
Abstract
Sexually transmitted infections (STIs) are associated with vaginal dysbiosis, and co-infections are common but understudied. In this study, 6217 reproductive-age women are recruited from 38 study centres across China at baseline and 2738 participants are followed up at 6 months. We profile the vaginal microbiota by 16S rRNA gene sequencing in conjunction with measurement of nine common STIs. The primary outcome of this study is STI status, and secondary outcome is the risk of cervical lesions. Mycoplasmas hominis (MH) far exceeds other STIs in the association with vaginal microbiota, whereases previously reported associations between Human papillomavirus (HPV) and vaginal dysbiosis might be confounded by the co-infected MH in this study. Both MH infection and increased bacterial diversity are independently associated with increased risk of cervical lesion in HPV-negative women (Shannon, OR (odds ratio) = 1.71, 95% CI (confidence interval) = 1.23-2.36; MH, OR = 2.42 95% CI = 1.36-4.30). These associations are also identified in longitudinal analyses (Shannon, HR (hazard ratio) = 1.72, 95% CI = 1.04-2.86; MH, HR = 2.37, 95% CI = 0.98-5.72). Our findings highlight the importance of considering MH status when studying vaginal microbiota in cervical lesions, and suggest the need for further investigation of microbiota-associated mechanisms in HPV-negative cervical lesions. (ClinicalTrials.gov. NCT04694495).
Similar content being viewed by others
Introduction
The composition of vaginal microbiota is critical to female reproductive health, and has been associated with fertility1,2, preterm birth3, sexually transmitted infections (STIs)4, and cervical lesions5. However, these findings have largely been established based on studies from US or European population, highlighting the need for broader geographical investigations and inclusion of more diverse populations in vaginal microbiome research. A healthy vaginal microbiota has been defined as a simple ecosystem dominated by Lactobacillus sp.6. Deeper understanding is still needed on the determinants of vaginal microbiota, which is crucial for women’s reproductive health.
STIs are one of major public health problems especially in developing countries7, with over 300 million infections estimated to occur annually8 and can lead to severe complications in women9,10. Various bacterial, viral and fungal pathogens, such as Chlamydia trachomatis (CT) and Human papillomavirus (HPV), are known to be transmitted through sexual contact, and co-infection were identified among certain STIs11,12,13, yet comprehensive description of the prevalence and co-infection patterns of common pathogens in Chinese women is still lacking. Several common STIs have been individually associated with the composition of vaginal microbiota, including HPV14,15,16, CT17,18, and Trichomonas vaginalis (TV)17,19. HPV is one of the most common STIs, and has been extensively studied in association with the vaginal microbiota1,15,16, due to the risk for cervical cancer. However, previous studies have reported contradictory results20,21, with different associated bacterial taxa and discordant statistical significance identified. Moreover, co-infection status has rarely been investigated to assess the effect of potential confounders in population-level studies.
Cervical cancer remains a significant global health burden, with ~600,000 new cases annually according to WHO. While HPV is the primary causative agent, only around 27% of women with persistent HPV infections will progress to cervical lesions, suggesting that additional factors influence disease development22. Although increasing evidence suggests that alterations in the vaginal microbiota, such as increased diversity and reduced relative abundance of Lactobacillus spp., are involved in the pathogenesis of HPV-related lesions23, some studies still struggle to demonstrate this association24. The independent and interactive effects of different STIs on vaginal microbiota composition and their collective impact on cervical lesions remain poorly understood.
In this study, we leverage a large prospective cohort of 6217 Chinese women from 38 centres, with a subset followed up at six months (CALM2004 project), to disentangle the complex relationships among STIs, the vaginal microbiota, and cervical lesions. Through both cross-sectional and longitudinal analyses, we profile the vaginal microbiome in relation to common STIs, co-infection patterns, and cervical lesions. Our findings highlight a central role of Mycoplasma hominis (MH) in vaginal dysbiosis, which may obscure associations between HPV and the vaginal microbiota through co-infections. Moreover, we demonstrate that vaginal dysbiosis and MH contribute to the progression of cervical lesions, underscoring the potential of microbiota-targeted preventive strategies for HPV-negative cervical lesions.
Results
Common STIs and co-infection patterns in Chinese women
Between November 2020, and October 2022, 7467 individuals were enrolled from 41 study centres across China at baseline. 1250 individuals were removed from our study due to exclusion criteria, or missing or low quality of data, resulting in a total of 6217 individuals from 38 study centres were included for downstream baseline analyses, with matched phenotype, STI testing results, and high quality 16S rRNA sequencing data (Fig. 1, Method). 5493 participants from 32 study centres were followed up at 6 months between May 2021 and June 2023, out of which 2738 (49.8%) finished the follow-up clinical examination and STI measurement, with a mean follow-up time of 239 days (Fig. 1).

HPV Human papillomavirus, STI sexually transmitted infection, CT Chlamydia trachomatis, MH Mycoplasma hominis, MG Mycoplasma genitalium, NG Neisseria gonorrhoeae, UU Ureaplasma urealyticum, HSV-2 Herpes simplex virus II, TV Trichomonas vaginalis, CA Candida albicans, TCT ThinPrep Cytology Test. Failure in 16S sequencing or quality control (unqualified samples for sequencing or low sequence depth (total reads <1000)).
