
Abstract
Myelin, along with the oligodendrocytes (OLs) that produce it, is essential for proper central nervous system (CNS) function in vertebrates. Although the accurate targeting of myelin to axons and its maintenance are critical for CNS performance, the molecular pathways that regulate these processes remain poorly understood. Through a combination of zebrafish genetics, mouse models, and primary OL cultures, we find that FBXW7, a recognition subunit of an E3 ubiquitin ligase complex, is a regulator of adult myelination in the CNS. Loss of Fbxw7 in myelinating OLs results in increased myelin sheath lengths with no change in myelin thickness. As the animals age, they develop progressive abnormalities including myelin outfolds, disrupted paranodal organization, and ectopic ensheathment of neuronal cell bodies with myelin. Through biochemical studies we find that FBXW7 directly binds and degrades the N-terminus of Myelin Regulatory Factor (N-MYRF), to control the balance between OL myelin growth and homeostasis.
Similar content being viewed by others


Introduction
In the vertebrate central nervous system (CNS), myelin is produced by specialized glial cells called oligodendrocytes (OLs). OLs wrap segments of axons, creating a multi-layered sheath that speeds the transmission of nerve impulses and provides critical support to axons1,2,3,4. The formation and targeting of myelin is influenced by external cues such as axonal caliber and neuronal activity, but is also tightly controlled by cell-intrinsic programs5,6,7,8,9,10,11,12. As OL precursor cells (OPCs) differentiate from a dynamic and proliferative state into a relatively stable post-mitotic myelinating OL, they reorganize and expand their cytoskeleton, cytoplasm, and membrane13,14,15, requiring significant transcriptional changes16,17,18,19. Although OLs are long-lived, surviving up to years in mice and decades in human white matter tracts, the myelin constituents themselves turn over comparatively rapidly, with a half-life of months20,21,22,23. Individual myelin sheaths can also be remodeled throughout life12,24,25. Therefore, understanding the molecular pathways and mechanisms that balance myelin growth and homeostasis is crucial for understanding myelin’s role in health, aging, and disease.
Previous work from our lab and others has identified F-box and WD repeat domain-containing protein 7 (FBXW7) as a key negative regulator of developmental myelination by both Schwann cells in the peripheral nervous system (PNS) of mice and OLs in the CNS of zebrafish26,27,28,29. Within the PNS, FBXW7 regulates Schwann cell numbers, along with their myelin thickness28. Surprisingly, loss of Fbxw7 also results in a breakdown of the normal 1:1 relationship between myelinating Schwann cells and axons, with individual Fbxw7 conditional knockout Schwann cells aberrantly myelinating multiple axons28. Within the CNS of zebrafish, fbxw7 regulates neural stem cell fate through Notch signaling, biasing the cells towards an OPC fate and increasing the pool of OL lineage cells in the spinal cord26. At later stages, loss of fbxw7 leads to increased myelin sheath length, attributed to dysregulation of mTOR signaling27. Fbxw7 encodes the F-box domain-containing recognition subunit of a SKP1-Cullin-Fbox (SCF) E3 ubiquitin ligase complex. It mediates its biological effects through targeting specific proteins for proteasomal degradation, thus controlling their total levels in the cell26,27,30,31,32. FBXW7 substrates are highly variable between cell types and have not yet been investigated in myelinating cells in an unbiased manner.
To interrogate the role of FBXW7 in the regulation of CNS myelination, we used a combination of zebrafish, primary mammalian OL cultures, and conditional knockout mouse models. We found that inactivation of fbxw7 in developing zebrafish resulted in enhanced OL maturation in the spinal cord. Conditional ablation of Fbxw7 in mature OLs in the adult mouse CNS increased myelin sheath lengths but also resulted in progressive myelin abnormalities including outfolds, disrupted paranodal organization, and ectopic ensheathment of neuronal cell bodies with myelin. We found that Fbxw7-deficient OLs had no changes in mTOR protein levels in primary mammalian OL cultures, suggesting the myelin phenotypes were not a consequence of dysregulated mTOR signaling. Previous work in hepatocarcinoma cells identified the pro-myelination transcription factor Myelin Regulatory Factor (MYRF) as a target of FBXW733. We demonstrate that the N-terminus of MYRF is a direct target of FBXW7 in OLs both in vitro and in vivo, with levels of N-MYRF protein and many of its transcriptional targets substantially increased in Fbxw7-deficient OLs. We also found that Myrf haploinsufficiency is sufficient to ameliorate both the increase in OL numbers in fbxw7 knockout zebrafish, as well as myelin outfolds and ectopic myelination of granule cell in the cerebellum in our conditional knock-out mouse models. Taken together, our findings demonstrate that FBXW7 is an evolutionarily conserved negative regulator of OL myelination and that its negative regulation of MYRF in the adult CNS is required for long-term myelin homeostatic maintenance.
Results
Fbxw7 regulates OPC specification and OL myelination in the zebrafish spinal cord
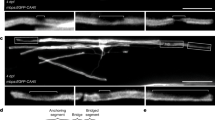
In zebrafish, global fbxw7 mutations cause hypermyelination in both the PNS and CNS26,27,28. Previously published zebrafish fbxw7 mutant alleles are late mutations within the region of the gene encoding the WD40 substrate recognition domain of the FBXW7 protein; therefore, existing mutants may not be complete loss-of-function alleles26,29. We used CRISPR-Cas9-mediated genome editing to create a new mutation, fbxw7vo86. This mutation introduces a frameshift and early stop codon in exon 5, which encodes the F-Box domain that allows FBXW7 to interact with its E3 complex (Supplementary Fig. 1a, b). Through in situ hybridization, live imaging, and qPCR, we found that the fbxw7vo86 mutation phenocopies the previously described N-ethyl-N-nitrosourea (ENU)-generated fbxwstl64 mutants, including an increase in myelin basic protein (mbp) driven GFP in the dorsal spinal cord of Tg(mbp:EGFP-caax) animals, and an increase in total mbp RNA by qRT-PCR (Fig. 1a, b and Supplementary Fig. 1c–e). Since fbxw7vo86 phenocopies the original ENU-generated mutation, we concluded that both of these mutations represent full loss-of-function alleles. As the fbxw7vo86 mutation disrupts the F-Box domain, the same region targeted in the Fbxw7fl/fl mouse line34 also used in our studies (described below), this allele was used for all subsequent analyses.

a Spinal cords of fbxw7vo86 and WT controls in (top) Tg(mbp:eGFP-caax), (middle) Tg(olig2:dsRED), and (bottom) Tg(mbp:nls-eGFP) transgenic backgrounds at 3 and 5 days post-fertilization (dpf; dotted-line delineates dorsal and ventral spinal cord tracts). b mbp RNA levels evaluated by qRT-PCR in 5 dpf fbxw7vo86 and WT control whole larvae. Mean ± SEM, N = 3 (larvae). Statistical significance determined by unpaired, two-tailed Student’s t test. c Quantification of olig2:dsRED+ cells/body segment in the dorsal spinal cord at 2, 3, and 5 dpf. Mean ± SEM, N = 7 (larvae). Statistical significance determined by two-way ANOVA. d Quantification of mbp:nls-eGFP+ OLs in the dorsal spinal cord/body segment at 3 dpf. Mean ± SEM, N = 7 (larvae). Statistical significance determined by unpaired, two-tailed Student’s t test. e mbp-nls:eGFP+ OLs from Fig. 1d were divided by the average number of olig2:dsRED+ cells/body segments in dorsal spinal cord at 3 dpf from Fig. 1c. Mean ± SEM, statistical significance determined by unpaired, two-tailed Student’s t test. f Spinal cords of fbxw7vo86 and WT control zebrafish in Tg(mbp:eGFP-caax) and Tg(mbp:nls-eGFP) transgenic backgrounds live-imaged at 3 dpf. Solid arrowheads denote OL somas, hollow arrowheads denote neuronal cell body wrapping. g Representative image of an fbxw7vo86 larva in Tg(mbp:eGFP-caax) (white) and Tg(nbt:dsRED) (magenta) backgrounds showing neuronal cell bodies in the spinal cord wrapped in mbp+ membrane. h Quantification of wrapped neuronal cell bodies in the spinal cord in both fbxw7vo86 and WT controls at 3 dpf. Mean ± SEM, N = 5 (larvae). Statistical significance determined by unpaired, two-tailed Student’s t test. i Representative images of individual labeled OLs from cell-type specific CRISPR-Cas9 knock-down of fbxw7 in sox10 expressing cells in the zebrafish spinal cord at 6 dpf. The UAS-nlscas9-P2A-TagRFPT-caax construct allows visualization of Cas9-expressing cells. j Quantification of average sheath length and k number per OL in controls and with fbxw7 knockdown. Mean ± SEM, Control N = 47 (cells from 40 animals), sgRNAfbxw7 N = 36 (cells from 29 animals). Statistical significance determined by unpaired, two-tailed Student’s t test. Created in BioRender. Emery, B. (2024) BioRender.com/o45h010.
In prior work, global disruption of fbxw7 in zebrafish led to enhanced OPC specification through disinhibition of Notch1 signaling, observed as an increase in mbp expression and numbers of olig2:dsRED-expressing cells26. Consistent with this, fbxw7vo86 mutants present with a significant increase in mbp:EGFP-caax expression and olig2:dsRED+ cell numbers in the developing dorsal spinal cord relative to wild-type controls at 2-, 3-, and 5-days post-fertilization (dpf) (Fig. 1a–c). To examine later stages of the OL lineage, we crossed fbxw7vo86 mutants into a transgenic Tg(mbp-nls:eGFP) line to label mature OL nuclei. Relative to wild-type clutchmates, the density of mbp-nls:eGFP-expressing cells was significantly increased in fbxw7vo86 mutants, even when normalized to the increased numbers of OL lineage cells (olig2:dsRED+) at 3 dpf (Fig. 1a, d, e). Additionally, we found that fbxw7vo86 mutant zebrafish displayed a significant number of neuronal cell bodies in the spinal cord wrapped in mbp+ membrane in the stable Tg(mbp-nls:EGFP); Tg(mbp:EGFP-caax) transgenic background at 3 dpf (Fig. 1f–h). While this phenotype may be due to the increase in OL numbers in the zebrafish spinal cord as previously reported35, it may also suggest that loss of Fbxw7 broadly disrupts OL myelin membrane organization and targeting. To determine whether Fbxw7 could regulate myelination via an OL intrinsic mechanism, we utilized a cell-specific CRISPR-Cas9-mediated gene disruption system our lab had previously developed36. We found that sox10-driven disruption of fbxw7 in sparsely targeted OL lineage cells resulted in a significant increase in myelin sheath lengths at 6 dpf, with no change in the number of sheaths formed per individual OL (Fig. 1i–k). These data suggest that Fbxw7 regulates OL myelination through a cell-autonomous mechanism in the developing zebrafish spinal cord.
FBXW7 regulates OL myelin sheath length, paranodal organization, and myelin homeostasis in both gray and white matter
Our findings in zebrafish indicated that Fbxw7 regulates key aspects of myelin growth early in development. Whether this role is conserved in mammalian CNS and whether FBXW7 regulates myelination past these early developmental stages has not been explored to date. To address this, we created an inducible Fbxw7 knockout (icKO) mouse by crossing Fbxw7fl/fl mice34 to the Plp1-CreERT line37, allowing for tamoxifen (TAM) -inducible knockout of Fbxw7 in mature OLs. Fbxw7fl/fl; Plp1-CreERT+ mice (Plp1-CreERT -Fbxw7fl/fl) and their CreERT negative littermate controls (Fbxw7fl/fl) were treated with TAM at 8 weeks of age, and tissue was taken 1-, 3-, and 6-months post-TAM for subsequent analyses (Fig. 2a).

