
Abstract
The critical protein Munc13 serves numerous roles in the docking and priming of synaptic vesicles. On the presynaptic plasma membrane, Munc13 is organized into nanoclusters corresponding to release sites where synaptic vesicles dock and fuse. However, it is currently not known whether there is any organization of Munc13 monomers within the nanoclusters. Recent work suggests that Munc13 may spontaneously self-organize into homo-oligomers, raising the possibility that synaptic nanoclusters comprise organized assemblies of Munc13. Here we investigate the functional impact of two distinct Munc13 core domain oligomers comprising C1-C2B-MUN-C2C both in vitro and in vivo. Interface mutations that specifically destabilized oligomeric assemblies of Munc13 disrupted vesicle docking, trans-SNARE formation, and Ca2+-triggered vesicle fusion in vitro and impaired neurotransmitter secretion and motor nervous system function in vivo. We suggest that a sequence of oligomeric Munc13 complexes rapidly couple vesicle docking to vesicle priming via the assembly of a precise number of SNAREs.
Similar content being viewed by others


Introduction
Chemical synaptic transmission is an orchestrated sequence of molecular events that endows synapses with a remarkably fast and tunable communication channel through precise regulation of synaptic vesicle (SV) fusion. The low basal rate of SV fusion at a release site in the absence of a Ca2+ trigger can jump by a factor of ten to one hundred million upon local Ca2+ elevation, and the latency between Ca2+ arrival and neurotransmitter release can be shorter than 100 µsec1,2,3. This extraordinary performance distinguishes SV exocytosis from other cellular trafficking events and requires specialized machinery. At its core, this process is driven by a tightly regulated assembly of SNARE proteins along with several critical SNARE-associated proteins, including Munc13-1, Munc18-1, Synaptotagmin 1 (Syt1), and Complexin, among others4. At the mechanistic level, many questions remain largely unanswered: How many SNAREs typically assemble to drive SV fusion, and how is this number supervised? How is a vesicle brought into proximity with the plasma membrane while full SNARE assembly and vesicle fusion are prevented in the absence of a calcium trigger? How do environmental signals such as residual Ca2+ and lipids influence the steps leading to SV priming and fusion? Residing at the hub of the presynaptic release apparatus, Munc13-1 is involved in nearly every aspect of presynaptic function, including release site building, synaptic vesicle docking and priming, SNARE assembly, SV fusion, and use-dependent plasticity4,5. Recent studies using electron microscopy and super-resolution imaging approaches demonstrate that Munc13-1 localizes to nanoclusters corresponding to individual release sites within the presynaptic terminal6,7,8,9,10. We recently uncovered two oligomeric assemblies of Munc13-1 formed in vitro and proposed a model based on their structures that provides insight into these questions and the distinct roles of Munc13-1 in promoting SV capture and the steps leading to vesicle priming11. Distinct homo-oligomeric assemblies of the Munc13-1 core domain (C1-C2B-MUN-C2C, hereon referred to as Munc13C) were observed in vitro via cryo-electron tomography (Cryo-ET) of two-dimensional protein crystals self-assembling between two phospholipid bilayers. These were proposed to correspond to sequential states of SV docking and pre-priming so that SVs are maintained at precise distances from target membranes while SNARE assembly is gated by the coordinated occlusion of SNARE-binding sites on the MUN domain11. In this scenario, the C-terminal end of Munc13C assists in SV capture and tethering while the N-terminal end interacts with the plasma membrane and responds to elevated DAG and intracellular Ca2+ to transition the upright trimer state (hexamer of trimers) to the lateral state (hexamer of monomers). These nanoclusters may represent local enrichments of independently functioning Munc13 monomers or Munc13 could be organized on the nanometer length scale into oligomers that function as a unit.
Several activities could be accomplished by this proposed Munc13C topology and sequence of events. We suggested that SVs are captured and tethered in the upright state only when a critical mass of Munc13C monomers have preassembled12. The upright state can then transition to the lateral state, bringing the SV nearer to the plasma membrane while also preventing MUN-catalyzed SNARE assembly due to occlusion of the MUN domain SNARE-binding sites by neighboring monomers. This transition is proposed to be promoted by a translocation of the C1 domain in the presence of diacylglycerol (DAG). Finally, ratcheting of the MUN domains down onto the plasma membrane (potentially assisted by Ca2+ ions binding the C2B domain) would catalyze the simultaneous assembly of precisely 6 SNAREpins (based on the six-fold symmetry of the lateral hexamer) while helping to bring the SV and plasma membranes into close proximity for SV priming. This model has been further elaborated recently to include an organized oligomeric assembly of VAMP2 and Synaptophysin on the SV as described elsewhere13,14.
In the present study, we designed and investigated point mutations predicted to disrupt the oligomeric interfaces and used a variety of structural, biochemical, and in vivo approaches to test this model. Disruptions in either a MUN-MUN interface in the trimer or in a MUN-C2C interface in the lateral hexamer led to pronounced structural alterations of Munc13C. Functionally, these mutations impaired vesicle docking, MUN-catalyzed SNARE assembly, and Ca2+-triggered fusion in vitro as well as impaired neurotransmitter secretion in vivo. Specifically, wild-type Munc13C promoted the rapid assembly of precisely six SNAREpins mirroring the six-fold symmetry of the lateral hexamer, whereas Munc13C interface-disrupting variants could trigger at most one or two stable SNAREpins to form. These observations highlight the importance of the two oligomeric Munc13 assemblies (i.e., upright trimer and lateral hexamer) in coupling vesicle docking to a fixed number of assembled SNAREpins and provide structural and functional insights into the kinetics and fidelity of vesicle priming.
Results
Disruption of the Munc13C trimer interface
In the upright trimer arrangement, trimers of Munc13C tethered two lipid bilayers with a spacing of 21 nm, consistent with a synaptic vesicle (SV) held in a pre-docked state with no SNARE assembly (Fig. 1A). Due to the low resolution of the 3D reconstruction in the trimer interface region, the specific amino acids that form this interface could not be identified with high confidence. However, the surface charge distribution suggested that the interface is electrostatic in nature, possibly corresponding to residues D1358 and D1369 in helix H12 of MUN-D subdomain and residues K1494, K1495, and K1500 in helix H15 of MUN-D subdomain (Fig. 1B) using the helix nomenclature of Rizo and colleagues and rat Munc13-1 amino acid numbering15. Protein sequence alignments for H12 and H15 across metazoa revealed conservation of the acidic residues in H12 as well as the lysines in H15 (Fig. 1C). To test the functional significance of the upright trimer interface, two Munc13C variants harboring the following charge reversals were designed to produce electrostatic repulsion within the interface: D1358R/D1369R (trimer 2xR) and K1494D/K1495D/K1500D (trimer 3xD). These protein variants were stable and well-folded. Notably, both the trimer 2xR and 3xD mutants disrupted continuous hexagonal lattice formation apparent in the CryoET top view (Fig. 1D).

A Surface representation of the Munc13C trimer (PDB: 7T7R). Domain color code: C1 (pink), C2B (blue), MUN (orange), and C2C (cyan). B Top view of the trimer interface. Green dashed lines show the proposed interaction between acidic (red) and alkaline (blue) residues within the MUN-D region of three Munc13C molecules. Putative residues involved in trimer formation are D1358 and D1369 (helix H12 of MUN domain) and K1494, K1495, and K1500 (helix H15 of MUN domain). C Sequence alignments for MUN H12 and H15 helices for a select group of metazoa with identical and similar residues highlighted in blue and red, respectively. The residues mutated in the trimer variants are highlighted in yellow. Protein sequences used for alignment: hydra = H. vulgaris (XP012567200), worm = C. elegans (NP001021874), human = H. sapiens (NP001073890), rat = R. rattus (XP032775345), fly = D. melanogaster (NP651949), aplysia = A. californica (XP035824675). D Top view of representative sections from cryo-electron tomograms showing the effect of mutations on crystal formation by Munc13C between lipid bilayers. Wild-type Munc13C (top left) forms a continuous hexagonal lattice (blue), whereas reversing polarity of K1494/K1495/K1500 (3xD top right), D1358/D1369 (2xR lower left) and K1495/K1500 (2xD lower right) disrupted the lattice. Isolated hexagons (red circles) are visible without forming the characteristic honeycomb pattern of wild-type Munc13C. Scale bars are 50 nm. Ten or more tomograms were collected for each variant using three separate sample preparations (see Methods for cryo-electron tomography details).
Isolated lateral hexagons were observed between two lipid bilayers along with scattered unorganized Munc13C monomers in the interface variants. Hexagonal lattices were rarely observed and only occurred in small patches, indicating that the lattice organization enabled by intact upright trimers was largely abolished. To better determine which basic residues were required for the trimer interface, two more Munc13C variants were created with only a pair of lysine charge reversals: K1495D/K1500D and K1494D/K1495D. Whereas the K1494D/K1495D variant had minimal impact, the K1495D/K1500D variant (hereon referred to as trimer 2xD) was similar to the 3xD variant, consistent with K1495 and K1500 participation at the trimer interface. The tomograms of all four Munc13C timer interface variants appeared similar in side-view cross section with a pronounced linear protein density centered between bilayers, consistent with the presence of a high number of intact lateral hexamers centered between the two membranes (Fig. S1B, C). Moreover, the bilayer separation within those regions was identical to wild type (21 nm), indicating the presence of a vertical conformation of Munc13C monomers. Hence, charge reversals in either H12 or H15 of the MUN domain selectively destabilized the upright trimer assembly while Munc13C hexamers still formed.
Disruption of the Munc13C lateral interface
The other Munc13C oligomeric arrangement observed by CryoET was a lateral hexamer, and the protein interface within this structure was formed by the C2C domain of one monomer and the MUN domain of its nearest neighbor (Fig. 2A)11. Previously, we posited two interface regions from this structure. First, a region of mixed hydrophobic and hydrophilic character was observed between MUN-C (several residues on MUN H7 and H8) and the C2C domain (residues on beta strands 2 and 8 as well as loop 2) (Fig. 2B). Second, a highly polar region was observed between MUN-B and C2C (D1039 near H3 and E1139 in H6 of the MUN domain and R1673 and R1678 on the C2C domain, Fig. 2B). Sequence alignments across metazoa indicated strong conservation of the MUN-C/C2C interface (Fig. 2C), whereas the electrostatic interface displayed higher variability across phylogeny (Fig. S1D, E). Two bulky residues (F1176 and I1215 in MUN H7 and H8, respectively) may interact with W1626 and W1684 of C2C. To destabilize this hydrophobic interface, F1176 and I1215 were mutated to asparagines (hereon referred to as the lateral FI/NN variant), and this variant was examined for disruptions in the hexagonal lattice via CryoET. For some in vitro experiments, we also generated a quadruple substitution including D1039R/E1139R (hereon referred to as the lateral quad variant). To examine the other side of the lateral hexamer interface, two C2C variants were generated: N1587A + W1626N + W1684N (hereon referred to as 3xNA), and N1687E + W1684E (hereon referred to as 2xNE) (Fig. S1D). Interestingly, the lateral FI/NN variant disrupted the Munc13C hexagonal lattice while also generating a rectangular crystal lattice (Fig. 2D). Qualitatively, the Munc13C density of the FI/NN variant was higher than wild type, with pronounced densities likely corresponding to vertical and lateral orientations of Munc13C molecules, respectively. The lateral hexagon oligomer was largely eliminated since there were no detectable hexagonal lattices in the CryoET top view (Fig. 2D, E), and lateral FI/NN crystals lacked the characteristic linear density in the side view (Fig. 2G compared to Fig. S1B). The inter-bilayer distance was 21 nm, similar to wild-type Munc13C, suggesting the presence of upright protomers in the new crystal. A preliminary low-resolution 3D reconstruction (Fig. 2F) reflected some features of the crystallographic pattern consistent with vertical assemblies of Munc13C monomers with twofold symmetry, but its resolution was not sufficient to permit a detailed Munc13C structural modeling. And finally, the C2C 3xNA and 2xNE variants eliminated both the hexagonal lattice and the linear intermembrane protein density as shown in a representative tomogram for 3xNA (Fig. S1F). These C2C mutations did not disrupt C2C-membrane interactions since pairs of lipid bilayers were observed to be stabilized at 21 nm separation in these variants, similar to wild-type Munc13C. Based on these observations, we inferred that the lateral FI/NN, quad, 3xNA, and 2xNE variants selectively disrupted the assembly of lateral hexamers. In summary, the collection of timer and lateral interface variants described here bolsters our proposed structural model of Munc13 oligomers and provides a useful set of reagents for exploring the functional implications of Munc13 oligomerization as detailed below.

