
Abstract
Oncogenic KRAS is amongst the key genetic drivers for initiation and maintenance of pancreatic ductal adenocarcinoma (PDAC). Here, we show that engineered exosomes with KrasG12D specific siRNA (iExoKrasG12D) reveal a biodistribution in pancreas with negligible toxicity in preclinical studies in mice and Rhesus macaques. Clinical testing of iExoKrasG12D in the iEXPLORE (iExoKrasG12D in Pancreatic Cancer) Phase I study employed a non-randomized single-arm classical 3 + 3 dose escalation design (Phase Ia), followed by an accelerated titration design (Phase Ib) (NCT03608631). The primary outcomes included safety, tolerability and target engagement, and the secondary outcomes aimed to assess disease control. Patients with advanced metastatic disease were enrolled after failure of multiple lines of therapy. iExoKrasG12D therapy was well-tolerated: the primary outcomes were met with iExoKrasG12D showing no dose-limiting toxicity. The maximum tolerated dose was not reached even at the highest dose. In some cases, iExoKrasG12D therapy was associated with stable disease response (secondary outcome). Downregulation of KRASG12D DNA and suppression of phospho-Erk was documented together with an increase in intratumoral CD8+ T cells following treatment. The CD8+ T cell recruitment priming by iExoKrasG12D informed on potential efficacy of immune checkpoint therapy and lead to validation testing in preclinical PDAC models. Combination therapy of iExoKrasG12D and anti-CTLA-4 antibodies, but not anti-PD1, revealed robust pre-clinical anti-tumor efficacy via FAS mediated CD8+ T cell anti-tumor activity. This first-in-human, precision medicine clinical trial and supporting preclinical functional studies offer new insights into priming of immunotherapy by oncogenic Kras inhibitor for future opportunistic combination therapy for PDAC patients.
Similar content being viewed by others


Introduction
Pancreatic ductal adenocarcinoma (PDAC) diagnosis is near uniformly lethal, with a median survival of less than 12 months1,2, despite the availability of combination chemotherapy regimens. The pivotal driver oncogenic event in PDAC is mutant KRAS, implicated in >90% of tumors, with KRASG12D found in half of PDAC cases with mutant KRAS3,4,5. Genetically engineered mice (GEM) modeling PDAC have demonstrated the prerequisite for mutant Kras in the initiation of PDAC, as well as the remarkable dependence of oncogenic Kras for cancer maintenance and extinction. In advanced stages of PDAC in mice, genetic extinction of mutant Kras resolves tumors and rescues mice from PDAC lethality6. We recently reported on the cancer cell expression of FAS death receptor and permissive FASL+CD8+ T cell-mediated anti-tumor response with Kras extinction in PDAC GEM6. Specifically, the suppression of oncogenic Kras in PDAC GEM relieved the epigenetic suppression of FAS death receptor on cancer cells, facilitating CD8+ T cell FASL-FAS mediated eradication of cancer cells6. Additionally, pharmacological inhibition of KrasG12D with an allele-specific small molecule inhibitor (MRTX1133) also enabled CD8+ T cell targeting of cancer cells re-expressing FAS, in both GEM and patient-derived organoids7. Combination therapy with MRTX113 and CTLA-4 antibodies showed significant survival advantage compared to controls, supporting synergistic anti-tumor efficacy for oncogenic Kras targeting with immune checkpoint blockade (ICB) therapy7. We and others also reported on the role of oncogenic Kras in shaping the suppressive metabolic and immune tumor microenvironment of PDAC8,9,10 generating hypotheses for possible synergistic opportunities with immunotherapies. Collectively, these studies offered insight into the failure of ICB in patients with PDAC – that the lack of clinical efficacy observed with ICB in patients with PDAC may be attributable to intrinsic and microenvironmental hurdles requiring specific combination therapy to sensitize PDAC to the therapeutic impact of ICB. A critical aspect in overcoming ICB ineffectiveness would include strategies of sustained and specific suppression of oncogenic Kras expression to generate lasting impact concurrent with ICB, while avoiding off-target collateral effects.
Although oncogenic Kras is recognized as a desirable therapeutic target in PDAC due to the near ubiquitous prevalence of “hotspot” mutations, it was dubbed the “undruggable” target due to its challenging chemistry11. More recently, groundbreaking oncogenic KRAS targeting with new direct small molecule inhibitors have made their way into clinical testing12, with long-awaited hope for enhancing PDAC survival. Despite the observed efficacy of monotherapy with available inhibitors, patients eventually develop resistance via pleiotropic mechanisms, and most experience at least some measure of treatment-related adverse events (most commonly, cutaneous and gastrointestinal)13,14. Keeping in mind the numerous challenges of targeting oncogenic Kras, we devised an alternative strategy that employed exosomes as a delivery vehicle for siRNA targeting. Exosomes or small extracellular vesicles (sEVs) are naturally produced lipid bilayer vesicles with the topography of their cell of origin, ranging in size from 40–200 nm, and with a regulated cargo that reflects cellular constituents15. Exosomes are a complex constituent of the systemic circulation and were demonstrated to likely play a role in inter-cellular communication15,16. The siRNA sequences used were previously reported to target oncogenic Kras rather than wild-type Kras, and we confirmed their specificity17. This effort was motivated by the observation that exogenously administered exosomes in mice accumulated in the pancreas and, when containing a KrasG12D siRNA payload, significantly suppressed Kras signaling, resulting in increased survival of PDAC GEM with KrasG12D mutation18. We coined these therapeutic exosomes “iExoKrasG12D”. We further demonstrated that iExoKrasG12D anti-tumor efficacy in mice relied on multiple aspects of exosomal biology, including their favored uptake in oncogenic Kras-expressing cells via macropinocytosis18. Our studies also identified that the CD47 “don’t eat me” signal on iExoKrasG12D enabled superior systemic half-life compared to synthetic particles, with an impact on the efficacy of the siRNA therapeutic payload in PDAC GEM18. Challenges for personalized therapy with exosomes included good manufacturing practice (GMP) and large-scale production of a clinically defined product. We reported on the feasibility of iExoKrasG12D GMP production for human testing19, which led to approval for first in human clinical testing.
Here, we tested the safety of iExoKrasG12D in murine and non-human primates (NHP) and we describe the results from the Phase I trial of iExoKrasG12D monotherapy in advanced PDAC. We designed a 3+ 3 clinical trial with escalating doses of iExoKrasG12D for metastatic PDAC with standard of care therapy failure (Phase Ia). Lack of dose-limiting toxicities (DLT) informed the follow-up accelerated dosing strategy, again yielding no measurable toxicity (Phase Ib). We also report on studies with patient-derived biopsies that informed on a potential synergistic combination of iExoKrasG12D with immune checkpoint blockade, specifically anti-CTLA-4, and subsequent murine studies validating this synergistic approach for Phase II trial design. Collectively, our preclinical studies, guided by the clinical trial, inform on a novel combination therapy of oncogenic Kras targeting with anti-CTLA-4 immune checkpoint blockade for robust anti-tumor response with potential to overcome resistance.
Results
GMP grade exosomes for siRNA delivery showed no toxicity in preclinical studies
The oncogenic Kras targeting approach we devised employed exosomes purified from the culture supernatant of bone marrow derived stromal cells from a healthy donor (Fig. 1a). The purified exosomes were subjected to electroporation for the incorporation of siRNA with a sequence targeting KRASG12D (Fig. 1A). The resulting exosomes were then processed for quality control (QC), which included size assessment by NanoSightTM (Supplementary Fig. 1A) and flow cytometry analyses for CD47 and putative exosomal marker CD63 (Supplementary Fig. 1B, C). Following electroporation with siRNA, the exosomes were referred to as ‘iExoKrasG12D’. Additional validation with exosomes markers, CD9 and CD81, are also provided (Supplementary Fig. 1C). The iExoKrasG12D were then quantified using microBCA assay to define the exosomal protein content used for dosing definition (discussed below). The siRNA content in the iExoKrasG12D equaled that of the exosomal protein content (1:1 by weight). We previously detailed the GMP, large-scale production of iExoKrasG12D, including quality control assessment and preclinical testing showing anti-tumor effect with increased survival in PDAC GEM19. Scanning electron microscopy of iExoKrasG12D showed a distribution of exosomes (Supplementary Fig. 1D) consistent with NanoSightTM size distribution measurements. Cryogenic electron microscopy (Cryo-EM) revealed iExoKrasG12D lipid bilayer vesicles, some of which showed multiple vesiculation (Fig. 1B, Supplementary Fig. 1E), consistent with recent reporting20. Keeping in mind the clinical utility of GMP iExoKrasG12D, we determined the toxicology profile of GMP iExoKrasG12D in mice and non-human primates (Rhesus macaques). Three cycles of iExoKrasG12D treatment administered intravenously, 3 doses per cycle for a total of 9 doses over the course of 6 weeks, was evaluated in healthy adult mice (Fig. 1C). A comprehensive toxicology profile was established, evaluating diluent for iExoKrasG12D (PlasmaLyte) and increasing iExoKrasG12D doses (1X, 2X, and 4X, Table 1). Body weight showed no changes with any of the dose levels (Fig. 1D, Supplementary Fig. 2A), and liver and kidney weights were unchanged (Supplementary Fig. 2B). Although a significant % change in spleen weight was registered for 1X iExoKrasG12D, no significant changes were noted for higher doses (Supplementary Fig. 2B). Comprehensive chemistry and hematology panels revealed no changes outside physiological ranges (Supplementary Fig. 2C, D). When significant changes were observed when compared to the control group, those changes also remained within normal physiological range (Supplementary Fig. 2C, D).