a Schematic of Fbxw7fl/fl and Plp1-CreERT-Fbxw7fl/fl mouse lines and experimental pipeline. b Representative images of myelin internodes in Fbxw7fl/fl and Plp1-CreERT-Fbxw7fl/fl 1-month post-tamoxifen (TAM) in mouse layer I of the primary somatosensory cortex (pSS), stained with MBP (magenta), CNP1 (green), and CASPR (cyan). Arrowheads show CASPR+ boundaries of internodes. c Quantification of sheath length from layer I pSS. Colored bolded dots represent average of ROI from individual biological replicates (N = 4 mice per genotype, average of 5 ROIs/mouse), small hollow dots represent values from individual sheaths (Fbxw7fl/fl N = 197, Plp1-CreERT-Fbxw7fl/fl N = 210). Statistical significance determined by unpaired, two-tailed Student’s t-test on ROI averages ± SEM. d Higher magnification images of myelin outfolds (arrowheads) in pSS cortex 6 months post-TAM. e Quantification of myelin outfolds in the pSS (N = 3 mice, average of 5 ROIs/mouse). Data shown as average ± SEM. Statistical significance determined by two-way ANOVA with Tukey’s multiple comparisons test. f Representative images of CASPR and ANKG stained longitudinal optic nerve sections from Fbxw7fl/fl and Plp1-CreERT-Fbxw7fl/fl mice at 6-months post-TAM. g Quantification of CASPR staining intensity as a function of distance from ANKG+ node. Thick lines represent mean for each genotype with > 5 individual heminodes quantified, thin lines represent individual heminode intensity histograms. Statistical significance calculated by nonparametric two-tailed Kolmogorov-Smirnov test (P < 0.0001). Fbxw7fl/fl total nodes= 27, Plp1-CreERT-Fbxw7fl/fl total nodes=32, averaged across 3 mice/genotype. Created in BioRender. Emery, B. (2024) BioRender.com/d36g650.
To determine if loss of Fbxw7 in mature OLs regulated myelin sheath maintenance, we sectioned cortical flat-mounts of layer I and performed immunofluorescence (IF) for (MBP; compact myelin), 2’,3’-cyclic nucleotide 3’ phosphodiesterase (CNP1; non-compact myelin), and contactin-associated protein (CASPR; paranodes). At 1-month post-TAM, Plp1-CreERT-Fbxw7fl/fl OLs showed a significant increase in myelin sheath length in the primary somatosensory cortex (pSS) (Fig. 2b, c), indicating that FBXW7 regulates myelin capacity in mature OLs. Additionally, we observed an increase in the number of MBP/CNP1+ focal hyperintensities that appeared to be myelin outfolds, which increased significantly as the animals aged (Fig. 2d, e). To investigate FBXW7’s role in white matter, we performed IF on Plp1-CreERT-Fbxw7fl/fl optic nerves at 6-months post-TAM. We found a pronounced breakdown of nodal organization with a significant (p < 0.0001) broadening in the distribution of Plp1-CreERT-Fbxw7fl/fl CASPR intensity at each heminode compared to controls (Fig. 2f, g). While Plp1-CreERT-Fbxw7fl/fl animals had no change in weight or general health (Supplementary Fig. 2a, b), we observed a significant reduction of OL numbers in the upper cortex (layers I-VI) at 6 months post-TAM in Plp1-CreERT-Fbxw7fl/fl animals relative to their age-matched control littermates (Supplementary Fig. 2c). This reduction was only observed above layer IV, with no change in OL numbers in deeper cortical layers or the corpus callosum (Supplementary Fig. 2d, e). Additionally, we found no change in OPC numbers at any of these timepoints or regions (Supplementary Fig. 2f–h), nor changes in astrocyte reactivity (Supplementary Fig. 2i). Collectively, these data suggest that FBXW7 functions in many aspects of OL biology, from early modulation of sheath lengths to long-term maintenance of paranode organization and myelin homeostasis in mammalian OLs.
Given that both gray and white matter tracts in Plp1-CreERT-Fbxw7fl/fl animals showed evidence of disrupted nodal organization and outfolds at 6-months post-TAM, we next wanted to assess the ultrastructure of the myelin. We therefore performed transmission electron microscopy (TEM) on optic nerves from Plp1-CreERT-Fbxw7fl/fl and Fbxw7fl/fl littermate controls 6 months post-TAM. Consistent with our observations in layer I of the cortex, we also found a significant increase in the number of myelin outfolds in Plp1-CreERT-Fbxw7fl/fl optic nerves (Fig. 3a, b). While control animals did have outfolds at low frequencies, as expected at 8 months of age25, the number and average length of outfolds in the Plp1-CreERT-Fbxw7fl/fl was significantly higher (Fig. 3b, c). Although myelin ultrastructure was disrupted in the optic nerve, we found no change in the proportion of axons myelinated or their corresponding g-ratios when severe outfolds were excluded from analyses (Fig. 3d, e). Along with outfolds, we also observed other myelin abnormalities throughout the optic nerve including myelin whorls, cellular accumulations, and double myelin sheaths (sheaths enveloped by an overlying sheath) (Fig. 3f–h). This double myelination was also observed by IF in layer I of the pSS cortex, where we found CASPR+ paranodes under MBP+ myelin sheaths in Plp1-CreERT-Fbxw7fl/fl mice, suggestive of double myelinated axons (Fig. 3i). The outfolds in the white matter tracts of the optic nerve and corpus callosum were so severe they were also visible at the light level by IF in Plp1-CreERT-Fbxw7fl/fl animals as hyperintense MBP+ puncta (Supplementary Fig. 3a–c). Additionally, we also observed myelin sheath blebbing and outfolds in fbxw7vo86 mutant zebrafish when injected with a sox10:eGFP-caax plasmid to label individual OLs at 5 dpf (Supplementary Fig. 3d). Taken together, these data show that FBXW7 is a conserved regulator of myelin sheath length, which was independent of myelin sheath thickness, as well as long-term maintenance of myelin homeostasis and nodal organization.

a Representative image from transmission electron microscopy (TEM) micrographs of optic nerves from Fbxw7fl/fl and Plp1-CreERT-Fbxw7fl/fl mice at 6-months post-TAM. Outfolds highlighted by arrow. b–e Quantification of number of outfolds (b), severity of outfolds (c), number of myelinated axons (d), and g-ratios (e). Fbxw7fl/fl (N = 4 mice), Plp1-CreERT-Fbxw7fl/fl (N = 3 mice). f Representative images of different myelin abnormalities in Plp1-CreERT-Fbxw7fl/fl optic nerves at 6-months post-TAM. g, h Quantification of OL myelin whorls in Fbxw7fl/fl (N = 4 mice), Plp1-CreERT-Fbxw7fl/fl (N = 3 mice) and double myelinated axons in Fbxw7fl/fl (N = 3 mice), Plp1-CreERT-Fbxw7fl/fl (N = 3 mice). All data displayed as mean ± SEM. Statistical significance determined by unpaired, two-tailed Student’s t test on animal averages. i Example image from pSS cortex of a CASPR+, CNP1+ paranode under MBP+ myelin sheath. Created in BioRender. Emery, B. (2024) BioRender.com/k49b495.
Loss of Fbxw7 in mature OLs results in ectopic ensheathment of neuronal cell bodies with myelin
Myelination within the CNS is highly targeted, with populations of axons displaying preferential degrees of myelination25,38,39 and OLs typically do not myelinate structures such as blood vessels or neuronal cell bodies40,41,42. There are also precise regional borders of myelination, as occurs in the cerebellum, which has distinct myelinated (granular layer) and non-myelinated (molecular layer) layers43. Exactly how this selective process is regulated is not well understood. We found that loss of Fbxw7 in mature OLs did not alter the tight delineation of myelination between the granular and molecular layers of the cerebellum, with the molecular layer remaining unmyelinated (Fig. 4a, Supplementary Fig. 4a). Unexpectedly, however, Plp1-CreERT-Fbxw7fl/fl animals displayed a significant number of granule cell bodies within the granular layer wrapped in MBP+ membrane starting at 3-months post-TAM, which drastically increased at 6-months post-TAM (Fig. 4a–d, and movie 1). This mistargeting of myelin appeared to be selective to the cerebellar granule cell population; we did not observe any neuronal cell bodies wrapped in myelin in the cortex, nor did we find other structures, such as blood vessels, wrapped in MBP+ membrane in any regions of the CNS analyzed. Additionally, we did not observe a change in the number of OLs in our Plp1-CreERT-Fbxw7fl/fl cerebellums compared to controls (Fig. 4e). It is important to note that Bergman glia in the cerebellum express Plp1 and, therefore, may have undergone Fbxw7 recombination in our Plp1-CreERT-Fbxw7fl/fl animals44. While we cannot exclude Fbxw7 KO Bergman glia as a contributing factor in our granule cell body ensheathment, we did not observe any obvious change in the number or morphology of the Purkinje cells, which are supported by Bergman glia, at 6 months post-TAM in the cerebellum (Supplementary Fig. 4a). Additionally, we found no change in GFAP expression (expressed by Bergman glia and astrocytes in the cerebellum) or reactivity of microglia (Supplementary Fig. 4b). To assess whether the myelin ensheathment was due to myelination of the cell bodies, or severe myelin outfolds that wrapped around the small somas of the granule cells, EM was performed of the cerebellum of the 6-month post-TAM animals. High-resolution IF imaging of MBP and NeuN (Fig. 4f) combined with TEM (Fig. 4g), showed that the observed ensheathment of the granule cells with myelin was likely due to excessive myelin outfolds in Plp1-CreERT-Fbxw7fl/fl animals. Since this ensheathment was not observed in any other area of the CNS analyzed, it is likely due to the high density and small size of the granule cell nuclei in the cerebellum. In addition to myelin outfolds, large cellular accumulations were also observed in putative Purkinje neuron axons in the Plp1-CreERT-Fbxw7fl/fl animals (Fig. 4h). These accumulations have been previously shown to occur in Purkinje axons after Purkinje cell-type specific deletions of cathepsin D (CTSD), which resulted in a significant increase in stressed and apoptotic Purkinje cells45. Taken together we have shown that loss of Fbxw7 in myelinating OLs of the cerebellum resulted in large outfolds that encircled and enwrapped the cell bodies of the granule cells, while also causing axon pathology.

a Representative images of anti-MBP stained cerebellums from Fbxw7fl/fl and Plp1-CreERT-Fbxw7fl/fl animals at 6-months post-TAM. Dotted line denotes the border of the granule layer. b Higher magnification image of MBP staining in the granular layer of a Plp1-CreERT-Fbxw7fl/fl animal showing cupped myelin structures, which surround NeuN+ nuclei (shown at higher magnification in c). d Quantification of NeuN+ cells wrapped in MBP positive membrane in the granular layer of the cerebellum of Fbxw7fl/fl (1 m: N = 4, 3 m: N = 3, 6 m: N = 4) and Plp1-CreERT-Fbxw7fl/fl mice (1 m: N = 4, 3 m: N = 3, 6 m: N = 4) at 1-, 3-, and 6-months post-TAM. e Quantification of CC1+ OLs in the white matter and granular layer of Fbxw7fl/fl (1 m: N = 4, 3 m: N = 3, 6 m: N = 4) and Plp1-CreERT-Fbxw7fl/fl mice (1 m: N = 4, 3 m: N = 3, 6 m: N = 4) at 1-, 3-, and 6-months post-TAM. Data shown as average ± SEM. Statistical significance determined by two-way ANOVA. f Image of MBP+ myelin outfold (white) around a granule cell (magenta) by IF at 6-months post-TAM reconstructed in Imaris. a and b dotted lines represent orientation of EM images in g. g Representative images of myelin outfolds around granule cell nuclei in the granule layer of the cerebellum at 6-months post-TAM. This was observed in all 4 mutant mice animals analyzed. h Images of severe cellular accumulations in the axons of putative Purkinje cells in Plp1-CreERT-Fbxw7fl/fl mice at 6-months post-TAM. This was observed in all mutant animals analyzed (N = 4). Created in BioRender. Emery, B. (2024) BioRender.com/o45h010/k49b495.
