
Abstract
Deep learning-based generative models hold significant promise for exploring the configuration space of crystalline materials, though their application remains in its early stages. In this study, we present CrystalFlow, a flow-based generative model designed to address the unique challenges of this domain. By combining Continuous Normalizing Flows and Conditional Flow Matching with a graph-based equivariant neural network and symmetry-aware data representations, CrystalFlow efficiently models lattice parameters, atomic coordinates, and atom types. This architecture enables data-efficient learning and the generation of high-quality crystal structures. Our results indicate that CrystalFlow achieves performance comparable to state-of-the-art models on established benchmarks while exhibiting versatile conditional generation capabilities (e.g., predicting structures under specific pressures or material properties), and is approximately an order of magnitude more efficient than diffusion-based models in terms of integration steps.
Similar content being viewed by others
Introduction
The prediction of the stable arrangement of atoms within a crystal, given specific chemical compositions and external conditions, is a longstanding challenge known as crystal structure prediction (CSP)1. This fundamental problem can be framed as a global optimization task on the potential energy surface (PES) of materials, which has far-reaching implications in the fields of physics, chemistry, and materials science, as the atomic structure of a crystal directly governs its physical and chemical properties2,3,4. During the past few decades, substantial advancements have been achieved in CSP, primarily attributed to the evolution of a diverse array of CSP methodologies and software, grounded in the integration of sophisticated structural sampling techniques, optimization algorithms, and quantum-mechanical calculations5,6,7,8,9,10,11. Today, these computational tools are indispensable in modern computational science, offering critical insights into the phase diagrams of condensed matter and facilitating the design of novel materials with tailored properties. This progress has led to numerous groundbreaking discoveries, such as the identification of high-pressure superhydride superconductors with record-breaking critical temperatures12,13,14,15.
Despite widespread successes, CSP remains inherently challenging. The dimensionality of the PES increases linearly with the number of atoms in the unit cell, while the number of local minima escalates exponentially16. These factors result in unfavorable scaling properties of CSP with respect to system size, and the pursuit of more efficient and robust methodologies for structure sampling in the vast crystal space remains a persistent endeavor within this domain.
Recent advancements in deep learning generative models have introduced transformative methodologies for understanding the underlying distributions of high-dimensional data and generating realistic samples. The remarkable capabilities of these generative models have been demonstrated in areas such as large language models17, image generation18, and protein design19. Coupled with the rapid expansion of comprehensive materials databases, these models present a promising strategy for efficiently sampling the vast crystal space, thereby helping to address the sampling challenges in CSP. Along this line, several generative models specifically designed for crystal structures have been proposed employing variational autoencoders20,21,22,23,24,25,26, generative adversarial networks27,28,29, diffusion models30,31,32,33,34,35,36, flow models37, stochastic interpolants38, and autoregressive models39,40,41,42,43,44,45,46,47,48,49. These advancements have led to improved generation performance in terms of metrics such as stability, newness, and uniqueness of the generated samples, as well as enhanced predictive power.
Most state-of-the-art crystal generative models can be broadly categorized into two main types based on the data representation: graph-based models and string-based models50,51. Graph-based models are frequently combined with diffusion/flow techniques and equivariant message passing networks. This combination is particularly effective in capturing the intrinsic symmetries of crystal structures, such as permutation, rotation and periodic translation. A prominent example in this category is the Crystal Diffusion Variational Autoencoder (CDVAE)24 which effectively integrates diffusion processes within the framework of a variational autoencoder. By utilizing SE(3) equivariant message-passing neural networks, CDVAE is able to account for key symmetries of crystals, demonstrating superior performance in terms of structural and compositional validity compared to previous models. Recent advancements along this line are represented by models such as DiffCSP30, MatterGen31, UniMat32, and FlowMM37, which jointly generate lattice, atomic coordinates, and/or atomic types, achieving steady improvements across various standard generative metrics. Furthermore, extensions of CDVAE have been developed for conditional generation based on specific material properties, broadening its applicability25,26. Conversely, string-based models utilized sequential tokenization of crystal structures39,40,41,42,43,44,45,46,47, such as standard Crystallographic Information Files (CIFs), or SLICES39. These models typically employ transformer to learn and process the sequential data, capturing the structural information encoded in these formats. Although these models generally do not explicitly consider the symmetry of crystals, they are well-suited for scaling up to work with much larger and more diverse training data, and enabling co-training with multimodal contents, in the same fashion as large language models.
Notably, space group symmetry, a critical inductive bias for modeling crystalline materials, has been introduced into both graph-based and string-based models, such as DiffCSP++33, SymmCD34, CrystalFormer42, CHGFlowNet52, WyFormer47, and SGEquiDiff53. This advancement mitigates the challenge of generating structures with high symmetry and enables more data- and compute-efficient learning. Recent advancements in this field have led to the emergence of unified generative frameworks that can simultaneously model molecules, crystals, and proteins within a single architecture, thereby exhibiting enhanced generalizability across a broad spectrum of chemical spaces36,48,49. Furthermore, the integration of structure generation and property prediction within a unified model has been shown to substantially improve both the fidelity and efficiency of generated structures, facilitating more accurate and versatile applications54.
In this study, we present CrystalFlow, an advanced generative model for crystalline materials that addresses key challenges in the rapidly evolving field of crystal generative modeling. Existing approaches, such as diffusion-based models, often require a large number of integration steps, leading to significant computational inefficiency, while string-based language models struggle to capture the intrinsic symmetries of crystals. To overcome these limitations, CrystalFlow employs Continuous Normalizing Flows (CNFs)55 within the Conditional Flow Matching (CFM) framework56,57, effectively transforming a simple prior density into a complex data distribution that captures the structural and compositional intricacies of crystalline material databases. This approach simultaneously generates lattice parameters, fractional coordinates, and atom types for crystalline systems, while establishing a symmetry-aware design through recent advancements in graph-based equivariant message-passing networks. By explicitly incorporating the fundamental periodic-E(3) symmetries of crystalline systems, CrystalFlow enables data-efficient learning, high-quality sampling, and flexible conditional generation. Our evaluation demonstrates that CrystalFlow achieves performance comparable to or surpassing state-of-the-art models across standard generation metrics when trained on benchmark datasets. Additionally, when trained with appropriately labeled data, CrystalFlow can generate structures optimized for specific external pressures or material properties, underscoring its versatility and effectiveness in addressing realistic and application-driven challenges in CSP.
Results
CrystalFlow model
The architecture of CrystalFlow is schematically illustrated in Fig. 1, with a detailed description of the methodology provided in the Methods section. Following established conventions in the crystal generative modeling community, a unit cell of a crystalline structure containing N atoms is represented as \({\mathcal{M}}=({\bf{A}},{\bf{F}},{\bf{L}})\). In this representation, \({\bf{A}}=[{{\bf{a}}}_{1},{{\bf{a}}}_{2},\ldots,{{\bf{a}}}_{N}]\in {{\mathbb{R}}}^{a\times N}\) encodes the chemical composition, where each atom type is mapped to a unique a-dimensional categorical vector. The fractional atomic coordinates within the unit cell are denoted by \({\bf{F}}=[{{\bf{f}}}_{1},{{\bf{f}}}_{2},\ldots,{{\bf{f}}}_{N}]\in {\left[0,1\right)}^{3\times N}\), and the lattice structure is described by the lattice matrix \({\bf{L}}=[{{\bf{l}}}_{1},{{\bf{l}}}_{2},{{\bf{l}}}_{3}]\in {{\mathbb{R}}}^{3\times 3}\).
