
Abstract
Anion‒water interactions in aqueous electrolytes can lower the freezing point for antifreezing applications, yet cation‒water interactions have usually been ignored. Here, we propose that the cation effect of Al3+ can simultaneously exert a deshielding effect on both H and O in water, which significantly weakens the water hydrogen bond and lowers the freezing point to −117 oC at a low salt concentration of 2.8 m. Additionally, dual-cation effects further optimize the ion diffusion kinetics and facilitate the formation of an Al‒Zn alloy layer for Zn electrode safeguarding. As demonstrated in batteries, the designed electrolyte enables the symmetrical Zn||Zn coin cell for 10,340 h of Zn plating/stripping and Zn||polyaniline pouch cell with a capacity retention of 100% after 500 cycles at 100 mA g-1 and −70 oC. Even at −80 oC, the Zn||polyaniline cell delivers a discharge capacity of 115.5 mA h g-1. The cation effect offers an effective strategy for regulating the water structure of low-temperature aqueous devices.
Similar content being viewed by others
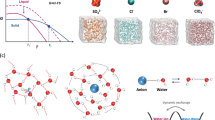

Introduction
Aqueous batteries have garnered significant attention as sustainable energy storage solutions, favored for their high safety, environmental sustainability, cost-effectiveness, and broad compatibility with large-scale applications1,2,3. However, water tends to freeze at low temperatures, which decreases the ionic conductivity and disrupts the interfacial wettability of aqueous electrolytes, ultimately restricting the operational temperature range of the battery. In particular, harsh environments such as frigid regions, outer space, and deep-sea conditions impose more stringent requirements on the operating temperature4,5,6. Therefore, exploring effective strategies to design electrolytes with high antifreezing performance is crucial for accelerating the practical application of aqueous batteries.
On the basis of the water cluster theory, all water molecules exist in the form of hydrogen bonds (HBs)7. The freezing of water is driven by the rearrangement of HBs between water molecules. Typically, HBs are formed through electrostatic interactions (O-H···O) between partially positively charged H atoms (HB donors) and partially negatively charged O atoms (HB acceptors)8,9. As the temperature decreases, initially disordered small water clusters gradually evolve into long-range ordered large HB networks through the formation of additional HBs10. Thus, disrupting these HBs can effectively impede water freezing. To date, several strategies have been developed to reconfigure the HB structure of water to reduce Tf through the organic cosolvent, additives, hydrogel electrolyte, molecular crowding effect, and the ion effect4,11,12. In particular, electrolyte design based on the ion effect has the advantages of simplicity, low toxicity, and high ionic conductivity. The ion effect primarily involves interactions between ions (anions/cations) dissociated from electrolyte salts and water molecules, which weakens HBs between water molecules. Currently, most studies have focused on the impact of the anion effect on the Tf of water5,13,14. Chaotropic anions can interact with H on water molecules to form additional HBs, thereby reducing the cluster size and increasing the tetrahedral entropy of water molecules to achieve a lower Tf5,14. However, the impact of cations, another concomitant product of salt dissociation, on electrolyte antifreezing has been relatively understudied.
The electric field generated by dissociated metal cations interacts with the highly electronegative O atoms of water molecules, leads to a reduction in the electron density around O (deshielding effect (DSE)) (Fig. 1a)10,15. This interaction confines water molecules within their solvated structures and drives the rearrangement of dipolar water molecules around the cations due to the electric field16,17. Recent studies have demonstrated that chaotropic cations and ions with high cationic potential are effective at realizing antifreezing performance1,18. However, chaotropic cations present weak interactions with water, which limit their regulatory effect on the HB structure. In addition, the unrestricted HB donors (H) still maintain high mobility and proton transfer frequency, leading to HB formation, especially as the temperature decreases significantly (Fig. 1b, c). For ions with high cationic potential, the regulatory mechanisms of cations on the HB structure remain unclear, and the crucial role of the cation in lowering Tf is still unknown. Therefore, revealing the effect of cations on the regulation of water structure to achieve low Tf is imperative yet still challenging.

a Relationship of the interaction between cations and O (in water) with respect to the number of HBs and Tt. b Relationship of the interaction between cations and H (in water) with respect to the number of HBs and Tt. c Design concept for different types of cations to interact with water and decrease the number of HBs among water molecules. d Cation effect diagram of different cations versus the 1H chemical shift. The fixed cation concentration is 0.9 m. e The q/r2 values of various cations. The inset images show the ESP distribution for the structure of the cation-water mixture. f Calculated average ΔHB numbers among different cation systems when the temperature decreases from 25 to 0 °C and further decreases to −20 °C. g DSC curve of 4Al. The inset shows optical photographs of different AlCl3 concentrations at 25 and −70 °C.
Owing to its high capacity and low redox potential, Zn metal stands out as an optimal choice for the negative electrode in aqueous batteries19,20. However, practical applications face challenges, including corrosion and dendrite formation, ascribed to the cation concentration polarization and active water molecules in the electrical double layer (EDL) on the zinc electrode surface21,22,23. When applied to aqueous Zn metal batteries, dynamic ion migration and a stable Zn electrode should be considered simultaneously, both of which can be effectively regulated through cation effects.
Here, we propose leveraging the cation effect in aqueous electrolytes to prevent electrolyte freezing with satisfactory ion diffusion kinetics and Zn metal electrode stability at low temperatures. Combined with spectral analysis, theoretical calculations, and electrochemical characterization, we clearly revealed the cation effects in reconfiguring the HBs between water molecules and cation solvation structures, highlighting their pivotal role in enhancing the thermodynamic and kinetic properties of the electrolyte. Specifically, the deshielding effect cation (DSEC) effectively inhibits the formation of HBs between water molecules (Fig. 1c), thereby increasing the entropy of the electrolyte and significantly lowering Tf to −117 °C at a low DSEC concentration of 2.8 m. The assembled aqueous Zn-based batteries demonstrate favorable rate capacities and long cycling performance at 50 ~ −80 °C. This study provides a concept for developing electrolytes in low-temperature aqueous systems.
Results
Cation effect on water structure for low Tf
To investigate the cation effect, an in-depth analysis was performed on electrolyte solutions containing various cations, including Li+, Na+, Mg2+, Zn2+, Ca2+ and Al3+, with Cl− as the constant anion. Nuclear magnetic resonance (NMR) spectroscopy, a powerful technique for characterizing molecular interactions and directly probing the changes in electron density at the oxygen (O) and proton (H) positions that participate in hydrogen bonding (HB), was conducted15,24. The δ(17OH2) and δ(1H2O) values in the NMR spectra, corresponding chemical shifts in 17O and 1H, correlate with the average number of HBs for each water molecule involved7. Compared with those in pure water, the 17O peaks in various cation electrolytes shift to lower fields due to the deshielding effect (DSE) (Supplementary Figs. 1 and 2). These results indicate that the electron density of O in water molecules decreases as a result of the interaction between positively charged cations and negatively charged O atoms, reducing the number of donor atoms involved in the formation of HBs. Additionally, the 17O peak in the Al3+ electrolyte results in the largest shift, suggesting the strongest interaction with O(H2O) and significant disruption of the HB network of water molecules25,26. For the 1H spectra, the 1H peaks present an upfield in the Li+, Na+, Mg2+, Zn2+, and Ca2+ electrolytes compared with those in water because of the shielding effect (SE) of these cations (Fig. 1d and Supplementary Figs. 3, 4). These cations are categorized as shielding effect cations (SECs). Notably, the 1H peak exhibits a notable downfield shift in the Al3+ electrolyte, indicating the deshielding effect of Al3+ (categorized as the deshielding effect cation (DSEC)). The strong electric field of DSEC induces electron density transfer from the H nucleus to the vacant orbitals of DSEC, leading to a reduction in electron density on H and a corresponding downshift in resonance frequency. This decreased shielding effect, arising from the interaction between DSEC and the H nucleus, weakens the ability of H to act as HB donor for the information of HB27. Conversely, when the shielding effect on the H nucleus is increased, the H resonance frequency remains higher (for SEC), thereby enabling H to function as the HB donor to participate in the formation of HB with O, particularly at significantly low temperatures. In general, the SE or DSE of cations on δ(17OH2) and δ(1H2O) is associated with the value of q/r2, where q and r are the charge density and ionic radius of ions, respectively15. A small ionic radius and higher charge density result in a strong electrical field (higher value of q/r2) to reconstruct the HB matrix among water molecules by reorienting and polarizing the water molecules around the ions, resulting in DSE for both O and H in water. Among these cations, Al3+ has the highest charge density and smallest radius (Supplementary Fig. 5), which results in the highest value of q/r2 (Fig. 1e), confirming the DSE of Al3+. In contrast, Ca2+ has a relatively low charge density and high cation radius, corresponding to the obvious SE on H. The order of DSE aligns with the order of q/r2 (Na+ <Ca2+ <Li+ <Zn2+ <Mg2+ <Al3+). The 1H peaks of Al-based salts with various anions all shift toward lower fields, providing further evidence for the DSE of Al3+ on H nuclei in water (Supplementary Fig. 6). For SEC, the disruption of HBs to O atoms in water molecules can affect the SE of H in the same water molecule. However, compared with SEC, DSEC has a greater direct effect on the H atom of water, which can further impair the formation of HBs between water molecules and reduce the average number of HBs for one water molecule. This greater destruction of HBs caused by DSEC results in a decrease in the size of water clusters and freezing suppression at low temperatures.