A Surface representation of the Munc13C lateral hexamer (PDB: 7T7C). Domain color code: C1 (pink), C2B (blue), MUN (orange), C2C (cyan). B Side view of the lateral interface. C2C (blue) and Munc13 MUN-C terminal helices (MCT, light pink) are positioned above the area of MUN-B and MUN-C domains (gray) of a neighboring Munc13C monomer. In detail, C2C contacts H7, H8 of MUN-C (green highlight) as well as H4, H6, and the loop between H3 and H4 of MUN-B Proposed residues involved in the lateral interface are labeled. C Sequence alignments for MUN H7 and H8 helices (top) as well as the C2C domain (bottom) with identical and similar residues highlighted in blue and red, respectively. The residues mutated in the lateral FI/NN, 3xNA, and 2xE variants are highlighted in yellow. Protein sequence accession numbers are given in Fig. 1. D Representative section of a cryo-electron tomogram with a top view of crystals formed by Munc13C F1176N/I1215N mutant between lipid bilayers (~50 tomographs in total were collected from three sample preparations for the FI/NN variant). E Fast Fourier transform (FFT) of the cryo-tomogram indicates a twofold rectangular symmetry for this crystal. F A slice through a 3D volume of the initial low-resolution reconstruction. G Section through a cryo-electron tomogram with a side view of the lateral FI/NN crystal. Note the absence of a characteristic density between the bilayers (Fig. S1B), indicating that the lateral hexamer is not present. Distance between bilayers is similar to wild type (~21 nm). Scale bars are 50 nm.
Munc13 interface variants impair vesicle capture and fusion in vitro
Several in vitro approaches have demonstrated roles for Munc13C in vesicle tethering and fusion12,16,17,18,19. We recently developed a reconstituted single-vesicle fusion assay that recapitulates both docking and priming aspects of Munc13 function in the context of several key synaptic proteins involved in SNARE assembly and fusion including Munc18-1/Syntaxin 1A, Synaptotagmin 1 (Syt1), and Complexin 1 (Cpx1) as illustrated in Fig. 3A20,21,22. Specifically, liposome attachment to the target membrane and subsequent Ca2+-evoked fusion are both strongly impaired and display slow kinetics in the absence of Munc13C20. In this assay, the pre-assembled Munc18-1/Syntaxin 1 A heterodimer and palmitoylated SNAP25 were reconstituted into fluorophore-labeled lipids (using ATTO465-DOPE) used to form giant unilamellar vesicles (GUVs), which were burst on silicon grids to form suspended lipid bilayers along with the soluble diacylglycerol mimetic 1,2-dihexanoyl glycerol (DHG) as described previously20. Fluorophore-labeled vesicles (ATTO647N-DOPE) containing VAMP2 and Syt1 were then added along with Cpx1 and Munc13C in the absence of Ca2+ while monitoring fluorescence at the suspended bilayer via confocal microscopy.

A Schematic of the single-vesicle docking/fusion assay using confocal microscopy. To reconstitute Munc13/Munc18-dependent vesicle priming and fusion, palmitoylated SNAP25, 1:1 Munc18-1/Syntaxin 1 complexin, and 1,2-hexanoyl-sn-glycerol (DHG) were incorporated into the suspended lipid bilayer (labeled with 1% ATTO465-DOPE). VAMP2 and Synaptotagmin 1 (Syt1) were incorporated into small-unilamellar vesicles (V) and labeled with 2% Atto647N-DOPE to visualize vesicles near the suspended bilayer. Munc13C and Complexin 1 (Cpx1) were added in solution. B Single vesicles were tracked by confocal microscopy and classified as docked or undocked based on their lateral mobility. Docked vesicles sometimes fused in the absence of Ca2+ (spontaneous fusion), whereas most vesicles fused upon addition of 100 µM Ca2+ (Ca2+-triggered fusion). C Munc13C variants harboring trimer interface mutations (trimer 2xR, 2xD, and 3xD, with N = 5, 4, and 4 replicates, respectively) were compared to wild-type Munc13C (WT, N = 12 replicates) by quantifying the docked vesicle fate in the absence of Ca2+ (D), and in response to 100 µM Ca2+ (E). F Lateral hexamer Munc13C variants (lateral FI/NN, Quad, 3xNA, and 2xE, with N = 5, 5, 4, and 4 replicates, respectively) were compared to wild-type Munc13 for vesicle fate (G) and response to 100 µM Ca2+ (H). After initial docking, vesicle fates were subdivided into three categories: vesicles remained docked (purple), spontaneously fused (green), or undocked (orange) (D, G). All data presented as mean ± SEM and each biological replicate comprised ~30 vesicles. Data were compared using one-way ANOVA and Tukey–Kramer analysis for multiple comparisons. **p < 0.01, *p < 0.05 compared to wild type and exact p values are indicated along with the biological replicate numbers in (E, H). The p values in (D, G) are given in the Methods section.
Using this approach with the trimer and lateral interface variants described above, several aspects of the role of Munc13 C in vesicle docking and fusion were investigated (Fig. 3B). Labeled vesicles diffused to the suspended bilayer and became tightly membrane-associated with minimal lateral mobility (hereon referred to as docking). Some vesicles fused spontaneously (confirmed with suspended membrane taking on vesicle membrane dye (ATTO647N) after fusion), while others disappeared without any observed fusion and were categorized as undocked23. The Munc13C trimer variants weakly impacted vesicle docking, and docked vesicles eventually undocked with similar probabilities as observed with wild-type Munc13C (Fig. 3C). Surprisingly, spontaneous fusion was significantly elevated by more than 3-fold in all trimer variants (Fig. 3C). Moreover, Ca2+-triggered fusion was impaired by about twofold in the 2xD and 3xD variants and by more than threefold in the 2xR variant (Fig. 3D). Thus, destabilizing the trimer interface both enhanced spontaneous fusion and impaired Ca2+-triggered fusion. These observations were compared with two MUN lateral variants (FI/NN and quad) as well as two C2C lateral variants (3xNA and 2xNE) (Fig. 3E). Both C2C lateral mutations were more similar to the trimer mutations with stable docking, enhanced spontaneous fusion, and moderately decreased Ca2+-triggered fusion (Fig. 3E, F). In contrast, the FI/NN and quad lateral variants displayed profound docking and Ca2+-triggered fusion defects (Fig. 3G, H). We next explored the possibility that the evident differences between the MUN and C2C lateral mutations could arise from downstream disruptions of Munc13 function beyond oligomerization.
Impact of Munc13 variants on soluble SNARE assembly in vitro
Some of the residues mutated in the Munc13C trimer and lateral interfaces have been implicated in SNARE-binding in previous studies. Specifically, two conserved residues in MUN helix H6 (asparagine and phenylalanine) were reported to impair Syntaxin 1 interactions24 while one of the residues mutated in the trimer 2xR (D1358) was proposed to interact with VAMP225. Thus, some aspects of impaired docking and fusion in the single-vesicle fusion assay may reflect inefficient catalysis of SNARE assembly by the mutated MUN domain in addition to destabilized Munc13C oligomerization. To assess the impact of both the trimer and lateral interface mutations on SNARE assembly, the Munc13C variants were incubated with stoichiometric amounts of soluble Munc18-1/Syntaxin 1 heterodimers together with Alexa Fluor (AF)647-labeled SNAP25 and AF568-labeled soluble VAMP2, and fluorescence resonance energy transfer (FRET) efficiency was monitored over time during Munc13-dependent SNARE assembly (Fig. 4A)26. FRET efficiency increased with mono-exponential kinetics upon addition of Munc18-1/Syntaxin 1 to the labeled SNAP25 and VAMP2, and this increase was accelerated in the presence of wild-type Munc13C at 0.5 µM (Fig. 4B). No increase in FRET efficiency was observed in the absence of the Munc18-1/Syntaxin heterodimer. As a further control for specificity of the FRET signal, the cumulative FRET signal in the absence of Munc13C was inhibited by around 75% in the presence of unlabeled VAMP2 cytoplasmic domain (CDV) (Fig. 4C, D). To quantify the enhancement in SNARE assembly rate catalyzed by the different Munc13 variants, we calculated the ratio of the SNARE assembly rate in the presence versus absence of Munc13C as summarized in Fig. 4E. Notably, the 2xR trimer variant as well as the FI/NN and quad lateral hexamer variants displayed significant impairment of SNARE assembly, whereas the remaining trimer variants (2xD, 3xD) and lateral variants (2xNE, 3xNA) all catalyzed SNARE assembly similar to wild-type Munc13C. Given that the 2xR, FI/NN, and quad variants also exhibited the largest impairments in Ca2+-triggered fusion (Fig. 3D, F), disruption of MUN-catalyzed SNARE assembly may have contributed to the severity of the fusion defect in those cases. However, the MUN H15 (2xD, 3xD) and C2C (3xNA, 2xNE) mutations impaired oligomerization, docking stability, and Ca2+-triggered fusion without compromising the SNARE assembly function of Munc13. Taken together, the single-vesicle docking/fusion and soluble SNARE assembly assays suggest that both the trimer and lateral interfaces are required for efficient Ca2+-triggered fusion. Moreover, because two MUN regions located at distinct oligomer interfaces play a role in catalyzing SNARE assembly, SNARE assembly may be directly regulated by Munc13 oligomerization.

A Solution FRET experiments measuring N-terminal SNARE assembly between SNAP25 (Q20C-AF647, red) and the cytoplasmic domain of VAMP2 (CDV) (S28C-AF568, green) were measured on a plate reader excited at 565 nm and emission recorded at 635 nm. Fluorescence was measured after the addition of Munc18-1/Syntaxin 1 (gray) in the presence and absence of Munc13C (blue). B Average FRET traces recorded over 90 min after the addition of Munc18-1/Syntaxin 1 protein in the presence (black) and absence (blue) of wild-type Munc13C. Traces are mean ± SEM for N = 6 replicates. No increase in FRET was observed in the absence of Munc18-1/Syntaxin 1 (red). C, D Inclusion of unlabeled CDV inhibited SNARE assembly by 76.4 ± 1.9% (green) with **p < 0.01 using the Student’s t-test (one-tailed). Traces are mean ± SEM (N = 6). SNAREs only (black) are the same data shown in (B) for—Munc13. E Average initial SNARE assembly rates for wild-type Munc13C and all trimer and lateral interface mutant variants normalized by SNARE assembly in the absence of Munc13C. Data were presented as a mean ± SEM from N = 6 experiments. Significance was tested using Tukey–Kramer to account for multiple comparisons. ** indicates a significant difference (p < 0.01) compared to the absence of Munc13C but not wild-type Munc13C. ## indicates a significant difference (p < 0.01) with wild-type Munc13C but not the absence of Munc13C (exact p values are indicated over the bars).
Munc13C assembles six stable SNAREpins under a clamped vesicle
Based on the observations described above, several questions remained unanswered. In particular, we wondered if stable assembled trans-SNARE complexes (hereon referred to as SNAREpins) were formed on docked vesicles prior to fusion. And if so, are both types of Munc13C oligomers prerequisite for initial vesicle capture, SNAREpin formation, or both? Does the underlying six-fold symmetry of the Munc13C oligomer constrain the number of SNAREpins assembled? We previously hypothesized that the lateral hexamer ratchets down onto the plasma membrane, allowing exposed MUN domains to catalyze formation of central SNAREpins while bringing the membranes into closer proximity11. Based on the in vitro vesicle fusion and SNARE assembly assays, we further hypothesize that forming and ratcheting down the lateral hexamer will expose regions of the MUN domain that are critical for binding SNAREs and catalyzing assembly. Destabilizing early steps in a sequential process could impair downstream SNAREpin assembly, thereby accounting for the poor performance of the trimer and lateral variants in promoting Ca2+-triggered fusion13.
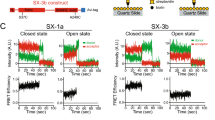
To answer these questions, we recently developed a TIRF microscopy approach to monitor vesicle docking onto suspended membranes on a silicon surface with single-molecule precision (Fig. 5A, B)14. To detect SNAREpin formation on docked vesicles, we exploited the fact that Cpx1 tightly binds the SNARE complex in a 1:1 molar ratio through its central helix27,28. Specifically, Cpx1 binds within a groove formed by VAMP2 and Syntaxin 1 A after the SNARE monomers have zippered partially together, and therefore, detection of a persistent fluorophore signal from a single labeled Cpx1 molecule at a docked vesicle within the TIRF field could be used as a proxy for a single stable SNAREpin. The sole natural cysteine in Cpx1 (C105) was tagged with Alexa 647 (Cpx1A647) and mixed with unlabeled Cpx1 (maintaining total Cpx1 concentration fixed at 2 µM) using several different concentrations of labeled Cpx ranging from 25 to 85%.