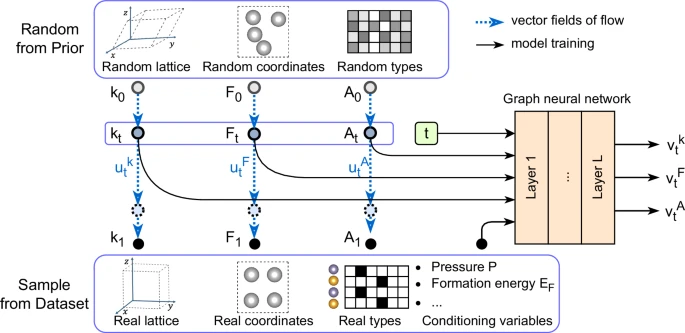
Random structures, represented by lattice representations k0, fractional coordinates F0, and atom types A0, are sampled from prior distributions. Real structures, characterized by k1, F1, and A1, are sampled from the dataset. Continuous normalizing flows are established between these two distributions, defined by vector fields \({{\bf{u}}}_{t}^{k}\), \({{\bf{u}}}_{t}^{F}\), and \({{\bf{u}}}_{t}^{A}\) at time t. Intermediate structure components kt, Ft, and At at a given time t, along with conditioning variables, serve as inputs to a graph neural network, which outputs vector fields \({{\bf{v}}}_{t}^{k}\), \({{\bf{v}}}_{t}^{F}\), and \({{\bf{v}}}_{t}^{A}\) (omitting model parameters θ). The model is trained by regressing the vector field v to match u. For crystal structure prediction tasks, A0 ≡ A1 is fixed as a conditioning variable.
To ensure rotational invariance in the lattice representation, an alternative parameterization of L is adopted using a rotation-invariant vector \({\bf{k}}\in {{\mathbb{R}}}^{6}\), derived via polar decomposition as \({\bf{L}}={\bf{Q}}\exp \left(\mathop{\sum }\nolimits_{i=1}^{6}{{\bf{k}}}_{i}{{\bf{B}}}_{i}\right)\). Here, Q is an orthogonal matrix representing rotational degrees of freedom, \(\exp (\cdot )\) denotes the matrix exponential, and \({\{{{\bf{B}}}_{i}\}}_{i=1}^{6}\in {{\mathbb{R}}}^{3\times 3}\) forms a standard basis of symmetric matrices33. This parameterization effectively decouples rotational and structural information, providing a compact and symmetry-preserving representation of the lattice.
In the context of CSP, the primary objective is to predict the stable structure for a given chemical composition A under specific external conditions, such as pressure P. To achieve this, we propose a generative model that learns the conditional probability distribution over stable or metastable crystal configurations, denoted as p(x∣y). Here, x = (F, L) represents the structural parameters, while y = (A, P) serves as the conditioning variables. In cases where the chemical composition A (i.e., atom types) is not pre-specified-a task referred to as de novo generation (DNG)-the model extends its scope to simultaneously predict not only the structural parameters (F, L) but also the atom types A.
The proposed framework, CrystalFlow, models the conditional probability distribution over crystal structures using a CNF approach55, which are trained using CFM techniques56,57. This advanced generative modeling technique establishes a mapping between the data distribution q(x1) and a simple prior distribution q(x0), such as a Gaussian, through continuous and invertible transformations. This formulation enables efficient sampling and exploration of complex, high-dimensional data spaces. The architecture employs a equivariant geometric graph neural network (GNN) to parameterize time-dependent vector fields for the lattice L, fractional atomic coordinates F, and atomic types A, which collectively define the flow transformations and explicitly preserves the intrinsic periodic-E(3) symmetries of crystals, including permutation, rotation, and periodic translation invariance.
During inference, random initial structures are sampled from simple prior distributions and evolved toward realistic crystal configurations through a learned conditional probability path. The model employs numerical ordinary differential equation (ODE) solvers to generate crystal structures, with adjustable integration steps to balance computational efficiency and sample quality. This capability allows for the efficient generation of stable and metastable crystal configurations, enabling the exploration of new materials and their properties.
We evaluate the performance of CrystalFlow on a diverse set of crystal generation tasks using datasets that span a broad range of compositional and structural diversity. The model’s effectiveness is systematically benchmarked against existing crystal generation methods using standard evaluation metrics. Furthermore, the quality of the generated structures is thoroughly analyzed through detailed density functional theory (DFT) calculations.
CSP performance on MP-20 and MPTS-52 datasets
We begin by evaluating the performance of CrystalFlow using two widely recognized benchmark datasets, MP-20 and MPTS-5258. The MP-20 dataset comprises 45,231 stable or metastable crystalline materials sourced from the Materials Project (MP), encompassing the majority of experimentally reported materials in the ICSD database with up to 20 atoms per unit cell. In contrast, MPTS-52 represents a more challenging extension of MP-20, containing 40,476 crystal structures with up to 52 atoms per unit cell, organized chronologically based on their earliest reported appearance in the literature. The datasets are divided into training, validation, and test subsets in a manner consistent with previous studies.
In accordance with standard practice, the predictive performance of the model is evaluated by calculating its match rate (MR) and the root mean squared error (RMSE) on the test set24,30,31,37. Specifically, for each structure in the test set, k candidate structures are generated using CrystalFlow, and it is determined whether any of the predicted structures match the ground truth structure. The MR is defined as the fraction of structures in the test set that are successfully predicted. To match structures and evaluate their similarity, we employ the StructureMatcher function from the Pymatgen library59. For consistency with prior studies, the same threshold parameters are used: ltol=0.3, stol=0.5, angle_tol=10 (see Methods for details). For matched structures, the RMSE between the positions of matched atom pairs is calculated and normalized by \({(\bar{V}/N)}^{1/3}\), where \(\bar{V}\) is the volume derived from the average lattice parameters. It is important to note that, while this benchmarking approach assumes known compositions, real-world CSP is often more general and challenging, as the precise stoichiometry may be unknown or only partially specified.
The MR and RMSE values for CrystalFlow at k = 1, 20, and 100 on the MP-20 and MPTS-52 datasets are presented in Table 1, alongside comparisons with other crystal generative models, including CDVAE, DiffCSP, and FlowMM. While k = 1 is conventionally used for benchmarking, practical CSP requires generating multiple candidate structures per composition to effectively explore complex energy landscapes and capture both stable and metastable (polymorphic) structures. Therefore, we report results at different k for a more realistic and comprehensive evaluation. The results indicate that CrystalFlow achieves performance that is comparable to or exceeds that of state-of-the-art models. On the MP-20 dataset, CrystalFlow demonstrates comparable MR and RMSE values to FlowMM, outperforming CDVAE and DiffCSP. On the more challenging MPTS-52 dataset, CrystalFlow achieves the best performance among all four models, highlighting its superior predictive capability. MR and RMSE values calculated using more stringent threshold parameters, as well as RMSE computed over all predictions (RMSE-all, including both matched and unmatched pairs), are presented in Supplementary S1. As anticipated, the application of stricter (i.e., smaller) tolerance values leads to a reduction in both match rate and RMSE, as only more closely matching structures are considered equivalent. Notably, the relative performance ranking among the evaluated methods remains largely unchanged under varying criteria.
