
Abstract
Extracellular vesicles (EVs) are promising biomarkers for cancer diagnosis and prognosis due to their ability to carry specific biomolecular cargo, including DNA. However, the clinical utility of DNA methylation-based liquid biopsies using EV-DNA remains underexplored. The low quantity and relatively long length of EV-DNA complicate whole-genome methylation profiling. To address this, we develop Tn5-assisted Enzymatic Methyl-sequencing with Post-conversion Tailing (TEMPT), a bisulfite-free whole-genome profiling method for EV-DNA. TEMPT employs single-adapter Tn5 tagmentation, enzymatic conversion of unmodified cytosines, and post-conversion tailing to generate high-depth whole-genome EV-DNA methylomes. We apply TEMPT to EV-DNA from 58 gastric cancer and polyp samples, generating methylomes from sub-nanogram inputs and identifying differentially methylated regions (DMRs) that distinguish cancer from controls. We identify potential cancer biomarkers through DMR-associated genes, highlighting the roles of EVs in cellular communication. Our findings suggest that immune cells may serve as an alternative source of EV-DNA. This approach holds significant promise for advancing EV-DNA research and its applications in early disease diagnosis.
Similar content being viewed by others


Introduction
Developing precise diagnostic technologies for early cancer diagnosis is essential for reducing mortality, improving patient survival rates, and alleviating patient burden. Liquid biopsy, which involves analyzing body fluids for early diagnosis, has gained widespread attention due to its lower invasiveness and greater flexibility compared to tissue biopsies1. Most liquid biopsy methods that analyze nucleic acid signatures rely on the detection of circulating cell-free DNA (cfDNA) from plasma2. While mutations in circulating tumor DNA (ctDNA) are associated with tumor progression, the lack of specificity makes it difficult to locate the tumor3. In contrast, epigenetic changes like DNA methylation provide an opportunity for cancer localization, making the detection of ctDNA methylation a central focus in early cancer screening4,5. However, the short half-life of ctDNA (5–150 min)2 and its fragmentated nature pose challenges for next-generation sequencing6, requiring rapid collection and advanced analytical techniques.
Extracellular vesicles (EVs), secreted by various cell types, play a crucial role in cell-to-cell communication during cancer progression. They carry biomolecules, including proteins, nucleic acids, and lipids, which contribute to tumor development and metastasis7. EVs also exhibit long-term stability in body fluids, facilitating the collection and analysis of biological samples8. EV-derived DNA primarily originates from two processes9,10. The first one is cell death, including apoptosis and necrosis. Apoptotic processes generate small DNA fragments (~160–180 base pairs), and necrosis produces larger DNA fragments, both subsequently incorporated into EVs. The second process is active secretion mechanisms that maintain cellular homeostasis under physiological conditions and contribute to disease progression in pathological states. However, the precise molecular basis of EV-DNA release remains partially understood and requires further investigation. Studies show that EV-DNA contains copy number variations (CNVs) and DNA methylation patterns that closely resemble those of the parent cells11,12, enhancing the potential of EVs as cancer biomarkers. Although earlier studies have explored differential EV-DNA methylation in cancer cell lines11,13, their investigations have been confined to in vitro models and predominantly relied on methylation arrays14 that require relatively high amounts of input DNA. These limitations highlight the need for the development of low-input approaches for EV-DNA methylation profiling. Existing genome-wide EV-DNA studies rely on methylation array that uses bisulfite conversion9,11,13, the gold standard method for DNA methylation profiling. Despite its accuracy, bisulfite treatment can cause DNA loss and requires hundreds of nanograms of DNA input15. Nevertheless, clinically feasible volumes of plasma typically contain less than 10 ng of cfDNA, and the EV-DNA content is about 20 times lower than that of cfDNA16, underscoring the need for efficient bisulfite-free methylome sequencing techniques. To avoid DNA damage caused by bisulfite treatment, researchers have developed several bisulfite-free methylation sequencing techniques17. TET-assisted pyridine borane sequencing (TAPS) combines ten-eleven translocation (TET) oxidation with mild chemical reactions to convert modified cytosines into dihydrouracil (DHU)18. Enzymatic methyl-seq (EM-seq) uses an entirely enzymatic conversion process, employing TET2 and β-glucosyltransferase (βGT) to protect 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC), followed by APOBEC to deaminate cytosine to uracil (U)19. These bisulfite-free methods minimize DNA loss, making them suitable for methylation profiling with low DNA inputs17. Among these techniques, EM-seq is commercially available and suitable for clinical use20,21.
Although third-generation sequencing technologies such as Oxford Nanopore22 and PacBio23 offer the advantage of directly sequencing long DNA fragments without methylation conversion, their substantial input DNA requirement render them less suitable for low-input samples, such as EV-DNA. In contrast, transposase-based library construction is particularly effective, as it facilitates both DNA fragmentation and adapter ligation in a single step, a process known as tagmentation24. Although the Tn5 transposome is highly efficient at DNA tagmentation, it only generates amplifiable libraries when different adapters are inserted at both ends of the DNA fragments, which occurs only about 50% of the time (Supplementary Fig. 1)25,26. To overcome this inefficiency, various single-adapter tagmentation strategies have been developed, such as random priming (sci-MET)27, single strand adapter ligation with 3’ random nucleotide overhangs (SALP)28, and locked nucleic acid-templated extension (s3)26. While these strategies have improved efficiency to some degree, they often require significant customization. Therefore, a simple and highly efficient library construction method is needed for low-input EV-DNA.
To address these challenges, we developed a method named TEMPT (Tn5-assisted Enzymatic Methyl-sequencing with Post-conversion Tailing) for whole-genome mapping of DNA methylation in EV-DNA. TEMPT utilizes single-adapter Tn5 tagmentation and post-conversion tailing for DNA library construction, overcoming the gene loss deficit of classical Tn5 transposome tagmentation. In addition, TEMPT employed enzymatic conversion of unmodified cytosine with EM-seq, which differs from bisulfite treatment and reduces DNA loss. Notably, TEMPT incorporates carrier RNA to improve EV-DNA recovery during purification and minimize external DNA contamination in the library. This technique enabled high-throughput methylome profiling of EV-DNA, uncovering potential biomarkers and paving the way for potential applications of EVs in cancer diagnosis and treatment.
Results
Overview of TEMPT for low-abundance EV-DNA methylome profiling
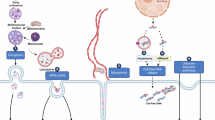
The design principle of TEMPT integrates molecular strategies (Fig. 1) to address the challenges posed by low concentrations of patient-derived EV-DNA, which often contains large double-stranded fragments29,30. Several key improvements were implemented: (1) Compared to cfDNA, EV-DNA is longer and present at much lower concentrations, making Tn5 transposome the optimal choice for fragmenting EV-DNA and adding adapters. To address the 50% inefficiency issue25, we first constructed Tn5 with a single adapter modified with cytosine methylation. Tagmentation was then carried out using this modified single-adapter Tn5, ensuring that all cleaved DNA fragments incorporated the same adapter at the 5’ end. (2) The enzymatic conversion of EV-DNA with 5’ adapters followed the methylation conversion process of EM-seq, utilizing mild conditions to prevent damage to the EV-DNA. The procedures protected 5mC and 5hmC through TET oxidation and T4-βGT glycosylation to form 5-carboxylcytosine (5caC) or 5-(β-glucosyloxymethyl) cytosine (5gmC), then deaminated unmodified C to U using APOBEC. However, since 5mC is much more prevalent than 5hmC in most cases, the combined levels of 5mC and 5hmC typically approximate the level of 5mC alone in most cell types31. (3) Next, we employed the tail-and-tag strategy to ligate adapters to the 3’ ends of the fragmented single-stranded DNA generated after enzymatic treatment32. This design prevented the formation of identical adapters on both ends, eliminating the 50% dual-adapter ligation issue and thereby enhancing efficiency. Briefly, the treated DNA was dephosphorylated and then extended with a poly C tail using the terminal deoxynucleotidyl transferase (TdT) enzyme. A splinted 3’ adapter with a 5’ protruding poly G end was then annealed to the tail to enable a highly efficient ligation step using Escherichia coli (E. coli) ligase. Next, the fragments of converted EV-DNA with adapters in both ends were amplified by PCR and sequenced. Raw data were processed for downstream analysis, including biomarker identification, model prediction, and gene oncology analysis. We evaluated the TEMPT recovery rate at each step, revealing that approximately 40.5% of DNA remained after the conversion step, indicating efficient DNA recovery in low-input EV-DNA samples (Supplementary Fig. 2). Moreover, we compared the TEMPT method with SALP and s3 for library preparation. While all three methods showed similar mapping rates, TEMPT produced higher library yields than SALP and s3, outperforming other methods for preparing sequencing libraries from low-input EV-DNA samples (Supplementary Fig. 3). The entire process achieved high-efficiency library construction and prevented DNA damage, enabling whole-genome methylome profiling for ultra-low input EV-DNA.

