
Abstract
Osteoporosis treatments commonly mitigate bone loss but rarely restore lost bone mass. Yes-associated protein (Yap) nuclear translocation is crucial for the osteogenic differentiation of bone marrow stromal cells (BMSCs), but is disrupted by many factors under osteoporotic conditions. Long non-coding RNAs (lncRNAs) regulate BMSCs differentiation and Yap localization across diseases, exhibiting tissue- and cell-specific effects. However, their role in aberrant Yap signaling within BMSCs under osteoporosis remains unclear. Here, we identify small nucleolar RNA host gene 18 (lnc-Snhg18), a functionally conserved lncRNA enriched in the osteolineage of leptin receptor–positive (LepR⁺) cells within bone, as a key regulator promoting osteogenesis. Mechanistically, lnc-Snhg18 directly binds Caveolin-1 (Cav1) and 14-3-3 eta protein (Ywhah), facilitating Cav1–Ywhah complex formation, thereby disrupting the Ywhah–Yap interaction and enabling Yap nuclear translocation. Knockout of lnc-Snhg18 in LepR⁺ cells accelerates bone loss and traps Yap in the cytoplasm, while its delivery restores bone mass and Yap signaling in osteoporosis models. These findings identify lnc-Snhg18 as a promising therapeutic target for osteoporosis and related disorders.
Similar content being viewed by others


Introduction
Osteoporosis is a prevalent global health issue characterized by reduced bone density and increased fracture risk due to an imbalance in bone remodeling1,2. Current therapeutic strategies primarily focus on slowing bone loss, yet they often fail to effectively restore already lost bone mass3. Bone marrow stromal cells (BMSCs) are key contributors to bone formation4, and their impaired osteogenic differentiation is a major factor in the pathogenesis of osteoporosis5,6.
Recent attention has focused on the Hippo signaling pathway, particularly the transcriptional coactivator Yes-associated protein (Yap), which is essential for regulating BMSCs osteogenic differentiation7,8. Nuclear localization of Yap is essential for its activation of osteogenic gene expression in BMSCs, and is modulated by various upstream signals. Among these, extracellular matrix (ECM) stiffness plays a particularly pivotal role9,10. Under osteoporotic conditions, reduced ECM stiffness11,12,13, along with other factors such as oxidative stress14,15 and hormonal imbalance16, can impair Yap nuclear translocation, thereby diminishing the osteogenic potential of BMSCs and contributing to progressive bone loss.
Long non-coding RNAs (lncRNAs) have emerged as critical regulators of gene expression and cellular differentiation17,18. Notably, they exhibit high tissue and cell-type specificity19,20, making them key players in disease-specific processes21,22. While lncRNAs have been implicated in the regulation of BMSCs differentiation23,24 and in influencing Yap’s subcellular localization in other disease contexts25,26,27,28, no studies have yet demonstrated their role in regulating abnormal Yap signaling in BMSCs during osteoporosis. Furthermore, species-specific differences and in vivo complexities pose challenges for lncRNA-based therapeutic development29. Several functionally conserved lncRNAs, including lnc-H1930 and lnc-DANCR31, have been implicated in bone metabolism, but their broad expression and diverse functions limit their suitability for bone-specific therapies. While lnc-ob1 is osteoblast-specific32, it primarily functions during late differentiation, offering limited regulation during the early stages of osteogenesis. Therefore, identifying functionally conserved lncRNAs that are specifically enriched in BMSCs or osteolineage cells, and investigating their potential to restore osteogenic capacity by modulating Yap signaling, holds significant research and clinical relevance in treating various forms of osteoporosis.
Addressing this gap, we identified lnc-Snhg18, a functionally conserved lncRNA enriched in LepR+ cells—a major subset of BMSCs and primary sources of osteoprogenitors, known for their critical role in bone regeneration and their responsiveness to mechanical forces—and osteogenic cells within bone tissue. Lnc-Snhg18, upregulating during osteogenesis and downregulating in osteoporosis, promotes BMSCs osteolineage commitment. Mechanistically, lnc-Snhg18 directly binds with Caveolin-1 (Cav1) and 14-3-3 eta protein (Ywhah), facilitating the Cav1–Ywhah complex, which disrupts Ywhah–Yap interaction and enhances Yap nuclear translocation. Knockout of lnc-Snhg18 in LepR+ cells accelerates bone loss and osteoporosis by increasing Yap cytoplasmic retention, whereas its overexpression via AAV mitigated bone loss in both OVX- and disuse-induced osteoporosis, significantly reducing Yap cytoplasmic retention. These findings provide insight into osteoporosis treatment by revealing how lncRNAs like lnc-Snhg18 can restore proper Yap signaling, crucial for enhancing BMSCs osteogenesis and recovering lost bone mass in osteoporosis models.
Results
Snhg18 in LepR⁺ MSCs and osteogenic cells reflects bone state
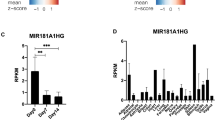
To identify osteogenesis-specific ncRNAs, we reanalyzed RNA-seq data from human mesenchymal stromal cells (MSCs) during adipogenesis and osteogenesis (GSE174794), identifying 189 ncRNAs significantly upregulated during osteogenesis (|logFC| > 1, p < 0.05). Analysis of ncRNA-seq data from an ovariectomy (OVX)-induced mouse osteoporosis model (GSE184226) revealed 5 ncRNAs with orthologous transcripts in humans and mice that were consistently downregulated in osteoporosis (|logFC| > 1, p < 0.05) (Fig. 1a). Among them, lnc-Snhg18/lnc-SNHG18 (ENSMUSG00000096956 in mouse, ENSG00000250786 in human)—a member of the small nucleolar RNA host gene (SNHG) family—stood out due to its orthologous relationship and the presence of the embedded, highly conserved snoRNA Snord123/SNORD123 (Fig. 1b; Supplementary Fig. 1a–d). This gene was consistently upregulated during osteogenesis and downregulated in osteoporosis. qRT-PCR confirmed that lnc-Snhg18 expression was significantly reduced in bone marrow stromal cells (BMSCs) from ovariectomized (OVX) mice compared to controls (Fig. 1c) and significantly upregulated in human BMSCs during osteogenesis (Fig. 1d), which were identified by flow cytometry analysis (Supplementary Fig. 2).

a Identification of osteogenesis-specific ncRNAs: 189 ncRNAs upregulated in human mesenchymal stromal cells during osteogenesis compared to adipogenesis (GSE174794), intersecting with 5 ncRNAs that are downregulated in mouse osteoporosis (GSE184226). Differentially expressed genes were identified using a linear model with empirical Bayes moderation and Benjamini–Hochberg correction for multiple comparisons (two-sided tests). b Genome browser view of lnc-Snhg18 in mouse and human. c qRT-PCR measurement of lnc-Snhg18 expression in mouse BMSCs (Sham vs. OVX). n = 4 biological replicates per group. d qRT-PCR measurement of lnc-Snhg18 expression in human BMSCs during osteogenesis. n = 3 biological replicates per group. e Time-course analysis of stress fracture dataset GSE152677. f qRT-PCR of lnc-Snhg18 expression in mouse skull defect tissue at different healing times. n = 6 biological replicates per time point. g Tissue-specific qRT-PCR of lnc-Snhg18 in various mouse tissues. n = 3 biological replicates per group. h Northern blot analysis of lnc-Snhg18 in multiple tissues and cell types. i Single-cell RNA-seq (scRNA-seq) analysis of mouse mandibular data (PRJNA697839) shows that lnc-Snhg18 is predominantly expressed in mesenchymal cells, particularly in MSCs and osteogenic subclusters. j Flow chart of preparation of scRNA-seq samples from mouse mandibular alveolar bone. Created in BioRender. Tu, D. (2025) https://BioRender.com/kdp734f. k UMAP and violin plots of lnc-Snhg18 expression across cell types and mesenchymal subclusters. The left panel shows UMAP clustering of various cell populations, with the middle violin plot highlighting lnc-Snhg18 expression, which is highest in mesenchymal cells. The right panel demonstrates that lnc-Snhg18 is highly expressed in MSCs and osteogenic subclusters. l Correlation heatmap of cell groups from two single-cell datasets. Statistical analyses were conducted as follows: Two-tailed Student’s t-test for (c, d); one-way ANOVA for (f). Data are presented as mean ± SD. Source data and exact p values are provided in the Source data file.
Further validation using fuzzy C-means33 clustering of gene expression during stress fracture healing (GSE152677) identified six temporal clusters, with lnc-Snhg18 in a cluster associated with extracellular matrix remodeling and osteogenesis, alongside key regulator Sp7 (Fig. 1e; Supplementary Fig. 3a, b), suggesting that the temporal expression of lnc-Snhg18 is closely related to osteogenesis. Consistently, lnc-Snhg18 expression peaked on day 7 in a mouse skull defect model and then declined (Fig. 1f).
Tissue-specific analysis showed that lnc-Snhg18 was predominantly expressed in bone, with lower levels in other tissues (Fig. 1g), which was confirmed by Northern blot (Fig. 1h). To further validate the specific expression pattern of lnc-Snhg18 in bone-related cell types, we performed a Northern blot analysis using four cell types: osteoclasts, macrophage, BMSCs, and osteoblasts. The results showed that lnc-Snhg18 was expressed at very low levels in osteoclasts and macrophage, while it was notably expressed in BMSCs and osteoblasts (Fig. 1h).
Single-cell RNA-seq analysis of mouse mandibular bone (PRJNA697839) further revealed that lnc-Snhg18 is enriched in mesenchymal cells, particularly in LepR+ MSCs, a major subset of BMSCs, and osteogenic subclusters (Fig. 1i). Additionally, we extracted mandibular bone tissue from mice and performed single-cell RNA-seq (Fig. 1j). Cells were divided into 11 subgroups based on classic cell surface markers (Supplementary Fig. 4a–c), with the mesenchymal cell population further subdivided into three subclusters (Supplementary Fig. 4d). Consistent with the Northern blot results, lnc-Snhg18 expression was enriched in the mesenchymal cell population, particularly in the LepR+ MSCs and osteogenic subclusters (Fig. 1k). Correlation heatmaps from two independently generated datasets confirmed the consistent expression of lnc-Snhg18 in these mesenchymal subpopulations (Fig. 1l).
These results suggest that lnc-Snhg18 is highly expressed in LepR+ MSC and osteogenic cells in bone tissue, becomes upregulated during osteogenesis, and is downregulated in conditions such as osteoporosis.
Snhg18/SNHG18 enhances BMSCs osteogenic differentiation
Given the prominence of LepR+ MSCs within the BMSCs pool, and the critical role in bone formation and their ability to differentiate into osteoblasts4, combined with the predominant expression of lnc-Snhg18 in these cells, we reanalyzed scRNA-seq data (GSE138689) from LepR-Cre-traced cells under adipogenic and osteo-chondrogenic stress. We found that lnc-Snhg18 is highly expressed in the osteogenic and periosteal lineages of LepR+ cells, while its expression is low in adipogenic and cycling populations (Fig. 2a). Pseudotime trajectory analysis of osteogenic lineage subsets showed a gradual upregulation of lnc-Snhg18 during osteolineage commitment (Fig. 2b). This was further confirmed by qRT-PCR, which showed increased lnc-Snhg18/lnc-SNHG18 expression in mouse and human BMSCs during osteogenesis (Figs. 1d and 2c), highlighting its role in promoting BMSC’s osteogenic commitment.