A direct comparison of inference times for CrystalFlow and other state-of-the-art models, all benchmarked on the same GPU device (NVIDIA A800), is provided in Table 1. These results clearly show that CrystalFlow is approximately an order of magnitude faster than the diffusion-based model DiffCSP, while maintaining comparable or superior generation quality. This substantial efficiency gain is primarily due to the significantly fewer integration steps required by flow-based models such as CrystalFlow and FlowMM. Here, the integration step refers to the number of discrete numerical steps used by the ODEs (in flow-based models) or stochastic differential equations (in diffusion-based models) solver during the generation process. Fewer integration steps not only accelerate sample generation but also reduce computational cost, making the model more practical for large-scale applications.
CSP performance on MP-CALYPSO-60 dataset
We further evaluate the performance of CrystalFlow using the extensive MP-CALYPSO-60 dataset described in ref. 26. This dataset is constructed by integrating two sources: (1) ambient-pressure crystal structures obtained from the MP database, and (2) crystal structures generated from previous CALYPSO60 CSP studies conducted over a wide pressure range, with the majority of structures corresponding to pressures between 0 and 300 GPa. See Supplementary S2 and ref. 26 for more details about the CALYPSO dataset. In contrast to the original dataset in ref. 26, structures containing more than 60 atoms per unit cell have been excluded. The resulting dataset comprises 657,377 crystal structures, spanning 86 elements and 79,884 unique chemical compositions. CrystalFlow, trained on this dataset, is conditioned on both chemical composition and external pressure, allowing it to generate crystal structures across a variety of pressure conditions. This capability is particularly important for simulating realistic conditions that materials may encounter in practical structure prediction applications.
We randomly selected 500 pairs of chemical compositions and pressures from the test set to serve as conditional inputs. For each conditional input, one structure was generated using CrystalFlow, as well as the previous developed Cond-CDVAE26 model, for comparative analysis. For CrystalFlow, integration steps S = 100, 1000, and 5000 were employed, whereas, for Cond-CDVAE, a diffusion-based model, a larger integration step of S = 5000 was used. To assess the quality of the generated structures, all samples were subjected to DFT single-point calculations and local optimizations at the corresponding target pressure by VASP package61 (see Sec. IV G for computational details). The relationship between the DFT-computed lattice stresses for the initial structures and the target pressures specified as generation conditions for every model is illustrated as scatter plots in Fig. 2a, while Fig. 2b presents the distributions of enthalpy differences between the structures generated by the two models, both before and after optimization.

CrystalFlow is trained on the MP-CALYPSO-60 dataset (see Results section C). Integration steps of S = 100, 1000, and 5000 are utilized for CrystalFlow, while S = 5000 is employed for Cond-CDVAE. a Hexagonal binned histograms of the relationship between the density functional theory (DFT) computed lattice stress and the target pressure for 500 structures generated by each model. The composition and target pressure are randomly sampled from the test set. b Distributions of enthalpy (H) differences for these structures before and after local optimization. c Average energy curves during local optimization for 200 SiO2 structures generated by each model at 0 GPa, with shaded areas denoting standard deviation. d Energy distributions of these SiO2 structures before and after local optimization. Source data are provided as a Source Data file.
As observed in Fig. 2a, with the exception of a few outliers, the structures generated by CrystalFlow across all integration steps exhibit significantly improved alignment with the target pressures compared to those generated by Cond-CDVAE. This underscores CrystalFlow’s superior ability to learn and incorporate the effects of pressure within the generative modeling process. Consequently, the majority of CrystalFlow-generated structures, even with integration steps as low as S = 100, exhibit lower enthalpy than those produced by Cond-CDVAE (Fig. 2b), suggesting that CrystalFlow generates more physically plausible lattice and geometric configurations. After optimization, the distribution of enthalpy differences between the two models narrows, as the optimization process mitigates the discrepancies between the initial structures. This suggests that both models are effective in learning and incorporating essential structural information from the dataset. An additional comparison with the known lowest-enthalpy reference structures in the dataset is provided in Supplementary S3. The results are consistent with those presented in Fig. 2b.
In practice, neither CrystalFlow nor other state-of-the-art generative models can guarantee that the initially generated structures correspond to true energy minima. Consequently, further quantum-mechanical geometry optimization (e.g., using DFT) is an essential step to ensure the local stability of these structures. Notably, this optimization step is typically the most computationally demanding part of the structure prediction workflow. To assess the computational efficiency and practical utility of generative models, two key metrics are considered: the convergence rate and the number of ionic steps required during local optimization. The convergence rate reflects the percentage of generated structures that successfully reach a local energy minimum, while the number of ionic steps indicates the number of iterations needed to relax atomic positions and minimize the total energy. As shown in Table 2, structures generated by CrystalFlow generally achieve a higher convergence rate compared to those generated by Cond-CDVAE. Additionally, the number of ionic steps required for CrystalFlow structures decreases with increasing integration steps, suggesting that higher integration steps improve the quality of the generated samples. At an integration step of S = 5000, CrystalFlow requires 39.82 average ionic steps, which is lower than the 45.91 steps needed for Cond-CDVAE, indicating a 13.3% reduction in computational cost.
Further analysis reveals that ~55.2% of structures generated by CrystalFlow (after local optimization), as determined by StructureMatcher, are new, i.e., not present in the training set. For comparison, the MatterGen31 model achieves a newness rate of 61% in the DNG task. Although differences in training sets and evaluation tasks preclude a direct comparison, these results demonstrate that CrystalFlow achieves a level of structural newness rate comparable to state-of-the-art generative models.
In the previous test, we generated one sample for each randomly selected chemical system from the test set. To further evaluate the model’s performance on a specific system, we conducted a case study using SiO2, a material known for its significant structural polymorphism. We generated 200 SiO2 structures at 0 GPa, each containing three formula units per unit cell (9 atoms), using both CrystalFlow with integration steps S = 100, 1000, and 5000, and Cond-CDVAE with S = 5000. The average energy curves, calculated relative to the corresponding local minimum for each structure during the optimization process, are presented in Fig. 2c. These curves provide a quantitative measure of both the initial deviation of the generated structures from their local minima and the efficiency of their relaxation toward locally stable configurations. As shown in the figure, CrystalFlow consistently yields lower energy curves compared to Cond-CDVAE, indicating that structures generated by CrystalFlow are initially closer to their local minima and require less relaxation to achieve stability. This result is further supported by the faster convergence rates as a function of ionic steps, as shown in Supplementary S4. It is noteworthy that, in the later stages of optimization (after 40–50 steps), Cond-CDVAE exhibits slightly lower average energies and reduced standard deviations. Our analysis reveals that most structures converge within 50 ionic steps, and the remaining uncertainties in the energy curves are primarily due to a small subset of structures that are more challenging to optimize. For these difficult cases, CrystalFlow-generated structures tend to have higher relative energies, resulting in increased standard deviations. The high quality of the structures generated by CrystalFlow, which is further supported by the energy distribution of structures (with S = 100) before and after optimization shown in Fig. 2d, as well as the convergence rate and number of ionic steps detailed in Table 2. With an integration step of S = 5000, CrystalFlow requires an average of 31.99 ionic steps, which is about 27.9% fewer than the 44.36 steps required by Cond-CDVAE.