A schematic illustration of the whole process for methylation profiling of EV-DNA samples. The whole process includes patient sample collection, EV extraction by ultracentrifugation and lysis, library construction by TEMPT, biomarker identification, model prediction, and gene oncology analysis. In brief, the first step in TEMPT involved fragmenting EV-DNA (gray) using single-adapter Tn5 tagmentation to add a 5’ adapter (orange), dark gray indicates the Mosaic End of the Tn5 adapter. Then, fragmented EV-DNA is enzymatically converted by TET, T4-βGT, APOBEC enzymes. Gray, blue, black, green, red balls indicate C, 5mC, 5hmC, 5caC/5ghmC, and U, respectively. After conversion, TdT adds dC nucleotides to the 3’ end of the DNA fragments, creating poly-C tail at the 3’ end. Then, a splinted adapter (light and dark blue) with a 5’ poly dG end is ligated using E. coli ligase. Finally, DNA fragments with adapters on both ends are amplified by PCR and sequenced. Purple and yellow indicate i5 and i7 indexes; green and amaranth correspond to i5 and i7 flow cell primers for amplification and sequencing. Raw data are processed for downstream analysis, including biomarker identification, model prediction, and gene oncology analysis.
Whole-genome methylation profiling of EV-DNA by TEMPT
Although ultracentrifugation (UC) has a moderate recovery rate, it delivers higher EV purity from plasma compared to other isolation methods33. We used UC to isolate EVs from plasma and subsequently characterized them using Transmission Electron Microscopy (TEM), Nanoparticle Tracking Analysis (NTA), and Western Blotting (WB). TEM and NTA results showed that the isolated EVs exhibit a distinct membrane structure and a size of 166.4 ± 71.2 nm, consistent with the relatively large EV subpopulation (80–200 nm) that contains internal DNA29,34,35 (Supplementary Fig. 4a, b). Additionally, we identified that the isolated EVs expressed the protein markers CD63, TSG101, and HSP70 while lacking the cellular marker protein calnexin, confirming that the isolated EVs possess exosomal characteristics (Supplementary Fig. 4c, d). Therefore, we continued using TEMPT for EV-DNA library preparation. EVs were extracted from about 1.5 mL plasma (about 3 mL whole blood) from 44 patients in the discovery cohort (Supplementary Data 1), and then EV-DNA was isolated and quantitatively assessed relative to the GAPDH gene using qPCR. The quantification concentration of EV-DNA in patients’ plasma ranged from 0.07 to 1.67 ng/mL, which was much lower than the concentration of cfDNA16. Prior to EV lysis, surface DNA was removed with thermolabile dsDNase to eliminate contamination. To assess processing efficiency, 20 ng of lambda DNA was spiked into plasma samples, and the same procedures were applied. Library construction resulted in 88.17% of reads mapping to the human genome, with only 2.66% mapping to lambda DNA (Supplementary Fig. 5). Consequently, we constructed libraries directly from the extracted and lysed EVs to prevent loss during the DNA extraction process (Supplementary Fig. 6a).
The libraries constructed from EV-DNA using TEMPT were sequenced at 30x depth and technically assessed (Fig. 2a). Coverage analysis revealed no significant differences in coverage ratio between the control and cancer groups (Supplementary Fig. 6b). Six samples with uneven coverage were excluded, leaving 38 samples for downstream analysis (Supplementary Data 2). In general, the libraries of 38 EV-DNA samples generated an average of 348 million read pairs per sample, and the average number of uniquely mapped read pairs was 308 million per sample (Fig. 2b). This indicated that TEMPT can effectively construct sequencing libraries for EV-DNA methylation, achieving high-quality data with minimal input. To accurately assess the quality of methylation conversion, we used two types of spike-in controls: one control with methylated cytosine and one control with unmethylated cytosine. Using CpG-methylated lambda DNA, we calculated that the overall non-conversion rate of 5mC across 38 samples was 1.27%. The conversion efficiency of unmodified 2 kb fragments was estimated at 99.73%, indicating a non-conversion rate of unmodified cytosines of 0.27% (Fig. 2c). These results confirmed that TEMPT achieved a high conversion rate and demonstrated robust reproducibility.

a Flowchart depicting the sequential filtering steps for clinical sample analysis (GP benign gastric polyp, GC gastric cancer). b Total read counts and uniquely mapped reads in EV-DNA TEMPT libraries from 38 clinical samples are shown, with SEM represented as error bars. c Cytosine conversion rate in EV-DNA TEMPT libraries from 38 clinical samples based on spike-in controls with unmodified or modified cytosines at the known positions. Each dot represents an individual clinical sample. d Distribution of benign and cancer samples across TNM stages (n = 38). e Genome-wide CpG modification levels of EV-DNA in benign controls and cancer samples. Each dot represents an individual clinical sample (n = 38). Each box represents the interquartile range. The lower and upper bounds of the box are the 25th and 75th percentiles of the data, respectively. The middle line indicates the median (50th percentile) and the whiskers are 1.5 × IQR. Unpaired Wilcoxon tests were performed. Source data are provided as a Source Data file (b–e).
Next, we performed a comprehensive methylome characterization on the 38 EV-DNA samples that passed the initial quality assessment, including 21 cases of gastric cancer at various TNM stages and 17 cases of gastric polyps (benign). The gastric cancer cases were further categorized into early (Tis, T1, 57%) and advanced stages (T2-T4)36 (Fig. 2d, Supplementary Fig. 7a). We chose patients with benign disease rather than healthy individuals because distinguishing between benign and cancer patients is more clinically relevant. To gain insight into the DNA methylation characteristics of EV in gastric cancer and polyps samples, we analyzed the average levels of CpG modification across the different sample groups: benign, early-stage cancer, and advanced-stage cancer. The average CpG methylation levels for the three groups were 74.43%, 72.83%, and 72.27%, respectively. The methylation level between the benign and advanced-stage cancer groups was significantly different (p-value = 0.011), while the difference between the benign and early-stage cancer groups was borderline significant (p-value = 0.057) (Fig. 2e). These findings suggest that global DNA hypomethylation becomes more evident as cancer progresses, even though it is not prominent at the early stage. Additionally, we observed increased variance in methylation levels within 1-Mb genomic windows when comparing the benign and cancer groups (Supplementary Fig. 7b, c). The distribution of EV-DNA methylation levels at CpG sites in both control and cancer groups was bimodal, with most CpG sites being either fully methylated or unmethylated (Supplementary Fig. 7d).