A Schematic representation of iExoKrasG12D GMP production. Conditioned media (CM) from mesenchymal stromal cells (MSC) cultured in GMP was centrifuged to remove debris, filtered through 0.2 μm filter, and ultracentrifuged. The exosomes pellet was resuspended, subjected to electroporation to incorporate siRNA targeting KRASG12D using a 1:1 ratio (by mass) of exosomal protein and siRNA, and evaluated using quality control (QC) analysis for size distribution (mean size <400 nm) and exosome surface detection of CD47 and CD63 by flow cytometry. B Representative picture of cryo-electron microscopy of GMP iExoKrasG12D. Scale bar: 100 nm. This experiment was performed once. C Schematic representation of a murine toxicology study using intravenous (i.v.) GMP iExoKrasG12D treatment (Rx) for three consecutive cycles. Each cycle represents two weeks (W1: week 1; W2: week 2, etc.) and three iExoKrasG12D treatments on days 1, 4, and 10. D Body weight measurements over time for the listed groups of mice, PT: pretreatment. 1X, 2X, and 4X exosomes reflect dosing corresponding to clinical trial dosing, see also Table 1, n = 10 mice per group. Data are presented as mean +/– SD. E Schematic representation of NHP toxicology study using intravenous (i.v.) GMP iExoKrasG12D treatment (Rx) for three consecutive cycles. Each cycle represents two weeks (W1: week 1; W2: week 2, etc.) and iExoKrasG12D treatments on the listed days, distributing three iExoKrasG12D treatments over the course of 2 weeks. F Body weight measurements over time for the listed NHP. For dosing details, see Table 1. G Schematic representation of NHP biodistribution study using intravenous (i.v.) or intraperitoneal (i.p.) GMP iExoKrasG12D that was labeled with PKH-67 or DiR dye. H, I Representative fluorescence images (H) of pancreas and liver sections from NHP following PKH-67+ iExoKrasG12D treatment and associated quantification (I) of the density of the ratio of PKH67 level over nuclear staining (DAPI) in the listed organs. Scale bar: 10 μm. This experiment was performed once. J Normalized relative luminescence unit (RLU) in the listed organs for the detection of DiR+ label following i.v. or i.p. DiR+ iExoKrasG12D treatment. CNS: central nervous system; GI: gastrointestinal; Med. oblongata: medulla oblongata; LN: lymph node. Data also presented in part in Supplementary Fig. 6B. Source data is provided as Source Data file.
Toxicology studies were also evaluated in NHP, following a similar dosing strategy as planned for clinical testing (Fig. 1E, Table 1). Three Rhesus macaques were treated with nine doses of GMP iExoKrasG12D over the course of 6 weeks (Fig. 1E, Table 1). Body weight measurements were unchanged (Fig. 1F, Supplementary Fig. 3A). Organ weight, chemistry, and hematological studies were minimally or insignificantly altered (Supplementary Fig. 3B, C). Gross examination was unremarkable, and urine analysis and a detailed anatomic pathology evaluation revealed no lesions or histopathological changes. Furthermore, we investigated the biodistribution of exogenously administered GMP iExoKrasG12D in Rhesus macaques 24 hours following intravenous (i.v.) or intraperitoneal (i.p.) injections (Fig. 1G, Table 1). In this experimental design, PKH-67+ labeled iExoKrasG12D-infused NHP (i.v.) served as a control for DiR+ labeled iExoKrasG12D (and vice versa). PKH-67+ exosomes were visualized in pancreas, liver, and brain tissue sections with confocal and high-resolution fluorescence microcopy, with minimal positive PKH-67+ exosomes noted in kidney, lung, skin, spleen, and tongue (Fig. 1I, J, Supplementary Figs. 4–5). Histopathological examination of pancreas, liver, and brain was unremarkable (Supplementary Fig. 6A). Whole tissue imaging of DiR+ fluorescent signal in NHP administered with DiR+ labeled iExoKrasG12D i.v. vs i.p. showed positive signal in central nervous system (CNS) tissues, gastrointestinal tract, and immune organs, including lymph nodes (Fig. 1J), with minimal positive foci noted in kidney, lung, skin, spleen, and tongue (Supplementary Fig. 6B). While liver capture of DiR+ signal was greater following i.v. administration of iExoKrasG12D, omentum capture of DiR+ signal was prominent following i.p. administration (Fig. 1J). In this experimental setting however, high background autofluorescence of certain control tissues, including occipital and parietal lobes of the cortical brain, pancreas, and prostate, prevented accurate capture of DiR+ signal (Supplementary Fig. 6B). Quantification for KRASG12D siRNA by digital PCR analysis showed detection of siRNA from iExoKrasG12D in liver, pancreas, and bone marrow (Supplementary Fig. 7A, B). Levels of siRNA were higher in the liver when iExoKrasG12D were administered i.v., with the highest level of siRNA captured in the pancreas of NHP following i.v. iExoKrasG12D, albeit RNA degradation prevented comparative analysis of siRNA in one i.v. treated NHP pancreas (Supplementary Fig. 7A, B). Taken together, the results support iExoKrasG12D administration into mice and NHP is safe, with a biodistribution indicative of iExoKrasG12D accumulation in the pancreas and liver.
Systemic iExoKrasG12D treatment showed no measurable toxicity in dose escalation clinical trial
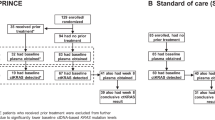
Clinical testing of iExoKrasG12D was initiated with a 3 + 3 dose escalation (Phase Ia) and enrolled 10 evaluable patients with metastatic pancreatic cancer at three dose levels (Fig. 2A-C). The three dose levels were equivalent to dose levels tested in toxicology studies in mice and NHP (Table 1). iExoKrasG12D dosing was defined using exosomal protein level (which contained an equivalent mass of siRNA payload [Table 1]). Patient eligibility included histologically confirmed metastatic PDAC with KRASG12D mutation (confirmed in tissue or blood), progression of disease on one or more line of systemic therapy in the metastatic setting (or stable disease with at least 4 months of chemotherapy with cytotoxic therapy), and Eastern Cooperative Oncology Group (ECOG) performance status of 0 and 1. Consented patients were at least 18 years of age, with negative pregnancy serum testing for women with childbearing potential and adequate contraception for males. Exclusion criteria included pregnancy, concurrent severe or uncontrolled stable angina, myocardial infarction within 6 months, symptomatic arrhythmia, uncontrolled diabetes, serious active infection, central nervous system disease (with the exception of treated brain metastasis). In Phase Ia, patients received 3 doses of iExoKrasG12D over the course of 2 weeks (1 cycle) for three consecutive cycles (Fig. 2D). Response was assessed by imaging using Response Evaluation Criteria in Solid Tumors (RECIST) 1.1, utilizing the Quantitative Imaging Analysis Core (QIAC), a centralized platform for diagnostic imaging at the MD Anderson Cancer Center. Dose-limiting toxicity grading followed the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE, Version 5.0). Six males and three females, with a range of 49 to 75 years of age, were enrolled (Table 2). All patients received multiple lines of prior systemic therapy and showed time from diagnosis to cancer-related death ranging from 1 year (or less) to 7 years (Table 2). Three patients, one each at dose level 1, 2, and 3, showed stable disease after 3 cycles and were enrolled in additional iExoKrasG12D treatment, per protocol (Table 2). One patient, enrolled in the dose 3 level, showed symptomatic progression of disease after receiving just one iExoKrasG12D infusion (less than 1 cycle) and was excluded from further analyses and replaced, given that rapid progression precluded continuation of 3 consecutive cycles (Table 2). All but one patient that completed at least 3 cycles of iExoKrasG12D therapy, including those with initial stable disease staging on iExoKrasG12D, eventually progressed with new lesions and/or target lesions (Table 2). S3D1 showed stable disease after completion of 6 cycles of iExoKrasG12D and was found subsequently to have progression of disease 3 months after completing the iExoKrasG12D trial. No related adverse events were noted for any patients at the dose levels tested in the 3 + 3 Phase Ia trial (Tables 2–3). Given lack of toxicity in all dose levels tested, a Phase Ib with accelerated dose titration was designed, with 3 additional patients enrolled at increased dose level for each patient (Fig. 2A, D). To ease patient infusion schedule, the accelerated titration Phase Ib study was revised with weekly administration, for a total of 6 infusions over the course of 6 weeks (Fig. 2D). Tissue biopsies were captured pre- and post- iExoKrasG12D treatment (Fig. 2D). Two females and one male in an age range of 48 to 76 years and with confirmed KRASG12D status were treated with increasing levels of iExoKrasG12D (Table 3). Timeline of disease progression, prior therapy, time of biopsy for mutation analysis, and visual display of target lesion monitoring indicated iExoKrasG12D therapy represented a short window of time compared to prior treatments (Fig. 3). All three patients showed stable disease and enrolled for additional iExoKrasG12D dosing before showing progression and growth of target lesions (Fig. 3, Table 4). Patient disease progression in this accelerated titration Phase Ib trial was between 1 (or less) to 2 years from cancer diagnosis to cancer related death (Table 3). Though patients demonstrated an ECOG status of 0 and 1 prior to enrollment, Phase Ib iExoKrasG12D treatment was administered to patients with 1-to-2 years disease progression from diagnosis to cancer related death (Fig. 3), and at 4-to-8-month range of time prior to cancer related death. In the Phase Ia/b clinical trial, iExoKrasG12D was well tolerated with no treatment-related adverse events reported and the maximum tolerated dose not reached (Tables 2, 4), thereby demonstrating a favorable safety profile. The highest dose level tested was 4.8 mg of iExoKrasG12D containing 4.8 mg of siRNA per dose, with 6 consecutive doses totaling 28.8 mg of iExoKrasG12D over the course of 12 weeks.