FBXW7 binds and degrades the N-terminus of MYRF
FBXW7 is a recognition subunit of the SKP1-Cullin-Fbox (SCF) E3 ubiquitin ligase complex. Its role is to recognize protein substrates following their phosphorylation at a phosphodegron motif, bringing them into the complex for ubiquitin tagging and subsequent proteasomal degradation30,31,34,46. This raised the question of which FBXW7 substrates are dysregulated in OLs after deletion of Fbxw7 to result in accelerated and ectopic myelin formation. As a preliminary analysis, we selected a set of known FBXW7 substrates including mTOR, JNK, and cJun that are also known to regulate myelination27,28,30,47 and screened them by western blot in siFbxw7-treated rat primary OL cultures. Surprisingly, we found no detectable changes in protein levels of mTOR, p-mTORser2448, JNK, or cJun (Supplementary Fig. 5a). To screen for FBXW7 substrates in OLs in an unbiased manner, we designed a dominant-negative FLAG-tagged version of FBXW7 missing its F-Box domain, driven under the CMV promoter (Fig. 5a). The F-Box domain of FBXW7 is required for its interaction with the SCF-E3 complex, allowing FBXW7 to disengage from its substrates33,46. Deletion of the F-Box domain while leaving the substrate recognition domains (WD40 repeats) intact results in a buildup of the 3xFLAG-FBXW7ΔF-Box protein bound to its substrates, allowing for effective protein-protein pulldown33. We electroporated primary rat OPCs with CMV-3xFLAG-FBXW7∆F-Box or pMax-GFP controls and differentiated them for 3 days (at which point approximately 60–70% of the Olig2+ cells are MBP+). We then lysed cells, performed co-immunoprecipitation (co-IP) with an anti-FLAG antibody and assessed the eluted proteins using unbiased Liquid Chromatography-Mass Spectrometry (LC-MS) (Fig. 5b). FBXW7 and MYCBP2, a known E3-independent negative regulator of FBXW748, were the most highly enriched proteins by LC-MS, validating the effectiveness of our pulldown. We also found that FBXW7ΔF-Box bound MAP1B (a microtubule-associated protein), RAE1 (an RNA export protein), MYRF (a pro-myelination transcription factor), and MYO1D (an unconventional myosin). Of these targets, MYRF seemed the best-placed to mediate the phenotypes seen following loss of Fbxw7, and so it became our subsequent focus.

a Schematic showing workflow of primary rat OPC isolation, expansion, and electroporation with either a dominant-negative Fbxw7 construct (CMV-3xFLAG-Fbxw7ΔFBox), pooled siRNAs against Fbxw7 or non-targeting controls (siControl). b LC-MS peptide counts for significant proteins enriched by anti-FLAG pull-down in 3xFlag-Fbxw7ΔFBox electroporated cells normalized to GFP electroporated controls. Mean ± SEM, (N = 4 independent cell isolations and IPs). Statistical significance determined by multiple unpaired, two-tailed Student’s t test. c IP-western blot for MYRF following pull down of FBXW7∆FBox from cultured rat OLs at 72 h differentiation. Representative blot shown from 3 independent experiments. d Western blot analysis of the N-MYRF cleavage product in siFbxw7 or siCont electroporated cells at 24 or 48 h differentiation. Western blots were repeated 7 times with 5 biological replicates. e Western blot of MYRF in siRNA electroporated OLs treated with cycloheximide (CHX) for 4 h. Relative intensity of the N-MYRF band quantified in f. Mean ± SEM, N = 4 independent cell isolations. Statistical significance determined by unpaired, two-tailed Student’s t test. g Representative images of MBP and MAG expression in cultured OLs differentiated for 48 or 72 h after electroporation with siControl or siFbxw7. h, i MBP+ and MAG+ OLs normalized to total OLIG2+ cells. Mean ± SEM, N = 6 independent cell isolations with 2 technical replicates (coverslips) per isolation. Statistical significance determined by two-way ANOVA. j qRT-PCR for myelin genes on siRNA treated cells. Mean ± SEM, N = 3 (independent cell isolations) with 2 technical replicates (qRT-PCR). Statistical significance determined by multiple unpaired, two-tailed Student’s t test. k Quantification of EdU incorporation in primary rat OPCs following siControl or siFbxw7 electroporation. Mean ± SEM, N = 3 independent cell isolations with 4 technical replicates (coverslips). Statistical significance determined by unpaired, two-tailed Student’s t test. l–n Proteins with +/- > 1.2 fold change were sorted by gene ontology (GO) terms “lipid metabolism,” “myelin,” and “adhesion”. N = 4 independent cell isolations with 2 technical replicates. o Table of the top 10 enriched proteins by TMT-LS/MS in siFbxw7 electroporated OLs relative to siControl electroporated OLs at 3 days differentiation. Proteins with 1 or more MYRF ChIP-Seq peaks within 50 kb of the transcription start site of their corresponding gene identified based on previously published data50. Created in BioRender. Emery, B. (2024) BioRender.com/w60g354.
MYRF is initially produced as an endoplasmic reticulum (ER)-bound transmembrane protein. It undergoes a self-cleavage event allowing the N-terminal cleavage product (N-MYRF) to translocate to the nucleus, where it works with SOX10 at the enhancers of many essential myelin genes to promote their transcription49,50,51,52. MYRF levels are tightly controlled within the OL lineage, and its expression is essential for OL differentiation as well as the production and maintenance of compact myelin17,53,54,55. Notably, MYRF has recently been reported as an FBXW7 target in hepatocarcinoma cells, suggesting the interaction may be broadly conserved across cell types33. To confirm the MYRF interaction with FBXW7 in primary OLs, we performed western blots on 3xFLAG-FBXW7∆F-Box co-IPs. Pull-down with anti-FLAG strongly enriched for endogenous N-MYRF, but not full-length MYRF (Fig. 5c). To determine the effects of this interaction on MYRF levels, we electroporated primary rat OPCs with siFbxw7 and siControl, differentiated them for 24–48 h, and blotted for endogenous N-MYRF. Knockdown of Fbxw7 led to a substantial increase in the levels of N-MYRF, consistent with FBXW7’s role in proteasomal degradation56 (Fig. 5d). To confirm that the increased N-MYRF levels were due to decreased degradation in the absence of FBXW7, we treated siControl and siFbxw7 OL cultures with cycloheximide (CHX) to inhibit protein translation. We found that after 4 h of CHX treatment, control cells had degraded the majority of both full-length MYRF and N-MYRF. In contrast, Fbxw7 knockdown cells showed little reduction in N-MYRF levels, but near complete loss of the full-length protein (Fig. 5e, f). Together, these findings strongly supported a role for FBXW7 in N-MYRF degradation.
When analyzing the N-MYRF blots we noticed two distinct molecular weights of N-MYRF separated by ~2 kDa, with the higher molecular weight band becoming more prevalent with Fbxw7 knockdown (Fig. 5d, e). Nakayama and colleagues found that phosphorylation of N-MYRF at serine 138 and 142 by GSK3β was required for FBXW7 to interact with N-MYRF33. Additional analyses of N-MYRF at serine 138 and 142 by sequence analyses predicted CDK5 as an additional potential mediator of N-MYRF phosphorylation. To determine whether build-up of a phosphorylated form of N-MYRF accounted for the observed change in molecular weight, control and siFbxw7 OL lysates were treated with a lambda phosphatase, and molecular weights were evaluated by western blot (Supplementary Fig. 5b). We found that treatment with phosphatase resulted in a near total loss of the larger molecular weight N-MYRF in both control and siFbxw7 OLs, consistent with phosphorylated N-MYRF constituting the majority of the increased N-MYRF in our Fbxw7 knockdown OLs. To determine if GSK3β or CDK5 were the kinase responsible for phosphorylating the phospodegron motif in N-MYRF to induce its interaction with FBXW7, we electroporated OPC cultures with pooled siRNAs against Gsk3b, Cdk5, or both, and differentiated them for 3 days. We found no change in the level of N-MYRF or corresponding myelin proteins (Supplementary Fig. 5c) in any of the knockdown conditions. While the GSK3β antibody showed a non-specific band around 50 kD, its predicted molecular weight band showed effective protein knockdown. Additionally, knockdown efficacy for siRNA against Fbxw7, Gsk3b, and Cdk5 was >60% as determined by qRT-PCR (Supplementary Fig. 5d). Together, these data suggest that although N-MYRF is phosphorylated in OLs and that this phosphorylated form is the target of FBXW7, this can occur through a GSK3β- and CDK5-independent mechanism.
Given MYRF’s well-defined role in OL differentiation and myelination we next wanted to investigate the functional consequences of elevated MYRF levels in Fbxw7 knockdown OLs. siFbxw7-electroporated OLs differentiated for 48–72 h showed a significant increase in the proportion of MBP+ and myelin-associated glycoprotein (MAG)+ cells compared to controls (Fig. 5g–i). In addition to an increased proportion of cells expressing myelin proteins, cultures also showed significant increases in Mbp, Mag, and Plp1 mRNA as assessed by qRT-PCR (Fig. 5j). To determine if loss of Fbxw7 in OPCs was affected proliferation, siFbxw7 and control OPCs were kept in proliferation media for 2 days after siRNA electroporation to allow for effective knockdown, then pulsed with 5-ethynyl-2’-deoxyuridine (EdU) for 6 h. Within that time, approximately 40% of OPCs had undergone a round of division, with no significant change seen in EdU incorporation between siFbxw7 or siControl treated OPCs (Fig. 5k), indicating that loss of Fbxw7 in OPCs was not sufficient to induce Myrf expression and the transition to a post-mitotic OL. These results indicate that once OPCs begin to differentiate and express Myrf, FBXW7 serves to regulate N-MYRF protein levels to control the balance and timing of OL myelination.
To further understand the consequences of loss of Fbxw7 on the OL proteome, we performed LC-MS on lysates from siFbxw7 and siControl electroporated rat OLs at 3 days of differentiation. Cell lysates were labeled with tandem mass tags (TMT), pooled, and run through LC-MS. Over 2700 proteins were sequenced with an R2 value of 0.99-1 within treatment groups. Select enriched proteins with validated antibodies were confirmed by western blot (Supplementary Fig. 5e). We found that Fbxw7 knockdown in primary OLs resulted in significant changes in 253 proteins with a false discovery rate (FDR) < 0.01 and 426 proteins with an FDR < 0.05. Within the 253 proteins with an FDR < 0.01, 158 proteins showed increased levels with Fbxw7 knockdown and 95 showed reduced levels (Data S1). Proteins with significant changes and a fold change greater than ±1.2 were sorted by Gene Ontology functions “lipid metabolism,” “myelin,” and “adhesion” (Fig. 5l–n) to provide a list of proteins with potential roles in FBXW7-dependent myelination.
During OL differentiation, N-MYRF directly binds the enhancer regions of genes underpinning myelination, with enrichment of N-MYRF chromatin-immunoprecipitation (ChIP) peaks seen within 50 kb of the transcription start sites of genes induced during OL differentiation50. Notably, when the proteins were ranked by fold change following Fbxw7 knockdown, 7 of the top 10 upregulated proteins and 73% of all proteins with fold change >1.5 had a predicted N-MYRF binding motif within 50 kb of the transcriptional start site of their corresponding gene (Fig. 5o). When we assessed RNA from corresponding samples by qPCR, we found that all the top 10 enriched proteins showed significant increases in their transcript levels with Fbxw7 knockdown, suggesting that many of the changes in protein abundance following loss of Fbxw7 were secondary to elevated MYRF transcriptional activity (Supplementary Fig. 5f). We observed that Myrf RNA was significantly down regulated in the siFbxw7 treated cells compared to controls over time, suggesting an additional mechanism(s) by which OLs regulate N-MYRF levels (Supplementary Fig. 5g).