At baseline, 2505 individuals were infected with at least one of the tested sexually transmitted pathogens (Table 1). STI-positive women were younger and had lower BMI than the STI-negative women, whereas higher proportion of STI-negative women were higher educated and more likely to take birth control measures including condom and intrauterine device (IUD). Human papillomavirus (HPV) was the most prevalent sexually transmitted pathogen with an overall prevalence of 19.5% (N = 1213), followed by Ureaplasma urealyticum (UU, 9.9%, N = 614), Candida albicans (CA, 8.9%, N = 556) and Mycoplasma hominis (MH, 7.9%, N = 490) (Supplementary Data 1, 2). The prevalence of STIs varied across study centres (Supplementary Data 3), therefore, study centre was further adjusted for in the subsequent analyses. Co-infection was common in the population, and 26.7% of STI-positive individuals were infected with two or more pathogens (Fig. 2A, Supplementary Fig. S1). Among STIs with high prevalence, co-infection was found in 36.9% HPV-positive individuals, 49% UU-positive individuals, and 59.5% MH-positive individuals. Co-infection of HPV with other STIs was the most common, including UU (N = 172), comprising 14.2% of HPV-infected subjects, followed by MH (N = 162), and CT (N = 99), whereas Mycoplasma genitalium (MG), Neisseria gonorrhoeae (NG) and Trichomonas vaginalis (TV) were not significantly associated with HPV infection.

A Prevalence and co-infection patterns of STIs. Co-infection patterns in sexually transmitted pathogens with at least 5 cases; a full figure provided in Supplementary Fig. S1. In the upset plot, the dots represent the infection status of STIs, a single dot indicates infection with one pathogen only, and multiple connected dots represent co-infections with the corresponding pathogens. The left bar plot (“Set size”) shows the total number of participants positive for each individual STI. The upper bar plot (Intersections) represents the number of participants infected with the pathogens indicated by the dots below. B Shannon diversity of participants with STIs. The colour indicates the significance of associations between STI and Shannon diversity by logistic regression model, including general infection (STI-free vs infection) and mono-infection (STI-free vs mono-infection), full summary statistics are provided in Supplementary Data 4. Boxes in plots visualise median and 25th and 75th percentiles, and whiskers mark range up to 1.5× inter-quartile ranges to show potential outliers. The number within each box indicates the sample size of the group. C The proportion of variance in microbiome composition that can be explained by each infection calculated by PERMANOVA. The colour indicates the significance of PERMANOVA. STI sexually transmitted infection, PERMANOVA permutational multivariate analysis of variance using distance matrix, HPV Human papillomavirus, CT Chlamydia trachomatis, UU Ureaplasma urealyticum, MH Mycoplasma hominis, MG Mycoplasma genitalium, HSV-2 Herpes simplex virus II, TV Trichomonas vaginalis, CA Candida albicans, NS not significant.
Composition of vaginal microbiome in Chinese reproductive-age women
A total of 173,582,142 sequencing reads (mean = 27,920.56 reads per sample, SD = 35,913.39) remained after quality control. After removing singletons, 2033 amplicon sequence variants (ASVs) were obtained, which were assigned to 445 species. We identified 68 core species with a relative abundance (RA) > 0.1% in at least 1% samples, and according to the rarefaction curve, the number of species identified did not increase after including samples from 10 study centres (Supplementary Fig. S2), indicating that our sample size was sufficient to identify the core species. The most abundant species was Lactobacillus iners (mean RA = 36.5%), followed by Lactobacillus crispatus (mean RA = 25.9%) and Gardnerella vaginalis (mean RA = 13.9%), whereas most other species had low abundance (Supplementary Fig. S3). Although the study centres covered different provinces across China, the overall composition patterns were consistent (Supplementary Fig. S3). Similar composition was identified among women without any of the tested STIs (Supplementary Fig. S4). In addition, community state type (CST)6,25 was also used to describe the vaginal microbiome compositions, and the most prevalent CST in this study was CST III (41.0%), followed by CST I (26.9%), and CST IV-B (21.2%), corresponding to the dominance of Lactobacillus iners, Lactobacillus crispatus and Gardnerella vaginalis respectively (Supplementary Fig. S5). The Lactobacillus dominance groups (CST I, II, III and V) was featured by lower Shannon diversity compared to the other groups (CST IV-A, IV-B, IV-C, Supplementary Fig. S5).
Association of STIs and co-infection with the baseline vaginal microbiome
Vaginal bacterial diversity and STIs
We found that most STIs, including MH, HPV, UU, CT and TV, were significantly associated with higher Shannon diversity, whereas CA infection had the opposite association (Fig. 2B, Supplementary Data 4). Among mono-infections, MH still showed the strongest association with Shannon diversity (p = 3.55 × 10−39), followed by CT (p = 3.17 × 10−4) and CA (p = 5.38 × 10−4) mono-infections. HPV and UU mono-infections were not significantly associated with Shannon diversity (p = 0.48 and 0.17, respectively). Additional co-infection analyses found significantly higher diversity in HPV or UU co-infected with MH (p = 2.39 × 10−22 and 2.46 × 10−12, respectively) compared with STI-free status (Supplementary Fig. S6, Supplementary Data 5).