a Expression levels of lnc-Snhg18 across different lineages of LepR⁺ cells, based on single-cell RNA-seq analysis (GSE138689), including 17,224 cells isolated from multiple mice across different conditions (Adipogenic, n = 14,075; Cycling, n = 428; Osteogenic, n = 1584; Periosteal, n = 1137). Each dot represents a single cell. Violin plots show the distribution of lnc-Snhg18 expression levels. The embedded box plots indicate the median (center line), the lower and upper quartiles (bounds of box), and the whiskers extend to the minima and maxima. b Pseudotime trajectory analysis of the osteogenic lineage cells (left) and expression dynamics of lnc-Snhg18 during the osteogenic differentiation continuum (right). c qRT-PCR of lnc-Snhg18 in mouse BMSCs at different osteogenesis time points. n = 3 biological replicates per time point. d Quantitative measurement of lnc-Snhg18 in cytoplasm and nuclear fractions in mouse BMSCs before and after OI by qRT-PCR. Gapdh served as cytoplasmic controls, U6 served as nuclear controls, Gas5 served as both a nuclear and cytoplasmic control. n = 3 biological replicates for both Control and OI conditions. e RNA–fluorescence in situ hybridization (FISH) staining of lnc-Snhg18 in mouse BMSCs before and after OI, with probe localization schematic and specificity verified using U6, 18S, and ASO controls. Scale bars, 50 µm. f qRT-PCR analysis of lnc-Snhg18, Sp7, Runx2 in mouse BMSCs transfected with control ASO, lnc-Snhg18 ASO, control vector or lnc-Snhg18 vector. n = 3 biological replicates for ASO experiments and n = 4 biological replicates for vector experiments. g Western blotting and quantification of Osterix (Sp7) expression in mouse BMSCs after 2 weeks of OI. n = 3 biological replicates per group. h Alkaline phosphatase, Alizarin red staining, and Von Kossa staining of mouse BMSCs transfected with control ASO, lnc-Snhg18 ASO, control vector, or lnc-Snhg18 vector. qRT-PCR analysis of lnc-SNHG18, SP7, RUNX2 in human BMSCs transfected with control ASO, lnc-SNHG18 ASO (i), with corresponding alkaline phosphatase and alizarin red staining (j). n = 3 biological replicates per group. k GSEA of extracellular matrix gene enrichment after lnc-Snhg18 knockdown. Normalized enrichment score (NES), p value, and false discovery rate (FDR) were shown. GSEA gene enrichment was assessed using the two-sided empirical permutation test with Benjamini–Hochberg correction for FDR. l GO enrichment analysis of differentially expressed genes in mouse BMSCs after lnc-Snhg18 knockdown. GO enrichment was assessed using the one-sided hypergeometric test with Benjamini–Hochberg correction for multiple comparisons. m qRT-PCR analysis of extracellular matrix-related genes in mouse BMSCs following lnc-Snhg18 knockdown. n = 3 biological replicates per group. n Masson’s trichrome staining of collagen matrix in mouse BMSCs following modulation of lnc-Snhg18 expression. For (e, h, n), experiments were conducted independently 3 times, consistently producing similar results. Statistical analyses were conducted as follows: One-way ANOVA for (c); two-tailed Student’s t-test for (f, g, i, m). Data are presented as mean ± SD. OI osteogenic induction. Source data and exact p values are provided in the Source data file.
To investigate the impact of lnc-Snhg18 on BMSCs osteogenic differentiation, we first confirmed its subcellular localization using nuclear-cytoplasmic separation assays (Fig. 2d) and RNA–fluorescence in situ hybridization (FISH) (Fig. 2e). Lnc-Snhg18 was detected in both the nucleus and cytoplasm, independent of osteogenic induction (OI). We then used Smart Silencers for knockdown and a lentiviral system for overexpression in mouse BMSCs. Overexpression of lnc-Snhg18 led to upregulation of Sp7 and Runx2 mRNA and increased Osterix (Sp7) protein levels, while knockdown produced the opposite effects (Fig. 2f, g). Alkaline phosphatase and alizarin red staining confirmed that lnc-Snhg18 knockdown inhibited extracellular matrix mineralization, whereas overexpression enhanced osteogenic mineralization (Fig. 2h). Similarly, knockdown of lnc-SNHG18 inhibited osteogenic differentiation in human BMSCs in vitro (Fig. 2i, j).
Bulk RNA-seq analysis revealed that genes associated with extracellular matrix organization and ossification were significantly affected by lnc-Snhg18 knockdown, as evidenced by GO and GSEA enrichment (Fig. 2k, l, Supplementary Data 1). Further validation by qRT-PCR confirmed the downregulation of key ECM-related genes (Col1a1, Col1a2, Col3a1, Postn, and Pdgfra) upon lnc-Snhg18 knockdown (Fig. 2m). Consistently, Masson staining showed reduced collagen deposition after lnc-Snhg18 knockdown and increased deposition after its overexpression, supporting its role in extracellular matrix formation (Fig. 2n).
In summary, our data indicate that lnc-Snhg18 regulates the osteolineage fate of BMSCs by modulating extracellular matrix organization and ossification.
Snhg18 directly binds Cav1 and Ywhah, enhancing interaction
Subsequently, we sought to identify specific protein interactors of lnc-Snhg18 and elucidate its role in regulating osteogenesis in BMSCs. To this end, we synthesized a biotin-labeled lnc-Snhg18 probe and performed RNA pull-down assays on mouse BMSCs undergoing osteogenic induction, followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of the retrieved proteins (Fig. 3a). Silver staining of the pull-down products revealed distinct differential bands within the 20–35 kDa range, suggesting the enrichment of specific protein partners (Fig. 3b). To refine the candidate list, we implemented a multi-step filtering strategy: only proteins uniquely detected in the sense strand pull-down (intensity > 0 in sense and = 0 in antisense) and identified by at least two iBAQ peptides were retained. Guided by the silver staining pattern, we further focused on proteins within the 20–35 kDa range. Western blot analysis ultimately confirmed that Cav1 and Ywhah were specifically enriched in the sense pull-down group, supporting their direct interaction with lnc-Snhg18 (Fig. 3c). The full dataset and screening workflow are detailed in the “Methods” section, Supplementary Fig. 5 and Supplementary Data 2.

a–c Identification of lnc-Snhg18 interacting proteins by RNA pull-down and mass spectrometry. a Mass spectrometry (MS) chromatograms of proteins pulled down by biotin-labeled sense (black, top) and antisense (red, bottom) lnc-Snhg18 transcripts in mouse BMSCs. b Silver staining of SDS-PAGE gel showing protein bands from input, sense (Sen.), and antisense (A.S.) pull-down samples. Specific enrichment in the sense group is indicated (red arrows). c Western blot validation of pull-down products confirming specific binding of Cav1 and Ywhah to lnc-Snhg18. Gapdh served as a negative control. RNA immunoprecipitation (RIP) assay for Cav1 (d) and Ywhah (e), followed by qRT-PCR analysis of lnc-Snhg18 in mouse BMSCs. n = 3 biological replicates per group. f Electrophoretic mobility shift assay (EMSA) was performed using biotin-labeled wild-type or mutant lnc-Snhg18 RNAs incubated with Ywhah–V5 and/or Cav1–Flag. Colocalization analysis of lnc-Snhg18 (purple) with Cav1 (green, g) and Ywhah (green, h) in mouse BMSCs, before and after OI, using RNA-FISH and IF staining. Scale bars, 20 µm and 200 µm. Colocalization analysis of lnc-Snhg18 with Cav1 (i) and Ywhah (j) in mouse BMSCs, before and after OI, quantified using Pearson’s R and Overlap R scores. n = 3 biological replicates for both Control and OI conditions. k Co-IP analysis of Cav1–Flag and Ywhah–V5 interaction in HEK293T cells transfected with control or lnc-Snhg18 overexpression vectors, with quantification of relative binding ratios of Cav1–Flag/Ywhah–V5. n = 3 biological replicates per group. For (b, c, f), experiments were conducted independently 3 times, consistently producing similar results. Statistical analyses were conducted as follows: Two-way ANOVA for (d, e);two-tailed Student’s t-test for (i, j, k). Data are presented as mean ± SD. OI osteogenic induction. Source data and exact p values are provided in the Source data file.
RNA immunoprecipitation (RIP) assays further confirmed the RNA pull-down results (Fig. 3d, e), supporting the interaction between lnc-Snhg18 and Cav1, as well as Ywhah. Additional electrophoretic mobility shift assays (EMSA) demonstrated that Ywhah and Cav1 specifically bind to lnc-Snhg18. Ywhah and Cav1 proteins formed complexes with biotin-labeled lnc-Snhg18 individually or together, resulting in band shifts in the gel, indicating protein–RNA interactions. The results of the competition assays showed that the binding of these proteins to lnc-Snhg18 is specific, as the band intensity significantly decreased in the presence of unlabeled lnc-Snhg18 as a competitor RNA, while non-competitor RNA did not affect the binding (Fig. 3f). To further confirm the structural basis of Snhg18–protein interactions, we predicted the secondary structure of lnc-Snhg18 using RNAfold and generated a mutant RNA by deleting three stable stem-loop regions (Supplementary Fig. 6a–d). EMSA using this mutant probe showed markedly reduced binding to Ywhah and Cav1, indicating that these interactions are dependent on the intact RNA structure of lnc-Snhg18 (Fig. 3f).
Additionally, colocalization results from cellular immunofluorescence (IF) and RNA in situ hybridization (RNA-FISH) further corroborated the interaction between lnc-Snhg18 and Cav1 and Ywhah, with significantly increased colocalization observed after osteogenic induction (Fig. 3g–j). Moreover, co-immunoprecipitation (Co-IP) results revealed an interaction between Cav1 and Ywhah, which was significantly enhanced upon overexpression of lnc-Snhg18 (Fig. 3k).
In conclusion, lnc-Snhg18 directly binds to Cav1 and Ywhah proteins, and in the presence of lnc-Snhg18, the interaction between Cav1 and Ywhah is enhanced.
Snhg18 promotes Ywhah–Yap disruption and Yap nuclear entry
Ywhah binds Yap, sequestering it in the cytoplasm and reducing its transcriptional activity34,35. However, Cav1 can disrupt this interaction, allowing Yap to translocate to the nucleus and exert its functions36. Since lnc-Snhg18 enhances the interaction between Cav1 and Ywhah, it may modulate Yap activity by facilitating Cav1’s disruption of the Ywhah–Yap complex, thereby promoting Yap’s nuclear localization. To explore the influence of lnc-Snhg18 on Yap and its interacting partners during osteogenesis, we transfected mouse MC3T3-E1 cells with plasmids and induced osteogenesis. Lnc-Snhg18 expression was controlled with a control and overexpression vector. We used Co-IP analysis to examine the interactions between Cav1, Yap, and Ywhah under these conditions. The Co-IP analysis showed that lnc-Snhg18 enhances the interaction between Cav1 and Ywhah while reducing the interaction between Yap and Ywhah (Fig. 4a, b).

