
Abstract
P-stereogenic organophosphorus compounds are a class of highly important compounds due to their potentials in asymmetric catalysis both as ligands or catalysts and medicinal chemistry. Herein, we report an efficient protocol under bimetallic catalysis of Pd/Cu for highly regio-, E- and enantioselective phosphinylation of allenylic acetates with racemic secondary phosphine oxides forming a wide range of versatile P-stereogenic 1,3-dienyl phosphine oxides with 86–95% ee. The regioselectivity is unique as compared to the traditional catalytic enantioselective allenylations.
Similar content being viewed by others

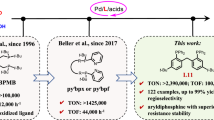

Introduction
Control of selectivity is an ever-lasting objective for the precise synthesis of covalent molecules in organic chemistry and medicinal chemistry. Allenes, due to their unique structure, exhibit intriguing and diverse reactivity patterns, thereby substantial efforts have been devoted to the selective transformation of allenes1,2,3,4. The Pd-catalyzed dynamic kinetic asymmetric transformation of racemic allenylic derivatives has emerged as a promising tool for the construction of axial chirality of allene with the efforts from Imada, Hayashi, Hamada, Trost, Ma, Zhang, and Shao, etc. with C- or N-nucleophiles (Fig. 1a)5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21. Triggered by the pivotal role of P-stereogenic organophosphorus compounds in asymmetric catalysis and medicinal chemistry22,23,24, we aimed to investigate the possible synthesis of P-stereogenic organophosphorus compounds via phosphinylation of allenylic esters with racemic secondary phosphine oxides (SPO) as the P-nucleophile (Fig. 1b). The challenge would be the precise control of the regio- and stereoselectivity to form either 2,3-allenyl phosphine oxides (allene-1–4) or 1,3-dienyl phosphine oxides (diene-1–4). The second challenge would be the control of diastereoselectivity of the 2,3-allenyl phosphine oxides referring to the allene axial chirality and the P-central chirality or the Z/E-selectivity of the C = C bond in the 1,3-diene products. Moreover, the reactivity of the chiral catalyst is another challenge due to the strong coordination ability of the secondary phosphine oxides via tautomerization to the trivalent phosphinous acid, which would competitively bind to the catalytic amount of metal catalyst, thus greatly reducing the catalytic activity25,26,27. Here we have successfully identified a palladium and copper co-catalytic recipe for the asymmetric phosphinylation of allenylic acetates providing P-stereogenic non-traditional 1,3-dienyl phosphine oxides (Fig. 1c). This transformation features a broad substrate scope and mild conditions with high regio-, E-, and enantioselectivity. Control studies revealed that CuCl and TMG are crucial for both reactivity and enantioselectivity.

a Pd-catalyzed dynamic kinetic transformation of racemic allenylic derivatives for chiral allene synthesis (previous work); b The challenges in Pd-catalyzed asymmetric reaction of racemic allenylic derivatives with racemic secondary phosphine oxides; c This work: Pd/Cu-cocatalyzed regio-, E- and enantioselective phosphinylation of allenylic esters with racemic alkyl aryl phosphine oxides.