Both general and mono-infection of MH explained the largest proportion of interindividual variation of beta diversity (R2 = 2.56% and 1.99% for general and mono-infection, respectively) among the tested STIs, followed by fungal infection (R2 = 0.49% and 0.44%), UU (R2 = 0.26% and 0.2%) and CT (R2 = 0.15% and 0.14%), which also explained significant proportions of the variance (p < 0.05, Fig. 2C, Supplementary Data 6). Although general infection of HPV, TV, MG and HSV-2 also explained the significant proportions, these STIs were no longer significant in the analysis of mono-infection. This pattern was also consistent across different CSTs (Supplementary Fig. S7, Supplementary Data 7), where MH showed the strongest associations with all CSTs except for CST II in both general- and mono-infection analysis. HPV general infection was associated with CST IV-A, -B and -C, but HPV mono-infection was only associated with CST IV-C. Taken these together, MH showed the strongest association with vaginal microbiome composition, while HPV infection was not associated with bacterial diversity when considering only the mono-infection status in this study.
MH exhibits the strongest correlation with vaginal microbiota at baseline
We next assessed the association between each bacterium and STIs by logistic regression model. In the analysis of general infection, we identified 175 significant bacteria-phenotype pairs from 56 species (FDR < 0.05, Fig. 3, Supplementary Data 8). The largest number of significant associations were identified for MH (N = 47, FDR < 0.05), followed by HPV and CT (N = 24 for both, FDR < 0.05). However, the number of significant associations was notably lower in the analysis of mono-infection compared to general infection, and only 52 significant pairs were identified (FDR < 0.05, Fig. 3, Supplementary Data 9). MH mono-infection was still associated with the most bacteria (N = 37, FDR < 0.05) among all STIs, including 25 positively associated species (e.g., Coriobacteriales bacterium, Prevotella buccalis, and Sneathia amnii) and 12 negatively associated species (Lactobacillus iners, Lactobacillus crispatus and Proteus mirabilis). Most of the identified taxa (35/37) were also consistently identified by the general infection model (FDR < 0.05, Supplementary Data 8, 9).

Associations of vaginal bacterial species with STIs under three different infection models. Significant positive (red) and negative (blue) associations of specific bacteria with STIs (p < 0.05), with non-significant associations indicated in white. “+” and “−” indicate positive and negative correlations that remain significant at FDR < 0.05. Three models from left to right: general infection model (infection-free vs infection), mono-infection model (infection-free vs mono-infection) and co-infection (infection-free vs co-infection (N > 30)). Full summary statistics can be found in Supplementary Data 8–10. MH Mycoplasma hominis, CT Chlamydia trachomatis, UU Ureaplasma urealyticum, HPV Human papillomavirus, HR.HPV high risk HPV, LR.HPV low risk HPV, TV Trichomonas vaginalis, MG Mycoplasma genitalium, HSV-2 Herpes simplex virus II, CA Candida albicans.
HPV-microbiota associations are confounded by MH co-infection
A dramatic decrease in the number of significant associations was identified for HPV, from 24 in the general-infection model to none in the mono-infection model after FDR correction (Fig. 3). Consistent with our diversity analyses (Fig. 2B, C), these results indicate that the association between HPV and vaginal microbiota might in fact be confounded by co-infection with other STIs. Thus, we performed logistic regression analysis of pairwise STIs co-infection and identified 110 significant associations from 41 species (FDR < 0.05, Fig. 3, Supplementary Data 10). The largest number of associations (N = 33) were identified for HPV-MH co-infection, with only a few significant associations were identified between HPV co-infection with other STIs. We further compared the significance and direction of associations among different models, and the results of HPV-MH co-infection were highly consistent with those of both MH mono-infection model and HPV general-infection model (Supplementary Fig. S8), indicating that the identified association between general HPV infection and vaginal bacteria was mainly confounded by the presence of MH in HPV-infected women in this study. Moreover, we analysed the associations of high-risk and low-risk HPV with vaginal microbiome. Most of the HPV-positive subjects (84.9%) were infected with high-risk HPV subtype, and the results of high-risk HPV were similar to those of the overall HPV (Fig. 3, Supplementary Data 8–10).
Complementary to the logistic regression, we also applied MaAsLin2 to investigate the associations between STIs and vaginal bacteria (Supplementary Data 11) as a cross-validation, and the results were consistent between two methods (Supplementary Figs. S9, 10). Moreover, analysis of variance explained indicated that MH accounted for the largest variation in Shannon diversity and in the abundance of most vaginal bacterial taxa among all tested STIs (Supplementary Fig. S11, Supplementary Data 12). Menstrual cycle was reported to be associated with vaginal microbiome, and we further performed sensitivity analysis in subjects with information of menstrual cycle (N = 4091, Supplementary Data 13). The results by additionally adjusting for menstrual cycle phase were comparable to the original model (Supplementary Data 14), and the stratified analysis also showed consistent association patterns with the original analysis (Supplementary Fig. S12, Supplementary Data 15).