A Schematic representation of dose escalation design. DL1: dose level 1; DL 2: dose level 2; etc. B CONSORT flow diagram adapted for the non-randomized Phase Ia trial. C CONSORT flow diagram adapted for the non-randomized Phase Ib trial. D Schematic representation of dosing schedule. PR/SD: partial response/stable disease; bx: biopsy. Tissue biopsy optional at end cycle 3in Phase Ib.

Schematic representation of treatment timeline for patients in DL 4: dose level 4, DL 5: dose level 5, and DL5, and DL6: dose level 6, depicting prior therapy, timeframe for iExoKrasG12D therapy, and target lesion progression; scale bar listed. C1D1: Cycle 1 Day 1; C2D1: Cycle 2 Day 1; etc. QW: weekly; Dx: diagnosis; mo: month; Dt: death; [CT]: clinical trial; **: mutation analysis.
Systemic iExoKrasG12D treatment showed target engagement and remodeled tumor immune microenvironment
We previously showed GMP iExoKrasG12D targeting KrasG12D in PDAC GEM synergized with gemcitabine chemotherapy to limit tumor growth and increase survival19. We first conducted preclinical studies to determine if serial liquid biopsies for circulating KrasG12D DNA would serve as a surrogate for tumor response. Following orthotopic cancer cell injection, mice were monitored for tumor burden by IVIS imaging. Prior to treatment start (pre-tx), blood was collected (Fig. 4A). The level of circulating KrasG12D DNA was compared in blood collected prior to iExoKrasG12D treatment and 9 days after treatment, with treatment groups including gemcitabine, GMP iExoKrasG12D, or a combination of iExoKrasG12D and gemcitabine treated mice, and with control group including mice treated with diluent or control, non-electroporated exosomes (CE: control exosomes)19. As cancer progressed in the control group, the relative level of KrasG12D DNA (expressed as a percent of KrasG12D copies over total Kras copies) increased with tumor burden (Fig. 4B). Suppression of tumor growth, with gemcitabine, iExoKrasG12D, or combination thereof, tracked with lower relative levels of circulating KrasG12D (Fig. 4B). iExoKrasG12D therapy and combination therapy with iExoKrasG12D showed lowest relative levels of circulating KrasG12D (Fig. 4B), congruent with target specific engagement and anti-tumor response19. Mimicking the preclinical data, assessment for relative levels of circulating KrasG12D in patients treated with iExoKrasG12D in the Phase I trial demonstrated reduced mutant allele fraction in several patients, including S2D2, S1D3, S4D3, as well as S1D5 (Fig. 4C, D).

A Schematic representation of murine experimental timeline for tumor inoculation, IVIS imaging of tumor burden, and blood collection. B Relative level of KrasG12D cfDNA (expressed as a percent of KrasG12D copies over total Kras copies) in pre-treatment (pre-tx) and post-treatment (post-tx) with iExoKrasG12D and/or gemcitabine. Each line represents a distinct mouse per the listed experimental group: Control n = 6; iExoKrasG12D n = 4; gemcitabine n = 6; iExoKrasG12D + gemcitabine n = 7 mice. The data presented in panels A-B are analyses of blood samples from our earlier study19. C, D Relative level of KRASG12D cfDNA in the listed patients over time in the 3 + 3 (C) and accelerated dose titration (D) studies. PRE: pre-treatment; POST: post-treatment, with cycle definition at each time point evaluated. E. Representative H&E-stained tumor section from metastases in the abdominal wall (abdomen) or liver pre- and post-treatment with iExoKrasG12D. Scale bar:100 μm. This experiment was performed once. F Representative pERK-stained tumor section from metastases in the abdominal wall of S1D4, scale bar listed. This experiment was performed once. Source data is provided as Source Data file.
Tissue biopsies pre- and post-treatment were captured in the accelerated titration Phase Ib trial, and sections comprising of cancer cells and stromal cells, indicated by H&E staining (Fig. 4E), were selected for further immunolabeling studies. Down regulation of phosphorylated ERK in post-treatment samples, an indicator of RAS pathway activity (Fig. 4F), was accompanied by a decrease the number of PanCK+ cancer cells, and a stable or relatively increased in αSMA+ stromal cells (Fig. 5A, B). Analysis of T cell infiltrates revealed an increase in intratumoral CD8+ T cells, CD4+ Foxp3+ Tregs and CD4+ Foxp3− T cells post-treatment compared to pre-treatment (Fig. 5C-D, Supplementary Fig. 8A). Taken together, these results highlight the consistent target engagement of iExoKrasG12D in human PDAC with remodeling of the tumor immune microenvironment that likely favors to anti-tumor response, similar to that observed in murine PDAC treated with iExoKrasG12D6,7.

A, B Representative images of immunolabeling for pERK, PanCK, αSMA (A), with quantification of percentage of pERK+ panCK+ cancer cells (out of panCK+ cells) and αSMA+ pixels (B) in paired biopsies. This experiment was performed once. C, D Representative images (C), and quantification (D) of CD8+ T cells infiltrates in paired biopsies. Data are presented as mean. Scale bar:100 μm. Source data is provided as Source Data file.
CD8+ T cell recruitment and CD4+ T cells or Tregs depletion favored iExoKrasG12D therapy in preclinical models of PDAC
Informed by the clinical data therein, we tested the efficacy of iExoKrasG12D therapy in combination with immune checkpoint blockade to enhance overall anti-tumor response. GMP-iExoKrasG12D downregulated KRASG12D transcript in Panc-1 cells (Supplementary Fig. 8B). Given the increase in intratumoral CD8+ T cells and CD4+ Foxp3+ Tregs in clinical trial biopsies following iExoKrasG12D therapy, we next tested the functional contribution of these cells in PDAC following KrasG12D inhibition with iExoKrasG12D. T cell infiltration following KrasG12D inhibition in established tumors of the autochthonous KTC (P48-Cre, LSL-KrasG12D/+, Tgfbr2F/F)18 PDAC GEM was measured (Fig. 6A). Analysis of the intratumoral T cell populations revealed an increase in infiltration of CD4+ and CD8+ T cells in the PDAC tumor microenvironment (TME) following iExoKrasG12D treatment (Fig. 6B, C), consistent with the findings in clinical samples (Fig. 5C-D, Supplementary Fig. 8A). Similar results were also noted in a second orthotopic KPC-689 PDAC model treated with iExoKrasG12D18 (Supplementary Fig. 9A), resulting in increased tumor infiltrating CD4+ and CD8+ T cells with iExoKrasG12D treatment (Supplementary Fig. 9B). To determine the functional contribution of the T cell infiltrates following KrasG12D inhibition, we crossed the KPC (Pdx1-Cre, LSL-KrasG12D/+, Trp53R172H/+) GEM with CD4−/− or CD8−/− mice and generated KPC mice depleted of CD4+ T cells or CD8+ T cells, respectively. KPC, KPC CD4−/− and KPC CD8−/− mice were treated with iExoKrasG12D and tumor burden was monitored by serial MRI (Supplementary Fig. 9C, D). The control (no iExoKrasG12D) KPC, KPC CD4−/− and KPC CD8−/− mice showed similar tumor progression and survival kinetics (Supplementary Fig. 9D-E). KPC mice treated with iExoKrasG12D demonstrated prolonged survival compared to control KPC mice (Supplementary Fig. 9E), consistent with our prior findings18. KPC CD4−/− mice treated with iExoKrasG12D demonstrated robust tumor growth inhibition by MRI and prolonged survival compared to control KPC CD4−/− mice and iExoKrasG12D treated KPC mice (Supplementary Fig. 9D, E). In contrast, KPC CD8−/− iExoKrasG12D treated mice showed no survival benefit compared to the control KPC CD8−/− mice (Supplementary Fig. 9D, E), indicating that CD8+ T cells are essential for KrasG12D inhibition-mediated suppression of PDAC7. Taken together, our data demonstrate that depletion of CD4+ T cells/T regs prime KrasG12D inhibition and effector CD8+ T cells are required for PDAC control with iExoKrasG12D therapy.