TEMPT robustly classifies cancer and benign cases
Given that cfDNA methylation signatures can differentiate cancer patients from healthy individuals, we investigated whether EV-DNA methylation patterns possess similar diagnostic potential. We first identified 647 differentially methylated regions (DMRs) between cancer and non-cancer samples, termed as cancer-related DMRs (Supplementary Data 3). Using these DMRs, we found distinct methylation patterns between the two groups (Fig. 3a), suggesting that the EV-DNA methylation characteristics could serve as biomarkers for cancer detection. Further, principal component analysis (PCA) using these cancer-related DMRs separated benign and cancer along the first principal component (PC1, Fig. 3b). Notably, the cancer-related DMRs were significantly enriched in regulatory regions, particularly enhancers (Fig. 3c). This enrichment suggests that the cancer-associated methylation features exhibited by EV-DNA may influence enhancer activity, potentially altering gene expression. Such changes could be associated with cancer development and provide potential biomarkers for liquid biopsy.

a Heatmap showing the correlation analysis results of clinical samples based on the methylation levels of 647 cancer-related DMRs, i.e., DMRs identified between benign and cancer samples (GP benign gastric polyp, GC gastric cancer). The color gradient in the heatmap represents the DNA methylation levels in the DMRs, with warmer colors indicating higher levels of methylation. Source data are provided as a Source Data file. b PCA plot of benign controls and cancer samples based on EV-DNA cancer-related DMRs (n = 38). Source data are provided as a Source Data file. c The enrichment analysis of cancer-related DMRs in genomic regulatory elements. Two-sided Fisher’s exact tests were performed. Source data are provided as a Source Data file. d ROC curve showing the classification performance of the random forest model based on DMRs identified in the discovery set between benign and cancer groups (n = 38). e ROC curve for the random forest model classifying benign and cancer groups (n = 20) based on DMRs in the validate cohort. f Heatmap showing the top twenty DMR-associated genes with most significantly differential methylation levels among 530 cancer-related DMR-associated genes. The x-axis indicates samples, and the y-axis indicates the corresponding DMR-associated genes. The color gradient in the heatmap represents the DNA methylation levels in the DMRs, with warmer colors indicating higher levels of methylation. Source data are provided as a Source Data file. g, h, Violin plots showing the differential methylation levels between cancer and benign samples (n = 38) for an intergenic DMR near MK167 g, and another intergenic DMR near UQCRFS1 h, box represents the interquartile range. The lower and upper bounds of the box are the 25th and 75th percentiles of the data, respectively. The middle line indicates the median (50th percentile), and the whiskers are 1.5 × IQR. Source data are provided as a Source Data file (g, h). i Venn diagram comparing annotated DMR-associated genes with DMR-associated genes annotated in gastric cancer cfDNA40.
In addition to differentiating between cancer and benign samples, we explored whether the EV-DNA methylation profiles could effectively predict cancer stages, specifically early stage (Tis, T1) and advanced stage (T2-T4). We identified stage-specific DMRs from cancer samples and performed a correlation analysis among these samples (Supplementary Data 4). The analysis revealed that the cancer EV-DNA samples clustered by early and advanced stages based on their stage-specific DMR profiles (Supplementary Fig. 8a). Furthermore, PCA using stage-specific DMRs demonstrated a clear partition between advanced-stage and early-stage samples along PC1 (Supplementary Fig. 8b). The functional enrichment analysis of these stage-specific DMRs revealed that, compared to the cancer-related DMRs, the stage-specific DMRs showed more significant enrichment in multiple genomic regions, including enhancers, promoters, and CTCF binding sites (Supplementary Fig. 8c). These findings suggested that EV-DNA methylation profiles were associated with cancer stages and possessed potential regulatory roles. Thus, they could be potentially used to monitor disease progression.
We next examined whether the EV-DNA methylation profiles by TEMPT can classify diseases. In doing so, we employed the machine learning model, the random forest, which was trained and evaluated through a five-fold cross-validation approach. Specifically, samples were randomly divided into five folds, with four-fold samples used for training and one-fold samples reserved as the testing set. Within each training set, DMRs were identified using Dispersion Shrinkage for Sequencing data (DSS) (Supplementary Fig. 9a, Supplementary Data 5). The analysis demonstrated that the EV-DNA methylation levels could robustly classify cancer and benign samples (AUC = 0.81, Fig. 3d, Supplementary Fig. 9b, c). For model evaluation, another 20 samples (Supplementary Data 6 and 7) were used as the validation set. The AUC for the validation cohort was 0.8, confirming the robustness and reproducibility of our model across gastric cancer samples (Fig. 3e, Supplementary Fig. 10, Supplementary Data 8 and Data 9).
TEMPT identifies potential markers for cancer development
Given the excellent performance of TEMPT in distinguishing between cancer and control groups, we proceeded with identifying potential biomarkers. We annotated 647 DMRs and identified 530 DMR-associated genes using Homer37 (Fig. 3f, Supplementary Fig. 11, Supplementary Data 10 and Data 11). We found that the top cancer-related DMRs were likely located near genes that have been reported to play significant roles in tumor development. For example, MK167 and UQCRFS1, which can promote tumor progression38,39, were annotated to the highly differential regions (Fig. 3g, h).
We next compared the EV-DNA DMR panel with the cfDNA-based DMRs for gastric cancer40. The analysis of DMR-associated genes revealed that only 20 DMR pairs (3.8%) shared the same associated genes between two datasets (Fig. 3i). Moreover, many of the DMR-associated genes that were unique in our data were related to EV-mediated intercellular communication. For example, one of these DMR-associated genes, YES1, expresses a cytoplasmic non-receptor tyrosine kinase of the Src family, is abnormally expressed in tumor tissues, and plays a role in transmitting extracellular signals through intracellular signaling pathways41. It indicates that the EV-DNA-based TEMPT characterized DNA methylation patterns that differ from cfDNA profiling. To directly compare DMRs between EV-DNA and cfDNA, paired samples from five patients were analyzed using methylation sequencing (Supplementary Data 6). Due to the higher concentration, cfDNA generated more sequencing reads (Supplementary Fig. 12a). We called cancer-specific DMRs for cfDNA and EV-DNA samples, respectively. Only 4.7% of DMRs overlapped between the cfDNA and EV-DNA, indicating unique methylation patterns in EV-DNA (Supplementary Fig. 12b, Supplementary Data 12 and 13). Gene Ontology (GO) enrichment analysis further supported these functional differences (Supplementary Fig. 12c, d), identifying EV-DNA-specific enrichment of DMRs in genes associated with cell communication, including FN142,43, SRC44, EPHA245, and ITGA546.
EV-DNA methylation profiles reveal cell migration and communication signatures in cancer patients
Unlike cfDNA, DNA encapsulated in EVs is more stable and can escape immune detection47, implying that EV DNA might serve special roles or carry unique information for cancer progression. While the biological functions of EV protein and miRNAs have been extensively studied30,48, the functional roles of DNA methylation carried by EVs remain elusive. Our analysis demonstrated that EV-DNA methylation could serve as the disease biomarker, and next, we investigated whether EV-DNA methylation profiles could provide insights into the biological functions of EVs.
To provide functional insights into EV-DNA methylation, we performed GO enrichment analysis of the 530 DMR-associated genes (False Discovery Rate (FDR)-adjusted p-value < 0.05 by the EnrichR49 analysis) (Fig. 4a). We found that the DMR-associated genes were enrichment in the biological processes related to cell migration, cell adhesion, and cell signaling (GO:0034329, GO:0031346, GO:0045582, and GO:0031345). We next looked into DMRs associated with these pathways and found that most of them were hypomethylated in tumor EV-DNA samples (Fig. 4b, Supplementary Data 14), suggesting a potential transcriptional activation of these genes in parent cells of cancer cases. For example, the DMR chr9: 111034953- 111035063 was located in the promoter region near LPAR1, which is critical for cancer cell motility and invasion (Fig. 4c)50. Our results align with previous findings that EVs can regulate cell migration by delivering diverse cargos, including those involved in cell signaling, polarization, and adhesion51. EVs have been reported to carry methylation characteristics similar to those of their parent cells9. Thus, we envisioned that DNA with methylation changes was encapsulated within EVs in parent cells, enabling the detection of these epigenetic alterations through EVs11,12.