Results
Optimization of reaction conditions
As shown in Table 1, the reaction conditions were optimized with racemic 4-cyclohexylbuta-2,3-dienyl acetate 1a and racemic methyl(phenyl)phosphine oxide 2a as the substrates. Initial studies were focused on the evaluation of chiral phosphine ligands (L1-L7, 12 mol%) with [Pd(allyl)Cl]2 (5 mol%) as the catalyst in the presence of TMG (2.0 equiv) in DCE (0.1 M) at 0 °C for 36 h. The use of P,N-bidentate ligand L1 or bidentate phosphine ligand L2 didn’t provide any product. To our surprise, this Pd-catalyzed phosphinylation of allenylic acetates with chiral biaryl bidentate phosphine ligand (L3, L4, L5) has shown exclusive regioselectivity providing the P-stereogenic 1,3-dienyl phosphine oxide 3a in 17–53% yields, without formation of the traditional axially chiral allenylic phosphine oxide 4a. However, the product was almost racemic (2–6% ee) presumably due to the poisoning effect on the catalyst of the secondary phosphine oxide. Changing the ligand to the MOP L6 or chiral phosphoramidite L7 failed to afford either 3a or 4a. The reaction was then investigated by adding Lewis acid to improve the reactivity of secondary phosphine oxide28 with chiral ligand L5. Indeed, the inclusion of FeCl2, CoCl2, or NiCl2 (0.5 equiv) improved the yield of 3a to 40–77%, but did not lead to any improvement of the enantioselectivity (4–7% ee, Table 1, entries 2–4), indicating the asymmetric phosphinylation of allenylic acetates with secondary phosphine oxides is indeed very challenging. Surprisingly, 3a was afforded in a high enantioselectivity of 90% ee and a moderate yield (48%) when CuCl2 was used (Table 1, entry 5). Encouraged by this result, other CuII and CuI salts were then evaluated and CuCl turned out to be the best, providing (RP,E)−3a in an excellent isolated yield of 95% and 89% ee with an excellent E-selectivity (Table 1, entry 10). It is worth noting that the counter anions of the copper additives have a great effect on the yield probably due to the influence of the counter anions on the Lewis acidity of the metals (Table 1, entries 8–10). A control experiment revealed that the reaction did not proceed without the palladium catalyst (Table 1, entry 11). The additional systematic evaluation of the reaction conditions including solvents and bases were also conducted but in futile (see Supplementary Table 1).
Reaction scope of enantioselective phosphinylation of allenylic acetates
With the optimal conditions in hand (Table 1, entry 10), we next explored the substrate scope of the methodology by examining 4-cyclohexylbuta-2,3-dien-1-yl acetate 1a with various racemic secondary phosphine oxides 2 (Fig. 2). Firstly, with Ar being fixed as the phenyl group, there is a very wide scope for the R2 group with C1-C3 carbon-chains and alkyl groups with a synthetically versatile methoxy and C = C bonds: the products (RP,E)-3a−3f were all formed with 80–98% yields and 87–90% ee. Furthermore, benzyl groups with diverse substitutes on the aryl group were also identified to be suitable R2 for this enantioselective phosphinylation reaction, affording the desired products (RP,E)-3g−3m in 70–91% yields and 92–95% ee. The secondary phosphine oxides with a variety of alkyl substituents at the para, meta, or ortho position of the aryl group (Ar) worked, affording the corresponding products (RP,E)-3n−3t in 63–97% yields with 86–94% ee. In addition, the para-Ph substituted product (RP,E)-3u was afforded in 71% yield and 93% ee. Introduction of etheric meta- or para-C − O bond to the aromatic ring of secondary phosphine oxides led to the formation of (RP,E)-3v−3y in high yields and high enantioselectivities. Electron-withdrawing group such as F, Cl, or CF3 in the para position of aryl phosphine oxide was well tolerated delivering (RP,E)-3z–3ab in 51–82% yields and 86–92% ee. The naphthalene and benzothiophene were also applicable, and the corresponding product (RP,E)-3ac–3ad were isolated in 76-80% yields with 91% ee. The absolute configuration of products has been successfully established with the single crystal X-ray diffraction analysis of (RP,E)-3g (CCDC 2325110).

Reaction conditions: 1 (0.20 mmol), 2 (2.0 equiv), [Pd(Allyl)Cl]2 (5 mol%), (R)-L5 (12 mol%), CuCl (0.5 equiv), TMG (2.0 equiv), and DCE (2.0 mL) at 0 °C for 34-43 h. The ee value was determined by chiral HPLC analysis. a the reaction was conducted at 10 °C for 60 h; b the reaction was conducted at –10 °C; c the reaction was conducted with CHCl3 as the solvent. TMG = Tetramethylguanidine.
We next sought to examine the scope of the allenylic acetates 1 with secondary phosphine oxide 2g (Fig. 2). The isopropyl substituted allenylic acetate delivered the desired (RP,E)-3ae in 81% yield and 95% ee. Tetrahydropyran and piperidine were also compatible affording (RP,E)-3af−3ag in 70–75% yields and 95% ee. It is worth noting that even the strained rings such as cyclopropane and cyclobutane survived and provided (RP,E)-3ai and (RP,E)-3aj in 92% ee. Furthermore, allenylic acetates bearing a linear C6 alkyl chain at the 4-postion also worked to afford the P-stereogenic product (RP,E)-3ak in 81% yield with 90% ee. A variety of functional groups such as chloro, bromo, phenyl, phthalylimide, and alkenyl could be introduced to the primary alkyl chain giving the products (RP,E)-3al−3aq in 87–91% ee. Moreover, Ph-substituted allenylic acetate was also compatible furnishing the corresponding product (RP,E)-3ar in moderate yield with 86% ee.