a Co-IP analysis of the interaction between Cav1, Yap, and Ywhah in mouse MC3T3-E1 cells transfected with plasmids in the presence or absence of lnc-Snhg18 after 24 h of OI. b Quantification of Co-IP analysis showing the relative binding ratios of Yap-HA/Ywhah–V5 (left) and Cav1–Flag/Ywhah–V5 (right) in the presence or absence of lnc-Snhg18 in mouse MC3T3-E1 cells. n = 3 biological replicates per group. c Co-IP analysis of the interaction between Yap, Cav1, and Ywhah in mouse bone tissue transfected with control or lnc-Snhg18 overexpression vectors. d Quantification of Co-IP analysis showing the relative binding ratios of Yap/Ywhah (left) and Cav1/Ywhah (right) in mouse bone tissue. n = 3 biological replicates per group. e Live-cell imaging of Yap-EGFP in mouse BMSCs transfected with control ASO or lnc-Snhg18 ASO, followed by OI for the indicated durations. Representative time-lapse fluorescence images show dynamic changes in Yap-EGFP subcellular distribution; nuclei are outlined by dashed circles. Scale bars, 10 µm (magnified) and 100 µm (overview). f Quantification of Yap-EGFP nuclear/cytoplasmic intensity ratio. n = 3 biological replicates per group. g Western blot analysis (upper panel) and quantification (lower panel) of Yap phosphorylation in mouse BMSCs transfected with control ASO, lnc-Snhg18 ASO after 24 h of OI. n = 3 biological replicates per group. h Schematic illustration of lnc-Snhg18–mediated promotion of Yap nuclear localization and osteogenesis. Statistical analyses were conducted as follows: Two-tailed Student’s t-test for (b, d, g); two-way ANOVA for (f). Data are presented as mean ± SD. OI osteogenic induction. Source data and exact p values are provided in the Source data file.
To further investigate whether this regulatory mechanism operates in vivo with comparable strength and specificity, we systemically administered AAV9 encoding the spliced transcript of lnc-Snhg18 (containing exon 1 and exon 2) into wild-type mice via tail vein injection. Seven weeks post-injection, a femoral bone defect model was established. Fourteen days after surgery, newly formed tissue from the defect site was harvested for tissue Co-IP analysis. Consistent with in vitro results, lnc-Snhg18 overexpression in vivo promoted Cav1–Ywhah complex formation while reducing the interaction between Ywhah and Yap (Fig. 4c, d).
Since Ywhah binding sequesters Yap in the cytoplasm and lnc-Snhg18 reduces this interaction, lnc-Snhg18 is likely to promote Yap nuclear translocation. Accordingly, the absence of lnc-Snhg18 leads to increased cytoplasmic retention of Yap. To dynamically monitor this effect, we performed live-cell imaging to track Yap dynamics at pre-treatment and at 1 h, 3 h, 5 h, and 7 h after osteogenic induction. In control group, Yap exhibited progressive nuclear accumulation over time. However, lnc-Snhg18 knockdown significantly impaired its translocation (Fig. 4e). Quantitative analysis of the nuclear-to-cytoplasmic fluorescence intensity ratio further confirmed this defect in Yap nuclear translocation (Fig. 4f). This increased cytoplasmic retention is accompanied by elevated Yap phosphorylation levels, as demonstrated by Western Blot analysis (Fig. 4g).
In summary, these findings demonstrate that lnc-Snhg18 plays a pivotal role in regulating Yap’s subcellular localization during osteogenesis by modulating the interactions between Cav1, Ywhah, and Yap. Specifically, lnc-Snhg18 enhances the interaction between Cav1 and Ywhah, facilitating Cav1’s ability to disrupt the Ywhah–Yap complex, thereby reducing the cytoplasmic sequestration of Yap. This reduction allows for increased nuclear translocation of Yap. Conversely, in the absence of lnc-Snhg18, the stronger interaction between Yap and Ywhah results in greater cytoplasmic retention and elevated phosphorylation of Yap (Fig. 4h). Together, these results underscore lnc-Snhg18’s function as a key mediator in the Cav1–Ywhah–Yap signaling axis, influencing Yap’s role in osteogenic differentiation.
Snhg18 knockout impairs bone homeostasis and Yap localization
Lnc-Snhg18 is predominantly expressed in LepR+ MSC lineage cells, which are crucial for maintaining bone homeostasis and responding to mechanical stress37,38. It influences their osteogenic commitment by modulating Yap activity. To further explore the role of lnc-Snhg18 in vivo, we generated LepR-Cre;Snhg18fl/fl conditional knockout (cKO) mice (Supplementary Fig. 7a, b), allowing for specific deletion of lnc-Snhg18 in LepR+ cells and assessment of its impact on bone density and osteoporosis process.
Adult male cKO mice exhibited reduced body weight compared to wild-type littermates (Fig. 5a). Micro-computed tomography (micro-CT) analysis showed significantly lower trabecular bone volume per tissue volume (BV/TV) and trabecular thickness (Tb.Th) in the femur of cKO mice compared to controls (Fig. 5b). Histological staining further showed reduced trabecular structure and collagen content in cKO femurs (Fig. 5c). Given the higher susceptibility of females to osteoporosis, a time-course micro-CT analysis was conducted in female cKO mice, revealing an earlier onset of osteoporotic features. Female cKO mice displayed significant reductions in trabecular BV/TV and Tb.Th as early as 12 weeks, while these changes only emerged in controls at 20 weeks. By 20 weeks, both BV/TV and Tb.Th remained significantly lower in cKO mice, indicating a lasting impact of lnc-Snhg18 deletion on bone microarchitecture (Fig. 5d, e)

a Representative images of Snhg18fl/fl and LepR-Cre;Snhg18fl/fl mice with body weight comparison. n = 8 mice per group. b Representative micro-CT images and quantitative analysis of distal femoral trabecular bone in 12-week-old male Snhg18fl/fl and LepR-Cre;Snhg18fl/fl mice. n = 8 and 6 mice per group. c Representative H&E and Masson’s trichrome staining of distal femurs from 12-week-old male Snhg18fl/fl and LepR-Cre;Snhg18fl/fl mice. Scale bars, 100 µm. d Time-course micro-CT analysis of femur in female Snhg18fl/fl and LepR-Cre;Snhg18fl/fl mice at 12, 16, and 20 weeks with quantification. n = 4 mice per group at 12w; n = 4 and 6 mice at 16w; n = 5 and 6 mice at 20w. e Representative H&E staining of femur structure in female Snhg18fl/fl and LepR-Cre;Snhg18fl/fl mice at 12, 16, and 20 weeks. f Mandibular bone structure and BV/TV analysis in control and unloaded conditions. n = 4 mice for Snhg18fl/fl, n = 5 mice for LepR-Cre;Snhg18fl/fl. g Immunofluorescence images and quantification of Yap (yellow) localization in LepR+ (purple) cells from Snhg18fl/fl and LepR-Cre;Snhg18fl/fl mice mandibles under control and unloaded conditions. The white arrow points to the nucleus of the LepR+ cell. Scale bars, 20 µm. n ≥ 100 cells per group from three independent experiments. h Masson’s trichrome staining of femoral bone defect areas in Snhg18fl/fl and LepR-Cre;Snhg18fl/fl mice. Scale bar, 500 µm. i Quantification of BV/TV and Tb.Th in femoral defects. n = 8 mice for Snhg18fl/fl, n = 7 mice for LepR-Cre;Snhg18fl/fl. j Immunofluorescence of Osterix (green), Col1a1(purple), and DAPI (blue) in femoral defects. Scale bars, 80 µm. For (c, e), experiments were conducted independently 3 times, consistently producing similar results. Statistical analyses were conducted as follows: Two-tailed Student’s t-test for (a, b, f (right), i); two-way ANOVA for (d, f (left), g). Data are presented as mean ± SD. BV/TV bone volume per tissue volume, Tb.Th trabecular thickness. Source data and exact p values are provided in the Source data file.
Lnc-Snhg18’s regulation of Yap—a key mechanical stress response mediator—is also linked to mandibular disuse osteoporosis38. In cKO mice, mandibular BV/TV was lower than that of controls under normal conditions, and this difference was further exacerbated upon unloading, with a significantly greater reduction in BV/TV (Fig. 5f). A similar phenotypic change was observed in the classic hindlimb unloading model, where cKO mice exhibited a more pronounced loss of bone mass compared to controls under the same unloading stress (Supplementary Fig. 8a, b), suggesting increased sensitivity to mechanical unloading. Furthermore, immunofluorescence revealed that Yap was primarily nuclear in control mice but increasingly cytoplasmic in cKO mice, especially under unloading (Fig. 5g). These findings align with previous reports of Yap cytoplasmic retention in LepR+ MSCs under mechanical unloading38, which may impair osteogenic function. Lnc-Snhg18 appears to counter this, promoting Yap nuclear localization.
We also assessed bone healing in femur defects. Two weeks post-surgery, histology revealed disorganized collagen fibers and delayed healing in cKO mice compared to controls (Fig. 5h). Micro-CT confirmed reduced BV/TV and Tb.Th, while immunostaining showed decreased Col1a1 and Osterix expression in cKO mice, further highlighting the negative impact of lnc-Snhg18 deletion on bone regeneration (Fig. 5i, j).
These results demonstrate that lnc-Snhg18 is critical for bone maintenance and repair, with its absence leading to reduced bone density, impaired healing, and increased Yap cytoplasmic retention, especially under mechanical unloading, worsening disuse osteoporosis.
Snhg18 rescue of bone loss is functionally independent
To further substantiate the functional role of lnc-Snhg18 in vivo, rescue experiments were performed using LepR-Cre;Snhg18fl/fl conditional knockout (cKO) mice. A spliced, mature transcript of lnc-Snhg18, comprising only exon 1 and exon 2, was administered intravenously via an AAV9 vector to restore lnc-Snhg18 expression in LepR⁺ BMSCs (Fig. 6a).

a RT-qPCR analysis of lnc-Snhg18 expression in BMSCs isolated from cKO mice 7 weeks after AAV injection. n = 6 mice per group. b Representative micro-CT images and quantification of femur BV/TV and Tb.Th in Snhg18fl/fl + NC, LepR-Cre;Snhg18fl/fl + NC and LepR-Cre;Snhg18fl/fl + Snhg18 groups 7 weeks after AAV injection. n = 5, 8, and 7 mice per group. c Representative H&E and Masson’s trichrome staining of distal femurs in Snhg18fl/fl + NC, LepR-Cre;Snhg18fl/fl + NC and LepR-Cre;Snhg18fl/fl + Snhg18 groups 7 weeks after AAV injection. Scale bars, 100 µm. Experiments were conducted independently 3 times, consistently producing similar results. d Representative micro-CT images, histological staining (H&E and Masson), and quantitative analysis of mandibles under unloading conditions in Snhg18fl/fl + NC, LepR-Cre;Snhg18fl/fl + NC, and LepR-Cre;Snhg18fl/fl + Snhg18 groups 8 weeks after AAV injection. Scale bars, 1 mm (Micro-CT), 100 µm (H&E and Masson). n = 5, 5, and 6 mice per group. e Immunofluorescence images and quantification of Yap localization in LepR+ MSCs in the mandibles, with DAPI (blue), Yap (yellow), and LepR (purple) staining, and AAV-driven fluorescence (green). The white arrow points to the nucleus of the AAV-targeted LepR+ MSCs. Scale bars, 20 µm. n ≥ 100 cells per group from three independent experiments. Statistical analyses were conducted as follows: Two-tailed Student’s t-test for (a); one-way ANOVA for (b, d, e). Data are presented as mean ± SD. BV/TV bone volume per tissue volume, Tb.Th trabecular thickness. Source data and exact p values are provided in the Source data file.