DNG performance on MP-20
Finally, we evaluate the DNG performance of CrystalFlow on the MP-20 dataset to assess its potential for inverse materials design tasks. Initially, we train CrystalFlow on MP-20 without conditioning, and compare its performance with other models using common DNG metrics, including structural and compositional validity, coverage, and property statistics. The structural validity is defined as the percentage of generated structures in which all pairwise atomic distances exceed 0.5 Å. The compositional validity, on the other hand, refers to the percentage of generated structures with an overall neutral charge, calculated using SMACT62. Coverage quantifies the structural and compositional similarity between the test set and the generated structures with a detailed definition given in Sec. IV H. For property statistics, we evaluate the similarity between the test set and the generated structures in terms of density ρ and number of elements Nel, using Wasserstein distances (wdist). We also measure the model’s ability to generate stable, diverse, new materials, denoted as SUN rate31. A structure is deemed stable (S.) if its energy above hull, relative to Matbench Discovery63 convex hull, is negative. Among these stable structures, one is considered new (N.) if it does not appear in the training set. Furthermore, take all stable and new structures, a structure is classified as unique (U.) if it is distinct from all others. The SUN rate was calculated after geometric optimization of the sampled structures via pre-relaxation with CHGNet64, followed by DFT relaxation using the same settings as prior studies.
The statistic results of 10,000 randomly generated structures are presented in Table 3, and demonstrate that CrystalFlow achieves a performance comparable to that of existing models across various metrics. Although its compositional validity is marginally lower than that of other approaches, CrystalFlow achieves the smallest wdist for density, underscoring its capability to generate structures with more physically reasonable lattice parameters. CrystalFlow achieves a competitive stable and SUN rate among state-of-the-art models. For example, the SUN rate of CrystalFlow (3.7%) is higher than that of DiffCSP (3.3%) and FlowMM (2.3%), but lower than FlowLLM (4.7%) and ADiT (4.7%). This comparison highlights both the strengths of our approach and potential avenues for further improvement, such as training on more comprehensive datasets (as in MatterGen), adopting advanced model architectures based on transformers (as in ADiT), or integrating large language models with diffusion/flow-based frameworks.
To demonstrate CrystalFlow’s capability to generate structures with targeted properties, the model is further trained on the MP-20 dataset with formation energy (EF) as a conditioning label. Subsequently, 10,000 structures are generated for each specified formation energy value, conditioned on EF = 0, − 1, − 2, − 3, and − 4 eV/atom, respectively. To facilitate efficient evaluation, energy calculations and geometric optimization are performed using CHGNet64, a universal and computationally efficient interatomic potential. Notably, after local optimization, ~93.5% of the generated structures were identified as new - i.e., absent from the training set as determined by StructureMatcher - highlighting CrystalFlow’s strong generative capability.
We compared the distributions of EF for the generated structures produced by CrystalFlow and Con-CDVAE25, a state-of-the-art model for conditional DNG tasks, as shown in Fig. 3. The generated structures exhibit EF distributions that are generally shifted in accordance with the specified target values; however, the centers of these distributions tend to be displaced toward higher-energy regions. Following geometric optimization, the distributions become more closely aligned with the target values. This improvement is particularly pronounced for target EF values that are well-represented in the training dataset. These results highlight the effectiveness of CrystalFlow in generating structures with targeted properties and its ability to leverage training data to improve the accuracy of conditional generation.

Distributions of formation energy (EF) for structures generated CrystalFlow, conditioned on target values of EF = 0, − 1, − 2, − 3, and − 4 eV/atom. For each target, 10,000 structures were generated. The dotted curve denotes the corresponding distribution of the training set. For comparison, results from Con-CDVAE trained on the MP-20 dataset using the less strategy (MP20-L) are shown in the lower panel, as reported in ref. 25. Source data are provided as a Source Data file.
A quantitative evaluation of model performance is presented in Supplementary S5, reporting the mean absolute error (MAE) and root mean square error (RMSE) between target and generated formation energies. While the generated energy distributions generally follow the target values, significant deviations persist, particularly for underrepresented targets in the training data. The MAE ranges from 0.67 to 2.02 eV/atom before optimization and improves to 0.17–0.82 eV/atom after optimization, underscoring the challenge of high-fidelity conditional generation with current crystal generative models. Notably, the Con-CDVAE model25 achieves superior performance, with formation energy distributions more tightly centered on target values, as shown in Fig. 3. This enhanced accuracy is attributed to both the advanced architectural design of Con-CDVAE and the incorporation of a property predictor, which filters latent variables for structure generation at a ratio of 20,000:200. These findings suggest that incorporating a property predictor could further enhance the conditional generation capability of CrystalFlow in practical material design applications.
Discussion
By integrating state-of-the-art generative modeling techniques with domain-specific knowledge in physical and material science, we introduce CrystalFlow, a crystal generative model specifically designed for the efficient exploration of the vast and complex materials space. CrystalFlow exhibits strong capabilities in generating high-quality crystal structures that adhere to fundamental physical principles and satisfy target constraints, as demonstrated by its superior performance across multiple benchmark datasets. These attributes position CrystalFlow as a highly promising and versatile structure-sampling tool, with significant potential for integration into a wide range of CSP methods and materials design workflows.
Flow-based models offer several advantages over diffusion-based approaches, including fewer integration steps and more flexible choices of prior distributions, making them particularly appealing for generative modeling in materials science. However, as this work represents an initial step in exploring these capabilities, the full potential of flow-based models remains to be comprehensively investigated. Future studies could focus on evaluating the influence of integration strategies, integration lengths, and prior distribution choices on the quality and diversity of generated structures. Moreover, incorporating fixed structural modifications or enforcing space group symmetries during generation could further enhance the model’s ability to produce realistic and physically meaningful crystal structures.
Assessing the performance of CrystalFlow on larger and more diverse datasets, as well as under multi-property constraints, would provide valuable insights into its scalability, robustness, and applicability to real-world materials discovery. Furthermore, integrating CrystalFlow into broader generative frameworks—such as hybrid architectures that combine flow-based models with autoregressive approaches—holds significant promise for further improving its generative performance. These directions represent exciting opportunities to advance the capabilities of crystal generative modeling.
We anticipate that this work will inspire further innovations at the intersection of artificial intelligence, condensed matter physics, and material science, ultimately advancing the discovery and design of next-generation materials.
Methods
Continuous normalizing flow
In CrystalFlow, we model the conditional probability distribution p(x∣y) over crystal structures in CSP/DNG tasks using CNF. This advanced generative modeling technique connects the data distribution q(x1) with a simple prior distribution q(x0), such as a Gaussian, through continuous and invertible transformations, enabling efficient sampling and exploration of complex data spaces.
The CNF framework55 is based on a smooth, time-dependent d-dimensional vector field \({\bf{u}}:[0,1]\times {{\mathbb{R}}}^{d}\to {{\mathbb{R}}}^{d}\), which defines an ODE:
The solution ϕt(x) of this ODE, starting from ϕ0(x) = x, describes the evolution of x over time. Modeling ut(x) with a neural network vt;θ(t, x) allows us to transform a simple prior density p0 into a complex target density p1 using the push-forward operation \({p}_{t}={[{\phi }_{t}]}_{\,{*}} \, {p}_{0}\) defined by: \({[{\phi }_{t}]}_{\,{*}} \, {p}_{0}({\bf{x}}):={p}_{0}\left({\phi }_{t}^{-1}\left({\bf{x}}\right)\right)\det | \partial_{t}^{{-1}}/\partial {\bf{x}}|\). The time-dependent density pt(x) is governed by the continuity equation: ∂p/∂t = − ∇ ⋅ (ptut), ensuring conservation of probability mass.