A Schematic of the experimental timeline using KTC GEM. iExoKrasG12D treatment was initiated at day 33 of age until day 44. B, C Representative images (B) and respective quantification (C) of CD4+ T cells (CD4), and CD8+ T cells (CD8) by immunolabeling in control and iExoKrasG12D treated KTC mice, n = 3 mice per group. D, E Representative images (D) and respective quantification (E) of CK19+ and Fas+ cells by immunolabeling in control and iExoKrasG12D treated mice, n = 3-4 mice per group. The data presented in panels A-E are analyses of tissues from our earlier study18. F Schematic of experimental timeline using orthotopic KPC mice. Treatment was initiated at day 12 post tumor implantation and mice euthanized 2 weeks post-treatment (day 26). G Tumor burden ((Pancreas or tumor weights/body weight) *100) of orthotopic KPC mice treated with iExoKrasG12D, control exosomes (CE), Isotypes control (Iso), αCTLA-4 and αPD-1, n = 5 mice per group. H, I Representative images (H), and quantification (I) of CK19 and pERK immunostainings in the indicated groups, n = 3-5 mice per group. J Representative image and respective quantification of CD8, CD19, CD31, and nuclear staining (DAPI) in tertiary lymphoid structure (TLS) in the tumors of mice in the indicated groups, n = 3-5 mice per group. Scale bar: 100 μm. Data are presented as mean +/– SD. Significance was determined by unpaired two-tailed t-test (C, E) or one-way ANOVA with Dunnett’s or Sidaak’s multiple comparisons test (G, I). nd: not detected; HPF, high-power filed; αCTLA-4, anti-CTLA-4; αPD-1, anti-PD-1. Source data is provided as Source Data file.
iExoKrasG12D efficacy relies on KrasG12D primed epigenetic regulation of Fas for Fas-FasL anti-tumor response by CD8+ T cells
Fas expression in pancreatic cancer cells following KrasG12D genetic suppression enables interaction with FasL-expressing CD8+ T cells and anti-tumor response6,7. CK19+ pancreatic cancer cells showed an increase in FAS expression (FAS+ CK19+) following iExoKrasG12D treatment in KTC GEM (Fig. 6D-E). In vitro, KPC-689 cells treated with iExoKrasG12D demonstrated upregulation of expression in Fas and the CD8+ T cell recruiter cytokine, IL-15 (Supplementary Fig. 9F). Mechanistically, the regulation of Fas by KrasG12D inhibition in pancreatic cancer cells included epigenetic remodeling, implicating methylation of the histones and recruitment of transcriptional repressor at the promoter site6,21,22. Chromatin immunoprecipitation (ChIP analysis) at the Fas promoter or transcriptional start site revealed that KrasG12D expression recruited DNA methyl transferase DNMT1 and histone methyl transferase EZH2, and iExoKrasG12D treatment inhibited DNMT1 and EZH2 recruitment (Supplementary Fig. 9G). EZH2 promotes trimethylation of histone H3 at the K27 site (H3K27me3)23 and iExoKrasG12D treatment downregulated H3K27me3 and upregulated acetylation of H3K27 within the Fas promoter (Supplementary Fig. 9G). Collectively, our data indicate that iExoKrasG12D treatment relieved KrasG12D-mediated epigenetic suppression of Fas in pancreatic cancer cells, enabling permissive immune tumor microenvironment (TME) priming to anti-tumor response via CD8+ T cell FASL-FAS-mediated eradication of cancer cells.
Clinical trial with iExoKrasG12D informed on preclinical combination therapy with immune checkpoint blockade
Based on the data collected, we next tested the efficacy of iExoKrasG12D therapy in combination with ICB to enhance anti-tumor response. Mice with orthotopic PDAC tumors (utilizing syngeneic cells derived from P48Cre, LSL-KrasG12D/+, Trp53F/+ GEM) were treated with iExoKrasG12D and ICB (Fig. 6F). Specifically, iExoKrasG12D treatment was combined with either anti-CTLA-4 or anti-PD-1 checkpoint immunotherapies (Fig. 6F). Age matched analysis of PDAC tissues revealed a decrease in tumor burden with iExoKrasG12D therapy, with further decrease in tumor burden following anti-CTLA-4 combination but not with anti-PD-1 combination (Fig. 6G). Immunolabeling of tumor sections revealed downregulation of pERK in all groups that received iExoKrasG12D (Fig. 6H, I). Analysis of CK19+ cells (used here as surrogate for quantification of pancreatic cancer cells) revealed a decrease in CK19+ cancer cells following iExoKrasG12D, with further decrease in mice treated with anti-CTLA-4 and iExoKrasG12D combination (Fig. 6H, I). Histological analysis of PDAC tissues demonstrated an increase in the presence of tertiary lymphoid structures (TLS) in mice treated with iExoKrasG12D (iExo) + anti-CTLA-4 with 100% penetrance (Supplementary Fig. 10A, B). The presence of TLS, characterized by formation of new lymphoid structures including B cells, CD8+ T cells, a newly developing vasculature with endothelial venules and antigen presenting cells24,25,26, is frequently associated with favorable responses to immunotherapy in the context of cancers such as melanoma, non-small cell lung carcinomas and better prognosis25,26. We performed TSA multiplex immunostaining analysis confirming the presence of CD8+ T cells, B cells, and endothelial cells in TLS accumulating in tumors of mice treated with iExoKrasG12D + anti-CTLA-4 (Fig. 6J). Collectively, these results support the superior anti-tumor response of iExoKrasG12D when combined specifically with anti-CTLA-4 therapy.
Discussion
Our study demonstrates the safety of iExoKrasG12D as personalized medicine in patients with advanced PDAC. To our knowledge, this is the first-in-human study using exosomes from an allogenic source of bone marrow derived mesenchymal stromal cells that were engineered to encapsulate a siRNA therapeutic payload (iExoKrasG12D) in a personalized medicine setting. The use of MSC derived exosomes in clinical testing was recently reviewed in27,28,29, with systemic delivery of bone marrow derived MSC exosomes used in few clinical instances. In both a compassionate case for refractory GvHD30 and in a phase 2 clinical trial in respiratory failure secondary to COVID19 infection31, no significant adverse events were associated with bone marrow derived MSC exosomes, though dosing, preparations, definition of the clinical product of these unmodified exosomes differed from those used in our study. Here we report on the lack of adverse events or toxicity of iExoKrasG12D in mice, NHP, and in PDAC patients. We also demonstrated the localization of exogenously administered exosomes to the liver and pancreas of NHP, as well as the target engagement of iExoKrasG12D in human PDAC. The target engagement results support the specificity of this approach to target oncogenes that are known drivers of cancer initiation and progression but for which pharmacological approaches are lacking.
Increase in dosing of systemic iExoKrasG12D in this Phase Ia/b clinical trial did not result in any adverse event and the maximum tolerated dose was not reached, suggesting iExoKrasG12D dosages may be further increased beyond the 28.8 mg/6 weeks dose tested here to reach clinical efficacy in the planned Phase II studies. The safety profile of the iExoKrasG12D compared to other nanolipid-based carriers of siRNA or other molecular therapeutic cargo may reflect the intrinsic properties of exosomes15. Specifically, their natural lipid composition and surface protein expression reflect, at least in part, that of their cell of origin, minimizing their immunogenicity. Despite their allogenic source, no adverse events, including immunological reactions, were observed. We chose bone marrow derived MSC as the cell source for the exosomes preparation and generation of iExosomes because of the clinical safety profile of bone marrow derived MSCs. It is unclear if the source of exosomes in this clinical trial had anti-tumor impact beyond their use as siRNA delivery nanovesicles. Both tumor promoting and tumor restraining functions of MSC derived exosomes have been reported32. It is possible that exosomes from MSCs of distinct bone marrow donors or tissue sources may leverage added benefit to the iExosomes therapy given their likely diverse immunosuppressive properties33, however more testing is required to define the natural properties of MSC exosomes in cancer therapy. The immune evasion of allogeneic and heterologous exosomes may rely on their surface expression of the CD47 don’t-eat-me signal, as previously reported18. The safety profile of iExoKrasG12D also lends itself to testing in combination with chemotherapy and immunotherapy. We previously reported on the synergistic impact of iExoKrasG12D in combination of gemcitabine in mice19.
We previously reported on the influx of intratumoral T cells following oncogenic Kras extinction and antitumor response mediated by re-expression of Fas on cancer cells, yielding anti-tumor response via its interaction with CD8+ T cells FasL6,7. Here, we demonstrated an influx of intratumoral CD8+ T cells in human biopsies following iExoKrasG12D therapy. Our data support not only target engagement by iExoKrasG12D, but also biological response with remodeling of the immune TME in clinical and preclinical studies. Our prior work demonstrated that although targeting of oncogenic Kras resulted in decrease in proliferation of cancer cells, their apoptosis was leveraged by the interaction of Fas on cancer cells and FasL in the tumor-infiltrating CD8+ T cells6. Further, the tumor-infiltrating CD4+ T cells following Kras targeting are predominantly Th2, Th17 and Tregs, all pro-tumorigenic inhibitory subsets with no Th1 infiltration6. Taken together, we speculate that a lack of Fas-FasL mediated apoptosis of the cancer cells, in combination with unchecked CD4-mediated inhibitory effects in the KPC CD8−/− mice, likely contributes to the lack of survival benefit and tumor inhibition in these mice. We showed synergistic anti-tumor response of iExosKrasG12D with anti-CTLA-4 in preclinical studies. The lack of impact on the survival of mice with PDAC lacking CD4+ T cells likely reflects that this approach (CD4 deletion) primarily depleted inhibitory CD4+ T cells and released the breaks on cytotoxic CD8+ T cells. We also showed that while anti-CTLA-4 therapy in combination with iExoKrasG12D lends itself to significant anti-tumor response, this was not observed when combined with anti-PD1 therapy. It may reflect a unique mechanism associated with the immune microenvironment remodeling by oncogenic Kras suppression following iExoKrasG12D treatment. The lack of additional benefit with anti-PD1 may reflect the terminal exhaustion of tumor infiltrating CD8+ T cells in PDAC8. While infiltration of Tregs following checkpoint inhibition therapy has poor prognostic value; in the context of KrasG12D inhibition however, the increased tumor-infiltrating Tregs enables effective priming of the immune microenvironment for anti-CTLA4 therapy. iExoKrasG12D combination with anti-CTLA-4 therapy leads to emergence of TLS, a positive prognostic signature for anti-tumor response to immune check point inhibitor therapy34,35,36. Leveraging the information from this clinical trial, the pre-clinical studies offer insights for the design of Phase II iExoKrasG12D combination trial with ipilimumab.