A Schematic for TIRF microscopy with single-molecule imaging. The same protein composition was used as in Fig. 3, except that both the suspended bilayer and vesicle were labeled with ATTO465-DOPE, and Cpx1 was labeled with Alexa Fluor 647 (Cpx1A647). The labeled Cpx1 was mixed with unlabeled Cpx1 at several concentrations (25, 50, 75, and 85% labeling) for a total concentration of 2 µM. B Two-color TIRF imaging was used to monitor vesicle arrival in the TIRF field as a rapid increase in fluorescence followed by a slow exponential decay due to bleaching (top gray curve) of immobile (clamped) vesicles. A small number of labeled Cpx1A647 molecules were monitored in the red channel while continuous bleaching produced step decays in Cpx1A647 fluorescence (middle orange curve) that were used to count Cpx1A647 molecules immobilized on the clamped vesicle. In some cases, immobile vesicle fluorescence would disappear within a single imaging frame (<100 msec, bottom gray curve), indicating that the vesicle either fused or undocked and left the TIRF field. C (Top) Representative trace for a clamped vesicle (gray) that captured two Cpx1A647 molecules (orange) using 25% labeled Cpx1. (Bottom) Representative trace for a clamped vesicle (gray) that captured three Cpx1A647 molecules (green) using 50% labeled Cpx1. Dashed lines indicate stepwise drops in fluorescence. D Histograms of Cpx1A647 counts were plotted for 161 docked vesicles across N = 3 experiments using 25% Cpx1A647 (orange) and for 117 docked vesicles across N = 3 experiments using 50% Cpx1A647 (green), generating an average of 1.6 and 3.0 Cpx1A647, respectively. Predicted histograms using N = 6 binding sites for Cpx1A647 were generated using a binomial distribution with p = 0.25 (black curve, top) or p = 0.5 (black curve, bottom). E The average number of Cpx molecules (mean ± SEM) associated with a clamped vesicle plotted versus the fraction of labeled Cpx1 (including the 25 and 50% labeling datasets as well as separate 75 and 85% datasets) was fit to a line with a slope of 5.5 (or 6.6 including 85% label data), indicating that 6 Cpx1 molecules stably associate with a clamped vesicle. The 75 and 85% labeling data were collected from N = 3 experiments with a total of n = 124 and n = 142 vesicles, respectively. F Cpx1A647 counting histograms were fitted to a series of binomial models varying N, and the total fit error was plotted against N increasing from 4 to 12 (see Methods for details). The best fits in three cases occurred with N = 6.
Fixing total Cpx1 at 2 µM matched the single-vesicle docking/fusion conditions described in Fig. 3, while varying the fraction of labeled Cpx1 allowed independent estimates of the total number of SNARE-bound Cpx1 molecules under a docked vesicle. Munc18-1/Syntaxin 1A complex and palmitoylated SNAP25 were reconstituted into GUVs, which were burst on a silicon surface to form a suspended bilayer as described earlier. Vesicles containing VAMP2 and Syt1 were fluorescently labeled using 1% ATTO465-DOPE lipids and added to the suspended bilayer together with the Cpx1/Cpx1A647 mixture, DHG, and wild-type Munc13C. In these TIRF experiments, the suspended bilayer also contained ATTO465-DOPE lipids to enable proper localization of the TIRF field, but during image acquisition, these fluorophores were bleached to generate a low fluorescence background required for detection of vesicle arrival.
Tightly docked vesicles were detected by a rapid increase in vesicle fluorescence intensity as the vesicle entered the TIRF field and became immobilized on the suspended bilayer followed by a slow exponential decay due to photobleaching of vesicular ATTO465 fluorophores (Fig. 5C). For wild-type Munc13, the majority of vesicles remained docked during the entire image acquisition time window before bleaching finally eliminated the vesicle fluorophores. Under these conditions, Cpx1A647 arrived either simultaneously with or rapidly after vesicle clamping (<100 msec, limited by image acquisition rate). The number of captured Cpx molecules for each docked vesicle was determined by analyzing the step-wise photobleaching time course (Fig. 5C). With 25% labeling, typically one or two Cpx1A647 molecules were captured immediately upon vesicle arrival, whereas with 50% labeling, three Cpx1A647 molecules were captured (Fig. 5C, D). As only a fraction of Cpx1 molecules were fluorescently tagged, the Cpx1A647 signal generally corresponded to a larger number of total bound Cpx1 molecules. For individual trials, however, binding was a stochastic process, and the precise number of total bound Cpx1 molecules could not be specified based solely on Cpx1A647 fluorescence. To more accurately determine the total number of bound Cpx1 (labeled and unlabeled), we fit a binomial distribution to the histogram of labeled Cpx1 observed for different labeling ratios and used the optimal values of the binomial fit to determine the average number of Cpx bound and therefore SNAREpins formed (Fig. 5D–F). Critically, the distributions of Cpx1 counts were well-described by binomial distributions with N = 6 ± 1 stable SNAREpins available to capture Cpx1 using 25%, 50%, 75%, and 85% labeling. Thus, using wild-type Munc13C, individual clamped vesicles rapidly sequestered Cpx1 onto six SNAREpins within 100 msec of vesicle docking.
Vesicle docking and the assembly of six SNAREpins require Munc13C oligomerization
To test the hypotheses that vesicle clamping and the subsequent assembly of six SNAREpins were catalyzed by oligomerized Munc13C, we applied the single-molecule TIRF Cpx1 counting assay to several trimer and lateral Munc13 variants. In striking contrast to wild-type Munc13C, many of the vesicles docked by the trimer and lateral variants rapidly undocked or fused within seconds after immobilizing (Fig. 6A, B). Specifically, more than 65% of the vesicles docked by the trimer variants were unstable, with vesicle fluorescence abruptly disappearing within 2–3 s of clamping, and the lateral FI/NN variant also exhibited a large increase in instability, although less than the trimer variants (Fig. 6A, B). Given the threefold enhancement of spontaneous fusion observed by confocal imaging using the 2xR and 2xD trimer variants, many of these unstable events in TIRF imaging likely corresponded to spontaneous fusion. However, the high undocking rate and poor fusogenicity observed in the lateral FI/NN variant by confocal imaging suggest that undocking accounted for most of the unstable events observed with lateral mutants. Another prominent difference observed with respect to wild-type Munc13C was a pronounced slowing of the vesicle capture rate, with both the trimer and lateral mutants requiring >15-fold longer time intervals to immobilize vesicles after they entered the TIRF field. Specifically, using wild-type Munc13C, vesicles entered the docked state in less than 100 msec of nearing the suspended bilayer, whereas clamping required ~1.5 s using the trimer and lateral variants (Fig. 6C, D). Unlike wild-type Munc13C where six stable SNAREpins were reliably and rapidly formed, vesicles docked by the trimer 2xR and 2xD variants as well as the lateral FI/NN and lateral quad variants formed only one or two stable SNAREpins based on 65–75% of observed docked vesicles failing to capture Cpx1 using 25% labeled Cpx1A647 (Fig. 6F–I). With destabilized trimers or lateral hexamers, the lower probability of forming and maintaining a functional oligomer prevented the stable assembly of 3 or more SNAREpins. In sum, irrespective of their differences, both classes of Munc13C mutations prolonged the time required to clamp vesicles and largely abolished stable SNAREpin formation, indicating requirements for both the trimer and lateral interfaces in these processes.

A Example trace for a stable docked vesicle (top, black) and an unstable docked vesicle (bottom, black). In both cases, no Cpx1A647 molecules were detected (red). B Vesicle instability was defined by the fraction of total vesicle docking events classified as unstable and plotted for experiments using wild-type Munc13C (WT, N = 5 replicates), two trimer variants (2xR and 2xD, N = 3 replicates, each), and one lateral hexamer variant (FI/NN, N = 3 replicates). Each replicate comprised ~30 vesicle docking events. All interface mutations destabilized vesicle docking by a factor of 15 to 35-fold. C For unstable docking events, the docking time course was partitioned into a capture phase as the vesicle entered the TIRF field but remained mobile (capture time) and a clamped phase where the vesicle was immobilized (dwell time). D The mean capture time is plotted for wild-type Munc13C (WT, n = 69) as well as three interface mutants (trimer 2xR, n = 27, trimer 2xD, n = 12, and lateral FI/NN, n = 13). WT Munc13C captures vesicles 15 to 30-fold faster than the interface mutants. E The mean dwell time is plotted for the same three interface mutants with n = 25, 15, and 12 docking events, respectively. Histograms for the number of Cpx molecules immobilized on clamped vesicles are shown for trimer 2xR (n = 147) (F), trimer 2xD (n = 143) (G), lateral FI/NN (n = 146) (H), and lateral quad (n = 207), where n = number of docked vesicles (I). Binomial distributions with N = 1 (red) and N = 2 (blue) are superimposed on all histograms for comparison. Data were presented as mean ± SEM and compared using one-way ANOVA and Tukey–Kramer analysis for multiple comparisons. **p < 0.01 compared to wild type, and n.s. p > 0.05. Individual p values are indicated above the bars.
In vivo impact of disrupting UNC-13 oligomeric interfaces
To further explore the functional impact of destabilized Munc13 oligomers in vivo, we examined the trimer and lateral interface variants using C. elegans as a model nervous system. Nematodes lacking the highly conserved Munc13-1 ortholog UNC-13 are immobile with essentially complete loss of chemical synaptic transmission, and expression of the long UNC-13 isoform containing an N-terminal C2A domain (UNC-13L) under a neuronal promoter has been shown to restore synaptic function29,30,31,32,33. To disrupt the homologous trimer interface in worm UNC-13, we generated rescuing trimer 2xR, 2xD, and 3xD UNC-13L constructs (Fig. S1A). For the lateral interface, where only one interface was conserved in C. elegans, the lateral FI/NN UNC-13L rescue construct contained I1283N/L1322N mutations (hereon referred to as lateral IL/NN). Likewise, the worm 2xE C2C variant contained N1659E and W1757E mutations (Fig. S1D). All of these constructs were expressed under a neuronal promoter in unc-13(s69) mutant animals to assess functional rescue of UNC-13. Transgenic animals expressing the UNC-13 variants were compared to unc-13(s69) and unc-13(s69) expressing wild-type UNC-13L as negative and positive controls, respectively. For each transgene, multiple independent transgenic lines were isolated and analyzed. All the trimer and lateral variants exhibited varying degrees of behavioral defects relative to the wild-type rescue strain. As a simple means of quantifying locomotory impairment, animals were placed in a liquid medium where movements of individual animals could be recorded and quantified (Fig. 7A). Wild-type animals transition from a crawling to thrashing mode of locomotion in a fluid environment, and the frequency of thrashes provides a straightforward quantitative assessment of locomotory motor function34. While unc-13(s69) animals were completely paralyzed, rescue with wild-type UNC-13L restored a robust trashing behavior of ~80 thrashes per minute (Fig. 7B). By contrast, all of the trimer and lateral interface variants thrashed at significantly reduced rates (Fig. 7B). In particular, the 2xR trimer mutant displayed the most severe defect with a ~90% decrease in thrashing relative to wild type.

A Animals will spontaneously swim or “thrash” rhythmically when transferred to a fluid medium. This form of locomotion requires a functional nervous system. B Average thrashing frequency for unc-13(s69) (orange, n = 43) or unc-13(s69) expressing a transgene encoding wild-type UNC-13L (gray, n = 39), UNC-13L(2xR) (red, n = 60), 2xD (blue, n = 53), 3xD (green), IL/NN (purple, n = 62), or 2xE (cyan, n = 46). C Cartoon depicting a C. elegans neuromuscular junction secreting ACh (red). Synaptic cleft acetylcholinesterase (green) hydrolyzes ACh and this process is blocked in the presence of aldicarb. D Paralysis time course on 1 mM aldicarb for wild type (black) and unc-13(s69) mutants expressing wild-type UNC-13L (gray), 2xR (red), or IL/NN (blue). Data were collected from N = 11, 9, 10, and 13 biological replicates, respectively. Each replicate comprised ~20 animals. Data were mean ± SEM. For statistical comparisons, data were compared using one-way ANOVA and Tukey–Kramer analysis for multiple comparisons. **p < 0.01 and * p < 0.05 compared to wild type. In B, the p values for wild type versus unc-13(s69), 2xR, 2xD, and IL/NN were <1 × 10−14. For wild type versus 3xD and 2xE, the p values are 1.6 × 10−11 and 1.5 × 10−8, respectively. See Methods for the individual p values in (D).