Theoretical calculations were then carried out to analyze the effects of cations on water HB structures. Density functional theory (DFT) calculations revealed that the binding energy of Al3+ to water is greater than that of water to water and other cations to water (Supplementary Fig. 7), and the distance between Al3+ and O is shorter than that between other cations (Supplementary Fig. 8), confirming the strong interactions between Al3+ and water. These results are consistent with the NMR results in Supplementary Fig. 1. With higher binding energy, solvation structure dissociation is hindered, leading to tighter binding between water molecules and cations. Therefore, it becomes more challenging to form an ordered HB network between water molecules as the temperature decreases, thus realizing optimized antifreezing performance for cation-enhanced electrolytes. The molecular electrostatic potentials (ESPs) for water‒water and cation‒water interactions indicate that Al3+ significantly influences the electron density of water molecules (insert images in Fig. 1e). The calculated ESP distributions and binding energies for solvation structures with different cations also support the above results (Supplementary Fig. 9). Additionally, the Hirshfeld charge distributions of the cation with water molecules reflect that both the O and H in the water molecule undergo greater electron transfer toward the Al3+ (Supplementary Fig. 10). Moreover, molecular dynamics (MD) simulation results indicate that the average number of HBs among water molecules in Al3+ systems experience minimal changes as the temperature decreases from 25 to 0 °C and further to −20 °C (Fig. 1f and Supplementary 11). All these results substantiate that when the O atom in a water molecule interacts with Al3+, the H in the same molecule has the weakest ability to form HBs with other water molecules. Al3+ can simultaneously and directly reduce the ability of both O and H in water to form HBs. The differential scanning calorimetry (DSC) measurements confirm that the Al3+ system renders antifreeze capability with the lowest freezing point (Supplementary Fig. 12).
To further investigate the impact of DSEC concentration on Tf, a series of AlCl3·6H2O-based electrolytes with concentrations of 1, 2, 3, 4, 5 and 5.3 m (the maximum solubility) (the actual concentrations of AlCl3 were 0.90, 1.63, 2.26, 2.80, 3.25 and 3.37 m, respectively) were prepared. As shown in Fig. 1g, only the 4 m AlCl3·6H₂O sample (2.80 m AlCl3, denoted as 4Al) remained in liquid state at −70 °C, whereas the others fully freezed. This antifreezing behavior is generally attributed to the high salt concentration, which destroys the HBs between water molecules. With increasing AlCl3 concentration, the HB numbers gradually decrease (Supplementary Fig. 13). However, in systems with lower AlCl3 concentrations (1–3 m), the number of HBs remains relatively high. This observation indicates that the insufficient Al3+ resulted in a limited deshielding effect on the O and H atoms of water molecules, leading to freezing when the temperature is significantly reduced. At higher concentrations, although the number of HBs between water molecules decreases significantly, there is a sharp increase in viscosity which promotes salt crystallization, thus impairing the antifreezing performance of the electrolyte (Supplementary Fig. 14)27. In contrast, the 4Al system achieves a balance between HB disruption and manageable viscosity, yielding antifreezing performance with a Tf as low as −117 °C (Fig. 1g).
To exclude the influence of anion concentration on Tf, comparative tests were performed under a fixed Cl− content (8.4 m). When the anion concentration was set to 8.4 m, the Al3+ system (−117 °C) was second only to that of the Li⁺ system (−123 °C), with the Al3⁺ concentration at 2.8 m, much lower than that of the Li⁺ system at 8.4 m. This indicates that the difference in Tf was primarily due to the cation effect rather than variations in Cl- concentration (Supplementary Fig. 15).
Water structure evolution
The evolution of the water structure was further investigated through spectral characterization and MD simulations. NMR spectra of the DSEC electrolytes at different concentrations were collected. The 17O peaks shift downfield with increasing AlCl3 concentration, demonstrating the DSE of the Al3+ for O atoms (Fig. 2a). An obvious downfield shift for 1H peaks is observed as the concentration increases from 1 m to 3 m (Fig. 2b). When the concentration further increases to 4 m, only a slight upfield shift occurs, suggesting that the deshielding effect reaches a maximum at this concentration. However, at 5 m and 5.3 m (saturation), a more noticeable upfield shift emerges, which can be attributed to the reorganization of the water coordination environment at high ion concentration28. The gradual increase in the chemical shift and broadening of the line width indicate enhanced interactions between Al3+ and water at higher AlCl3 concentrations. As more water is confined by Al3+, more HB networks are destroyed, frustrating the freezing of the electrolyte. The reduced intensity of the NMR spectra for 17O and 1H is related to the increased viscosity (Supplementary Fig. 14). Furthermore, a shift toward higher fields with increasing AlCl3 content can be observed in the 27Al NMR spectra of the electrolytes (Fig. 2c), indicating increased electron density around Al3+ due to Al3+‒water interactions.

a 17O NMR, b 1H NMR and c 27Al NMR spectra of different AlCl3 concentration electrolytes. d Raman spectra of H2O and different AlCl3 concentrations. e Proportion of IW, MW and NW areas. f Superimposed 2D LF 1H T1‒T2 mapping of different AlCl3 concentrations. g Calculated HB number between water molecules. h Calculated MSD versus time profiles of the water.
NMR spectra provide an average overview of the water structure, and more detailed insights into the water structure can be obtained from Raman spectra7. As Fig. 2d shows, the v-O-H peaks of water are decomposed into three components, each corresponding to three types of water molecules: network water (NW) at ~3210 cm−1, intermediate water (IW) at ~3410 cm−1 and multimer water (MW) at ~3630 cm−1. NWs exhibit strong HBs between water molecules, forming tetrahedral structures that favor the formation of larger water clusters; IWs have moderate HBs, allowing connections with other water molecules and leading to medium-sized aggregates; MWs feature weaker HBs between water molecules, exist in free or oligomeric forms and lack the characteristic formations of HB clusters29,30. With increasing AlCl3 concentration, the O-H peaks provide a clear blue shift and peak narrowing, indicating progressive weakening of HBs and a transition from NW to MW. Quantitative analysis (Fig. 2e) shows that higher salt concentrations increase the MW fraction while reducing NW and IW populations, reflecting increased HB disruption and higher entropy of the electrolyte. According to thermodynamic theory, a high-entropy electrolyte is conducive to achieving a low Tf1. As demonstrated from the MD simulations, compared to pure water, the coordination environment in AlCl3 solutions becomes more complex, corresponding to the higher entropy in Al3+ containing electrolytes (Supplementary Fig. S16). These simulation results corroborate the Raman results, indicating a reduced fraction of network water and a higher degree of disorder in the electrolyte, consistent with an elevated entropy state and a lower Tf.