Synthetic utility
To elucidate the synthetic potential of this method, further transformations of the P-stereogenic products were investigated (Fig. 3). Initially, the reaction of allenylic acetate 1a with 2g was successfully scaled up to 5.0 mmol scale, affording the corresponding P-stereogenic phosphine oxide (RP,E)-3g with a comparable yield and enantioselectivity (1.4812 g, 85% yield and 92% ee, Fig. 3a). Then oxidative Heck reaction of (RP,E)-3g with 4-methylbenzeneboronic acid could provide P-stereogenic 1,3-dienyl phosphine oxide (RP,E,E)-4 in 63% yield29. In addition, reduction of (RP,E)-3a was realized to afford the phosphine-BH3 adduct 5 in 60% yield and 90% ee (Fig. 3b). Furthermore, the P-stereogenic cyclic products (RP,E)-6 and (RP,E)-7 could be furnished without the erosion of ee via the intramolecular olefin metathesis reaction under the catalysis of the Grubbs (II) catalyst (Fig. 3c)30.

a Scale-up reaction and Heck reaction; b Reduction to phosphine-borane adducts; c Grubbs reaction.
Mechanism study and DFT calculation
To unveil the mechanistic nature of this Pd-catalyzed phosphinylation of allenylic acetates, we first examined the impact of the amount of CuCl to unveil the crucial role of CuCl (Fig. 4a). When the amount of CuCl was reduced from 50 mol% to 20 mol%, the yield of (RP,E)-3a dropped dramatically from 99% to 59% with a slightly decreased enantioselectivity (89% ee vs. 84% ee). Further reducing the amount of CuCl to 10 mol% resulted both in a sharply decreased yield and enantioselectivity (25% yield and 42% ee). The enantioselectivity of the product (RP,E)-3a was merely 6% ee when CuCl was omitted. Further increasing of the amount of CuCl to 1.0 equivalent resulted in a lower yield and same ee. These results strongly suggest a critical role of CuCl for control of the enatioselectivity and the reactivity of secondary phosphine oxide. The Pd-catalyzed phosphinylation of racemic substrate 1a was also performed at partial conversion, and the unreacted allenylic acetate 1a was recovered in only 7% ee (Fig. 4b). Furthermore, the reaction of 1a with 2a in the presence of enantiomeric ligand of (S)-L5 under the optimized conditions afforded the enantiomer (SP,E)-3a in 82% yield with 90% ee, indicating that the absolute configuration of the P-stereogenic center has been dictated by that of the ligand and the E-selectivity has nothing to do with the absolute configuration of the ligand (Fig. 4c). In addition, we prepared the enantioenriched (SP)-2a31 and subjected it to the standard reaction conditions with (R)-L5 as the ligand and the reaction led to the perfect matched formation of the product (RP, E)-3a with a higher enantioselectivity of 95% ee (Fig. 4d, Eq. 1) while the same reaction of (SP)-2a with (S)-L5 as the ligand (to mimic the reaction of (RP)-2a with (R)-L5 as we could not get the (RP)-2a in high ee), the enantiomer (SP,E)-3a was obtained in comparable yield with a much lower enantioselectivity of 81% ee (Fig. 4d, Eq. 2). Interestingly, when the reaction of 1a with (SP)-2a (90% ee) was conducted without CuCl, the same product (RP,E)-3a was afforded in a much lower yields of 29-32% with 90% ee and 91% ee with (R)-L5 or (S)-L5 as the ligand (Fig. 4d, Eq. 3 and 4). Then we subjected (SP)-2a to the reaction with various conditions (Fig. 4e): No racemization was observed in 12 h under the conditions with either TMG or the Pd catalyst, while a very low racemization was observed in the presence of CuCl or the Pd/TMG. However, (SP)-2a was almost completely racemized within 2 h in the presence of both CuCl and TMG, implying that CuCl and TMG worked together for the rapid racemization of secondary phosphine oxides. Those results indicated that this Pd-catalyzed enantioselective phosphinylation reaction should be mostly a dynamic kinetic transformation process.