Micro-CT analysis demonstrated that lnc-Snhg18 significantly ameliorated the bone loss phenotype observed in cKO mice, as indicated by increased BV/TV and Tb.Th (Fig. 6b). Corresponding improvements in bone architecture were corroborated by histological staining (Fig. 6c). In femoral sections, the cKO group displayed disorganized and sparse collagen matrix distribution, characterized by reduced blue staining signal; conversely, overexpression of lnc-Snhg18 restored dense and continuous collagen deposition, indicative of enhanced osteoid formation (Fig. 6c). In the mandibular unloading model, lnc-Snhg18 overexpression similarly rescued mandibular bone mass in cKO mice, resulting in improved matrix organization and collagen fiber content compared to negative control (NC)-treated mice, thereby partially reversing unloading-induced matrix degeneration (Fig. 6d).
Immunofluorescence staining further showed that lnc-Snhg18 expression restored nuclear localization of Yap in LepR+ cells (Fig. 6e). These results collectively support the notion that the mature lnc-Snhg18 transcript plays a critical role in maintaining bone mass and regulating Yap signaling activity.
Furthermore, considering that Snord123 and lnc-Snhg18 are located at the same genomic locus (Fig. 1b), we conducted additional experiments to elucidate their distinct functions. In cKO mice, we employed a similar strategy to deliver Snord123 via AAV9, which successfully restored Snord123 expression (Supplementary Fig. 9a, b). Notably, Snord123 overexpression did not restore femoral bone mass in lnc-Snhg18 cKO mice and did not mitigate mandibular bone loss in the unloading model (Supplementary Fig. 9c, d). To investigate the broader therapeutic potential of Snord123, we overexpressed it in wild-type mice subjected to either an OVX-induced osteoporosis model or a tooth extraction-induced mandibular unloading model. In both models, Snord123 did not significantly affect bone mass (Supplementary Fig. 9e, f).
Consistent with the in vivo results, in vitro overexpression of Snord123 in LepR+ BMSCs did not significantly alter the expression of key osteogenic transcription factors Runx2 and Sp7 (Supplementary Fig. 9g), nor did Von Kossa staining show a notable effect on mineralized nodule formation (Supplementary Fig. 9h).
Collectively, these results suggest that Snord123 does not play an independent or essential role in the mechanism through which lnc-Snhg18 regulates bone regeneration. Conversely, the reintroduction of the spliced lnc-Snhg18 transcript markedly restored bone mass and osteogenic activity, underscoring lnc-Snhg18 as the principal and functionally dominant effector in driving osteogenesis at this locus.
Snhg18 delivery mitigates bone loss in osteoporosis models
Finally, to evaluate the therapeutic potential of lnc-Snhg18 in preventing bone loss in vivo, we systemically delivered a spliced, mature lnc-Snhg18 transcript (containing exon 1 and exon 2) via AAV9 vectors in both OVX-induced and disuse-induced osteoporosis models. Previous studies have demonstrated that systemically administered AAV9 exhibits bone-targeting properties39,40. To confirm this in our study, we examined EGFP fluorescence driven by AAV expression using fluorescence microscopy. The results showed that EGFP was most abundant in bone tissue, moderately expressed in the heart and liver, and nearly undetectable in the lungs, kidneys, and spleen (Supplementary Fig. 10a, b). Moreover, qPCR analysis confirmed significant overexpression of lnc-Snhg18 in bone tissue (Supplementary Fig. 10c), indicating that the AAV-based delivery strategy achieved effective and bone-specific expression.
The construction of both osteoporosis models was described in detail in the “Methods” section. Animals were divided into three groups: (1) Control group, including the OVX sham group and contralateral mandibular bone without tooth extraction (Control), receiving control AAV9 administration; (2) OVX + NC/ Unload + NC group, subjected to OVX or tooth extraction-induced unloading, followed by control AAV9 administration; (3) OVX + Snhg18/Unload + Snhg18 group, subjected to the same surgical procedure, followed by AAV9-mediated overexpression of lnc-Snhg18.
In the OVX model, we adopted the same dosing regimen as previously reported39 (Fig. 7a). Seven weeks post-AAV injection, the OVX + Snhg18 group showed a significantly higher femoral mineral apposition rate (MAR) compared to OVX + NC (Fig. 7b). Micro-CT analysis revealed that lnc-Snhg18 overexpression significantly improved trabecular bone architecture in the femur, including increased BV/TV and trabecular thickness (Fig. 7c, d). Von Kossa staining further confirmed the restoration of mineralized trabecular structure in the OVX + Snhg18 group (Fig. 7e). As revealed by Masson staining, the OVX + Snhg18 group demonstrated trabecular thickness and volume closely resembling those of the sham group. Notably, collagen staining signals were similarly restored, further supporting the rescue effect of lnc-Snhg18 overexpression (Fig. 7f). In addition, previous studies have reported cytoplasmic retention of Yap in osteoporotic conditions12,13. Immunofluorescence analysis revealed that lnc-Snhg18 overexpression promoted nuclear localization of Yap in AAV-targeted LepR+ MSCs, with a significantly higher percentage of Yap detected in both the nucleus and cytoplasm compared to the predominantly cytoplasmic distribution observed in the OVX + NC group (Fig. 7g), suggesting that lnc-Snhg18 helps restore proper Yap signaling dynamics.

a Study and treatment methods diagram in OVX-induced osteoporosis model with AAV9 injection (i.v., 4 × 10¹¹ GC). Seven weeks later, mice received subcutaneous calcein and alizarin injections at 6-day intervals for dynamic histomorphometry. b Double calcein/alizarin labeling showing new bone formation rates in femurs, with MAR quantification. Scale bars, 100 µm. n = 6 mice per group. c Representative micro-CT images of the femur and mandible in Sham + NC, OVX + NC, and OVX + Snhg18 groups. Scale bars, 1 mm. d Quantitative analysis of BV/TV and Tb.Th in femur. n = 6, 8, and 8 mice per group. e Von Kossa staining of femur. Scale bars, 1 mm. f Masson’s trichrome staining of the femur in Sham + NC, OVX + NC, and OVX + Snhg18 groups. Scale bars, 100 µm. Experiments were conducted independently 3 times, consistently producing similar results. g Immunofluorescence images and quantification of Yap localization in LepR+ MSCs in the femur, with DAPI (blue), Yap (yellow), and LepR (purple) staining, and AAV-driven fluorescence (green). The white arrow points to the nucleus of the AAV-targeted LepR+ MSCs. Scale bars, 20 µm. n ≥ 100 cells per group from three independent experiments. h Study and treatment timeline in disuse osteoporosis model with AAV9 injection (i.v., 4 × 10¹¹ GC). Created in BioRender. Tu, D. (2025) https://BioRender.com/kdp734f. i Masson’s trichrome staining of mandible with BV/TV and Tb.Th quantification. Scale bars, 500 µm. n = 6 mice per group. j Immunofluorescence images and quantification of Yap localization in LepR+ MSCs in the mandible, with DAPI (blue), Yap (yellow), and LepR (purple) staining, and AAV-driven fluorescence (green). The white arrow points to the nucleus of the AAV-targeted LepR+ MSCs. Scale bars, 50 µm. n ≥ 100 cells per group from three independent experiments. Statistical analyses were conducted as follows: One-way ANOVA for (b, d, g, i, j). Data are presented as mean ± SD. MAR mineral apposition rate, BV/TV bone volume per tissue volume, Tb.Th trabecular thickness. Source data and exact p values are provided in the Source data file.
In the disuse-induced osteoporosis model, intervention was performed according to the experimental timeline (Fig. 7h). In this model, lnc-Snhg18 overexpression significantly alleviated mandibular bone loss (Fig. 7i) and reduced cytoplasmic retention of Yap within AAV-targeted LepR+ MSCs (Fig. 7j), further supporting its role in regulating Yap activity and improving the osteogenic microenvironment.
Taken together, our findings indicate that lnc-Snhg18 effectively mitigates bone loss and restores Yap signaling dynamics in both OVX-induced and disuse-induced osteoporosis models, highlighting its therapeutic potential.
Discussion
Bone marrow stromal cells (BMSCs) are the primary source of osteogenesis, and their osteogenic differentiation is critical in the treatment of osteoporosis5,6. The nuclear localization of Yap determines the osteogenic potential of BMSCs7,8, and lncRNAs have been found to precisely regulate Yap localization25,26,27,28, though challenges in efficacy arise from species-specific differences and the complexity of in vivo functional studies29. In adults, LepR+ MSCs maintain bone homeostasis and serve as the primary source of osteoprogenitor cells4. Therefore, identifying highly expressed and functionally conserved lncRNAs in these cells that regulate Yap localization holds significant therapeutic potential for targeted treatment, markedly improving the efficacy of osteoporosis therapy.
In our study, we identified lnc-Snhg18 (human ortholog: lnc-SNHG18), a member of the small nucleolar RNA host gene (SNHG) family, as a key regulator of osteogenesis. Lnc-Snhg18 is highly enriched in LepR⁺ mesenchymal stromal cells and their osteogenic descendants, and its expression is essential for maintaining osteogenic potential. Similar to other SNHG family members, lnc-Snhg18 harbors an intronic snoRNA gene, Snord123. Through both pathological and in vitro models, we dissected the functional contributions of the lnc-Snhg18 locus. Our findings demonstrate that the bone phenotypes observed in conditional knockout mice are attributable to the loss of lncRNA transcript rather than the snoRNA component. Notably, overexpression of Snord123 alone failed to restore bone mass or osteogenic gene expression. These findings are reminiscent of a recent report on the SNHG26 locus41, where only the lncRNA, not the co-encoded snoRNA SNORD93, was required for functional rescue in wound healing. Together, these results suggest that lncRNAs and their associated snoRNAs may have independent biological functions despite being transcribed from the same genomic locus.
Further investigations revealed that the knockout of lnc-Snhg18 in LepR⁺ lineage cells leads to significant bone loss and exacerbated osteoporotic features. Conversely, AAV-mediated overexpression of lnc-Snhg18 promotes bone formation and alleviates bone loss in both OVX- and disuse-induced osteoporosis models. These findings highlight lnc-Snhg18 as an independent and positive regulator of bone homeostasis. Interestingly, the magnitude of bone loss observed in lnc-Snhg18 conditional knockout mice compared to controls tends to diminish with age. Although statistically significant differences persist, this observation suggests the emergence of age-related compensatory mechanisms in the bone marrow niche that may partially buffer the osteogenic defects caused by lnc-Snhg18 deficiency in LepR⁺ MSCs.
Enhancing Yap nuclear translocation has emerged as a promising therapeutic strategy for osteoporosis12,42. In our study, we found that Yap’s nuclear localization in BMSCs correlates with the expression level of lnc-Snhg18, and elevated levels of lnc-Snhg18 markedly promote Yap nuclear translocation. Mechanistically, lnc-Snhg18 directly binds Cav1 and Ywhah, facilitating the formation of the Cav1–Ywhah complex while diminishing the interaction between Ywhah and Yap. This disruption reduces Yap’s cytoplasmic sequestration and promotes its translocation to the nucleus. In vivo, lnc-Snhg18 overexpression enhanced Yap nuclear accumulation in LepR⁺ cells and alleviated bone loss phenotypes in both hormone-deficient and mechanical unloading models. Previous studies, including those by Moreno-Vicente et al.36, have shown that Cav1 modulates the Ywhah–Yap interaction in response to mechanical cues. Our research further elucidates the distinctive and central role of lnc-Snhg18 within this critical interaction framework.