Loss of Fbxw7 in OLs increases nuclear MYRF levels in vivo
We next sought to determine whether the myelin changes seen in Plp1-CreERT-Fbxw7fl/fl mice may be mediated by elevated MYRF levels. Since the antibody we used to detect MYRF recognizes an epitope within the N-terminal 100 amino acids of MYRF, it recognizes both the full-length form, which is bound to the ER, and the N-terminal cleavage product, which is translocated to the nucleus50,51. To determine the abundance of each, we use the cellular localization of the cytoplasmic (full-length) or nuclear (N-terminal) for quantification of MYRF levels in Plp1-CreERT-Fbxw7fl/fl and control optic nerve OLs. In Plp1-CreERT-Fbxw7fl/fl animals, we found a significant increase in the levels of nuclear-MYRF relative to their control littermates at 1- and 3-months post-Fbxw7 deletion (Fig. 6a, b, d). Subsequently, the levels of nuclear MYRF in Plp1-CreERT-Fbxw7fl/fl optic nerves returned to control levels by 6 months post-TAM. This was associated with a significant reduction in the ratio of cytoplasmic to total (nuclear + cytoplasmic) MYRF levels (Fig. 6e). Hypothesizing that this reduced ratio of full-length MYRF to nuclear MYRF may represent a homeostatic response by the OL to control MYRF protein levels in the absence of FBXW7 by down-regulating Myrf transcription, we first performed qRT-PCR for Myrf transcripts in the siFbxw7 electroporated rat OLs in vitro, finding a significant reduction relative to siControl electroporated cells from 5 days post-knock down (Supplementary Fig. 5g). To determine whether Myrf mRNA was also reduced in vivo, we isolated from optic nerves from 1- and 12-months post-TAM animals. At 1-month post-TAM there was a 62.7% reduction in Myrf transcript levels compared to controls. This reduction persisted at 12-months post-TAM, which showed a 61.5% reduction in Myrf levels compared to age-matched controls (Fig. 6f).

a Representative images of optic nerves from Fbxw7fl/fl and Plp1-CreERT-Fbxw7fl/fl mice at 1-, 3-, and 6-months post-TAM stained for MYRF. b High-resolution images showing cytoplasmic localization of MYRF in the optic nerve OLs of 6-months post-TAM animals. Arrowheads indicate cytoplasmic localization of MYRF (likely uncleaved precursor) in the Fbxw7fl/fl control. c Representative images of cerebellums from Fbxw7fl/fl and Plp1-CreERT-Fbxw7fl/fl mice at 6-months post-TAM stained for MYRF. d Quantification of nuclear MYRF intensity in Plp1-CreERT-Fbxw7fl/fl (1 m: N = 3, 3 m: N = 3, 6 m: N = 3) optic nerves normalized to controls (1 m: N = 3, 3 m: N = 3, 6 m: N = 4) at 1-, 3-, and 6-months post-TAM. Data shown as mean ± SEM. Statistical significance determined by two-way ANOVA with Tukey’s multiple comparisons test. e Ratio of cytoplasmic relative to total (cytoplasmic and nuclear) MYRF at 6-months post-TAM in Fbxw7fl/fl (N = 4) and Plp1-CreERT-Fbxw7fl/fl mice (N = 3). Data shown as mean ± SEM, statistical significance determined by unpaired, two-tailed Student’s t test. f qRT-PCR quantification of Myrf RNA fold change in the optic nerve from of Fbxw7fl/fl (1 m: N = 3, 12 m: N = 3) and Plp1-CreERT-Fbxw7fl/fl mice (1 m: N = 3, 12 m: N = 6) at 1- and 12- months post-TAM. Data shown as mean ± SEM. Statistical significance determined by two-way ANOVA with Šídák’s multiple comparisons test. g Quantification of nuclear MYRF intensity in Plp1-CreERT-Fbxw7fl/fl cerebellums normalized to controls at 1-, 3-, and 6-months post-TAM. For both genotypes, 1 m (N = 4), 3 m (N = 4), 6 m (N = 4). Data shown as mean ± SEM. Statistical significance determined by two-way ANOVA with Tukey’s multiple comparisons test. h Quantification of NeuN+ cells wrapped in MBP positive membrane in the granule cell layer of the cerebellum in 6- and 12-month post-TAMed Plp1-CreERT-Fbxw7fl/fl (1 m: N = 4, 12 m: N = 3) and control animals (1 m: N = 4, 12 m: N = 3). Data shown as mean ± SEM. Statistical significance determined by two-way ANOVA with Šídák’s multiple comparisons test. i qRT-PCR quantification of Myrf RNA fold change in the cerebellum from Fbxw7fl/fl (1 m: N = 4, 12 m: N = 5) and Plp1-CreERT-Fbxw7fl/fl (1 m: N = 3, 12 m: N = 5) mice at 1- and 12- months post-TAM. Data shown as mean ± SEM. Statistical significance determined by two-way ANOVA with Tukey’s multiple comparisons test. Created in BioRender. Emery, B. (2024) BioRender.com/o45h010/k49b495.
To determine if the increase in MYRF protein levels were seen throughout the CNS, we next evaluated MYRF levels in the cerebellum. We found significantly increased MYRF levels at 3- and 6-months post-TAM (Fig. 6c, g). Since MYRF levels remained elevated at 6-months post-TAM in the cerebellum, we wondered if this would lead to a continued accumulation of myelin ensheathed granule cells in our aged 12-month post-TAMed animals. Indeed, we found that there was a 4-fold increase in the number of ensheathed granule cell bodies between 6- and 12-months post-TAM in Plp1-CreERT-Fbxw7fl/fl animals (Fig. 6h). As in the optic nerve, there was also a significant decrease in Myrf transcript levels in the cerebellum of Plp1-CreERT-Fbxw7fl/fl animals at both 1- and 12-months post-TAM compared to age matched controls. Taken together these data demonstrate that FBXW7 negatively regulates MYRF protein levels within OLs across the life span of the animal, and that loss of FBXW7, and corresponding increases in MYRF protein levels, triggers an additional mechanism(s) that downregulates the transcription of Myrf in an attempt to balance the levels of this important transcription factor.
Myrf haploinsufficiency rescues myelin phenotype in zebrafish and mouse models
The elevation of N-MYRF protein levels in Plp1-CreERT-Fbxw7fl/fl animals, combined with the significant downregulation of Myrf RNA in vivo and in vitro, strongly suggested that elevated N-MYRF levels may underlie the aberrant myelination seen following loss of Fbxw7. Since Fbxw7 loss of function OLs appear to utilize an additional mechanism(s) to down regulate Myrf RNA, we wondered if crossing our Plp1-CreERT-Fbxw7fl/fl mutant animals into a Myrf heterozygous background would reduce the burden of N-MYRF protein levels and rescue Fbxw7 KO myelin phenotypes. To address this we crossed our Plp1-CreERT-Fbxw7fl/fl animals into the established Myrffl/fl line17 to create Plp1-CreERT-Fbxw7fl/fl; Myrffl/wt mice line along with their CreERT negative littermate controls (Fbxw7fl/fl; Myrffl/wt). TAM was administered at 8 weeks of age, and tissue was harvested at 1-, 3-, and 6-months post-TAM for subsequent analyses.
At 3-months post-TAM, when we observed the largest increase in MYRF protein levels in Fbxw7 icKO optic nerves, we observed a significant decrease in MYRF intensity in Plp1-CreERT-Fbxw7fl/fl; Myrffl/wt animals compared to icKO animals (Fig. 7a, e). We next evaluated Myrf levels in the cerebellum. Again, we found that Plp1-CreERT-Fbxw7fl/fl;Myrffl/wt animals had a significant decrease in MYRF staining intensity at 6-month post-TAM (Fig. 7b, f). To determine if an additional floxed allele in our Plp1-CreERT animals reduced Fbxw7 recombination, we performed qRT-PCR on the optic nerves in our Plp1-CreERT-Fbxw7fl/fl;Myrffl/wt 1-month post-TAM animals. We found no significant change in Fbxw7 expression between Fbxw7 icKO and Plp1-CreERT-Fbxw7fl/fl;Myrffl/wt animals at 1-month post-TAM (Supplementary Fig. 6a). We next wanted to assess how Myrf haploinsufficiency and normalization of MYRF protein levels would influence the myelin phenotypes observed in Fbxw7 icKO mice. We found a significant decrease in granule cell bodies ensheathed by myelin outfolds in Plp1-CreERT-Fbxw7fl/fl;Myrffl/wt animals compared to Fbxw7 icKO animals in the cerebellum at 6-months post-TAM (Fig.7b, h). Additionally, deletion of one allele of Myrf was sufficient to prevent the increase in myelin outfolds present in layer I of the pSS in Fbxw7 icKO animals at 6-months post-TAM by IF (Fig. 7c, g).

a Representative images of optic nerves from Fbxw7fl/fl, Plp1-CreERT-Fbxw7fl/fl, and Plp1-CreERT-Fbxw7fl/fl; Myrf fl/wt mice at 3-months post-TAM stained for MYRF. b Representative images of cerebellums from Fbxw7fl/fl, Plp1-CreERT-Fbxw7fl/fl, and Plp1-CreERT-Fbxw7fl/fl; Myrf fl/wt mice at 6-months post-TAM stained for MYRF, MBP, and NeuN. c Representative images of myelin internodes in layer I of the pSS in Fbxw7fl/fl, Plp1-CreERT-Fbxw7fl/fl, and Plp1-CreERT-Fbxw7fl/fl; Myrf fl/wt mice at 6-months post-TAM stained with MBP (magenta), CNP1 (green), and CASPR (cyan). d Representative images of the spinal cords of fbxw7vo86/vo86 and fbxw7vo86/vo86; myrfue70/WT zebrafish on Tg(mbp:eGFP-caax) and Tg(mbp:nls-eGFP) transgenic backgrounds at 3 dpf. e Quantification of nuclear MYRF intensity in Fbxw7fl/fl (N = 4), Plp1-CreERT-Fbxw7fl/fl (N = 3), and Plp1-CreERT-Fbxw7fl/fl; Myrf fl/wt (N = 3), normalized to controls at 3-months post-TAM in the optic nerve. Data shown as mean ± SEM. Statistical significance determined by two-way ANOVA with Tukey’s multiple comparisons test. f Quantification of nuclear MYRF intensity in Fbxw7fl/fl (N = 4), Plp1-CreERT-Fbxw7fl/fl (N = 3), and Plp1-CreERT-Fbxw7fl/fl; Myrf fl/wt (N = 4), normalized to controls at 6- months post-TAM in the cerebellum. For both e and f, small dots represent individual cell intensities, larger dots represent animal means ± SEM with Tukey’s multiple comparisons test. Statistical significance determined by one-way ANOVA on animal means. g Quantification of myelin outfolds in layer I of the pSS in Fbxw7fl/fl (N = 6), Plp1-CreERT-Fbxw7fl/fl (N = 6), and Plp1-CreERT-Fbxw7fl/fl; Myrffl/wt (N = 4) mice at 6-months post-TAM. Data shown as mean ± SEM, each datapoint represents mean from an individual animal. Statistical significance determined by two-way ANOVA, with Tukey’s multiple comparisons test. h Quantification of NeuN+ cells wrapped in MBP+ membrane in the granule layer of the cerebellum of Fbxw7fl/fl and Plp1-CreERT-Fbxw7fl/fl mice at 3- and 6- months post-TAM. Fbxw7fl/fl (3 m: N = 7, 6 m: N = 5), Plp1-CreERT-Fbxw7fl/fl (3 m: N = 6, 6 m: N = 6), and Plp1-CreERT-Fbxw7fl/fl; Myrffl/wt (3 m: N = 2, 6 m: N = 4). Data shown as mean ± SEM. Statistical significance determined by two-way ANOVA with Tukey’s multiple comparisons test. i Quantification of mbp:nls+ OLs in the zebrafish dorsal spinal cord of each genotype. WT (N = 5), myrfue70/WT (N = 9), fbxw7vo86/WT (N = 6), fbxw7vo86/vo86 (N = 6), fbxw7vo86/vo86; myrfue70/WT (N = 4). Mean ± SEM, statistical significance determined by two-way ANOVA with Tukey’s multiple comparisons test.
To test if the epistatic relationship between myrf and fbxw7 is evolutionarily conserved in zebrafish OLs, we crossed the fbxw7vo86/vo86 line to a myrf mutant line (myrfue70/WT)57 to determine if reducing myrf levels could suppress fbxw7 mutant phenotypes in vivo. At 3 dpf, we observed a significant reduction in the number of mbp:nls-EGFP+ OLs in the spinal cord of fbxw7vo86/vo86, myrfue70/WT animals compared to fbxw7vo86/vo86 mutants alone (Fig. 6d, i) as well as neuronal cell body myelin ensheathment (Supplementary Fig. 6b). These data, along with our work in primary OL cultures and conditional KO mouse models show that FBXW7 is an evolutionarily conserved negative regulator of N-MYRF in OLs required for many facets of OL biology, from OL sheath length, paranodal organization, to long-term homeostatic maintenance of myelin.