Methods
The study was conducted according to the principles of Good Clinical Practice with prior approvals obtained from the Institutional Review Board (IRB) of MD Anderson’s Human Research Protection Program (HRPP) and with investigational new drug (IND) authorization granted by the Food and Drug Administration (FDA). For animal studies, all procedures were reviewed and approved by the Institutional Animal Care and use Committee (IACUC) at MD Anderson Cancer Center (MDACC)d Mice were housed in AAALAC-accredited facility at MDACC; NHPs were housed in AAALAC-accredited facility at Michale E. Keeling Center for Comparative Medicine and Research (KCCMR) at Bastrop, TX.
Exosomes source and production
GMP iExoKrasG12D were derived from the culture supernatant of human bone marrow mesenchymal stromal cells. The procedures to produce GMP-grade iExoKrasG12D were detailed previously19. Briefly, conditioned media from human bone marrow mesenchymal stromal cells grown in a bioreactor was subjected to filtration and centrifugation. The exosomes were characterized by NanoSightTM NS300 (Malvern) and flow cytometry analyses, and subsequently electroporated with siRNA19. MicroBCA analysis was used to define GMP iExoKrasG12D doses, expressed as mass of exosomal protein. Flow cytometry analyses of GMP iExoKrasG12D was also detailed in prior report19, and gating strategy is shown in Supplementary Fig. 1B. Briefly, exosomes were bound to aldehyde/sulfate beads (10 μl, Life Technologies), saturated with 1 M glycine, washed and blocked with 10% Bovine Serum Albumin (BSA) in PBS. The exosomes on beads were labeled with anti-CD47 (eBioscience, catalog 14-0479), anti-CD63 (BD Biosciences, catalog 556019), anti-CD-81 (BD Biosciences, catalog 555675), anti-CD9 (MilliporeSigma, catalog SAB4700092), or Mouse IgG1, κ isotype control antibody (BD Biosciences, catalog 555746), washed, and incubated with secondary antibody (Invitrogen, catalog A21202). For analyses, the LSR Fortessa X-20 cell analyzer was used, and data were analyzed using FlowJo software (TreeStar Inc.).
Clinical study design and patient population
The iEXPLORE (iExoKrasG12D in Pancreatic Cancer, NCT03608631) clinical trial is a first-in-human, open-label, single-center Phase I study of mesenchymal stromal cells-derived exosomes with KRASG12D siRNA evaluating the safety, tolerability, and target engagement in patients with metastatic pancreatic ductal adenocarcinoma harboring the KRASG12D mutation. Bone marrow-derived MSCs were obtained from a single male donor (age range 20–25 years old)_and the GMP production of iExosomes was previously detailed19.
The study enrolled patients at MD Anderson Cancer Center from 2021 to 2023 with pre-defined criteria, including 18 years of age on the day of consenting to the study, and histologically confirmed metastatic PDAC harboring KRASG12D mutation, as informed from any previous routine molecular profiling (e.g. Foundation One) of tissue or blood. Patients had documented progression of disease on one or more lines of systemic therapy. If with stable disease, the patient must have completed at least 4 months of chemotherapy with cytotoxic therapy. Patients presented with an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 and adequate organ functions. The study enrolled both males and females (defined as sex assigned at birth) and did not focus on the impact of treatment or disease progression in a gender specific matter. There was no plan for gender specific analyses of the data the given small sample size of the study. Women of childbearing potential (WOCBP, defined as not post-menopausal for 12 months or no previous surgical sterilization) had a negative serum pregnancy test within one week prior to initiation of treatment and requested to be using an adequate method of contraception to avoid pregnancy throughout the study and for up to 12 weeks after the last dose of study drug to minimize the risk of pregnancy. Exclusion criteria included concurrent severe and/or uncontrolled medical conditions that could compromise participation in the study, such as unstable angina, myocardial infarction within 6 months, unstable symptomatic arrhythmia, uncontrolled diabetes, serious active or uncontrolled infection, pregnancy or lactation, known CNS disease (except for treated brain metastasis, unless the patient was treated by neurosurgical resection or brain biopsy taken within 3 months prior to Day 1). Patient demographic data are detailed in Fig. 2C and Fig. 3A.
The clinical product starting dose of 0.150 mg of exosomal protein (which included an equivalent 0.150 mg of siRNA) was informed by non-toxic dosing in preclinical studies and applying conversion to human equivalent dosing (Table 1). iExoKrasG12D were generated by the Stem Cell Transplantation and Cellular Therapy Laboratory at MD Anderson according to Good Manufacturing Practice (GMP) and clinical grade siRNA was purchased from City of Hope RNA synthesis core. The iExoKrasG12D diluent was PlasmaLyte. Infusion rate of iExoKrasG12D was set at 2 mL/min, with infusion time ranging from 5 min to 64 min for increasing dose levels. Each iExoKrasG12D infusion was followed by PlasmaLyte infusion (100 mL over the course of approximately 30 min). The initial Phase Ia study enrolled 3 patients at the dose levels (Dose Levels 1, 2, and 3) listed in Fig. 2A. Given that no toxicity was observed, the Phase Ib study enrolled patients in an accelerated titration design, with a single patient at each of the increasing dose levels (Dose Levels 4, 5, 6). Phase Ib enrollment included capture of pre- and post-treatment tissue biopsies (Fig. 2B). Toxicity scoring followed the Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0 for toxicity and adverse event reporting. Dose limiting toxicity (DLT) were defined as grade 3 hematologic toxicity with bleeding, grade 4 thrombocytopenia, grade 4 neutropenia of ≥7 days duration or ≥ grade 3 neutropenia of any duration with fever ≥ 38.5 °C, grade 2 or greater autoimmune reaction, grade 2 or higher hypersensitivity reaction, grade 4 or higher infusion-related reaction, grade 3 or 4 events, excluding alopecia, nausea, vomiting, and/or diarrhea, unless these occur despite maximal prophylaxis and/or treatment. No DLT were encountered, and maximum tolerated dose (MTD) was not reached.
Clinical response was measured with CT imaging according to RECIST 1.1 criteria, approximately at every 3 cycles, as shown in Fig. 2B. Target lesions and non-target lesions were ascertained, with Partial Response (PR) defined as at least a 30% decrease in the sum of diameters of target lesions (taking as reference the baseline sum diameters); Progressive Disease (PD) was defined by at least a 20% increase, and Stable Disease (SD) defined as neither sufficient shrinkage to qualify for PR nor sufficient increase to qualify for PD. Appearance of new lesion(s) (NL) also defined PD.
Non-human primate (NHP) preclinical studies
Two preclinical studies are reported, using GMP iExoKrasG12D, and prospectively written protocols were followed. Experiment 1 studied the toxicology of GMP iExoKrasG12D (Fig. 1E). Experiment 2 studied the biodistribution of GMP iExoKrasG12D (Fig. 1G).
Experiment 1: GMP-iExoKrasG12D was administered to adult female and male Rhesus macaques (Macaca mulatta) of Indian origin (4.6 to 6.1 kg body weight, 4-5 years old). Rhesus macaques were transferred from the KCCMR Rhesus Macaque Breeding and Research Resource Colony (RMBRR) to study housing acclimation prior to initiation of the pre-clinical study and were randomly assigned to 2 groups. NHP (13-069) in group 1 received PlasmaLyte (diluent, control), and NHPs in group 2 received 0.15 mg GMP iExoKrasG12D. All doses were administered in 2 ml volume, intravenously at a peripheral venous access point, and flushed with 0.5 ml volume of PlasmaLyte. A total of 9 doses were administered over the course of 42 days (see Fig. 1E). The NHPs were euthanized one week after the final dose. Body weight (longitudinal and at experimental endpoint), organ weights, chemistry and hematology panels were completed. Gross necropsy findings and histopathological examination of tissues were performed following the proceeding standardized by the KCCMR.
Experiment 2: DiR and PKH-67 labeled GMP-iExoKrasG12D were administered to adult (approximately 7 years of age) male Rhesus macaques (Macaca mulatta) of Indian origin (~6 kg body weight). For DiR labeling of GMP-iExoKrasG12D, exosomes were diluted to a final concentration of 5 billion exosomes per 1 ml of PBS and 1 µl of XenoLight DiR (Perkin Elmer, mg/mL) added. The samples were incubated for 15 min at 37 °C followed by incubation at 4 °C for 5 min. Labeled exosome samples were then diluted in 10 ml of PBS and centrifuged in a SW41Ti rotor (Beckman Coulter) at 200,000 × g for 3 h at 4 °C. PKH-67 labeling was performed according to manufacturer’s instructions (Sigma Aldrich). Briefly, exosomes were diluted to a final concentration of 5 billion exosomes per 1 ml in diluent C and 4 µl of PKH-67 added. Exosomes were incubated for 5 min at room temperature followed by addition of 2 ml of 1% BSA in water. Samples were then diluted in 8 ml PBS and centrifuged in a SW41Ti rotor at 200,000 × g for 3 h at 4 °C. Following centrifugation, the supernatant was removed, the exosome pellet resuspended in PlasmaLyte, and exosome concentration evaluated by NanosightTM NS300 for both DiR and PKH labeled samples.