As a more specific assay of neuromuscular synaptic function, sensitivity to the cholinesterase inhibitor, aldicarb, was monitored in the transgenic animals. Acetylcholine (ACh) is rapidly cleared from the neuromuscular junction (NMJ) by synaptic acetylcholinesterase enzymes (AChE), and pharmacological inhibition of AChE causes a buildup of ACh, leading to paralysis with a characteristic time course (Fig. 7C). Previous work has demonstrated that defects in synaptic transmission cause slower paralysis kinetics and resistance to aldicarb32,35,36,37,38,39. Consistent with the thrashing assay as well as in vitro measures of Munc13C function in the trimer 2xR variant, trimer 2xR transgenic animals were markedly resistant to aldicarb, exhibiting little to no paralysis following 100 minutes of aldicarb exposure (Fig. 7D). The lateral IL/NN transgenic animals also displayed profound resistance to aldicarb treatment, similar to the trimer 2xR animals (Fig. 7D). Moreover, even a single point mutation in helix H7 (I1283N) was sufficient to produce a significant impairment in ACh secretion by this assay (Fig. S2A, B). The 3xD trimer mutant animals displayed a more modest but significantly slower paralysis on aldicarb whereas the 2xD and 2xE mutants were indistinguishable from wild type by this assay (Fig. S2C, D). Spontaneous locomotion on agar plates in the absence of food was also quantified for the trimer and lateral mutant animals after 1 h of starvation as an additional metric of nervous system dysfunction. Both the 2xR and IL/NN mutants displayed marked reductions in average speed relative to wild type (Fig. S2E). Overall, the locomotion and aldicarb sensitivity assays indicated that disruptions in either the trimer or lateral interface were sufficient to impair neurotransmitter secretion at the C. elegans NMJ and bolster the conclusions drawn from our in vitro assays of homo-oligomeric Munc13C function.
Impact of Munc13 oligomerization on the efficiency of vesicle priming
Several previous studies examining the distribution of Munc13 within presynaptic boutons have concluded that Munc13 localizes in nanoclusters6,8,9,10,40. Could there be an advantage of Munc13 oligomerization within these clusters for catalyzing vesicle priming? We explored the theoretical impact of clustering and oligomerization on a minimal stochastic model of SNARE assembly that assumes Munc13 monomers catalyze the assembly of a single SNAREpin with some unknown probability p. Based on previous studies, we also assumed that efficient vesicle priming (and ultimately fusion) would only occur when three or more SNAREs are assembled41,42,43. If Munc13 monomers were randomly distributed on the AZ membrane at even fairly high densities, the failure probability of achieving three or more successful SNARE assembly events under a single docked vesicle would remain high (Fig. 8A, B). For example, even at a high density of 1000 Munc13 monomers/µm2 and a probability p = 1 for successful SNAREpin assembly, the failure probability remains about 70% for achieving 3 or more assembled SNAREpins.

A The distribution of Munc13 monomers under a 50 nm vesicle (gray) assuming a uniform random two-dimensional distribution of Munc13 on the plasma membrane with various densities as indicated. B Failure rate (defined as the probability of fewer than three SNAREpins forming under a vesicle) is plotted versus the probability of an individual SNAREpin successfully assembling on a Munc13 (p). The density corresponding to 50–100 copies of Munc13 per AZ is highlighted (magenta). C. Clustering of Munc13 at a fixed AZ density of 500 per µm2 was modeled by associating a range of binding energies (εB in units of kBT) with Munc13 monomers under a vesicle as indicated. These distributions were used to model Munc13 clustering observed at presynaptic sites7,8,40. D Failure rate versus SNAREpin probability for the clustered Munc13 distributions. E Probability of being in the loosely docked state versus tightly docked state as a function of Munc13 monomer number under a vesicle. The baseline energy penalty of transiting to the tightly docked state was set to 35 kT and the docking energy contribution per Munc13 monomer (εZ) was varied between 1 and 10 kT as indicated. F Failure rate versus SNAREpin probability for clustered Munc13 distributions using a binding energy εB = 10 kT and varying the docking energy εZ as indicated. G An equilibrium reaction scheme for individual SNAREs (S, orange and blue) binding to an oligomer of Munc13 (M, yellow) to form assembled SNAREpins where M is composed of N Munc13 monomers. H Failure rate versus SNAREpin probability for oligomers of size N = 3 (red), N = 6 (black), and N = 8 (green) using several cooperativity factor values: γ = 1 (no cooperativity), 2, 4, and 6. Oligomers of N = 6 with modest cooperativity can essentially eliminate the chance of failure to form three SNAREpins over a broad range of individual SNAREpin probabilities. See Methods for additional details.
If a small number of Munc13 monomers were tightly clustered and localized in proximity to SVs in accord with previous observations, this failure probability would drop substantially depending on how many Munc13 monomers localize within a nanocluster. We modeled this scenario by adding a binding energy term to the Munc13 density assuming that Munc13 monomers interact with something correlated to docked vesicles such as other AZ proteins, lipids, or vesicle-associated proteins (Fig. 8C, D). For instance, assigning a modest binding energy of 2 kT per Munc13 monomer with an active zone density of 500 Munc13/µm2, the p = 1 failure probability drops to about 20%. Tight SV docking requires Munc13 activity, and formation of assembled SNAREpins requires SVs to be positioned within a few nanometers of the plasma membrane44. We modeled vesicle docking as a stochastic two-state process (loosely versus tightly docked) and included a term for the energy provided by each Munc13 monomer to stabilize the tightly docked state (Fig. 8E, F). This minimal model illustrates the steep dependence of efficient SNARE assembly on both the clustering and docking properties of Munc13. By contrast, oligomerization of Munc13 monomers with positive cooperativity between monomers readily localizes to vesicles and efficiently catalyzes three or more SNAREs to assemble under a vesicle (Fig. 8G, H). Even at a relatively low value of p = 0.4, the failure probability for a Munc13 hexamer with moderate cooperativity (γ = 4) is less than 5%. This simple model does not take into account mechanical and kinetic factors that may account for further advantages of Munc13 acting in oligomeric structures45. Notably, we do not include potential contributions by Synaptotagmin 1 or other SV-associated proteins that could reinforce the mechanical tethering of SVs to the plasma membrane46. Thus, Munc13 homo-oligomers provide an elegant means of localizing Munc13 and boosting the probability of SNARE assembly while maintaining a relatively low abundance of Munc13 at the AZ.
Discussion
We previously identified two Munc13C oligomers (i.e. upright trimer and lateral hexamer) and proposed that these distinct assemblies reflect a key feature of the topological organization of the release apparatus. Here, we designed several Munc13C variants to disrupt these oligomeric states as monitored by CryoET through point mutations in the MUN domain or C2C domain while leaving the overall protein stability intact. A distinct Munc13C oligomer with a twofold symmetry emerged in the lateral FI/NN variant based on cryoEM tomography, while other oligomeric mutations disrupted the hexameric crystal symmetry observed for the wild-type Munc13C protein. In vitro, perturbations in both oligomer types markedly slowed entry into the clamped-vesicle state and impaired calcium-triggered fusion of docked vesicles. In addition, the faithful assembly of six stable SNAREpins on clamped vesicles observed with wild-type Munc13C was abolished in the oligomer mutants. In vivo, both sets of oligomer mutations in the worm ortholog UNC-13 disrupted normal locomotor behavior and decreased ACh secretion. Significant differences between the mutated interface variants were also observed. Notably, mutations in MUN H7/H8 designed to disrupt the lateral hexamer interface as well as mutations in MUN H12 designed to disrupt the upright trimer interface impaired the SNARE assembly function of Munc13 in vitro, possibly due to reduced binding by Syntaxin 1 and VAMP2, respectively. These two sets of mutations also displayed more severe phenotypes in vivo, consistent with the possibility that UNC-13 oligomerization and SNARE assembly were both impacted. In sum, these findings bolster the viewpoint that a precisely arranged assembly of Munc13 molecules coordinates discrete events, including vesicle capture, tight docking, and SNAREpin assembly and that each of these steps is required for efficient calcium-triggered fusion.
Evidence for Munc13 multimerization
The results presented here bolster the hypothesis that Munc13 functions as a multimeric complex at release sites to promote SV docking and priming. Several in vitro studies of Munc13 have provided evidence for Munc13 oligomerization. Munc13-1 can homodimerize through its N-terminal C2A domain47, but this region of the full-length protein was not included in the current study, indicating that even lacking this dimerization domain, the core domains of Munc13-1 can oligomerize. We have previously shown that Munc13C spontaneously clusters on membranes in vitro12. Moreover, Munc13C can capture vesicles on supported bilayers with much higher efficiency when clusters of six or more monomers form, consistent with a cooperative process as well as with the six-fold symmetry of the Munc13C oligomers explored here11,12,48. Finally, the in vitro dependence of Munc13-1 concentration on catalysis of trans-SNARE assembly using membrane-anchored SNAREs displayed a high degree of cooperativity with a Hill coefficient of 4.5, consistent with an arrangement of five or more SNARE complexes cooperatively organized by Munc13-149.
At synapses, several studies have described clustering of presynaptic Munc13-1 within the active zone. In particular, super-resolution imaging of Munc13-1 in hippocampal cultures revealed nanoassemblies of Munc13-1 containing an estimated 10-12 labeled monomers corresponding to a single release site40. STED microscopy approaches visualized presynaptic Munc13-1 nanoclusters and estimated a cluster radius of about 23 nm7. These Munc13-1 nanoclusters align with postsynaptic PSD-95 nanoclusters, suggesting participation of Munc13-1 in nanocolumnar organization within the synapse6. Freeze-fracture replica labeling in mouse cerebellar cortex and hippocampus also revealed similar small clusters of Munc13-1 associated with release sites8,9. Interestingly, super-resolution imaging of the fly Munc13-1 ortholog Unc-13 in larval NMJ active zones detected prominent clustering with some evidence for subclusters containing three Unc-13 monomers, perhaps reflecting the homotrimeric state investigated here10. Despite the daunting technical challenges of counting individual proteins on the tens of nanometers length scale within synaptic boutons, these studies collectively strengthen the viewpoint that multiple Munc13 monomers coalesce at single release sites, perhaps to form functional units.
Functional implications of Munc13 oligomers at release sites
Vesicle tethering and catalysis of SNARE assembly are two major roles previously proposed for Munc13, and both tasks are likely to profit from cooperativity and precision within a multi-subunit assembly4,5,50,51. Membrane-bound Munc13C clusters in vitro capture and retain pure phospholipid vesicles only when six or more monomers are present, and this steplike increase in avidity depends on the membrane-binding C-terminal C2C domain, likely due to lashing several low-affinity sites together into an effectively high-affinity surface12. Perhaps more critical than enhancing membrane binding, the ability to supervise a precise number of assembled SNAREpins may be the true functional advantage of Munc13 oligomers.
Work from many labs over the past two decades has demonstrated that only a small number of SNAREs are required to achieve membrane fusion and subsequent fusion pore expansion, and yet the plasma membrane and vesicle membrane are inundated with a high density of SNARE proteins41,42,43,52,53,54,55,56. Specifically, membrane fusion can be initiated by a single SNAREpin, while at least three SNAREpins are thought to be required for successful pore expansion41,42,43. FRET-based measurements of SNAP25 in chromaffin cell vesicles suggested that six to seven copies of SNAP25 under conformational changes during vesicle priming, consistent with the formation of a small number of SNAREpins57. The unique performance demands placed on the presynaptic fusion apparatus may have selected for a relatively sophisticated molecular machine that can constrain and template a precise number of SNARE copies and integrate chemical information from multiple presynaptic proteins, small molecules, lipids, and calcium ions, all while positioning these components at fixed locations between the plasma and vesicle membranes. Given the likely scenario that any one SNARE assembly event may fail to produce an effective SNAREpin due to the stochastic behavior of single proteins, there is an obvious benefit of simultaneously templating six SNAREs within a single complex in terms of error tolerance. For instance, to achieve a 95% chance of successful assembly of three SNAREpins when only three SNAREs are independently templated, the individual SNAREpins must each assemble successfully with a probability greater than 98% using binomial statistics as described in the model in Fig. 8. By contrast, six independent sites for SNAREpin assembly drops this required probability down to about 70%. Furthermore, with modest positive cooperativity between assembling SNAREs due to their mechanical coupling within the Munc13 oligomer, the tolerance further drops to around 40–50% for individual SNAREpins (see Methods for a more detailed treatment of this equilibrium cooperativity model). Compared to clusters of independent Munc13 monomers, cooperative hexamers of Munc13 can thus dramatically improve the robustness and performance of SNAREpin assembly.