1H low-field nuclear magnetic resonance (1H LF‒NMR) provides insights into the relaxation dynamics of water molecule mobility by monitoring H activity in water13,31. As the AlCl3 concentration increased, the T2 value gradually decreased, indicating that the activity of the water was restricted (Supplementary Fig. 17). Superimposed 2D LF 1H T1‒T2 maps were further examined, and the ratio of T1 to T2 was calculated to investigate the mobility of water molecules32,33. A higher T1/T2 value indicates lower mobility. The pure water solution has equal values of T1 and T2 at the diagonal position, with a T1/T2 value of 1. Notably, the value of T1/T2 increased with increasing AlCl3 concentration, reflecting enhanced binding of water molecules by Al3+ and reduced molecular mobility (Fig. 2f). This suppresses the rearrangement of water molecules to form a continuous HB network with a much lower Tf. Additionally, temperature-dependent 1H LF‒NMR tests track dynamic changes in water molecules in situ, highlighting the antifreezing performance of AlCl3 (Supplementary Fig. 18).
MD simulations were conducted to assess the impact of Al3+ on the HB structure and diffusion kinetics of water molecules. The average number of HBs among water molecules is significantly reduced with Al3+, confirming disrupted HB networks and a decrease in water cluster size (Fig. 2g). The mean square displacement (MSD) of water in 4Al shows a striking decrease within 100 ns, demonstrating the low diffusion kinetics of the water molecules (Fig. 2h). The combination of the above experimental spectral and theoretical calculation analyses verified the mechanism of the cation effect for low Tf. The introduced DSEC can lead to DSE on O and H atoms synchronously, which greatly destructs the HBs between water molecules, reduces the size of the water cluster and increases the electrolyte entropy, thus achieving a low Tf at a relatively low salt concentration.
Dual-cation competition and co-deposition effect
For designing low-temperature electrolytes, the high antifreezing property is not the only factor crucial for battery operation at low temperatures, and dynamic ion migration also needs to be considered. As NMR spectroscopy and DFT calculations reveal, the strong interactions between trivalent Al3+ and water in the DSEC system effectively lower the freezing point (Tf). However, these strong interactions concurrently hinder ion migration and increase the energy barrier for desolvation.
As reported, dual-cation electrolytes can promote ion diffusion kinetics34. The introduced secondary cation can form a competitive relationship with the first cation to coordinate the solvent molecule for cation effect optimization. Therefore, to further increase the ion migration rate and lower the desolvation barrier of the cation, ZnCl2 was introduced as a secondary salt into 4Al to form dual-cation electrolytes (DCEs). To regulate the cation-solvent interactions, different concentrations of ZnCl2 were added at 1 m, 2 m and 3 m, denoted as DCE41, DCE42 and DCE43, respectively. The corresponding total ion concentrations in these systems are 3.8, 4.8, and 5.8 m. Before delving into the discussion of regulated interactions in DCEs, the antifreezing capability was first determined. All three DCEs remained unfrozen after 12 h at −70 °C (Supplementary Fig. 19a, b), demonstrating significant antifreezing performance. DSC results (Supplementary Fig. 19c) further confirm that DCE41, DCE42 and DCE43 maintain low Tf values of −111 °C, −107 °C, and −105 °C, respectively, slightly higher than that of 4Al (−117 °C).
The reconfigured electrolyte structure in DCEs was further investigated. 1H NMR spectra of pure ZnCl2 are recorded in Supplementary Fig. 20. Compared with those of the pure water solution, the 1H chemical shift gradually decreased with increasing ZnCl2 concentration, indicating the SE. In contrast, the 1H peaks exhibit persistent downfield shifts in DCEs (Fig. 3a). This reveals that DSEC (Al3+) rather than SEC (Zn2+) is the main contributor to the breaking of HBs between water molecules in DCEs. Furthermore, compared with those of 4Al, the 1H peaks of the DCEs shift downfield with increasing DSE concentration, illustrating that the HB structure and interactions of Al3+‒H2O are altered by the introduction of ZnCl2. Distinct from single salt systems, three additional single peaks are detected in DCEs, indicating that a solvation structure of [Al‒Zn(H2O)x]5+ was formed35,36. With the addition of ZnCl2, the electrolyte structure is restructured through the competitive coordination effect between the dual cations. This could regulate the cation‒water interactions and improve the ion desolvation kinetics during the electrochemical process. Moreover, in DCEs, the incorporation of Zn2+ further diversifies the coordination environment by introducing Zn‒Ow interactions, resulting in even higher structural disorder relative to the 4Al system (Supplementary Fig. 21). Rheological measurements showed that the viscosity of DCE41 slightly decreases compared to 4Al, and remains relatively stable across increasing ZnCl2 concentrations (Supplementary Fig. 22). These suggest that the introduction of ZnCl2 does not significantly affect the electrolyte viscosity, which ensures the high ionic conductivity of the DCEs37. The ionic conductivities of the 4Al and DCEs were then characterized at −80 to 25 °C (Supplementary Fig. 23). The DCEs deliver high ionic conductivity when the temperature drops below −70 °C. As shown, DCE41 and DCE42 exhibit the highest ionic conductivities of 1.16 mS cm−1 and 0.12 mS cm−1 at −70 °C and −80 °C, respectively. Therefore, DCE41 was employed for testing at temperatures above −70 °C, whereas DCE42 was selected for electrochemical tests at −80 °C.

a 1H NMR spectra of 4Al and DCEs. b LF 1H NMR curves of 4Al and DCEs. c Calculated Ea values of 4Al and DCE41. Snapshots of the MD simulation cell and the desolvation processes of cations for d 4Al and e DCE41.
Cation‒water interactions in DCEs were also determined via 1H LF‒NMR tests. The T2 peak shifts to longer relaxation times in DCEs, indicating the relatively high mobility of water molecules (Fig. 3b). Additionally, the 1H LF‒NMR curves of the ZnCl2 aqueous solution were analyzed. The T2 peaks shifted to lower values for ZnCl2 solutions than for water, demonstrating that the higher T2 values in DCEs are attributed to the reduced interactions between Al3+ and water after the introduction of ZnCl2 (Supplementary Fig. 24). The desolvation kinetics of the cations were then assessed by the activation energy (Ea) and calculated via the Arrhenius equation20. The Ea values are 70.0 and 60.4 kJ mol−1 in 4Al and DCE41, respectively (Fig. 3c), confirming the low desolvation energy barrier in DCE41 due to improved water mobility caused by interaction regulation in DCEs. Moreover, in situ electrochemical impedance spectroscopy (EIS) combined with distribution of relaxation time (DRT) analysis revealed that the interfacial charge transfer resistance in the dual-cation system remains consistently lower than that of 4Al across the temperature range from 25 to −60 °C (Supplementary Fig. 25). This clearly demonstrates faster interfacial charge transfer kinetics in DCE41, which plays a critical role in maintaining robust electrochemical performance at subzero temperatures38.
To elucidate the water molecule configuration and cation solvation structure evolution in different electrolytes, MD simulations were further conducted. In pure aqueous solution, water molecules present a typical tetrahedral structure due to HB interactions (Supplementary Fig. 26). This HB structure tends to form an extended and ordered HB network at lower temperatures, resulting in a higher Tf. Nevertheless, in 4Al, Al3+ coordinates with water molecules and Cl‒ ions in the first solvation shell (Fig. 3d). Upon introducing of ZnCl2, the snapshot reveals that Al3+ is coordinated by water molecules and Cl‒ ions, whereas Zn2+ is coordinated by water molecules and Cl‒ ion. Nevertheless, the Cl‒ in the Zn solvation shell interacts with the H2O in the Al3+ solvation shell, resulting in a dual-cation coordination solvation structure (Fig. 3e), in accordance with the 1H NMR results in Fig. 3a. Radial distribution functions (RDFs) and coordination number distribution functions (CNDFs) further validate this structure. Two sharp peaks can be obtained at 1.85 Å and 2.36 Å for Al‒O and Al‒Cl in 4Al from the RDF curves (Supplementary Fig. 27). Additionally, the average coordination numbers (ACNs) for water molecules and Cl‒ in the Al3+ solvation structure are 4.52 and 1.48 for 4Al, respectively. For DCE41, the ACNs of Al‒O slightly decrease from 4.52 to 4.36, and those of Al‒Cl increase from 1.48 to 1.64, which is attributed to the interactions between Cl‒ (in the Zn2+ solvation shell) and water (in the Al3+ solvation shell). The MD simulation results for 1Zn are presented in Supplementary Figs. 28 and 29. As shown, in the Zn2+ solvation shell, the ACN of water molecules decreases from 5.64 (1Zn) to 5.23 (DCE41), whereas the ACN of Cl‒ increases from 0.36 (1Zn) to 0.77 (DCE41). This indicates a regulated solvation shell for Zn2+, which is beneficial for faster desolvation dynamics and improved side reaction inhibition on the Zn electrode.