Discussion
Here we present evidence that once cells are committed to the OL lineage, FBXW7 regulates OL myelin capacity, organization, and homeostasis, in part through the negative regulation of the N-MYRF transcription factor. With the temporal control of inducible conditional knock-out mice we found that loss of Fbxw7 in myelinating OLs resulted in increased myelin sheath length, severe myelin outfolds, disorganized paranodes, and surprisingly, wrapping of neuronal cell bodies in the cerebellum by severe outfolds. These phenotypes are particularly striking since Fbxw7 was targeted in mature OLs, indicating that inactivation of FBXW7 reinitiates aspects of myelin growth in the adult CNS. While myelin sheath plasticity has been reported in the context of axonal activity7,36,58, the underlying mechanisms that regulate these changes have not been fully characterized. Because loss of Fbxw7 results in changes to many adhesion and cytoskeletal proteins, it is possible that FBXW7-mediated negative regulation of OL proteins may represent one of these underlying mechanisms of plasticity, potentially through its regulation of N-MYRF and its transcriptional targets.
Ensheathment of neuronal cell bodies occurs in zebrafish when OL numbers exceed their normal balance to receptive axons35. It is entirely possible that the increase in ensheathed neuronal cell bodies we observe in the fbxw7vo86 zebrafish is a consequence of increased OPC specification and OL numbers26. In contrast, myelin outfolds that ensheathed the cerebellar granule cell bodies of Plp1-CreERT-Fbxw7fl/fl mice were not accompanied by an increase in the density of OLs, so are unlikely to be mediated by a mismatch between the myelinating cells and their targets. We did not observe myelin outfolds wrapping cell bodies or other structures in the cerebral cortex of Plp1-CreERT-Fbxw7fl/fl animals, although outfolds were present. Why cerebellar granule cell bodies were the only observed neurons wrapped in myelin outfolds in the Fbxw7 conditional knockouts is puzzling. Perhaps the density and/or size of cerebellar granule cells make them especially susceptible to ensheathment by redundant myelin outfolds. Indeed, ensheathment of granule cell bodies has also been observed in the normal development of the toad59 and squirrel monkey cerebellum60, following N-WASP inactivation within the OL lineage cells61, and with over-expression of the extracellular domain of Cadm4 adhesion protein in OLs62. While aberrant cell body myelination has also been observed in the spinal cord of global KO of myelin-guidance protein JAM2, this was believed to be driven by neuronal loss of JAM2. Since the granule layer of the cerebellum is the primary region where cell body myelination is observed when OLs are targeted, it suggests that the unique architecture of this region is highly prone to ectopic myelin placement. Additionally, our findings that Fbxw7 loss of function myelin causes putative Purkinje axon pathology also leads to the interesting question of how inappropriate myelin may affect the health and function of the underlying neurons and circuitry.
In Schwann cells, FBXW7 regulates myelin sheath thickness, with no obvious change to sheath length28. In OLs these roles seem to be reversed, with FBXW7 regulating myelin sheath length but not thickness. Although this may be due to inherent differences in the biology of these two cell types, it may also be due to the differences in the tools used to evaluate its function. In our prior Schwann cell work, Fbxw7 was constitutively deleted in development using Desert hedgehog Cre (Dhh-Cre), which is expressed in Schwann cell precursors as early as E12.528,63. In contrast, here we used a TAM-inducible system to delete Fbxw7 from myelinating OLs in 8-week-old Plp1-CreERT mice. Whether the effects of Fbxw7 on myelination would change depending on the timing of OL deletion remains unclear but is an exciting proposition for future work.
Our studies and others highlight the complicated role FBXW7 plays in myelinating cell biology across species and cell types. Fbxw7 is widely expressed in most, if not all cell types in the CNS, and its biological functions depend on the available substrates within each cell46,56,64. Therefore, FBXW7 is likely to have distinct targets and diverging roles at different stages of the OL lifespan. For instance, FBXW7 negatively regulates NOTCH levels in neural precursor cells (NPCs) to control OPC specification in zebrafish26. The same group also found that in later stages of the OL lineage, FBXW7 negatively regulates mTOR to control myelination in the spinal cord of zebrafish. Likewise, we previously showed that in the mammalian PNS, FBXW7 regulates early Schwann cell numbers, axonal ensheathment, and myelin thickness in an mTOR-dependent manner28. In contrast, here we present evidence that in mammalian OLs, mTOR was not a direct target of FBXW7, with knockdown of Fbxw7 resulting in no detectable change in mTOR protein abundance, phosphorylation, or downstream signaling. Using an unbiased pull-down and LC-MS approach, we instead identified several direct FBXW7-interacting proteins in primary OLs: MYCBP2, MAP1B, RAE1, and MYRF. Of these, MYCBP2 was a previously identified FBXW7 interactor that likely inhibits its activity independent of the E3 complex48. Although we focus here on MYRF, MAP1B and RAE1 also represent intriguing FBXW7 targets within myelinating cells for future studies.
As a critical regulator of OL differentiation and myelination, MYRF levels appear to be tightly regulated within the OL lineage. Not only is MYRF subject to tight transcriptional regulation by SOX10 and ZFP2465, but its mRNA is subject to negative regulation by miR-145-5p in OPCs, presumably to discourage premature differentiation66. While we have shown that loss of FBXW7 results in an increase in the abundance of the N-terminal MYRF cleavage product, this increase in protein levels seems to have engaged a compensatory mechanism resulting in a decrease in Myrf transcript levels, presumably in an attempt to return MYRF to normal levels. Our MS data from Fbxw7 knock-down cells showed that ZFP24 was significantly downregulated compared to controls, presenting a possible link to the change in Myrf transcription65 and an unexplored mechanism that OLs may use to regulate Myrf mRNA levels and myelin homeostasis. Additionally, while we showed Myrf mRNA was significantly downregulated in both the ON and cerebellum at later timepoints, it’s interesting to note that this transcriptional downregulation was only sufficient to reduce nuclear MYRF protein levels in the ON but not the cerebellum. This raises an intriguing question: does the underlying physiology of the myelinated circuit affect OLs requirement and expression of MYRF? While we have shown that FBXW7 binds N-MYRF to control its levels both in vitro and in vivo, this does not preclude the possibility that additional mechanisms are involved in MYRF protein stability, cleavage, and function.
The full-length MYRF protein trimerizes and self-cleaves to enable release of the active transcription factor50,51,67. Although this process is negatively regulated by TMEM98, the protein product of one of MYRF’s own target genes67,68, the degree to which TMEM98 negatively regulates the production of the N-MYRF transcription factor at endogenous levels remains unclear. FBXW7 negatively regulates N-MYRF levels in primary OLs, corroborating the finding that FBXW7 targets N-MYRF in mHepa cells33 and suggesting a conserved regulatory mechanism. Indeed, the fact that myelin and OL abnormalities could be suppressed in Plp1-CreERT-Fbxw7fl/fl mice and fbxw7vo86 fish by Myrf haploinsufficiency highlights N-MYRF as a central FBXW7 target within the OL lineage. In contrast to mHepa cells, however, in primary OLs, the FBXW7 interaction does not seem to depend on the phosphorylation of the phosphodegron motif by GSK3β or CDK5. The intracellular pathways and mediating kinases that initiate N-MYRF turnover by FBXW7 will be important to determine in future work.
Notably, the dysregulated proteins seen in Fbxw7 knockdown OLs included many cell adhesion and cell surface proteins. Previous research has highlighted the connection between adhesion molecules and proper myelination. For instance, when contactin-associated protein (Caspr), contactin-1 (Cntn1), neurofascin (Nfasc155), and myelin-associated glycoprotein (Mag) were globally deleted in both the zebrafish and mouse CNS, similar myelin phenotypes were observed as in Plp1-CreERT-Fbxw7fl/fl animals69. Specifically, loss of these adhesion proteins resulted in outfolds, double myelinated axons, and disorganized paranodal loops as well as neuronal cell body ensheathment in the zebrafish spinal cord. It is exciting that within our model, where Fbxw7 deletion is restricted to myelinating OLs, we see such similar phenotypes when compared to global disruption of adhesion proteins in the CNS.
In summary, we have shown that FBXW7 is an evolutionarily conserved regulator of OL myelin capacity and homeostasis. We found that FBXW7 regulates myelination by controlling sheath elongation independent of myelin wraps and is required for long-term maintenance of myelin integrity and paranodal organization within the adult CNS, in part, through its negative regulation of N-MYRF.
Methods
Zebrafish husbandry
All zebrafish experiments were performed in compliance with the institutional ethical regulations for animal testing and research at Oregon Health & Science University (OHSU, protocol TR02_IP00001148). fbxw7stl64, fbxw7vo86, and myrfue70 zebrafish were maintained as heterozygotes. Experimental larvae were generated by incrosses to yield wild-type, heterozygous, and homozygous zebrafish. To create fbxw7vo86/vo86; myrfue70/WT animals, fbxw7vo86/WT; myrfue70/WT zebrafish were outcrossed to fbxw7vo86/WT. Zebrafish larvae are fed a diet of rotifers and dry food (Gemma 75) from 5 days post-fertilization (dpf) until 21 dpf. From 21 dpf until 3 months, fish are fed using rotifers and dry food (Gemma 150). Adult fish are maintained and fed with brine shrimp and dry food (Gemma 300). For larval zebrafish studies, sex cannot be considered as a biological variable as sex has not yet been determined.
Generation of fbxw7 vo86 zebrafish mutants
CRISPR/Cas9-mediated genome editing was used to generate genetic mutants in zebrafish. The CHOPCHOP web tool74 was used to select target sites, and individual sgRNAs were synthesized using the MEGAshortscript T7 Transcription kit (Thermo Fisher). The sgRNA 5-GATGTAATCCGTCGTCTCTGTGG-3 was mixed with Cas9 Nuclease (Integrated DNA Technologies) to a final concentration of 50 ng/mL sgRNA and 1 mg/mL of Cas9 protein and injected into one-celled zygotes at a volume of 1-2 nL. Progeny of injected F0 generation animals were screened for the presence of inherited indels resulting in frameshifts or truncations by PCR, and these F1 progenies were used to establish stable mutant lines. Genotyping for both larval and adult zebrafish was performed by digesting tissue in Tris buffer with Proteinase-K overnight at 55 °C. PCR was performed with GoTaq DNA Polymerase (Promega, M300A). For fbxw7vo86 genotyping, PCR with (F 5-AAAATAGGGGCTTGCTCTGG-3, R 5-AAGTCCAGTTAAATTGAGAAGCC-3) was used to amply a 530 bp region around indels. PCR products were digested with 10U of BsmBI-v2 (NEB, R0580) at 55 °C overnight and resolved on a 2% agarose gel. The uncleaved fbxw7vo86 product size was 530 bp, and WT cleaved product sizes were 294 and 236 bp.
Zebrafish mosaic cell labeling
For mosaic labeling of oligodendrocytes (OLs), fbxw7vo86/WT zebrafish were incrossed, and fertilized one-cell zygotes were injected with 1–2 nl of a solution containing 10 ng of sox10:EGFP-caax plasmid, 25 ng of Tol2 transposase mRNA, 0.02% phenol red and 0.2 M KCl. Embryos were genotyped after imaging as described above.
Cell-type specific CRISPR-Cas9 gene disruption in zebrafish
For cell-type specific CRISPR-Cas9-mediated gene disruption we utilized methods as previously described36. Briefly, sgRNAs targeting fbxw7 exon 5 (5-GATGTAATCCGTCGTCTCTGTGG-3) and exon 7 (5-GCTGCCTGAAGCAGATCCTTTGG-3) were cloned into 10xUAS:myrmScarlet-p2A-Cas9, U6:sgRNA1;U6:sgRNA2 backbones and injected into Tg(sox10:Kalta4)75 fertilized embryos at the one-cell stage. Empty backbones were used as controls. At desired timepoints, fish were anesthetized with 600 μM tricaine (TRS5, Pentair), screened for fluorescence, embedded laterally in 1.5% low-melting-point agarose (A9414, Sigma), and imaged with a 20x dipping objective on a ZEISS LSM 980 with Airyscan 2. Sheaths were analyzed using ImageJ36.
