
Abstract
Growth plate (GP), a critical cartilaginous structure in amniotes, underpins longitudinal bone growth, yet the intricate mechanisms behind its polarized mineralization during evolution remain unclear. Herein, employing high-resolution analytical techniques, we reveal that the GP-epiphysis interface displays a sharp transition in tissue modulus, acting as a protective shell for the underlying GP, whereas the GP-metaphysis interface exhibits a gradual modulus increase, enabling efficient load redistribution to the metaphysis. This mechanical microenvironment contributes to unique microstructural and compositional transformations from GP to epiphysis and metaphysis. Notably, the GP-epiphysis interface acts as a mineralization inhibition zone while the GP-metaphysis serves as a mineralization promotion zone, orchestrated by a complex network of proteins. Proteins such as secreted phosphoprotein 1 (SPP1) and alpha-2-HS-glycoprotein (AHSG) at the GP-epiphysis interface inhibit mineralization, forming a defense line; while ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) and alkaline phosphatase, biomineralization associated (ALPL), coexisting with SPP1 and AHSG, promote a sequential nucleation and assembly of calcium phosphate minerals at the GP-metaphysis. Such polarized mineralization patterns maintain the homeostasis of GPs and drive polarized bone elongation. Replicating this process in vitro, we synthesize stable amorphous calcium phosphate which shows highly controlled transformation into hydroxyapatite. This work provides a more comprehensive view of the structural integrity of human bone in development and offers strategies for controlled biomineralization.
Similar content being viewed by others

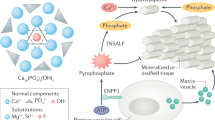

Introduction
Organisms have developed their own mechanisms to regulate mineral assemblies, essential for maintaining various physiological functions across different wildlife forms1,2. Growth plates (GPs), as a bone growth structure unique to vertebrates, provide an important model to understand mineralization mechanisms of hard tissues3. Calcium phosphate (CaP), as the primary component of vertebrate hard tissues, has been the subject of extensive research due to its manipulable assembly properties4,5. Current studies have focused on controlling CaP assembly through ions6,7, peptides8,9, proteins10, DNA11 and RNA12. However, the complex phase transition processes involved in non-classical nucleation render CaP assembly uncontrollable. Although bone mineralization pathways during the growth of developmental bones have been explored13,14,15, and amorphous calcium phosphate (ACP) particles have been observed in rapidly forming long bones16, the mineralization of GP responsible for long bone growth remains largely unexplored.
The developmental GP-bone interfaces serve as an ideal template for studying human mineral growth because of their unique polarized mineralization pattern. Following the formation of the secondary ossification center, the GPs are separated from the articular cartilage by the bony epiphysis and situated between the epiphysis and metaphysis17 (Fig. S1). Longitudinal bone growth is attributable to the continuous mineralization of the hypertrophic zone of the growth plate into the metaphysis via endochondral ossification18,19,20. In contrast to the actively mineralized GP-metaphysis interface, the GP cartilage does not undergo continuous mineralizing into bone tissue at the GP-epiphysis interface21. This phenomenon maintains the polarized nature of bone elongation, progressing from the GP towards the metaphysis. Previous studies have extensively revealed the unique contribution of disparate cell types within the GP in guiding long-term bone elongation22,23. However, the polarized mineralization patterns of GPs and their associated regulatory mechanisms have rarely been explored from a materials science perspective.
GPs are located in a unique hard-soft-hard mechanical environment between the epiphysis and metaphysis. The separation of articular cartilage and GP evolves as a mechanism to shield the GP from the intense mechanical forces related to weight-bearing in a terrestrial environment for amniotes24. Numerous studies have investigated the mechanical properties of zonal GP tissues or performed in situ testing to map these properties across the GP25,26,27. However, the relationship between localized mechanical properties and biochemical transitions at the cartilage-bone interfaces, which are key to understanding the polarized biomineralization patterns of GPs, remains poorly characterized. Moreover, under physiological conditions, bone tissue exhibits remarkable adaptation to the mechanical environment, dynamically responding to biomechanical stress and generating mineralized structures and compositions optimized for the mechanical response28,29. Understanding the interplay between localized mechanical responses and the corresponding microstructural and compositional transitions at the two key cartilage-bone interfaces can help explain the polarized biomineralization pattern in GPs.
The process of biological mineralization is a finely regulated process orchestrated by cells, involving the transient stabilization of ACP by biomacromolecules2, transportation of ACP to the extracellular matrix (ECM)30, ACP nucleation31, crystal growth and assembly32. Mineralization inhibitors at the GP-epiphysis interface effectively deter the precipitation of abundant calcium and phosphate ions within the ECM10,33, while at the GP-metaphysis region, the inhibitory effect of mineralization is neutralized by biomineralization promoters like enzymes34,35, allowing calcium and phosphate ions to form mineral precursors for further crystallization5,36,37. These processes are believed to be controlled by a complex network of proteins comprising inhibitors and promoters that regulate mineralization dynamics at the GP interfaces. Thus, a comprehensive understanding of the underlying biomechanical, biochemical, structural and biomolecular mechanisms involved in the polarized mineralization patterns at the GP-bone interfaces is of great significance in fields such as controlled mineral assembly, bone growth and development studies, and bone tissue engineering.
Herein, we collected human GP samples from varying developmental stages (phalange GPs: 0–5 years old; tibia GPs: 6–14 years old) (Table S1) to reveal the polarized bone lengthening, focusing on mechanical microenvironment, microstructural and compositional transformation, as well as nanoscale crystal assembly of the key transitional GP-epiphysis and GP-metaphysis regions by multiple high-resolution imaging technologies (Fig. S2). Our results confirmed the existence of a mineral precursor phase during GP biomineralization and revealed a series of proteins that regulate polarized GP mineralization. These findings enabled us to stabilize ACP and control its transformation into hydroxyapatite (HAp) in vitro.
Results
Mechanical characteristics of the GP-epiphysis/metaphysis interface
The in-situ biomechanical performance of tibia GP tissue, including epiphysis and metaphysis, under physiological loading was evaluated under physiological loading using a synchrotron X-ray microscope (XRM) at a high resolution of 5.64 μm per voxel (Fig. 1A). After compressive loading, the sample exhibited deformation (Fig. S3A). We reconstructed the 3D structure of the tissue before and after loading by Micro-computed tomography (micro-CT) and applied Digital Volume Correlation (DVC) to quantify local displacement and strain38 (Fig. S3B). The strain was mapped from the epiphysis, across GP tissue, to the metaphysis. The GP-metaphysis interface showed more significant load-induced displacements compared to the GP-epiphysis interface (Fig. 1B, Movie S1–3). A sharp increase in local strain was observed at the transition from the GP to the epiphysis, whereas the transition from the GP to the metaphysis exhibited a more gradual rise in strain level (Fig. 1C).

A Schematic illustration of the in situ micro-CT imaging of the whole GP sample via synchrotron XRM; B The displacement magnitude images via DVC processing of GP tissue under compression in the (i) XZ, (ii) YZ and (iii) XY plane, The arrows point to the interfaces with significant displacement changes, scale bar = 1 mm; C Views of (i) 3D strain distribution of GP tissue when compression is applied, the arrows represent the displacement direction, scale bar = 1 mm, and expanded views of the (ii) GP-epiphysis interface, scale bar = 200 μm, and (iii) GP-metaphysis interface, scale bar = 200 μm, processed by DVC; D Schematic of AFM analysis of the GP-Epiphysis and GP-metaphysis interface; E, F The 3D modulus distribution map of all selected areas from GP to epiphysis/metaphysis. Source data are provided as a Source Data file.
Next, we evaluated the modulus transition from the GP to the epiphysis/metaphysis using atomic force microscopy (AFM) (Fig. 1D). The tissue modulus increased sharply from 130.70 ± 36.56 MPa in the resting zone of the GP to 11.92 ± 6.60 GPa in the epiphysis (Fig. 1E, Figs. S4A, B, E–G, S5). In contrast, the GP-metaphysis interface demonstrated a relatively progressive increment from 416.20 ± 107.18 MPa in the hypertrophic zone of the GP to 3.22 ± 1.53 GPa in the metaphysis (Fig. 1F, Figs. S4C, D, E–G, S5). The distinct modulus transition patterns were consistent with the differential mechanical responses at the GP-epiphysis and GP-metaphysis interfaces.