The desolvation energies, derived from the molecular geometries (Fig. 3d, e), are assessed for the step-by-step desolvation processes. For 4Al, high desolvation energies are imperative for removing both the first and the last coordinated water molecules from hydrated Al3+. In contrast, the desolvation energies are lower in the dual-cation coordinated solvation environment, implying facilitated desolvation processes in DCEs. This finding is consistent with the lower activation energy (Ea) observed in Fig. 3c. A schematic summary of the DCE interactions is presented in Supplementary Fig. 30, depicting the enhanced thermodynamics and kinetics of the electrolyte with high entropy, significantly low Tf, and improved ion deposition behavior.
Electrochemical properties of DCE for Zn
The electrochemical properties of DCEs were evaluated at low temperatures. Zn metal (~ 22 µm) was employed as the negative electrode owing to its low cost and high compatibility with aqueous electrolytes. Thus far, the side reactions at the Zn/electrolyte interface and Zn dendrite growth degrade the cycle time of Zn‒based batteries. Symmetrical Zn cells and asymmetrical Zn||Cu cells were assembled to assess the impact of DCEs on the long-term Zn plating/stripping stability of Zn metal. At −60 °C, the symmetrical cell can operate for more than 3100 h with a stable voltage at a current density of 0.2 mA cm−2 and an area capacity of 0.2 mA h cm−2, indicating the favorable stability of Zn metal in collaboration with DCE41 (Supplementary Fig. 31). The symmetrical cells were also performed at higher current density and area capacity. With DCE41, the symmetrical cell remained stable for more than 718 h (0.4 mA cm−2, 0.4 mA h cm−2), 1014 h (0.2 mA cm−2, 0.6 mA h cm−2) and 740 h (0.2 mA cm−2, 0.8 mA h cm−2), respectively (Supplementary Fig. S32). Even at a high current density of 1 mA cm−2 and an area capacity of 0.25 mA h cm−2, the symmetrical cell can still function for more than 540 h (Supplementary Fig. 33). Additionally, the symmetrical battery exhibits stable voltage curves at various current densities, and the voltage can be recovered immediately when the current density returns from 1 to 0.2 mA cm−2, verifying the favorable rate performance and improved kinetic behavior of DCE41 (Supplementary Fig. 34). The positive effect of the DCEs on the long-term stability of Zn metal was also proven by shelving-recovery experiments, in which the symmetrical cell was operated at 0.2 mA cm−2 and 0.2 mA h cm−2 with a 20 h shelve for every 10 Zn plating/stripping cycles to simulate practical conditions (Fig. 4a). The cell was stable for more than 10,340 h (over 430 days), confirming that DCE41 can stabilize Zn metal and inhibit electrochemical corrosion, static corrosion and dendrite growth in practical applications. The asymmetrical Zn | |Cu cell was further assembled to accurately quantify the reversibility of the Zn metal with DCE41 during the plating/stripping process. As shown in Fig. 4b, the asymmetrical cell with DCE41 can exhibit a high average Coulombic efficiency (CE) of 99.56% and long Zn plating/stripping stability for more than 1280 cycles (>2560 h). Furthermore, owing to the strong antifreezing properties of DCE41, the symmetrical battery can exhibit Zn plating/stripping performance for more than 1100 h at −70 °C (Fig. 4c). The symmetrical battery performance is also assessed at 25 °C (Supplementary Fig. 35). At the high current density and area capacity of 3 mA cm−2, 3 mA h cm−2 and 5 mA cm−2, 5 mA h cm−2, the cells with DCE41 present reversible Zn plating/stripping for more than 270 h and 150 h, realizing the high depth of discharge (DOD) of Zn metal for 23.3% and 38.8%, respectively. In terms of four critical parameters, including areal capacity, cumulative capacity, DOD and operating temperatures, the metrics of DCE41 have exceeded those of most aqueous Zn-based systems reported to date (Supplementary Fig. 36 and Table 3)18,39,40,41,42.

a Galvanostatic voltage profiles of the symmetrical Zn cells at 0.2 mA cm−2 and 0.2 mA h cm−2 with a 20 h shelve for every 10 cycles at −60 °C. b CE performance of the asymmetrical Zn | |Cu cell at 0.2 mA cm−2 and 0.2 mA h cm−2 at −60 °C. c Galvanostatic voltage profiles of the symmetrical Zn cells at 0.2 mA cm−2 and 0.2 mA h cm−2 at −70 °C. d Cross-sectional TEM image of Zn metal after 100 plating/stripping cycles at 0.2 mA cm−2 and 0.2 mA h cm−2 with the deposited capacity of 20 mA h cm−2 at −60 °C in DCE41. The associated e line-scan profiles and f EDS maps of Al and Zn. g The associated HRTEM images of the Al‒Zn alloy and Zn metal. XPS spectra of h Al 2p and i Zn 2p for the Zn metal after 100 plating/stripping cycles at 0.2 mA cm−2 and 0.2 mA h cm−2 with the deposited capacity of 20 mA h cm−2 at −60 °C in DCE41.
Interfacial chemistry of Zn metal cycled in DCE
The mechanism underlying Zn metal stabilization was investigated by analyzing the morphological and structural evolution of Zn after Zn plating/stripping in DCE41. Transmission electron microscopy (TEM) revealed that a dense and uniform interphase layer with a thickness of ~ 200 nm can be observed on the Zn metal after 100 cycles (Fig. 4d). Close contact and obvious phase boundaries are maintained between the interphase layer and the Zn metal. The line-scan profiles and corresponding energy-dispersive X-ray spectroscopy (EDS) maps show that the deposition layer was composed of uniformly distributed Al and Zn (Fig. 4e, f and Supplementary Fig. 37), illustrating the formation of the Al‒Zn alloy on the Zn metal. High-resolution TEM (HRTEM) images revealed the amorphous Al‒Zn alloy phase near the phase boundary (Supplementary Fig. 38). The lattice spacing of the Al‒Zn alloy layer was 0.329 nm, which differed from the 0.250 nm spacing of the (101) plane of the fine lattice fringes in the Zn phase (Fig. 4g). Additionally, the selected area electron diffraction (SAED) pattern for the deposition layer shows broad and weak diffusion rings, verifying the formation of the Al‒Zn alloy amorphous phase during Zn plating/stripping (Supplementary Fig. 39). Field emission scanning electron microscopy (FESEM) images also revealed a relatively smooth metal deposition morphology and uniform Al and Zn distributions on the cycled Zn metal (Supplementary Fig. 40)43. Comparative tests with 4Al confirmed that DCE41 facilitates the formation of a more uniform and stable Al–Zn alloy layer (Supplementary Figs. 41–43), highlighting the advantage of the dual-cation strategy.
X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD) were used to further characterize the structure of the Al‒Zn alloy on the Zn metal after Zn plating/stripping with DCE41. The XPS spectra clearly show the existence of Al on the cycled Zn metal, demonstrating the alloying process during Zn plating/stripping (Fig. 4h). Furthermore, the Zn 2p peaks of cycled Zn metal shift to lower binding energies than those of bare Zn, suggesting that the electronic states of Zn change during the alloying process with Al (Fig. 4i)43. Additionally, the peaks shift, and the I(002)/I(100) growth in the XRD patterns of the cycled Zn metal confirms the formation of an Al‒Zn alloy (Supplementary Fig. 44)44. The uniform Al‒Zn alloy layer enhances resistance to corrosion reactions and dendrite growth, thereby improving the long-term stability of the Zn metal.