Mouse husbandry and tamoxifen (TAM) administration
All mice were housed in OHSU animal facilities, maintained in a pathogen-free temperature and humidity-controlled environment on a 12-h light/dark cycle. All procedures were approved by the OHSU Institutional Animal Care and Use Committee (protocols TR01_IP00001328 and TR02_IP00001148). Fbxw7fl/fl mice34 were previously purchased from Jackson Laboratories (B6;129-Fbxw7tm1Iaai/J, JAX: 017563) and crossed to Plp1-CreERT mice (B6.Cg-Tg[Plp1-cre/ERT]3Pop/J, JAX: 005975). Myrffl/fl mice17, maintained on a C57BL/6 N background, were crossed into the Plp1-CreERT-Fbxw7fl/fl animals to create Plp1-CreERT-Fbxw7fl/fl;Myrffl/wt animals. CreERT negative littermates served as controls. Genotypes were determined by PCR analysis using established primers for each line and were revalidated at experimental endpoints. Genotyping primers below. All experiments were conducted in both male and female mice. For TAM injection, 8-week-old mice were dosed with 100 mg/kg tamoxifen (Sigma T5648, dissolved at 20 mg/ml in corn oil) for five consecutive days via intraperitoneal injection.
Gene | 5-Forward primer | Reverse primer-3 | Size |
Fbxw7fl/fl | GGCTTAGCATATCAGCTATGG | ATAGTAATCCTCCTGCCTTGGC | 497 bp |
Fbxw7wt/wt | GGCTTAGCATATCAGCTATGG | ATAGTAATCCTCCTGCCTTGGC | 315 bp |
Plp1-CreERT | ATACCGGAGATCATGCAAGC | GGCCAGGCTGTTCTTCTTAG | 200 bp |
Cre control | CTAGGCCACAGAATTGAAAGATCT | GTAGGTGGAAATTCTAGCATCATCC | 324 bp |
Myrfwt/wt | GGGAGGGGGCTTCAAGGAGTGT | CCCCCAGCATGCCGATGTACAC | 460 bp |
Myrffl/fl | GGGAGGGGGCTTCAAGGAGTGT | CCTTTCGCCAGGGGGATCTTG | 668 bp |
Tissue processing
Mice were terminally anaesthetized with ketamine (400 mg/kg) and xylazine (60 mg/kg) before being transcardially perfused with 20 mL of phosphate buffered saline (PBS) and 40 mL of 4% paraformaldehyde (19210, Electron Microscopy Sciences) in PBS. For immunofluorescence (IF), tissues were post-fixed in 4% paraformaldehyde in PBS (2 h for optic nerves, overnight for brains) at 4 °C and cryopreserved in 30% sucrose for at least 72 h at 4 °C. Cortical flat mounts were processed as previously described39. Cryopreserved tissue was embedded in OCT (4583, Sakura), frozen on dry ice, and stored at −80 °C until sectioning on a cryostat (Leica CM3050-S). Cryosections (12 μm thickness for brain, 16 µm for optic nerve) were mounted on Superfrost Plus slides (1255015, Fisher Scientific) and stored at −80 °C. Tissue for transmission electron microscopy was post-fixed in 2% paraformaldehyde (15710, Electron Microscopy Sciences) with 2% glutaraldehyde (16310, Electron Microscopy Sciences).
Immunofluorescence
Slides stored in −80 °C were air dried for at least 2 h before being rehydrated in 1x PBS. For MBP staining, tissue was delipidated by treating slides with ascending and descending ethanol solutions (50%, 75%, 85%, 95%, 100%) before being washed 3x in 1x PBS. Slides were blocked for 1 h at room temperature with 10% fetal calf serum (SH30910.03, Cytiva) with 0.2% Triton X-100 (10789704001, Sigma). Primary antibodies were applied overnight in 1x PBS, 5% fetal calf serum and 0.2% Triton X-100 in a sealed humidified container at room temperature. The following primary antibodies were used: chicken anti-MBP (1:500; MBP, Aves), mouse anti-CC1 monoclonal (1:500; OP80, Millipore), goat anti-PDGFRα (1:500; AF1062, R&D Systems), rabbit anti-Iba1 (1:1000; 019-19741, Wako), rabbit anti-GFAP (1:1000; Z0334, Dako), rabbit anti-MYRF (1:500; A16355, ABclonal), mouse anti-Calbindin1 (1:500; C9848, Sigma). Following incubation with primary antibodies, slides were washed 3x in 1 × 0.2% Triton X-100 PBS before appropriate Alexa Fluor 488, 555 or 647 secondary antibodies (1:1,000; Invitrogen) were applied for one h at room temperature. Slides were then again washed 3x with 1 × 0.2% Triton X-100 PBS, washed in Milli-Q H2O, air dried, then coverslipped with Fluoromount G (0100-01, Southern Biotech). Images were acquired with in 2 weeks.
Primary rat OLs were cultured on glass coverslips, fixed for 8 min in 4% PFA in PBS and stained in 24-well plates as described above. We used the following primary antibodies: chicken anti-MBP (1:500; MBP, Aves), rabbit anti-OLIG2 (1:500; AB9610, Millipore), and mouse anti-MAG (1:500; AB1567, Millipore). Coverslips were mounted with ProLong Diamond (P36965, Thermo Fisher) on Superfrost Plus slides.
Cortical flatmounts were sectioned at 40 μm and stored in PBS with 0.02% NaN3 (sodium azide) at 4 °C for 1 week–2 months. For staining, tissue was blocked in 10% fetal calf serum with 0.2% Triton X-100 for 2 h with agitation at room temperature. Flatmounts were then incubated with primary antibodies at room temperature with agitation in 1x PBS 0.2% Triton X-100 for 4 days. We used the following primary antibodies: chicken anti-MBP (1:200; MBP, Aves), rabbit anti-CASPR (1:500; 34151-001, Abcam), and mouse anti-CNPase1 (1:500; MAB326, Millipore). Tissue was washed 3 × 20 min 1 × 0.2% Triton X-100 PBS. Alexa Fluor 488, 555 and 647 secondary antibodies (1:1,000; Invitrogen) were applied for two days at 4 °C protected from light. Tissue was washed 3 × 20 min 1 × 0.2% Triton X-100 PBS, slide mounted, rinsed in water, air dried, and coverslipped with Fluoromount G (0100-01, Southern Biotech). For quantification of sheath length from layer I pSS, 280 μm × 280 μm × 30 μm images were taken of the pSS and 2 random ROIs were generated. All sheaths that passed through the ROIs were measured using ImageJ NeuroTracer in 3D.
Immunofluorescence from tissue was acquired on a ZEISS LSM 980 with Airyscan 2. Immunofluorescence from cultured OLs were imaged on Zeiss ApoTome2 at 20x. All cell counts and fluorescence intensities were quantified using ImageJ.
Isolation and electroporation of primary rat OPCs
Rat OPCs were isolated from P6-8 Sprague Dawley rat pups as previously described76. OPCs from each animal were expanded in 3 × 175 cm2 flasks for 3–4 days in the presence of 10 ng/mL platelet derived growth factor-aa (PDGFAA, Peprotech 100-13 A). Cells were harvested fresh for each round of experiments and used at the time of first passage. OPCs were electroporated with Amaxa Basic Nucleofector Kit for Primary Mammalian Glial Cells (VPI-1006, Lonza) with 20 nM siRNAs/5 million OPCs or 4 μg of plasmid/5 million OPCs. siRNA pools for rat Fbxw7 (L-115782-00-0005, Horizon), Gsk3b (L-080108-02-0005, Horizon), Cdk5 (L-094624-02-0005, Horizon), or non-targeting controls (D-001810-10-05, Horizon) were used. Primary rat OLs were plated at 20k/coverslip for staining, 250k cells/well of a 6-well plate for RNA, and 1 million cells/60 × 15 mm plate for protein isolation.
EdU incorporation and cycloheximide (CHX) treatment
Primary rat OPCs were expanded and electroporated with siRNAs and replated into proliferation media containing PDGFAA for 48 h. Cells were pulsed with 10 μM 5-ethynyl-2’-deoxyuridine (EdU) for 6 hours. Cells were fixed with 4% PFA in PBS for 8 min at room temperature. Cells were stained with Click-iT EdU Cell Proliferation Kit with Alexa Fluor 647 dye (C10340, Thermo Fisher). siRNA electroporated OLs were differentiated for 3 days and treated with cycloheximide (CHX; 239763-M, Sigma) at 100 μg/mL for 4 hours to stop protein translation. Cell lysates were processed for Western blot analyses as described below.
Transmission electron microscopy (TEM)
Following post-fixation in 2% paraformaldehyde (15710, Electron Microscopy Sciences) with 2% glutaraldehyde (16310, Electron Microscopy Sciences), optic nerves and cerebellums were stored in a buffer of 1.5% paraformaldehyde, 1.5% glutaraldehyde, 50 mM sucrose, 22.5 mM CaCl2 2H2O in 0.1 M cacodylate buffer for at least seven days. Tissue was then infiltrated with 2% osmium tetroxide (19190, Electron Microscopy Sciences) using a Biowave Pro+ microwave (Ted Pella)54 before dehydration in acetone and embedding in Embed 812 (14120, Electron Microscopy Sciences). 0.4 µm sections were cut on an ultramicrotome and stained with 1% Toluidine Blue (T161-25, Fisher Scientific) with 2% sodium borate (21130, Electron Microscopy Sciences). 60 nm sections were mounted on copper grids (FCF100-Cu-50, Electron Microscopy Sciences) and counterstained with UranyLess for 5 min followed by 3% lead citrate (22409, 22410, Electron Microscopy Sciences) for 5 min. Grids were imaged at 4800x on an FEI Tecnai T12 transmission electron microscope with a 16 Mpx camera (Advanced Microscopy Techniques Corp). For g-ratio analysis, 5–8 images per animal were used. Outer myelin and axon diameters for g-ratio analyses were manually traced using ImageJ.
Cloning of dominant-negative 3xFLAG- FBXW7∆F-Box
Fbxw7 coding sequence missing the F-Box domain with upstream 3x FLAG tags (3xFLAG- Fbxw7∆F-Box) was purchased as a double stranded gBlocks gene fragment from Integrated DNA Technologies (IDT) with KpnI + HindIII restriction enzyme overhangs. pCMV-SPORT6 backbone and 3xFLAG- FBXW7∆F-Box inserts were digested with KpnI + HindIII restriction enzymes, gel purified, and ligated with T4 DNA ligase (EL0011, Thermo Fisher). Constructs were transformed into DH5α one-shot competent cells (12297016, Thermo Fisher) and purified with a PureLink HiPure plasmid maxiprep kit (K0491, ThermoFisher). pmaxGFP vector (Lonza) was used as a control for co-IP experiments.
Immunoprecipitation of 3xFLAG- FBXW7∆F-Box in cultured OLs
4 μg of CMV-3xFLAG-FBXW7∆F-Box and pmaxGFP were electroporated into rat OPCs. Cells were differentiated for 3 days and then lysed with cell lysis buffer (20 mM Tris pH7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA) with cOmplete Mini Protease Inhibitor Cocktail (11836153001, Millipore) for 30 min at 4 °C with rotation. Lysates were spun at 4 °C for 10 min at 9800 × g. 5% of lysates were frozen for input controls. 4 μg of mouse anti-FLAG M2 antibody (F3165, Millipore) was added to lysates and rotated at 4 °C for 2 h. 40 μL Dynabeads Protein G (10003D, Thermo Fisher) was added to lysates and rotated for 1 hour at 4°C. Beads were collected with a magnetic rack and washed 5x with cell lysis buffer with rotation at 4 °C for 5 mins. Proteins were released from Dynabeads Protein G beads for LC-MS by boiling in 1% SDS. Proteins for Western blots were boiled with 1x Laemmli buffer.