Distinct CaP mineralization patterns at two interfaces
We then observed the structural and compositional transition at the soft-hard interfaces, including the GP-epiphysis and GP-metaphysis interfaces (Fig. 2A–G, Fig. S6A), using scanning electron microscopy (SEM), density-dependent color-SEM (DDC-SEM), focused ion beam-SEM (FIB-SEM) and energy-dispersive X-ray spectroscopy (EDX). At the GP-epiphysis interface, the porous and loose GP cartilage in the resting zone underwent a sharp transformation into dense and mature epiphyseal tissue (Fig. 2H, Fig. S6B). Meanwhile, at the GP-metaphysis interface, densely packed collagen fibers in calcified zones gradually mineralized into bone tissue, with minerals transitioning from immature spherical minerals to crystal platelets (Fig. 2I, Fig. S6C). FIB-SEM also showed extrafibrillar spherical minerals at the mineralization front, which fused across the interfibrillar space and permeated the collagen fibrils to transform into a fully mineralized metaphysis region (Fig. S7). EDX line scans and mapping of calcium (Ca) and phosphorus (P) distribution further highlighted the differences between the sharp elemental transformation across the GP-epiphysis interface (Figs. 2J, L) and the gradual transition across the GP-metaphysis interface occurring over a longer distance (Figs. 2K, M). Moreover, the minerals in the epiphysis region exhibited Ca/P ratio analogous to mature HAp crystals, and this value remained consistent throughout the epiphysis (zone 1, 1.763 ± 0.067; zone 2, 1.693 ± 0.050; zone 3, 1.660 ± 0.043) (Figs. S5D, S6F). In contrast, the Ca/P ratios of the metaphysis tissue near the mineralization front (zone 1, 2.205 ± 0.190; zone 2, 1.930 ± 0.072) were significantly higher than those far from the front (zone 3, 1.612 ± 0.085) (Fig. S6E, G).

A The schematic illustration of human tibia growth plate, with resting GP-epiphysis interface and active GP-metaphysis interface; B X-ray image of human knee joint with GP; C Representative photograph of GP, epiphysis and metaphysis in tibia; D SO staining of GP, scale bar = 200 μm, (n = 3/group from 10 donors); E The DDC-SEM micrograph of GP with epiphysis and metaphysis tissue, scale bar = 100 μm; F, G Enlarged DDC-SEM micrographs of the GP-epiphysis interface and GP-metaphysis interface, scale bar = 10 μm; H, I Representative enlarged SEM images from the GP to epiphysis and the GP to metaphysis, blue arrows pointing to the sharp transformation from the GP to epiphysis in (H) and mineral particles in (I), scale bar = 400 nm; J, K The EDX line scanning from (J) the epiphysis to GP and (K) the GP to metaphysis, showing Ca and P distribution across the interfaces, the red dotted line marked the interface region; L, M Enlarged SO staining image and SEM image of (L) the GP-epiphysis interface and (M) the GP-metaphysis interface, as well as corresponding EDX mapping of Ca, P and O, the dotted lines showing the regions of elemental transition, scale bar = 100 μm; SO, Safranin-O staining; (n = 3/group from 9 donors in D–I, L, M). Source data are provided as a Source Data file.
We performed stimulated Raman scattering (SRS) imaging to further evaluate the mineral phase and the spatial distribution of components such as glycosaminoglycans (GAGs), proteins, lipids and especially minerals across the two interfaces (Fig. S8). The abrupt increase of PO43− v1 symmetric stretching peak at 960 cm−1 indicated that the GP-epiphysis interface underwent a sharp transition from non-mineralized GP cartilage to fully mineralized epiphysis occupied by carbonated crystalline HAp (Fig. 3A, B, Fig. S9A). In contrast, the GP-metaphysis interface displayed a distinct pattern: a noticeable shift in the broad peak of the PO43− v1 band occurred at 950 cm–1 and 955 cm–1, which finally transformed into the narrow peak at 960 cm−1 (Fig. 3C, Fig. S9B), indicating the possible existence of an amorphous phase and the gradual maturation of mineral deposits from amorphous to crystalline minerals across this interface39,40,41,42. The appearance of the peak at 955 cm–1 may be caused by overlapping peaks of ACP at 950 cm–1 and HAp at 960 cm–142. Additionally, SRS mapping validated the presence of a region enriched with ACP at the frontier of the GP-metaphysis interface, which subsequently mineralized into HAP (Fig. 3D). These results align with the observed spherical structures in SEM and high Ca/P ratios at the GP-metaphysis interface. Closer inspection of the mineral spatial distribution showed an increase in HAp crystallinity, substituted carbonate content and mineral-to-collagen ratio, as well as a decrease in CO32–/PO43– ratio across the GP- epiphysis interface (Fig. 3E–G). In contrast, a more gradual trend was observed from the GP to the metaphysis (Fig. 3H–J).

A, C Raman spectra collected in the 300 ~ 1300 cm−1 from GP to (A) epiphysis and (C) metaphysis at different area (zone 1–6) in (B) and (D), The information of marked Raman shifts: PO43− v1 band at 950 ~ 960 cm−1, PO43− v2 band at 430 ~ 433 cm−1 and PO43− v4 band at 573 ~ 590 cm−1 and CO32–v1 band at 1071 cm–1; B, D The maps of ACP (950 cm-1), ACP/HAp interfering peaks (955 cm–1), HAp (960 cm–1) and GAG (1410 cm–1) in B) GP-epiphysis interface and D) GP-metaphysis interface by SRS imaging, scale bar = 10 μm; E, H Raman peak intensity maps of PO43– v1 band at 960 cm–1 and corresponding maps of full-widths at half-maximum (FWHM) of the peak at 960 cm-1 at (E) GP-epiphysis interface and (H) GP-metaphysis interface, scale bar = 5 μm; F, I The maps of CO32– v1 band at 1071 cm–1 and CO32–/ PO43– ratio at (F) GP-epiphysis interface and (I) GP-metaphysis interface, revealing the ionic substitution of minerals in both interfaces, scale bar = 5 μm; G, J The mineral to collagen maps (960 cm–1 to 1595 ~ 1700 cm–1) of G) GP-epiphysis interface and (J) GP-metaphysis interface, revealing relative content and distribution of HAp, scale bar = 5 μm. Source data are provided as a Source Data file.
Disparate chemical environments contribute to distinct CaP transformation patterns at two interfaces
To uncover the nature of transitioning minerals at the two interfaces, we investigated nanoscale CaP mineral assembly by cryo-transmission electron microscopy (cryo-TEM) and selected area electron diffraction (SAED). The nanocrystals with diameters ranging from 5–10 nm appeared at the frontier of the GP-epiphysis interface (Fig. 4Ai, Bi), which gradually transformed into bulk platelets (Fig. 4A–C, Figs. S10, S11, S12A, S13). Differently, at the mineralization front of the GP-metaphysis interface, amorphous clusters with total dimensions of 150–200 nm, consisting of nanometer-sized building blocks, strands of spherical units, and nodules, were observed (Fig. 4Di, Ei, Figs. S14A, S15A, S16A, B and Fig. S17A)6,31. These structures corresponded to the spherical structures and amorphous phase shown in Figs. 2,3, respectively. As mineralization progressed, the ACP-like clusters mineralized into poorly crystalline nanoparticles with diameters of 5–10 nm, which continued to grow by oriented particle attachment, a process frequently found in the early stages of crystallization in the non-classical mineralization pathway31,32,43 (Fig. 4Dii, Eii, F, Figs. S12B, S14B–D, S15B, C). These nanoparticles eventually transformed into single bulk crystals. The crystal platelets then aggregated into mineral spherulites (Fig. 4Diii, Figs. S14E, S15D, E, S16C, D), which spread across the collagen fibrils and grew larger until complete mineralization of the fibrils (Fig. 4Div, Eiv, Figs. S14F, G, S15F, S16E–G). Similar mineral assembly processes at the GP-epiphysis and GP-metaphysis interfaces were also observed in GP samples of human phalange tissue (Figs. S13, S16, S17).

A, B Representative cryo-TEM images from GP to epiphysis and reconstructed tomographic pictures of cryo-TEM images, scale bar = 100 nm; C Schematic illustration of crystal assembly process at the GP-epiphysis interface; D, E Representative cryo-TEM images from GP to metaphysis and reconstructed tomographic pictures of cryo-TEM images, scale bar = 100 nm, (n = 3/group from 7 donors in (A, B, D, E); F Schematic illustration of the crystal assembly process at the GP-metaphysis interface; G, H EELS spectra taken at P L23-edge, C K-edge, Ca L23-edge, N K-edge and O K-edge collected from different mineral particles in zone 1-4 in (I) and (J); I, J EELS maps corresponding to the spatial distribution of Ca in magenta (collected at Ca L23-edge), O in yellow (collected at O K-edge) and N in cyan (collected at N K-edge) of selected HAADF-STEM images in Fig. S18, from (I) GP-epiphysis and (J) metaphysis, scale bar = 20 nm. Source data are provided as a Source Data file.
To further elucidate the local chemical microenvironment of each mineral phase at two GP interfaces, an analysis using high-angle annular dark-field-scanning transmission electron microscopy (HAADF-STEM) equipped with electron energy loss spectroscopy (EELS) was performed (Table S2). The signal intensity of calcium (Ca L23 edge) and phosphate (P L23 edge) increased across both interfaces, showing the increasing inorganic mineral content (Fig. 4G, H). Simultaneously, the carbonate and nitrogen signals were more intense at the front of both soft-hard interfaces and became weaker as the mineral matured (Fig. 4G, H). Moreover, carbonyl groups (peak B) and nitrogen (peak B) signals were interspersed in ACP-like structures and poorly crystalline minerals at the GP-metaphysis interface (Fig. 4I, J, Figs S18, 19), which may be attributed to the presence of proteins either within or surrounding the minerals39. Thus, disparate chemical environments, such as proteins and GAGs, may contribute to the distinct CaP transformation patterns dat two interfaces.