Rhesus macaques were transferred from the KCCMR RMBRR to study housing acclimation prior to initiation of the pre-clinical study and were randomly assigned to 3 groups. NHP (13-048) in group 1 received GMP iExoKrasG12D (siRNA KrasG12D) labeled with PKH-67, intravenously, NHP (13-052) in group 2 received GMP iExoKrasG12D (siRNA KrasG12D) labeled with DiR, intravenously, and NHP (13-072) in group 3 received GMP iExoKrasG12D (siRNA KrasG12D) labeled with DiR, intraperitoneally. All doses for the biodistribution studies contained approximately 70 mg of exosomal protein (or approximately 70 billion exosomes) administered in 2.5 mL volume (Table 1). Rhesus macaques were euthanized 24 h post dose with urine, blood, and organs collected. Organs were processed for formalin fixing and paraffin embedding, snap frozen tissues, OCT-embedded tissues, and fresh tissue for IVIS imaging. For H&E analysis of paraffin tissues, 5 µm sections were cut and stained for H&E using ST Infinity H&E staining system (Leica) and Leica Autostainer XL according to the manufacturer’s instructions.
NHP tissue analyses
For tissue analyses of DiR+ GMP iExoKrasG12D biodistribution in NHP, approximately 0.2 to 1 cm3 NHP tissue pieces were evaluated for DiR+ GMP iExoKrasG12D accumulation using In Vivo Imaging System (IVIS Spectrum, Perkin Elmer). Tissues were imaged 24 h post injection using a 710 nm excitation filter and 780 nm emission filter with background fluorescence removed by subtracting the background measurements at time of imaging. Tissues were compared to the negative control (NHP 13-048, injected with PKH-67 labeled exosomes).
For microscopic analyses of PKH-67+ GMP iExoKrasG12D biodistribution in NHP, frozen sections (5 µm) were cut from tissues embedded in optimal cutting temperature (OCT) medium. Sections were stained with 0.25 µg/ml DAPI in TBS for 5 min then mounted with coverslips using Fluoroshield (Sigma). Slides were imaged on a Keyence BZ-X710 fluorescent microscope and images stitched together in BZ-X Analysis software (Keyence) to generate a whole tissue image. Threshold values for green fluorescence were established based on negative control slides (NHP 13-052, injected with DiR-labeled exosomes) for each tissue and applied to stitched images in ImageJ. After applying the established threshold values to images, green fluorescence integrated density was quantified within the tissue area determined by the DAPI-positive area. The integrated density of control slides was subtracted to calculate a normalized integrated density for each tissue.
For high resolution imaging of PKH-67 labeled exosomes, DAPI labeled sections (as described above) were imaged with a Zeiss LSM800 confocal microscope equipped with a 63x Plan-Apochromat objective. For the images presented in Supplementary Fig. 5B, a z-stack spanning the entire tissue thickness at 1 µm intervals was acquired at 20× magnification, and the images were presented as a maximum intensity projection of the entire z-stack.
Murine preclinical studies
All mice received LabDiet 5053 ad libitum and were housed at 21–23 °C, 40–60% humidity and 12 h light/dark cycle. Two preclinical studies were reported. The toxicology study (Fig. 1A) employed GMP iExoKrasG12D administered intravenously to adult wild-type C57BL/6 albino females (age 25-30 weeks). Mice were purchased from Jackson Laboratories and allowed to acclimate for 2 weeks or more prior to initiation of the preclinical study. Ten mice were randomly assigned to 4 groups each. Mice in group 1 received PlasmaLyte (diluent, control), and mice in group 2 to 4 received increasing dose of GMP iExoKrasG12D. Group 2 was administered 0.66 µg of iExoKrasG12D per dose, Group 3 was administered 1.32 µg of iExoKrasG12D per dose, and Group 4 was administered 2.65 µg of iExoKrasG12D per dose. All doses were administered in 50 µl volume, intravenously via the retro-orbital plexus. A total of 9 doses were administered over the course of 6 weeks (see Fig. 1C). The mice were euthanized within one week after the final dose and postmortem pathology investigations were performed by a veterinary pathologist blinded to the treatment dose of these animals. Body weight (longitudinal and at experimental endpoint), organ weights, chemistry and hematology panels were completed. Gross necropsy findings and histopathological examination of tissues were performed following the standardized procedures by the Department of Veterinary Medicine and Surgery, Division of Veterinary and Comparative Pathology at MDACC.
P48-Cre, LSL-KrasG12D/+, Tgfbr2F/F (KTC) GEM tumor tissue sections used for analysis in Fig. 6A-D were obtained from a previously reported study18. The blood of KPC-689 (derived Pdx1-Cre, LSL-KrasG12D/+, Trp53R172H/+ female mouse) orthotopic pancreatic tumor-bearing mice treated with gemcitabine with and without iExoKrasG12D was obtained from a previously reported study19 (Fig. 4A, B). For studies with T cell depletion mice, we crossed KPC (Pdx1-Cre, LSL-KrasG12D/+, Trp53R172H/+)37 mice with CD4−/− (Cd4tm1Mak)38 depleted of CD4+ T cells or CD8+ T cells CD8−/− (Cd8tm1Mak)39. iExoKrasG12D treatment was initiated when mice reached 12 weeks of age (Supplementary Fig. 9C–E). Tumor tissues from mice with orthotopic pancreatic tumor (KPC-689 cells, derived Pdx-1Cre, LSL-KrasG12D/+, Trp53R172H/+ mice, were injected in the tail of the pancreas of C57BL6/J mice (0.5 ×106 cells in 20 ml PBS using 27-gauge Hamilton syringe), and treated with iExoKrasG12D from our prior study18 were analyzed for T cell infiltration (Supplementary Fig. 9A, B; F, G). The combination preclinical study using GMP iExoKrasG12D and ICB antibodies utilized orthotopic PKC-Hy19636 (derived from the tumor of a P48Cre, LSL-KrasG12D/+, Trp53F/+ female mouse)40 cells that were injected in the tail of the pancreas of C57BL6/J mice (0.5 × 106 cells in 20 ml PBS using 27-gauge Hamilton syringe, Fig. 6F–J, Supplementary Fig. 10B, later referred to as ‘KPC orthotopic’). Both male and female adult (6-10 weeks of age) mice were used and purchased from Jackson laboratories. For checkpoint blockade immunotherapy experiments, anti-CTLA-4 (BioXcell, Clone 9H10) or anti-PD-1 (BioXcell, Clone 29F1.A12) were injected three times per week (first dose 200 µg, followed by 2 doses of 100 µg intraperitoneal injections) in a final volume of 100 µL diluted in PBS. The control mice received a cocktail of isotype antibodies (Rat IgG2a (BioXcell, Clone 2A3) and Syrian hamster IgG (BioXcell, BE0087)) and control exosomes (non-electroporated with siRNA) in the same dosage, route and frequency as the treatment groups. For iExoKrasG12D treatments, 109 GMP iExoKrasG12D (equivalent to approximately 1 µg) were injected everyday intraperitoneally in the orthotopic KPC mice and every other day in the KTC GEM study. The investigator was not blinded to the treatment groups for analyses. For assessments of tumor volumes, mice were imaged at indicated time points using Bruker 7 T MRI at the MD Anderson Small Animal Imaging Facility (SAIF). The maximal tumor burden was reviewed and approved by the Institutional Animal Care and use Committee (IACUC) at MD Anderson Cancer Center (MDACC) and the maximal tumor burden was not exceeded. The mice were euthanized at the indicated time points for age-matched analysis.
Immunolabeling studies
For samples from the clinical trial, serial sections were cut through the block to collect 5 μm formalin fixed paraffin embedded (FFPE) sections. H&E analysis was performed on slides every 100 μm to select serial sections with optimal tumor tissue for immunostaining analysis. Immunohistochemistry for phosphorylated ERK (pERK, Fig. 4F) were carried on 5 μm thick FFPE sections using an automated protocol (Leica Bond RX; Leica Biosystems Nussloch GmbH). The tissue sections were deparaffinized and rehydrated following the Leica Bond protocol. Antigen retrieval was performed for 20 min with Bond Solution #1 (Leica Biosystems, equivalent to citrate buffer, pH 6.0). The primary antibody [Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP® Rabbit monoclonal antibody (Cell signaling, Cat#4370), dilution 1:400, Cell signaling, Cat#4370] was incubated for 15 min at room temperature. The primary antibody was detected using the Bond Polymer Refine Detection kit (Leica Biosystems) with DAB as chromogen. The slides were counterstained with hematoxylin, dehydrated, and coverslipped.
Immunofluorescence analysis was performed on 5 μm thick FFPE sections using Tyramide signal amplification (TSA) after deparaffinization and hydration, following which antigen retrieval was performed for 20 min at 98 °C in Tris-EDTA buffer (pH-8.6) in a steamer. Next, the sections were incubated in 3% H2O2 for 10 min at room temperature, 1.5% bovine serum albumin (BSA) for 1 h and primary antibody at indicated concentrations for 3 h (Supplementary Table 1). Following addition of the indicated secondary polymer (30 min at RT), opal reagents were used at a 1:100 concentration (30 min at RT) for TSA staining. Subsequently, antigen retrieval was performed before use of the next primary antibody. The slides were mounted using FluoroshieldTM with DAPI (Sigma Aldrich F6057). For single stained IHCs, DAB reagent (5 to 15 min) was added following incubation with the secondary polymer, counterstained with hematoxylin, dehydrated and mounted for analysis. For quantification of immune infiltrates, representative images were taken from 3-5 high power fields (200× magnification) per biological replicate to count the number of immune cells and average of multiple images is plotted as a replicate. We used Zeiss Axio Imager Z.2 for IF imaging. For quantification of CK19 and pERK immunostainings, average pixels positivity of 5 high power field images were used. Leica microsystems DFC295 microscope for brightfield imaging.