An additional advantage of sequestering a small number of SNARE proteins into a highly confined and organized arrangement is a dramatic acceleration of SNARE assembly speed compared to an unorganized diffusion-limited alternative45. The TIRF SNAREpin counting experiments presented here indicate that six SNAREpins are assembled in less than 100 msec upon vesicle docking, consistent with the observed rapid replenishment of synaptic release sites during activity that have an estimated time interval of 250 msec at the calyx of Held58. And finally, the synchronicity of cooperative SNAREpin assembly can supply a greater force to bring membranes into close proximity and drive the fusion apparatus into a primed state59. We propose that a succession of Munc13 oligomers guides a fixed number of SNAREs into assembled SNAREpins to achieve vesicle priming with both high speed and high fidelity. This molecular precision ensures a reliable and stereotyped fusion process within a synapse faced with a potentially broad range of activity levels. In addition to the impact of Munc13 oligomerization, we have recently reported another SNAREpin templating mechanism dependent on the SV tetraspanin Synaptophysin (Syp) operating in the absence of Munc13 to stabilize 12 SNAREpins14. The interrelationship between the Munc13 and Syp templating mechanisms requires further exploration, but the existence of multiple mechanisms for constraining the number of SNAREpins assembled under a vesicle highlights the centrality of this concept to the process of regulated exocytosis.
What is the sequence of events leading to SNARE assembly?
Mutations that disrupt either of the two distinct Munc13C assemblies were found to impair calcium-triggered fusion in vitro and neurotransmitter secretion in vivo. Thus, both oligomers reflect functionally important states of the pre-primed fusion machine and may also indicate specific steps in a stereotyped progression of Munc13 rearrangements that coordinate membrane positioning with SNARE templating. Although the experiments presented here do not specify a unique order to these steps, vesicle binding to the upright trimer followed by a transition to the lateral hexamer and ending with a flattening of the hexamer represents the most parsimonious ordering based on synaptic ultrastructural evidence for SVs moving from loosely tethered (20 nm gap between SV and plasma membrane) to tightly docked locations at the active zone (<10 nm gap) and the critical role played by Munc13 in these studies44,60.
Numerous detailed sequential models of vesicle docking and SNARE assembly have been developed from biochemical, genetic, and physiological data, and they are generally compatible with an oligomeric framework for Munc13 function4,17,50,60,61,62,63. One important feature of any sequential model is its treatment of directionality and reversibility as vesicles are moved through a series of states into a relatively stable primed state. SNARE zippering is likely to supply much of the free energy required to stabilize the primed state in all of these models, but SNARE proteins may promiscuously assemble in an unregulated fashion to trigger fusion while bypassing this state, a process partially antagonized by NSF/αSNAP-catalyzed SNARE disassembly64,65. We noticed that the lateral hexagon interface occludes regions on the MUN domain indicated in Syntaxin 1 interactions proposed to expose its H3 SNARE domain and initiate SNARE assembly24, and the MUN FI/NN variant disrupted MUN-catalyzed SNARE assembly in vitro. Additionally, the trimer 2xR mutations in the MUN domain alter an aspartate proposed to interact with VAMP225 while similarly disrupting SNARE assembly in vitro. In this scenario, Munc13 oligomers will exert further control over SNARE assembly by coordinately hiding or exposing catalytic regions of the MUN domain. Furthermore, SNARE-binding after transitioning out of an inhibited state would antagonize the reverse reaction since the interface residues would be occupied by SNARE interactions. Finally, the fate of Munc13 after ratcheting down to the plasma membrane remains unclear. We did not observe a flattened oligomeric state under our in vitro cryoEM conditions, and recent cryoEM tomographic studies at the synapse have observed a variety of protein densities associated with tightly docked SVs60,66. Perhaps further rearrangements or disassembly of the oligomeric structure occur as SVs mature into a tightly docked or primed state. As the molecular details of SV docking, priming, and fusion continue to be elucidated, we anticipate that sequential and cooperative interactions between SNARE proteins and assemblies of core fusion proteins, including Munc13, will play a key part in accounting for the remarkable performance of the synaptic fusion apparatus.
Potential roles for Munc13 oligomerization in synaptic plasticity and diversity
While the fundamental molecular mechanisms underlying synaptic transmission are largely identical across synapses within a nervous system as well as between the nervous systems of most animals, detailed studies of synaptic properties and their use-dependence have revealed that features such as synaptic strength and plasticity vary broadly across the mammalian brain9,67,68,69,70. Numerous studies have implicated Munc13 isoforms as contributing to synaptic diversity and short-term presynaptic plasticities such as post-tetanic potentiation, augmentation, and use-dependent SV pool replenishment31,32,58,71,72,73,74,75. Studies exploring presynaptic long-term potentiation in hippocampal mossy fibers and homeostatic plasticity in the fly NMJ reported changes in Munc13 distribution and abundance following plasticity induction10,76. Karlocai and colleagues correlated Munc13-1 abundance and active-zone distribution with synaptic properties across a population of hippocampal synapses, revealing a broad heterogeneity of Munc13-1 abundance even within active zones possessing the same number of release sites9. And Neher and colleagues hypothesized that shifts in a dynamic equilibrium between loosely docked and tightly docked states of SVs could account for some aspects of synaptic diversity and use-dependent plasticity70,77. The central role of Munc13 in SV docking and its sensitivity to Ca2+ and lipids such as PI(4,5)P2 and DAG support the notion that Munc13 could play a part in multiple forms of presynaptic plasticity5,70,78,79. And if Munc13 functions in larger homo-oligomeric complexes as proposed here, changes in monomer abundance as well as lipid and Ca2+ effects on oligomerization dynamics would be expected to impact synaptic strength via changes in SV docking, priming, and fusion. Notably, a human point mutation in the hinge region between the C2B and MUN domains of the human ortholog UNC13A was found to have a dominant gain-of-function synaptic phenotype, perhaps resulting from destabilizing an autoinhibited state32,80. Destabilizing the hinge region in a few monomers within a larger oligomeric UNC13A may be sufficient to release the autoinhibition and boost neurotransmitter release, possibly reflecting a mechanism normally used by Munc13 for use-dependent enhancement of synaptic strength. Several neuronal Munc13 isoforms are expressed throughout the rodent nervous system, and there is some evidence that different Munc13 isoforms contribute to distinct synaptic properties81,82,83. Indeed, functional synaptic differences in Unc-13 family isoforms has been observed outside of vertebrates as well84,85,86. Perhaps heteromultimers of distinct Munc13 isoforms will provide a means of modulating the docking and priming properties of Munc13 and furnish mechanistic insight into the impact of Munc13 isoform differences on functional synaptic diversity.
Conclusion and outlook
In this study, we investigated the functional significance of two previously characterized Munc13C oligomers identified by cryo-electron tomography. Although it remains uncertain whether these exact molecular arrangements exist at synaptic release sites, our results strongly suggest that Munc13 multimerization substantially impacts synaptic performance. Given the complexity and molecular crowding at synapses, interactions with other active-zone proteins and synaptic vesicle components could alter the specific oligomeric structures and SNARE stoichiometries observed. Indeed, heteromultimeric assemblies involving Munc13 and proteins such as RIM1 may occur within this environment. Such assemblies would continue to serve as mechanisms for counting SNAREs, facilitating rapid and precise SNAREpin formation, and efficiently coordinating the priming machinery. Future cryo-electron tomography experiments examining Munc13 assemblies in combination with other active-zone components will be essential to further clarify these organizational principles. Additionally, ongoing genomic analyses in patients with neurological impairments may pinpoint critical regions and residues within human neuronal Unc-13 proteins80,87,88,89. Integrating these genetic findings into the structural context of oligomeric protein assemblies could offer valuable insights into synaptic dysfunction underlying these conditions. Continued structural studies at synaptic release sites, along with more comprehensive in vitro fusion assays incorporating realistic synaptic vesicle and active-zone compositions, will progressively refine our understanding of these fundamental mechanisms.
Methods
Protein constructs
The following cDNA constructs previously described20,26 were used in this study: full-length VAMP2 (mouse His6-SUMO-VAMP2, residues 1-116); full-length SNAP25 (mouse His6-SNAP25b, residue 1-206); Synaptotagmin (rat Synaptotagmin 1-His6, residues 57–421); Complexin (human His6-Complexin 1, residues 1–134) and Munc13 C1-C2B-MUN-C2C domain (rat His12-Munc13-1, residues 529-1735 with residues 1408–1452 replaced by the sequence EF and 1532–1550 deleted). We utilized an expression clone in the pET-duet vector to express and purify the Munc18-1/Syntaxin 1 A complex (rat His6-SUMO Munc18/Syntaxin 1). Phusion High Fidelity Mastermix (New England Biolabs, Ipswich, MA) was used to generate all interface mutations in Munc13C.
Lipids, including 1, 2-dioleoyl-sn-glycero-3-phosphocholine DOPC, 1, 2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), L-α-phosphatidylinositol-4, 5-bisphosphate (Brain PIP2) and 1, 2-dioctadecanoyl-sn-glycerol (DAG) were purchased from Avanti Polar Lipids (Alabaster, AL). Fluorescent lipids ATTO465-DOPE and ATTO647N-DOPE were purchased from ATTO Tec (Siegen, Germany). 1, 2-dihexanoyl-sn-glycerol (DHG) was purchased from Cayman Chemicals (Ann Arbor, MI). Tris (2-carboxyethyl) phosphine hydrochloride (TCEP HCl) was purchased from Thermo Fisher (Waltham, MA).
Protein expression and purification
All SNARE and associated proteins were expressed and purified as described previously22,26. Briefly, proteins were expressed in Escherichia coli strain BL21(DE3) or Rosetta 2 (Novagen, Darmstadt, Germany) and cells were lysed with a cell disruptor (Avestin, Ottawa, Canada) in high salt buffer containing 400 mM KCl, 50 mM HEPES, 10% glycerol, pH 7.4, supplemented with 2% Triton X-100, 1 mm TCEP, and 1 mm phenylmethylsulfonyl fluoride. Samples were clarified using a Type 45 Ti rotor (Beckman Coulter, Brea, CA, USA) for 30 min at 35,000 rpm and incubated with Ni-NTA beads (Thermo Fisher, Waltham, MA, USA) for 4–16 h at 4 °C. The resin was subsequently washed (two column volumes) in the wash buffer (high salt buffer supplemented with 1 mM TCEP, 32 mM imidazole) with no detergent (Complexin and SNAP25) or 1% Octylglucoside (VAMP2 and Synaptotagmin) or 1% Triton X-100 (Munc18/Syntaxin). The proteins were either eluted with 300 mM Imidazole (Synaptotagmin), cleaved off the resin with Thrombin (SNAP25 and Complexin) or SUMO protease (VAMP2 and Munc18/Syntaxin) in high salt buffer for 2 h at room temperature. SNAP25 and Complexin proteins were further purified using gel filtration (Superdex75 Hi-load column, Cytiva, Marlborough, MA), and Synaptotagmin 1 protein was subjected to an anion exchange column (MonoS, GE Healthcare, Chicago, IL) to remove nucleotide contaminants. The peak fractions were pooled and concentrated using filters of appropriate cutoffs (EMD Millipore, Burlington, MA, USA). SNAP25 was then palmitoylated using a 20-fold excess of Palmitoyl Coenzyme A (Avanti Research) in HEPES buffer supplemented with 1% Triton X-100 for 30 min at room temperature with gentle mixing.