The biomolecule-based regulatory mechanism of GP-guided polarized long bone elongation
Next, we performed liquid chromatography-tandem mass spectrometry (LC-MS/MS) to reveal regulatory proteins at the two interfaces (Fig. 5A, Fig. S20). Among the 4678 proteins detected, 33 were significantly enriched at the GP-epiphysis interface compared to GP, and 18 exhibited significantly higher expression levels compared to epiphysis. These enriched proteins at the GP-epiphysis interface are mainly grouped under cartilage matrix and ECM modulators and mineralization inhibition regulators, playing a critical role in maintaining cartilage matrix homeostasis and delaying mineralization (Fig. S21, Table S3). For the GP-metaphysis interface, 15 proteins were significantly enriched compared to GP and 10 proteins compared to metaphysis, indicating that the GP-metaphysis interface serves as a key region for balancing cartilage cytoskeletal maintenance and the regulation of mineralization (Fig. S22, Table S4).

A Experimental procedure for LC-MS/MS. The GP samples from 3 biological replicates were divided into five groups: epiphysis (E), GP-epiphysis (GP-E), GP, GP-metaphysis (GP-M) and metaphysis (M) tissue; B, C Volcano plot showing proteins with differential expression between interfaces and GP tissue/bone tissue (epiphysis and metaphysis). Dotted lines indicate FDR < 0.05 and |log₂FC | > 1. Two-sided moderated t-tests were performed using the limma package with Benjamini–Hochberg correction; SPP1, AHSG, ENPP1 and ALPL have been highlighted in the volcano plot; D Representative images of GP samples immunostained for AHSG (magenta) /calcium (cyan) and SPP1(magenta) /calcium (cyan) at the GP-epiphysis interface; E Representative images of GP samples immunostained for AHSG/calcium, SPP1/calcium, ENPP1 /calcium and ALPL/calcium at the GP-metaphysis interface, scale bar = 20 μm, (n = 3/group from 3 donors in (D, E); F Summary diagram of the macromolecule-based regulatory mechanism of GP-guided polarized long bone elongation; G Representative cryo-TEM and SAED images of prepared ACP after 1 d and 42 d of reaction at 37 °C, scale bar = 200 nm; H Representative images of STEM images, EDS mapping images of Ca, P, N, O and SAED pattern of the ACP particles after 1 d and 42 d of reaction at 37 °C, scale bar = 100 nm/50 nm, (n = 3 per time point). Source data are provided as a Source Data file.
Next, we mainly focused primarily on proteins associated with mineralization regulation. Notably, secreted phosphoprotein 1 (SPP1) and alpha 2-HS glycoprotein (AHSG) were significantly enriched at the GP-epiphysis interface compared to the GP (Fig. 5B), which was further confirmed by immunofluorescence staining (Fig. 5D, Fig. S23A, B). SPP1 and AHSG are able to chelate Ca ions to inhibit HAp nucleation and growth44, leading to the suppression of mineralization at the GP-epiphysis interface. Enrichment of SPP1 and AHSG was also found at the GP-metaphysis interface compared to GP tissue (Fig. 5C, E, Fig. S23C, D). Besides, Ecto-nucleotide pyrophosphatase phosphodiesterase 1 (ENPP1) and alkaline phosphatase (ALPL) are highly expressed at the front of the GP-metaphysis interface (Figs. 5C, E, Fig. S23E, F). ENPP1 can hydrolyze adenosine triphosphate (ATP), generating adenosine monophosphate (AMP) and pyrophosphate (PPi), which is then hydrolyzed by ALPL to inorganic phosphate (Pi), thus providing phosphate sources to facilitate the mineralization process at the GP-metaphysis interface45. In coordination with Ca ions provided by SPP1 and AHSG, proteins expressed at the GP-metaphysis interface provide a biomineralization-promoting condition for rapid bone lengthening.
In addition to the biomacromolecules, ions such as iron (Fe2+) and strontium (Sr2+), detected by FIB-SEM equipped with time-of-flight secondary ion mass spectrometry (TOF-SIMS), were only enriched in a 10 μm-wide region at the front of the GP-epiphysis interface compared to the GP-metaphysis interface (Fig. S24).
Based on published studies and our findings, we outline the macromolecule-based regulatory mechanism that governs GP-guided polarized long bone elongation (Figs. 5F, 6) and attempt to manipulate the mineralization process, including maintaining a stable ACP phase and controlling ACP-HAp transformation in vitro, using these regulatory proteins. ATP was added to provide the phosphorous source in this system. After sufficient reaction of ATP and ENPP1, ALPL was added to the solution to produce Pi by hydrolysis of PPi. ASHG was added to the CaCl2 solution to form calciprotein particles and regulate calcium-phosphate deposition. Subsequently, the two solutions were mixed to produce calcium phosphate precursors (Fig. S25A). After 5 min of reaction at 37 °C, clusters with obvious contrast were observed by cryo-TEM, which revealed spherical amorphous particles with a diameter of 50–150 nm after 30 min and exhibited typical ACP morphologies46 after 2 h (Fig. S25B). SAED and EDS mapping further confirmed the non-crystalline state of the prepared ACP particles. In this system, the biomolecule-stabilized ACP precursors can maintain their amorphous feature for at least 35 days at 37 °C (Fig. S26). After 42 days of reaction at 37 °C, ACP precursors underwent phase transition and precipitated into HAp crystals, producing needle-like and platelet-like mature morphologies (Fig. 5G, H). These outcomes collectively reveal that ACP was successfully fabricated by proteins found at the GP-metaphysis interface in the human knee joint.

The inhibitory proteins, SPP1 and AHSG, form a line of defense against mineralization in GP-epiphysis interface. When combining with the mineralization-promoting enzymes, ENPP1 and ALPL at the GP-metaphysis interface, these proteins accelerate the formation of ACP and its transformation to HAp and facilitating the progression of the mineralization front.
To assess the ACP’s ability to transform into HAp on-demand, we introduced additional ALPL into the stabilized ACP to produce sufficient Pi (Fig. S27), thus facilitating further mineralization. HR-TEM results revealed that ACP began forming a crystalline structure within 7 days and fully transformed into HAp within 14 days, demonstrating its ability to transition from storage to functional material when needed (Fig. S28).
These results highlight the translational potential of our highly stable ACP as a bioinspired material for bone regeneration. Its mild synthesis conditions, extended stability, and ability to transition into functional HAp make it an ideal candidate for on-demand biomedical applications.
Discussion
Previous research highlighted the adaptive nature of bone tissue structure and mineralization in response to external loading28,29,47,48. In the case of GPs, studies have shown that the mechanical properties of different zones contribute significantly to tissue adaptation and growth25,26. In this study, our results show that the GP-epiphysis interface undergoes a sudden change in mechanical properties while the GP-metaphysis interface exhibits a gradient transition. The epiphysis, with its remarkable stiffness, likely acts as a protective shell and prevents catastrophic damage to the resting zone in GPs upon impact, thus ensuring the survival of the stem cell reservoir and facilitating bone development48. The stiffness gradient from the GP to the metaphysis facilitates load redistribution, which has been reported to be beneficial for biomineralization during bone lengthening49,50. During long bone lengthening, bone tissue dynamically responds to mechanical stress and optimizes its structures and compositions28,29. Our research extends this understanding by examining how the different mechanical properties of the epiphysis and metaphysis contribute to distinct adaptive responses towards mechanical loading. These responses lead to variations in structural transformations and biomineralization performance at these two soft-hard interfaces, suggesting that applying different patterns of external mechanical loading could be a promising approach for regulating mineralization in vitro.
Previous studies have observed CaP particles and their assembly in the ECM within GPs15,51,52, which share a remarkable similarity to the minerals observed in this study. However, the nanoscale mineral assembly has not been addressed in prior research. The fractal-like hierarchical architecture of minerals at the nanoscale in mature human bone tissue has been well investigated53. By demonstrating the nano HAp crystal assembly during biomineralization in developing human bone, our research complements the existing knowledge and provides a more comprehensive view of the structural integrity of human bone tissue (Fig. 4). Moreover, Raman results (Fig. 3) revealed two different mineralization distribution patterns at the GP-epiphysis interfaces (sharp HAP distribution) and the GP-metaphysis interface (gradual ACP-HAp transformation), which were largely associated with their distinct mechanical microenvironments (Fig. 1) and chemical environments (Figs. 2, 4).