Electrochemical performance in full batteries
To evaluate the electrochemical properties of DCEs at low temperatures, polyaniline (PANI) was selected as the positive electrode material since its charge storage is dominated by surface redox reactions rather than bulk Zn2+ intercalation. This feature makes the electrochemical behavior relatively less dependent on ion diffusion, thereby reducing temperature sensitivity compared with conventional inorganic positive electrodes27. As Supplementary Fig. 45 shows, the redox peak positions and shapes of the CV curves in DCE41 closely resemble those in the 4Al electrolyte rather than in 1Zn. To quantitatively assess ion diffusion, diffusion coefficients were calculated with Randles–Sevcik equation45. Comparing to 4Al, the diffusion coefficients of the corresponding ions (peak 1) in DCE41 were increased from 3.61 × 10−8 to 6.00 × 10−8 cm2 s−1. A trend that is further corroborated by the GITT results (Supplementary Fig. 46). To more directly differentiate the diffusion behaviors of the ions, MD simulations were performed (Supplementary Fig. 47). In the presence of Zn2+ (DCE41 electrolyte), the diffusion coefficient of Al3+ is increased compared to the 4Al system. This observation is consistent with the CV and GITT conclusions and further corroborates that the dual-cation electrolyte design effectively enhances the diffusion dynamics of Al3+.
The assembled Zn | |PANI full battery with DCE41 delivers high discharge specific capacities of 174.5, 160.5, 144.5, 121.0, 100.7 and 66.9 mA h g−1 at 0.2, 0.5, 1, 2, 3 and 5 A g−1, respectively, at −60 °C (Fig. 5a). Furthermore, the specific capacity of 171.8 mA h g−1 can be recovered when the specific current immediately returns from 5 A g−1 to 0.2 A g−1, indicating favorable rate performance (Supplementary Fig. 48). In contrast, the Zn | |PANI battery with 4Al exhibits specific capacities of 164.3, 71.8, 40.5, 21.4, 7.8, and 3.9 mA h g−1 at the same specific currents, respectively. Additionally, when the specific current is cycled back from 5 A g−1 to 0.2 A g−1, only 100.4 mA h g−1 is retained, demonstrating poor rate performance (Supplementary Fig. 49). The long-term cycling performance was further tested. At 0.5 A g−1, the full battery with DCE41 presents prominent cycling stability and delivers a high specific capacity of 113.8 mA h g−1 after more than 750 cycles, with a high CE of 99.5% (Supplementary Fig. 50). More strikingly, the DCE41-assisted full battery can achieve high capacity retentions of 95.0% and 80.0% after 2000 and 12,000 cycles, even at 5 A g−1 (Fig. 5b). However, due to the higher desolvation energy barrier, the capacity at 5 A g−1 for the 4Al electrolyte is only 2.9 mA h g−1, much lower than that of DCE41 (Supplementary Fig. 51). In addition, the battery is stable for many cycles at −70 °C, and 100% capacity retention can be achieved after 700 cycles at 0.5 A g−1 (Supplementary Fig. 52). The Zn | |PANI pouch battery was tested to further assess the advantages of the designed DCE. A pouch cell with a high PANI loading of 7.0 mg cm−2 can deliver stable cycling performance with a high capacity of 1.74 mA h after 500 cycles at 100 mA g−1 at −70 °C (Fig. 5c). At an low temperature of −80 °C, the battery with DCE42 presented a high specific capacity of 115.5 mA h g−1 at 20 mA g−1 after 85 cycles (Supplementary Fig. 53). The full battery also demonstrated long cycling performance over a wide temperature range. At 25 °C and 50 °C, DCE41 enables batteries with high capacities of 112.5 mA h g−1 and 127.9 mA h g−1 after 1000 and 200 cycles, respectively (Supplementary Fig. 54). These long cycling performances can be attributed to the cation effect, which results in the strong antifreezing ability of the electrolyte, improved desolvation kinetics of the cations, and the formation of a uniform Al‒Zn alloy layer in situ. Furthermore, the universality of the dual-cation competition strategy was validated in the Zn | |Zn0.25V2O5 battery (Supplementary Fig. 55). Compared with the reported low‒temperature aqueous battery systems, the full battery performance under the operation temperatures in this work is competitive (Fig. 5d and Supplementary Table 4)1,11,12,13,18,27,29,38,39,40,41,42,46,47,48,49,50,51,52,53,54,55,56,57,58, and shows potential for practical applications in cold conditions, such as in North and South Poles environments.

a Galvanostatic charge/discharge curves at different specific currents at −60 °C. b Cycling performance at 5 A g−1 at −60 °C. c Pouch battery cycling performance at 100 mA g−1 at −70 °C. d Comparisons of the aqueous Zn-based battery performance in this work with that of previously reported electrolytes at low temperatures (≤−40 °C).
Discussion
In this paper, we propose the cation effect for designing aqueous electrolytes that achieve high low-temperature tolerance and good electrochemical performance. We highlight the interaction regulation behavior between cations and water molecules, which enables both the O and H atoms in water to simultaneously experience the deshielding effect, thereby significantly reducing the formation of HBs between water molecules and lowering the Tf of the electrolyte. An low Tf of −117 °C can be achieved in an AlCl3 aqueous solution with a low electrolyte salt concentration of 2.8 m. Moreover, the introduction of a second cation can further regulate the interaction between cations and water molecules to optimize ion deposition kinetics and promote the formation of in situ corrosion-resistant alloy layers on the Zn metal surface. As a proof of concept, the Zn-based battery composed of a cation effect-based DCE demonstrated a long Zn metal lifespan (>10,340 h, over 430 days) at −60 °C, favorable stable cycling performance for the Zn | |PANI pouch battery (>500 cycles) at −70 °C and a high specific capacity (115.5 mA h g−1) at −80 °C. Freeze-resistant aqueous electrolytes based on the cation effect provide an effective strategy for the application of low-cost, highly safe aqueous battery energy storage systems in harsh environments.
Methods
Materials, electrolytes and positive electrodes
Aluminum chloride hexahydrate (AlCl3·6H2O, 97.0%) and zinc chloride (ZnCl2, 99.0%) were purchased from Macklin. Lithium chloride (LiCl, 99%), sodium chloride (NaCl, 99.5%), calcium chloride (CaCl2, 98%) and magnesium chloride (MgCl2, 99.0%) were purchased from Shanghai Titan Technology Co., Ltd. Deionized water was obtained from the Millipore reverse osmosis water purification system. AlCl3 electrolytes with different concentrations were obtained by dissolving a specified quantity of AlCl3·H2O in deionized water at an ambient temperature of 23 ± 2 °C, followed by continuous stirring until complete dissolution and subsequent resting for 6 h prior to testing. The DCEs were prepared by adding a specified amount of ZnCl2 salt to a 2.8 m (m: mol kg−1) AlCl3 electrolyte. Zn foils (>99.99% purity, thickness: 22 μm; Qingyuan Metal Co., Ltd.) were used as the negative electrodes, and Cu foils (>99.99% purity, thickness: 9 μm; Canrd Technology Co., Ltd.) were employed for asymmetrical cells assembly. The positive electrode of polyaniline (PANI) was prepared via in situ polymerization method59. Aniline monomer (Sigma-Aldrich Co., Ltd., 99.5%) was dissolved in 1 M hydrochloric acid (HCl, Sinopharm, 36.0 ~ 38.0%) solution and cooled in an ice bath. Ammonium persulfate (APS, Sigma-Aldrich Co., Ltd., 98%) was added as the oxidizing initiator under stirring for 1 h, followed by centrifugation to collect the precipitate. The product was dried at 60 °C for 12 h to obtain PANI powder. The Zn0.25V2O5 powder was prepared by hydrothermal method60. V2O5 (Sinopharm, 99.99%) was dispersed in a mixed solution of deionized water and acetone, followed by the addition of zinc acetate (Aladdin, 99.0%). The mixture was sealed in a Teflon-lined autoclave and heated at 180 °C for 24 h. The product was washed with deionized water and isopropanol (Sinopharm, 70%), then dried at 60 °C for 24 h. Both PANI and Zn0.25V2O5 electrodes were prepared by mixing the active material, acetylene black (Guangdong Canrd New Energy Technology Co., Ltd., 99.9%), and polyvinylidene fluoride (PVDF, Canrd Technology Co., Ltd., 99.5%, molecular weight 1,200,000) at a mass ratio of 6:3:1 in N-methyl-2-pyrrolidone (NMP, Sigma-Aldrich Co., Ltd., 99.5%) to form a homogeneous slurry. The slurry was coated onto carbon cloth (Ce-Tech Co., Ltd., W0S1011, thickness: 0.35 mm) using an automatic coating machine (Pushen Testing Instrument (Shanghai) Co., Ltd., model: AFA-l Automatic Film Applicator), dried at 60 °C for 12 h, and cut into 1 cm × 1 cm squares using a paper cutter (Deli Group Co., Ltd., model: 8016) for cell assembly. The prepared PANI and Zn0.25V2O5 electrodes were stored in sealed glass bottles at 23 ± 2 °C prior to use.