Munc13C was purified as described previously11. Briefly, Munc13C was expressed in ExpiHEK-293 cell cultures using ExpiFectamine as a transfection reagent (Thermo Fisher, Waltham, MA). Pellets were resuspended in lysis buffer and lysed using a Dounce homogenizer. The sample was clarified using a Type 45 Ti rotor (Beckman Coulter, Brea, CA) at 35,000 rpm for 30 min at 4 °C, and the clarified supernatant was incubated overnight with Ni-NTA beads, in the presence of DNAse 1, RNAse A, and Benzonase to remove nucleotide contamination. The protein was further washed in high salt buffer without Triton X-100 before being cleaved with PreScission protease for 2 h at room temperature. The cleaved proteins were further purified via gel filtration (Superdex 200, Cytiva, Marlborough, MA). In all cases, the protein concentration was determined using a Bradford Assay (Bio-Rad, Hercules, CA, USA), with BSA as a standard, and protein purity was verified using SDS/PAGE analysis with Coomassie stain. All proteins were flash-frozen and stored at −80 °C for long-term storage.
Lipid membrane preparation for cryo-electron tomography
Vesicles were prepared with a lipid composition consisting of DOPC/DOPS/PIP2 in a molar ratio of 14/80/6. The lipid stocks were mixed in chloroform with the addition of 20 μL methanol to dissolve PIP2, and the solvent was evaporated under N2 gas, followed by vacuum drying for 1 h. The resulting dried lipid film was rehydrated for 1 h at room temperature with constant vortexing in buffer containing 20 mM MOPS, pH 7.4, 150 mM KCl, 1 mM EDTA, and 0.5 mM TCEP at a final lipid concentration of 1 mM. After rehydration, the mixture was sonicated for 5 min using a bath sonicator (Branson Ultrasonics). To remove large aggregates of lipid membranes, the prepared solution was stored overnight at 4 °C. Vesicles were separated from sedimented aggregates and used for crystallization experiments. The protocol resulted in unilamellar vesicles with a broad range of sizes. Crystals were formed immediately before freezing by mixing 1 μM Munc13C with 100 μM lipid membranes in a 1:1 (vol/vol) ratio, total volume of 20 μL. Once mixed, samples were incubated at room temperature for 5 min before freezing.
Electron microscopy sample preparation and data acquisition
Samples were vitrified using a Vitrobot Mark IV (Thermo Fisher Scientific) held at 8 °C with 100% humidity. Samples (2.5 μL) were applied to freshly glow-discharged 200 mesh Lacey Formvar/carbon grids directly in the blotting chamber, and grids were blotted for 5 s with blot force −1 and then plunge frozen in liquid ethane cooled by liquid nitrogen. Samples were screened with a Glacios Cryo TEM 200 kV (Thermo Fisher Scientific) equipped with a K2 Summit direct electron detector (Gatan) operated in super-resolution counting mode. Dark and gain reference images were acquired prior data acquisition session. Cryo-electron tilt series were acquired using SerialEM with a bidirectional scheme ranging from −48° to +48° in 3° increments centered at 0°. This corresponded to a total of 33 movie frames with a cumulative electron dose of 82.5 e-/Å2. The nominal magnification was 13,500×, resulting in a physical pixel size of 3.018 Å. The nominal defocus range was set from −4 to −5 μm. Collected super-resolution tilt movies were initially aligned and 2x binned (resulting in a pixel size of 3.018 Å) using alignframes from the IMOD software package90.
The resulting tilt series were further aligned and reconstructed into 6x binned (18.1 Å/pixel) tomograms using AreTomo91. Ten tomograms were selected for further processing based on the visual quality of the crystal lattice, and corresponding tilt series movies were reprocessed using Warp 1.0.992. Super-resolution movies of individual tilts were 2x binned and aligned with simultaneous defocus estimation. These tilt series were exported from WARP and individually aligned using AreTomo. The alignment parameters were then imported back into Warp to create exposure-filtered tilt series, followed by tomogram reconstruction at 6x binned pixel size (18.1 Å/pixel). Approximately 500 particles were manually picked from two tomograms and used to generate an initial model in RELION 493. This model was used as a template for particle picking across selected tomograms in Warp. A total of 3035 particles were picked, and subtomograms were extracted using a 48-pixel box size at 18.1 Å/pixel and imported into RELION 4 for 3D refinement. The resulting 3D reconstruction is available in the EMDB under accession number EMD-71039. For Figs. 1, 2, the Munc13 protein oligomeric organization was visualized using the molecular solvent-excluded surface display function in ChimeraX of the corresponding structure atomic coordinates. The ribbon representation was rendered using ChimeraX94.
Suspended bilayer and vesicle preparation
For the suspended bilayer, we approximated the presynaptic membrane physiological composition with 81% DOPC, 15% DOPS, 3% PIP2, and 1% ATTO465-PE for visualization as previously described20. The lipids were mixed and dried under N2 gas and desiccated under vacuum. Bilayer samples were rehydrated with Munc18/Syntaxin 1 and palmitoylated SNAP25 (1:1600 protein: lipid input ratio) in 5x buffer (125 mM HEPES, 600 mM KCl, 1 mM TCEP, pH 7.4) supplemented with 2% Triton X-100 for 30 min. Samples were then mixed directly with SM-2 Biobeads (Bio-Rad, Hercules, CA) for 30 min with gentle agitation. The samples were removed from the beads and further dialyzed overnight in 5X buffer without detergent using a regenerated cellulose membrane (Spectrum Labs) on a flow dialysis with a 6–8 kDa molecular weight cut off.
Lipid bilayers were created by drying and rehydrating membranes to form GUVs as previously described20. Briefly, 4 µL drops of dialyzed proteoliposomes with Munc18-1/Syntaxin 1 + palmitoylated SNAP25 were air-dried on a clean Mattek dish and rehydrated twice. In the second rehydration, the sample was diluted 5X to 20 µL with distilled water and then added to a cleaned silicon chip containing 1x buffer (25 mM HEPES, 120 mM KCl, 1 mM TCEP, and pH 7.4) supplemented with 5 mM Mg2+. After a 20-min interval to allow bilayer formation, the bilayer was extensively washed with 1x buffer supplemented with 1 mM Mg2+, and the fluidity of the lipid bilayer was verified using fluorescence recovery after photobleaching (FRAP) using the ATTO465 fluorescence.
Small-unilamellar vesicles were prepared using 83% DOPC, 15% DOPS, 2% ATTO647N-PE as previously detailed20. The samples were dried under N2 gas and desiccated under vacuum. Lipids were rehydrated with VAMP2 (1:100) and Synaptotagmin 1 (1:250) in buffer (140 mM KCl, 50 mM HEPES, 1 mM TCEP, pH 7.4) supplemented with 1% octyl β-glucoside. After 30 min of mixing, samples were rapidly diluted 3X below CMC and allowed to sit for another 30 min before being dialyzed overnight in buffer without detergent. The samples were subjected to additional purification on a discontinuous Nycodenz gradient.
For TIRF experiments, a few modifications were made to the protocol above. The fluorescent lipid for the suspended bilayer was reduced to 0.1% while the liposome fluorophore was changed to 2% ATTO465. The suspended bilayer was prepared on a silicon chip, with buffer supplemented with 45% OptiPrep gradient media for index matching as previously reported95. OptiPrep on the top side was washed out with 1X buffer after bilayer formation. Bilayers contained 0.1% ATTO465 and were photobleached with 100% laser power after verifying they had formed.
Confocal single-vesicle fusion assays
Single vesicle assays were performed as described previously with a few modifications20. DHG was added to the pre-formed bilayer and incubated for 5 min. Vesicles (0.5 µM lipids) were added from the top using a pipette and allowed to interact with the bilayer for 3 min. We used ATTO647N-labeled DOPE fluorescence to track the fate of the individual vesicles. All vesicles that attached to the suspended bilayer during the 3-min observation period were identified as docked. Initially, all docked vesicles exhibited diffusive mobility and subsequently transitioned to an immobile, tightly docked state. Among these mobile and immobile vesicles, some underwent spontaneous fusion, evident by a burst of fluorescence intensity followed by a rapid decrease. Furthermore, we observed instances of undocking, where vesicle fluorescence disappeared without a fusion burst, primarily during the mobile phase. Undocking events were infrequent once vesicles reached the immobile state. After the initial 3 min interaction phase, the excess vesicles in the chamber were removed by buffer exchange (3X buffer wash) and CaCl2 was added from the top to a final concentration of 100 µM to monitor the fusion of docked vesicles.
TIRF single-vesicle Cpx-counting assays
The Cpx1 counting experiments underneath docked vesicles were performed with TIRF microscopy (Nikon) on a reconstituted planar suspended bilayer on the silicon surface. The method to create a suspended bilayer compatible with single-molecule imaging was described in detail elsewhere95. Before adding any vesicles, the bilayer was completely bleached, followed by the addition of 2 µM total Cpx1 (a varying mixture of labeled and unlabeled protein). Both the vesicle and labeled complexin were simultaneously monitored with 488 and 633 nm solid-state lasers, respectively, using a dual viewer 2 (DV2, Photometrics). All data were analyzed with ImageJ. The number of labeled complexin molecules underneath a vesicle was calculated from the measured step-wise photobleaching. Cpx1A647 counting distributions were compared to binomial distributions: probability \({p}_{k}=\left(\begin{array}{c}N\\ k\end{array}\right){f}^{k}{\left(1-f\right)}^{N-k}\) where N is the number of binding sites (SNAREpins) and f is the fraction of total Cpx1 labeled with a fluorophore. The total error between the measured distribution dk and a binomial distribution pk for f = 0.25, 0.5, 0.75, and 0.85 over a range of values for N was computed as \(E\left(N\right)=\sqrt{{\sum }_{k=0}^{N}{\left({d}_{k}-{p}_{k}\right)}^{2}}\) (Fig. 5F).
FRET-based soluble SNARE assembly assay
Assembly of the soluble SNARE domains was monitored using FRET between the cytoplasmic domain of VAMP2 (CDV) and SNAP25 following the addition of the Munc18-1/Syntaxin 1 heterodimer. For site-specific labeling with fluorophores, native cysteines (SNAP25 C85, 88, 90, 92G, VAMP2 C103S) were substituted and N-terminal cysteines were introduced into SNAP25 (residue Q20) and VAMP2 CDV (residue S28) as detailed before26 and labeled overnight at 4 °C with gentle agitation with a fivefold excess of thiol-reactive fluorescent probes Alexa Fluor 647 maleimide (SNAP25) and Alexa Fluor 568 maleimide (VAMP2 CDV). Excess dye was removed using the Thermo Fisher dye-removal columns, and labeling efficiency was determined by calculating concentrations using extinction coefficients for protein and dye at 280 nm and 576/651 nm, respectively. Labeling efficiency was typically greater than 90%. 1.5 µM labeled SNAP25 and VAMP2 were premixed with or without 0.5 µM Munc13C in FRET buffer (140 mM KCl, 50 mM HEPES, and pH 7.4) supplemented with 1 mM MgCl2 and 1 mM TCEP and incubated for 10 min at room temperature. About 1.5 µM of Munc18-1/Syntaxin-1 heterodimer (at room temperature) was added to a final reaction volume of 50 µL to start the reaction. Samples were excited at 568 nm and emission fluorescence was monitored at 680 nm in a plate reader (Molecular Devices) for 90 min with 30-s intervals. In one set of control experiments, no Munc18-1/Syntaxin-1 was added to monitor the baseline stability of the background FRET signal. In another set of controls, 2 µM unlabeled cytoplasmic domain of VAMP2 (CDV) was added to compete with labeled VAMP2. The rate of Munc13-catalyzed acceleration of SNARE assembly was quantified both by fitting the background-subtracted fluorescence traces to an exponential of the form: \(F\left(t\right)={F}_{\max }\cdot \left(1-{e}^{-{kt}}\right)\), where Fmax was determined for traces collected with wild-type Munc13C (providing the most accurate estimate of the true Fmax) and then held constant for all other fits except when using unlabeled VAMP2 CDV. This approach was necessary since the reaction was incomplete at 90 min and could not be measured on longer time scales due to technical limitations such as bleaching and evaporation. The impact of Munc13C variants was quantified by the ratio of fitted rate constants relative to the absence of Munc13C: \(R({\mathrm{variant}})=\left\langle {k}_{{\mathrm{variant}}}\right\rangle /\left\langle {k}_{-{\mathrm{Munc}}13}\right\rangle\). SNARE assembly in the absence of Munc13C was quantified in the presence and absence of unlabeled VAMP2 CDV by unconstrained estimates of Fmax in both cases.