Western blots
For western blots on cultured OLs, plates were washed 3x with cold DPBS and lysed with RIPA buffer (50 mM Tris-HCL pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% Sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 0.5 mM EGTA) with complete protease inhibitors (11836153001, Roche), and phosphatase inhibitors (04906837001, Roche) before being spun at 13,000 × g at 4 °C. Protein lysate was removed and frozen at −80 °C. Lysates were boiled at 98 °C in 1x Laemmli buffer for 5 min and run on Bis-Tris-gel (NP0335BOX, Invitrogen). To transfer proteins to PVDF membranes (IPVH00010, Thermo Scientific), transfer cassettes were assembled (A25977, Thermo Fisher Scientific) and filled with transfer buffer (NP0006-01, Thermo Scientific) containing 10% methanol and transferred at 20 V for 1 hour. Following transfer, blots were rinsed in 1x TBS with 0.1% Tween-20 (TBST) before blocking in 1x TBST with 5% milk powder for one hour at room temperature. Blots were probed with antibodies against MYRF (16355, ABClonal), mTOR (2983, Cell Signaling), Phospho-mTOR (Ser2448; 5536, Cell Signaling), cJun (9261 L, Cell Systems), TMEM98 (14731-1-AP, Proteintech), MYO1D (ab70204, Abcam), GSK3β (ab32391, abcam), DPYSL5 (CRMP5; ab36203, Abcam). All antibodies for western blots were used at a concentration of 1:1000. Blots were incubated in primary antibodies diluted in 2.5% BSA (BP9706-100, Fisher Scientific) with 1% NaN3 in TBST overnight at 4 °C. After overnight incubation, blots were washed in 3x TBST and incubated with appropriate HRP-conjugated secondary (Goat anti-rat 7077, Cell Signaling; Goat anti-mouse 7076, Cell Signaling, Goat anti-rabbit 7074, Cell Signaling) at 1:5000 for two hours with 2.5% milk powder in TBST. Immunoreactivity was visualized using chemiluminescence (34080, Thermo Fisher Scientific) and imaged on a Syngene GBox iChemiXT. Blots were then re-probed with β-actin-HRP (1:5000; A3854, Sigma). Densitometric analyses were performed in ImageJ by quantifying the intensity of bands relative to the ACTB loading control and then normalized to background.
Mass spectrometry and analysis
Bulk mass spectrometry
OLs electroporated with pooled siRNAs targeting Fbxw7 or non-targeting controls were differentiated for 72 hours and lysed with eFASP buffer (4% SDS, 0.2% DCA, 100 mM TEAB), frozen at −80 °C, and submitted to the OHSU proteomic core. Samples were then sonicated using Bioruptor Pico (30 s on 30 s off, 10 cycles), heated to 90 °C for 10 mins, cooled, centrifuged, and protein concentration was determined by BCA. 55 μg of protein/sample were digested with eFASP and measured by peptide assay. 18 μg peptides/sample were labeled with TMT 11-plex, normalized, and pooled. Pooled TMT samples were then run on an 18 fraction 2dRPRP LC/MS on Orbitrap Fusion. Data was then analyzed with COMET/PAWS pipeline in edgeR.
Immunoprecipitation mass spectrometry
OL samples from IP were prepared for proteomics analysis. Samples were dried on a SpeedVac and resuspended in 5% SDS, 50 mM TEAB pH 8. Samples were reduced with dithiothreitol and alkylated with iodoacetamide. Proteins were bound to S-trap columns (Protifi, Inc.) where they were washed before being digested with trypsin. A Quantitative Colorimetric Peptide Assay (23275, Pierce) was performed. Peptides were analyzed by LC-MS on a Dionex Ultimate HPLC to an Orbitrap Q-Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA).
qRT-PCR
RNA was isolated from primary rat OPC/OLs with the RNeasy Mini Kit (74104, QIAGEN) and stored at −80 °C. RNA from optic nerves and cerebellums were isolated by homogenizing tissue in 700 μl of TRIzol (15596026, Thermo Fisher). 140 μl of chloroform (J67241.K4, Thermo Fisher) was added to lysate and shaken for 30 seconds and left at room temperature for 5 min. Reactions were then centrifuged for 15 min at 12,000 × g at 4 °C. The upper, colorless, aqueous phase containing RNA was then purified further with RNeasy UCP Micro (optic nerves) and Mini (cerebellums) Kits (73934, 74104, QIAGEN) and stored at −80 °C. cDNA was generated with SuperScript III First-Strand Synthesis (18080400, Thermo Fisher) and stored at −20 °C. qPCR was performed with PowerUP SYBR Green (A25742, Thermo Fisher) on a QuantStudio 6 Flex Real-Time PCR System (4485691, Thermo Fisher). qRT-PCR primers were designed on the Integrated DNA Technologies (IDT) PrimerQuest program.
qRT-PCR | Primers | ||
Gene | Forward | Reverse | Species |
Myrf | CTTCCTTCTGGTCATCTCCTTG | GAGAGTGAAGGTGGGCAAATA | Mouse |
Actb | GAGGTATCCTGACCCTGAAGTA | CACACGCAGCTCATTGTAGA | Mouse |
Mbp | AGTCGCAGAGGACCCAAGAT | ACAGGCCTCTCCCCTTTC | Rat |
Plp1 | CACTTACAGCAGGTGATTAGAGG | AAACAAGAGATAAACAACTGGGA | Rat |
Mog | GTCTATCGGCGAGGGAAAGGT | CACGGCGGCTTCTTCTTGGT | Rat |
Myrf | CAGCAGTGGGAAAGGGAATAA | AACTCAGCTCCCGATAGAGAT | Rat |
Tmem98 | CAGTGTAACACCACGTCTACC | CTCGATTCTTAGAGGGCAACTC | Rat |
Itgb3 | GCTGTCCTGTATGTGGTAGAAG | CAGAGTAGCAAGGCCAATGA | Rat |
Dpysl5 | TCCATACCCACACCCTCATA | ACTCTCTCCTACTCTCCCTTTC | Rat |
Myo1d | ACTTCAAGCGCCTCATGTATAA | CCTCTTGGTCACCTCTGTAATG | Rat |
Fam107b | GAAGCACGAGAGCCGATTATAG | TCAAGGGCAAGCCATCTTAC | Rat |
Vsnl1 | TGTCTGGCCCACATACAATAC | CGAGAGGTTACAAATGAGGTAAGA | Rat |
Pea15 | ATGAAGACACAGGAGAGAGAGA | GTGAGTGTATTAGGGCAGGTTAG | Rat |
Tppp | ACTGGGCAGAACTCAGAATG | CAATGAATCACGGCCCAAAC | Rat |
Rpl13A | CTCATGATGACTGCAGCAAACC | GGATCCCTCCAC CCTATGACA | Rat |
mbp | AATCAGCAGGTTCTTCGGAGGAGA | AAGAAATGCACGACAGGGTTGACG | Zebrafish |
actb | CGAGCTGTCTTCCCATCCA | TCACCAACGTAGCTGTCTTTCTG | Zebrafish |
Quantification and statistical analysis
Myelin outfolds in the corpus callosum were quantified in ImageJ by image thresholding and particle analyses. MYRF IF in the optic nerve and cerebellum was quantified in ImageJ by overlaying a DAPI mask on MYRF images followed by particle analyses for mean gray values. CC1+ cell in the cortex and corpus callosum were quantified manually. MBP+ wrapped NeuN+ cell bodies were quantified manually in the cerebellum. Western blots were quantified by densitometry in ImageJ. All bands were normalized to blot background and bands from proteins of interest were normalized to loading controls. Statistical analyses were conducted with Prism 10 (Graphpad). In all cases the figure legend indicates the statistical test used and P values are presented in figures. Sample size is stated in figure legends. Animals were assigned to group based on genotype by random selection and analyses were conducted blinded to genotype.
Mass spectrometry data was processed using the PAW pipeline77 with the Comet search engine (version 2016.03)78 searching versus the Rattus Norvegicus Uniprot database (UP000002494, Taxon ID:10116, one protein sequence per gene). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD06357079,80,81.
Availability of materials
Fish lines and reagents generated in this study including plasmids will be made available on request to the corresponding authors (monk@ohsu.edu and emeryb@ohsu.edu).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper and mass spectrometry data is available via ProteomeXchange with identifier PXD063570: (https://www.ebi.ac.uk/pride/archive/projects/PXD063570). Source data are provided with this paper.
References
Waxman, S. G. & Bennett, M. V. L. Relative conduction velocities of small myelinated and non-myelinated fibres in the central nervous system. Nat. N. Biol. 238, 217–219 (1972).
Ritchie, J. M. et al. Physiological Basis of Conduction in Myelinated Nerve Fibers. in Myelin (ed. Morell, P.) 117–145 (Springer US, Boston, MA, 1984).
Nave, K.-A. & Trapp, B. D. Axon-glial signaling and the glial support of axon function. Annu. Rev. Neurosci. 31, 535–561 (2008).
Huxley, A. F. & Stämpfli, R. Direct determination of membrane resting potential and action potential in single myelinated nerve fibres. J. Physiol. 112, 476–495 (1951).
Raff, M. C., Miller, R. H. & Noble, M. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature 303, 390–396 (1983).
Lee, S. et al. A culture system to study oligodendrocyte myelination-processes using engineered nanofibers. Nat. Methods 9, 917–922 (2012).
Gibson, E. M. et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344, 1252304 (2014).
Mitew, S. et al. Pharmacogenetic stimulation of neuronal activity increases myelination in an axon-specific manner. Nat. Commun. 9, 306 (2018).
Czopka, T., ffrench-Constant, C. & Lyons, D. A. Individual oligodendrocytes have only a few hours in which to generate new myelin sheaths in vivo. Dev. Cell 25, 599–609 (2013).
Zhou, Q., Choi, G. & Anderson, D. J. The bHLH transcription factor Olig2 Promotes oligodendrocyte differentiation in collaboration with Nkx2.2. Neuron 31, 791–807 (2001).
Hughes, E. G., Kang, S. H., Fukaya, M. & Bergles, D. E. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 16, 668–676 (2013).
Hughes, E. G., Orthmann-Murphy, J. L., Langseth, A. J. & Bergles, D. E. Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat. Neurosci. 21, 696–706 (2018).
Saher, G. et al. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 8, 468–475 (2005).
Nawaz, S. et al. Actin filament turnover drives leading edge growth during myelin sheath formation in the central nervous system. Dev. Cell 34, 139–151 (2015).
Zuchero, J. B. et al. CNS myelin wrapping is driven by actin disassembly. Dev. Cell 34, 152–167 (2015).
Cahoy, J. D. et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. J. Soc.28, 264–278 (2008).
Emery, B. et al. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 138, 172–185 (2009).
Elbaz, B. & Popko, B. Molecular control of oligodendrocyte development. Trends Neurosci. 42, 263–277 (2019).
Marques, S. et al. Transcriptional convergence of oligodendrocyte lineage progenitors during development. Dev. Cell 46, 504–517.e7 (2018).
Maggie, S.Y. et al. Dynamics of oligodendrocyte generation and myelination in the human brain. Cell 159, 766–74 (2014).
Sabri, M. I., Bone, A. H. & Davison, A. N. Turnover of myelin and other structural proteins in the developing rat brain. Biochem. J. 142, 499–507 (1974).
Saher, G. & Simons, M. Cholesterol and Myelin Biogenesis. in Cholesterol Binding and Cholesterol Transport Proteins: Structure and Function in Health and Disease (ed. Harris, J. R.) 489–508 (Springer Netherlands, Dordrecht, 2010).
Tripathi, R. B. et al. Remarkable stability of myelinating oligodendrocytes in mice. Cell Rep. 21, 316–323 (2017).
Auer, F., Vagionitis, S. & Czopka, T. Evidence for myelin sheath remodeling in the CNS revealed by in vivo imaging. Curr. Biol. 28, 549–559.e3 (2018).
Hill, R. A., Li, A. M. & Grutzendler, J. Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat. Neurosci. 21, 683–695 (2018).
Snyder, J. L., Kearns, C. A. & Appel, B. Fbxw7 regulates Notch to control specification of neural precursors for oligodendrocyte fate. Neural Dev. 7, 15 (2012).