Characterizations
Unless otherwise specified, all measurements were conducted at an ambient temperature of 23 ± 2 °C. 1H NMR spectra were collected with AVANCE III 400 MHz equipment via the external standard method: a coaxial double tube was employed, where 1000 µL of D2O was added to the inner tube for field frequency locking for NMR testing, and an electrolyte sample of 500 µL was added to the outer tube as the test object. 17O and 27Al NMR spectra were also recorded on AVANCE III 400 MHz equipment without field locking, in which 600 µL of electrolyte was added to a single tube with no deuterated solvents. Raman spectra were measured on a Thermo Fisher Scientific DXR2xi instrument with 532 nm excitation, and the electrolyte samples were sealed in capillary tubes for testing. LF 1H NMR spectra and 2D LF 1H T1‒T2 mapping were conducted on a VTMR20-010 V-I NMR analyzer with a 1H NMR probe (Suzhou Niumag Analytical Instrument Corporation). In situ temperature-dependent T2 relaxation spectra were obtained from 25 to −80 °C. DSC was carried out on a differential scanning calorimeter (204F1, Netzsch) at 25 ~ −150 °C at a cooling rate of 10 K min−1. The viscosity of different electrolytes was evaluated using a Rotary Rheometer (MARS 60). XPS spectra were recorded on an Escalab 250Xi. XRD patterns were determined by an X-ray diffractometer (Rigaku D/max-2550VB) with Cu Kα radiation. High-resolution TEM images, corresponding EDS maps, line-scan profiles and SAED patterns were obtained via field-emission electron microscopy (FEI, Tecnai G2 F20 S-Twin), and the tested Zn metal samples were prepared via ultramicrotomy. FESEM and EDS mapping images were acquired by scanning electron microscopy (Hitachi SU8230).
Electrochemical measurements
CR2016 coin-type symmetrical Zn cells and asymmetrical Zn | |Cu cells were assembled with one sheet of glass fiber A (GF/A, Whatman, lateral dimension: φ = 19 mm, 260 µm, average pore size: 1.6 µm, porosity: >90%) as the separator. The cell case and spring (CR20 type; diameter: 15.4 mm, thickness: 1.2 mm) were fabricated from stainless steel. Electrodes (Zn or Cu; 1 cm × 1.00 cm) were prepared in single pieces. Electrolyte (90 μL) was dispensed using a calibrated micropipette (DRAGONLAB) with polypropylene tips. After assembly at an ambient temperature of 23 ± 2 °C, the cells were sealed using a hydraulic crimping machine (MTI, MSK-110) and rested for 4 h at the corresponding testing temperature prior to electrochemical measurements. The Zn | |PANI or Zn | |Zn0.25V2O5 full batteries were assembled via Swagelok blocking cells with an inner diameter of 14 mm, a polytetrafluoroethylene (PTFE) body (length: 76 mm), and titanium rods (99.5% purity, φ = 14 mm) as current collectors. Each cell contained one single-side-coated positive electrode (cut into φ = 14 mm discs) and one Zn electrode. The separator was a φ = 14 mm GF/A disc with 70 μL electrolyte. For Zn | |PANI pouch battery, one Zn electrode (2.45 cm × 3.45 cm) and one PANI electrode (2.45 cm × 3.45 cm, single-side-coated on carbon cloth) were used, with a GF/A separator (2.50 cm × 3.50 cm). The components were stacked in the order Zn electrode–GF/A–PANI electrode, with the carbon cloth serving as the current collector for the positive electrode. A total of 300 μL of electrolyte was injected before sealing with an MSK-115A-MS vacuum sealer (Shenzhen Kejing Star Technology Co., Ltd.). The cell was rested at 25 °C for 4 h, then at −70 °C for 4 h before testing. No external pressure was applied during cycling. The discharge‒charge profiles of all the batteries were obtained on a Neware battery test system (CT-4008Tn) in a test chamber (YDC-300HT) at 25 °C; a high- and low-temperature test chamber (BTC-706) at 50 °C, −60 °C and −70 °C; and low-temperature refrigerators (Jie Sheng, DW-86W28) at −80 °C. The ionic conductivities of the electrolytes were measured via EIS curves in symmetrical Ti Swagelok-type cells at Autolab 204 (Metrohm) at 25 ~ −80 °C and were calculated via Eq. (1):
where l is the electrolyte thickness, R corresponds to the high-frequency intercept of the EIS curves, and S represents the electrolyte area.
EIS measurements were performed on a CHI 660 electrochemical workstation. Prior to testing, the cells were rested for 30 min at the target measurement temperature to reach a stable open-circuit condition. A perturbation with an amplitude of 5 mV was applied over a frequency range of 0.1–100,000 Hz. A total of 73 data points were collected across the scanned frequency range. EIS curves of the Zn cells with different electrolytes were also obtained to calculate the Arrhenius activation energy (Ea) with Eq. (2):
where Rct is the interfacial transfer resistance; A and R are the frequency factor and gas constant, respectively; and T is the Kelvin temperature.
Computational methods
The density functional theory (DFT) calculations were performed in ORCA package with the B3LYP functional and Grimme’s D4 dispersion correction61,62,63,64,65,66,67. The def2-TZVP basis set was employed for all atoms, along with the def2/J auxiliary basis set for Coulomb fitting and the RIJCOSX approximation for efficient exchange integrals68,69. The geometry optimization and frequency calculations were conducted with very tight SCF convergence criteria70. The convergence criteria were set to normal. The conductor-like polarizable continuum model (CPCM) with the SMD solvation model was used to account for solvent effects, with water selected as the solvent.
The binding energy (ΔEbind) and desolvation energy (ΔEdesol) were calculated via the following Eq. (3):
where EAB represents the total energy of the cation-water molecules, EA represents the energy of the cation, and EB represents the energy of the water molecules.
The solvation energy (ΔGsol) and the desolvation energy (ΔGdesol) were calculated via the following Eq. (4):
For solvation energy, GAB represents the total Gibbs free energy of the solvation structure, GA represents the energy of the cation, and GB represents the Gibbs free energy of the water molecules.
For desolvation energy, GAB represents the total Gibbs free energy of the initial solvation structure, GA represents the Gibbs free energy of final solvation structure, and GB represents the Gibbs free energy of the water molecules in the absence of the cation.
Molecular electrostatic potential (ESP) was calculated and visualized using Gaussian16 software package71. The coordinate file of different ions with H2O is in Supplementary Data 1–5.
