
Abstract
Tandem catalysis is an effective approach to achieve highly selective and high-rate multi-electron/proton transfer reactions, such as nitrate electroreduction, which are important for various physicochemical and biological processes. However, present tandem catalysts suffer from uncontrollable interface, limited crystal phase, and complex synthesis protocols. Here, we report facile seed-mediated synthesis of unconventional phase 4H/fcc Au−Cu heterostructures with a unique beaded-bracelet nanostructure (BBN). Importantly, the exposed Au/Cu interface density can be continuously tuned by modulating discrete Cu domain density on Au nanowires. As a proof-of-concept application, 4H/fcc Au−Cu BBN demonstrates high catalytic performance in nitrate electroreduction to ammonia, with a yield rate and partial current density of 116.2 mg h-1 cm-2 and 1652.0 mA cm-2, respectively. In-situ and theoretical investigations suggest that the unconventional 4H phase and tandem catalysis between Au and Cu domains account for the superior electrocatalytic performance. Besides, this method can be extended to synthesize other unconventional phase heteronanostructures.
Similar content being viewed by others


Introduction
With increasing availability of low-cost renewable electricity, electrocatalytic multi-electron/proton transfer reactions are making great strides towards net-zero sustainable development with closed nitrogen and carbon cycles1,2,3,4,5,6,7,8,9. For instances, electrocatalytic reduction of nitrate (NO3−) to ammonia (NH3), involving the transfer of 8 e− and 9 protons, is a carbon-free approach for NO3− removal in wastewater and simultaneous NH3 production10,11,12,13,14,15,16,17; electrocatalytic carbon dioxide reduction, involving the transfer of 2 to 18 e− along with protons, can convert over-accumulated carbon dioxide to value-added chemicals/fuels18,19,20,21,22,23,24. However, multi-electron/proton transfer processes typically involve multi-steps and multi-pathways, which are difficult to control selectively and efficiently by catalysts of a single-active component25,26,27. For example, although copper (Cu) catalysts are a promising type of single-active component catalysts in nitrate reduction reaction (NO3RR)28,29,30,31,32, they are still constrained in some key steps of NO3RR, i.e., insufficient active hydrogen (*H) supply at a relatively low reducing potential and weak NO3− adsorption/activation under a relatively high reducing potential33,34,35,36. To overcome such limitations, tandem catalysts stand out as an ideal solution37,38,39,40,41,42. A tandem catalyst integrates two or more active components of complementary activities in one structure43,44,45,46, to promote different steps/pathways sequentially at each of their optimal efficiency and thus cooperatively achieve the ultimate catalytic performance as a whole.
Although great progress has been made, currently developed metal tandem catalysts still encounter three critical issues. Firstly, the interface density between different components is relatively low, which limits intermediate transfer between different active components and inhibits the overall catalytic performance45,46,47. For example, in the reports of silver (Ag)–Cu37,48, palladium (Pd)–Cu46, and gold (Au)–Cu Janus nanocrystals48,49, each Ag/Pd/Au domain only pairs with one Cu domain with one interface, leading to inefficient transfer of most intermediates, especially those generated far from the interface. Secondly, the crystal phase of existing metal tandem catalysts is almost limited to their thermodynamically stable phase, which is usually not very active. Unconventional phase tandem catalysts should be promising, because unconventional phase metals/alloys have been observed to demonstrate much better catalytic performance in different types of reactions than the common counterparts50,51,52,53,54,55,56. Thirdly, current synthesis routes of such delicate metal tandem catalysts are very complex, time-consuming, and involve multiple processes and facilities45,46,48,57, which are undesirable for practical applications. Therefore, it is of great significance to develop a versatile synthesis approach to generate unconventional phase tandem catalysts with a high density of exposed interfaces for their complementary activities.
In this work, we report a facile seed-mediated method of synthesizing unconventional phase 4H/fcc Au–Cu beaded-bracelet nanostructures (BBNs), where Cu grows as “beads” on a 4H/fcc Au nanowire (NW) “chain.” Significantly, the Cu domain density on 4H/fcc Au NWs can be continuously tuned, resulting in a wide range of the exposed Au/Cu interface densities. As a proof-of-concept application, the as-synthesized 4H/fcc Au–Cu BBNs with the highest exposed Au/Cu interface density, achieved in this study, exhibit excellent catalytic performance in NO3RR, achieving a high Faradaic efficiency (FE) of 98.9% and an optimum yield rate of 116.2 mg h−1 cm−2 with a partial current density of 1652.0 mA cm−2 towards NH3. In situ attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy and in situ differential electrochemical mass spectrometry (DEMS) unveil that Cu and Au domains play complementary roles in nitrate electroreduction to NH3, respectively, and the shortest NO3RR pathway dominates over the 4H/fcc Au–Cu BBNs. Density functional theory (DFT) calculations suggest that the tandem catalysis between Au and Cu domains and the unconventional 4H phase simultaneously contribute to the superior NO3RR performance. This method can be well extended to the synthesis of other catalytic heteronanostructures, such as 4H/fcc Au@Ag–Cu with exposed Ag–Cu interfaces.
Results
Synthesis and structural characterization
The heterophase 4H/fcc Au–Cu BBNs were synthesized using a facile seed-mediated method, by growing Cu on 4H/fcc Au NWs (Supplementary Figs. 1–3 and Supplementary Note 1), as illustrated in Fig. 1a. Briefly, after the as-prepared high-purity 4H/fcc Au NWs were well dispersed in the mixture of oleylamine (OAM) and dodecylamine (DDA), copper acetylacetonate (Cu(acac)2), 1,2-butanediol (BDO) and hexadecyltrimethylammonium chloride (CTAC) were added and then heated at 140 °C, in which BDO was the reductant and CTAC was used to control reduction rate (see “Methods” for more details). Transmission electron microscopy (TEM) and high-angle annular dark-field scanning TEM (HAADF-STEM) images with different magnifications show that the obtained 4H/fcc Au–Cu BBNs have high purity and uniform size distribution (Fig. 1b–d). The diameter of Au NWs is 12.3 ± 3.1 nm (Supplementary Fig. 4a, b), the same as the bare Au NWs before growing Cu (Supplementary Fig. 1c). The Cu domains have a diameter of 33.7 ± 6.3 nm (Supplementary Fig. 4a, c). Figure 1d shows that the Cu beads are evenly distributed on the Au chains. The Au/Cu atomic ratio is around 65.5/34.5 in 4H/fcc Au–Cu BBNs (Supplementary Fig. 5).

a Schematic illustration of the synthesis of 4H/fcc Au–Cu BBNs. HAADF-STEM (b, d) and TEM (c) images of 4H/fcc Au–Cu BBNs. e Spherical aberration-corrected atomic-resolution HAADF-STEM image of the representative Au/Cu interface in 4H/fcc Au–Cu BBNs. The white dashed lines are used to distinguish different crystal phases. f Schematic illustration of the unit cell and atomic model of 4H phase viewed from [110] direction. g HAADF-STEM image and the corresponding EDS elemental mappings of a typical 4H/fcc Au–Cu BBN.
The atomic structure of 4H/fcc Au–Cu BBNs was unveiled by the aberration-corrected HAADF-STEM (Fig. 1e, Supplementary Fig. 6 and Supplementary Fig. 7). It shows that Cu follows the epitaxial growth Au seeds. Significantly, the “ABCB” characteristic stacking sequence is identified in the Cu domain, indicating the formation of unconventional 4H Cu. To better elucidate this point, the unit cell and atomic arrangement viewed along the [110] direction of 4H phase are schematically illustrated in Fig. 1f. The fcc phase coexists with 4H one in the Cu domain (Fig. 1e and Supplementary Fig. 8). The measured close-packed interplanar spacing of 4H Cu is 2.03 Å, similar to that of fcc Cu (2.02 Å). The HAADF-STEM image and corresponding energy-dispersive X-ray spectroscopy (EDS) elemental mappings reveal that discrete Cu domains are formed and evenly distributed on 4H/fcc Au NWs (Fig. 1g). In general, it forms a “beaded bracelet”-like nanostructure, in which Au NWs act as the chain and Cu domains serve as the bead (right panel of Fig. 1a).