a The impact of the amount of CuCl on the reaction; b The partial conversion experiments; c Synthesis of the enantiomer; d Reactions with chiral secondary phosphine oxides; e Racemization study of the SPO (the equivalent of reagents was based on (Sp)−2a); f Proposed reaction mechanism.
Based on these data we proposed the catalytic cycle depicted in Fig. 4f. The oxidative addition of PdL* with 1a would generate the η1-1,3-dienyl-Pd species E-A32. (SP)-2a would replace OAc- to coodinate with Pd atom in E-A, yielding complex E-B. Deprotonation of E-B with TMG leads to the formation of intermediate E-C. The nucleophilic substitution of phosphine on the palladium produces the phosphoryl palladium intermediate E-D, which is followed by reductive elimination to produce the final product (RP,E)-3a. At the meantime, the slowly reacting SPO (RP)-2a would undergo rapid racemization to form (SP)-2a by pyramidal inversion33,34 in the presence of CuCl and TMG.
To further clarify the reaction mechanism as well as the critical role played by CuCl, density functional theory (DFT) calculations were carried out to probe the reaction between 1a and 2a, utilizing (R)-L5 as the ligand (for details, see Supplementary Information). As illustrated in Fig. 5a, the formation of complex Int1, which was selected as the free energy reference, involves the coordination of Pd atom with the C2 = C3 double bond in (R)-1a. The subsequent oxidative addition proceeds irreversibly via transition state TS1 with a free energy barrier of 5.9 kcal/mol, providing the η1-1,3-dienyl-Pd intermediate Int2. The enantiomer (S)-1a could also undergo oxidative addition to produce Int2, with a slightly higher energy barrier of 8.1 kcal/mol via TS1’. (SP)-2a participates in the reaction through ligand exchange with OAc– in Int2, providing Int3_1 with an endergonicity of 13.9 kcal/mol. The subsequent deprotonation of Int3_1 by TMG proceeds through TS2_1 with an activation barrier of 27.7 kcal/mol (TS2_1 relative to Int2) to yield Int4_1, simultaneously facilitating the conversion of pentavalent to trivalent phosphine. On the other hand, the involvement of CuCl would greatly facilitate the deprotonation step by the coordination of copper with the oxygen of SPO, reducing the free energy barrier to only 10.2 kcal/mol. Notably, CuCl also stabilizes both intermediates Int3 and Int4, and a Gibbs free energy change of −4.2 kcal/mol indicates that this deprotonation step is irreversible. Subsequently, the nucleophilic substitution of phosphine on the palladium center in Int4 requires a free energy of only 6.2 kcal/mol (TS3), resulting in the formation of a phosphoryl palladium intermediate Int5, which is exergonic by 12.0 kcal/mol (relative to Int4). The reductive elimination of Int5 produces the final product dienyl phosphine oxide (RP,E)-3a, which features an energy barrier of 22.0 kcal/mol (TS4) and is the rate limiting step of the whole process.

a Free energy profiles (∆Gsol in kcal/mol) for the reaction of 1a and 2a. b The origin of the regioselectivity. c The origin of the enantioselectivity.