Digital PCR analyses KrasG12D siRNA
NHP tissue RNA was extracted using Zymo Research Corporation Direct-zol™ RNA MiniPrep kit according to the manufacturer’s direction. As previously described, Custom TaqMan Small RNA Assay kit (Applied Biosystems) was used to detect the sense strand of the KrasG12D siRNA. Briefly, the manufacturer’s protocol was followed using 5 μl of RNA and the result of the dPCR was expressed as number of positive wells (detecting the siRNA) normalized to the total RNA concentration.
Digital PCR analyses for circulating KrasG12D DNA
We previously reported on the anti-tumor efficacy of GMP iExoKrasG12D with and without gemcitabine in an orthotopic model of pancreatic cancer19. The blood of mice from the previously published experiment (Reference19) was analyzed for the detection of KrasG12D mutation in circulating DNA. Briefly, blood samples were collected from tumor bearing mice (KPC-689 orthotopic tumors with KrasG12D mutation) one day prior to the start of treatment (pre-treatment/pre-tx) and 9 days after treatment was started (post-treatment/post-tx) (see Fig. 4A). Murine blood samples were collected and left at room temperature for 4 h followed by centrifugation at 3,000 x g for 5 min to collect serum. cfDNA from the serum for dPCR were extracted by using QIAamp DNA Micro Kit and DNA concentration was measured by Qubit dsDNA HS kit according to manufacturer’s instructions. The probe assay ID used was AH6R5PI (KRAS G12D c35G>A (WT:C — > MUT:T). 6.5 µl of DNA was added regardless of DNA concentration.
Approximately 1.5 to 4 ml plasma samples for patients on the clinical trial were also analyzed. ctDNA isolated from 0.5 ml of patient plasma using the cfPure V2 Cell Free DNA Extraction Kit (Biochain) according to manufacturer’s instructions. TaqMan Liquid Biopsy dPCR Assay (Invitrogen A44177, Assay ID Hs000000051_rm) in conjunction with Quant Studio 3D dPCR 20 K was used to amplify 1 or 2 ng of ctDNA and quantify (averaging values for 1 and 2 ng) KRASG12D and KRAS (wildtype/unmutated allele) in ctDNA.
The analysis of the digital PCR data was performed with the manufacturer’s software (QuantStudio 3D Analysis Suite). The setting of detection limit used was the same as previously reported41. The results were expressed as a percent of the ratio of Target/Total (KrasG12D/(KrasG12D+KrasWT)).
Quantitative RT-PCR
For in vitro experiments to determine KrasG12D knockdown efficacy of iExoKrasG12D, 105 Panc-1 cells (from the American Type Culture Collection, ATCC, tested negative for mycoplasma) were seeded in 6 well plates (ThermoFisher, 140685) in 10% FBS DMEM with PS in a final volume of 2 ml per well overnight. Subsequently, 109 iExoKrasG12D were added in serum free DMEM in a final volume of 1 ml and RNA was extracted from the cell lines using PureLinkTM RNA Mini Kit (Invitrogen, 12183025) as per the manufacturer’s instructions at indicated time points for downstream analysis. RNA concentrations were determined using spectrophotometer (Nanodrop 2000) and 500 μg to1 mg RNA was used for reverse transcription using High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific, 4368814). The cDNA was further diluted 1:20 for qPCR analysis with SYBRTM green master mix (Applied BiosystemsTM, A25742). Primers used for qPCRs include:
KrasG12D
F: 5’-ACTTGTGGTGGTTGGAGCAGA-3’
R: 5’-TAGGGTCATACTCATCCACAA-3’
Fas
F: 5’-TATCAAGGAGGCCCATTTTGC-3’
R:5’-TGTTTCCACTTCTAAACCATGCT-3’
Il15
F: 5’-ACATCCATCTCGTGCTACTTGT-3’
R: 5’-GCCTCTGTTTTAGGGAGACCT-3’
18 s
F: 5’-GTAACCCGTTGAACCCCATT-3’
R: 5’-CCATCCAATCGGTAGTAGCG-3’
Gapdh
F: 5’-AGGTCGGTGTGAACGGATTT-3’
R: 5’-TGTAGACCATGTAGTTGAGGTCA-3’
Gene expression for listed genes was normalized to 18 s or Gapdh and fold change was determined by ratios of 2-dCt values. Statistical comparisons were performed on dCt values.
Chromatin immunoprecipitation
For ChIP experiments, 0.5 to 1 ×106 cells per immunoprecipitation (IP) condition from iExo and ScrExo [2 doses (109 exosomes/dosage) administered 24 h apart] of KPC-689 cells (established from the pancreas tumors of Pdx1cre/+;LSL-KrasG12D/+;LSL-Trp53R172H/+ (KPC) as previously described18) were collected on day 3 by trypsinization. Cross-linking was performed by shaking cells in a suspension of 1% methanol free formaldehyde (ThermoFischer, Cat# 28908) in complete medium at room temperature for 10 to 12 min. Subsequently, the solution was mixed with 125 mM glycine and incubated at room temperature for 5 min. The cells were washed twice with ice cold PBS (with 1 mM PMSF) and the resulting cell pellets were stored at –80 °C for downstream analysis. ChIP assays were performed at the MD Anderson’s Epigenomics Profiling Core with some modifications to previously described high-throughput ChIP protocol42. For histone ChIPs, H3K27ac, Abcam – Cat# ab4729; H3K27me3, Diagenode – Cat# C15410195 and H3, Abcam – Cat# ab1791 were used. For ChIPs for non-histone antibodies (DNMT1, Novus Biologicals, Cat# NB100- 56519 and EZH2, Cell Signaling Technology, Cat# 5246S) were used. Subsequently, qPCR assays were performed using primers for the promoter/transcription start site using the respective primers (FAS TSS (Histone ChIP)- F: 5’- CTGCCTCTGGTAA GCTTTGG-3’ R: 5’- CAGCCACATCTGGAATCTCA-3’; FAS promoter site (DNMT1, EZH2 ChIP)- F: 5’-CCC TGTATTCCCATTCATCG-3’ and R: 5’-ACTAGGGG AGGGGACAGAAA-3’). Normalized transcript levels (to IgG or histone H3) were used for statistical comparisons.
Electron microscopy
Scanning electron microscopy
GMP iExoKrasG12D was submitted to the High Resolution Electron Microscopy Facility for processing and imaging. Fixed samples were washed in 0.1 M sodium cacodylate buffer and treated with 0.1% Millipore-filtered cacodylate buffered tannic acid, postfixed with 1% buffered osmium, and stained en bloc with 1% Millipore-filtered uranyl acetate. The samples were dehydrated in increasing concentrations of ethanol, infiltrated, and embedded in LX-112 medium. The samples were polymerized in a 60 °C oven for approximately 3 days. Ultrathin sections were cut in a Leica Ultracut microtome (Leica, Deerfield, IL), stained with uranyl acetate and lead citrate in a Leica EM Stainer, and examined in a JEM 1010 transmission electron microscope (JEOL, USA, Inc., Peabody, MA) at an accelerating voltage of 80 kV. Digital images were obtained using AMT Imaging System (Advanced Microscopy Techniques Corp, Danvers, MA). All pictures were captured with the help of the facility.
Cryo-electron microscopy
GMP iExoKrasG12D was submitted to the CryoEM Core at Baylor College of Medicine for processing and imaging. The surfaces of QUANTIFOIL holey carbon grids were plasma cleaned for 20 seconds inside an Ernest Fullam Glow Discharge. The grids were transferred to a VitroBot Mark IV plunge freezer where 2.5 µl of the iExoKrasG12D sample and 1 µl of AURION 10 nm gold tracers were applied to the carbon-side of the grid before immediately being plunged into liquid nitrogen. The vitrified sample was transferred to a JEOL 2200FS operating at 200 kV equipped with a Direct Electron DE-64 detector. Micrographs were collected at 8k × 8k resolution with a final A/pixel of 2.18. The approximate total electron dose per micrograph was 20 e-/A−2. The micrograph movies were motion corrected and gain-normalized using EMAN243.
Graph generation and statistical analyses
Graphs were generated using GraphPad Prism Version 10.4.1. Statistical analyses were performed using this software, and tests and p-values are listed in the figure legend and figures, respectively.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data for each figure is provided. Data are located in controlled-access data storage at the MD Anderson Cancer Center. For the clinical study, individual participant data that underlie the results reported in this manuscript and study protocol will be made available indefinitely to investigators whose proposed use has been approved by an independent review committee and upon request to the corresponding author. Source data are provided with this paper.
References
Society, A. C. Cancer Facts &Figures 2024. Atlanta: American Cancer Society (2024).
Siegel, R. L., Kratzer, T. B., Giaquinto, A. N., Sung, H. & Jemal, A. Cancer statistics, 2025. CA Cancer J. Clin. 75, 10–45 (2025).
Ying, H., et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 30 (2024).
Biankin, A. V. et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405 (2012).
Waters, A. M. & Der, C. J. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold spring harb perspect med 8 (2018).
Mahadevan, K. K. et al. Elimination of oncogenic KRAS in genetic mouse models eradicates pancreatic cancer by inducing FAS-dependent apoptosis by CD8(+) T cells. Dev. Cell 58, 1562–1577 e1568 (2023).
Mahadevan, K. K. et al. KRAS(G12D) inhibition reprograms the microenvironment of early and advanced pancreatic cancer to promote FAS-mediated killing by CD8(+) T cells. Cancer Cell 41, 1606–1620 e1608 (2023).