Importantly, the Cu coverage on Au NWs can be continuously tuned by simply adjusting CTAC amount, obtaining a series of 4H/fcc Au–Cu heteronanostructures, as schematically illustrated in Fig. 2a. In this process, the Cl− in CTAC is believed to play a vital role by coordinating with Cu precursors and thus affecting their reduction kinetics. When a large amount of CTAC (0.73 mg) was used, the reduction speed of Cu precursors was very low, leading to the formation of 4H/fcc Au–Cu BBNs with low density of Cu beads (4H/fcc Au–Cu BBNs-l; Fig. 2b–d). When the amount of CTAC was decreased to 0.48 mg and 0.21 mg, the reduction speed of Cu precursors was improved accordingly, thereby obtaining 4H/fcc Au–Cu BBNs with moderate and high densities of Cu beads, respectively (4H/fcc Au–Cu BBNs-m, Fig. 2e–g; 4H/fcc Au–Cu BBNs-h, Fig. 2h–j). When CTAC was not added, the extremely high reduction rate of Cu resulted in the generation of 4H/fcc Au–Cu core-shell nanostructures (4H/fcc Au–Cu CSNs, Fig. 2k–m). It is worth noting that the reduction kinetics of metal precursors affects the nucleation and growth modes of Cu, and balances the Cu growth and the Au aggregation47,58,59. TEM images show the good purity of as-prepared four kinds of heteronanostructures (Fig. 2b, c, e, f, h, i, k, l). HAADF-STEM images and corresponding EDS mappings indicate the BBN or CSN of as-synthesized 4H/fcc Au–Cu heteronanostructures (Fig. 2d, g, j, m). The detailed change of Cu coverages and the distances between Cu domains are presented in Supplementary Fig. 9. It is worth noting that with the continuous modulation of Cu coverage on 4H/fcc Au NWs, the density of exposed Au/Cu interface (pointed out by the black arrow in Fig. 2a and further defined in Supplementary Fig. 10a, b), which is important for catalytic applications45,60,61,62, also has a noticeable change. In this case, the as-prepared Au–Cu heteronanostructures can be used to study the impact of Cu coverage and corresponding exposed Au/Cu interface densities on their catalytic performance, which will be discussed later. Detailed control experiments suggest that the good dispersion of 4H/fcc Au NWs and the use of Cl− is critically important for the successful synthesis of 4H/fcc Au–Cu BBNs (Supplementary Figs. 11–13 and Supplementary Notes 2 and 3).

a Schematic illustration for increasing the Cu coverage in 4H/fcc Au–Cu heteronanostructures by decreasing CTAC amount. TEM images (b, c) and HAADF-STEM image and the corresponding EDS elemental mapping (d) of 4H/fcc Au–Cu BBNs with low Cu domain density. TEM images (e, f) and HAADF-STEM image and the corresponding EDS elemental mapping (g) of 4H/fcc Au–Cu BBNs with moderate Cu domain density. TEM images (h, i) and HAADF-STEM image and the corresponding EDS elemental mapping (j) of 4H/fcc Au–Cu BBNs with high Cu domain density. TEM images (k, l) and HAADF-STEM image and the corresponding EDS elemental mapping (m) of 4H/fcc Au–Cu CSNs. The EDS elemental mappings in (d–m) share the same scale bars with their corresponding HAADF-STEM images in (d–m).
X-ray spectral analysis
The crystal structures of 4H/fcc Au–Cu heteronanostructures were investigated by X-ray diffraction (XRD). The fcc Cu nanoparticles (NPs) were synthesized as a control sample to better understand the properties of 4H/fcc Au–Cu BBNs (Supplementary Fig. 14). As shown in Supplementary Fig. 15, the characteristic peaks of (100)4H, (101)4H, and (102)4H and (200)f are clearly observed in the XRD patterns of 4H/fcc Au–Cu BBNs-h and CSNs, suggesting that Au NWs still maintain their 4H/fcc heterophase after Cu overgrowth. Besides, the peak positions of (100)4H, (101)4H, (102)4H, and (200)f do not show observable shifts compared to the pristine 4H/fcc Au NWs, suggesting that Cu is grown on the surface of Au NWs instead of forming alloy with Au. The chemical state of 4H/fcc Au–Cu heteronanostructures was examined by X-ray photoelectron spectroscopy (XPS; Supplementary Fig. 16). The XPS peak positions of Au 4f indicate the metallic state of Au in 4H/fcc Au–Cu heteronanostrucrures. Similarly, Cu also dominantly stays in the metallic state in 4H/fcc Au–Cu heteronanostructures, and the observed tiny satellite peaks of copper oxide could originate from the inevitable oxidation of Cu in the air.
X-ray absorption spectroscopy was further used to study the electronic structure and coordination environment of 4H/fcc Au–Cu heteronanostructures. The Cu K-edge X-ray absorption near-edge structure (XANES) spectra of 4H/fcc Au–Cu BBNs-h, 4H/fcc Au–Cu CSNs, and fcc Cu NPs are similar (Fig. 3a). The white line positions of them are near and located between Cu foil and CuO. In the extended X-ray absorption fine structure (EXAFS) spectra of Cu K-edge, the peaks at ~1.5 and ~2.1 Å are ascribed to Cu–O and Cu–Cu paths, respectively (Fig. 3b). According to the fitted results (Supplementary Fig. 17 and Supplementary Table 1), the coordination number (CN) of Cu–O path is obviously lower than that of Cu–Cu path in 4H/fcc Au–Cu BBNs-h, 4H/fcc Au–Cu CSNs and fcc Cu NPs, implying the dominant chemical state of Cu is metallic. It is in good agreement with the XPS results (Supplementary Fig. 16). Moreover, the Cu–Cu bonds in 4H/fcc Au–Cu BBNs-h, 4H/fcc Au–Cu CSNs, and fcc Cu NPs have a similar bond length of ca. 2.55 Å to that of Cu foil. The Cu–Cu coordination number of 4H/fcc Au–Cu BBNs-h (4.3) is slightly higher than that of 4H/fcc Au–Cu CSNs (3.0), which could arise from their different Cu distribution patterns on 4H/fcc Au NWs. In the wavelet transform (WTs) of Cu K-edge EXAFS spectra (Fig. 3c), the intensity maxima of Cu–Cu of 4H/fcc Au–Cu BBNs-h, 4H/fcc Au–Cu CSNs, and fcc Cu NPs are very similar to that of Cu foil, further highlighting that metallic Cu is dominant in the as-prepared heteronanostructures.

a Normalized Cu K-edge XANES spectrum of the as-prepared 4H/fcc Au–Cu heteronanostructures, fcc Cu NPs, CuO, and Cu foil. b Fourier transform of Cu K-edge EXAFS spectra of 4H/fcc Au–Cu heteronanostructures, fcc Cu NPs, CuO, and Cu foil. c Wavelet transform of Cu K-edge EXAFS spectra for 4H/fcc Au–Cu heteronanostructures, fcc Cu NPs, and Cu foil. d Normalized Au L3-edge XANES spectra of 4H/fcc Au–Cu heteronanostructures, 4H/fcc Au NWs and Au foil. e Fourier transform of Au L3-edge EXAFS spectra of 4H/fcc Au–Cu heteronanostructures, 4H/fcc Au NWs, and Au foil. f Wavelet transform of Au L3-edge EXAFS spectra for 4H/fcc Au–Cu heteronanostructures, 4H/fcc Au NWs, and Au foil. Source data are provided as a Source data file.
In terms of Au L3-edge, the white line positions in XANES spectra are almost identical for 4H/fcc Au–Cu BBNs-h, 4H/fcc Au–Cu CSNs, 4H/fcc Au NWs, and Au foil (Fig. 3d), implying further the metallic state of Au in these 4H/fcc Au–Cu heteronanostructures. According to the fitting results of Au L3-edge EXAFS spectra, the Au–Cu path exists in 4H/fcc Au–Cu BBNs-h and 4H/fcc Au–Cu CSNs with the coordination number of 1.9 and 1.8, respectively, which should be derived from the Au/Cu interface (Supplementary Fig. 18, Supplementary Table 2 and Fig. 1e). In addition, the Au–Au bond lengths of 4H/fcc Au–Cu BBNs-h and 4H/fcc Au–Cu CSNs are close to each other but smaller than that of bare Au NWs, indicating the similar Cu-induced strain effect on the Au lattice in these two samples. In the WTs of Au L3-edge EXAFS spectra, the observed similar intensity maximums of 4H/fcc Au–Cu BBNs-H, 4H/fcc Au–Cu CSNs, 4H/fcc Au NWs, and Au foil suggest that growing Cu on Au NWs does not generate any discernible impact on Au chemical state (Fig. 3f).
Electrochemical NO3RR measurement
To verify whether the materials design is effective in electrocatalytic multi-electron/proton transfer reactions, the performance of the as-prepared 4H/fcc Au–Cu BBNs was evaluated for NO3RR as a proof-of-concept. The measurement was first performed in a typical three-electrode H-type cell using alkaline electrolyte containing 1 M KOH and 0.1 M KNO3, in which the catalyst-modified glass carbon electrode (GCE) was used as the working electrode. It should be noted that the potentials reported here are not iR corrected in order to reflect the actual energy consumption and avoid unnecessary errors introduced by iR correction.