a Gene Ontology enrichment of genes associated with DMRs between benign controls and cancer samples in EV-DNA using EnrichR49. The top ten enriched GO categories are ranked by the combined score calculated by EnrichR. The color indicates Enrichment FDR, and the dot size indicates the number of genes per GO term. b Heatmap showing the differential methylation levels between benign controls and cancer samples across DMR-associate genes related to key biological processes. The x-axis represents samples, and the y-axis represents DMR-associated genes. The color gradient in the heatmap represents the DNA methylation levels in the DMRs, with warmer colors indicating higher levels of methylation. c, d, Violin plot depicting methylation differences between benign and cancer samples (n = 38) for DMRs near LPAR1 (c) FN1 (d), box represents the interquartile range. The lower and upper bounds of the box are the 25th and 75th percentiles of the data, respectively. The middle line indicates the median (50th percentile), and the whiskers are 1.5 × IQR. e Stacked bar plots displaying average tissue contributions in benign and cancer samples (n = 38), estimated by NNLS. Contributions below 1.5% are grouped as “Other”. Source data are provided as a Source Data file (a–e).
We identified several genes related to cell migration that may potentially undergo changes during disease progression. Additionally, our analysis suggests the possibility of key genes involved in EV-mediated cell signaling within relevant pathways. For example, FN1 was annotated to be near the DMR chr2: 215491107-215491218, which was hypomethylated in cancer samples (Fig. 4d, Supplementary Fig. 13). This gene encodes the cell adhesion protein fibronectin, which plays a crucial role in cell junction assembly and cell growth. Previous studies also showed that tumor-derived microvesicles carry fibronectin, which can bind to integrins on cancer-associated fibroblasts (CAFs), promoting cell anchorage and growth and potentially transforming healthy cells42,43. Additionally, SLIT2 and ROBO1, which are associated with the GO term positive regulation of cell projection organization and the Slit/Robo signaling pathway, were also annotated near the significantly hypomethylated regions in cancer, suggesting a potential involvement in EV-mediated signaling during cancer progression (Supplementary Fig. 14). The Slit/Robo signaling pathway is involved in tumor progression, driving cancer cell migration and metastasis by reducing cell adhesion, reassembling the cytoskeleton, and modulating other related processes52. Similarly, DMRs of EV-DNA between cancer and benign samples suggested that EVs play a significant role in cell-cell communication.
Moreover, stage-specific DMRs further highlighted the involvement of EVs in this process. We found that the DMR-associated genes between early and advanced cancer samples were enriched in biological processes related to cell adhesion and migration (GO:0030334, GO:0098742, GO:0007043, GO:0034329) (Supplementary Fig. 15). A deeper investigation into these DMRs revealed that most were hypomethylated in advanced-stage EV-DNA samples (Supplementary Fig. 16, Supplementary Data 15 and 16). EVs are crucial in regulating communication between cancer cells and the tumor microenvironment during cancer progression53. Notably, we identified DMR-associated genes such as EGFR and CXCR4 (Supplementary Fig. 17), which are potentially linked to the GO term regulation of cell migration and might play a role in tumor progression. It has been reported that cancer-derived EVs can transfer oncogenic Epidermal Growth Factor Receptor (EGFR) to endothelial cells, leading to autocrine reprogramming54. Additionally, our finding aligns with the previous observation that cancer cells can enhance their invasive and metastatic capabilities by secreting EVs containing CXC motif chemokine receptor 4 (CXCR4)55.
Overall, our analysis of cancer-related and stage-specific DMRs revealed that the DNA methylation pattern in EVs of cancer patients is potentially associated with cell communication and migration. This demonstrated that the analysis of EV-DNA methylation differences using TEMPT identified tumor-associated methylation markers. It also implies the potential biological functions of methylated regions in EV-DNA, which is of particular interest for future studies.
TEMPT identifies immune cells as a major EV origin in gastric cancer samples
Immune cells can secrete various types of EVs for both innate and adaptive immunity56. For example, T cells can secrete EVs containing genomic and mitochondrial DNA, which are transferred to dendritic cells for pre-activation57. Our functional enrichment analysis revealed that both cancer-specific and stage-specific DMRs were associated with genes enriched in immune-related pathways, suggesting that immune cells are one of the sources of EV-DNA (Fig. 4a, Supplementary Fig. 15). To test this hypothesis and map the cellular sources of EV-DNA, we predicted the tissue contributions to EV-DNA by deconvolving the EV-DNA whole-genome methylation profiles using non-negative least squares regression (NNLS)5,58 and the tissue methylation reference map (Supplementary Data 17)59.
We found that the primary contributors of EV-DNA were neutrophils, similar to cfDNA59, and that the overall tissue contribution patterns were similar between the cancer and benign groups (Fig. 4e, Supplementary Data 18). Importantly, we found that a few immune cell types differentially contributed to cancer and benign EV-DNA samples. There were mild changes in contributions from monocytes, macrophages, plasma cells, and naive T cells. Specifically, the contribution levels of monocytes and naive T cells decreased in cancer EV-DNA samples, while the contribution levels of macrophages and plasma cells increased compared to benign samples (Supplementary Fig. 18a, b). The contribution from macrophages was 2.3 times greater in GC samples compared to GP samples, and the contribution from plasma cells was 2.8 times higher in GC samples. However, the changes were not statistically significant due to the small sample size and the large variation in the cancer group (p-value = 0.27 for macrophage, 0.108 for plasma cell by Wilcoxon-test; Supplementary Fig. 18c, d).
We investigated whether certain DMRs were associated with immune-related functions. In addition to the aforementioned SLIT2 and ROBO1, involved in the Slit/Robo signaling pathway that regulates T cell chemotaxis52, we found that the Src family kinase FYN was also annotated to a stage-specific DMR chr6: 11958745-11958796 (Supplementary Fig. 19). FYN plays a role in T cell development, maintenance, and activation60. Overall, our analysis unraveled that immune cells were one of the key contributors to EV-DNA and the carried methylation information. It also implied that the sources of EV-DNA change along with cancer development.
Discussion
Whole-genome methylation analysis of EV-DNA in clinical samples has not been systematically studied, and several challenges remain unaddressed. A key challenge is the very low quantity of EV-DNA available for analysis. Furthermore, the relatively longer length of EV-DNA compared to cfDNA necessitates additional fragmentation to meet the library preparation requirements for next-generation sequencing (NGS). Our TEMPT approach utilizes single-adapter Tn5 tagmentation and post-conversion tailing for DNA library construction. This strategy enhances the molecular utilization of the DNA template and avoids DNA loss typically associated with traditional bisulfite conversion. Unlike previous approaches using Illumina’s methylation array11,13, our approach offers more comprehensive whole-genome information from minimal amounts of EV-DNA. Additionally, compared to other single-adapter Tn5 approaches, our TEMPT method does not rely on extensive protocol optimization or customized reagents, yet achieves high library yield and mapping efficiency. This makes it particularly well-suited for whole-genome methylation profiling of low-input EV-DNA samples. However, TEMPT has several inherent limitations when compared to other low-input methylation sequencing approaches. A primary drawback is its throughput, which is considerably lower than that of highly scalable single-cell methylation sequencing techniques, making it less suitable for large-scale cohort analyses. The methodology is also prone to data bias from the Tn5 transposase, which has a known insertion preference that can lead to the under-representation of GC-rich regulatory regions such as CpG islands. Furthermore, the significant number of PCR cycles required for library amplification from ultra-low input DNA can introduce amplification bias, potentially skewing the quantitative accuracy of the final methylation data.