ACP is widely accepted as the vital precursor and intermediate phase during biomineralization. Although the presence of ACP in ECM vesicles of developing bone has been reported a few decades ago15,46, the limited availability of samples and characterization methods have hindered the full investigation of the ACP-like phase in human bone tissue. While previous studies have provided quantitative and qualitative insights into collagen, proteoglycans, mineral composition, carbonate levels, and crystallinity in the GP and surrounding bone tissues by Raman and Fourier transform infrared (FTIR) spectroscopy54,55,56, they have not reported the detection of an amorphous phase during the mineralization process, especially in human samples. In this study, we confirmed the existence and transformation of ACP as the mineralization precursor during human bone lengthening by multiple high-resolution imaging technologies (Figs. 3, 4). It’s widely recognized that ACP precursors are released from intracellular vesicles via exocytosis and organelles, with mitochondria30 and lysosomes57. One limitation of our research is the lack of investigation into the connection between ACP precursors and cellular behavior during GP mineralization.
Non-collagenous proteins (NCPs) have been regarded as key factors in stabilizing ACP and regulating biomineralization58,59. However, the specific proteins involved and their cooperative mechanisms during biomineralization remain unclear. Based on published studies and our findings, we propose a macromolecule-based regulatory mechanism that governs GP-guided polarized long bone elongation (Figs. 5F, 6). At the GP-epiphysis interface, the abundant calcium and phosphate ions in the ECM are prevented from precipitation by biomineralization inhibitors like SPP1 and AHSG44,60. Moreover, the ion distribution, such as Fe2+ and Sr2+ around the GP-epiphysis interface, may also play a role in inhibiting crystal growth6,61 and maintaining the inhibition state at this interface. Together, these factors establish an additional line of defense against mineralization. While at the GP-metaphysis interface, enrichment of SPP1 and AHSG likely contributes to the transient stabilization of ACP and poorly crystalline minerals, preventing prematurely rapid HAp formation during GP mineralization. Furthermore, the inhibitory effect of these two proteins on calcium phosphate mineralization is abolished by the enzymatic activity of the resident cells36,60. PPi produced by hydrolysis of ATP by ENPP145,62 at the beginning of GP mineralization could maintain an extracellular bioreservoir of phosphate. As the mineralization progresses, PPi is actively enzymatically cleaved by ALPL into Pi63,64. The enzymatic activity of ALPL on PPi not only abolishes the inhibitory effect on biomineralization but also generates phosphate ions to promote mineralization. PPi or Pi-packed granules chelate calcium ions and form the disordered calcium phosphate as mineral precursors and promote intrafibrillar mineralization of bone tissue. Nevertheless, several limitations exist in our study. The manual dissection process may introduce residues from adjacent tissues into the interface, which could mask proteomics data variations and potentially account for the differences in replicates. Moreover, the regulatory mechanism of polarized GP mineralization proposed in our study is speculative. To further elucidate the in situ protein distribution in their biological context and their interaction with resident cells at high resolution, advanced technologies such as correlated light microscopy and electron microscopy (CLEM) need to be employed in multiple replicate samples in the future.
In recent years, constructing ACP to achieve biomimetic mineralization has been a hot topic in the field of bone tissue engineering. ACP has been successfully stabilized by proteins8,10, polymers59,65, small molecules66 and ions6,67. However, the reconstructed ACP stabilized by protein segments and polymers can only maintain its metastable state for several hours or days8,68. In addition, the biocompatibility of the ACP is limited by the acidic condition or toxic stabilizing agents69. In this study, based on the speculative mineralization mechanism at the GP interfaces, we fabricated highly-stable ACP that maintained its amorphous state for over 35 days at 37 °C with pH of 7.0–7.4, through strategic protein combination. Stabilizing ACP under mild physiological conditions for such a long period advances our understanding of controlled mineralization processes, preventing pathological mineralization and developing biomedical materials for in bone regenerative medicine and tissue engineering. By adding ALPL protein, the ACP particles were transformed into HAp in vitro. By adjusting the concentration and combination of these proteins and different ions, we aim to create ACPs with adaptable sizes, structures, assemblies and orientations, tailored for diverse applications in future bone regenerative medicine.
Methods
Sample preparation
Ethical approval for this study was obtained from the Second Affiliated Hospital of Zhejiang University Ethics Committee (2022LSYD0923) and Second Hospital of Shanxi Medical University Ethics Committee (Ethics No. 2019YX260). Written informed consent was obtained from the legal guardians of all donors, in accordance with institutional guidelines. The growth plate (GP) samples were procured from patients undergoing amputation surgery due to osteosarcoma or trauma (n = 6) or from individuals with polydactyly (n = 4). All data were fully anonymized prior to analysis. Only sex and age ranges were used in reporting (Table S1), with no inclusion of personal identifiers, clinical centers, or rare disease identifiers that would allow participant re-identification. All specimens were carefully selected to ensure the absence of pathological tissue, and a small section was extracted for histological examination to confirm their normalcy. The GPs, along with the epiphyseal and metaphyseal tissues, were meticulously harvested, washed with sterile phosphate-buffered saline (PBS), and stored at -80 °C for subsequent analysis. Each tibial sample was divided into four portions: one for histology and immunofluorescence staining, another for high-resolution analyses (including SEM, Cryo-TEM, FIB, AFM, and others), a third for XRM, and the final portion for LC-MS analysis (Fig. S29). Due to size limitations, phalange samples were divided into only two parts: one for histology and the other for high-resolution analyses such as SEM and TEM.
Histology and immunofluorescence staining of GP, GP-epiphysis interface and GP-metaphysis interface
To prepare for histological and immunofluorescence staining, GP samples underwent fixation in 4% (w/v) paraformaldehyde for 24 to 48 h. For Safranin O staining (Solarbio), the samples required decalcification in ethylenediaminetetraacetic acid (EDTA) for 3 weeks, followed by dehydration in graded ethanol, clearing in xylene, and paraffin embedding. Samples were sliced (7 μm) using a Leica slicer. For IF staining, non-decalcified samples were cryo-sectioned (10 μm) and blocked in 5% bovine serum albumin (BSA) for 1, 2 h. Then, the samples were left to incubate with primary antibodies overnight at 4 °C, followed by PBS washing, and then incubated with fluorescein-conjugated secondary antibodies (Abcam) for 1.5 h at 37 °C. DAPI (Beyotime) was used for visualizing the cell nuclei. Observation was done with a Zeiss LSM 880 confocal microscope. Primary antibodies used were: SPP1 (Santa, sc-21742), AHSG (Proteintech, 66094-1-Ig), ALPL (Proteintech, 11187-1-AP), ENPPI (Abcam, ab223268). Mineralized tissue was visualized with 10 μM calcein (DOJINDO) for 20 min at room temperature, which enabled clear observation of the mineralized structures under confocal imaging conditions.
XRM
As described previously, the GP samples (n = 2, 0.5 mm long × 0.5 mm wide × 1 cm height, with GP, epiphysis and metaphysis tissue) were obtained from the healthy tissue of osteosarcoma patients. The samples were trimmed into a cube with plain epiphysis and metaphysis surfaces. Subsequently, the samples underwent 1% compressive strain in the axial direction, and the sample were scanned by an X-ray Microscope (Xradia 620 Versa, Zeiss) before and after straining, obtaining three-dimensional structural information of the sample. To ensure the integrity of the samples and maintain their biomechanical properties, XRM imaging was conducted under fully hydrated conditions. During the scanning process, the samples were immersed in PBS at room temperature. This immersion was maintained for the entire duration of the scan to prevent dehydration. Scan parameter settings are listed as follows: The scan field of view was 11.28 mm high and 11.28 mm wide. Images were collected before and after the GP sample was compressed by 105 μm under a preload of 4.25 N, waiting 15 mins for load relaxation and then micro-CT scanning (5.64 μm per voxel) was performed with the stabilized sample. The total scan time was 2.1 h. The 3D datasets of static and strained sample were visualized, processed and analyzed using the Digital Volume Correlation (DVC) module in Amira 6.5 (Thermo Fischer). Spatial information of local displacements and strain magnitude was calculated and presented in correlation with the sample morphology according to the software manual.
Methods for 3D DVC analysis using amira software
DVC analysis was conducted with Amira software in accordance with the guidelines offered by Thermo Fisher Scientific (https://www.youtube.com/watch?v=mQdvNcLKRig&list=PLoxdPzacxPYjDVMD4tPCaVbuQjxYizr_g&index=3). Initially, the 3D data of both undeformed and deformed samples underwent processing. Non-sample components were eliminated, and the volumes of interest, such as those containing the GP, epiphysis, and metaphysis tissues, were extracted. The average displacement level was determined by comparing 2D slices of the undeformed and deformed volumes. Subsequently, a global DVC mesh was constructed from the undeformed volume data, with the cell size configured to approximately four times the maximum displacement. The DVC global approach was then initiated, employing the reference mesh of the undeformed volume data and correlating it with the deformed volume. Regularization was incorporated, and the max iteration was set to 20 while the convergence criteria were set to 0.001. The displacement was visualized by enabling the Displacement View module, presenting displacement data alongside sample slices. The strain along the z-axis (Ezz) was extracted and displayed in conjunction with undeformed sample slices and the 3D sample surface view. Additionally, a cube was sectioned from the interior to exhibit the strain distribution on the xz and yz planes, accompanied by a colormap legend where positive values denote tensile strain and negative values denote compressive strain.