Notably, in dissecting the interaction between lnc-Snhg18 and Ywhah, we observed a clear disparity in their intracellular abundance. This “stoichiometrically asymmetric” mode of regulation is not unprecedented. For instance, although the expression level of the lncRNA NEAT1 is relatively low compared to the glycolytic enzymes PGK1, PGAM1, and ENO1, it can still function as a scaffold to coordinate their complex assembly43. This phenomenon may be explained by the distinctive molecular characteristics of lncRNAs: compared to peptides of similar length, RNA molecules possess extended backbones, intricate secondary structures, and multivalent interaction surfaces that collectively enhance their protein-binding capacity44,45. Lnc-Snhg18 may further exert its regulatory function through subcellular enrichment, enabling localized scaffolding of protein complexes. Consistent with this, transcriptome-wide analyses have shown that highly structured RNAs tend to interact with a broader range of proteins, underscoring the critical role of RNA architecture in shaping binding multiplicity and specificity45.
This principle is also exemplified in our study. Cav1, a membrane-associated protein, is involved in sensing and transducing cellular signals46. Although Cav1 lacks canonical RNA-binding domains, RNA pull-down, RNA immunoprecipitation, and EMSA assays consistently demonstrated a direct interaction between Cav1 and lnc-Snhg18. Subsequent functional analyses revealed that this interaction is highly dependent on the intact structural conformation of lnc-Snhg18. This structure-driven protein–RNA interaction highlights the importance of RNA folding in mediating recognition by noncanonical RNA-binding proteins and provides a mechanistic basis for understanding the regulatory specificity of such interactions.
Comprehensive evidence supports a critical role for lnc-Snhg18 in modulating Yap nuclear translocation and activity via the Cav1–Ywhah interaction model. In line with findings by Zhang et al.10, which reported a marked increase in bone marrow stiffness between 2 and 12 months of age, our data suggest that enhanced extracellular matrix (ECM) stiffness, a key biophysical cue promoting Yap nuclear localization7, may act as a compensatory mechanism that mitigates age-associated impairments in marrow-resident stromal cells. This mechanistic insight provides a plausible explanation for the diminished bone loss disparity observed between control and lnc-Snhg18 knock-out mice over time, reinforcing the view that Yap is the key downstream effector of lnc-Snhg18 in the regulation of skeletal homeostasis.
To further explore the translational relevance of the lnc-Snhg18-Cav1–Ywhah–Yap interaction model, we analyzed genome-wide association study (GWAS) data to identify single-nucleotide polymorphisms (SNPs) associated with bone-related traits. Notably, SNPs in YAP (e.g., rs7931899 associated with heel bone mineral density and rs10895278 associated with birth weight) and CAV1 (e.g., rs6466578 linked to body height) showed significant associations. In contrast, no SNPs linked to bone phenotypes were identified for SNHG18 or YWHAH (Supplementary Table 1). These findings highlight the evolutionary conservation of the mechanotransduction axis in skeletal regulation and suggest that lncRNAs such as SNHG18 may influence this mechanosignal transduction via mechanisms not captured by common SNP variation.
Despite these promising results, several limitations should be acknowledged. Although AAV9 vectors provided high transduction efficiency in bone tissue39,40, off-target expression was also observed. Developing next-generation delivery systems with enhanced tissue specificity remains an important goal. Additionally, conserved sequence motifs within lncRNAs may act as modular functional domains47,48,49, future efforts should aim to map these regions within lnc-Snhg18. Finally, while our mechanistic model is well-supported by molecular biology approaches, higher-resolution structural evidence suitable for long non-coding RNAs will be essential to definitively delineate the interaction interfaces within the lnc-Snhg18–Cav1–Ywhah complex.
In conclusion, our study demonstrates that lnc-Snhg18 is a functionally conserved and abundantly expressed long non-coding RNA in LepR⁺ MSC lineage cells, capable of autonomously promoting osteogenic differentiation by regulating Yap subcellular localization. This lncRNA emerges as a promising therapeutic target for osteoporosis induced by hormonal deficiency or mechanical unloading. Given the broad involvement of Yap signaling in diverse pathological conditions50, the potential of lnc-Snhg18 as a regulatory molecule in other disease contexts merits further investigation. Overall, this study expands our understanding of lncRNA biology and uncovers an additional regulatory layer within the Cav1–Ywhah–Yap mechanotransduction axis, highlighting the translational potential of lnc-Snhg18 in various pathological conditions.
Methods
Animal models
All mice were housed at the Laboratory Animal Center of Tongji University with constant temperature (22 ± 2 °C) and humidity (55 ± 10%) in a 12-h light cycle. All animal studies were approved by the Institutional Animal Care and Use Committee of Tongji University (approval number: [2022]-DW-10). The C57BL/6Smoc-Snhg18em1(flox)Smoc (Snhg18fl/fl) (NM-CKO-2114159) mouse with C57BL/6J background was purchased from the Shanghai Model Organisms Center, China. The B6.129(Cg)-LepRtm2(cre)Rck/J(LepR-Cre) (Strain #:008320) mouse with C57BL/6 was purchased from the Jackson Laboratory. Snhg18fl/fl mice were crossed with LepR-Cre mice to generate LepR-Cre;Snhg18fl/fl (cKO) mice (Supplementary Fig. 7a, b). Genotyping of Snhg18fl/fl mice was performed by PCR using primers listed in Supplementary Data 3. The expected PCR product sizes were 321 bp for the wild-type allele, 375 bp for the floxed allele, and both bands for heterozygous mice.
The femurs of 12-week-old male cKO mice and littermate controls were used for bone mass measurements and histological experiments. Similarly, femurs from 12-week-old, 16-week-old, and 20-week-old female cKO mice and littermate controls were also used for these analyses. Twelve-week-old male cKO and littermate control mice underwent left maxillary first molar extraction, and 4 weeks postoperatively, the left mandible (unloaded group) and right mandible (control group) were harvested for bone mass measurements and histological experiments, using methods previously described51. For the femur bone defect model, using methods previously described52, 12-week-old male cKO and littermate control mice underwent defect surgery on the left femurs, and 14 days post-surgery, the left femurs were collected for analysis. For the skull defect model, using methods previously described53, 12-week-old C57BL/6J mice underwent the creation of a circular defect with a diameter of 2 mm in the skull using a dental drill under sterile conditions. At 7-, 14-, and 21-day post-surgery, newly formed bone tissue within a 2 mm margin surrounding the cranial defect was carefully harvested for further analysis. For the hindlimb unloading model, 12-week-old male cKO and littermate control mice were subjected to modeling for 21 days using a previously described method54. In rescue experiments based on cKO mice, 8-week-old male Snhg18fl/fl and cKO mice received AAV via tail vein, and femoral bone phenotypes were analyzed 7 weeks later. For the mandibular unloading model, 8-week-old Snhg18fl/fl and cKO mice were injected with AAV, followed by bilateral maxillary first molar extraction 4 weeks later; mandibles were collected 4 weeks post-surgery. In wild-type mice, 12-week-old males underwent molar extraction to induce mandibular unloading, followed by AAV injection 4 weeks later and analysis at 7 weeks post-injection. For the OVX model, using methods previously described39, 12-week-old females were ovariectomized, injected with AAV 6 weeks later, and analyzed 7 weeks post-injection. The total number of animals used per group is provided in the figure legends and Source data. Male and female mice were used as appropriate according to the standard of each model; sex-specific effects were not analyzed.
Human specimen
This research was reviewed and approved by the Ethics Committee (approval number: [2022]-SR-55) of the Affiliated Stomatological Hospital of Tongji University. All procedures were performed in accordance with the Declaration of Helsinki. Human mandibular bone tissues from three male donors (aged 24, 27, and 32 years), all in good general health, were collected during orthognathic surgeries. Sex was assigned based on clinical records at the time of surgery and was not self-reported. Informed consent was obtained from all participants after a detailed explanation of the study procedures. No financial or other compensation was provided to the participants. Sex or gender was not considered as a variable in the study design, and no sex- or gender-based analysis was conducted due to the limited sample size and the specific purpose of the study.
Isolation and characterization of mesenchymal stromal cells (MSCs) from human mandible
Human mandible mesenchymal stromal cells were isolated as described previously55, briefly by collecting mandibular cancellous bone during surgery, flushing bone marrow from bone fragments using α-MEM, isolating adherent cells by the whole bone marrow adherence method, and expanding them in culture. Cells were characterized by flow cytometry. The following antibodies were used at a dilution of 1:100: anti-CD73 (APC) (51-9007649, BD, clone: AD2), anti-CD90 (FITC) (51-9007657, BD, clone: 5E10), anti-CD105 (PerCP-CyTM5.5) (51-9007648, BD, clone:266) to positively select MSCs, and anti-CD34 (PE) (550761, BD, clone: 563), anti-CD45 (BV510) (563204, BD, clone: HI30), anti-CD14 (FITC) (abs180059, Absin, clone: 2K14-6), and anti-CD19 (FITC) (abs180030, Absin, clone: CB19) to exclude non-MSC populations. To control for non-specific binding, isotype controls corresponding to each fluorochrome were used. After antibody staining, cells were washed and resuspended in 500 μL PBS. Flow cytometry was performed on a BD FACSAria III (BD Biosciences) equipped with the corresponding lasers for the chosen fluorochromes. Data were analyzed using FlowJo software (v10.8.1). MSCs were identified as CD73⁺CD90⁺CD105⁺CD34−CD45⁻CD14⁻CD19⁻ cells. Sorted cells were collected for subsequent downstream applications, such as RNA extraction or further cell culture.
Isolation of mouse bone marrow stromal cells
Mouse BMSCs were isolated as described previously56. Briefly, bone marrow cells were flushed from the femurs and tibiae and directly plated without filtration in α-MEM complete medium lacking exogenous cytokines. Spindle-shaped adherent cells were collected and passaged on day 5. All in vitro experiments were performed using passage 3 cells.
Cell transfection
To knock down lnc-Snhg18/lnc-SNHG18, mouse BMSCs and human MSCs were transfected with Smart Silencers (lnc3211124101938, lnc3250718102929, RiboBio) using RiboFECT™ CP (C10502, RiboBio) every 3 days during osteogenic differentiation. For overexpression, mouse BMSCs were transduced with lentivirus at MOI 100 in the presence of 8 µg/mL polybrene (TR1003, Sigma). The overexpression construct consisted of the spliced form of mouse lnc-Snhg18, which includes exon 1 and exon 2. Cells were incubated for 72 h post-transduction before osteogenic differentiation. HEK293T (OriCell®293T, H4-1401, Cyagen Biosciences) cells were transfected with plasmids using Lipo8000™ (C0533, Beyotime) according to the manufacturer’s instructions. HEK293F (A14527, Thermo Fisher) cells were transfected with plasmids using Lipo293F™ Plus Transfection Reagent (C0519, Beyotime) according to the manufacturer’s instructions. MC3T3-E1 cells (OriCell®MC3T3-E1 Subclone 14, M7-0201, Cyagen Biosciences) were transduced with lentivirus at an MOI of 50 in the presence of 8 µg/mL polybrene (TR1003, Sigma), selected with 10 µg/mL puromycin (P8833, Sigma) for 14 days with medium replacement every 2–3 days, and subsequently transfected with plasmids using Lipo8000™ (C0533, Beyotime) during osteogenic differentiation.