Our study demonstrated that TEMPT EV-DNA methylation profiles could robustly classify cancer, as evidenced by their high performance in both discovery and validation cohorts. While conventional methods have predominantly relied on cfDNA to investigate methylation patterns, cfDNA’s short half-life and high fragmentation lead to a limited sampling window2. Previous studies have reported that mutations in EV-DNA can differentiate between healthy and diseased states61. Our application of TEMPT for whole-genome deep sequencing of EV-DNA significantly complements existing research on cfDNA and EV-DNA mutations. Through gene annotation near DMRs, we identified several tumor-related biomarkers, suggesting that EV-DNA has the potential to reflect the methylation status of the parent cells. Additionally, our study highlighted the role of EVs in cellular communication, as evidenced by the DMR-associated genes. GO analysis further revealed that EV-DNA features are associated with immune cells, underscoring that immune cells contribute to the pool of EV-DNA.
Moreover, we demonstrated the utility of the TEMPT method in cellular deconvolution, revealing previously unreported insights into the tissue of origin (TOO) and immune cell composition of EV-DNA. The TEMPT data enabled the deconvolution of EV-DNA from different cellular origins and allowed for the assessment of relative fold changes based on methylation characteristics. Given the limitation of ultracentrifugation-based isolation methods, some of the EV-DNA may derive from peripheral blood mononuclear cells56,62,63. In line with previous findings9,10, our TOO analysis implies that EV-DNA can carry methylation signatures from various cell types, including tumor cells and leukocytes or lymphocytes. Yet, we cannot confirm whether these immune-related signatures are derived from the tumor-filtrating immune cells or immune cells from other tissues or bloodstream64,65.
Our study presents promising findings, yet certain aspects require further investigation. The cohort size limits statistical power in assessing associations between EV-DNA biomarkers and clinical factors such as age, tumor stage, and treatment response. Expanding the cohort size would strengthen the EV-DNA methylation reference database and enhance computational models for more accurate profiling. It would also enable direct comparisons with tumor biopsies and support longitudinal analyses aimed at identifying EV-DNA methylation signatures relevant to early cancer detection and metastatic monitoring. Post-treatment profiling could further clarify methylation dynamics following therapy. Additionally, addressing EV heterogeneity, including diverse sources and in situ subpopulation profiling66, will enhance the accuracy of EV-DNA analysis. Furthermore, the integration of our TEMPT method with protein A-Tn567 offers the potential for simultaneous exploration of histone modifications on EV-DNA68. These advancements will ultimately refine its clinical utility and broaden potential applications.
Methods
Sample acquisition
The study was conducted among 36 patients with TNM stage Tis-T4 gastric cancer and 30 patients with gastric polyps. The blood samples were obtained from Zhongnan Hospital of Wuhan University, Wuhan, China. The detailed diagnostic information for all samples was included in Supplementary Data 1 and 6. No sex-stratified analyses were performed.
Isolation of EVs from plasma
The experimental details of the EV process were comprehensively presented as recommended69. Blood samples of patients with gastric cancer or gastric polyps were obtained from blood draws. Approximately 3 mL peripheral blood was collected in a vacuum negative pressure tube containing EDTA. Blood samples were centrifuged at 2000 × g for 20 min at 4 °C within 4 h of collection to obtain cell-free plasma, which was then stored at −80 °C. The plasma samples were diluted in filtered PBS (1:1) and centrifuged at 16,500 g for 45 min at 4 °C (Eppendorf). The collected supernatants were filtered through a 0.22 μm filter (Millipore, Billerica, MA) and transferred to ultracentrifuge tubes. Then, the tubes were filled with filtered PBS and centrifuged at 100,000 × g for 2 h at 4 °C (Beckman Coulter, TLA120). The pellets were resuspended in filtered PBS and centrifuged at 100,000 × g for 2 h at 4 °C. The obtained EVs were resuspended in 10 μL 1.25 × TAPS buffer (12.5 mM TAPS buffer, pH 8.5, 6.25 mM MgCl2).
Transmission electron microscope (TEM) analysis of EVs
Plasma-EVs were diluted to an appropriate ratio, and then 10 μL of the sample was added to a carbon retaliation copper mesh that was pretreated by a glow discharge apparatus. After incubating for 1 min, the remaining liquid was blotted with filter paper. Added 10 μL uranyl acetate solution and dried it immediately, then added 10 μL of uranyl acetate solution again and stained for about 10 min. After drying, the copper mesh was loaded into the TEM instrument (JEM-1400Plus) for imaging using Gatan Microscopy Suite software (GMS 3).
Nanoparticle tracking analysis (NTA) of EVs
Plasma-EVs were diluted 100-fold with 1×PBS before being analyzed. For each run, 200 µL of the prepared sample was injected into the sample chamber of the NanoSight NS300 instrument (Malvern Panalytical) with a blue 488 nm laser at room temperature. The sample was performed for three independent duplicates, and each duplicate was recorded for 60 seconds in a 25.0 fps video by using a sCMOS camera. Dynamic monitoring NTA acquisition was performed using a syringe loading system (the speed was 50), and the threshold was set to 5. After that, the data capturing and analysis were performed on NTA analytical software (the version is NTA 3.4 Build 3.4.4).
Western blotting
Plasma-EVs were dissolved in 1×PBS and quantified with BCA Protein Assay Kit (Biosharp), then boiled with 4×SDS loading buffer for 15 min at 95 °C. About 10 μg EV-samples and 4 μL protein ladder (Thermo Scientific) were loaded on 12% polyacrylamide gels and separated by electrophoresis. The following step is to transfer the proteins in gels to PVDF membranes (Millipore). For CD63 and TSG101, the transfer process lasted 90 min; for HSP70, 100 min; and for calnexin, 120 min. The membranes were blocked using 5% BSA in 1×TBST for 1 h at 4 °C and then incubated with primary antibodies overnight at 4 °C. The primary antibodies used for the western blotting are as follows: CD63 (ABclonal, catalog number A19023, clone ARC51703, lot number 3522110801, 1:500), TSG101 (ABclonal, catalog number A5789, clone ARC0853, lot number 4000000853, 1:1000), HSP70 (ABclonal, catalog number A23457, clone ARC53082, lot number 5500012382, 1:1000), calnexin (ABclonal, catalog number A4846, clone ARC0648, lot number 4000000648, 1:1000). After that the membranes were washed four times with 1×TBST and incubated with HRP-labeled secondary antibodies, goat anti-rabbit IgG (proteintech, catalog number, SA00001-2, 1:5000) for 1 h at 4 °C, then washed the membranes for five times. Finally, the signals were developed by chemiluminescence substrate (Beyotime) and captured using the bio-imaging system (Biorad).
Lysis of EVs and DNA quantification of EV-DNA
The collected EV solutions were added 1 μL Thermoliable dsDNase (Byotime) and reacted at 37 °C for 20 min, and the Thermoliable dsDNase was inactivated at 55 °C for 10 min. This step is to remove the dsDNA on the EV surface. After the removal of surface dsDNA, the EV solutions were added 1 μL Thermolabile Proteinase K (Thermo Scientific). The EVs were lysed at 37 °C for 3 h, 55 °C for 20 min, and 80 °C for 10 min. The relative quantity of EV-DNA was quantified by quantitative PCR (qPCR) using the GAPDH gene. The forward primer of GAPDH is 5’-TGCACCACCAACTGCTTAGC-3’, and the reverse primer of GAPDH is 5’-GGCATGGACTGTGGTCATGAG-3’.