Association of STIs and vaginal microbiome with cervical lesion at baseline
Vaginal pathogens and bacteria associated with TCT results
ThinPrep Cytology Test (TCT) is an important examination in cervical cancer screening. In our cohort, 450 out of 4689 women had abnormal TCT results, including 257 of grade ASC-US (atypical squamous cells of undetermined significance), and 110 of LSIL (low-grade squamous intraepithelial lesion). No significant differences were identified between normal and abnormal TCT groups based on age, BMI or education level, whereas women with abnormal TCT were less likely to take birth control measures (Supplementary Data 16).
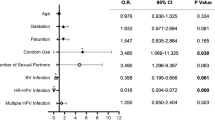
HPV infection showed the strongest correlation with a higher risk of abnormal TCT (OR = 8.74, 95% CI = 6.70–11.40, Supplementary Data 17, Fig. 4A), followed by MH (OR = 2.49, 95% CI = 1.77–3.50), CT (OR = 1.67, 95% CI = 1.06–2.62) and UU (OR = 1.46, 95% CI = 1.05–2.03). HPV accounted for the largest proportion of variance in abnormal TCT results (Supplementary Fig. S11). Shannon diversity (p = 1.79 × 10−3, Supplementary Data 18) and three taxa were significantly associated with abnormal TCT results (FDR < 0.05, Supplementary Fig. S13, Supplementary Data 19), including Anaerococcus tetradius, Dialister micraerophilus, as well as Veillonella montpellierensis which has been reported to induce moderate cytotoxicity to cervical epithelial cells26. Sub-group analysis of TCT grades suggested that the ASC-US group had higher alpha diversity than the control group (p = 0.035), while no significant differences were identified for LSIL (Supplementary Data 18). Five taxa (Prevotella buccalis, Prevotella disiens, Mageeibacillus indolicus, Anaerococcus tetradius, and Veillonella montpellierensis) were significantly associated with ASC-US and three (Prevotella bivia, Prevotella colorans, angd Peptoniphilus genus) were associated with LSIL (p < 0.05), although none remained significant after FDR correction (Supplementary Fig. S13, Supplementary Data 20).

The OR with 95% CI and p value are plotted for each feature, including STI test in this study, Shannon diversity and vaginal microbiota species that were associated with TCT results (p < 0.05). Analysis was first performed in all samples (A), and further stratified analysis was performed in HPV negative subjects (B) and HPV positive subjects (C). Results with p < 0.05 are highlighted. Logistic regression models were conducted using baseline samples only. Sample sizes (N) for each analysis are indicated in the figure panels. Full summary statistics can be found in Supplementary Data 19, 21, 16. Error bars represent the 95% CI. OR odds ratio, CI confidence interval, HPV Human papillomavirus, HR.HPV high risk HPV, LR.HPV low risk HPV, MH Mycoplasma hominis, HSV-2 Herpes simplex virus II, CT Chlamydia trachomatis, TV Trichomonas vaginalis, UU Ureaplasma urealyticum, MG Mycoplasma genitalium, CA Candida albicans.
Vaginal dysbiosis and MH infection are associated with higher risk of HPV-negative abnormal TCT
Given the large effect of HPV infection on abnormal TCT, we further analysed the samples stratified by HPV status. In the HPV negative group, MH infection and Shannon diversity were significantly associated with higher risk of abnormal TCT (MH, OR = 2.42, 95% CI = 1.36–4.30; Shannon, OR = 1.71, 95% CI = 1.23–2.36; Fig. 4B, Supplementary Data 18). Furthermore, in HPV-negative women, MH and Shannon diversity accounted for a greater proportion of the variance in cervical cytological abnormalities compared to other STIs, although the total variance explained (pseudo-R²) was modest (Supplementary Fig. S11). 13 taxa were associated with abnormal TCT (p < 0.05, Supplementary Fig. S13, Supplementary Data 21), including positive associations with Fannyhessea vaginae, Prevotella bivia and Anaerococcus tetradius, which have been previously associated with adverse outcome including preterm birth, and cervical intraepithelial neoplasia3,27,28. These association results could lead to the hypothesis that MH and vaginal microbiota may also be risk factors for abnormal TCT in HPV-negative women.
In the HPV-positive group, none of other STIs were associated with TCT results (Fig. 4C). Six bacteria were associated with TCT (p < 0.05, Supplementary Fig. S13, Supplementary Data 21), suggesting that the positively associated taxa might also be risk factors for abnormal TCT in addition to HPV infection. Notably, there were no overlapping taxa associated with abnormal TCT in the HPV positive and negative groups, suggesting different roles of vaginal microbiota based on HPV infection status or co-exclusion with HPV. The overall association patterns in the sensitivity analyses of samples stratified by total STI status were consistent with that of the HPV-stratified analyses (Supplementary Fig. S13, Supplementary Data 22).