Total RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cell and tissue samples using TRIzol reagent (15596018, Invitrogen) according to the manufacturer’s instructions. First-strand cDNA synthesis was performed using the RevertAid First-Strand cDNA Synthesis Kit (K1622, Thermo Scientific). Quantitative real-time PCR (qRT-PCR) was performed using PowerTrack™ SYBR Green Master Mix (A46109, Applied Biosystems) on a QuantStudio 7 Flex (Thermo Scientific), and qRT-PCR data were analyzed using QuantStudio Software (v1.7.2, Thermo Scientific). Actb/ACTB was used as an internal control. Primer sequences are provided in Supplementary Data 3.
Western blotting
For Western blotting, cells were lysed in RIPA lysis buffer supplemented with protease and phosphatase inhibitors (P1005, Beyotime). Protein concentrations were determined using the BCA Protein Assay Kit (A55860, Thermo Scientific). Equal amounts of protein samples were separated by SDS-PAGE and transferred onto PVDF membranes (IPVH00010, Millipore). Membranes were blocked with 5% non-fat milk in TBST for 1 h at room temperature, followed by overnight incubation at 4 °C with the following primary antibodies: Rabbit anti-Osterix (ab209484, Abcam, clone: EPR21034, 1:1000), Rabbit anti-Phospho-Yap (Ser127) (13008, CST, clone: D9W2I, 1:1000), Rabbit anti-Yap (14074, CST, clone: D8H1X, 1:1000), Rabbit anti-Gapdh (5174, CST, clone: D16H11, 1:1000) and Mouse anti β-Actin (3700, CST, clone: 8H10D10, 1:1000) as loading control. After washing, membranes were incubated with HRP-conjugated Goat anti-Rabbit secondary antibody (A0208, Beyotime, 1:1000), HRP-conjugated Goat anti-Mouse secondary antibody (A0216, Beyotime, 1:1000) for 1 h at room temperature. Western blots were detected using Amersham Imager 680 Analysis Software.
Von Kossa and Masson’s trichrome staining of cultured cells
For mineralization analysis, Von Kossa staining was performed on LepR⁺ BMSCs after 14 days of osteogenic induction. Cells were fixed in 4% paraformaldehyde for 30 min, rinsed with distilled water, and treated with 5% silver nitrate (G1043, Solarbio) under ultraviolet light for 20 min. Unreacted silver was removed using 5% sodium thiosulfate for 5 min. Mineralized nodules were imaged under a light microscope (Nikon). For collagen evaluation, Masson’s trichrome staining was conducted.
Public data analysis
Public bulk RNA-seq and single-cell RNA-seq datasets were sourced from GEO and SRA databases. The bulk RNA-seq datasets included transcriptional profiles of hMSCs during adipogenesis and osteoblastogenesis (GSE174794), long non-coding RNA profiles in C57BL/6 mice after ovariectomy (GSE184226), and bone healing profiles following ulnar stress fracture (GSE152677). Differential expression analysis was conducted using the “limma” package57 (v3.64.1; https://bioconductor.org/packages/release/bioc/html/limma.html) in R, with thresholds set at p value < 0.05 and |logFC| > 1. Gene clustering related to bone healing was performed using the Mfuzz package33,58 (v2.68.0; https://bioconductor.org/packages/release/bioc/html/Mfuzz.html) with fuzzy c-means clustering. For single-cell RNA-seq analysis, scRNA-seq data from mouse mandibular tissue (PRJNA697839) were utilized to examine lnc-Snhg18 expression across different bone cell types. Data processing followed previously established methods59, including quality control, dimensionality reduction, and clustering using the Seurat R package60 (v4.0.2; https://satijalab.org/seurat/). Variable genes were identified with the FindVariableGenes function, followed by PCA for initial dimensionality reduction and UMAP for visualization and cell-type annotation based on specific markers. To confirm lnc-Snhg18 expression across various LepR+ cell lineages, additional scRNA-seq data (GSE138689) were analyzed using the same procedures61, which included quality control, normalization, and identification of highly variable genes using Seurat60 (v4.0.2; https://satijalab.org/seurat/). Dimensionality reduction was performed with t-SNE, revealing distinct cell populations such as adipogenic, cycling, osteogenic, and periosteal clusters. Pseudotime analysis with the Monocle R package62,63 (v2.22.0; http://cole-trapnell-lab.github.io/monocle-release/) was used to map differentiation trajectories, particularly within osteogenic clusters.
Experimental data analysis
For bulk RNA-seq, BMSCs isolated from 4-week-old C57BL/6J were transfected with control ASO or lnc-Snhg18 ASO during osteogenic induction for 7 days, then the BMSCs were collected, and a TRIzol reagent kit (15596018, Invitrogen) was used to obtain total RNA. The isolated RNA was purified with an RNA purification kit (DP419, Tiangen). Sequencing libraries were prepared using the Next® Ultra™ RNA Library Prep Kit for Illumina® (E7530, NEB), with index codes incorporated to link sequences to corresponding samples. The libraries were then sequenced on an Illumina HiSeq platform. Differential expression analysis was conducted using DESeq264 (v1.38.3; https://bioconductor.org/packages/release/bioc/html/DESeq2.html), applying a threshold of log2 fold change > 1 or <−1, with an adjusted p value < 0.05. Gene Ontology (GO) Biological Processes enrichment analysis was performed using DAVID Bioinformatics Resources (v6.8; https://david.ncifcrf.gov/) with GO terms from the Gene Ontology database (http://geneontology.org/). Gene Set Enrichment Analysis (GSEA; v4.0.3; https://www.gsea-msigdb.org/gsea/) was performed using gene sets from the Molecular Signatures Database (MSigDB; v2025.1.Mm; https://www.gsea-msigdb.org/gsea/msigdb/mouse/).
For single-cell RNA sequencing, mandibular bone tissues were harvested from six 8-week-old male C57BL/6J wild-type mice (Shanghai Biomodel Organism Co.). After tissue digestion and filtration, red blood cells were lysed using ACK buffer, and the remaining cells were resuspended in 0.4% BSA PBS. Approximately 15,000 cells were loaded for cDNA library construction using Chromium single-cell v3.0 reagents and subsequently sequenced on the Illumina NovaSeq platform. The raw sequencing data were aligned to the mouse genome (mm10) and processed with Cellranger (v3.1, 10x Genomics) to generate a single-cell gene expression matrix. This matrix was imported into the Seurat package (v4.0.2; https://satijalab.org/seurat/)57 for downstream analysis. Quality control was performed by filtering cells based on the following thresholds: UMI counts >2000, gene counts >1000, and mitochondrial gene content <10% of total gene expression. Variable genes were identified using the FindVariableGenes function in Seurat. Principal component analysis (PCA) was then conducted, followed by UMAP for dimensionality reduction and visualization based on the PCA results. Cell-type annotation for different subclusters was performed according to the expression of specific marker genes (Supplementary Fig. 4a–c). The identified cell populations were annotated as follows: epithelial cells (Col17a1), mesenchymal cells (Dcn), vascular mural cells (Cdh5), T cells (Cd3d), basophils (Mcpt8), dendritic cells (Ccr9), erythroid cells (Car2), B cells (Cd79a), granulocyte-macrophage progenitors (GMPs) (Elane), monocytes/macrophages (Ccr2), and neutrophils (Camp). Further subclustering of the mesenchymal cells revealed distinct subpopulations (Supplementary Fig. 4d), including Asporin-positive fibroblasts (Aspn), mesenchymal stromal cells (LepR), and osteogenic cells (Bglap), based on their unique gene expression profiles.
Northern blot
Total RNA was extracted from samples using TRIzol reagent, followed by quantification. DIG-labeled DNA probes were prepared using the Roche DIG High Prime DNA Labeling and Detection Kit II (1585614, Roche), where 200 ng of denatured template DNA was incubated with DIG-11-dUTP for 1 h at 37 °C. For Northern blot analysis, 15–20 µg of RNA was separated on a 1% denaturing agarose gel and transferred onto a nylon membrane by capillary blotting. The membrane was hybridized overnight with the DIG-labeled probe at 50 °C in DIG Easy Hyb solution, then blocked and incubated with an anti-DIG-alkaline phosphatase conjugate. Chemiluminescent detection was performed using CSPD substrate, and the signal was visualized on X-ray film. The primer sequences used for generating the probes are given in Supplementary Data 3.
Electrophoretic mobility shift assay (EMSA)
V5-tagged Ywhah and Flag-tagged Cav1 were overexpressed in HEK293F cells and captured using anti-V5 magnetic beads (M215-11, MBL) and anti-Flag magnetic beads (Bimake, B26101). Elution was performed with V5 or Flag peptides (P9813, P9801, Beyotime). Full-length wild-type and mutant lnc-Snhg18 RNAs were transcribed in vitro (AM1334, Thermo Scientific), biotin-labeled, and purified by phenol-chloroform extraction. The mutant RNA was generated by deleting three structurally stable stem-loop regions as predicted by RNAfold (see Supplementary Fig. 6a–d). EMSA was performed using the LightShift™ Chemiluminescent RNA EMSA Kit (20158, Thermo Scientific) following the manufacturer’s instructions. For binding reactions, 10 pmol of biotin-labeled RNA was incubated with 10 or 20 µg of V5-tagged Ywhah or Flag-tagged Cav1 proteins in a 20 µl reaction containing the supplied binding buffer, 5% glycerol, 0.25 mg/ml BSA, 1 mM MgCl₂, and 0.5 mM DTT. After 30 min of incubation at room temperature, samples were separated on 4–6% native polyacrylamide gels pre-run in 0.5× TBE buffer at 100 V, and transferred onto a nylon membrane. The membrane was cross-linked using UV light at 254 nm (120 mJ/cm²), blocked, and probed with the stabilized streptavidin-HRP conjugate provided in the kit. Detection of biotin-labeled RNA-protein complexes was performed using enhanced chemiluminescence according to the kit protocol.
Cytosolic and nuclear fractionation and RNA isolation
After collecting the cells, they were harvested on ice, and ~1 × 10⁶ cells were used for the nuclear and cytoplasmic fractionation experiment. The PARIS Kit (AM1921, Thermo Scientific) was then employed according to the manufacturer’s protocol to isolate the cytoplasmic and nuclear fractions, which were immediately subjected to RNA purification and subsequent qPCR analysis.
RNA pull-down assay and mass spectrometry
LncRNA-Snhg18 and antisense lncRNA-Snhg18 plasmids containing T7 promoter were synthesized by RiboBio (Shanghai). In detail, the sequence of interest was placed downstream from the T7 promoter. Oligos are designed for PCR templates for in vitro transcription. The PCR primers of lnc-Snhg18-sense and lnc-Snhg18-antisense are given in Supplementary Data 3. Purify the PCR product using a commercial PCR clean-up kit, DNA TIANgel Purification kit (DP219, Tiangen), then the Transcript Aid T7 High Yield Transcription Kit (AM1334, Thermo Scientific) was used for in vitro transcription. The PierceTM RNA 3′ End Desthiobiotinylation Kit (20163, Thermo Scientific) was used to label biotinylated nucleotides to the RNA strand. Thirty picomoles of biotinylated RNA was incubated with 30 μl streptavidin magnetic beads for 30 min at room temperature with rotation. Then, 500 μg BMSCs lysate at 14 days after osteogenic differentiation was added to the RNA-bound beads and incubated at 4 °C for 6–8 h with rotation. The protein–RNA-bead complex was washed twice with 200 μl wash buffer, then added SDS loading buffer to the beads and boiled at 100 °C for 5 min.