AFM
The Cypher atomic force microscope (Oxford Instruments Asylum Research, USA) was performed to describe the micromechanical properties of both GP-bone interfaces (n = 3 for each interface). The 150 μm cryo-sectioned samples underwent washing and immersion in PBS for subsequent measurement. A grid of 32 × 32 pixels in 5 × 5 μm area was measured by silicon nitride cantilevers (AC160TS-R3, Olympus) with a tip radius of 9 ± 2 nm and a spring constant of 26 N/m. The speed of ramping was set at 3 μm/s until reaching a force of 6 μN, followed by retracting the tip at the same speed. The Hertz model with a conical tip was applied to analyze the Force-Displacement (FD) data for fitting Young’s modulus. The Origin software was used to analyze the data and real map images. 4 areas were measured across each interface.
SEM, DDC-SEM and EDX analyses
For scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) analysis, cryo-sectioned GP samples (30 μm), including continuous epiphysis, epiphysis-GP interface, GP, GP-metaphysis interface and metaphysis tissue were rinsed with double distilled water to remove optimal cutting temperature compound (OCT) and then dehydrated by a graded ethanol (20% to 100%) for 30 min in each solution. Next, the samples underwent gold sputtering before observation (HITACHI SU5000), 5 kV accelerating voltage for collecting secondary electrons (SE) and observation of microstructure. Density-dependent color SEM (DDC-SEM) images were acquired at 10 kV, employing both backscattered electron (BSE) mode and SE mode. DDC-SEM images were processed using Image J software, with the green for SE images, the red for BSE images, and two channels stacked to produce single images. Additionally, EDX spectra were collected in point, line, and mapping modes to analyze the elemental compositions of the interfaces. EDX spectra were collected in point, line, and mapping modes to analyze the elemental compositions of the interfaces.
Sample preparation, data acquisition, processing, and three-dimensional image reconstruction of FIB-SEM
The frozen samples of the GP-metaphysis interface (2 mm × 2 mm × 2 mm) were fixed in a 2.5% (w/v) glutaraldehyde solution for over 12 h at 4 °C. Following this, the samples underwent a series of treatments: rinsing in PBS three times for 15 min each, immersion in a solution comprised of 2% osmium tetroxide and 3% potassium ferrocyanide (mixed in a 1:1 ratio) for 1 h at 4°C, followed by triple rinsing in double-distilled water for 10 min each. Subsequently, the samples were treated with a 1% (w/v) thiocarbohydrazide solution for 20 min, followed by triple rinsing in double-distilled water for 10 min each. After fixation in a 2% (w/v) osmium tetroxide solution for a duration of 30 min at ambient temperature and another round of triple rinsing in double-distilled water, the samples were submerged in a 1% (w/v) uranyl acetate solution for an overnight period (over 12 h) at 4 °C. After washing with double-distilled water for 10 min, 3 times, the samples underwent dehydration using a series of ethanol concentrations (30%, 50%, 70%, 90%, and 100% twice), with each solution applied for 30 min. Then the samples were moved into 100% acetone solution for 20 min, twice. Next, the samples underwent gradient penetration in epoxy resin and were embedded in resin.
The resin blocks were trimmed by ultramicrotome (Leica) until the surface of the sample in the resin blocks became visible. SEM imaging (Thermo Fisher, Teneo VS) was utilized to locate the region of interest, followed by imaging with a dual beam SEM (Thermo Fisher, FIB Helios G3 UC) once the area of interest was identified. After identifying the area of interest, the serial-surface view mode was employed with a slice thickness of 5 nm at 30 keV and 0.79 nA. In each serial face, backscatter mode (BSE) imaging was conducted using a 2 kV acceleration voltage and a current of 0.2 nA, employing an IVD detector. The resolution of each image was 3072 × 2048 pixels, with 15 μs and 4.25 nm per pixel.
The image stacks were first carefully aligned and denoised using the anisotropic diffusion function available in Amira Software 2019 (Thermo Fisher). Following this, a threshold-based segmentation method for volume rendering was applied to ensure the accurate representation of collagen and mineral structures. For the analysis of mineral tesselles, label analysis tools within Amira were used to measure their aspect ratios. In addition, to achieve clear 3D visualization, the image stacks underwent consistent alignment and processing, with threshold parameters tailored to ensure the best possible distinction between relevant structures.
Stimulated Raman scattering microscopy (SRS)
SRS was conducted in liquid state with a commercial SRS microscope (Multimodal Nonlinear Optical Microscopy System, UltraView, Zhendian (Suzhou) Medical Technology Co., Ltd, China), equipped with the InSight X3 (Spectra-Physics/Newport; pulse width, <120 femtoseconds; tuning range, 680 to 1300 nm) femtosecond laser as light source, supplying tunable pimp beam and fixed Stokes beam. The tested samples (n = 3) were observed by a microscope equipped with 20 X NA 0.8 objective (Olympus) lens and an SRS detection module. The pixel count of each image was 512 × 512. The mapping of ACP (950 cm−1), ACP/HAp compounds (955 cm−1), HAp (960 cm−1), GAG (1410 cm−1), protein (1660 cm−1 and 2925 cm−1), lipid (2850 cm−1) were analyzed by ImageJ and customized software (SpecFinder). And the final exhibited images were cropped from the original 512 × 512-pixel images.
Raman spectroscopy
Raman spectroscopy was performed under liquid conditions. The samples (n = 3) were cryo-sectioned into 30 μm without fixation. After washing with double distilled water to remove OCT, the samples underwent observation utilizing a confocal Raman microscope (LabRAM Odyssey) that was outfitted with a 532 nm excitation wavelength and a grating with 1800 grooves per millimeter. The scanning was conducted over a 20 μm × 20 μm square area with 1600 points. The spectra were gathered within the range of 200 ~ 1800 cm−1 utilizing an electron multiplying charge-coupled device (EMCCD) detector, featuring a spectral resolution of approximately 0.5 cm−1. The mapping images were obtained by continuous scanning of 1600 points in a 20 μm region for each image with an accumulation time of 0.5 s per point. The HAp contents (960 cm−1), CO32− substitution (1071 cm−1), mineral crystallinity (full width at half maximum, FWHM of 960 cm−1peak), and CO32−/PO43− ratios in mapping images were analyzed by LabSpec software.
Cryo-TEM observation and tomographic reconstruction
For Cryo-TEM observation and tomographic reconstruction, the GP-epiphysis interface and GP-metaphysis interface samples (about 2 mm each sample, and 3 samples for each interface) were prepared by high-pressure freezing (HPF) combined with freeze substitution (a mixture of 0.1% osmium tetroxide, 0.1% uranyl acetate, 0.5% glutaraldehyde, 1.5% H2O and 100% acetone). The high-pressure freezing (HPF) was carried out using a Leica EM ICE system. The samples were placed into carriers (Leica 16770141, Type A) with 1-hexadecene as the cryoprotectant. Freeze substitution was conducted using the Leica EM AFS2 apparatus with a substitution mixture. After substitution, the samples were embedded in Spurr resin for further analysis. Then the specimens were sectioned to 150 nm-thick onto bare 100-mesh copper grids by an ultramicrotome (Leica EM UC7) with a cryo-chamber to maintain the samples under −150 °C. The slices were observed in a cryo-TEM (FEI Talos F200C 200 kV). For tomographic reconstruction, regions of interest were imaged by tilting the grid in 2° steps from 56° to −56°. The weighted back-projection method was utilized for tomographic reconstruction. Segmentation and 3D visualization were performed using Amira 2019.1 (Visage Imaging Inc., Andover, MA, USA).
TEM, HR-TEM, SAED, STEM and EELS analyses
For TEM, HR-TEM, STEM, EELS and SAED analyses, the sample preparation was the same as Cryo-TEM. The samples were sectioned to 100 nm-thick onto bare 100-mesh copper grids by an ultramicrotome (Leica EM UC7) for further observation. Slices containing GP-epiphysis and GP-metaphysis were prepared separately. The slices underwent imaging in a transmission electron microscope (TEM) with spherical aberration correction (FEI Titan G2 80–200), which was furnished with an EELS detector, operating at 80 kV. Regions of interest underwent TEM, HR-TEM, and SAED pattern analyses. The areas for EELS analysis were localized by HAADF-STEM imaging. For EELS mapping, a whole EELS spectrum was acquired at each area. EELS spectra were acquired within the range of 200 ~ 600 eV to investigate the characteristic edges of elements (P, C, Ca, N, O). Each EELS mapping image contained about 20000 spectra. Principal component analysis (PCA) was applied for spectrum calibration, normalization, background subtraction, and processing. Gatan Digital Micrograph software was employed for the analysis of STEM images and EELS.