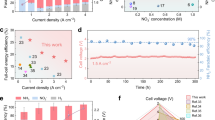
The linear sweep voltammetry (LSV) curves, acquired with a rotating disk electrode (RDE) at 1600 rpm to alleviate the slow diffusion issue of NO3− in the electrolyte and access most closely the intrinsic activity of the catalyst, suggest that the 4H/fcc Au–Cu BBNs-h show a more positive onset potential than the control samples of 4H/fcc Au NWs and fcc Cu NPs (Fig. 4a). In addition, the 4H/fcc Au–Cu BBNs-h demonstrate a much higher current density than both 4H/fcc Au NWs and fcc Cu NPs in the whole potential range, with the highest current density of 642.9 mA cm−2. To investigate the ability of converting NO3− to NH3, the NH3 FE and yield rate were also measured for 4H/fcc Au–Cu BBNs-h (Fig. 4b, c and Supplementary Figs. 19 and 20). The analysis of by-products in NO3RR and the cross-validation of the quantification methods of NH3 were also conducted to ensure the accuracy of the electrochemical measurements (Supplementary Figs. 21–25). The 4H/fcc Au–Cu BBNs-h exhibit the highest FENH3 of 98.6% at −0.6 V (vs. RHE), and show FENH3 of above 85.0% in a wide potential window from −0.4 to −1.0 V (vs. RHE). In contrast, the highest FENH3 of the fcc Cu NPs and the 4H/fcc Au NWs is only 89.4% at −0.4 V (vs. RHE) and 70.8% at −0.6 V (vs. RHE), respectively. Significantly, the highest NH3 yield rate of 4H/fcc Au–Cu BBNs-h is 58.4 mg h−1 mg−1catalyst (23.4 mg h−1 cm−2), which is 1.6 times that of fcc Cu NPs (37.6 mg h−1 mg−1catalyst) and 1.8 times that of 4H/fcc Au NWs (32.5 mg h−1 mg−1catalyst). The 15N-labeling experimental results confirm that the generated NH3 is exclusively derived from the nitrate source (i.e., KNO3) rather than the catalysts or environmental contaminants (Supplementary Fig. 26). The as-prepared 4H/fcc Au–Cu BBNs-h also presents better NO3RR performance than the physical mixture of 4H/fcc Au NWs and fcc Cu (denoted as 4H/fcc Au/Cu catalysts, Supplementary Fig. 27) and the physical mixture of fcc Au NPs and fcc Cu NPs (denoted as fcc Au/Cu catalysts, Supplementary Fig. 28). Moreover, the as-prepared 4H/fcc Au–Cu BBNs-h also exhibit good NO3RR performance at a relatively low nitrate concentration (Supplementary Figs. 29 and 30). Such performance values clearly indicate that the 4H/fcc Au–Cu BBNs-h are among the best NO3RR catalysts to date (Supplementary Table 3).

a LSV curves of 4H/fcc Au–Cu BBNs-h, 4H/fcc Au NWs, and fcc Cu NPs with RDE at 1600 rpm. Faradaic efficiency (b) and yield rate (c) of NH3 obtained on 4H/fcc Au–Cu BBNs-h, 4H/fcc Au NWs, and fcc Cu NPs. d ECSA comparison between 4H/fcc Au–Cu BBNs-h, 4H/fcc Au NWs, and fcc Cu NPs. e Comparison of NO2− Faradaic efficiency between 4H/fcc Au NWs and fcc Cu NPs. f The effect of Cu domain density in 4H/fcc Au–Cu heteronanostructures on NO3RR performance. g Schematic illustration of the NO3RR measurements of 4H/fcc Au–Cu BBNs in flow cell, where the fast electrolyte flowing ensures the abundant NO3− supply and helps to refresh the electrode surface. h, Flow cell performance of 4H/fcc Au–Cu BBNs-h: the partial current and Faradaic efficiency of NH3 obtained in the electrolyte containing 1 M KOH and 1 M KNO3 under different potentials. Where error bars are shown, values are means, and error bars indicate s.d. (n = 3 replicates). Source data are provided as a Source data file.
To understand the reasons why 4H/fcc Au–Cu BBNs-h exhibit higher activity, NH3 FE, and yield rate than the single-component nanostructures, i.e., fcc Cu NPs and 4H/fcc Au NWs, several in-depth analyses were carried out. First, the electrochemically active surface area (ECSA) was determined. The results show that 4H/fcc Au–Cu BBNs-h possess an electrochemical double-layer capacitance (Cdl) of 3.4 mF cm−2, which is 1.3 and 8.5 times that of 4H/fcc Au NWs (2.6 mF cm−2) and fcc Cu NPs (0.4 mF cm−2), respectively (Fig. 4d and Supplementary Fig. 31). This result suggests that more active sites are generated when combining Au and Cu together. Subsequently, the huge difference between Au and Cu in producing NO2−, a very critical intermediate for NH3 generation, was noticed. It was identified that the 4H/fcc Au NWs can efficiently promote the conversion of NO3− to NO2− with an FE several times higher than the fcc Cu NPs, especially at a more negative potential (Fig. 4e and Supplementary Fig. 32). Therefore, it is believed that the produced NO2− on the Au domain could spill over to the Cu domain across the Au/Cu interface and thus enhance NH3 generation on the Cu domain26,36,63. The fcc Cu NPs were further observed to be able to convert NO2− to NH3 with a higher FE compared to the 4H/fcc Au NWs (Supplementary Fig. 33). These observations can account for the superior NO3RR performance of 4H/fcc Au–Cu BBNs to that of the single-component fcc Cu NPs at the corresponding potentials. Furthermore, the impact of Au/Cu interface on NO3RR performance was systematically studied using as-prepared 4H/fcc Au–Cu heteronanostructures with different Au/Cu interface densities. Both NH3 FE and yield rate show “volcano-shaped” curves with respect to the Cu coverage on the 4H/fcc Au NWs, and the optimal NO3RR performance appears at the highest Au/Cu interface density (Fig. 4f and Supplementary Figs. 34–36).
The catalytic stability of the 4H/fcc Au–Cu BBNs was evaluated by consecutive-cycle electrolysis. After electrolysis in the electrolyte containing 1 M KOH and 0.1 M KNO3 for 16 cycles, 4H/fcc Au–Cu BBNs-h had a steady FE of above 95% towards NH3, and the NH3 yield rate showed no obvious decline (Supplementary Fig. 37). TEM images and composition analysis reveal that the 4H/fcc Au–Cu BBNs-h still maintain their heteronanostructure and show neglectable composition change after the durability test (Supplementary Fig. 38a–c). The XPS, XRD, and HAADF-STEM characterizations confirm that the surface states and crystal phase can be well maintained (Supplementary Fig. 38d–f and Supplementary Fig. 39). These results indicate the high stability of 4H/fcc Au–Cu BBNs in electrochemical NO3RR.
To scale up and overcome the limited mass transfer of NO3− in an H-type cell, a flow cell with a fast flow rate of electrolyte was further constructed to evaluate the NO3RR performance of 4H/fcc Au–Cu BBNs (Fig. 4g). When using an aqueous solution of 1 M KOH and 0.1 M KNO3 as the electrolyte, the optimal potential of 4H/fcc Au–Cu BBNs-h is positively shifted 400 mV compared with that in H-type cells, with a NH3 FE of 98.8% and yield rate of 21.22 mg h−1 cm−2 at −0.2 V (vs. RHE) (Supplementary Fig. 40). The partial current density towards NH3 is improved from 42.4 to 267.2 mA cm−2 at −0.2 V (vs RHE). 4H/fcc Au–Cu BBNs-h were also evaluated in the nuclear wastewater-grade electrolyte containing 1 M NO3−. As shown in Fig. 4h, in the aqueous solution of 1 M KOH and 1 M KNO3, an ampere-level partial current (above 1 A) of NH3 was obtained when the potential was more negative than −0.4 V (the electrode area is 1 cm), and the highest partial current reaches up to 1.652 A cm−2 at −0.8 V (vs. RHE). The 4H/fcc Au–Cu BBNs-h exhibit the highest FENH3 of 98.9% at −0.6 V (vs. RHE) and the highest NH3 yield rate of 116.2 mg h−1 cm−2 at −0.8 V (vs. RHE, Supplementary Fig. 41). These results reveal the great potential of 4H/fcc Au–Cu BBNs for NO3RR towards practical scenarios.