Furthermore, computational investigations were conducted to elucidate the regioselectivity of the reaction. Due to the facile σ-π-σ tautomerization, η1-1,3-dienyl-Pd intermediate Int4 could convert to η1-allenyl-Pd intermediate Int7 via the π-allyl Pd intermediate Int6. The final (RP,E)-3a product may also be achieved through the nucleophilic attack of the phosphine atom on the central carbon C1 of allene in Int7 via TS5, a pathway rendered less favorable by 11.1 kcal/mol in comparison to TS4. The phosphine atom directly attacks the terminal carbon C3 of allene in Int4 to yield the allenylic phosphine oxide 4a via TS6. However, owing to the 12.0 kcal/mol higher in free energy of TS6 compared to TS4, no allene products were observed in the experiments. In the Pd-O intermediate Int4, the trivalent phosphine can undergo intramolecular nucleophilic attack at three possible sites: the Pd center, the central carbon (C1), or the terminal carbon (C3). Natural Population Analysis (NPA) reveals that for the the Pd atom carries positive charge (+0.083), while C1 and C3 are negatively charged (−0.190 and −0.381, respectively), strongly favoring nucleophilic attack at the Pd site (Fig. 5b). To further support this, we computed the local electrophilicity index (ωk), which shows that the Pd center has the highest electrophilicity (0.032 eV), surpassing those of C1 (0.025 eV) and C3 (0.024 eV). These findings align with the observed activation barriers: TS3 (Pd path) is significantly lower in energy than TS5 (C1 path, +14.9 kcal/mol) and TS6 (C3 path, +15.8 kcal/mol), confirming that the Pd site is the most favorable for nucleophilic attack. Furthermore, the phosphoryl palladium intermediate Int5 undergoes reductive elimination via two competing pathways: (1) C1-P bond formation through a 3-membered cyclic TS4, or (2) C3-P bond formation via a 5-membered cyclic TS7. The latter pathway is energetically disfavored, with TS7 exhibiting a 38.9 kcal/mol barrier (16.9 kcal/mol higher than TS4). This energy difference originates from greater structural distortion in TS7, as evidenced by bond angle comparisons (Fig. 5b): while TS4 (α = 112.9°, β = 125.4°) shows minimal deviation from Int5 (α = 115.0°, β = 127.0°), TS7 (α = 106.0°, β = 123.2°) displays significant angular strain. The enhanced distortion in TS7 accounts for both its higher energy and the observed regioselectivity for the reductive elimination of Int5.
Additionally, computational analyses were undertaken to elucidate the observed enantioselectivity. The deprotonation step of Int3 to Int4 is computed to be irreversible and the subsequent nucleophilic substitution and reductive elimination both preserve the enantioselectivity. Thus, the deprotonation step is considered as the enantioselectivity-determining step. The deprotonation transition state TS2’ for (RP)-2a is disfavored due to an increased Gibbs free energy of 2.7 kcal/mol compared to the transition state TS2 associated with (SP)-2a, thereby elucidating the enantioselectivity of the reaction (Fig. 5c). The IGMH35 analysis of TS2 identifies two stabilizing C–H···π interactions (2.50 Å and 2.79 Å) between SPO (SP)-2a and the PdL*-allyl moieties, which contributed to the stabilization the TS2 structure. In contrast, TS2’ lacks comparable non-covalent interactions. Instead, the shortest H···H contact (2.02 Å) between the cyclohexyl and methyl groups indicates significant steric repulsion. The combination of stabilizing C-H···π interactions in TS2 and destabilizing steric clash in TS2’ rationalizes the 2.7 kcal/mol energy difference between the two transition states.
Discussion
In this work, we have developed a highly regio-, E-, and enantioselective catalytic phosphinylation of allenylic acetates forming a chiral phosphine center within the 1,3-dienyl phosphine oxide skeleton. The roles of CuCl and TMG for rapid racemization of SPO and deprotonation have been unveiled and validated by DFT studies, which also rationalize the origin of the observed regio- and enantiocontrol.
Methods
Materials
Unless otherwise noted, materials were purchased from commercial suppliers and used without further purification. All the solvents were treated according to general methods. Flash column chromatography was performed using 200–300 mesh silica gel. See Supplementary Methods for experimental details.
Procedure for regio- and enantioselective phosphinylation of allenylic acetates
To an oven dried 10 mL Schlenk tube was added CuCl (0.1 mmol, 0.5 equiv), [Pd(allyl)Cl]2 (0.01 mmol, 5 mol%), chiral phosphine ligand (R)-L5 (0.024 mmol, 12 mol%), DCE (2.0 mL), TMG (0.4 mmol, 2.0 equiv), allene 1 (0.2 mmol), and phosphine oxide 2 (0.4 mmol, 2.0 equiv) sequentially under nitrogen. The Schlenk tube was then sealed and stirred for 36 h at 0 °C. The reaction mixture was then filtered through a pad of celite eluenting with CH2Cl2/EtOAc (20 mL). The filtrate was concentrated, and the residue was purified by silica gel chromatography to afford the corresponding product.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information file. For the experimental procedures, data of NMR and HPLC analysis, see Supplementary Methods and Charts in Supplementary Information file. The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Center ((RP,E)-3g: CCDC 2325110). These data could be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. Data supporting the findings of this manuscript are also available from the corresponding author upon request.