Conditional flow matching
In many scenarios, the vector field ut(x) is intractable, posing significant challenges for analysis and computation. To address this issue, CFM56,57 presents a simulation-free training strategy that incorporates an additional conditioning variable z. This methodology is effective provided that a tractable conditional vector field ut(x∣z) can be defined. Suppose that the marginal probability path pt(x) is a mixture of probability paths pt(x∣z) that vary with some conditioning variable z, that is,
where q(z) is some distribution over the conditioning variable. If pt(x∣z) evolves under a vector field ut(x∣z) from initial conditions p0(x∣z), then the vector field:
generates the probability path pt(x) from initial conditions p0(x). The CFM training objective becomes ut(x∣z).
This framework can be seamlessly extended to conditional generation with respect to a conditioning variable y in a manner of classifier-free guidance65. In practical, we adopted a guidance factor of 1 in our conditional generation, meaning no empty-condition part was used. In this case, the evolution of the system is described by:
And the training objective is still ut(x∣z).
Here, we utilize the independent coupling variant of the CFM (I-CFM)66,67 framework, where the conditioning variable z is defined by the pair (x0, x1), which represent the initial and terminal points. pt(x∣z) is a probability path interpolating between x0 and x1. The boundary conditions of Eq. (5) satisfy p0(x∣y) ≈ q(x0∣y) = q(x0) being the prior distribution and p1(x∣y) ≈ q(x1∣y) being target distribution. The prior q(x0), conditional probability path pt(x∣z) and the conditional vector field ut(x∣z) are detailed in Sec. IV C. Finally, the training objective is written as:
where vt;θ(t, x, y) is a time-dependent vector field parametrized as a neural network with parameters θ.
Joint equivariant flow
In the context of crystal generative modeling, it is crucial to incorporate the fundamental periodic-E(3) symmetries of crystalline systems, namely, permutation invariance, rotation invariance, and periodic translation invariance, to enable data-efficient learning, computational efficiency, and high-quality sampling. To ensure that the push-forward distribution remains invariant under these symmetry transformations, it is generally necessary to employ an equivariant flow in conjunction with an invariant prior with respect to these symmetries68,69. In practice, achieving this invariance can be made more tractable by carefully selecting the representation of the crystal structure \({\mathcal{M}}\) and designing an appropriate model architecture. A GNN is employed to intrinsically account for permutation invariance. By utilizing a rotation-invariant representation of the lattice and the fractional coordinate system, we render the periodic-E(3) invariance tractable by fulfilling the periodic translation invariance with respect to F.
In CSP scenarios, CrystalFlow generates crystalline materials by concurrently evolving the lattice representation k and the fractional atomic coordinates F. Let the prior distribution be q(x0) = q(k0)q(F0), where k0 and F0 denote the respective components of the initial state. The conditional probability path \({p}_{t}({\bf{x}}| {\bf{z}}):={p}_{t}^{k}({\bf{k}}| {\bf{z}}){p}_{t}^{F}({\bf{F}}| {\bf{z}})\) is determined by the tuple \({{\bf{u}}}_{t}({\bf{x}}| {\bf{z}}):=({{\bf{u}}}_{t}^{k}({\bf{k}}| {\bf{z}}),{{\bf{u}}}_{t}^{F}({\bf{F}}| {\bf{z}}))\). In the following, we derive the conditional probability path and the corresponding vector field for each component.
Flow on lattice
This choice of invariant lattice representation k provides significant flexibility in defining both the prior distribution and the conditional probability path. For simplicity, we adopt a Gaussian prior \(q({{\bf{k}}}_{0})={\mathcal{N}}({{\bf{k}}}_{0};{{\boldsymbol{\mu }}}_{0}^{k},{{\boldsymbol{\sigma }}}_{0}^{k})\), where the mean and standard deviation are set as \({{\boldsymbol{\mu }}}_{0}^{k}=(0,0,0,0,0,1)\) and \({{\boldsymbol{\sigma }}}_{0}^{k}=0.1\), respectively. The probability path is defined with a time-dependent mean \({{\boldsymbol{\mu }}}_{t}^{k}\) determined by linear interpolation between the initial and terminal points, given by \({{\boldsymbol{\mu }}}_{t}^{k}=t{{\bf{k}}}_{1}+(1-t){{\bf{k}}}_{0}\), and a constant standard deviation \({{\boldsymbol{\sigma }}}_{t}^{k}={{\boldsymbol{\sigma }}}^{k}\). The conditional probability path and vector field are formulated as:
By setting σk = 0, the conditional probability path reduces to a deterministic interpolation, consistent with the Rectified Flow framework66.
Flow on fractional coordinates
As previously discussed, the push-forward distribution \({p}_{t}^{F}({\bf{F}}| {\bf{y}})\), defined in Eq. (5), must satisfy periodic translation invariance. According to Theorem D.1 in ref. 37, this requirement can be fulfilled by employing an invariant q(z∣y) in conjunction with a pairwise invariant conditional probability path \({p}_{t}^{F}({\bf{F}}| {\bf{z}})\). We achieve this objective by, first, employing a uniform prior distribution \(q({{\bf{F}}}_{0})={\mathcal{U}}({{\bf{F}}}_{0};0,{\bf{I}})\) guarantees q(z∣y) is invariant, and second, establishing the conditional probability path as a wrapped Gaussian distribution bridging F0 and F1. The mean of the wrapped Gaussian, \({{\boldsymbol{\mu }}}_{t}^{F}\), is determined using minimum image convention and linear interpolation, expressed as: \({{\boldsymbol{\mu }}}_{t}^{F}={{\bf{F}}}_{0}+t(w({{\bf{F}}}_{1}-{{\bf{F}}}_{0}-0.5)-0.5)\), where w(x) = x − ⌊x⌋ represents the wrapping operation that ensures periodicity. The standard deviation is taken to be constant, \({{\boldsymbol{\sigma }}}_{t}^{F}={{\boldsymbol{\sigma }}}^{F}\). The conditional probability path is then expressed as:
where \({{\mathcal{N}}}_{w}\) represents the wrapped Gaussian distribution defined as:
This ensures the probability distribution keeps the same over any period with interval 1, maintaining the crystal periodicity. Note that the choice of \({{\mathcal{N}}}_{w}\) also makes it unnecessary to add wrapping operation on \({{\boldsymbol{\mu }}}_{t}^{F}\).
Let the flow be the following simplest form: \({\phi }_{t}^{F}({\bf{F}})=w({{\boldsymbol{\sigma }}}_{t}^{F}({\bf{z}}){\bf{F}}+{{\boldsymbol{\mu }}}_{t}^{F}({\bf{z}}))\). Given that \({{\boldsymbol{\sigma }}}_{t}^{F}\) is constant, and neglecting the singular points, the unique vector field is derivated as:
By construction, the conditional vector field \({{\bf{u}}}_{t}^{F}({\bf{F}}| {\bf{z}})\) is pairwise invariant under periodic translations, resulting in the conditional probability path \({p}_{t}^{F}({\bf{F}}| {\bf{z}})\) being pairwise periodic translation invariant, as demonstrated in Sec. IV I. We set σF = 0 to be consistent with the Rectified Flow framework66.
Loss function
Regressing the vector fields \({{\bf{u}}}_{t}^{k}\) in Eq. (9) and \({{\bf{u}}}_{t}^{F}\) in Eq. (12) gives rise to the total loss function \({\mathcal{L}}\) used for training CrystalFlow, expressed as:
where λk and λF are weights for the respective loss terms, x represents the interpolating structure (kt, Ft), and y represents the chemical composition A and external pressure P. The vector fields \({{\bf{v}}}_{t;\theta }^{k}\) and \({{\bf{v}}}_{t;\theta }^{F}\) are parameterized by neural networks with learnable parameters θ.