The protein products pulled down by lnc-Snhg18 and antisense were subjected to mass spectrometry (MS) analysis (n = 1 per group, control and experimental). Peptides were enzymatically hydrolyzed, extracted, and purified, then separated on an EASY-nLC 1000 system (Thermo Scientific) using an analytical C18 column (1.9 µm, 75 µm × 20 cm) at a flow rate of 200 nL/min. MS analysis was performed on an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific) in data-dependent acquisition (DDA) mode. Raw data files were analyzed using Proteome Discoverer 2.4 software (Thermo Scientific) with the following search parameters: database, SwissProt; taxonomy, Mus musculus; enzyme, trypsin; fixed modification, carbamidomethyl (C); variable modifications, acetyl (protein N-term), Met-loss, Met-loss+acetyl, oxidation (M); maximum missed cleavages, 2; target FDR (strict), 0.01; target FDR (relaxed), 0.05; peptide mass tolerance, 10ppm; MS/MS tolerance, ±0.6 Da.
To identify specific lnc-Snhg18-interacting proteins, a multi-step filtering strategy was applied: proteins were retained only if exclusively detected in the sense pull-down (intensity > 0 in the sense and = 0 in the antisense), and only candidates with at least two iBAQ peptides were considered. Based on silver staining results, we further narrowed the candidate list by focusing on proteins in the 20–35 kDa range where distinct differential bands were observed (seven proteins). Ultimately, Western blot validation was performed using Rabbit anti-Cav1 (3267, CST, clone: D46G3, 1:1000) and Rabbit anti-Ywhah (15222-1-AP, Proteintech, clone: AG7435, 1:1000), confirming that Cav1 and Ywhah specifically interacted with lnc-Snhg18. Additional candidates, including Sfn, Prdx6, Lgals3, Phb2, and Hnrnpc, were excluded by Western blotting using Rabbit anti-Sfn (BA3752, Boster, 1:1000), Rabbit anti-Prdx6 (13585-1-AP, Proteintech, clone: AG4512, 1:1000), Mouse anti-Lgals3 (60207-1-Ig, Proteintech, clone: AG6753, 1:1000), Rabbit anti-Phb2 (12295-1-AP, Proteintech, clone: AG2977, 1:1000), and Rabbit anti-Hnrnpc (11760-1-AP, Proteintech, clone: AG2356, 1:1000), respectively. The full list of identified proteins is provided in Supplementary Data 2.
RNA–fluorescence in situ hybridization (FISH) and immunofluorescence (IF)
Fluorescein-labeled probes targeting U6 snRNA, 18S rRNA, and lnc-Snhg18 were designed and synthesized by RiboBio (Shanghai). The sequences of the lnc-Snhg18 FISH probes are provided in Supplementary Data 3. FISH detection was performed using a Fluorescence In Situ Hybridization Kit (C10910, RiboBio) according to the manufacturer’s instructions. The fluorescence probe-labeled cells were then stained with Rabbit anti-Cav1 antibody (CST, 3267, clone: D46G3), Rabbit anti-Ywhah antibody (CST, 9640) at a dilution of 1:100, and DAPI (C1002, Beyotime), followed by fixation with anti-fade mounting medium (0100-01, SouthernBiotech). Images were captured using a Nikon laser confocal microscopy system. Data analysis was conducted with ImageJ software (National Institutes of Health, USA, V1.8.0.112).
RNA-binding-protein immunoprecipitation assay
Mouse BMSCs were collected and lysed, after which immunoprecipitation was carried out using specific antibodies against Rabbit anti-Cav1 (3267, CST, clone: D46G3, 1:50) and Rabbit anti-Ywhah (15222-1-AP, Proteintech, clone: AG7435, 1:50). A normal IgG antibody (2729, CST, 1:50) served as the control to evaluate the specificity of the RNA-protein interactions. The RIP procedure followed the guidelines provided by the manufacturer (17700, Millipore). In brief, cell lysates were incubated with magnetic beads conjugated to the antibodies overnight at 4 °C. Following extensive washes, RNA-protein complexes were eluted, and the associated RNA was isolated using TRIzol reagent. Quantitative real-time PCR (qPCR) was then performed to quantify lnc-Snhg18 RNA in the immunoprecipitated samples, with results compared to the IgG control to determine the specificity of the interactions between lnc-Snhg18 and Cav1 or Ywhah.
Immunoprecipitation (IP) and western immunoblotting (WB)
For both cell and tissue IP assays, the Pierce Classic Magnetic IP/Co-IP Kit (88804, Thermo Scientific) was used following the manufacturer’s protocol. For cell-based experiments, cells were lysed in pre-chilled IP lysis buffer and incubated for 5 min at 4 °C. Lysates were centrifuged at 13,000 × g for 10 min at 4 °C, and the supernatant was collected for protein quantification. The supernatants were then incubated with anti-V5 magnetic beads (M215-11, MBL) for 4–6 h at 4 °C. After washing, immunoprecipitated complexes were analyzed by Western blotting using Rabbit anti-FLAG (14793, CST, clone: D6W5B, 1:1000), Rabbit anti-V5 (13202, CST, clone: D3H8Q, 1:1000), and Rabbit anti-HA (3724, CST, clone: C29F4, 1:1000). For tissue-based Co-IP validation, AAV9 vectors encoding the spliced lnc-Snhg18 transcript were administered to wild-type mice 7 weeks prior to establishing a femoral defect model. Fourteen days post-surgery, newly formed bone tissues were harvested and homogenized in pre-chilled IP lysis buffer using a tissue grinder on ice. Homogenates were centrifuged at 13,000 × g for 10 min at 4 °C, and the supernatants were collected for protein analysis. Co-IP was then performed using Rabbit anti-Ywhah (15222-1-AP, Proteintech, clone: AG7435, 1:50) for immunoprecipitation. Immunoprecipitated complexes were subsequently analyzed by Western blotting with Rabbit anti-Cav1 (3267, CST, clone: D46G3, 1:1000), Rabbit anti-Ywhah (15222-1-AP, Proteintech, clone: AG7435, 1:1000), and Rabbit anti-Yap (14074, CST, clone: D8H1X, 1:1000). For the detection of immunoprecipitated proteins, the IP-specific secondary antibody VeriBlot for IP Detection Reagent (ab131366, Abcam) was applied at a 1:200 dilution.
Live-cell imaging of Yap dynamics during osteogenic induction
For live-cell confocal imaging, primary mouse BMSCs were seeded onto 35-mm glass-bottom dishes (801001, Nest) and transfected with a Yap-EGFP expression plasmid using Lipo8000™ transfection reagent (C0533, Beyotime), following the manufacturer’s protocol. Eighteen hours after transfection, the medium was replaced with osteogenic induction medium (α-MEM supplemented with 10% FBS, 50 μg/mL ascorbic acid, and 10 mM β-glycerophosphate). Live-cell time-lapse imaging was performed using a Nikon Ti2 inverted fluorescence microscope equipped with an environmental chamber maintained at 37 °C and 5% CO₂, using a 40×/0.95 NA objective. Time-lapse sequences were acquired every 5 min for a total of 7 h to monitor dynamic changes in Yap subcellular localization during osteogenic induction. Z-stack images (3 optical sections) were collected with a step size of 0.4μm using NIS-Elements software (Nikon, v4.5000.1117.0). Image processing and analysis were performed using NIS-Elements Advanced Research module.
Micro-computed tomographic (μCT) analyses
Bones from mice were fixed in 4% PFA for 48 h at 4 °C and scanned with a μCT-50 (Scanco Medical) at a 14 μm resolution and 70 kVp voltage. The images were reconstructed in 3D with a threshold value set from 212 to 1000. Representative X-ray images were taken in the sagittal plane. For trabecular architecture analysis, 100 slices were reconstructed. For alveolar bone mass, 20 slices from the mandibular first molar root furcation were analyzed. Femur bone defect analysis used a cylindrical VOI of 1200 μm, and 85 slices were reconstructed. Bone-related parameters included BV/TV and Tb.Th.
In vivo AAV9 administration
To achieve in vivo overexpression, AAV9 carrying either the spliced lnc-Snhg18 transcript (containing only exon 1 and exon 2) or the Snord123 sequence was systemically administered. Viruses were diluted in 150 μl PBS and delivered via tail vein injection at a dose of 4 × 10¹¹ genome copies per mouse (2 × 10¹³ GC/kg). Mice were sacrificed for endpoint analyses 7–8 weeks after injection, as specified in the figure legends.
Hard tissue sectioning and histological analysis
Femurs from Sham + NC, OVX + NC, and OVX + Snhg18 groups were collected and fixed in 4% PFA for 24 h, followed by embedding in light-cured resin (EXAKT) for 13.5 h. Sections (15–20 μm) were obtained using an EXAKT micro section and grinding system. For Von Kossa staining, Calcium Stain Kit (G1043, Solarbio) was utilized following the manufacturer’s instructions. For dynamic histomorphometric analysis, mice received subcutaneous injections of 25 mg/kg calcein (C0875, Sigma) and 50 mg/kg alizarin-3-methyliminodiacetic acid (A5533, Sigma) 6 days apart. Samples were fixed in 4% PFA, embedded in resin for 12 h, and sectioned longitudinally at 15–20 μm.
Histological and immunofluorescence staining
Mouse femur and mandible samples were fixed in 4% paraformaldehyde for 24 h, decalcified in 10% EDTA, and embedded in paraffin. Sections (5 μm) were cut, deparaffinized, rehydrated. Hematoxylin and Eosin (H&E) staining was performed using a commercial kit (C0105, Beyotime) following the manufacturer’s instructions. Masson’s trichrome staining was conducted using a staining kit (D026, Nanjing Jiancheng Bioengineering Institute) according to the manufacturer’s protocol to evaluate collagen matrix deposition. For immunofluorescence staining, antigen retrieval was achieved with hyaluronidase at 37 °C for 1 h, followed by permeabilization with 0.2% Triton X-100 in PBS for 15 min. Sections were blocked using blocking buffer (P0260, Beyotime) for 10 min and incubated with primary antibodies of Rabbit anti-Yap (14074, CST, clone: D8H1X, 1:200), Goat anti-LepR (AF497, R&D, clone: Q3US58,1:100), Rabbit anti-Osterix (ab209484, Abcam, clone: EPR21034, 1:200), Mouse anti-Col1a1 (67288-1-Ig, Proteintech, clone: 1E9A7, 1:200) overnight at 4 °C. Then, slides were incubated with fluorophore-conjugated secondary antibodies: Donkey anti-Rabbit 488 (A21206, Invitrogen, 1:2000), Goat anti-Rabbit 594 (A21207, Invitrogen, 1:2000), Goat anti-Mouse 594 (A32744, Invitrogen, 1:2000), Donkey anti-Goat 647 (A21447, Invitrogen, 1:2000) at room temperature for 1 h. Nuclei were counterstained with DAPI for 5 min, followed by fixation with anti-fade mounting medium (0100, SouthernBiotech). Images were captured using a Nikon laser confocal microscopy system. Data analysis was conducted with ImageJ software (National Institutes of Health, USA, V1.8.0.112).