C. elegans strains
Strains were maintained and genetically manipulated as previously described96. Animals were raised at 20 °C on nematode growth media seeded with OP50. Primary strains used: N2, unc-13(s69). JSD1392: tauEx574;unc-13(s69) = Psnb-1::UNC-13L wild-type rescue construct. JSD1385 (2xR): tauEx571;unc-13(s69) = Psnb-1::UNC-13L(D1459R;E1470R). JSD1413: tauEx579;unc-13(s69) = Psnb-1::UNC-13L(L1322N). JSD1414: tauEx580;unc-13(s69) = Psnb-1::UNC-13L(I1283N). JSD1415 (IL/NN): tauEx581;unc-13(s69) = Psnb-1::UNC-13L(I1283N;L1322N). JSD1444 (2xD): tauEx585;unc-13(s69) = Psnb-1::UNC-13L(K1585D;K1590D). JSD1455 (3xD): tauEx588;unc-13(s69) = Psnb-1::UNC-13L(K1584D;K1585D;K1590D). JSD1479 (2xE): tauEx590;unc-13(s69) = Psnb-1::UNC-13L(N1659E;W1757E).
Thrashing assay
Adult animals (N = 10 to 20) were transferred to a small drop of M9 saline solution on a glass coverslip and one-minute videos of the thrashing worms were captured using an ORCA-05G CCD camera (Hamamatsu) and an Olympus SZX16 stereomicroscope at 10 frames per second. The number of thrashes executed by each animal during the recording was counted manually offline. Thrash rates for each genotype were pooled and analyzed by one-way ANOVA and post hoc analysis across all genotypes.
Aldicarb resistance assay
To measure aldicarb sensitivity in C. elegans, ~20 animals were placed on agar plates containing 1 mM aldicarb (CarboSynth) as described previously37,97. Animals were scored for paralysis every 10 min for 2 h for each genotype. The experimenter was blind to the genotype, and each genotype was assayed at least ten times. Paralysis curves for each genotype were generated by averaging time courses.
Spontaneous locomotion
For C. elegans locomotion, ~40 worms of each genotype were moved to agar plates with no bacterial food using M9 buffer 1 h before tracking. One-minute videos of the spontaneous movement were captured as described above. The movement was tracked by a custom object tracking software on Matlab98. Worms that moved 30 s or less were not included in the data set. The data set for each genotype comprised between 80 and 300 individual worms except for the 2xE strain, for which data from 31 worms were collected. Speeds for each genotype were pooled and analyzed by one-way ANOVA followed by a post hoc significance test across all genotypes.
Clustering and oligomerization models of SNARE assembly
If individual Munc13 monomers each catalyze the assembly of a single SNARE complex with some probability p, then the probability of fewer than k assembled SNAREs out of N possible SNAREpins is given by
If Munc13 is distributed in a presynaptic AZ with density ρ0, binds to a synaptic vesicle (or some neighboring protein or lipid) with energy εB, and the synaptic vesicles cover a fraction f of the AZ surface, then the mean Munc13 density below a synaptic vesicle is:
The probability of N Munc13 monomers localizing under a 50 nm diameter vesicle corresponding to area A is given by a Poisson distribution:
For a given density ρ0, one can combine Eqs. (1) and (2) to compute the failure to assemble three or more SNAREpins as a function of p. The scenario where Munc13 is randomly distributed with no binding (εB = 0) is presented in Fig. 8A, B. The highly nonuniform clustered distributions of Munc13 that have been experimentally observed were modeled by increasing the binding energy, εB, from 0 to 10 kT for ρ0 = 500 Munc13/µm2 and f = 0.1 as depicted in Fig. 8C, D.
Successful SNAREpin assembly requires vesicles to adopt a tightly docked state within 5 nm of the plasma membrane, and Munc13 is required to achieve this state44. We incorporated this contribution of Munc13 by assuming vesicles stochastically move between a loosely docked state and a tightly docked state with an equilibrium energy difference of 35 kT (εloose) such that in the absence of Munc13, no vesicles would transit to the tightly docked state. Each Munc13 monomer under a vesicle contributes energy (εZ) towards stabilizing the tightly docked state using the following equilibrium relationship for the effective probability of SNAREpin formation:
where p is the probability of successful SNAREpin formation when the vesicle is tightly docked. This relationship and its impact on the failure probability are shown in Fig. 8E, F. Finally, to model the impact of oligomers of size N on failure probability, a simple modification of the binomial model including positive cooperativity between Munc13 monomers was utilized as follows:
\(\Pr \left\{ < k\right\}=1/Z+{\sum }_{j=1}^{k-1}\left(\begin{array}{c}N\\ j\end{array}\right){r}^{j}\cdot {\gamma }^{j-1}/Z\), where \(Z=1+{\sum }_{j=1}^{N}\left(\begin{array}{c}N\\ j\end{array}\right){r}^{j}\cdot {\gamma }^{j-1}\) and γ > 1 is the cooperativity parameter (Fig. 8G, H). For γ = 2 (representing a moderate amount of positive cooperativity), a failure probability of less than 0.05 would require only p > 0.57 for N = 6. A higher cooperativity using γ = 4, p > 0.4, suffices to achieve a 95% success rate for three or more SNAREpins assembling. Thus, even with relatively poor SNAREpin assembly efficiency, a hexamer of Munc13 could provide a robust safety factor to guarantee a sufficient number of assembled SNAREpins in this simplified model of vesicle priming.
Statistics and reproducibility
For the statistical comparisons presented in Fig. 3D, G, the following p values were generated by Tukey–Kramer. Vesicle Docking: WT vs 2xR = 2.8 × 10−7, WT vs 2xD = 3.3 × 10−5, WT vs 3xD = 6.1 × 10−8, WT vs FINN = 9.6 × 10−12, WT vs Quad = 7.7 × 10−13, WT vs 3xNA = 1.2 × 10−4, WT vs 2xE = 7.1 × 10−9. Spontaneous Fusion: WT vs 2xR = 1 × 10−2, WT vs 2xD = 1.2 × 10−3, WT vs 3xD = 9.6 × 10−6, WT vs FINN = 0.95, WT vs Quad = 0.53, WT vs 3xNA = 2.3 × 10−6, WT vs 2xE = 6.1 × 10−14. Vesicle Undocking: WT vs 2xR = 1 × 10−2, WT vs 2xD = 0.76, WT vs 3xD = 0.4, WT vs FINN = 4.4 × 10−10, WT vs Quad = 2.1 × 10−10, WT vs 3xNA = 0.52, WT vs 2xE = 0.9. For the statistical comparisons presented in Fig. 7D, the following p values were generated by Tukey–Kramer for wildtype versus UNC-13L, 2xR, and IL/NN variants, respectively: (0.6, 0.9, 0.9) for t = 60 min, (0.1, 0.5, 0.3) for t = 70 min, (0.3, 0.01, 0.004) for t = 80 min, (0.8, 1.8 × 10−5, 4.6 × 10−6) for t = 90 min, (0.97, 2.8 × 10−9, 1 × 10−9) for t = 100 min, (0.75, 1.4 × 10−13, 1.3 × 10−13) for t = 110 min, and (0.002, 1.3 × 10−13, 1.3 × 10−13) for t = 120 min.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw cryoelectron data have been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-71039. The datasets generated for this study are available in the Figshare repository [https://doi.org/10.6084/m9.figshare.29392028]. Two previous Munc13-1 oligomeric structures were utilized in Figs. 1, 2: 7T7R and 7T7C. Source data are provided with this paper.
References
Neher, E. & Sakaba, T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59, 861–872 (2008).
Sabatini, B. L. & Regehr, W. G. Timing of neurotransmission at fast synapses in the mammalian brain. Nature 384, 170–172 (1996).
Dittman, J. S. & Ryan, T. A. The control of release probability at nerve terminals. Nat. Rev. Neurosci. 20, 177–186 (2019).
Rizo, J. Molecular mechanisms underlying neurotransmitter release. Annu. Rev. Biophys. 51, 377–408 (2022).
Dittman, J. S. Unc13: a multifunctional synaptic marvel. Curr. Opin. Neurobiol. 57, 17–25 (2019).
Dharmasri, P. A., Levy, A. D. & Blanpied, T. A. Differential nanoscale organization of excitatory synapses onto excitatory vs. inhibitory neurons. Proc. Natl Acad. Sci. USA 121, e2315379121 (2024).
Emperador-Melero, J. et al. Distinct active zone protein machineries mediate Ca. Nat. Neurosci. 27, 1680–1694 (2024).
Rebola, N. et al. Distinct nanoscale calcium channel and synaptic vesicle topographies contribute to the diversity of synaptic function. Neuron 104, 693–710.e9 (2019).
Karlocai, M. R. et al. Variability in the Munc13-1 content of excitatory release sites. Elife 10, e67468 (2021).
Dannhäuser, S. et al. Endogenous tagging of Unc-13 reveals nanoscale reorganization at active zones during presynaptic homeostatic potentiation. Front. Cell Neurosci. 16, 1074304 (2022).
Grushin, K., Kalyana Sundaram, R. V., Sindelar, C. V. & Rothman, J. E. Munc13 structural transitions and oligomers that may choreograph successive stages in vesicle priming for neurotransmitter release. Proc. Natl Acad. Sci. USA 119, e2121259119 (2022).
Li, F. et al. Vesicle capture by membrane-bound Munc13-1 requires self-assembly into discrete clusters. FEBS Lett. 595, 2185–2196 (2021).
Rothman, J. E., Grushin, K., Bera, M. & Pincet, F. Turbocharging synaptic transmission. FEBS Lett. 597, 2233–2249 (2023).
Bera, M. et al. Synaptophysin chaperones the assembly of 12 SNAREpins under each ready-release vesicle. Proc. Natl Acad. Sci. USA 120, e2311484120 (2023).
Xu, J. et al. Mechanistic insights into neurotransmitter release and presynaptic plasticity from the crystal structure of Munc13-1 C1C2BMUN. Elife 6, e22567 (2017).
Quade, B. et al. Membrane bridging by Munc13-1 is crucial for neurotransmitter release. Elife 8, e42806 (2019).
Camacho, M. et al. Control of neurotransmitter release by two distinct membrane-binding faces of the Munc13-1 C. Elife 10, e72030 (2021).
Shu, T., Jin, H., Rothman, J. E. & Zhang, Y. Munc13-1 MUN domain and Munc18-1 cooperatively chaperone SNARE assembly through a tetrameric complex. Proc. Natl Acad. Sci. USA 117, 1036–1041 (2020).
Liu, X. et al. Functional synergy between the Munc13 C-terminal C1 and C2 domains. Elife 5, e13696 (2016).
Sundaram, R. V. K. et al. Novel roles for diacylglycerol in synaptic vesicle priming and release revealed by complete reconstitution of core protein machinery. Proc. Natl Acad. Sci. USA 120, e2309516120 (2023).
Ramakrishnan, S. et al. High-throughput monitoring of single vesicle fusion using freestanding membranes and automated analysis. Langmuir 34, 5849–5859 (2018).
Ramakrishnan, S., Bera, M., Coleman, J., Rothman, J. E. & Krishnakumar, S. S. Synergistic roles of synaptotagmin-1 and complexin in calcium-regulated neuronal exocytosis. Elife 9, e54506 (2020).
Kalyana Sundaram, R. V. et al. Roles for diacylglycerol in synaptic vesicle priming and release revealed by complete reconstitution of core protein machinery. Proc. Natl Acad. Sci. USA 120, e2309516120 (2023).
Yang, X. et al. Syntaxin opening by the MUN domain underlies the function of Munc13 in synaptic-vesicle priming. Nat. Struct. Mol. Biol. 22, 547–554 (2015).
Wang, S. et al. Munc18 and Munc13 serve as a functional template to orchestrate neuronal SNARE complex assembly. Nat. Commun. 10, 69 (2019).
Kalyana Sundaram, R. V. et al. Munc13 binds and recruits SNAP25 to chaperone SNARE complex assembly. FEBS Lett. 595, 297–309 (2021).
Chen, X. et al. Three-dimensional structure of the complexin/SNARE complex. Neuron 33, 397–409 (2002).
Pabst, S. et al. Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J. Biol. Chem. 275, 19808–19818 (2000).
Richmond, J. E., Davis, W. S. & Jorgensen, E. M. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat. Neurosci. 2, 959–964 (1999).
Madison, J. M., Nurrish, S. & Kaplan, J. M. UNC-13 interaction with syntaxin is required for synaptic transmission. Curr. Biol. 15, 2236–2242 (2005).