References
Ma, S. Transition metal-catalyzed/mediated reaction of allenes with a nucleophilic functionality connected to the α-carbon atom. Acc. Chem. Res. 36, 701–712 (2003).
Ma, S. Some typical advances in the synthetic applications of allenes. Chem. Rev. 105, 2829–2871 (2005).
Blieck, R., Taillefer, M. & Monnier, F. Metal-catalyzed intermolecular hydrofunctionalization of allenes: easy access to allylic structures via the selective formation of C−N, C−C, and C−O bonds. Chem. Rev. 120, 13545–13598 (2020).
Pagès, L., Abdine, R. A. A., Monnier, F. & Taillefer, M. Transition metal-catalyzed intermolecular hydroarylation of allenes. Eur. J. Org. Chem. 2022, e20220072 (2022).
Ogasawara, M., Ikeda, H., Nagano, T. & Hayashi, T. Palladium-catalyzed asymmetric synthesis of axially chiral allenes: a synergistic effect of dibenzalacetone on high enantioselectivity. J. Am. Chem. Soc. 123, 2089–2090 (2001).
Trost, B. M., Fandrick, D. R. & Dinh, D. C. Dynamic kinetic asymmetric allylic alkylations of allenes. J. Am. Chem. Soc. 127, 14186–14187 (2005).
Imada, Y., Nishida, M., Kutsuwa, K., Murahashi, S.-I. & Naota, T. Palladium-catalyzed asymmetric amination and imidation of 2,3-allenyl phosphates. Org. Lett. 7, 5837–5839 (2005).
Nemoto, T., Kanematsu, M., Tamura, S. & Hamada, Y. Palladium-catalyzed asymmetric allylic alkylation of 2,3-allenyl acetates using a chiral diaminophosphine oxide. Adv. Synth. Catal. 351, 1773–1778 (2009).
Wan, B. & Ma, S. Enantioselective decarboxylative amination: synthesis of axially chiral allenyl amines. Angew. Chem. Int. Ed. 52, 441–445 (2013).
Song, S., Zhou, J., Fu, C. & Ma, S. Catalytic enantioselective construction of axial chirality in 1,3-disubstituted allenes. Nat. Commun. 10, 507 (2019).
Zha, T. et al. Direct catalytic asymmetric and regiodivergent N1- and C3- allenylic alkylation of indoles. Angew. Chem. Int. Ed. 62, e202300844 (2023).
Li, Q., Fu, C. & Ma, S. Catalytic asymmetric allenylation of malonates with the generation of central chirality. Angew. Chem. Int. Ed. 51, 11783–11786 (2012).
Li, Q., Fu, C. & Ma, S. Palladium-catalyzed asymmetric amination of allenyl phosphates: enantioselective synthesis of allenes with an additional unsaturated unit. Angew. Chem. Int. Ed. 53, 6511–6514 (2014).
Liu, H.-C., Hu, Y.-Z., Wang, Z.-F., Tao, H.-Y. & Wang, C.-J. Synergistic Cu/Pd-catalyzed asymmetric alenylic alkylation of azomethine ylides for the construction of α-allene-substituted nonproteinogenic α-amino acids. Chem. Eur. J. 25, 8681–8685 (2019).
Dai, J., Duan, X., Zhou, J., Fu, C. & Ma, S. Catalytic enantioselective simultaneous control of axial chirality and central chirality in allenes. Chin. J. Chem. 36, 387–391 (2018).
Trost, B. M., Schultz, J. E., Chang, T. & Maduabum, M. R. Chemo‑, regio‑, diastereo‑, and enantioselective palladium allylic alkylation of 1,3-dioxaboroles as synthetic equivalents of α‑hydroxyketones. J. Am. Chem. Soc. 141, 9521–9526 (2019).
Zhang, J. et al. Enantio- and diastereodivergent construction of 1,3-nonadjacent stereocenters bearing axial and central chirality through synergistic Pd/Cu catalysis. J. Am. Chem. Soc. 143, 12622–12632 (2021).
Dai, J. et al. Construction of acyclic all-carbon quaternary stereocenters and 1,3-nonadjacent stereoelements via organo/metal dual catalyzed asymmetric allenylic substitution of aldehydes. Angew. Chem. Int. Ed. 62, e202300756 (2023).