Baseline MH and vaginal microbiota are associate with an increase risk in abnormal TCT onset
We next performed a longitudinal analysis to test the association between MH and vaginal microbiota and their role in the progress of abnormal TCT. We identified that higher baseline Shannon diversity was significantly associated with an increased risk of both new-onset infection (HR = 1.72, 95% CI = 1.07–2.77) and persistent infection (HR = 2.08, 95% CI = 1.29–3.36) of MH, but was not significantly associated with HPV progress (Fig. 5B, Supplementary Data 23). The results remained consistent after additional adjustment for baseline and follow-up HPV or MH infection status in the Cox regression model, suggesting that the association between Shannon diversity and MH progression is independent of HPV co-infection (Supplementary Data 23).

A Definition of outcomes in the longitudinal analysis, persistent negative vs new-onset is marked as (N) and transient vs persistent positive in marked as (P) in lower panels. B Baseline Shannon diversity and the progress of STIs (MH and HPV). C Baseline STI/ Shannon diversity and the progress of TCT. The analysis was performed in all samples (upper), HPV-negative samples (middle) and HPV-positive samples (bottom). The HR with 95% CI and p value are plotted for each association by cox-regression model. Results with p < 0.05 are highlighted. Full summary statistics can be found in Supplementary Data 23. Error bars represent the 95% CI. HR hazard ratio, CI confidence interval, MH Mycoplasma hominis, HPV Human papillomavirus, P persistent infection, N new-onset, TCT ThinPrep Cytology Test.
At 6-months follow-up, 178 subjects had an abnormal TCT result, including 55 persistent and 123 new-onset subjects (Supplementary Data 24). Baseline infection of HPV and MH was significantly associated with an increased risk of new-onset abnormal TCT (HR = 5.30, 96% CI = 3.43–8.19 for HPV; HR = 1.91, 95% CI = 1.09–3.36 for MH), while only HPV was significantly associated with an increased risk of persistent abnormal TCT (HR = 2.7, 95% CI = 1.14–6.40) (Fig. 5C, Supplementary Data 23). Although baseline Shannon diversity was not significantly associated with TCT progress, the association with new-onset abnormal TCT reached marginal significance (HR = 1.42, 95% CI = 1.00–2.04).
In women without HPV infection at baseline, higher baseline Shannon diversity was associated with an increased risk of new-onset abnormal TCT (HR = 1.72, 95% CI = 1.04–2.86), but was not associated with persistent TCT (Fig. 5C, Supplementary Data 23). Moreover, baseline MH was associated with an increased risk of new onset TCT with marginal significance (HR = 2.37, 95% CI = 0.98–5.72), suggesting that baseline MH might also be a risk factor for cervical lesion in HPV-negative women, but this finding still needs further confirmation. In HPV-positive women, we did not identify a significant association for both baseline Shannon diversity and MH on TCT progress (Fig. 5C, Supplementary Data 23). Taking these together, both cross-sectional and longitudinal analyses revealed that MH infection and vaginal microbiota might also be risk factors for abnormal TCT in women without HPV infection.
Discussion
To the best of our knowledge, this is the largest observational study to date investigating the association between vaginal microbiota and STIs in China based on both cross-sectional and longitudinal analyses. Surprisingly, and contrary to previous report, HPV infection did not show strong associations with vaginal microbiota. Instead, MH infection was predominantly associated with vaginal dysbiosis, suggesting that it might confound the associations between vaginal microbiota and other pathogens through co-infection. Furthermore, both the cross-sectional and longitudinal results revealed that MH and vaginal microbiota may be potential risk factors for cervical lesion in HPV-negative women.
Our study highlights an important role of MH in vaginal dysbiosis, which has been previously understudied. Although a significant correlation between MH and G. vaginalis has been previously reported29, our study identifies that MH shows the strongest association with overall microbiota composition among all STIs examined, and co-infection with MH is a critical factor to understand the interactions between vaginal microbiota and STIs. We found that that MH was negatively associated with four dominant vaginal Lactobacillus species (L. crispatus, L. iners, L. gasseri and L. jensenii), driving CSTs that characterize healthy vaginal communities6, while MH was positively associated with multiple anaerobes including G. vaginalis, P. amnii, F. vaginae, S. sanguinegens, that were previously linked to adverse outcomes. MH has been reported to be resistant to multiple antibiotics30, due to the genetic alterations in DNA gyrase and/or the topoisomerase IV complex, which impair the efficacy of commonly used antibiotics31, highlighting the need for further studies on its relation with vaginal microbiota that might provide novel insights for MH treatment. Consistent with our cross-sectional results, the longitudinal analysis also revealed that baseline vaginal microbiota dysbiosis increased the risk of MH persistent and new-onset infection, indicating that keeping a healthy vaginal microbiota is crucial in the prevention and treatment of MH.