Hu, Z., Vashlishan-Murray, A. B. & Kaplan, J. M. NLP-12 engages different UNC-13 proteins to potentiate tonic and evoked release. J. Neurosci. 35, 1038–1042 (2015).
Michelassi, F., Liu, H., Hu, Z. & Dittman, J. S. A C1-C2 Module in Munc13 inhibits calcium-dependent neurotransmitter release. Neuron 95, 577–590.e5 (2017).
Liu, H. et al. The M domain in UNC-13 regulates the probability of neurotransmitter release. Cell Rep. 34, 108828 (2021).
Pierce-Shimomura, J. T. et al. Genetic analysis of crawling and swimming locomotory patterns in C. elegans. Proc. Natl Acad. Sci. USA 105, 20982–20987 (2008).
Rand, J. B. & Russell, R. L. Molecular basis of drug-resistance mutations in C. elegans. Psychopharmacol. Bull. 21, 623–630 (1985).
Miller, K. G. et al. A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc. Natl Acad. Sci. USA 93, 12593–12598 (1996).
Nurrish, S., Segalat, L. & Kaplan, J. M. Serotonin inhibition of synaptic transmission: Galpha(0) decreases the abundance of UNC-13 at release sites. Neuron 24, 231–242 (1999).
Martin, J. A., Hu, Z., Fenz, K. M., Fernandez, J. & Dittman, J. S. Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr. Biol. 21, 97–105 (2011).
Wragg, R. T. et al. Evolutionary divergence of the C-terminal domain of complexin accounts for functional disparities between vertebrate and invertebrate complexins. Front. Mol. Neurosci. 10, 146 (2017).
Sakamoto, H. et al. Synaptic weight set by Munc13-1 supramolecular assemblies. Nat. Neurosci. 21, 41–49 (2018).
Shi, L. et al. SNARE proteins: one to fuse and three to keep the nascent fusion pore open. Science 335, 1355–1359 (2012).
Bao, H. et al. Dynamics and number of trans-SNARE complexes determine nascent fusion pore properties. Nature 554, 260–263 (2018).
Heo, P., Coleman, J., Fleury, J. B., Rothman, J. E. & Pincet, F. Nascent fusion pore opening monitored at single-SNAREpin resolution. Proc. Natl Acad. Sci. USA 118, e2024922118 (2021).
Imig, C. et al. The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron 84, 416–431 (2014).
Mion, D., Bunel, L., Heo, P. & Pincet, F. The beginning and the end of SNARE-induced membrane fusion. FEBS Open Bio 12, 1958–1979 (2022).
Berns, M. M. M., Yildiz, M., Winkelmann, S. & Walter, A. M. Independently engaging protein tethers of different length enhance synaptic vesicle trafficking to the plasma membrane. J. Physiol. (2025).
Lu, J. et al. Structural basis for a Munc13-1 homodimer to Munc13-1/RIM heterodimer switch. PLoS Biol. 4, e192 (2006).
Li, F., Grushin, K., Coleman, J., Pincet, F. & Rothman, J. E. Diacylglycerol-dependent hexamers of the SNARE-assembling chaperone Munc13-1 cooperatively bind vesicles. Proc. Natl Acad. Sci. USA 120, e2306086120 (2023).
Bhaskar, B. R. et al. Differential SNARE chaperoning by Munc13-1 and Munc18-1 dictates fusion pore fate at the release site. Nat. Commun. 15, 4132 (2024).
Rothman, J. E., Krishnakumar, S. S., Grushin, K. & Pincet, F. Hypothesis - buttressed rings assemble, clamp, and release SNAREpins for synaptic transmission. FEBS Lett. 591, 3459–3480 (2017).
Padmanarayana, M. et al. A unique C2 domain at the C terminus of Munc13 promotes synaptic vesicle priming. Proc. Natl Acad. Sci. USA 118, e2016276118 (2021).
Hua, Y. & Scheller, R. H. Three SNARE complexes cooperate to mediate membrane fusion. Proc. Natl Acad. Sci. USA 98, 8065–8070 (2001).
Han, X., Wang, C. T., Bai, J., Chapman, E. R. & Jackson, M. B. Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science 304, 289–292 (2004).
Domanska, M. K., Kiessling, V., Stein, A., Fasshauer, D. & Tamm, L. K. Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J. Biol. Chem. 284, 32158–32166 (2009).
van den Bogaart, G. et al. One SNARE complex is sufficient for membrane fusion. Nat. Struct. Mol. Biol. 17, 358–364 (2010).
Mohrmann, R., de Wit, H., Verhage, M., Neher, E. & Sorensen, J. B. Fast vesicle fusion in living cells requires at least three SNARE complexes. Science 330, 502–505 (2010).
Zhao, Y. et al. All SNAP25 molecules in the vesicle-plasma membrane contact zone change conformation during vesicle priming. Proc. Natl Acad. Sci. USA 121, e2309161121 (2024).
Lipstein, N. et al. Munc13-1 is a Ca. Neuron 109, 3980–4000.e7 (2021).
Manca, F. et al. SNARE machinery is optimized for ultrafast fusion. Proc. Natl Acad. Sci. USA 116, 2435–2442 (2019).
Papantoniou, C. et al. Munc13- and SNAP25-dependent molecular bridges play a key role in synaptic vesicle priming. Sci. Adv. 9, eadf6222 (2023).
Richmond, J. E., Weimer, R. M. & Jorgensen, E. M. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature 412, 338–341 (2001).
Ma, C., Su, L., Seven, A. B., Xu, Y. & Rizo, J. Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339, 421–425 (2013).
Baker, R. W. et al. A direct role for the Sec1/Munc18-family protein Vps33 as a template for SNARE assembly. Science 349, 1111–1114 (2015).
Prinslow, E. A., Stepien, K. P., Pan, Y. Z., Xu, J. & Rizo, J. Multiple factors maintain assembled trans-SNARE complexes in the presence of NSF and αSNAP. Elife 8, e38880. (2019).
Brunger, A. T. et al. The pre-synaptic fusion machinery. Curr. Opin. Struct. Biol. 54, 179–188 (2019).
Held, R. G., Liang, J. & Brunger, A. T. Nanoscale architecture of synaptic vesicles and scaffolding complexes revealed by cryo-electron tomography. Proc. Natl Acad. Sci. USA 121, e2403136121 (2024).
Dittman, J. S., Kreitzer, A. C. & Regehr, W. G. Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J. Neurosci. 20, 1374–1385 (2000).
O’Rourke, N. A., Weiler, N. C., Micheva, K. D. & Smith, S. J. Deep molecular diversity of mammalian synapses: why it matters and how to measure it. Nat. Rev. Neurosci. 13, 365–379 (2012).
Burkhardt, P. & Sprecher, S. G. Evolutionary origin of synapses and neurons - Bridging the gap. Bioessays 39 (2017).
Lin, K. H., Taschenberger, H. & Neher, E. A sequential two-step priming scheme reproduces diversity in synaptic strength and short-term plasticity. Proc. Natl Acad. Sci. USA 119, e2207987119 (2022).
Junge, H. J. et al. Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell 118, 389–401 (2004).
Lipstein, N. et al. Nonconserved Ca(2+)/calmodulin binding sites in Munc13s differentially control synaptic short-term plasticity. Mol. Cell Biol. 32, 4628–4641 (2012).
Lipstein, N. et al. Dynamic control of synaptic vesicle replenishment and short-term plasticity by Ca2+-calmodulin-Munc13-1 signaling. Neuron 79, 82–96 (2013).
Lee, J. S., Ho, W. K., Neher, E. & Lee, S. H. Superpriming of synaptic vesicles after their recruitment to the readily releasable pool. Proc. Natl Acad. Sci. USA 110, 15079–15084 (2013).
Taschenberger, H., Woehler, A. & Neher, E. Superpriming of synaptic vesicles as a common basis for intersynapse variability and modulation of synaptic strength. Proc. Natl Acad. Sci. USA 113, E4548–E4557 (2016).
Fukaya, R. et al. Increased vesicle fusion competence underlies long-term potentiation at hippocampal mossy fiber synapses. Sci. Adv. 9, eadd3616 (2023).
Lin, K. H. et al. Number and relative abundance of synaptic vesicles in functionally distinct priming states determine synaptic strength and short-term plasticity. J Physiol. (2025).
Silva, M., Tran, V. & Marty, A. Calcium-dependent docking of synaptic vesicles. Trends Neurosci. 44, 579–592 (2021).
Kim, O. et al. Presynaptic cAMP-PKA-mediated potentiation induces reconfiguration of synaptic vesicle pools and channel-vesicle coupling at hippocampal mossy fiber boutons. PLoS Biol. 22, e3002879 (2024).
Lipstein, N. et al. Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder. J. Clin. Invest. 127, 1005–1018 (2017).
Augustin, I. et al. The cerebellum-specific Munc13 isoform Munc13-3 regulates cerebellar synaptic transmission and motor learning in mice. J. Neurosci. 21, 10–17 (2001).
Rosenmund, C. et al. Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron 33, 411–424 (2002).
Chen, Z., Cooper, B., Kalla, S., Varoqueaux, F. & Young, S. M. The Munc13 proteins differentially regulate readily releasable pool dynamics and calcium-dependent recovery at a central synapse. J. Neurosci. 33, 8336–8351 (2013).
Fulterer, A. et al. Active zone scaffold protein ratios tune functional diversity across brain synapses. Cell Rep. 23, 1259–1274 (2018).
Pooryasin, A. et al. Unc13A and Unc13B contribute to the decoding of distinct sensory information in Drosophila. Nat. Commun. 12, 1932 (2021).
Hu, Z., Tong, X. J. & Kaplan, J. M. UNC-13L, UNC-13S, and Tomosyn form a protein code for fast and slow neurotransmitter release in Caenorhabditis elegans. Elife 2, e00967 (2013).
Ma, X. R. et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 603, 124–130 (2022).
Brown, A. L. et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603, 131–137 (2022).
Wang, J. et al. UNC13B variants associated with partial epilepsy with favourable outcome. Brain 144, 3050–3060 (2021).
Kremer, J. R., Mastronarde, D. N. & McIntosh, J. R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996).
Zheng, S. et al. AreTomo: an integrated software package for automated marker-free, motion-corrected cryo-electron tomographic alignment and reconstruction. J. Struct. Biol. X 6, 100068 (2022).
Tegunov, D. & Cramer, P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152 (2019).
Zivanov, J. et al. A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0. Elife 11, e83724 (2022).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Kalyana Sundaram, R. V. et al. Native planar asymmetric suspended membrane for single-molecule investigations: plasma membrane on a chip. Small 18, e2205567 (2022).
Brenner, S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974).
Dittman, J. S. & Kaplan, J. M. Behavioral impact of neurotransmitter-activated G-protein-coupled receptors: muscarinic and GABAB receptors regulate Caenorhabditis elegans locomotion. J. Neurosci. 28, 7104–7112 (2008).
Ramot, D., Johnson, B. E., Berry, T. L. Jr, Carnell, L. & Goodman, M. B. The parallel worm tracker: a platform for measuring average speed and drug-induced paralysis in nematodes. PLoS ONE 3, e2208 (2008).
Acknowledgements
We thank Dr. Shyam Krishnakumar for helpful discussions and members of the Rothman and Dittman labs for critically reading the manuscript. This work was supported by grants from NIH DK 027044 (JER), NIH NS 130894 (JSH), and NIH NS 116747 (JSD).
Author information
Authors and Affiliations
Contributions
M.B., K.G., R.V.K.S., J.E.R., and J.S.D. conceived the study. M.B., K.G., R.V.K.S., J.S.H., J.E.R., and J.S.D. designed the experiments. K.G. and A.R. performed the protein structural experiments. M.B. and R.V.K.S. performed the reconstituted vesicle docking, SNAREpin counting, and fusion experiments. J.S.H., J. Chen, M.P., and J.S.D. performed the C. elegans experiments. R.V.K.S. performed the solution-state FRET experiments. F.P. and J.S.D. performed the numerical modeling studies. A.C., S.L., and J. Coleman contributed to the protein biochemistry. M.B., K.G., R.V.K.S., J.S.D., and J.E.R. contributed to the interpretation of the data and the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Huan Bao, Jiajie Diao, and Jun Yao for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bera, M., Grushin, K., Kalyana Sundaram, R.V. et al. Two successive oligomeric Munc13 assemblies scaffold vesicle docking and SNARE assembly to support neurotransmitter release. Nat Commun 16, 7222 (2025). https://doi.org/10.1038/s41467-025-62420-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62420-7