Sample preparation for proteomics
Entire bulk growth plate tissue (n = 3) was isolated to accurately segment the growth plate regions for proteomics analysis, as depicted in Fig. S30. From the visual structure of the growth plate, the epiphysis and metaphysis were easily distinguishable and were separated for proteomic analysis. Then samples were cryo-sectioned into 100 μm, and after sectioning, the slices were segmented by scalpel blade under a stereomicroscope into five parts: epiphysis tissue, epiphysis-GP interface tissue, GP tissue, GP-metaphysis tissue and metaphysis tissue (Fig. S31). About six 100 μm-thick slices were required for each group. The cryo-sectioned samples were segmented manually using a stereoscope, which enables discrimination of five types of tissues. For digestion, the samples of each group were transferred to 0.6 mL Ep tube and then diluted with 20 μL of 100 mM NH4HCO3 for 10 min at 95 °C. Next, 1 μL of trypsin (1 μg/μL) was added to the samples for overnight digestion (12 h) at 37 °C. Following digestion, any remaining debris was eliminated via centrifugation at 14,000 g for 15 min at 4 °C. The supernatant containing peptides was collected for further experiments. Peptides were quantified using a Nanodrop spectrophotometer (ND-2000C, Thermo) and equal amounts of peptides were taken for desalting. The pH of the peptide solution was adjusted to 2–3 by the addition of 20% trifluoroacetic acid (TFA) (Macklin). Desalting was performed using 1.9 μm Reprosil-Pur C18 beads (Dr. Maisch, Ammerbuch, Germany) according to the manufacturer’s instructions, with equilibration by 20 μL of 0.1% TFA. After equilibrating by 20 μL of 0.1% TFA, the samples were eluted with 0.1% TFA in 80% acetonitrile (Thermo) and subsequently dried using a vacuum concentrator for further analysis.
LC-MS/MS analysis
For LC-MS/MS analysis, tryptic peptides were solubilized in 0.1% formic acid (Thermo Fisher) and immediately introduced onto a specialized reversed-phase analytical column filled with 1.9 μm Reprosil-Pur C18 beads (Dr. Maisch, Ammerbuch, Germany). During the process, the gradient elution involved a gradual rise from 3% to 8% solvent (0.1% formic acid in 98% acetonitrile) over 3 min, followed by increases to 20% over 37 min, then to 30% over 12 min, and finally reaching 80% over 4 min, maintaining this level for the last 4 min. This process was conducted at a consistent flow rate of 300 nL/min using an UltiMate 300 nanoLC system. Next, the peptides underwent NSI source initiation, followed by tandem mass spectrometry analysis using the Orbitrap Exploris 480 (Thermo Fisher), which was integrated with the Ultra Performance Liquid Chromatography (UPLC) system for online coupling. The electrospray voltage was adjusted to 2.0 kV. The full scan mass-to-charge range spanned from 400 to 1200, with intact peptides detected in the Orbitrap at a resolution of 60,000. Peptides were subsequently chosen for LC-MS/MS analysis, employing a normalized collision energy (NCE) setting of 27, and ensuing fragments were identified in the Orbitrap with a resolution of 15,000. A data-dependent approach was employed, alternating between a single MS scan and 20 MS/MS scans with a dynamic exclusion of 30 s. Automatic gain control (AGC) was configured at 5E4. The compensation voltages for FAIMS were adjusted to −45V and −65V.
Database search for proteomics
The LC-MS/MS data were processed using the MaxQuant search engine (version 1.6.15.0). The tandem mass spectra were compared against the Uniprot Human database concatenated with the reverse decoy database. Trypsin was designated as the cleavage enzyme, permitting a maximum of 2 missed cleavages. The mass deviation for precursor ions was defined as 20 ppm during the initial search and 5 ppm during the primary search, while the mass deviation for fragment ions was set at 0.02 Da. A fixed modification of carbamidomethyl on Cys and a variable modification of oxidation on Met were stipulated. The label-free quantification approach (LFR) was employed, with the Benjamini–Hochberg FDR adjusted to below 1%. Peptides were required to achieve a minimum score exceeding 40.
Proteomic analysis
The LC-MS/MS data was processed using the MaxQuant search engine (version 1.6.15.0). Initially, the tandem mass spectra were aligned with the Uniprot Human database in conjunction with a reversed decoy database. Trypsin/P was employed as the enzyme for protein cleavage, permitting up to 2 potential missed cleavages. During the initial search, the precursor ion mass tolerance was established to 20 ppm, while for the main search, it was tightened to 5 ppm. Additionally, the tolerance for fragment ion mass was set at 0.02 Da. A fixed modification of carbamidomethyl on cysteine was designated, while oxidation on methionine was treated as a variable modification. The LFQ method was applied for label-free quantification, with the FDR adjusted to <1%, and a minimum peptide score threshold of > 40 was established.
The DEP package (version 1.20.0) was employed for downstream analysis in R (version 4.3.3). Protein intensities were first filtered to remove: (1) reverse database matches, (2) common contaminants (from MaxQuant’s contaminant database), and (3) proteins identified solely by site-specific modifications. Proteins with at least two valid intensity values in ≥50% of samples within at least one experimental group were retained. Missing values (defined as intensities below the detection limit in technical replicates) were imputed using the K-nearest neighbors algorithm (k = 10, Euclidean distance metric, with 100 iterations and a fixed random seed set.seed = 123). Variance-stabilizing normalization (VSN) was applied to reduce technical variation, as it effectively stabilizes variance across a wide range of intensity values and minimizes heteroscedasticity commonly observed in label-free quantification datasets. Differential expression analysis was performed using the limma-voom framework with empirical Bayes moderation. Significant proteins were defined as those with an adjusted p-value < 0.05 (Benjamini-Hochberg FDR) and an absolute log2 fold change > 1. For visualization, the top 500 proteins were selected based on the highest absolute moderated t-statistic values to maximize group discrimination. Volcano plots were generated with FDR-adjusted p-values on the −log₁₀ scale and log₂ fold changes on the x-axis, using ggplot2 to highlight proteins meeting significance criteria in red. Heatmaps were clustered using Euclidean distance and complete linkage clustering for both samples and proteins, with row-wise Z-score normalization to visualize expression patterns. PCA plots were constructed from variance-stabilized intensities of the top 500 discriminatory proteins, with samples colored by tissue type and ellipses representing 95% confidence intervals. Quality control metrics included: (1) Pearson correlation matrices of sample intensities to assess technical replicate consistency, (2) distribution plots of missing values per sample to identify outliers, and (3) principal component analysis of all detected proteins to verify biological group separation.
TOF-SIMS analysis
The sample preparation for TOF-SIMS mirrors that of FIB-SEM. Utilizing FIB-SEM combined with TOF-SIMS (Thermo Fisher, Helios 5 UX), the distribution and relative abundance of chemical constituents within the samples were analyzed. Throughout the examination, the samples’ surfaces encountered pulses of gallium ion beams. The resulting secondary ions were extracted at a voltage of 10 kV, and a reflection mass spectrometer was utilized to gauge their time of flight from the samples to the detector. Each region measured 100 × 100 μm, comprising 256 × 256 pixels, with 500 scans conducted per area. Both positive and negative ion mass spectra were acquired. The two-dimensional chemical heatmaps with a color-coded scale, showing the intensities of detected secondary ion signals and indicating the relative ion abundance of the scanned area, were analyzed and obtained by TOF-SIMS Explore software.
Preparation of biomolecule-stabilized ACP
Buffer solution 1 was prepared by dissolving 140 mM NaCl (Aladdin) and 50 mM Tris in deionized water, and the pH of buffer solution 1 was adjusted to 7.4 with HCl (Diamond). Calcium solution was prepared by dissolving 40 mM CaCl2 (Aladdin) in buffer solution 1. Buffer solution 2 was prepared by dissolving 100 mM Tris-HCl (Diamond), and 5 mM MgCl2 (Sigma-Aldrich) in deionized water, and the pH of buffer solution 2 was adjusted to 9.0 with NaOH (Sigma-Aldrich). Thereafter, all approaches were conducted in a clean bench. Buffer solution 1, calcium solution, and buffer solution 2 were sterilized by percolating the solutions through a 0.22 µm membrane filter. AHSG solution as well as ALPL solution were prepared by dissolving 2 mg/mL active recombinant human fetuin A/AHSG protein, and 1 mg/mL recombinant human ALPL protein in buffer solution 1, respectively. ENPP1 solution was prepared by dissolving 0.5 mg/mL recombinant human ENPP1 protein in buffer solution 2. AHSG, ALPL and ENPP1 were purchased from ABclonal.
In reaction solution A, 5 µL of 100 mM ATP solution (Novoprotein), 32 uL ENPP1 solution, and 3 uL buffer solution 2 were mixed and incubated at 37 °C for 1 h. Then 10 uL of ALPL solution was added, followed by a 10-min incubation. In reaction solution B, 5 µL AHSG solution, 25 µL calcium solution, and 20 µL buffer solution 1 were mixed and incubated at 37°C for 10 min. The 50 µL reaction solution A and reaction solution B were mixed in a 1:1 ratio and incubated at 37 °C. At 5 min, 15 min, 30 min, 1 h, 2 h, 12 h, 1 d, 3 d, 5 d, 7 d, 10 d, 14 d, 21 d, 28 d, 35 d and 42 d, samples of the mixture were collected with a 300-mesh gold support grid (Zhongjingkeyi Technology Co., China) for TEM.