Flow on chemical composition
While CrystalFlow is primarily designed for CSP, it can be readily extended to address inverse materials design problems, where the goal is to generate materials with specific target properties. This process is described by the generative model p(x∣y), where x = (F, L, A) and y denotes the desired properties, such as formation energy. In DNG scenarios where A (atom types) is not fixed, an additional CNF is required to generate A alongside the other structural components.
To generate A, a one-hot encoding of atomic types is employed. To reduce the dimensionality of this representation while preserving the relationships among chemical species, the periodic table is reorganized into a grid of 13 rows and 15 columns, with each species assigned a unique row-column position (detailed in Supplementary S6). The row and column indices are encoded as one-hot vectors and concatenated to represent each atomic type: (arow, acolumn). This results in a compact 28-dimensional vector representation for each atomic type. Since A is an invariant representation, the CNF applied to A is analogous to that used for the lattice k. Specifically, we employed a Gaussian prior \(q({{\bf{A}}}_{0})={\mathcal{N}}({{\bf{A}}}_{0};0,{\bf{I}})\in {{\mathbb{R}}}^{28}\). The conditional probability path and vector field are formulated as:
where σA is also set to 0.
In comparison to the CSP task, an additional loss term for A is introduced, defined as:
where \({{\bf{v}}}_{t;\theta }^{A}\) is the predicted vector field. The total loss is given by:
where λk, λF, and λA are weights. The vector field \({{\bf{v}}}_{t;\theta }^{A}\) is parameterized by neural networks.
Model architecture
A GNN is employed to predict the vector fields \({{\bf{v}}}_{t;\theta }^{k}\) and \({{\bf{v}}}_{t;\theta }^{F}\) for CSP, as well as an additional vector field \({{\bf{v}}}_{t;\theta }^{A}\) for DNG. Note that the pairwise invariance of \({{\bf{u}}}_{t}^{F}(\cdot | \cdot )\) under periodic translation should be satisfied, which is equivalently described as a requirement for a velocity-like vector field. Specifically, we adopted the GNN architecture implemented in DiffCSP30. The GNN consists of L consecutive layers, where \({{\bf{H}}}^{(l)}=\{{{\bf{h}}}_{i}^{(l)}\,| \,i=1,\ldots,N\}\) denotes the node representations at the l-th layer. The input features for node i are defined as \({{\bf{h}}}_{i}^{(0)}={\varphi }_{0}({f}_{t}(t),{f}_{A}({{\bf{a}}}_{i}))+{\varphi }_{y}({f}_{y}({\bf{y}}\setminus {\bf{A}}))\), which simultaneously takes in conditioning variables. Here, y⧹A represents user-specified conditions excluding the atomic composition A. The functions ft, fA, and fy correspond to sinusoidal positional encoding, atomic embedding, and Gaussian basis encoding, respectively. The updates for the node feature \({{\bf{h}}}_{i}^{(l)}\) and the edge feature \({{\bf{m}}}_{ij}^{(l)}\) between nodes i and j at the l-th layer are computed as follows:
where φ□ represent multi-layer perceptrons (MLPs). \({{\bf{F}}}_{ij}^{\text{FT}}\) denotes the Fourier transform of the relative fractional coordinate fj − fi, which is periodic translation invariant. After L layers of message passing of fully connected graphs, the vector field is read out by:
where φk, φF and φA are also MLPs. See Supplementary S7 for other hyperparameters.
Model inference
During the inference stage, a random initial state x0 — comprising the lattice parameters, fractional atomic coordinates, and, in the case of DNG, atom types — is sampled from the prior distribution defined in Sec. IV C. The final structure is subsequently determined by solving Eq. (4) over the interval t ∈ [0, 1] using the trained vector field with a numerical ODE solver, as expressed by:
where \(s(t):=1+{s}^{{\prime} }t\) is a scaling term introduced as part of the anti-annealing numerical scheme37, and Δt = 1/S, with \(S\in {\mathbb{{Z}^{+}}}\) representing the number of integration steps. Unless stated otherwise, we set S = 100, corresponding to a step size of Δt = 1/100. The anti-annealing parameter is set to \({s}^{{\prime} }=5\) for CSP tasks, where it is applied exclusively to F. For DNG tasks, the parameter is set to \({s}^{{\prime} }=0\), effectively disabling the anti-annealing adjustment.
Structure matching
We utilized the StructureMatcher function in the Pymatgen library to match structures and measure their similarity. The matching criteria are governed by the following parameters:
-
ltol (lattice tolerance) controls the relative tolerance for comparing the lattice lengths of two structures. Two lattices are considered matching if their lattice vector magnitudes differ by less than ltol relative to the compared lattice length.
-
stol (site tolerance) sets the maximum allowed distance, expressed a fraction of the average interatomic spacing ((V/N)1/3, where V is the cell volume and N is the number of atoms), for matching atomic sites between two structures. If the distance between corresponding atoms is less than stol times this average spacing, those sites are considered matched.
-
angle_tol (angle tolerance) specifies the maximum allowed difference (in degrees) between the lattice angles of two structures during alignment, used in conjunction with ltol.
Unless otherwise specified, the default tolerance parameters ltol=0.2, stol=0.3, angle_tol=5 are used throughout this work.
DFT computational details
DFT calculations were performed with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional70 and all-electron projector-augmented wave method71, as implemented in the VASP code61. For calculations of high-pressure structures in Results section C, an energy cutoff of 520 eV and a Monkhorst–Pack k-point sampling grid spacing of 0.25 Å−1 were used to ensure the convergence of the total energy. The default settings of PBE functional, Hubbard U corrections, and ferromagnetic initializations in Pymatgen59MPRelaxSet function were employed. The maximum optimization ionic step and the maximum running time are constrained to 150 steps and 20 hours, respectively.
Evaluation metrics for DNG tasks
Following previous works, we employ the coverage metric to measure the structural and compositional similarity between the test set \({{\mathcal{S}}}_{t}\) and the generated structures \({{\mathcal{S}}}_{g}\). Specifically, let \({d}_{S}({{\mathcal{M}}}_{1},{{\mathcal{M}}}_{2})\) and \({d}_{C}({{\mathcal{M}}}_{1},{{\mathcal{M}}}_{2})\) denote the L2 distances of the CrystalNN72 structural fingerprints and the normalized Magpie73 compositional fingerprints. The Coverage Recall is defined as:
where δS and δC are pre-defined thresholds. The Coverage Precision is defined similarly by swapping \({{\mathcal{S}}}_{t}\) and \({{\mathcal{S}}}_{g}\). The recall metrics measure how many ground-truth materials are correctly predicted, while the precision metrics measure how many generated materials are of high quality.
To evaulate the stability, uniqueness, and newness rates, we perform a two-step geometric optimization on each structure using CHGNet followed by DFT relaxation. The energy above hull is computed against the Matbench Discovery convex hull. These setups are the same as ref. 36. A structure is considered stable if its energy above hull is negative. The uniqueness and novelty are compared on self-to-self and all structures in MP-20 dataset.
Proof for the invariance of the vector field
We first demonstrate that the conditional velocity field \({{\bf{u}}}_{t}^{F}\), as defined in Eq. (12), is pairwise invariant under periodic translations. This invariance ensures that the conditional probability path \({p}_{t}^{F}({\bf{F}}| {\bf{z}})\), as defined in Eq. (10), is also pairwise invariant under periodic translations.