In situ mechanism investigation
To probe in detail the reaction mechanism and the roles of Au and Cu in the 4H/fcc Au–Cu BBNs during NO3RR, in situ ATR-FTIR spectroscopy was used to monitor the adsorbed intermediates on the surface of as-prepared catalysts in electrocatalytic nitrate reduction. As shown in Fig. 5a–c, the peak intensity of *NH2 intermediate (3212 and 3133 cm−1)12,64 on the fcc Cu NPs is much stronger than that (3245 and 3180 cm−1) on the 4H/fcc Au NWs, indicating that Cu is more active in the hydrogenation of N-species than Au. Furthermore, the intermediates of *NO (1545 cm−1) and *NO2 (1271 cm−1)12,65 in the ATR-FTIR spectra of the 4H/fcc Au NWs are observed, especially when the potential is more negative than −0.3 and −0.7 V (vs. RHE), respectively. On the contrary, there hardly exist any intermediates of *NO and *NO2 in the ATR-FTIR spectra of the fcc Cu NPs. This phenomenon suggests that Au is capable of converting NO3− to *NO and *NO2 intermediates while Cu promotes the hydrogenation of *NO and *NO2 towards NH3, thereby implying a tandem catalytic process between Au and Cu in the 4H/fcc Au–Cu BBNs-h. This is highly consistent with the electrochemical measurement results (Fig. 4b, c, e).

In situ ATR-FTIR spectra of NO3RR on 4H/fcc Au–Cu BBNs-h (a), fcc Cu NPs (b), and 4H/fcc Au NWs (c) at different potentials. d In situ DEMS patterns of NO3RR on 4H/fcc Au–Cu BBNs-h. e Peak intensity comparison between the intermediates of *N, *NHOH, and *NOH. f Analysis of NO3RR pathways on 4H/fcc Au–Cu BBNs-h. Source data are provided as a Source data file.
In situ DEMS was applied to determine the NO3RR pathway on 4H/fcc Au–Cu BBNs-h (Fig. 5d–f and Supplementary Fig. 42). Note that there are four common pathways for NO3− reduction to NH3, and the key differences among those are whether the intermediates of *NOH, *NHOH, or *NH2OH are generated or not34. As shown in Fig. 5d, nearly all the NO3RR intermediates are detected, suggesting that there is more than one specific type of NO3RR pathway on 4H/fcc Au–Cu BBNs-h. Interestingly, when further comparing the peak intensities of *N, *NH2OH and *NOH intermediates in Fig. 5e, it is noted that the *N peak has an extremely higher intensity than those of *NH2OH and *NOH peaks, indicating that the shortest reaction pathway, i.e., *NO3 → *NO2 → *NO → *N → *NH → *NH2 → *NH3 (Pathway 1), is dominant over the 4H/fcc Au–Cu BBNs-h (Fig. 5f).
Theoretical calculations
To further reveal the NO3RR mechanism on the 4H/fcc Au–Cu BBNs, DFT calculations were employed to investigate the influences of asymmetric Au–Cu configurations and the unconventional 4H phase from the aspects of electronic structures and binding energies. Details of the calculations are provided in “Methods,” Supplementary Information and Supplementary Data 1. Bader charge transfer (Q) between adsorbates and catalysts (4H/fcc Au and 4H/fcc Cu domains) is compared in Fig. 6a, where a similar trend of charge transfers is observed for 4H/fcc Au and 4H/fcc Cu domains. Specifically, the charge transfer first experiences a decrease from *NO3 to the next two intermediates (*NO2 and *NO), then a sharp increase from *NO to *N, and finally a gradual reduction until the final product, NH3. Importantly, 4H/fcc Au domains exhibit much weaker electronic interactions than 4H/fcc Cu domains throughout the whole process. To further identify this trend, the charge density interaction between the key intermediate (*NO2) and the catalysts (4H/fcc Au and 4H/fcc Cu) was also investigated (Fig. 6b), where charge density increases in the yellow zone and decreases in the blue zone after adsorption. Although the 4H/fcc Au and 4H/fcc Cu domains demonstrate similar electron interaction tendencies with *NO2, the 4H/fcc Cu domains show greater scope of participation in charge transfer than the 4H/fcc Au domains, implying that 4H/fcc Cu domains have a more intense charge exchange with the reaction intermediates. These results imply that the 4H/fcc Cu domains are more active than 4H/fcc Au domains in NO3RR. Besides the electronic structures of the metal sites, the density of states (DOSs) of key adsorbates for the formation of NH3 were also studied (Fig. 6c). For the 4H/fcc Cu surface, there is a good linear correlation from *NO3 to *N and the following hydrogenation to *NH3, demonstrating the smooth conversion process of intermediates, which is much weakened on the 4H/fcc Au surface, especially from *NH2 to *NH3. The above results together indicate that the 4H/fcc Cu domains have a better capability for hydrogenation of N-species in NO3RR, agreeing with the results of in situ ATR-FTIR and in situ DEMS studies.

a Bader charge transfer between the adsorbates and catalysts (4H/fcc Au and 4H/fcc Cu). b The charge density between the intermediates and the catalysts. The isosurface level is set as 0.0015 e/Bohr3. c DOSs of key adsorbates for the formation of NH3 on 4H/fcc Au and 4H/fcc Cu. d The reaction trends of converting NO3− to NO2− and NO2− to NH3 on the 4H/fcc Au and 4H/fcc Cu. e The comparison of *NO2 adsorption energy on Au domain, Au/Cu interface, and Cu domain. Source data are provided as a Source data file.
To further understand the role of Au and Cu domains during NO3RR in detail, the overall reaction energetics for NH3 generation are compared for the 4H/fcc Au and the 4H/fcc Cu domains (Fig. 6d and supplementary Figs. 43 and 44). It is noted that for generating the key intermediate, i.e., from NO3− to NO2−, the 4H/fcc Au domains exhibit stronger conversion trend (−1.96 eV) than the 4H/fcc Cu domains (−1.46 eV), following by a much-alleviated energy barrier of 1.22 eV to release NO2−, while the 4H/fcc Cu domains show a relatively large energy barrier of 1.90 eV. In contrast, for converting NO2− to NH3, the 4H/fcc Cu domains exhibit an energetically favorable trend than the 4H/fcc Au domains. These results indicate that a tandem catalytic mechanism exists in the 4H/fcc Au–Cu BBNs, where the 4H/fcc Au domains favor the reduction of NO3− to NO2− and the 4H/fcc Cu domains facilitate NO2− conversion to NH3.
As the *NO2/NO2− transfer is the key step for the tandem catalysis mechanism of NO3RR, the adsorption of *NO2 on different components of the 4H/fcc Au–Cu BBNs was further analyzed (Supplementary Fig. 45). As shown in Fig. 6e, the Cu domains show the tightest adsorption towards *NO2 compared to the Au domains and the Au/Cu interfaces, no matter in the 4H Au–Cu or the fcc Au–Cu, suggesting a strong likelihood of *NO2 transfer from the Au to the Cu domains. Moreover, the 4H Au–Cu shows a linear correlation with a higher slope than the fcc Au–Cu towards *NO2 adsorption from the Au to the Cu domain, which supports a fast conversion process with small energy barriers during NO3RR. The above results further reveal the existence of a tandem catalysis mechanism on the 4H/fcc Au–Cu BBNs towards NO3RR.
The influences of 4H and fcc phases on NO3RR were also investigated in detail. By means of the electron localization function (ELF), it is identified that the 4H phase exhibits more non-localized electrons (green zone) than the fcc one, indicating a better capability of electron transfer over the 4H phase (Supplementary Fig. 46). For the overall energy trend in Supplementary Fig. 47 and Supplementary Fig. 48, from *NO3 to *NH, the 4H phase shows a more apparent downhill trend than the fcc phase. The 4H phase also shows a much larger energy gap than the fcc phase in the last step from *NH2 to *NH3, making the 4H phase more energetically favorable in the whole NO3RR process.
Extended synthesis
The 4H/fcc Au@Ag−Cu BBNs were also synthesized via a slight modification of the developed method, as schematically illustrated in Fig. 7a. A thin layer of Ag was first deposited on 4H/fcc Au NWs to synthesize 4H/fcc Au@Ag core-shell nanostructure, which is confirmed by TEM, HAADF-STEM, and EDS elemental mappings (Fig. 7b, c). Then, using the as-prepared 4H/fcc Au@Ag nanostructure as seeds, Au@Ag−Cu BBNs were further synthesized. TEM image suggests the high purity and good uniformity of the obtained 4H/fcc Au@Ag–Cu BBNs (Fig. 7d). HAADF-STEM image and the corresponding EDS elemental mappings of a representative 4H/fcc Au@Ag–Cu BBN show that after the growth of Cu, Au and Ag are still uniformly distributed and maintain their core (Au)-shell (Cu) structure (Fig. 7e). In contrast, Cu element is segregated as Cu beads, suggesting the successful formation of Au@Ag–Cu BBNs.