Regulation of ACP to HAp
The regulation process was based on the above-described preparation of biomolecule-stabilized ACP. After mixing reaction solution A and reaction solution B at a 1:1 ratio, an additional volume of ALPL solution was added to achieve a final ALPL concentration of 200 µg/mL in the entire system. The mixture was then incubated at 37 °C. Samples were collected at 1, 4, 7, and 14 days and subsequently characterized using Cryo-TEM, SAED and EDS mapping to observe the phase transformation from ACP to HAp.
Cryo-TEM, STEM, SAED and EDS mapping of ACP
The samples of reconstructed ACP for Cryo-TEM, STEM imaging and EDS mapping were prepared as said above. The ACP was collected in 300-mesh gold support grids for further cryo-TEM and STEM observation. The ACP samples were observed in a cryo-TEM (FEI Talos F200C) at 200 kV. The samples for STEM were imaged in a field emission TEM (JEOL JEM-F200) at 80 kV. STEM-EDX mapping (Ca, P, N, C) and SAED were performed in the areas of interest.
Phosphate ion release assay
5 uL of 100 mM ATP solution was mixed with 32 uL 0.5 mg/mL ENPP1 solution and 3uL buffer solution (140 mM NaCl and 50 mM Tris, pH = 7.4). After 1 h incubation at 37 °C, 10 uL of 1 mg/mL ALPL was added into the reaction solution, and phosphate release was measured after 2 h, 4 h, and 6 h. Phosphate concentration was quantified with a Phosphate Assay Kit (MAK308-1KIT, Sigma-Aldrich) following the protocol. In brief, the reaction solution was diluted with DI water by gradient, and mixed with the Malachite Green Reagent by the volume ratio of 1:2. After 30 min of incubation at room temperature, the absorbance at 620 nm was measured with a spectrophotometer (NanodropTM One, Thermo Scientific). The phosphate concentration of the reaction solution was calculated by contrasting the absorbance to the standard curve. Theoretically, 10 mM ATP should produce 20 mM Pi; however, 25.4 mM Pi was observed. This suggests that a certain concentration of phosphate may be present in the lyophilized protein powder, likely originating from the buffer in the original protein solution.
Statistics & reproducibility
Investigators were blinded to group allocation throughout data collection and analysis. Unless specified otherwise in figure legends, all experiments, including those yielding representative images such as micrographs, were independently repeated at least three times with similar results using three biological replicates. Data are presented as mean ± SD. Statistical differences between group means were analyzed using Origin software (Version 2024). For comparisons involving more than two groups, a one-way ANOVA with Tukey’s post hoc test was used. Comparisons between two groups were performed using two-way ANOVA followed by Tukey’s post hoc test. A p-value < 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated in this study are provided in the Supplementary Information file/Source Data file. The mass spectrometry proteomics data generated in this study have been deposited in the iProX database (http://www.iprox.org) under accession code IPX0010900000. Source data are provided with this paper.
References
Arnold, A. et al. Hormonal regulation of biomineralization. Nat. Rev. Endocrinol. 17, 261–275 (2021).
Gal, A. et al. Macromolecular recognition directs calcium ions to coccolith mineralization sites. Science 353, 590–593 (2016).
Estefa, J. et al. New light shed on the early evolution of the limb-bone growth plate and bone marrow. Elife 10, 1–30 (2021).
Dey, A. et al. The role of prenucleation clusters in surface-induced calcium phosphate crystallization. Nat. Mater. 9, 1010–1014 (2010).
Lotsari, A., Rajasekharan, A. K., Halvarsson, M. & Andersson, M. Transformation of amorphous calcium phosphate to bone-like apatite. Nat. Commun. 9, 4170 (2018).
Chen, S. et al. Formation of amorphous iron-calcium phosphate with high stability. https://doi.org/10.1002/adma.202301422 (2023).
Golafshan, N. et al. Tough magnesium phosphate-based 3D-printed implants induce bone regeneration in an equine defect model. Biomaterials 261, 120302 (2020).
Chang, R. et al. Phosphorylated and phosphonated low-complexity protein segments for biomimetic mineralization and repair of tooth enamel. Adv. Sci. 9, 1–16 (2022).
Wang, D. et al. Controlling enamel remineralization by amyloid-like amelogenin mimics. Adv. Mater. 32, 1–13 (2020).
Jahnen-Dechent, W. & Pasch, A. Solving the insoluble: calciprotein particles mediate bulk mineral transport. Kidney Int. 103, 663–665 (2023).
Shen, M. et al. Extracellular DNA: a missing link in the pathogenesis of ectopic mineralization. Adv. Sci. 9, 1–13 (2022).
Shen, M. et al. Multifunctional nanomachinery for enhancement of bone healing. Adv. Mater. 34, 1–12 (2022).
Mahamid, J. et al. Bone mineralization proceeds through intracellular calcium phosphate-loaded vesicles: Aa cryo-electron microscopy study. J. Struct. Biol. 174, 527–535 (2011).
Fernández-Iglesias, Á. et al. The formation of the epiphyseal bone plate occurs via combined endochondral and intramembranous-like ossification. Int. J. Mol. Sci. 22, 1–16 (2021).
Varsano, N. et al. Characterization of the growth plate-bone interphase region using cryo-FIB SEM 3D volume imaging. J. Struct. Biol. 213, 107781 (2021).
Kerschnitzki, M. et al. Bone mineralization pathways during the rapid growth of embryonic chicken long bones. J. Struct. Biol. 195, 82–92 (2016).
Behrooz, A., Kask, P., Meganck, J. & Kempner, J. Automated quantitative bone analysis in in vivo x-ray micro-computed tomography. IEEE Trans. Med. Imaging 36, 1955–1965 (2017).
Ağirdil, Y. The growth plate: a physiologic overview. EFORT Open Rev. 5, 498–507 (2020).
Tiffany, A. S. & Harley, B. A. C. Growing Pains: the need for engineered platforms to study growth plate biology. Adv. Healthc. Mater. 11, 1–19 (2022).
Melrose, J., Shu, C., Whitelock, J. M. & Lord, M. S. The cartilage extracellular matrix as a transient developmental scaffold for growth plate maturation. Matrix Biol. 52–54, 363–383 (2016).
Voller, T., Cameron, P., Watson, J. & Phadnis, J. The growth plate: anatomy and disorders. Orthop. Trauma 34, 135–140 (2020).
Mizuhashi, K. et al. Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature 563, 254–258 (2018).
Newton, P. T. et al. A radical switch in clonality reveals a stem cell niche in the epiphyseal growth plate. Nature 567, 234–238 (2019).
Xie, M. et al. Secondary ossification center induces and protects the growth plate structure. Elife 9, 1–32 (2020).
Radhakrishnan, P., Lewis, N. T. & Mao, J. J. Zone-specific micromechanical properties of the extracellular matrices of growth plate cartilage. Ann. Biomed. Eng. 32, 284–291 (2004).
Amini, S. et al. Stress relaxation of swine growth plate in semi-confined compression: depth dependent tissue deformational behavior versus extracellular matrix composition and collagen fiber organization. Biomech. Model. Mechanobiol. 12, 67–78 (2013).
Sergerie, K., Lacoursière, M. O., Lévesque, M. & Villemure, I. Mechanical properties of the porcine growth plate and its three zones from unconfined compression tests. J. Biomech. 42, 510–516 (2009).
Chen, J. H., Liu, C., You, L. & Simmons, C. A. Boning up on Wolff’s Law: mechanical regulation of the cells that make and maintain bone. J. Biomech. 43, 108–118 (2010).
Christen, P. et al. Bone remodelling in humans is load-driven but not lazy. Nat. Commun. 5, 4855 (2014).
Pei, D. et al. Contribution of mitophagy to cell-mediated mineralization: revisiting a 50-year-old conundrum. Adv. Sci. 5, 1800873 (2018).
Habraken, W. J. E. M. et al. on-association complexes unite classical and non-classical theories for the biomimetic nucleation of calcium phosphate. Nat. Commun. 4, 1507 (2013).
De Yoreo, J. J. et al. Crystallization by particle attachment in synthetic, biogenic, and geologic environments. Science 349, aaa6760 (2015).
Fleisch, H., Russell, R. G. G. & Straumann, F. Effect of pyrophosphate on hydroxyapatite and its implications in calcium homeostasis. Nature 212, 901–903 (1966).
Novais, E. J. et al. A new perspective on intervertebral disc calcification—from bench to bedside. Bone Res. 12, 1–12 (2024).
Goettsch, C. et al. TNAP as a therapeutic target for cardiovascular calcification: A discussion of its pleiotropic functions in the body. Cardiovasc. Res. 118, 84–96 (2022).