Specifically, let \({\boldsymbol{\tau }}:=\hat{{\boldsymbol{\tau }}}{{\bf{1}}}^{\top }\), where \(\hat{{\boldsymbol{\tau }}}\in {{\mathbb{R}}}^{3\times 1}\) is an arbitrary translation vector and \({\bf{1}}\in {{\mathbb{R}}}^{N\times 1}\) is a column vector of ones. The periodic translation operation applied to the fractional coordinates F is then expressed as g∘F ≔ w(F + τ). We now demonstrate the pairwise periodic translation invariance of \({{\bf{u}}}_{t}^{F}\) under this operation as follows:
Next, we demonstrate the pairwise periodic translation invariance of the conditional probability path \({p}_{t}^{F}({\bf{F}}| {\bf{z}})\), as defined in Eq. (10). Reformulating \({p}_{t}^{F}({\bf{F}}| {\bf{z}})\) as:
where \({{\mathcal{N}}}_{w}\) denotes the wrapped normal distribution, we proceed with the proof as follows:
Thus, the conditional probability path \({p}_{t}^{F}({\bf{F}}| {\bf{z}})\) is shown to be invariant under pairwise periodic translations. This result relies on the properties of the wrapped normal distribution, as established in Lemma 3 of Jiao et al.30.
Data availability
The authors declare that the main data supporting the findings of this study are contained within the paper and its associated Supplementary Information. MP-20 and MPTS-52 datasets and the trained model checkpoints are available at Zenodo74 and GitHub (https://github.com/ixsluo/CrystalFlow). Source Data and structures generated by CrystalFlow are available at Figshare75 under CC BY 4.0 license.
Code availability
The CrystalFlow source code is available at Zenodo74 and GitHub (https://github.com/ixsluo/CrystalFlow) under the MIT license.
References
Olson, G. B. Designing a new material world. Science 288, 993–998 (2000).
Wang, Y. & Ma, Y. Perspective: crystal structure prediction at high pressures. J. Chem. Phys. 140, 040901 (2014).
Oganov, A. R., Pickard, C. J., Zhu, Q. & Needs, R. J. Structure prediction drives materials discovery. Nat. Rev. Mater. 4, 331–348 (2019).
Wang, Y., Lv, J., Gao, P. & Ma, Y. Crystal structure prediction via efficient sampling of the potential energy surface. Acc. Chem. Res. 55, 2068–2076 (2022).
Goedecker, S. Minima hopping: an efficient search method for the global minimum of the potential energy surface of complex molecular systems. J. Chem. Phys. 120, 9911–9917 (2004).
Glass, C. W., Oganov, A. R. & Hansen, N. USPEX-evolutionary crystal structure prediction. Comput. Phys. Commun. 175, 713–720 (2006).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 82, 094116 (2010).
Pickard, C. J. & Needs, R. J. Ab initio random structure searching. J. Phys. Condens. Matter 23, 053201 (2011).
Oakley, M. T., Johnston, R. L. & Wales, D. J. Symmetrisation schemes for global optimisation of atomic clusters. Phys. Chem. Chem. Phys. 15, 3965–3976 (2013).
Wang, J. et al. MAGUS: machine learning and graph theory assisted universal structure searcher. Natl Sci. Rev. 10, nwad128 (2023).
Gusev, V. V. et al. Optimality guarantees for crystal structure prediction. Nature 619, 68–72 (2023).
Wang, H., Tse, J. S., Tanaka, K., Iitaka, T. & Ma, Y. Superconductive sodalite-like clathrate calcium hydride at high pressures. Proc. Natl Acad. Sci. USA 109, 6463–6466 (2012).
Liu, H., Naumov, I. I., Hoffmann, R., Ashcroft, N. W. & Hemley, R. J. Potential high-Tc superconducting lanthanum and yttrium hydrides at high pressure. Proc. Natl Acad. Sci. USA 114, 6990–6995 (2017).
Peng, F. et al. Hydrogen clathrate structures in rare earth hydrides at high pressures: possible route to room-temperature superconductivity. Phys. Rev. Lett. 119, 107001 (2017).
Drozdov, A. P. et al. Superconductivity at 250 K in lanthanum hydride under high pressures. Nature 569, 528–531 (2019).
Stillinger, F. H. Exponential multiplicity of inherent structures. Phys. Rev. E 59, 48–51 (1999).
Brown, T. B. et al. Language models are few-shot learners. In Advances in Neural Information Processing Systems (NeurIPS, 2020).
Rombach, R., Blattmann, A., Lorenz, D., Esser, P. & Ommer, B. High-Resolution Image Synthesis with Latent Diffusion Models. In 2022 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), vol. 00, 10674–10685 (CVPR, 2022).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Hoffmann, J. et al. Data-driven approach to encoding and decoding 3-D crystal structures. Preprint at https://arxiv.org/abs/1909.00949 (2019).
Noh, J. et al. Inverse design of solid-state materials via a continuous representation. Matter 1, 1370–1384 (2019).
Court, C. J., Yildirim, B., Jain, A. & Cole, J. M. 3 D inorganic crystal structure generation and property prediction via representation learning. J. Chem. Inf. Model. 60, 4518–4535 (2020).
Ren, Z. et al. An invertible crystallographic representation for general inverse design of inorganic crystals with targeted properties. Matter 5, 314–335 (2022).
Xie, T., Fu, X., Ganea, O.-E., Barzilay, R. & Jaakkola, T. Crystal diffusion variational autoencoder for periodic material generation. In International Conference on Learning Representations (ICLR, 2022).
Ye, C.-Y., Weng, H.-M. & Wu, Q.-S. Con-CDVAE: a method for the conditional generation of crystal structures. Comput. Mater. Today 1, 100003 (2024).
Luo, X. et al. Deep learning generative model for crystal structure prediction. npj Comput. Mater. 10, 254 (2024).
Nouira, A., Sokolovska, N. & Crivello, J.-C. CrystalGAN: learning to discover crystallographic structures with generative adversarial networks. In AAAI 2019 Spring Symposium on Combining Machine Learning with Knowledge Engineering (AAAI-MAKE 2019).
Kim, B., Lee, S. & Kim, J. Inverse design of porous materials using artificial neural networks. Sci. Adv. 6, eaax9324 (2020).
Zhao, Y. et al. Physics guided deep learning for generative design of crystal materials with symmetry constraints. npj Comput. Mater. 9, 38 (2023).
Jiao, R. et al. Crystal structure prediction by joint equivariant diffusion. In Advances in Neural Information Processing Systems (NeurIPS, 2023).
Zeni, C. et al. A generative model for inorganic materials design. Nature 639, 624–632 (2025).
Yang, M. et al. Scalable diffusion for materials generation. In International Conference on Learning Representations (ICLR, 2024).
Jiao, R., Huang, W., Liu, Y., Zhao, D. & Liu, Y. Space group constrained crystal generation. In International Conference on Learning Representations (ICLR, 2024).
Levy, D. et al. SymmCD: symmetry-preserving crystal generation with diffusion models. In AI for Accelerated Materials Discovery Workshop (NeurIPS, 2024).
Han, X.-Q. et al. InvDesFlow: an AI-driven materials inverse design workflow to explore possible high-temperature superconductors. Chin. Phys. Lett. 42, 047301 (2025).
Joshi, C. K. et al. All-atom diffusion transformers: unified generative modelling of molecules and materials.In Workshop on AI for Accelerated Materials Discovery (ICLR, 2025).
Miller, B. K., Chen, R. T. Q., Sriram, A. & Wood, B. M. FlowMM: generating materials with Riemannian flow matching. In Proceedings of the 41st International Conference on Machine Learning, vol. 235, 35664–35686 (PMLR, 2024).