a Schematic illustration of the synthesis of 4H/fcc Au@Ag−Cu BBNs. TEM image (b), HAADF-STEM image, and the corresponding EDS elemental mappings (c) of 4H/fcc Au@Ag CSNs. TEM image (d), HAADF-STEM image, and corresponding EDS elemental mappings (e) of 4H/fcc Au@Ag–Cu BBNs. f Spherical aberration-corrected atomic-resolution HAADF-STEM image of a representative Au–Ag–Cu interface marked in (e). g Zoom-in atomic-resolution HAADF-STEM image and the corresponding atomic-resolution EDS elemental mappings of the Au–Ag–Cu interface marked in (f).
The aberration-corrected HAADF-STEM image of the marked area with white dashed rectangle in Fig. 7e shows that at the interface, it forms an Au–Ag–Cu sandwich-layered structure with an epitaxial-growth relationship (Fig. 7f). Significantly, 4H Cu also extensively exists in the Cu domain. The zoom-in HAADF-STEM image and atomic-resolution EDS elemental mappings further confirm the Au–Ag–Cu sandwich-layered structure and the coherent lattice fringes of 4H phase from Au to Ag to Cu (Fig. 7g). These results indicate that we can also change the exposed component from Au–Cu asymmetric nanostructures to Ag–Cu asymmetric nanostructures while maintaining their unconventional phase (Supplementary Fig. 49). It is believed that this method can also be extended to the synthesis of other types of unconventional phase heteronanostructures with tunable exposed interfaces, which hold great potential to engineer high-performance catalysts towards many other multi-electron/proton transfer reactions.
Discussion
In summary, we have successfully synthesized unconventional phase 4H/fcc Au–Cu BBNs by a facile seed-mediated method. Significantly, by simply adjusting the amount of CTAC, the Cu domain density on 4H/fcc Au NWs can be continuously tuned, resulting in the delicate modulation of the exposed Au/Cu interface density. For electrochemical NO3RR, the as-synthesized 4H/fcc Au–Cu BBNs with the highest Au/Cu interface density demonstrated the best catalytic performance, with the optimal NH3 FE and yield rate of 98.9% and 116.2 mg h−1 cm−2 in a flow cell with an aqueous electrolyte of 1 M KOH and 1 M KNO3. In situ ATR-FTIR studies indicated that the Au domains favor the conversion of NO3− to *NO/*NO2 intermediates, while the Cu domains facilitate the hydrogenation of *NO/*NO2 intermediates to NH3. In situ DEMS investigations unveiled that NO3RR on 4H/fcc Au–Cu BBNs mainly proceeds via the shortest reaction pathway, i.e., *NO3 → *NO2 → *NO → *N → *NH → *NH2 → *NH3. DFT calculations suggested the existence of a tandem catalytic mechanism between Au and Cu domains, and the 4H phase also plays a key role in enhancing the NO3RR performance. The developed synthesis approach can be further extended to the synthesis of other unconventional phase tandem catalysts. This work highlights the significance of designing unconventional phase tandem catalysts with unique nanostructures to control challenging multi-electron/proton transfer reactions.
Methods
Materials
Gold (III) chloride hydrate (HAuCl4·xH2O, ~50% Au basis), potassium nitrate (KNO3, ACS reagent, ≥99.0%), potassium nitrite (KNO2, ACS reagent, ≥96.0%), and Nafion (117 solution, 5 wt.%) were purchased from Sigma-Aldrich. Oleylamine (OAM, 80-90%), dodecylamine (DDA, 98%), 1,2-butanediol (BDO, >98% (GC)), 1,2-hexanediol (HDO, 98%), copper acetylacetonate (Cu(acac)2, 97%), hexadecyltrimethylammonium chloride (CTAC, ≥97%), ammonium chloride (NH4Cl, GR, >99.8%), potassium hydroxide (KOH, ACS), maleic acid (AR, ≥ 99% (HPLC)) and cyclohexane (>99%) were purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd. N-ethylcyclohexylamine (>99% (GC)) and the solution of sodium hypochlorite (NaClO, 0.1 M) were purchased from Macklin. Ethanol (ACS Grade, absolute) and n-hexane (ACS Grade, 99%) were purchased from Anaqua Global International Inc. Limited. All the chemical reagents and materials were used as received without any further purification. Deionized (DI) water with a resistance of 18.2 MΩ cm was used in the experiments.
Synthesis of 4H/fcc Au NWs
The 4H/fcc Au NWs were synthesized using our previously reported method with some modifications66. In specific, 15 mg of HAuCl4·xH2O was first swiftly added into 10 mL of the mixture of OAM and n-heptane (v/v = 1/1), followed by adding 200 µL of N-ethylcyclohexylamine into the mixed solution. After quickly capping and sealing the reaction bottle, the obtained solution was slightly sonicated until entirely dissolving HAuCl4·xH2O. Then, the reaction vial was placed into an oil bath and heated at 68 °C for 48 h. After the reaction, the obtained products were washed with the mixture of cyclohexane and ethanol (v/v = 5/1) 5 times by sonication and centrifugation. The obtained 4H/fcc Au NWs were finally redispersed into 5 mL of n-hexane and kept in a refrigerator for further use.
Synthesis of 4H/fcc Au–Cu BBNs
The 4H/fcc Au–Cu BBNs with high Cu domain density were synthesized by a seed-mediated method. Firstly, 0.5 mL of the stoking solution of 4H/fcc Au NWs was centrifugated, and the supernatant was discarded with only the precipitation left in the centrifugation tube. Then, 400 µL of the mixture of OAM and DDA (v/v = 2/3) was added to the precipitation. After sonicating the obtained solution for 1 h, 200 µL of BDO solution containing 0.21 mg of CTAC and 625 µL of Cu(acac)2 solution (1 mg mL−1 in OAM) were added into the above solution. After further sonicating for 1 h, the reaction solution was transferred into a 4 mL glass vial and then placed into an oil bath. After heating at 140 °C for 20 min, the reaction solution was cooled down with an ice bath. Then, the obtained products were washed by cyclohexane three times via sonication and centrifugation. The obtained 4H/fcc Au–Cu BBNs were finally redispersed in 1 mL of n-hexane and stored in a refrigerator for further use after purging N2 for 5 min.
Synthesis of 4H/fcc Au–Cu BBNs with different Cu domain density
The synthesis of 4H/fcc Au–Cu BBNs with moderate Cu domain density and low Cu domain density was similar to that of 4H/fcc Au–Cu BBNs, except that the CTAC amount was changed to 0.48 mg and 0.73 mg, respectively. The obtained 4H/fcc Au–Cu BBNs-m and 4H/fcc Au–Cu BBNs-l were dispersed in 1 mL n-hexane and stored in a refrigerator for further use after purging N2 for 5 min.
Synthesis of 4H/fcc Au–Cu CSNs
The synthesis of 4H/fcc Au–Cu CSNs was similar to that of 4H/fcc Au–Cu BBNs, except that no CTAC was added in the growth solution. The obtained 4H/fcc Au–Cu CSNs were dispersed in 1 mL of n-hexane and stored in a refrigerator for further use after purging N2 for 5 min.
Synthesis of fcc Cu NPs
The fcc Cu NPs were synthesized by using a one-pot wet-chemical method. Specifically, 15.71 mg of Cu(acac)2 were first added into 14.30 mL of the mixture of OAM and DDA (v/v = 2/3). After sonicating for 15 min, 0.72 mL of HDO was added to the above solution. After thoroughly mixing, the reaction vial was placed into an oil bath and heated under 170 °C for 3 h. After the reaction, the products were washed with cyclohexane 3 times by sonication and centrifugation. The fcc Cu NPs were finally redispersed into 3 mL of n-hexane and stored in a refrigerator for further use after purging N2 for 5 min.
Synthesis of 4H/fcc Au/Cu catalyst
The 4H/fcc Au/Cu catalyst was prepared by physically mixing the 4H/fcc Au NWs and the fcc Cu NPs under intensive sonication.
Synthesis of fcc Au/Cu catalyst
The fcc Au/Cu catalyst was prepared by physically mixing the fcc Au NPs and the fcc Cu NPs under intensive sonication.