The assembly of 5mC-Tn5 with symmetric adapter
The adapter sequence with a Mosaic End that can be recognized by transposase was synthesized with all cytosines methylated. The sequences are as follows: MErev, 5’- [phos] CTGTCTCTTATACACATCT-3’; 5mC-ME-A 5’- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3’. The two oligonucleotides were annealed at 40 μM in 1 × annealing buffer in a program of 95 °C for 2 min, then slowly ramped down to 25 °C at a rate of −1.5 °C/min. After that, 8.25 μL Tn5 transposase (Abclonal) and 2.62 μL of the annealed mix were mixed in assemble buffer and incubated at 35 °C for 2 h. The assembled 5mC-Tn5 was stored at −20 °C.
Preparation of spike-in DNAs in EV-DNA sequencing
The positive control of spike-in DNA was prepared by methylation of unmethylated λ-DNA (Promega) in a 50 μL reaction. Briefly, 1 μg unmethylated λ-DNA was added 1 μL S-adenosylmethionine (SAM, 32 mM), 2 μL CpG Methyltransferase (M.SssI, NEB M0226S) in 1 × Mg2+ free buffer. This reaction was incubated at 37 °C for 4 hours, and 1 μL SAM and 0.5 μL M.SssI were added midway through the reaction. After the reaction, the λ-DNA was purified using 1.0 × VAHTS DNA clean beads (Vazyme). The above steps were repeated once, and the full CpG methylated λ-DNA was quantified by Qubit dsDNA HS Assay (Thermo Scientific). The negative control of spike-in DNA was prepared by PCR amplification. The pNIC28-Bsa4 plasmid was used as a template for a 2 kb unmodified amplicon. The forward primer is 5’-CACAGATGTCTGCCTGTTCA-3’, and the reverse primer is 5’-AGGGTGGTGAATGTGAAACC-3’.
Enzymatic conversion of EV-DNA and library preparation
Due to the low DNA concentration in EVs, the EV lysate was directly used for library construction to ensure optimal DNA utilization.
-
(1)
The tagmentation reaction was carried out in a 20 μL reaction. The mixture contained 14 μL EV lysate, 4 μL 40% PEG8000, and 1 μL 5mC-Tn5 (20-fold diluted). To measure the conversion rate of the reaction, 1 μL spike-in DNAs with positive control and negative control were added to the reaction, with final concentrations of 1% and 0.025% (w/w), respectively.
-
(2)
The tagmentation reaction was incubated at 55 °C for 10 min. Then, the 5mC-Tn5 was removed by adding 1 μL Thermolabile Proteinase K (Thermo Scientific) and reacted at 37 °C, 10 min, 55 °C 10 min. The fragmented DNA was purified using 1.8 × VAHTS DNA clean beads. To increase the recovery rate of DNA during purification, 100 ng carrier RNA was added.
-
(3)
The enzymatic conversion of EV-DNA was performed using an EpiArt DNA enzymatic methylation kit (Vazyme, EM301). The DNA with 5’-end tags was oxidized in 50 μL reaction with 5 μL oxidation reaction buffer, 1 μL oxidation reactions, 1 μL oxidation reagent, 10 μL oxidation enzyme 1, 1 μL oxidation enzyme 2, and 5 μL Fe2+ solution at 37 °C for 1 h. The reaction was terminated using 1 μL stop reagent and incubated at 37 °C for 30 min. Then, the DNA was purified with 1.8 × beads and eluted in 16 μL RNase-free H2O. The oxidated DNA was denatured by adding 4 μL 0.1 M NaOH and incubated at 50 °C for 10 min, and the tube was placed on ice for 2 min after heating to maintain the DNA in a single-stranded state. The deamination reaction was performed in 100 μL reaction with 10 μL DEA reaction buffer, 1 μL BSA, and 1 μL DEA enzyme mix at 37 °C for 3 h. The reaction was purified using 1.8 × beads and eluted in 10 μL RNase-free H2O.
-
(4)
The 3’ adapter was ligated to converted DNA (Vazyme, NE103). The ligation of the 3’ adapter was performed in a 20 μL reaction with 5 μL ligation buffer, 2.5 μL ligation enzyme mix, and 2.5 μL 3’ adapter at 37 °C for 15 min and 95 °C for 2 min. Then, 2.5 μL extension primer and 17.5 μL extension enzyme mix were added to the solution following the program of 98 °C 1 min, 62 °C 2 min, and 65 °C 5 min. Afterwards, the DNA was purified with 1.2 × beads and eluted in 22 μL RNase free H2O.
-
(5)
The PCR amplification (16-17 cycles) of converted DNA was performed in a 50 μL reaction with 25 μL VAHTS HiFi amplification mix V3, 2 μL N5XX (Vazyme, TD202), and 1 μL DM7XX (Vazyme, N321). The DNA was purified with 0.8 × beads twice. The final concentration was quantified using Qubit dsDNA HS Assay (Thermo Scientific).
Deep sequencing of EV-DNA library
The amplified DNA libraries were qualified by Tapestation 4200 (Agilent) with the average size between 400–500 bp. Considering the DNA libraries were base unbalanced; the pools were sequenced on an Illumina NavoPE150 System with a 10% PhiX spike-in to improve the base complexity.
Sequencing data preprocessing
The raw sequencing data underwent quality control using FastQC (version 0.12.1, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Subsequently, the raw sequenced reads were processed using Trim Galore (version 0.6.10, https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) to trim low-quality bases and adapters, employing the following parameters: --paired --length 35 --clip_R1 15 --clip_R2 10 --three_prime_clip_R1 10 --three_prime_clip_R2 10. Bismark70 (version 0.24.2, https://www.bioinformatics.babraham.ac.uk/projects/bismark/) was used to align the trimmed reads to the reference genome, including spike-in sequences and the human reference genome (GRCh38). Next, deduplication was performed using the Bismark Deduplicate function. CpG methylation sites were computed and extracted from the deduplicated BAM files using bismark_methylation_extractor with default parameters.
Technical quality control
As a positive control (lambda DNA), the non-conversion rate was calculated using the formula T/(C + T). For the negative control (2 kb), the conversion rate was calculated using the formula T/(T + C). Samples with a conversion rate exceeding 99% (2 kb) and a non-conversion rate less than 4% (lambda DNA) were kept for downstream analysis.
Differentially methylated region (DMR) calling
To detect differentially methylated loci, differentially methylated regions (DMRs) between cancer and benign samples in the discovery and validation cohorts were identified using the DSS71 R package (version 2.46.0). Our discovery cohort consists of 21 cancer samples and 17 benign samples, and our validation cohort has 11 cancer and 9 benign samples (Supplementary Data 1 and 6). Regions were selected based on a delta threshold of 0.15 and a p-value cutoff of 0.05 between the two groups. The callDMR function from the DSS package was used to identify statistically significant CpG sites and merge adjacent loci to form DMRs. No minimum CpG coverage filter was applied, as DSS inherently accounts for sequencing depth when calculating differentially methylated loci.
Correlation on DNA methylation and feature enrichment
The methylation levels of each DMR were calculated by dividing the total number of reads with methylated CpG sites by the total number of reads in this region. To compare the methylation profiles between samples, we calculated the Pearson correlation coefficients between the DMR methylation levels of each sample pair. To identify the potential regulatory roles of the cancer-specific or stage-specific DMRs, we mapped them to genomic features and conducted the Fisher test. We used two sets of annotations of genomic features. The first set containing regulatory elements was downloaded from the Ensembl72 website (ftp://ftp.ensembl.org/pub/release97/regulation/homo_sapiens/homo_sapiens.GRCh38.Regulatory_Build.regulatory_features.20190329.gff.gz). The second set contains CpG islands, downloaded from the UCSC73 website (http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/cpgIslandExt.txt.gz).
Cancer state prediction using DMR methylation signatures
For the discovery cohort, the predictive model was trained and evaluated using five-fold cross-validation. Samples were randomly divided into five folds, with onefold designated as the testing set and the remaining folds used for model training. DMRs were called using DSS in the training set. The predictive model was then fitted on these DMRs using random forest and tested on the rest folds. This process was repeated five times. Prediction scores of test samples from the random forest model in five-fold cross-validation were collected, and receiver operating characteristic (ROC) curves and AUC were calculated using R package ROCR74. For the independent validation cohort, the predictive model was trained and evaluated based on nine-fold cross-validation due to the smaller sample size.
