 120–128 (2007).
Ishida-Yamamoto, A. et al. Lamellar granule secretion starts before the establishment of tight junction barrier for paracellular tracers in mammalian epidermis. PLoS ONE 7, e31641 (2012).
Kuroda, S. et al. Perturbation of lamellar granule secretion by sodium caprate implicates epidermal tight junctions in lamellar granule function. J. Dermatol. Sci. 59, 107–114 (2010).
Zaver, S. A. et al. Targeting SERCA2 in organotypic epidermis reveals MEK inhibition as a therapeutic strategy for Darier disease. JCI Insight 8, https://doi.org/10.1172/jci.insight.170739 (2023).
Odland, G. F. A submicroscopic granular component in human epidermis*. J. Invest. Dermatol. 34, 11–15 (1960).
Caulfield, J. B. & Wilgram, G. F. An electron-microscope study of dyskeratosis and acantholysis in darier’s disease. J. Invest. Dermatol. 41, 57–65 (1963).
Peck, G. L., Kraemer, K. H., Wetzel, B., Klingler, W. G. & Cohen, I. K. Cornifying darier disease— A unique variant: I. Report of a case. Arch. Dermatol. 112, 495–503 (1976).
Dickson, M. A. et al. Human keratinocytes that express hTERT and also bypass a p16INK4a-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell Biol. 20, 1436–1447 (2000).
Smits, J. P. H. et al. Investigations into the FLG null phenotype: Showcasing the methodology for CRISPR/Cas9 editing of human keratinocytes. J. Invest. Dermatol. 143, 1520–1528 (2023).
Rios, A. C. et al. Intraclonal plasticity in mammary tumors revealed through large-scale single-cell resolution 3D imaging. Cancer Cell 35, 618–632 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS: A J. Integr. Biol. 16, 284–287 (2012).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Bolte, S. & Cordelières, F. P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 (2006).
Acknowledgements
We thank Veit Flockerzi at Saarland University for providing the FWE antibody used in this study, and Aaron Mohammed from the Innovative Genomics and Bioinformatics Core (IGBC) facility at Creighton University for assistance with RNA-seq analysis. We acknowledge additional support provided by members of the Advanced Microscopy, Flow Cytometry, and IGB core facilities and the Integrated Biomedical Imaging Facility (IBIF) at Creighton University. This work was supported by the State of Nebraska LB595, LB506 and LB606 (L.A.H.). This work also utilized Core Services and Pilot or Feasibility project funding supported by grant UM-SBDRC 1P30AR075043 of NIH/NIAMS to the University of Michigan. S.Y.W. was supported by an R01 grant (R01-AR080654) from the NIH.
Author information
Authors and Affiliations
Contributions
J.C.R. and L.A.H. conceptualized the project and designed the experiments J.C.R. performed experiments in Figs. 1, 2a, e–f, 3a, b, 4–7 and Supplementary Figs. 1, 2, 3a–c, 4a–c, 5–9 with technical and analytic help from P.T.K., R.E.J., L. MN.M., G.L.P., P.O.H., and J.A.G. J.PH.S. performed experiments or generated samples used in Figs. 2, 3c and Supplementary Figs. 3d, 4a–d, 5b with assistance from I.M.J.J.VVW. under the supervision of E.H.VDB. K.S. performed and analyzed LFQ-MS in Fig. 4 under the supervision of V.K. in the UNMC Mass Spectrometry and Proteomics Core Facility. K.N.S. performed experiments supporting Supplementary Fig. 7d under the supervision of C.L.S. M.K.S. contributed to the design of the hFWE KO strategy for development of N/TERT-2G cell lines with support from J.E.G. J.E.G. and J.G.M. obtained clinical samples used in Figs. 1, 3, 4, 7, and Supplementary Fig. 5. J.C.R. wrote the manuscript, which was then reviewed and edited by L.A.H., J.A.G., J.E.G., S.Y.W., and C.L.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rudd, J.C., Smits, J.P.H., Kuwong, P.T. et al. Flower dependent trafficking of lamellar bodies facilitates maturation of the epidermal barrier. Nat Commun 16, 6892 (2025). https://doi.org/10.1038/s41467-025-62105-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62105-1