HPV has received significant attentions on its potential interactions with the vaginal microbiota, due to its high prevalence and the potential risk for cervical cancer32. It is worth noting that previous reports have found inconsistent associations between HPV infections and vaginal microbiota, with different bacteria identified by different studies15,33. For example, Sneathia and Prevotella have been reported to be more abundant in HPV-positive women15, while another study identified Bacteroides, Acinetobacter, Faecalibacterium, Streptococcus, Finegoldia, and Moryella as differential taxa associated with HPV status16. Moreover, some studies reported no significant association between HPV-infection and vaginal microbiota regarding CST and alpha diversity16,21,34. It should be noted that the CST are developed based on a US population and the application to Chinese population should be interpretate with caution. Our results suggest that, beyond geographical or population-specific effects, prior contradictory findings could be partially attributed to the lack of consideration on the co-infection status of HPV with other STIs, especially MH. The results of longitudinal analysis were also unsupportive of the influence of baseline vaginal microbiota in the progress of HPV. Taken these together, our results stressed the importance of considering other STIs, especially MH, in the study of HPV and vaginal microbiome. These findings also suggest that more attention should be paid to understanding microbe–microbe interactions in women’s reproductive health.
Although we present different vaginal microbiome patterns of general, mono- and co-infection of STIs, the causal relationship between STIs and vaginal microbiome has not been fully elucidated. Vaginal microbial dysbiosis is associated with increased inflammation, which may in turn compromise the epithelial barrier and facilitate infection by sexually transmitted pathogens35,36. On the other hand, the infection of pathogens may induce the immune responses and change the vaginal microenvironment37. Further validation from different populations, prospective studies and investigations into the molecular mechanisms of STI-microbiota interactions are required to establish causality. Moreover, the co-infection observed in this study represents only a concurrent state. Future research is deserved to determine the causal relationships between co-infected STIs and to elucidate the underlying mechanisms of their interactions.
Although HPV is known as a major cause of cervical cancer, increasing studies have provided evidence for HPV-negative cervical cancer, with prevalence varying from 3% to 52%, and with different histologic types and prognosis compared to HPV-positive cervical cancer38,39. Part of the HPV-negative cancer cases were attributed to the missing of testing on certain HPV types in the screening40, and in our study, we attempted to minimize this risk including 21 common HPV types, however, we cannot rule out the possibility of false-negative results due to untested or rare genotypes. We confirmed previous results that HPV has the strongest effect on both baseline and progress of abnormal TCT; however, 33% of subjects with abnormal TCT were HPV-negative in our cohort. In this specific group of HPV-negative women, MH was the strongest risk factor compared to other STIs for the abnormal TCT onset, emphasizing the importance of regular screening and treatment of MH in the prevention of cervical lesions. Moreover, we also identified increased Shannon diversity, as well as the higher abundance of dysbiosis-related species, including Streptococcus5, Sneathia sanguinegens27, and Anaerococcus tetradius27, that were associated with abnormal TCT in HPV-negative women, indicating that targeting the vaginal dysbiosis may also benefit for the prevention of cervical lesions. Taken together, we provided new insights into the HPV-negative cervical lesion by stressing the risk of MH infection and vaginal microbiota dysbiosis on abnormal TCT, which may be additional risk factors for cervical lesions when HPV was negative, suggesting that the HPV-negative women may benefit from additional screening for MH and assessment of vaginal microbiome composition, particularly in the context of abnormal cytology.
MH has traditionally been regarded as a commensal bacterium within the vaginal microenvironment, and previous studies did not recommend routine testing or treatment for MH41,42. However, emerging research has increasingly highlighted its associations with adverse outcomes, including HPV infection43,44, BV45,46, pelvic inflammatory disease47, preterm birth48, and postpartum endometritis49. Despite these findings, the causal relationship between MH and these outcomes, as well as its role in cervical lesions, remains insufficiently explored. In this study, we provided important insight that MH is associated with vaginal dysbiosis and the progress of cervical lesions from a Chinese multi-centre cohort, which calls for more attention and investigations on MH in vaginal health. MH has been associated with the vaginal dysbiosis, due to its ability in metabolizing arginine into ammonia, which elevates vaginal pH and potentially facilitate the growth of G.vaginalis -- a key player in BV pathogenesis50. Additionally, MH expresses lipoproteins that stimulate interleukin-23 (IL-23) production by dendritic cells and macrophages, eliciting a local pro-inflammatory immune response51. Although direct evidence linking MH to cervical lesions is limited, one study has reported that co-infection with TV and MH was associated with an elevated risk of cervical abnormalities, possibly by facilitating epithelial invasion or enhancing local inflammation51. Nevertheless, the further application of these findings in clinical practice requires additional studies with higher level of evidence, focusing specifically on the progression of MH and its role in clinical outcomes. Intervention studies, including randomized controlled trials, as well as molecular investigations, are essential to elucidate the effects of MH treatment and clearance on cervical lesion progression, and provide more insights into the clinical applications.