Molecular dynamics (MD) simulations were performed using the GROMACS package72. For the MD simulations of different cation systems, the Amber99SB-ILDN force field was employed in conjunction with the TIP4P-Ew water model73,74. For ions not covered by the Amber99SB-ILDN force field75,76,77, the missing parameters were defined and supplemented using the General Amber Force Field (GAFF) force field75. The simulation systems consisted of 4000 H2O molecules with 80 AlCl3 (or 80 ZnCl2, 80 MgCl2, 80 CaCl2, 80 LiCl, or 80 NaCl) or 3200 H2O molecules with 160 AlCl3 (or 40 ZnCl2, or 160 AlCl3 + 40 ZnCl2) in a 5 × 5 × 5 nm3 cubic supercell. Before initiating the MD simulation, the initial models were relaxed using the steepest descent algorithm for energy minimization, with a convergence criterion of 1000.0 kJ/mol nm (emtol) and a maximum step size of 0.002 nm (emstep) over 2,500,000 steps (nsteps). Following minimization, an isothermal-isobaric (NPT) ensemble simulation was conducted to equilibrate the system. The NPT simulation utilized the molecular dynamics algorithm with a total of 5,000,000 steps and a time step of 2 fs (dt), corresponding to a total simulation time of 10 ns. Temperature was controlled at 293.15 K using the V-rescale thermostat with a coupling time constant of 0.1 ps, and pressure was maintained at 1.0 bar using the Berendsen barostat with a time constant of 1.0 ps and a compressibility of 4.5 × 10−5 bar−1. Electrostatic interactions were treated using the particle mesh Ewald (PME) method with a Fourier spacing of 0.12 nm, and van der Waals (vdW) interactions were modeled with a cutoff of 1.0 nm. Periodic boundary conditions (PBC) were applied in all directions. Notably, the density obtained from NPT simulations exhibits good agreement with experimental measurements. The experimentally determined density of 4Al is 1.27 g cm−3, while the simulated density is 1.21 g cm−3. Subsequently, a canonical (NVT) ensemble simulation was performed to further equilibrate the system. The NVT simulation ran for 50,000,000 steps (totaling 100 ns) with a time step of 2 fs. The system temperature was maintained at 293.15 K using the V-rescale thermostat with a coupling time constant of 0.1 ps. A grid-based neighbor list approach was employed, with a neighbor list cutoff of 1.0 nm and updates performed every 20 steps. The hydrogen bonds were recognized if the donor‒acceptor distance was ≤0.35 nm and the donor‒hydrogen‒acceptor angle was ≥30° in the simulated systems12. A 10 ns statistical analysis was conducted for different cationic systems. The same method was applied at 253.15 K and 273.15 K. The same simulation protocol was applied at additional temperatures of 253.15 K and 273.15 K to investigate temperature-dependent behavior. The corresponding initial and final configurations of different ions systems are in Supplementary Data 6–12.
Data availability
The datasets produced in this study are available in this article and in the Supplementary Information. Additional data or analyses are available from the corresponding authors upon request. Theoretical computational data files are also provided. Source data are provided with this paper.
References
Jiang, L. et al. Rational design of anti-freezing electrolytes for extremely low-temperature aqueous batteries. Nat. Energy 9, 839–848 (2024).
Yang, C. et al. All-temperature zinc batteries with high-entropy aqueous electrolyte. Nat. Sustain 6, 325–335 (2023).
Chao, D. et al. Roadmap for advanced aqueous batteries: from design of materials to applications. Sci. Adv. 6, eaba4098 (2020).
Jiang, L., Hu, Y.-C., Ai, F., Liang, Z. & Lu, Y.-C. Rational design of anti-freezing electrolyte concentrations via freeze concentration process. Energy Environ. Sci. 17, 2815–2824 (2024).
Zhang, Q. et al. Chaotropic anion and fast-kinetics cathode enabling low-temperature aqueous Zn batteries. ACS Energy Lett. 6, 2704–2712 (2021).
Liu, S. et al. From room temperature to harsh temperature applications: fundamentals and perspectives on electrolytes in zinc metal batteries. Sci. Adv. 8, eabn5097 (2022).
Li, R., Jiang, Z., Shi, S. & Yang, H. Raman spectra and 17O NMR study effects of CaCl2 and MgCl2 on water structure. J. Mol. Struct. 645, 69–75 (2003).
Sun, T., Nian, Q., Ren, X. & Tao, Z. Hydrogen-bond chemistry in rechargeable batteries. Joule 7, 2700–2731 (2023).
Emamian, S., Lu, T., Kruse, H. & Emamian, H. Exploring nature and predicting strength of hydrogen bonds: a correlation analysis between atoms-in-molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory. J. Comput. Chem. 40, 2868–2881 (2019).
Marcus, Y. Effect of ions on the structure of water: structure making and breaking. Chem. Rev. 109, 1346–1370 (2009).
Sun, T., Zheng, S., Du, H. & Tao, Z. Synergistic effect of cation and anion for low-temperature aqueous zinc-ion battery. Nano Micro Lett. 13, 204 (2021).
Qiu, M. et al. Tailoring water structure with high-tetrahedral-entropy for antifreezing electrolytes and energy storage at -80 °C. Nat. Commun. 14, 601 (2023).
Zhang, Q., Lu, Y., Liu, X., Xie, W. & Chen, J. Nonaggregated anions enable the undercooled aqueous electrolyte for low-temperature applications. J. Am. Chem. Soc. 146, 12743–12749 (2024).
Huang, S., Hou, L., Li, T., Jiao, Y. & Wu, P. Antifreezing hydrogel electrolyte with ternary hydrogen bonding for high-performance zinc-ion batteries. Adv. Mater. 34, e2110140 (2022).
Mäemets, V. & Koppel, I. Effect of ions on the 17O and 1H NMR chemical shifts of water. J. Chem. Soc. Faraday Trans. 94, 3261–3269 (1998).
Ohtaki, H. & Radnai, T. Structure and dynamics of hydrated ions. Chem. Rev. 93, 1157–1204 (1993).
Bylaska, E., Valiev, M., Rustad, J. & Weare, J. Structure and dynamics of the hydration shells of the Al3+ ion. J. Chem. Phys. 126, 104505 (2007).
Shi, M. et al. Super hydrous solvated structure of chaotropic Ca2+ contributes superior anti-freezing aqueous electrolytes and stabilizes the Zn anode. Angew. Chem. Int. Ed. 63, e202407659 (2024).
Yu, X. et al. Unlocking dynamic solvation chemistry and hydrogen evolution mechanism in aqueous zinc batteries. J. Am. Chem. Soc. 146, 17103–17113 (2024).
Shi, X. et al. A weakly solvating electrolyte towards practical rechargeable aqueous zinc-ion batteries. Nat. Commun. 15, 302 (2024).
Zhang, S. J. et al. pH-triggered molecular switch toward texture-regulated Zn anode. Angew. Chem. Int. Ed. 62, e202301570 (2023).
Chen, R. et al. Rational design of an in-situ polymer-inorganic hybrid solid electrolyte interphase for realising stable Zn metal anode under harsh conditions. Angew. Chem. Int. Ed. 63, e202401987 (2024).
Cai, Z., Wang, J. & Sun, Y. Anode corrosion in aqueous Zn metal batteries. eScience 3, 100093 (2023).
Svishchev, I. & Kusalik, P. Proton chemical shift of water in the liquid state computer simulation results. J. Am. Chem. Soc. 115, 8270–8274 (1993).
Li, R., Jiang, Z., Yang, H. & Guan, Y. Effects of ions in natural water on the 17O NMR chemical shift of water and their relationship to water cluster. J. Mol. Liq. 126, 14–18 (2006).
Reuben, J. Hydrogen-bonding effects on oxygen-17 chemical shifts. J. Am. Chem. Soc. 91, 5725–5729 (1969).
Zhang, Q. et al. Modulating electrolyte structure for ultralow temperature aqueous zinc batteries. Nat. Commun. 11, 4463 (2020).
Zhao, Z. et al. Solvation structure regulation for highly reversible aqueous Al metal batteries. J. Am. Chem. Soc. 146, 2257–2266 (2024).
Zhang, R. et al. Weakly solvating aqueous-based electrolyte facilitated by a soft co-solvent for extreme temperature operations of zinc-ion batteries. Energy Environ. Sci. 17, 4569–4581 (2024).
Brubach, J. Dependence of water dynamics upon confinement size. J. Phys. Chem. B 105, 430–435 (2001).
Liu, Y., Song, Y. & Wu, P. Self-evolving hierarchical hydrogel fibers with circadian rhythms and memory functions. Adv. Mater. 36, 2404506 (2024).
Conte, P. Effects of ions on water structure: a low-field 1H T1 NMR relaxometry approach. Magn. Reson. Chem. 53, 711–718 (2015).
McConville, P. & Pope, J. 1H NMR T2 relaxation in contact lens hydrogels as a probe of water mobility. Polymer 42, 3559–3568 (2001).