Characterizations
The TEM images were taken on a FEI Tecnai T12 TEM operated at 120 kV. The high-resolution TEM (HRTEM) images were collected on a JEOL 2100F operated at 200 kV. Aberration-corrected scanning transmission electron microscopy (STEM) measurements were conducted on a cold-field-emission spherical aberration-corrected transmission electron microscope (Thermo Fisher Scientific, Spectra 300) operated at 200 kV. High-angle annular dark field (HAADF)-STEM images were collected with a convergence angle of 25 mrad, a probe current of 25 pA, and a collection semi-angle range between 62 and 200 mrad. Some key parameters of the atomic STEM-EDX mapping: screen current 95 pA, image size 109 × 215, pixel size 21.95 pm, dispersion 5 eV; four symmetric EDX detectors (Super-X G2), frame time 1.28 s, total acquisition time 435 frames, and dwell time 40 µs, Gaussian blur post-filtering applied. The scanning electron microscopy (SEM) images and the related EDS spectra were obtained by a Thermo Scientific Quattro S operated at 20 kV. XRD patterns were collected by SmartLab 9 kW (Rigaku) with a Cu Kα radiation (λ = 1.5406 Å) source. XPS tests were performed in a VG ESCALAB 220i-XL surface analysis system with a base vacuum lower than 10−9 Torr. XAS characterizations were conducted on the beamline 1W1B Beijing Synchrotron Radiation Facility in the transmission mode. Data processing was performed by using the Athena and Artemis software packages67. The loading amount of Cu was confirmed by an inductively coupled plasma optical emission spectrometry (ICP-OES, PerkinElmer, Optima 8000).
Electrochemical NO3RR measurement in H-type cell
Preparation of the working electrode
The as-synthesized heteronanostructures (or catalysts) were first transferred from n-hexane to ethanol. Then 400 µg of catalyst in ethanol (the concentration was confirmed by ICP-OES) were centrifugated, and the obtained precipitate was redispersed into 97 µL of ethanol, followed by the addition of 3 µL of Nafion solution (5 wt.%, Chemours). After sonicating the above solution for 15 min to get a homogeneous catalyst ink, 50 μL of the catalyst ink were dropped onto the glass carbon electrode (GCE, GaossUnion) with a diameter of 8 mm, making the loading amount of 400 µg cm−2. After the electrode was dried, the as-prepared working electrode was kept in a vacuum oven before the electrocatalytic test.
NO3RR test
All the electrochemical measurements were conducted in a three-electrode system by an Ivium-n-Stat electrochemical workstation. The catalysts were tested in a two-chamber H-type cell (C007-10, GaossUnion) separated by an ion exchange membrane (Nafion 212, Chemours). The Ar-saturated solution containing 1 M KOH and 0.1 M KNO3 was used as the electrolyte, which was prepared before the electrochemical experiments by dissolving the salts in deionized water and stored in a sealed container. Typically, both anode and cathode chambers were filled with 15 mL of electrolyte. The catalyst-modified GCE, Hg/HgO electrode (GaossUnion), and Pt plate (GaossUnion) were applied as the working, reference, and counter electrodes, respectively. The reference electrode is regularly calibrated every month. The applied potentials were converted to the RHE scale based on the equation of E (vs RHE) = E (vs Hg/HgO) + 0.197 + 0.0591 × pH. All the applied potentials in this work were recorded without iR correction. The Faradaic efficiency was calculated by the equation of FE = (n × F × C × V)/Q × 100%, where n is the number of electrons required per molecule of product, F is Faraday constant, C is the concentration of the product, V is the volume of the electrolyte, and Q is the total charge passed during electrolysis. All the electrochemical tests were performed under room temperature.
Electrochemical NO3RR measurement in the flow cell
Preparation of the working electrode
Typically, 3 mg of catalysts were added into 1 mL of ethanol, followed by adding 25 µL of Nafion solution (5 wt.%, Chemours). After sonicating for 15 min to form a homogenous catalyst ink, 0.83 mL of catalyst ink was dropped uniformly onto a gas diffusion electrode (GDE; size: 1.0 × 2.5 cm, Sigracet 29BC) to ensure the catalyst loading amount of around 1.0 mg cm−2. The accurate loading is calculated by weighing the electrode before and after the dropping of the catalysts. After the catalyst-modified GDE was naturally dried, it was stored in a vacuum oven for further use.
NO3RR test
The electrochemical NO3RR measurement was performed in a two-channel flow cell (101017-1.2, GaossUnion) composed of an anode chamber and cathode chamber. An anion exchange membrane (Fumasep, FAA-3-PK-130) was used to separate the anode and cathode chambers. The catalyst-modified GDE, saturated Hg/HgO (GaossUnion), and Ni foam (Sinero) were used as working, reference, and counter electrodes, respectively. The reference electrode is regularly calibrated every month. Gaskets were used in the flow cell to seal the cell, and the exposed cathode area between the gasket is 1 cm2. The Ar-saturated solution containing 1 M KOH and 0.1 M KNO3 (or 1 M KOH and 1 M KNO3) was used as both anolyte and catholyte. The electrolytes were prepared before the electrochemical experiments by dissolving the salts in deionized water and stored in a sealed container. The catalyst loading for the test in 1 M KOH and 0.1 M KNO3 is 0.96 mg cm−2, while that for the test in 1 M KOH and 1 M KNO3 is 0.93 mg cm−2. The electrolytes in the cathode and the anode chambers were independently circulated by a two-channel peristaltic pump (LongerPump, BT100-2J) with a flow rate of 50 mL min−1. The solution resistance in the H-type cell and flow cell was measured using electrochemical impedance spectroscopy (EIS) in the frequency range of 0.1 Hz to 100 kHz.
15N-labeling control experiments
The 15N-labeling control experiments were conducted by replacing the reaction solution of 1 M KOH + 0.1 M K14NO3 with the solution of 1 M KOH + 0.1 M K15NO3 or with the solution without any NO3− (just 1 M KOH). The other electrochemical reaction conditions were kept the same as the ordinary NO3RR experiments.
Detection and quantification of the NO3RR products
NH3/NH4+
The NH3/NH4+ was detected by the indophenol blue method10,68,69 and the 1H nuclear magnetic resonance (NMR) method. In specific, 1 mL of electrolyte was taken out after the test and diluted with DI water to suitable folds. Firstly, 2.5 mL of solution A (composed of 0.625 M NaOH, 0.36 M salicylic acid, and 0.17 M sodium citrate) were added. Then 300 μL of solution B (sodium nitroferricyanide, 10 mg mL−1) and 150 μL of solution C (NaClO, active chlorine 6–14 wt.%) were added, successively. After mixing homogeneously, the solution was kept without disturbance for 2 h in a dark environment. Then, the absorbance values at 660 nm of these mixed solutions were analyzed by UV-vis spectrophotometer (Shimadzu-UV1700), and the NH3 concentrations can be determined according to the calibration curves. For the 1H NMR method, 400 μL of the tested electrolyte were first mixed with 100 μL of DMSO-d6 (with maleic acid as the internal standard, 10 mM). Then, 200 μL of 4 M HCl were further added to the solution to acidify it. The standard curves of NH4+ with different concentrations were first made by both indophenol blue method and 1H NMR method, and then used to quantify the concentration of NH4+ in the electrolyte after NO3RR.
NO2 −
The NO2− was detected by UV-vis spectrophotometry according to the standard method70. In specific, the coloring reagent was first prepared. 4 g of p-aminobenzenesulfonamide, 0.2 g of N-(1-Naphthyl) ethylenediamine dihydrochloride, and 10 mL of phosphoric acid (density = 1.70 g mL−1) were added to 50 mL of DI water and then fully sonicated. After that, 5 mL of diluted electrolyte were mixed with 0.1 mL of the above coloring reagent. After 20 min, the absorption spectrum was taken at the wavelength of 540 nm to analyze the concentration of NO2−. The standard curves of NO2− with different concentrations were first made by UV-vis spectrophotometry method and then used to quantify the concentration of NO2− in the electrolyte after NO3RR.
Hydrazine
Hydrazine (N2H4) in the electrolyte was determined by the Watt and Chrisp method11. Specifically, 5.99 g of 4-(dimethylamino)benzaldehyde, 30 mL of hydrochloric acid, and 300 mL of ethanol were thoroughly mixed and used as the color reagent. Then, 2.5 mL of electrolyte were collected from the cathode chamber and mixed with 2.5 mL of the above color reagent. After being kept in the dark for about 20 min at room temperature, the absorbance of the obtained solution at a wavelength of 457 nm was measured using the UV-vis spectrophotometer (Shimadzu-UV1700). The standard curves of N2H4 with different concentrations were first made and then used to quantify the concentration of N2H4 in the electrolyte after NO3RR.
NH2OH
NH2OH was detected by 1H NMR, where NH2OH was captured by an excess amount of C2H2O3 through oximation. Specifically, 0.4 mL of the catholyte after 1-h electrolysis was mixed with 0.1 mL of DMSO-d6 and 12.5 µL of 50 wt.% C2H2O3.