Functional analysis of DMRs
DMRs were annotated to the nearby genes using Homer (version 4.11)37, and the Gene transfer format (GTF) file was filtered to keep only protein-coding genes. Gene Ontology (GO) enrichment analysis was performed on these annotated genes using the EnrichR49 online tool.
Tissue deconvolution analysis
To investigate the tissue origin of EV-DNA methylation signature, we performed the deconvolution analysis using NNLS75. We used a reference matrix that captured the tissue-specific DNA methylation patterns, emphasizing enhancer regions. This reference matrix was constructed from CpG methylation data derived from 144 publicly available Whole Genome Bisulfite Sequencing (WGBS) samples from tissues and blood cells6. These samples in the CpG methylation data were categorized into 32 physiologically distinct tissue and blood cell types.
Given the tissue reference matrix59 \(A\) and a vector of observed methylation ratios \({y}_{s}\) in a sample \(s\), the tissue contribution \(x\) was estimated by solving the following minimization problem
subject to \(x\ge\)0. To quantify the cell-type-specific relative abundance, we calculated the average proportions cross GP or GC samples for each cell type. We calculated the contribution changes by dividing the average contribution in GC samples by that in GP samples.
Statistics and reproducibility
No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Ethics approval and consent to participate
This study was conducted in accordance with all relevant ethical regulations and was approved by the Medical Ethics Committee of Zhongnan Hospital of Wuhan University, No. 2022245. Informed consent was obtained from all participants prior to their inclusion in the study, and participants were made aware of the inclusion of three or more indirect identifiers in the Supplementary Data.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequencing data generated in this study have been deposited in the Genome Sequence Archive76 at the National Genomics Data Center77, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human) under accession code HRA008519 (https://ngdc.cncb.ac.cn/gsa-human/). Access to these data was permitted by the Ministry of Science and Technology of China (Registration Number: 2025BAT00885). The raw sequencing data are available under restricted access due to consent limitations. Data access requests should be directed to Zhiyuan Hu (zhiyuan.hu@whu.edu.cn). Applicants must have obtained ethics approval from their institutional ethics committees. Access is typically granted within approximately one month, and there are no restrictions on the duration of access once granted. The processed data generated in this study are provided in the Supplementary Data and the Source Data file. Source data are provided with this paper.
Code availability
The code of the current study is available through zenodo78: https://doi.org/10.5281/zenodo.16624305.
References
Ignatiadis, M., Sledge, G. W. & Jeffrey, S. S. Liquid biopsy enters the clinic — implementation issues and future challenges. Nat. Rev. Clin. Oncol. 18, 297–312 (2021).
Song, P. et al. Limitations and opportunities of technologies for the analysis of cell-free DNA in cancer diagnostics. Nat. Biomed. Eng. 6, 232–245 (2022).
Cohen, J. D. et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926–930 (2018).
Shen, S. Y. et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563, 579–583 (2018).
Moss, J. et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 9, 5068 (2018).
Mouliere, F. et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 10, eaat4921 (2018).
Xu, R. et al. Extracellular vesicles in cancer — implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 15, 617–638 (2018).
Geeurickx, E. et al. The generation and use of recombinant extracellular vesicles as biological reference material. Nat. Commun. 10, 3288 (2019).
Malkin, E. Z. & Bratman, S. V. Bioactive DNA from extracellular vesicles and particles. Cell Death Dis. 11, 584 (2020).
Tsering, T., Nadeau, A., Wu, T., Dickinson, K. & Burnier, J. V. Extracellular vesicle-associated DNA: ten years since its discovery in human blood. Cell Death Dis. 15, 668 (2024).
Maire, C. L. et al. Genome-wide methylation profiling of glioblastoma cell-derived extracellular vesicle DNA allows tumor classification. Neuro Oncol. 23, 1087–1099 (2021).
Yokoi, A. et al. Mechanisms of nuclear content loading to exosomes. Sci. Adv. 5, eaax8849 (2019).
Kim, K.-A. et al. Genome-wide methylation profiling reveals extracellular vesicle DNA as an ex vivo surrogate of cancer cell-derived DNA. Sci. Rep. 14, 24110 (2024).
Moran, S., Arribas, C. & Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 8, 389–399 (2016).
Clark, S. J., Statham, A., Stirzaker, C., Molloy, P. L. & Frommer, M. DNA methylation: bisulphite modification and analysis. Nat. Protoc. 1, 2353–2364 (2006).
Casanova-Salas, I. et al. Circulating tumor extracellular vesicles to monitor metastatic prostate cancer genomics and transcriptomic evolution. Cancer Cell 42, 1301–1312.e7 (2024).
Du, Y. et al. Bisulfite-free mapping of DNA cytosine modifications: challenges and perspectives. Sci. CHINA Chem. 66, 3044–3053 (2023).
Liu, Y. et al. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 37, 424–429 (2019).
Sun, Z. et al. Nondestructive enzymatic deamination enables single-molecule long-read amplicon sequencing for the determination of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Genome Res. 31, 291–300 (2021).
Vaisvila, R. et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res. 31, 1280–1289 (2021).
An, Y. et al. DNA methylation analysis explores the molecular basis of plasma cell-free DNA fragmentation. Nat. Commun. 14, 287 (2023).
Eisenstein, M. Oxford Nanopore announcement sets sequencing sector abuzz. Nat. Biotechnol. 30, 295–296 (2012).
Rhoads, A. & Au, K. F. PacBio sequencing and its applications. Genomics Proteom. Bioinform. 13, 278–289 (2015).
Adey, A. & Shendure, J. Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing. Genome Res. 22, 1139–1143 (2012).
Adey, A. C. Tagmentation-based single-cell genomics. Genome Res. 31, 1693–1705 (2021).
Mulqueen, R. M. et al. High-content single-cell combinatorial indexing. Nat. Biotechnol. 39, 1574–1580 (2021).
Mulqueen, R. M. et al. Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol. 36, 428–431 (2018).
Wu, J., Dai, W., Wu, L. & Wang, J. SALP, a new single-stranded DNA library preparation method especially useful for the high-throughput characterization of chromatin openness states. BMC Genomics 19, 143 (2018).
Liu, H. et al. Analysis of extracellular vesicle DNA at the single-vesicle level by nano-flow cytometry. J. Extracell. Vesicles 11, e12206 (2022).
Möller, A. & Lobb, R. J. The evolving translational potential of small extracellular vesicles in cancer. Nat. Rev. Cancer 20, 697–709 (2020).
Cao, Y. et al. Single-cell bisulfite-free 5mC and 5hmC sequencing with high sensitivity and scalability. Proc. Natl Acad. Sci. USA 120, e2310367120 (2023).
Liang, N. et al. Ultrasensitive detection of circulating tumour DNA via deep methylation sequencing aided by machine learning. Nat. Biomed. Eng. 5, 586–599 (2021).
Tian, Y. et al. Quality and efficiency assessment of six extracellular vesicle isolation methods by nano-flow cytometry. J. Extracell. Vesicles 9, 1697028 (2020).
Jeppesen, D. K. et al. Reassessment of exosome composition. Cell 177, 428–445.e18 (2019).
Guo, S. et al. The detection, biological function, and liquid biopsy application of extracellular vesicle-associated DNA. Biomark. Res. 12, 123 (2024).
Li, L. et al. Integrative proteogenomic characterization of early esophageal cancer. Nat. Commun. 14, 1666 (2023).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Xiong, D.-D., Zeng, C.-M., Jiang, L., Luo, D.-Z. & Chen, G. Ki-67/MKI67 as a predictive biomarker for clinical outcome in gastric cancer patients: an updated meta-analysis and systematic review involving 53 studies and 7078 patients. J. Cancer 10, 5339–5354 (2019).