Zhang, J. et al. Synergistic Pd/Cu-catalyzed 1,5-double chiral inductions. J. Am. Chem. Soc. 146, 9241–9251 (2024).
Gong, B. et al. Enantioselective synthesis of axially chiral alkylidenecycloalkanes via copper-catalyzed functionalization of acyl allenols. ACS Catal. 15, 2351–2358 (2025).
Gao, L. et al. Nickel-catalyzed enantioselective synthesis of dienyl sulfoxide. Angew. Chem. Int. Ed. 63, e202317626 (2024).
Pradere, U., Garnier-Amblard, E. C., Coats, S. J., Amblard, F. & Schinazi, R. F. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem. Rev. 114, 9154–9218 (2014).
Xu, G., Senanayake, C. H. & Tang, W. P‑chiral phosphorus ligands based on a 2,3-dihydrobenzo[d][1,3]oxaphosphole motif for asymmetric catalysis. Acc. Chem. Res. 52, 1101–1112 (2019).
Imamoto, T. P‑Stereogenic phosphorus ligands in asymmetric catalysis. Chem. Rev. 124, 8657–8739 (2024).
Ackermann, L. Air- and moisture-stable secondary phosphine oxides as preligands in catalysis. Synthesis 2006, 1557–1571 (2006).
Dubrovina, N. V. & Börner, A. Enantioselective catalysis with chiral phosphine oxide preligands. Angew. Chem. Int. Ed. 43, 5883–5886 (2004).
Shaikh, T. M., Weng, C.-M. & Hong, F.-E. Secondary phosphine oxides: Versatile ligands in transition metal-catalyzed cross-coupling reactions. Coord. Chem. Rev. 256, 771–803 (2012).
Cai, B. et al. Asymmetric hydrophosphinylation of alkynes: facile access to axially chiral styrene-phosphines. Angew. Chem. Int. Ed. 62, e202215820 (2023).
Song, E., Park, J., Oh, I.-K., Jung, H. M. & Lee, S. Ligand-free palladium-catalyzed Mizoroki-Heck-type reaction of arylboronic acids and alkenes using silver cation. Bull. Korean Chem. Soc. 31, 1789–1792 (2010).
Scholl, M., Ding, S., Lee, C. W. & Grubbs, R. H. Synthesis and activity of a new generation of Ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org. Lett. 1, 953–956 (1999).
Xu, Q., Zhao, C.-Q. & Han, L.-B. Stereospecific nucleophilic substitution of optically pure H-phosphinates: a general way for the preparation of chiral P-stereogenic phosphine oxides. J. Am. Chem. Soc. 130, 12648–12655 (2008).
Ogasawara, M. et al. Synthesis, structure, and reactivity of (1,2,3-η3-butadien-3-yl) palladium complexes. Organometallics 26, 5025–5029 (2007).
Reichl, K. D., Ess, D. H. & Radosevich, A. T. Catalyzing pyramidal inversion: configurational lability of P‑stereogenic phosphines via single electron oxidation. J. Am. Chem. Soc. 135, 9354–9357 (2013).
Wang, X. et al. Enantioselective synthesis of five to eight-membered P-stereogenic benzo-fused heterocycles via copper-catalyzed dynamic kinetic resolution. Adv. Synth. Catal. 366, 2285–2291 (2024).
Lu, T. & Chen, Q. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 43, 539–555 (2022).
Acknowledgments
We are grateful for financial support from National Natural Science Foundation of China (grant No. 92156022 for J.Y., 22001008 for Q.L.), Anhui Provincial Natural Science Funds (grant No. 2308085Y13 for Q.L.), and Anhui Agricultural University.
Author information
Authors and Affiliations
Contributions
Q.L. and S.M. conceived and directed the project. G.L. performed the reactions and control experiments. S.H. and X.Z. performed the DFT calculations. X.L., W.Z., C.Y., and Y.Y. helped with the collection of new compounds and data analysis. H.J. and J.Y. helped to discuss the results and commented on the manuscript. Q.L. wrote the paper with input from all other authors. G.L. and S.H. contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, G., Hong, S., Li, X. et al. P-stereogenic centre via Pd/Cu-bimetallic catalytic selective phosphinylation of allenylic acetates. Nat Commun 16, 6916 (2025). https://doi.org/10.1038/s41467-025-62204-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62204-z