Reznikov, N., Steele, J. A. M., Fratzl, P. & Stevens, M. M. A materials science vision of extracellular matrix mineralization. Nat. Rev. Mater. 1, 16041 (2016).
Nudelman, F. et al. The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat. Mater. 9, 1004–1009 (2010).
Madi, K. et al. In situ characterization of nanoscale strains in loaded whole joints via synchrotron X-ray tomography. Nat. Biomed. Eng. 4, 343–354 (2020).
Nitiputri, K. et al. Nanoanalytical electron microscopy reveals a sequential mineralization process involving carbonate-containing amorphous precursors. ACS Nano 10, 6826–6835 (2016).
Stammeier, J. A., Purgstaller, B., Hippler, D., Mavromatis, V. & Dietzel, M. In-situ Raman spectroscopy of amorphous calcium phosphate to crystalline hydroxyapatite transformation. MethodsX 5, 1241–1250 (2018).
Akiva, A. et al. On the pathway of mineral deposition in larval zebrafish caudal fin bone. Bone 75, 192–200 (2015).
Akiva, A. et al. Mineral formation in the larval zebrafish tail bone occurs via an acidic disordered calcium phosphate phase. J. Am. Chem. Soc. 138, 14481–14487 (2016).
Penn, R. L. & Banfield, J. F. Imperfect oriented attachment: Dislocation generation in defect-free nanocrystals. Science 281, 969–971 (1998).
Jahnen-Dechent, W. & Smith, E. R. Nature’s remedy to phosphate woes: calciprotein particles regulate systemic mineral metabolism. Kidney Int. 97, 648–651 (2020).
Kato, K. et al. Crystal structure of Enpp1, an extracellular glycoprotein involved in bone mineralization and insulin signaling. Proc. Natl Acad. Sci. USA 109, 16876–16881 (2012).
Chen, S. et al. Formation of amorphous iron-calcium phosphate with high stability. Adv. Mater. 35, 1–11 (2023).
Cusack, M. & Freer, A. Biomineralization: elemental and organic influence in carbonate systems. Chem. Rev. 108, 4433–4454 (2008).
Huang, W. et al. A natural impact-resistant bicontinuous composite nanoparticle coating. Nat. Mater. 19, 1236–1243 (2020).
Kim, S. et al. In situ mechanical reinforcement of polymer hydrogels via metal-coordinated crosslink mineralization. Nat. Commun. 12, 1–10 (2021).
Milazzo, M., Jung, G. S., Danti, S. & Buehler, M. J. Mechanics of mineralized collagen fibrils upon transient loads. ACS Nano 14, 8307–8316 (2020).
Haimov, H. et al. Mineralization pathways in the active murine epiphyseal growth plate. Bone 130, 115086 (2020).
Wu, L. N. Y., Genge, B. R., Kang, M. W., Arsenault, A. L. & Wuthier, R. E. Changes in phospholipid extractability and composition accompany mineralization of chicken growth plate cartilage matrix vesicles. J. Biol. Chem. 277, 5126–5133 (2002).
Reznikov, N., Bilton, M., Lari, L., Stevens, M. M. & Kröger, R. Fractal-like hierarchical organization of bone begins at the nanoscale. Science 360, eaao2189 (2018).
Eckstein, K. N. et al. The heterogeneous mechanical properties of adolescent growth plate cartilage: a study in rabbit. J. Mech. Behav. Biomed. Mater. 128, 105102 (2022).
Paschalis, E. P., Jacenko, O., Olsen, B., Mendelsohn, R. & Boskey, A. L. Fourier transform infrared microspectroscopic analysis identifies alterations in mineral properties in bones from mice transgenic for type X collagen. Bone 19, 151–156 (1996).
Bozkurt, O., Bilgin, M. D., Evis, Z., Pleshko, N. & Severcan, F. Early alterations in bone characteristics of type i diabetic rat femur: a fourier transform infrared (FT-IR) imaging study. Appl. Spectrosc. 70, 2005–2015 (2016).
Iwayama, T. et al. Osteoblastic lysosome plays a central role in mineralization. Sci. Adv. 5, eaax0672. (2019).
Thrivikraman, G. et al. Rapid fabrication of vascularized and innervated cell-laden bone models with biomimetic intrafibrillar collagen mineralization. Nat. Commun. 10, 3520 (2019).
Wu, H. et al. Hyaluronic acid-mediated collagen intrafibrillar mineralization and enhancement of dentin remineralization. Carbohydr. Polym. 319, 121174 (2023).
Depalle, B. et al. Osteopontin regulates type I collagen fibril formation in bone tissue. Acta Biomater. 120, 194–202 (2021).
Bussola Tovani, C. et al. Formation of stable strontium-rich amorphous calcium phosphate: possible effects on bone mineral. Acta Biomater. 92, 315–324 (2019).
Li, L. et al. Hydrolysis of 2'3'-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol. 10, 1043–1048 (2014).
Somerman, M. J. et al. Counter-regulatory phosphatases TNAP and NPP1 temporally regulate tooth root cementogenesis. Int. J. Oral Sci. 7, 27–41 (2014).
Andleeb, H., Hussain, M., Abida, S. & Sevigny, J. Bioorganic Chemistry Synthesis and computational studies of highly selective inhibitors of human recombinant tissue non-specific alkaline phosphatase (h- TNAP): a therapeutic target against vascular calcification. Bioorg. Chem. 101, 103999 (2020).
Hao, J. et al. A seminal perspective on the role of chondroitin sulfate in biomineralization. Carbohydr. Polym. 310, 120738 (2023).
Shao, C. et al. Citrate improves collagen mineralization via interface wetting: a physicochemical understanding of biomineralization control. Adv. Mater. 30, 1–7 (2018).
Li, Y. et al. Mechanically reinforced artificial enamel by Mg2+-induced amorphous intergranular phases. ACS Nano 16, 10422–10430 (2022).
He, J. et al. Polyzwitterion manipulates remineralization and antibiofilm functions against dental demineralization. ACS Nano 16, 3119–3134 (2022).
Yao, S. et al. Osteoporotic bone recovery by a highly bone-inductive calcium phosphate polymer-induced liquid-precursor. Adv. Sci. 6, 1900683 (2019).
Acknowledgments
The authors acknowledged the financial support from the National Key Research and Development Program of China (2023YFB3813000), and the National Natural Sciences Foundation of China (No. T2121004, 82394441, 92268203, 32371411), and the Key Research and Development Program of Zhejiang (2024SSYS0026). The authors would extend their gratitude to Mr. Jiadan Wu and Ms. Junyan Xie (The Second Affiliated Hospital, Zhejiang University) for their assistance on growth plate samples collection. The authors would like to thank Mr. Lu Lan and Mr. Shoupu Yi (Multimodal Nonlinear Optical Microscopy System, UltraView, Zhendian, Suzhou) for their assistance on stimulated Raman scattering microscopy. The authors also thank Mr. Jiansheng Guo (Center of Cryo-Electron Microscopy, Zhejiang University) for his assistance with FIB-SEM, Ms. Lingyun Wu (Center of Cryo-Electron Microscopy, Zhejiang University) for her assistance with Cryo-TEM and Beibei Wang for her assistance with TEM and ultrathin slicing. The authors would like to thank Ms. Guoqing Zhu from the Center of Electron Microscopy of Zhejiang University for her technical assistance on spherical aberration corrected TEM (FEI Titan G2 80-200) characterization and Ms. Qingyun Lin from the Center of Electron Microscopy of Zhejiang University for her technical assistance on F20 TEM characterization. The authors also thank Mr. Pengda Zou and Ms. Minghui Li (Mass Spectrometry Core Facilities, The First Affiliated Hospital, Zhejiang University School of Medicine) for their assistance with LC-MS. The authors also thank Mrs. Chunjie Cao, Ms. Biyu Chen and Ms. Xi Lin from Carl Zeiss AG for their assistance in XRM and data analysis. The authors also thank Ms. Pei Sheng and Mr. Yangjian Lin (Instrumentation and Service Center for Physical Sciences, Westlake University) for their assistance in TOF-SIMS and data analysis.
Author information
Authors and Affiliations
Contributions
C.X. and W.L. contributed equally to this work. Conceptualization: C.X., X.W., and H.O. Methodology: C.X., W.L., B.W., J.F., R.L., J.Z., H.M., and Y.Y. Investigation: C.X., W.L., X.Y., and B.W. Resources: W.D., Y.W., X.D. Visualization: C.X., W.L., X.Y., B.W., R.M., and Y.Y. Supervision: H.O. and X.W. Writing (original draft): C.X., W.L., X.Y., and B.W. Writing (review & editing): C.X., X.Z., X.W., and H.O. All the authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, C., Li, W., Yao, X. et al. Physical and chemical niche of human growth plate for polarized bone development. Nat Commun 16, 7328 (2025). https://doi.org/10.1038/s41467-025-62711-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62711-z