Dey, P. et al. Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer cells utilizes cytokines from the tumor microenvironment. Cancer Discov. 10, 608–625 (2020).
Ozdemir, B. C. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734 (2014).
Ying, H. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670 (2012).
Asimgil, H., et al. Targeting the undruggable oncogenic KRAS: the dawn of hope. JCI Insight 7 (2022).
Aguirre, A. J., Stanger, B. Z. & Maitra, A. Hope on the horizon: a revolution in kras inhibition is creating a new treatment paradigm for patients with pancreatic cancer. Cancer Res 84, 2950–2953 (2024).
Chung, V., Mizrahi, J. D. & Pant, S. Novel therapies for pancreatic cancer. JCO Oncol Pract, OP2400279 (2024).
Hong, D. S. et al. KRAS(G12C) Inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med 383, 1207–1217 (2020).
Kalluri, R. & LeBleu, V. S. The biology, function, and biomedical applications of exosomes. Science 367 (2020).
Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Invest 126, 1208–1215 (2016).
Rejiba, S., Wack, S., Aprahamian, M. & Hajri, A. K-ras oncogene silencing strategy reduces tumor growth and enhances gemcitabine chemotherapy efficacy for pancreatic cancer treatment. Cancer Sci. 98, 1128–1136 (2007).
Kamerkar, S. et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 546, 498–503 (2017).
Mendt, M. et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 3, e99263 (2018).
Kapoor, K. S. et al. Single extracellular vesicle imaging and computational analysis identifies inherent architectural heterogeneity. ACS Nano 18, 11717–11731 (2024).
Mou, H. et al. Genetic disruption of oncogenic kras sensitizes lung cancer cells to fas receptor-mediated apoptosis. Proc. Natl Acad. Sci. USA 114, 3648–3653 (2017).
Wajapeyee, N., Malonia, S. K., Palakurthy, R. K. & Green, M. R. Oncogenic RAS directs silencing of tumor suppressor genes through ordered recruitment of transcriptional repressors. Genes Dev. 27, 2221–2226 (2013).
Duan, R., Du, W. & Guo, W. EZH2: a novel target for cancer treatment. J. Hematol. Oncol. 13, 104 (2020).
Sautes-Fridman, C. et al. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol. 7, 407 (2016).
Sautes-Fridman, C., Petitprez, F., Calderaro, J. & Fridman, W. H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 19, 307–325 (2019).
Schumacher, T. N. & Thommen, D. S. Tertiary lymphoid structures in cancer. Science 375, eabf9419 (2022).
Lotfy, A., AboQuella, N. M. & Wang, H. Mesenchymal stromal/stem cell (MSC)-derived exosomes in clinical trials. Stem Cell Res Ther. 14, 66 (2023).
Tan, F. et al. Clinical applications of stem cell-derived exosomes. Signal Transduct. Target Ther. 9, 17 (2024).
Lee, B. C., Kang, I. & Yu, K. R. Therapeutic features and updated clinical trials of mesenchymal stem cell (MSC)-derived exosomes. J Clin Med 10 (2021).
Kordelas, L. et al. MSC-derived exosomes: a novel tool to treat therapy-refractory graft-versus-host disease. Leukemia 28, 970–973 (2014).
Lightner, A. L. et al. Bone marrow mesenchymal stem cell-derived extracellular vesicle infusion for the treatment of respiratory failure from COVID-19: A randomized, placebo-controlled dosing clinical trial. Chest 164, 1444–1453 (2023).
Zhao, R., Chen, X., Song, H., Bie, Q. & Zhang, B. Dual role of MSC-derived exosomes in tumor development. Stem Cells Int 2020, 8844730 (2020).
Naji, A. et al. Biological functions of mesenchymal stem cells and clinical implications. Cell Mol. Life Sci. 76, 3323–3348 (2019).
Di Caro, G. et al. Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin. Cancer Res 20, 2147–2158 (2014).
Lehmann, J. et al. Tertiary lymphoid structures in pancreatic cancer are structurally homologous, share gene expression patterns and b-cell clones with secondary lymphoid organs, but show increased T-cell activation. Cancer Immunol. Res 13, 323–336 (2025).
Vanhersecke, L. et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat. Cancer 2, 794–802 (2021).
Hingorani, S. R. et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 (2003).
Chambers, C. A., Kuhns, M. S., Egen, J. G. & Allison, J. P. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev. Immunol. 19, 565–594 (2001).
Rahemtulla, A. et al. Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature 353, 180–184 (1991).
Deng, Y. et al. Glucocorticoid receptor regulates PD-L1 and MHC-I in pancreatic cancer cells to promote immune evasion and immunotherapy resistance. Nat. Commun. 12, 7041 (2021).
Yang, S. et al. Detection of mutant KRAS and TP53 DNA in circulating exosomes from healthy individuals and patients with pancreatic cancer. Cancer Biol. Ther. 18, 158–165 (2017).
Blecher-Gonen, R. et al. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat. Protoc. 8, 539–554 (2013).
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007).
Acknowledgements
The work was partially supported by the Cancer Prevention and Research Institute of Texas. The iExoKrasG12D preclinical studies and Phase I trial were supported by the MD Anderson Moon Shot Program and in part by the Translational Molecular Pathology-Immunoprofiling Lab (TMP-IL) Moon Shots Platform. AM is supported by the Khalifa bin Zayed Foundation. RK is also supported by NIH R35CA263815 for work related to exosomes and cancer. The work was done using the High-Resolution Electron Microscopy and the Small Animal Imaging Facility Core Facilities at MDACC. The CryoEM data were collected at the Baylor College of Medicine CryoEM ATC, subsidized by CPRIT Core Facility Award RP190602 which also supported acquisition of CryoEM equipment used in this study. R35GM151999 and R01GM080139 to SJL in support of CryoEM data collection and analysis. AKJ is partially supported by MD Anderson institutional support to Epigenomics Profiling Core. We thank Kenneth Dunner Jr. (MD Anderson Cancer Center, Department of Cancer Biology) for help with the electron microscopy analyses; Xunian Zhou (MD Anderson Cancer Center, Department of Cancer Biology) for flow cytometry analyses; Swati Gite, Leticia Campos Clemente, Mei Jian, Wei Lu and Khaja Khan (all from MD Anderson Cancer Center, Department of Translational Molecular Pathology) for coordination of tissue sample and analysis of chromogenic IHC data; Charles Kingsley, Jorge Delacerda and Houra Taghavi (all from MD Anderson Cancer Center, Small Animal Imaging Facility Core Facilities) for assistance with MRI imaging. The authors acknowledge the contributions of Dr. Pamela Papadopoulos in MD Anderson Moon Shots Operations for Program Management support and Dr. Mark Hurd in the Ahmed Center (MD Anderson Cancer Center) for biospecimen collection support.
Author information
Authors and Affiliations
Contributions
V.S.K., M.L.K., K.K.M., K.M.M., S.Y., H.S., A.S.M., A.J.K. and M.G. contributed to the murine preclinical studies, and V.S.K., M.L.K., K.M.M., S.Y., H.S., R.F. also contributed to the NHP preclinical studies. A.C.F. and S.J.L. contributed to data acquisition and analysis. V.S.K., K.K.M., A.S.M., L.M.S.S., C.H., and M.E.S. contributed to human sample analyses. V.S.K., B.G.S., J.J.L., G.V., R.T.S., A.M., M.M., E.S., and S.P. contributed to the clinical trial design and execution. V.S.K., K.K.M., R.K., B.G.S., S.P., A.M., and E.S. conceptually designed the strategy for this study, provided intellectual input, and contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
AM is a consultant for Tezcat Biosciences and is named on a patent that has been licensed to Thrive Earlier Detection (an Exact Sciences company). CH reports research funding to the institution from Sanofi, Avenge, Iovance, KSQ, Theolytics, BTG, Novartis, 280Bio, Astrazeneca, EMD Serono, Takeda, Obsidian, Genentech, BMS, Summit Therapeutics, Artidis, and Immunogenesis, and personal fees from Regeneron and stock options from Briacell outside the submitted work. LMSS reports research support from Theolytics, advisory role/consulting fees from BioNTech, and travel support from 10x Genomics, all outside the scope of this work. MD Anderson Cancer Center, VSK, ES, MM, RK hold a patent in the area of exosome biology. Patents related to EVs and Exosomes have been licensed to PranaX, Inc. VSK and RK are stock equity holders in PranaX, Inc. VSK and RK are founders, stock owners, and scientific consultants to PranaX, Inc. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Oscar Wiklander and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kalluri, V.S., Smaglo, B.G., Mahadevan, K.K. et al. Engineered exosomes with KrasG12D specific siRNA in pancreatic cancer: a phase I study with immunological correlates. Nat Commun 16, 8696 (2025). https://doi.org/10.1038/s41467-025-63718-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63718-2
This article is cited by
-
Identification of HSPE1 as a new actionable cancer vulnerability leads to an innovative and effective combination therapy for pancreatic ductal adenocarcinoma
Molecular Cancer (2026)
-
The avatar principle: exosomal dynamics guiding tumor adaptation and next-generation therapeutic strategies
Journal of Nanobiotechnology (2026)
-
Bioengineering of extracellular vesicles with scaffold proteins for drug delivery
Journal of Nanobiotechnology (2026)
-
The status of extracellular vesicles as drug carriers and therapeutics
Nature Reviews Bioengineering (2026)