Jun, K. H., Kim, S. Y., Yoon, J. H., Song, J. H. & Park, W. S. Amplification of the UQCRFS1 gene in gastric cancers. J. Gastric Cancer 12, 73–80 (2012).
Kandimalla, R. et al. EpiPanGI Dx: a cell-free DNA Methylation fingerprint for the early detection of gastrointestinal cancers. Clin. Cancer Res. 27, 6135–6144 (2021).
Garmendia, I., Redin, E., Montuenga, L. M. & Calvo, A. YES1: a novel therapeutic target and biomarker in cancer. Mol. Cancer Ther. 21, 1371–1380 (2022).
Sung, B. H., Ketova, T., Hoshino, D., Zijlstra, A. & Weaver, A. M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 6, 7164 (2015).
Purushothaman, A. et al. Fibronectin on the surface of myeloma cell-derived exosomes mediates exosome-cell interactions. J. Biol. Chem. 291, 1652–1663 (2016).
Ye, C. et al. Src family kinases engage differential pathways for encapsulation into extracellular vesicles. J. Extracell. Biol. 2, e96 (2023).
Takasugi, M. et al. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 8, 15729 (2017).
Xiao, Y. et al. Myofibroblast-derived extracellular vesicles facilitate cancer stemness of hepatocellular carcinoma via transferring ITGA5 to tumor cells. Mol. Cancer 23, 262 (2024).
Jin, Y. et al. DNA in serum extracellular vesicles is stable under different storage conditions. BMC Cancer 16, 753 (2016).
Kinoshita, T., Yip, K. W., Spence, T. & Liu, F.-F. MicroRNAs in extracellular vesicles: potential cancer biomarkers. J. Hum. Genet. 62, 67–74 (2017).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–W97 (2016).
Geraldo, L. H. M. et al. Role of lysophosphatidic acid and its receptors in health and disease: novel therapeutic strategies. Signal Transduct. Target. Ther. 6, 45 (2021).
Sung, B. H., Parent, C. A. & Weaver, A. M. Extracellular vesicles: critical players during cell migration. Dev. Cell 56, 1861–1874 (2021).
Tong, M., Jun, T., Nie, Y., Hao, J. & Fan, D. The role of the slit/robo signaling pathway. J. Cancer 10, 2694–2705 (2019).
Xu, J., Liao, K. & Zhou, W. Exosomes regulate the transformation of cancer cells in cancer stem cell homeostasis. Stem Cells Int. 2018, 4837370 (2018).
Al-Nedawi, K., Meehan, B., Kerbel, R. S., Allison, A. C. & Rak, J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl Acad. Sci. USA 106, 3794–3799 (2009).
Rodríguez, M. et al. Exosomes enriched in stemness/metastatic-related mRNAS promote oncogenic potential in breast cancer. Oncotarget 6, 40575–40587 (2015).
Buzas, E. I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol. 23, 236–250 (2023).
Torralba, D. et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat. Commun. 9, 2658 (2018).
Cheng, A. P. et al. A cell-free DNA metagenomic sequencing assay that integrates the host injury response to infection. Proc. Natl Acad. Sci. USA 116, 18738–18744 (2019).
Siejka-Zielińska, P. et al. Cell-free DNA TAPS provides multimodal information for early cancer detection. Sci. Adv. 7, eabh0534 (2021).
Pellicci, D. G., Koay, H.-F. & Berzins, S. P. Thymic development of unconventional T cells: how NKT cells, MAIT cells and γδ T cells emerge. Nat. Rev. Immunol. 20, 756–770 (2020).
Elzanowska, J., Semira, C. & Costa-Silva, B. DNA in extracellular vesicles: biological and clinical aspects. Mol. Oncol. 15, 1701–1714 (2021).
Shi, A. et al. Plasma-derived extracellular vesicle analysis and deconvolution enable prediction and tracking of melanoma checkpoint blockade outcome. Sci. Adv. 6, eabb3461 (2020).
Wen, C. et al. Biological roles and potential applications of immune cell-derived extracellular vesicles. J. Extracell. Vesicles 6, 1400370 (2017).
Karimi, N., Dalirfardouei, R., Dias, T., Lötvall, J. & Lässer, C. Tetraspanins distinguish separate extracellular vesicle subpopulations in human serum and plasma – Contributions of platelet extracellular vesicles in plasma samples. J. Extracell. Vesicles 11, e12213 (2022).
Nieuwland, R. & Siljander, P. R. A beginner’s guide to study extracellular vesicles in human blood plasma and serum. J. Extracell. Vesicles 13, e12400 (2024).
Feng, X. et al. High-throughput capture and in situ protein analysis of extracellular vesicles by chemical probe-based array. Nat. Protoc. 20, 1057–1081 (2025).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Ghanam, J. et al. Extracellular vesicles transfer chromatin-like structures that induce non-mutational dysfunction of p53 in bone marrow stem cells. Cell Discov. 9, 12 (2023).
Welsh, J. A. et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 13, e12404 (2024).
Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011).
Feng, H. & Wu, H. Differential methylation analysis for bisulfite sequencing using DSS. Quant. Biol. 7, 327–334 (2019).
Zerbino, D. R., Wilder, S. P., Johnson, N., Juettemann, T. & Flicek, P. R. The ensembl regulatory build. Genome Biol. 16, 56 (2015).
Fujita, P. A. et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 39, D876–D882 (2011).
Sing, T., Sander, O., Beerenwinkel, N. & Lengauer, T. ROCR: visualizing classifier performance in R. Bioinformatics 21, 3940–3941 (2005).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Chen, T. et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics Proteom. Bioinform. 19, 578–583 (2021).
CNCB-NGDC Members and Partners Database resources of the National Genomics Data Center, China National Center for Bioinformation in 2024. Nucleic Acids Res. 52, D18–D32 (2024).
Jiao, Z. & Hu, Z. zhiyuan-hu-lab/TEMPT-paper: The first release of TEMPT paper. Zenodo https://doi.org/10.5281/ZENODO.16624305 (2025).
Acknowledgements
This work was supported by the National Key R&D Program of China (2023YFC3402200 to X.Z.), National Natural Science Foundation of China (22307101 to B.L.), National Key R&D Program of China (2022YFA1303500 to X.Z.), National Natural Science Foundation of China (92153303 to X.Z., 22307099 to Y.L.), Zhongnan Hospital of Wuhan University Science and Technology Achievement Transformation Fund, Project 202225KJCGZH to H.W., Fundamental Research Funds for the Central Universities (2042025kf0022, 2042022dx0003) to Z.H. We would like to thank Dr. Jingfei Cheng from University of Oxford for sharing the tissue reference data of matrix A.
Author information
Authors and Affiliations
Contributions
B.L. designed the study, performed the experiments, and analyzed the data. Z.J. carried out the bioinformatic analysis and contributed to data interpretation. S.D. collected the samples, contributed to validation. B.L., Z.J., and S.D. wrote the manuscript. W.Y., J.J., and Y.D. assisted with methodology development and data curation. X.W. and H.W. contributed to validation and data processing. Z.H. supervised the bioinformatic analysis and provided resources. Y.L. and X.Z. conceived the study and supervised the work. All authors contributed to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
X.Z., B.L., Y.L., Z.H., and W.Y. are inventors on a Chinese patent application related to the method described in this study (application number: 202411361596.8). The patent application was filed by the authors and is currently pending. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Abu Ali Ibn Sina and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, B., Jiao, Z., Dong, S. et al. Whole-genome methylation profiling of extracellular vesicle DNA in gastric cancer identifies intercellular communication features. Nat Commun 16, 8084 (2025). https://doi.org/10.1038/s41467-025-63435-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63435-w