H2
The gas product of H2 was detected by an online gas chromatography (GC). Specifically, during the NO3RR test, the cathode was sealed and connected to an online GC. At the same time, Ar gas with a flow rate of 10 sccm was continuously purged in from the inlet of the cell to the electrolyte and then flows out from the outlet to the online GC. Therefore, the flowing Ar gas can carry any generated H2 into the GC to be analyzed.
In situ ATR-FTIR test
The in situ ATR-FTIR measurement was performed in the absorption mode on a Nicolet iS50 IR spectrometer, on which the detector is a liquid-nitrogen-cooled mercury-cadmium-telluride (MCT) detector. The Au-coated Si hemispherical prism (20 mm in diameter, MTI Corporation) loaded with catalysts was used as the working electrode. The ATR-FTIR spectra were collected at a resolution of 8 cm−1 with a scan number of 32. The LSV curve was acquired at a scan rate of 5 mV s−1 in the potential window of 0.3 to −1.0 V (vs RHE). All spectra were subtracted with the background.
In situ DEMS test
The in situ DEMS measurement was conducted on a Linglu DEMS system. The signal was collected during linearly sweeping the working electrode from 0.2 to −1.0 V (vs RHE) at a scanning rate of 5 mV s-1 in the Ar-saturated solution containing 1 M KOH and 0.1 M KNO3. A home-made electrochemical cell was used for the experiments. The carbon paper loaded with the catalyst, Pt wire, and Hg/HgO were used as the working, counter, and reference electrodes, respectively. A polytetrafluoroethylene membrane was used to separate the gas and electrolyte.
Calculation details
All DFT calculations in this work were performed with the Vienna Ab Initio Simulation Package (VASP)71. The main exposed (101)fcc and (110)4H surfaces of Cu and Au were used in the simulations. Perdew–Burke–Ernzerhof (PBE) of generalized gradient approximation was used to treat electron–electron exchange–correlation functionals72,73. The cutoff energy of 400 eV was utilized for the plane-wave basis. All atoms were relaxed to energy and force tolerance less than 10−4 eV and 0.02 eV Å−1, respectively. A 3 × 3 × 1 k point mesh was used for the 4H/fcc Au–Cu BBNs, and a 15 Å vacuum space along the Z direction was set to ensure that there is no interaction between adjacent layers74. The Bader charge partition analysis was employed for the charge transfer between the adsorbates and the 4H/fcc Au–Cu BBNs75. Bader charge transfer of the adsorbed molecules was defined as ΔQmolecule = Qadsorbed molecule−Qisolated-molecule, in which Qadsorbed molecule and Qisolated molecule represent the total Bader charge of all the atoms of the molecules in adsorption system and in unadsorbed state. The adsorption energy of the adsorbed molecules was defined as Eads = Etotal−(Emolecule + Esubstrate), in which Emolecule and Esubstrate represent the energy of the specified atoms or molecules and the pristine system, respectively, and Etotal is the total energy of the corresponding system after specified atoms or molecules adsorbed on the substrate. The solvation effect was incorporated throughout all calculations using VASPsol implicit solvation method, with a dielectric constant of 78.4 for water.
Data availability
The data that supports the findings of this study are included in the published article and its Supplementary Information. Source data are provided with this paper.
References
De Luna, P. et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes?. Science 364, eaav3506 (2019).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, eaad4998 (2017).
Zhang, G. et al. Ammonia recovery from nitrate-rich wastewater using a membrane-free electrochemical system. Nat. Sustain. 7, 1251–1263 (2024).
Hepburn, C. et al. The technological and economic prospects for CO2 utilization and removal. Nature 575, 87–97 (2019).
Liang, J. et al. Metal bond strength regulation enables large-scale synthesis of intermetallic nanocrystals for practical fuel cells. Nat. Mater. 23, 1259–1267 (2024).
Luo, M. et al. PdMo bimetallene for oxygen reduction catalysis. Nature 574, 81–85 (2019).
Fang, J.-Y. et al. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat. Commun. 13, 7899 (2022).
Fang, W. et al. Durable CO2 conversion in the proton-exchange membrane system. Nature 626, 86–91 (2024).
Tan, Y., Fu, J., Luo, T., Liu, K. & Liu, M. Theoretical insights into the selectivity of single-atom Fe–N–C catalysts for electrochemical NOx reduction. J. Am. Chem. Soc. 147, 4937–4944 (2025).
Chen, F.-Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759–767 (2022).
Chen, G.-F. et al. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper–molecular solid catalyst. Nat. Energy 5, 605–613 (2020).
Han, S. et al. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism. Nat. Catal. 6, 402–414 (2023).
Guo, W. et al. Accelerating multielectron reduction at CuxO nanograins interfaces with controlled local electric field. Nat. Commun. 14, 7383 (2023).
Tang, S. et al. General synthesis of high-entropy single-atom nanocages for electrosynthesis of ammonia from nitrate. Nat. Commun. 15, 6932 (2024).
Lim, J., Cullen, D. A., Stavitski, E., Lee, S. W. & Hatzell, M. C. Atomically ordered PdCu electrocatalysts for selective and stable electrochemical nitrate reduction. ACS Energy Lett. 8, 4746–4752 (2023).
Gao, Q. et al. Synthesis of core/shell nanocrystals with ordered intermetallic single-atom alloy layers for nitrate electroreduction to ammonia. Nat. Synth. 2, 624–634 (2023).
Liao, W. et al. Boosting nitrogen activation via Ag nanoneedle arrays for efficient ammonia synthesis. ACS Nano 17, 411–420 (2023).
Li, F. et al. Molecular tuning of CO2-to-ethylene conversion. Nature 577, 509–513 (2020).
Huang, J. E. et al. CO2 electrolysis to multicarbon products in strong acid. Science 372, 1074–1078 (2021).
Shin, H., Hansen, K. U. & Jiao, F. Techno-economic assessment of low-temperature carbon dioxide electrolysis. Nat. Sustain. 4, 911–919 (2021).
Wu, H. et al. Selective and energy-efficient electrosynthesis of ethylene from CO2 by tuning the valence of Cu catalysts through aryl diazonium functionalization. Nat. Energy 9, 422–433 (2024).
Belsa, B. et al. Materials challenges on the path to gigatonne CO2 electrolysis. Nat. Rev. Mater. 9, 535–549 (2024).
Yu, S. et al. CO2-to-methanol electroconversion on a molecular cobalt catalyst facilitated by acidic cations. Nat. Catal. 7, 1000–1009 (2024).
Ma, Y. et al. Surface modification of metal materials for high-performance electrocatalytic carbon dioxide reduction. Matter 4, 888–926 (2021).
Morales-Guio, C. G. et al. Improved CO2 reduction activity towards C2+ alcohols on a tandem gold on copper electrocatalyst. Nat. Catal. 1, 764–771 (2018).
Liu, Y. et al. Efficient tandem electroreduction of nitrate into ammonia through coupling Cu single atoms with adjacent Co3O4. Nat. Commun. 15, 3619 (2024).
Zeng, T., Chen, J., Yu, Z. H. & Tse, E. C. M. CuFe cooperativity at the membrane-electrode interface elicits a tandem 2e–+2e– mechanism for exclusive O2-to-H2O electroreduction. J. Am. Chem. Soc. 146, 31757–31767 (2024).
Xiong, Y. et al. Electrochemical nitrate reduction: ammonia synthesis and the beyond. Adv. Mater. 36, 2304021 (2024).
Xu, Y. et al. Sulphur-boosted active hydrogen on copper for enhanced electrocatalytic nitrate-to-ammonia selectivity. Angew. Chem. Int. Ed. 63, 202400289 (2024).
Zhang, B. et al. Defect-induced triple synergistic modulation in copper for superior electrochemical ammonia production across broad nitrate concentrations. Nat. Commun. 15, 2816 (2024).
Carvalho, O. Q. et al. Role of electronic structure on nitrate reduction to ammonium: a periodic journey. J. Am. Chem. Soc. 144, 14809–14818 (2022).
Zhang, J. et al. Evidence for distinct active sites on oxide-derived Cu for electrochemical nitrate reduction. J. Am. Chem. Soc. 146, 30708–30714 (2024).
Fu, Y. et al. Enhancing electrochemical nitrate reduction to ammonia over Cu nanosheets via facet tandem catalysis. Angew. Chem. Int. Ed. 62, 202303327 (2023).
Zhang, K. et al. Cu-based catalysts for electrocatalytic nitrate reduction to ammonia: fundamentals and recent advances. EES Catal. 2, 727–752 (2024).
Bai, L. et al. Electrocatalytic nitrate and nitrite reduction toward ammonia using Cu2O nanocubes: active species and reaction mechanisms. J. Am. Chem. Soc. 146, 9665–9678 (2024).