The main strength of this study is our large sample size in a multicentre cohort, and the simultaneous measurement of multiple common STIs and vaginal microbiota using consistent protocols. Moreover, the longitudinal design enabled us to disentangle the role of vaginal microbiota and STI in the progress of cervical lesions. Limitations include potential biases due to patients lost to follow-up, and a limited follow-up time, which may impact our estimates of HPV progression and cervical lesions. Thus, we compared the baseline characteristics between participants who completed the follow-up and those who were lost to follow-up (Supplementary Data 25). Participants retained in the follow-up cohort were more likely to be highly educated than those lost to follow-up, but the baseline prevalence of STIs, cervical lesions, and CSTs were comparable between the two groups, suggesting that the loss to follow-up was not systematically associated with our primary outcomes. However, we cannot completely rule out the influence of potential unmeasured confounders, including changes in sex partners, medication use, treatment, or personal health-seeking behaviour during follow-up. This limits our ability to assess the impact of clinical interventions on STI progression or microbiota dynamics. Although infections were defined as persistent or new-onset based on PCR positivity, we cannot completely exclude the possibility of reinfection with a different strain or subtype. Moreover, we used 16S rRNA gene sequencing, which limits our ability to identify strain-level taxa and functional mechanisms.
In this population-level study, we provided a profile of vaginal microbiome in association with extensive common STIs, co-infection patterns and cervical lesions by both cross-sectional and longitudinal analyses. The results suggested a central role of MH in the associations between STIs and vaginal dysbiosis, which could have obscured the associations between HPV and vaginal microbiota through co-infections, and draw attention to the potentials of regular screening and treatment of MH in clinical practice. Moreover, our data indicated an important contribution of vaginal dysbiosis and MH in the progress of cervical lesions, enlightening the microbiota-targeted development of preventive approaches for HPV-negative cervical lesions.
Methods
Study design and participants
This observational cohort study was performed within the CALM 2004 project. This study has been registered online at ClinicalTrials.gov (NCT04694495) and China Human Genetic Resources Management Office (2021SLCJ0955). The study was approved by the ethics committee of ZhuJiang Hospital of Southern Medical University (NO.2020-KY-071-01) and all participating centres. Written informed consent was obtained from all participants. Standardized protocols and training ensured consistency across centres. Study protocols including participant recruitment, sample size calculation, and sample collection, processing and testing have been reported previously52. In brief, between November 2020 and October 2022, around 7300 adult premenopausal women from 41 study centres across China were enrolled at baseline. In the present study, we first excluded individuals: 1) had ineligible samples; 2) who requested withdrawal from the study; and 3) had duplicate samples. Thus, in this study, 7467 individuals were enrolled initially and further excluded if they 1) >55 years old (28 excluded), 2) had sexual behaviour, vaginal medication or douching within 72 h prior to sampling (123 excluded), 3) took antibiotics or antifungal drugs in past one month (122 excluded), 4) had a history of cervical tumour or surgery (266 excluded). Additional 360 subjects without clinical records, and 58 without results from STI test were further excluded. In the quality control of 16S data, 293 samples were excluded because of unqualified samples for sequencing or low sequence depth (total reads <1000). Sample size of each study centre included in the final analysis of this study was listed in Supplementary Data 26.
Follow-up
Participants from 32 centres were followed up at around six months. Vaginal and cervical samples were collected with the sample protocol during both baseline and the follow-up visit (between May 2021 and June 2023). Clinical examination including ThinPrep Test (TCT), HPV genotypes and other STIs were recorded at both baseline and follow-up using the uniformed methodology below.
Sample collection and preparation
Participants underwent cervical screening in each study centre using uniformed protocols, with vaginal swabs and cervical exfoliated cells collected at both baseline and follow-up visit. Three vaginal secretion samples were collected by fully inserting a swab (Jiangsu Kangjie Medical Devices Co., Ltd., China) into the posterior vaginal fornix and rotating clockwise 10 times exactly 360° by the clinician. The swabs were then placed into a storage tube and immediately stored at −80 °C. One swab was used for bacterial DNA extraction, one for clinical laboratory examination, and the last one for cryogenic storage at −80 °C. Two tubes of cervical exfoliated cells were collected from the cervical canal by inserting plastic brushes and then rotating clockwise 5 times exactly 360°. Immediately after collection, the cytobrush will be placed in ThinPrep Pap Test PreservCyt Solution (Hybribio, China), and immediately stored at 4 °C for cytology and STI measurement within 24 h. The reserved frozen samples and the samples used for nucleic acid extraction and 16S rRNA sequencing were transported to the Zhujiang Hospital through the cold chain from each research centre for the unified follow-up processing.