Statistics and reproducibility
No methods were used to predetermine sample size. No data were excluded from the analyses, and no blinding or randomization was employed for data analysis. Data were presented as means ± SD, with the number of independent experiments indicated in the legends. Two-tailed unpaired Student’s t-test, one-way ANOVA, or two-way ANOVA was applied to analyze the data after determining whether the data were normally distributed. SPSS 26.0 (IBM) was used for all statistical calculations. Significant differences were considered at p values below 0.05 and were indicated in the figures.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The scRNA-seq and bulk RNA-seq data generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) database under accession code PRJNA1172916. The mass spectrometry data from RNA pull-down assays have been deposited in the iProX database affiliated to the ProteomeXchange Consortium under accession code PXD064764. Other published datasets applied in this study are as follows: GSE184226, GSE174794, GSE152677, PRJNA697839, and GSE138689. All other data generated in this study are provided in the Supplementary Information/Source data file. Source data are provided with this paper.
References
Lane, N. E. Epidemiology, etiology, and diagnosis of osteoporosis. Am. J. Obstet. Gynecol. 194, S3–11 (2006).
Wright, N. C. et al. The recent prevalence of osteoporosis and low bone mass in the United States based on bone mineral density at the femoral neck or lumbar spine. J. Bone Min. Res. 29, 2520–2526 (2014).
Liang, B., Burley, G., Lin, S. & Shi, Y. C. Osteoporosis pathogenesis and treatment: existing and emerging avenues. Cell. Mol. Biol. Lett. 27, 72 (2022).
Zhou, B. O., Yue, R., Murphy, M. M., Peyer, J. G. & Morrison, S. J. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 15, 154–168 (2014).
Pino, A. M., Rosen, C. J. & Rodriguez, J. P. In osteoporosis, differentiation of mesenchymal stem cells (MSCs) improves bone marrow adipogenesis. Biol. Res. 45, 279–287 (2012).
Rodriguez, J. P., Montecinos, L., Rios, S., Reyes, P. & Martinez, J. Mesenchymal stem cells from osteoporotic patients produce a type I collagen-deficient extracellular matrix favoring adipogenic differentiation. J. Cell. Biochem. 79, 557–565 (2000).
Dupont, S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011).
Pan, J. X. et al. YAP promotes osteogenesis and suppresses adipogenic differentiation by regulating beta-catenin signaling. Bone Res. 6, 18 (2018).
Ma, Y. et al. Extended preconditioning on soft matrices directs human mesenchymal stem cell fate via YAP transcriptional activity and chromatin organization. APL Bioeng. 7, 016110 (2023).
Zhang, X. et al. Harnessing matrix stiffness to engineer a bone marrow niche for hematopoietic stem cell rejuvenation. Cell Stem Cell 30, 378–395 e378 (2023).
Zhang, D. et al. Osteoporosis-decreased extracellular matrix stiffness impairs connexin 43-mediated gap junction intercellular communication in osteocytes. Acta Biochim. Biophys. Sin.52, 517–526 (2020).
Zheng, C. et al. Targeting sulfation-dependent mechanoreciprocity between matrix and osteoblasts to mitigate bone loss. Sci. Transl. Med. 15, eadg3983 (2023).
Li, Z. et al. The PCK2-glycolysis axis assists three-dimensional-stiffness maintaining stem cell osteogenesis. Bioact. Mater. 18, 492–506 (2022).
Jin, J. et al. Oxidative stress-CBP axis modulates MOB1 acetylation and activates the Hippo signaling pathway. Nucleic Acids Res. 50, 3817–3834 (2022).
Zhang, W. et al. Synthetic nanoparticles functionalized with cell membrane-mimicking, bone-targeting, and ROS-controlled release agents for osteoporosis treatment. J. Control. Release 378, 306–319 (2025).
Wei, Y. et al. YAP/TAZ: molecular pathway and disease therapy. MedComm4, e340 (2023).
Flynn, R. A. & Chang, H. Y. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell 14, 752–761 (2014).
Statello, L., Guo, C. J., Chen, L. L. & Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 22, 96–118 (2021).
Cabili, M. N. et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927 (2011).
Mercer, T. R., Dinger, M. E. & Mattick, J. S. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159 (2009).
Dong, K. et al. CARMN is an evolutionarily conserved smooth muscle cell-specific LncRNA that maintains contractile phenotype by binding myocardin. Circulation 144, 1856–1875 (2021).
Hosono, Y. et al. Oncogenic role of THOR, a conserved cancer/testis long non-coding RNA. Cell 171, 1559–1572 e1520 (2017).
Ju, C. et al. Mesenchymal stem cell-associated lncRNA in osteogenic differentiation. Biomed. Pharmacother. 115, 108912 (2019).
Silva, A. M. et al. Long noncoding RNAs: a missing link in osteoporosis. Bone Res. 7, 10 (2019).
Qu, L. et al. A feed-forward loop between lncARSR and YAP activity promotes expansion of renal tumour-initiating cells. Nat. Commun. 7, 12692 (2016).
Ni, W. et al. Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m(6)A reader YTHDF3. Mol. Cancer 18, 143 (2019).
Li, R. H. et al. A phosphatidic acid-binding lncRNA SNHG9 facilitates LATS1 liquid-liquid phase separation to promote oncogenic YAP signaling. Cell Res. 31, 1088–1105 (2021).
He, X. Y. et al. LncRNA modulates Hippo-YAP signaling to reprogram iron metabolism. Nat. Commun. 14, 2253 (2023).
Meng, F. et al. A potential therapeutic drug for osteoporosis: prospect for osteogenic LncRNAs. Front. Endocrinol.14, 1219433 (2023).
Zhou, Z., Hossain, M. S. & Liu, D. Involvement of the long noncoding RNA H19 in osteogenic differentiation and bone regeneration. Stem Cell Res. Ther. 12, 74 (2021).
Wang, C. G., Hu, Y. H., Su, S. L. & Zhong, D. LncRNA DANCR and miR-320a suppressed osteogenic differentiation in osteoporosis by directly inhibiting the Wnt/beta-catenin signaling pathway. Exp. Mol. Med. 52, 1310–1325 (2020).
Sun, Y. et al. The long noncoding RNA lnc-ob1 facilitates bone formation by upregulating Osterix in osteoblasts. Nat. Metab. 1, 485–496 (2019).
Kumar, L. M EF. Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2, 5–7 (2007).
Kanai, F. et al. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 19, 6778–6791 (2000).
Elbediwy, A. et al. Enigma proteins regulate YAP mechanotransduction. J. Cell Sci. 131, jcs221788 (2018).
Moreno-Vicente, R. et al. Caveolin-1 modulates mechanotransduction responses to substrate stiffness through actin-dependent control of YAP. Cell Rep. 26, 1679–1680 (2019).
Zhang, D. et al. Lepr-expressing PDLSCs contribute to periodontal homeostasis and respond to mechanical force by Piezo1. Adv. Sci.10, e2303291 (2023).
Wang, F., Wang, H., Zhang, H., Sun, B. & Wang, Z. A novel mechanism of MSCs responding to occlusal force for bone homeostasis. J. Dent. Res. 103, 642–651 (2024).
Yang, Y. S. et al. Bone-targeting AAV-mediated silencing of Schnurri-3 prevents bone loss in osteoporosis. Nat. Commun. 10, 2958 (2019).
Yang, Y. S. et al. Suppression of heterotopic ossification in fibrodysplasia ossificans progressiva using AAV gene delivery. Nat. Commun. 13, 6175 (2022).
Li, D. et al. The lncRNA SNHG26 drives the inflammatory-to-proliferative state transition of keratinocyte progenitor cells during wound healing. Nat. Commun. 15, 8637 (2024).
Liu, R. X. et al. Trim21 depletion alleviates bone loss in osteoporosis via activation of YAP1/beta-catenin signaling. Bone Res. 11, 56 (2023).
Park, M. K. et al. NEAT1 is essential for metabolic changes that promote breast cancer growth and metastasis. Cell Metab. 33, 2380–2397 e2389 (2021).
Chujo, T., Yamazaki, T. & Hirose, T. Architectural RNAs (arcRNAs): a class of long noncoding RNAs that function as the scaffold of nuclear bodies. Biochim. Biophys. Acta 1859, 139–146 (2016).
Sanchez de Groot, N. et al. RNA structure drives interaction with proteins. Nat. Commun. 10, 3246 (2019).
Parton, R. G. & del Pozo, M. A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 14, 98–112 (2013).
Lin, N. et al. An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Mol. Cell 53, 1005–1019 (2014).
Huarte, M. et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142, 409–419 (2010).
Jin, F. et al. A functional motif of long noncoding RNA Nron against osteoporosis. Nat. Commun. 12, 3319 (2021).
Franklin, J. M., Wu, Z. M. & Guan, K. L. Insights into recent findings and clinical application of YAP and TAZ in cancer. Nat. Rev. Cancer 23, 512–525 (2023).
Chen, Y. et al. Occlusal force maintains alveolar bone homeostasis via type H angiogenesis. J. Dent. Res. 102, 1356–1365 (2023).
Monfoulet, L., Rabier, B., Chassande, O. & Fricain, J. C. Drilled hole defects in mouse femur as models of intramembranous cortical and cancellous bone regeneration. Calcif. Tissue Int. 86, 72–81 (2010).
Behr, B., Panetta, N. J., Longaker, M. T. & Quarto, N. Different endogenous threshold levels of Fibroblast Growth Factor-ligands determine the healing potential of frontal and parietal bones. Bone 47, 281–294 (2010).
Swift, J. M., Nilsson, M. I., Hogan, H. A., Sumner, L. R. & Bloomfield, S. A. Simulated resistance training during hindlimb unloading abolishes disuse bone loss and maintains muscle strength. J. Bone Min. Res. 25, 564–574 (2010).
Zong, C., Zhao, L., Huang, C., Chen, Y. & Tian, L. Isolation and culture of bone marrow mesenchymal stem cells from the human mandible. J. Vis. Exp. 182, e63811 (2022).
Huang, S. et al. An improved protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. J. Orthop. Transl. 3, 26–33 (2015).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Futschik, M. E. & Carlisle, B. Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol. 3, 965–988 (2005).
Lin, W. et al. Mapping the immune microenvironment for mandibular alveolar bone homeostasis at single-cell resolution. Bone Res. 9, 17 (2021).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Mo, C. et al. Single-cell transcriptomics of LepR-positive skeletal cells reveals heterogeneous stress-dependent stem and progenitor pools. EMBO J. 41, e108415 (2022).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
This study was sponsored by the Original Exploration Program of the National Natural Science Foundation of China (82350001 to Z.W.), the Young Elite Scientist Sponsorship Program by CAST (2023QNRC001 to Y.W.), the Young Scientist Fund of the National Natural Science Foundation of China (82201033 to Y.W.), and the Chenguang Program of Shanghai Education Development Foundation and Shanghai Municipal Education Commission (23CGA23 to Y.W.). Figures 1j and 7h were created with BioRender (https://app.biorender.com/illustrations/687b2f125f8d171ace55d11a and https://app.biorender.com/illustrations/687b40d1ce799aa4cb71d55a).
Author information
Authors and Affiliations
Contributions
Z.W. conceived the project, designed the study, supervised the research, and wrote the manuscript. J.H., Y.W., and Y.F. performed the majority of the experiments, advised on experimental design, edited the manuscript, and created the schematic diagrams. D.W. and Y.C. performed experiments and analyzed data. Z.L. analyzed scRNA-seq and bulk RNA-seq data. X.J. and H.W. assisted in generating the mouse models. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Noriaki Ono and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, J., Weng, Y., Feng, Y. et al. Snhg18 regulates Yap subcellular localization to maintain bone homeostasis. Nat Commun 16, 7543 (2025). https://doi.org/10.1038/s41467-025-62838-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62838-z