He, W. et al. Splicing the active phases of copper/cobalt-based catalysts achieves high-rate tandem electroreduction of nitrate to ammonia. Nat. Commun. 13, 1129 (2022).
Ma, Y. et al. Confined growth of silver-copper Janus nanostructures with {100} facets for highly selective tandem electrocatalytic carbon dioxide reduction. Adv. Mater. 34, 2110607 (2022).
Zhang, G. et al. Tandem electrocatalytic nitrate reduction to ammonia on mbenes. Angew. Chem. Int. Ed. 62, 202300054 (2023).
Wu, S. et al. Unraveling the tandem effect of nitrogen configuration promoting oxygen reduction reaction in alkaline seawater. Adv. Energy Mater. 14, 2400183 (2024).
Li, P. et al. Pulsed nitrate-to-ammonia electroreduction facilitated by tandem catalysis of nitrite intermediates. J. Am. Chem. Soc. 145, 6471–6479 (2023).
Wang, P. et al. Boosting electrocatalytic CO2–to–ethanol production via asymmetric C–C coupling. Nat. Commun. 13, 3754 (2022).
Zhang, S. et al. Fe/Cu diatomic catalysts for electrochemical nitrate reduction to ammonia. Nat. Commun. 14, 3634 (2023).
Yamada, Y. et al. Nanocrystal bilayer for tandem catalysis. Nat. Chem. 3, 372–376 (2011).
Chen, P.-C. et al. Heterostructured Au–Ir catalysts for enhanced oxygen evolution reaction. ACS Mater. Lett. 3, 1440–1447 (2021).
Huang, J., Mensi, M., Oveisi, E., Mantella, V. & Buonsanti, R. Structural sensitivities in bimetallic catalysts for electrochemical CO2 reduction revealed by Ag-Cu nanodimers. J. Am. Chem. Soc. 141, 2490–2499 (2019).
Lyu, Z. et al. Kinetically controlled synthesis of Pd-Cu Janus nanocrystals with enriched surface structures and enhanced catalytic activities toward CO2 reduction. J. Am. Chem. Soc. 143, 149–162 (2021).
Qiu, J., Nguyen, Q. N., Lyu, Z., Wang, Q. & Xia, Y. Bimetallic Janus nanocrystals: syntheses and applications. Adv. Mater. 34, 2102591 (2022).
Ma, Y. et al. Site-selective growth of fcc-2H-fcc copper on unconventional phase metal nanomaterials for highly efficient tandem CO2 electroreduction. Adv. Mater. 36, 2402979 (2024).
Jia, H. et al. Symmetry-broken Au–Cu heterostructures and their tandem catalysis process in electrochemical CO2 reduction. Adv. Funct. Mater. 31, 2101255 (2021).
Chen, Y. et al. Phase engineering of nanomaterials. Nat. Rev. Chem. 4, 243–256 (2020).
Lu, Q. et al. Crystal phase-based epitaxial growth of hybrid noble metal nanostructures on 4H/fcc Au nanowires. Nat. Chem. 10, 456–461 (2018).
Fan, Z. et al. Heterophase fcc-2H-fcc gold nanorods. Nat. Commun. 11, 3293 (2020).
Zhou, X. et al. Preparation of Au@Pd core–shell nanorods with fcc-2H-fcc heterophase for highly efficient electrocatalytic alcohol oxidation. J. Am. Chem. Soc. 144, 547–555 (2022).
Gloag, L. et al. Synthesis of hierarchical metal nanostructures with high electrocatalytic surface areas. Sci. Adv. 9, eadf6075 (2023).
Yin, J. et al. A new hexagonal cobalt nanosheet catalyst for selective CO2 conversion to ethanal. J. Am. Chem. Soc. 143, 15335–15343 (2021).
Zhang, Q. et al. Crystal structure control of binary and ternary solid-solution alloy nanoparticles with a face-centered cubic or hexagonal close-packed phase. J. Am. Chem. Soc. 144, 4224–4232 (2022).
Huang, X. et al. Regioselective epitaxial growth of metallic heterostructures. Nat. Nanotechnol. 19, 1306–1315 (2024).
Zeng, J. et al. Controlling the nucleation and growth of silver on palladium nanocubes by manipulating the reaction kinetics. Angew. Chem. Int. Ed. 51, 2354–2358 (2012).
Zhu, C. et al. Kinetically controlled overgrowth of Ag or Au on Pd nanocrystal seeds: from hybrid dimers to nonconcentric and concentric bimetallic nanocrystals. J. Am. Chem. Soc. 134, 15822–15831 (2012).
Zhang, J. et al. In-situ electrochemical reconstruction and modulation of adsorbed hydrogen coverage in cobalt/ruthenium-based catalyst boost electroreduction of nitrate to ammonia. Nat. Commun. 15, 8583 (2024).
Gao, X. et al. Intermediate-regulated dynamic restructuring at Ag-Cu biphasic interface enables selective CO2 electroreduction to C2+ fuels. Nat. Commun. 15, 10331 (2024).
Zhu, C. et al. Dynamic restructuring of epitaxial Au–Cu biphasic interface for tandem CO2-to-C2+ alcohol conversion. Chem 8, 3288–3301 (2022).
Liu, L., Zheng, S.-J., Chen, H., Cai, J. & Zang, S.-Q. Tandem nitrate-to-ammonia conversion on atomically precise silver nanocluster/MXene electrocatalyst. Angew. Chem. Int. Ed. 63, 202316910 (2024).
Liao, W. et al. Sustainable conversion of alkaline nitrate to ammonia at activities greater than 2 A cm−2. Nat. Commun. 15, 1264 (2024).
Huang, Y. et al. Pulsed electroreduction of low-concentration nitrate to ammonia. Nat. Commun. 14, 7368 (2023).
Chen, Y. et al. High-yield synthesis of crystal-phase-heterostructured 4H/fcc Au@Pd core–shell nanorods for electrocatalytic ethanol oxidation. Adv. Mater. 29, 1701331 (2017).
Newville, M. IFEFFIT: interactive XAFS analysis and FEFF fitting. J. Synchrotron Radiat. 8, 322–324 (2001).
Zhao, Y. et al. Revealing ammonia quantification minefield in photo/electrocatalysis. Angew. Chem. Int. Ed. 60, 21728–21731 (2021).
Chen, D. et al. Synergistic modulation of local environment for electrochemical nitrate reduction via asymmetric vacancies and adjacent ion clusters. Nano Energy 98, 107338 (2022).
Wang, Y., Yu, Y., Jia, R., Zhang, C. & Zhang, B. Electrochemical synthesis of nitric acid from air and ammonia through waste utilization. Natl Sci. Rev. 6, 730–738 (2019).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Henkelman, G., Arnaldsson, A. & Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 36, 354–360 (2006).
Acknowledgements
This work was supported by grants (Project No. 22175148 and 52173222) from National Natural Science Foundation of China, grants (Project No. 21309322, 17300424, E-HKU701/23 and T23-713-22R) from Research Grants Council of Hong Kong, grant (Project No. JCYJ20220530140815035 and JCYJ20230807111605012) from Shenzhen Science and Technology Program, grants (ECF 2021-152 and 2021-141) from the HK Environment and Conservation Fund, ITC via Hong Kong Branch of National Precious Metals Material Engineering Research Center, and grants (Project No. 9610480, 9610663, 7006007, 7020103 and 9680301) from City University of Hong Kong. Open Access made possible with partial support from the Open Access Publishing Fund of the City University of Hong Kong. The authors thank the computational resources from the BigData Computing Center of Southeast University.
Author information
Authors and Affiliations
Contributions
Z.F. proposed the research direction and guided the project. Y.M., Li.G., L.C., W.G., L.G., Z.G. and Z.F. analyzed and discussed the experimental results and drafted the manuscript. Y.M. conducted the materials synthesis and characterization. L.C. and L.G. performed the aberration-corrected STEM characterization. S.C. conducted the XAS measurement. K.Q. and Y.M. analyzed the XAS data. Y.M. and W.G. performed the electrochemical NO3RR tests. T.Z., F.H., W.S. and Y.M. conducted the in situ ATR-FTIR and in situ DEMS tests. Li.G., Q.L., and C.L. performed the DFT calculations. J.Z., G.W., M.S., Ji.Y., J.Y., Y.W., F.L. and A.Z. carried out some supporting experiments. All authors checked the manuscript and agreed with the content. Our work meets the Research ethics and Reporting standards of the journal.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Junwei Fu, Min Liu, Yuhuan Cui, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, Y., Guo, L., Chang, L. et al. Unconventional phase metal heteronanostructures with tunable exposed interface for efficient tandem nitrate electroreduction to ammonia. Nat Commun 16, 7632 (2025). https://doi.org/10.1038/s41467-025-63013-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63013-0