Hoellmer, P. et al. Open materials generation with stochastic interpolants. In Workshop on AI for Accelerated Materials Discovery (ICLR, 2025).
Xiao, H. et al. An invertible, invariant crystal representation for inverse design of solid-state materials using generative deep learning. Nat. Commun. 14, 7027 (2023).
Antunes, L. M., Butler, K. T. & Grau-Crespo, R. Crystal structure generation with autoregressive large language modeling. Nat. Commun. 15, 10570 (2024).
Sriram, A., Miller, B. K., Chen, R. T. Q. & Wood, B. M. FlowLLM: flow matching for material generation with large language models as base distributions. In Advances in Neural Information Processing Systems (NeurIPS, 2024).
Cao, Z., Luo, X., Lv, J. & Wang, L. Space group informed transformer for crystalline materials generation. Preprint at https://arxiv.org/abs/2403.15734 (2024).
Chen, Y. et al. MatterGPT: a generative transformer for multi-property inverse design of solid-state materials. Preprint at https://arxiv.org/abs/2408.07608 (2024).
Wang, B. et al. SLICES-PLUS: a crystal representation leveraging spatial symmetry. Mater. Des. 253, 113856 (2025).
Choudhary, K. AtomGPT: atomistic generative pretrained transformer for forward and inverse materials design. J. Phys. Chem. Lett. 15, 6909–6917 (2024).
Gruver, N. et al. Fine-tuned language models generate stable inorganic materials as text. In International Conference on Learning Representations (ICLR, 2024).
Kazeev, N. et al. Wyckoff transformer: generation of symmetric crystals. Prepring at https://arxiv.org/abs/2503.02407 (2025).
Zhang, G. et al. UniGenX: unified generation of sequence and structure with autoregressive diffusion. Preprint at https://arxiv.org/abs/2503.06687 (2025).
Lu, S. et al. Uni-3DAR: Unified 3D generation and understanding via autoregression on compressed spatial tokens. Preprint at https://arxiv.org/abs/2503.16278 (2025).
Wang, Z., Hua, H., Lin, W., Yang, M. & Tan, K. C. Crystalline material discovery in the era of artificial intelligence. Preprint at https://arxiv.org/abs/2408.08044 (2024).
Wang, Z. et al. Advances in high-pressure materials discovery enabled by machine learning. Matter Radiat. Extremes 10, 033801 (2025).
Nguyen, T. M. et al. Hierarchical GFlownet for crystal structure generation. In Advances in Neural Information Processing Systems (NeurIPS, 2023).
Chang, R. et al. Space group equivariant crystal diffusion. Preprint at https://arxiv.org/abs/2505.10994 (2025).
Wu, L. et al. Siamese foundation models for crystal structure prediction. Preprint at https://arxiv.org/abs/2503.10471 (2025).
Chen, R. T. Q., Rubanova, Y., Bettencourt, J. & Duvenaud, D. Neural ordinary differential equations. In Advances in Neural Information Processing Systems (NeurIPS, 2018).
Lipman, Y., Chen, R. T. Q., Ben-Hamu, H., Nickel, M. & Le, M. Flow matching for generative modeling. In International Conference on Learning Representations (ICLR, 2023).
Tong, A. et al. Improving and generalizing flow-based generative models with minibatch optimal transport. In Transactions on Machine Learning Research (TMLR, 2024).
Jain, A. et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Ong, S. P. et al. Python materials genomics (pymatgen): a robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. CALYPSO: a method for crystal structure prediction. Comput. Phys. Commun. 183, 2063–2070 (2012).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Davies, D. et al. SMACT: semiconducting materials by analogy and chemical theory. J. Open Source Softw. 4, 1361 (2019).
Riebesell, J. et al. Matbench Discovery – A framework to evaluate machine learning crystal stability predictions. Preprint at https://arxiv.org/abs/2308.14920 (2023).
Deng, B. et al. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat. Mach. Intell. 5, 1031–1041 (2023).
Zheng, Q. et al. Guided flows for generative modeling and decision making. Preprint at https://arxiv.org/abs/2311.13443 (2023).
Liu, X., Gong, C. & Liu, Q. Flow straight and fast: learning to generate and transfer data with rectified flow. In International Conference on Learning Representations (ICLR, 2023).
Albergo, M. S. & Vanden-Eijnden, E. Building normalizing flows with stochastic interpolants. In International Conference on Learning Representations (ICLR, 2023).
Klein, L., Krämer, A. & Noé, F. Equivariant flow matching. In Advances in Neural Information Processing Systems (NeurIPS, 2023).
Köhler, J., Klein, L. & Noé, F. Equivariant flows: exact likelihood generative learning for symmetric densities. In Proceedings of the 37th International Conference on Machine Learning (PMLR, 2020).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Zimmermann, N. E. R. & Jain, A. Local structure order parameters and site fingerprints for quantification of coordination environment and crystal structure similarity. RSC Adv. 10, 6063–6081 (2020).
Ward, L., Agrawal, A., Choudhary, A. & Wolverton, C. A general-purpose machine learning framework for predicting properties of inorganic materials. npj Comput. Mater. 2, 16028 (2016).
Luo, X. et al. Crystalflow: a flow-based generative model for crystalline materials. Zenodo, https://doi.org/10.5281/zenodo.16908358 (2025).
Luo, X. et al. Crystalflow: a flow-based generative model for crystalline materials. Figshare Dataset. Source structures data generated by CrystalFlow. https://doi.org/10.6084/m9.figshare.29978608.v3 (2025).
Acknowledgements
X.S. wants to thank the National Science and Technology Innovation 2030 Major Program from the Ministry of Science and Technology of China (No. 2024ZD0606900). J.L. wants to thank the National Key Research and Development Program of China (Grant No. 2022YFA1402304), the National Natural Science Foundation of China (Grants Nos. 12034009, 12374005, 52288102, 52090024, and T2225013), the Fundamental Research Funds for the Central Universities, the Program for JLU Science and Technology Innovative Research Team. Y.W. wants to thank the National Natural Science Foundation of China (Grants Nos. T2521005, and T2225013). L.W. wants to thank the National Natural Science Foundation of China (Grants Nos. T2225018, 92270107, and 12188101). Z.W. wants to thank the Postdoctoral Fellowship Program of CPSF (Grant No. GZC20252243). We want to thank “Changchun Computing Center" and “Eco-Innovation Center" for providing inclusive computing power, and technical support of Huawei AI for Science Lab during the completion of this paper and the support provided by MindSpore Community. Part of the calculation was performed in the high-performance computing center of Jilin University. We also want to thank all CALYPSO community contributors for providing the structural data to construct the dataset.
Author information
Authors and Affiliations
Contributions
J.L., L.W., Y.W., and Y.M. designed the research. X.L. and Q.W. wrote the code and trained the model. Z.W. and X.L. collected the dataset. X.L., Z.W. and Q.W. conducted the calculations. X.L., Z.W., Q.W., X.S., J.L., L.W., Y.W., and Y.M. analyzed and interpreted the data, and contributed to the writing of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Benjamin Kurt Miller and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Luo, X., Wang, Z., Wang, Q. et al. CrystalFlow: a flow-based generative model for crystalline materials. Nat Commun 16, 9267 (2025). https://doi.org/10.1038/s41467-025-64364-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64364-4
This article is cited by
-
Materials discovery acceleration by using conditional generative methodology
npj Computational Materials (2025)
-
Generative AI for crystal structures: a review
npj Computational Materials (2025)