Tian, H. et al. Three-dimensional Zn-based alloys for dendrite-free aqueous Zn battery in dual-cation electrolytes. Nat. Commun. 13, 7922 (2022).
Qiao, Y., Pedersen, C. M., Wang, Y. & Hou, X. NMR insights on the properties of ZnCl2 molten salt hydrate medium through its interaction with SnCl4 and fructose. ACS Sustain. Chem. Eng. 2, 2576–2581 (2014).
Chen, C. et al. NMR insights into the unexpected interaction of SnCl4 with D-glucosamine and its effect on 5-HMF preparation in ZnCl2 molten salt hydrate medium. ChemistrySelect 1, 6540–6545 (2016).
Zhu, Y. Concentrated dual-cation electrolyte strategy for aqueous zinc-ion batteries. Energy Environ. Sci. 14, 4463–4473 (2021).
Yao, L. et al. Reconstruction of zinc-metal battery solvation structures operating from -50 ~ +100 °C. Nat. Commun. 15, 6249 (2024).
Bu, F. et al. Bio-inspired trace hydroxyl-rich electrolyte additives for high-rate and stable Zn-ion batteries at low temperatures. Angew. Chem. Int. Ed. 63, e202318496 (2024).
Zha, Z. et al. Electrolyte design via cation-anion association regulation for high-rate and dendrite-free zinc metal batteries at low temperature. J. Am. Chem. Soc. 146, 31612–31623 (2024).
Xu, M. et al. Bicontinuous-phase electrolyte for a highly reversible Zn metal anode working at ultralow temperature. Energy Environ. Sci. 17, 8966–8977 (2024).
You, C. et al. An inexpensive electrolyte with double-site hydrogen bonding and a regulated Zn2+ solvation structure for aqueous Zn-ion batteries capable of high-rate and ultra-long low-temperature operation. Energy Environ. Sci. 16, 5096–5107 (2023).
Yan, C. et al. Architecting a stable high-energy aqueous Al-ion battery. J. Am. Chem. Soc. 142, 15295–15304 (2020).
Dou, Q. et al. Unveiling solvation structure and desolvation dynamics of hybrid electrolytes for ultralong cyclability and facile kinetics of Zn-Al alloy anodes. Energy Environ. Sci. 15, 4572–4583 (2022).
Gong, Q., Hou, L., Li, T., Jiao, Y. & Wu, P. Regulating the molecular interactions in polymer binder for high-performance lithium-sulfur batteries. ACS Nano 16, 8449–8460 (2022).
Yao, J. et al. Atomic level-macroscopic structure-activity of inhomogeneous localized aggregates enabled ultra-low temperature hybrid aqueous batteries. Angew. Chem. Int. Ed. 63, e202409986 (2024).
Sun, T. et al. An ultralow-temperature aqueous zinc-ion battery. J. Mater. Chem. A. 9, 7042–7047 (2021).
Shi, Y. et al. An anti-freezing hydrogel electrolyte for flexible zinc-ion batteries operating at -70 °C. Adv. Funct. Mater. 33, 2214546 (2023).
Cao, L. et al. Highly reversible aqueous zinc batteries enabled by zincophilic-zincophobic interfacial layers and interrupted hydrogen-bond electrolytes. Angew. Chem. Int. Ed. 60, 18845–18851 (2021).
Liu, D. S. et al. Regulating the electrolyte solvation structure enables ultralong lifespan vanadium-based cathodes with excellent low-temperature performance. Adv. Funct. Mater. 32, 2111714 (2022).
Cheng, M. et al. “Anions-in-colloid” hydrated deep eutectic electrolyte for high reversible zinc metal anodes. Angew. Chem. Int. Ed. 63, e202410210 (2024).
Zhang, L. et al. Aqueous zinc-ion batteries with boosted stability and kinetics under a wide temperature range. Angew. Chem. Int. Ed. 64, e202500434 (2025).
Xie, D. et al. Co-solvent and additive joint engineering enable long-life and wide-temperature Zn metal battery. Energy Storage Mater. 70, 103524 (2024).
Chen, Y. et al. Regulating interfacial reactions through electrolyte chemistry enables an anion-rich interphase for wide-temperature zinc metal batteries. Energy Environ. Sci. 18, 713–727 (2025).
Yan, C., Wang, Y., Deng, X. & Xu, Y. Cooperative chloride hydrogel electrolytes enabling ultralow-temperature aqueous zinc ion batteries by the hofmeister effect. Nano Micro Lett. 14, 98 (2022).
Xue, R. et al. Enhancing temperature adaptability of aqueous zinc batteries via antifreezing electrolyte and site-selective ZnSe-Ag interface layer design. ACS Nano 17, 17359–17371 (2023).
Xie, Z. et al. Carbonate-assisted chaotropic electrolyte for zinc ion battery with wide temperature operation. ACS Energy Lett. 9, 3380–3390 (2024).
Wang, Y. et al. Solvent control of water O−H bonds for highly reversible zinc ion batteries. Nat. Commun. 14, 2720 (2023).
Wan, F. et al. An aqueous rechargeable zinc-organic battery with hybrid mechanism. Adv. Funct. Mater. 28, 1804975 (2018).
Pan, Y., Zuo, Z., Jiao, Y. & Wu, P. Constructing lysozyme protective layer via conformational transition for aqueous Zn batteries. Adv. Mater. 36, 2314144 (2024).
Neese, F. Software update: the ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 12, e1606 (2022).
Garcia-Rates, M. & Neese, F. Efficient implementation of the analytical second derivatives of hartree-fock and hybrid DFT energies within the framework of the conductor-like polarizable continuum model. J. Comput. Chem. 40, 1816–1828 (2019).
Neese, F. Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 8, e1327 (2017).
Neese, F. et al. and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree−Fock exchange. Chem. Phys. 356, 98–109 (2009).
Izsak, R. & Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 135, 144105 (2011).
Izsak, R., Neese, F. & Klopper, W. Robust fitting techniques in the chain of spheres approximation to the Fock exchange: the role of the complementary space. J. Chem. Phys. 139, 094111 (2013).
Helmich-Paris, B., de Souza, B., Neese, F. & Izsak, R. An improved chain of spheres for exchange algorithm. J. Chem. Phys. 155, 104109 (2021).
Schäfer, A., Huber, C. & Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 100, 5829–5835 (1994).
Schäfer, A., Horn, H. & Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577 (1992).
Neese, F. Approximate second-order SCF convergence for spin unrestricted wavefunctions. Chem. Phys. Lett. 325, 93–98 (2000).
Frisch, M. J. et al. Gaussian 16 Rev. B.01. (2016).
Berendsen, H., van der Spoel, D. & Drunen, R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 (1995).
Götz, A. W. et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J. Chem. Theory Comput. 8, 1542–1555 (2012).
Salomon-Ferrer, R., Götz, A. W., Poole, D., Le Grand, S. & Walker, R. C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh ewald. J. Chem. Theory Comput. 9, 3878–3888 (2013).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
Han, P., Nie, W., Zhao, G. & Gao, P. Theoretical investigation on the structure and physicochemical properties of choline chloride-based deep eutectic solvents. J. Mol. Liq. 366, 120243 (2022).
Sousa da Silva, A. W. & Vranken, W. F. ACPYPE-antechamber PYthon parser interfacE. BMC Res. Notes 5, 367 (2012).
Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (NSFC) (Nos. 52473207 received by Y.J., 52433003 received by P.W.), Research Funds for the Central Universities (No. 2232023A-06 received by Y.J.), and Natural Science Foundation of Shanghai (24ZR1401100 received by Y.J.).
Author information
Authors and Affiliations
Contributions
Y.J. and P.W. designed this project. D.F. performed the experiments, analyzed the data and wrote the paper. Y.X. performed the theoretical calculations and analyzed the data. Y.J. and P.W. supervised the project and co-wrote the manuscript. All the authors discussed the results and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dacheng Kuai and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Feng, D., Xie, Y., Jiao, Y. et al. Leveraging cation effect for low temperature aqueous Zn-based batteries. Nat Commun 16, 9254 (2025). https://doi.org/10.1038/s41467-025-64278-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64278-1