Kearns, C. A., Ravanelli, A. M., Cooper, K. & Appel, B. Fbxw7 limits myelination by inhibiting mTOR signaling. J. Neurosci. 35, 14861–14871 (2015).
Harty, B. L. et al. Myelinating Schwann cells ensheath multiple axons in the absence of E3 ligase component Fbxw7. Nat. Commun. 10, 2976 (2019).
Sanchez, N. E. et al. Whole genome sequencing-based mapping and candidate identification of mutations from fixed zebrafish tissue. G3(Bethesda) 7, 3415–3425 (2017).
Nateri, A. S., Riera-Sans, L., Costa, C. D. & Behrens, A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science 303, 1374–1378 (2004).
Ye, X. et al. Recognition of Phosphodegron Motifs in Human Cyclin E by the SCFFbw7 Ubiquitin Ligase*. J. Biol. Chem. 279, 50110–50119 (2004).
Yada, M. et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23, 2116–2125 (2004).
Nakayama, S., Yumimoto, K., Kawamura, A. & Nakayama, K. I. Degradation of the endoplasmic reticulum-anchored transcription factor MyRF by the ubiquitin ligase SCFFbxw7 in a manner dependent on the kinase GSK-3. J. Biol. Chem. 293, 5705–5714 (2018).
Thompson, B. J. et al. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J. Exp. Med. 205, 1395–1408 (2008).
Almeida, R. G. et al. Myelination of neuronal cell bodies when myelin supply exceeds axonal demand. Curr. Biol. 28, 1296–1305.e5 (2018).
Li, J., Miramontes, T. G., Czopka, T. & Monk, K. R. Synaptic input and Ca2+ activity in zebrafish oligodendrocyte precursor cells contribute to myelin sheath formation. Nat. Neurosci. 27, 219–231 (2024).
Doerflinger, N. H., Macklin, W. B. & Popko, B. Inducible site-specific recombination in myelinating cells. Genes. N. Y. N. 2000 35, 63–72 (2003).
Orthmann-Murphy, J. et al. Remyelination alters the pattern of myelin in the cerebral cortex. eLife 9, e56621 (2020).
Call, C. L. & Bergles, D. E. Cortical neurons exhibit diverse myelination patterns that scale between mouse brain regions and regenerate after demyelination. Nat. Commun. 12, 4767 (2021).
Almeida, R. G. The Rules of attraction in central nervous system myelination. Front. Cell. Neurosci. 12, 367 (2018).
Klingseisen, A. et al. Oligodendrocyte neurofascin independently regulates both myelin targeting and sheath growth in the CNS. Dev. Cell 51, 730–744.e6 (2019).
Redmond, S. A. et al. Somatodendritic expression of JAM2 inhibits oligodendrocyte myelination. Neuron 91, 824–836 (2016).
Barron, T., Saifetiarova, J., Bhat, M. A. & Kim, J. H. Myelination of purkinje axons is critical for resilient synaptic transmission in the deep cerebellar nucleus. Sci. Rep. 8, 1022 (2018).
Chung, S.-H., Guo, F., Jiang, P., Pleasure, D. E. & Deng, W. Olig2/Plp-positive progenitor cells give rise to Bergmann glia in the cerebellum. Cell Death Dis. 4, e546 (2013).
Koike, M. et al. Purkinje cells are more vulnerable to the specific depletion of cathepsin D than to that of Atg7. Am. J. Pathol. 187, 1586–1600 (2017).
Winston, J. T., Koepp, D. M., Zhu, C., Elledge, S. J. & Harper, J. W. A family of mammalian F-box proteins. Curr. Biol. 9, 1180–S3 (1999).
Zhang, J. et al. Rack1 protects N-terminal phosphorylated c-Jun from Fbw7-mediated degradation. Oncogene 31, 1835–1844 (2012).
Richter, K. T., Kschonsak, Y. T., Vodicska, B. & Hoffmann, I. FBXO45-MYCBP2 regulates mitotic cell fate by targeting FBXW7 for degradation. Cell Death Differ. 27, 758–772 (2020).
Hornig, J. et al. The transcription factors Sox10 and Myrf define an essential regulatory network module in differentiating oligodendrocytes. PLOS Genet 9, e1003907 (2013).
Bujalka, H. et al. MYRF is a membrane-associated transcription factor that autoproteolytically cleaves to directly activate myelin genes. PLOS Biol. 11, e1001625 (2013).
Li, Z., Park, Y. & Marcotte, E. M. A bacteriophage tailspike domain promotes self-cleavage of a human membrane-bound transcription factor, the myelin regulatory factor MYRF. PLOS Biol. 11, e1001624 (2013).
Aprato, J. et al. Myrf guides target gene selection of transcription factor Sox10 during oligodendroglial development. Nucleic Acids Res 48, 1254–1270 (2020).
Hartley, M. D. et al. Myelin repair stimulated by CNS-selective thyroid hormone action. JCI Insight 4, e126329 (2019).
Duncan, G. J. et al. Remyelination protects neurons from DLK-mediated neurodegeneration. Nat. Commun. 15, 9148 (2024).
Koenning, M. et al. Myelin gene regulatory factor is required for maintenance of myelin and mature oligodendrocyte identity in the adult CNS. J. Neurosci. 32, 12528–12542 (2012).
Jin, J., Ang, X. L., Shirogane, T. & Wade Harper, J. Identification of Substrates For F-box Proteins. in Methods in Enzymology. 399, 287–309 (Academic Press, 2005).
Madden, M. E. et al. CNS hypomyelination disrupts axonal conduction and behavior in larval zebrafish. J. Soc. Neurosci. 41, 9099–9111 (2021).
Hines, J. H., Ravanelli, A. M., Schwindt, R., Scott, E. K. & Appel, B. Neuronal activity biases axon selection for myelination in vivo. Nat. Neurosci. 18, 683–689 (2015).
Rosenbluth, J. Redundant myelin sheaths and other ultrastructural features of the toad cerebellum. J. Cell Biol. 28, 73–93 (1966).
Cooper, M. H. & Beal, J. A. Myelinated granule cell bodies in the cerebellum of the monkey (Saimiri sciureus). Anat. Rec. 187, 249–255 (1977).
Katanov, C. et al. N-wasp regulates oligodendrocyte myelination. J. Neurosci. 40, 6103–6111 (2020).
Elazar, N. et al. Axoglial adhesion by Cadm4 regulates CNS myelination. Neuron 101, 224–231.e5 (2019).
Jaegle, M. et al. The POU proteins Brn-2 and Oct-6 share important functions in Schwann cell development. Genes Dev. 17, 1380–1391 (2003).
Kipreos, E. T. & Pagano, M. The F-box protein family. Genome Biol. 1, 3002 (2000).
Elbaz, B. et al. Phosphorylation state of ZFP24 controls oligodendrocyte differentiation. Cell Rep. 23, 2254–2263 (2018).
Kornfeld, S. F., Cummings, S. E., Fathi, S., Bonin, S. R. & Kothary, R. MiRNA-145-5p prevents differentiation of oligodendrocyte progenitor cells by regulating expression of myelin gene regulatory factor. J. Cell. Physiol. 236, 997–1012 (2021).
Garnai, S. J. et al. Variants in myelin regulatory factor (MYRF) cause autosomal dominant and syndromic nanophthalmos in humans and retinal degeneration in mice. PLOS Genet 15, e1008130 (2019).
Huang, H. et al. Interactive repression of MYRF self-cleavage and activity in oligodendrocyte differentiation by TMEM98 protein. J. Neurosci. 38, 9829–9839 (2018).
Djannatian, M. et al. Two adhesive systems cooperatively regulate axon ensheathment and myelin growth in the CNS. Nat. Commun. 10, 4794 (2019).
Almeida, R. G., Czopka, T., ffrench-Constant, C. & Lyons, D. A. Individual axons regulate the myelinating potential of single oligodendrocytes in vivo. Development 138, 4443–4450 (2011).
Kim, H. et al. Notch-regulated perineurium development from zebrafish spinal cord. Neurosci. Lett. 448, 240–244 (2008).
Karttunen, M. J., Czopka, T., Goedhart, M., Early, J. J. & Lyons, D. A. Regeneration of myelin sheaths of normal length and thickness in the zebrafish CNS correlates with growth of axons in caliber. PLOS ONE 12, e0178058 (2017).
Peri, F. & Nüsslein-Volhard, C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 133, 916–927 (2008).
Labun, K. et al. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res 47, W171–W174 (2019).
Almeida, R. G. & Lyons, D. A. Intersectional gene expression in zebrafish using the split KalTA4 system. Zebrafish 12, 377–386 (2015).
Dugas, J. C. & Emery, B. Purification of oligodendrocyte precursor cells from rat cortices by immunopanning. Cold Spring Harb. Protoc. 2013, pdb.prot070862 (2013).
Wilmarth, P. A., Riviere, M. A. & David, L. L. Techniques for accurate protein identification in shotgun proteomic studies of human, mouse, bovine, and chicken lenses. J. Ocul. Biol. Dis. Info 2, 223–234 (2009).
Eng, J. K., Jahan, T. A. & Hoopmann, M. R. Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24 (2013).
Perez-Riverol, Y. et al. The PRIDE database at 20 years: 2025 update. Nucleic Acids Res 53, D543–D553 (2024).
Deutsch, E. W. et al. The ProteomeXchange consortium at 10 years: 2023 update. Nucleic Acids Res 51, D1539–D1548 (2023).
Perez-Riverol, Y. et al. PRIDE inspector toolsuite: moving toward a universal visualization tool for proteomics data standard formats and quality assessment of ProteomeXchange datasets. Mol. Cell. Proteom. MCP 15, 305–317 (2016).
Acknowledgements
We would like to thank current and past members of the Emery and Monks labs, particularly Suhail Akram, Emma Brennan, Austin Forbes, Tia Perry, and Adriana Reyes for excellent animal care. We would especially like to thank Dr. Breanne Harty of the Monk lab for her work on first characterizing FBXW7 in the zebrafish CNS and the mouse PNS. We would also like to thank Dr. Ronald Waclaw at Cincinnati Children’s Hospital for alerting us to the anti-MYRF antibody. We would like to thank Dr. Ashok Reddy of the OHSU Proteomic Core for his comments and suggestions while designing our proteomic experiments. Imaging work was supported by the OHSU Advanced Light Microscopy Core (ALMC, RRID: SCR_009960). The research reported in this publication used computational infrastructure of OHSU's Advanced Computing Center (ACC, RRID: SCR_009959) supported by NIH S10OD034224. Mass spectrometric analysis was performed by the OHSU Proteomics Shared Resource (RRID: SCR_009991) with partial support from NIH core grants P30EY010572, P30CA069533, and S10OD012246. This project was supported by the National Multiple Sclerosis Society (RG-1901-33272) and the National Institute of Neurological Disorders and Stroke (R01NS138188) to B.E. and K.R.M. and the Herbert R. & Jeanne C. Mayer Foundation and the National Institutes of Health - National Institute of Neurological Disorders and Stroke (F31NS122433) to H.Y.C. B.E. was supported by an endowment from the Warren family.
Author information
Authors and Affiliations
Contributions
H.Y.C., T.S., K.R.M., and B.E. conceived of the project. H.Y.C designed, performed, and analyzed all experiments with the following exceptions: J.L generated the vo86 zebrafish line (H.Y.C performed all validation experiments and analyses). J.L. performed and analyzed the fbxw7 cell-specific CRISPR-Cas9-mediated gene disruption in Fig. 1f–h. R.A.D. processed and imaged optic nerve and cerebellum TEMs in Figs. 3 and 4. D.A.L., J.E.E. and M.E.M. generated the myrf ue70 zebrafish line in Fig. 7d, I, and Supplementary Fig. 6b, which D.A.L. provided prior to its publication. H.Y.C., K.R.M., and B.E. wrote the manuscript. All authors provided feedback on the manuscript and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Collins, H.Y., Doan, R.A., Li, J. et al. FBXW7 regulates MYRF levels to control myelin capacity and homeostasis in the adult central nervous system. Nat Commun 16, 7822 (2025). https://doi.org/10.1038/s41467-025-62715-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62715-9