Assessment of STIs
Cervical exfoliated cells were used for test of HPV genotypes and other sexually transmitted pathogens detection. HPV and subtypes were measured by HPV GenoArray diagnostic kit (Hybribio, China). We tested 21 HPV subtypes, including 13 high risk subtypes (HPV16, HPV18, HPV31, HPV33, HPV35, HPV39, HPV45, HPV51, HPV52, HPV56, HPV58, HPV59, HPV68), and eight low risk subtypes (HPV6, HPV11, HPV42, HPV43, HPV44, HPV53, HPV66, CP8304). Other STIs, including Neisseria gonorrhoeae (NG), Chlamydia trachomatis (CT), Ureaplasma urealyticum (UU), Mycoplasma hominis (MH), Mycoplasma genitalium (MG), Herpes simplex virus type II (HSV-2) and Trichomonas vaginalis (TV), were measured by the STIs detection kit (Hybribio, China) according to the manufacturer’s instructions. Initially, 0.5-1.0 mL of cell preservation solution, containing cervical cells, was centrifuged at 13,000 rpm for 5 min with the supernatant subsequently discarded. Following this, 0.5 mL of cell preservation solution was added, the cells were centrifuged at 13,000 rpm for 1 min, and the supernatant was disposed of, ensuring minimal moisture residue. Each sample was then treated with 50 μL of cell lysate, the cell resuspension was agitated, and then boiled for 10 min. After centrifugation at 13,000 rpm for 10 min, the supernatant was preserved. A volume of 3.0 μL of the extracted DNA was employed as the template for the PCR amplification. DNA fragments from HPV and other STIs were subsequently amplified by PCR, using biotin-labelled primers, prior to the flow-through hybridization of amplification products with nylon membranes tagged with probes on a flow-through hybridization platform (Hybribio, China). Analysis of the results was accomplished using a chemical colour development technique. The presence of Candida albicans (CA) was determined by the wet film technique on vaginal secretions and enzymatic testing on the vaginal swabs, and positive results further confirmed by culturing of the vaginal secretion samples.
Cervical cytology by the ThinPrep Cytologic Test
Another sample of cervical exfoliated cells were placed in ThinPrep Pap Test PreservCyt Solution (Hybribio, China) and further analysed using the Bethesda system (TBS) for the ThinPrep Test (TCT). The results were classified based on the TBS grading system, including negative for intraepithelial lesion or malignancy (NILM), atypical squamous cells (ASC), atypical glandular cells (AGC), low-grade squamous intraepithelial lesion (LSIL), high-grade squamous intraepithelial lesion (HSIL), and squamous cervical cancer (SCC).
16S rRNA amplicon sequencing and processing
DNA was extracted from vaginal secretions collected at baseline using the QIAamp DNA Micro Kit (QIAGEN, Germany) following the manufacturer’s instructions. To achieve high species-level resolution of vaginal microbiota, we applied an optimized taxonomic classification pipeline previously developed and validated by our group, which has demonstrated high accuracy in vaginal bacterial species identification53. The V1-V3 hypervariable regions of the 16S rRNA gene were amplified by PCR, followed by pair-ended sequencing using the Illumina Novaseq PE250 platform. The raw sequencing data were demultiplexed into paired-end FASTQ files using unique barcodes, and adaptor/primer sequences were removed using a customized Perl script to retain reads with a minimum length of 100 bp. The forward reads were denoised by DADA2 denoise-single plugin (Version 2021.11), which includes quality filtering (--p-trunc-len 223), dereplication and chimera removal to generate amplicon sequence variant (ASV) table and the representative sequences54. The taxonomic classification was performed following our pipeline determined optimal for the species-level classification of vaginal microbiota53: the representative sequences were annotated by QIIME2 using the pre-fitted scikit-learn taxonomy55 classifier based on the combination of Greengenes256 (Release 2022.10), SILVA57 (Version 138) and RDP58 (Release 11.5). Negative controls were included to minimize potential contamination. Decontamination was assessed using the R package decontam59, combined with manual inspection of putative contaminating ASVs, according to the bacterial density and frequency, as well as the black list that reported known reagent and laboratory contamination60,61. Samples were rarefied at 1000 mapped reads, those with low sequence depth (total reads <1000) were excluded from the downstream analyses.
Statistical analysis
Descriptive statistics included frequencies (proportions) for categorical variables, and mean with standard deviation for continuous variables. The χ2 test or Wilcoxon test was used for the inter-group comparison. We filtered rare taxa and only focused on bacteria that were present with a relative abundance >0.01% in at least 1% participants. Microbiome distance was calculated using Bray-Curtis dissimilarity followed by a Principal Coordinate Analysis (PCoA). The proportion of variance in microbiome composition that can be explained by each infection was calculated by permutational multivariate analysis of variance using distance matrix (PERMANOVA) using the function “adonis” in R package “vegan”, with 1000 times of permutation. We performed this analysis on two types of STI phenotype, one for general infection (STI-free vs infection of each STI) and another for mono-infection (STI-free vs mono-infection of each STI). We performed the analysis for each STI respectively.
Shannon diversity was calculated using R package “microbiome”, and phylogenetic diversity was calculated using package “picante”, as a supplementary analyses to Shannon diversity. The association between alpha diversity and STI was performed by logistic regression models adjusting for potential confounders, including Model1(STI (STI-free vs infection) ~alpha diversity + age + BMI + centre), and Model2 (STI (STI-free vs mono-infection) ~alpha diversity + age + BMI + centre.
Community state type (CST) was calculated using the VALENCIA (VAginaL community state typE Nearest CentroId clAssifier) method25. The associations between each CST type (one type vs other types) and STI (STI-free vs infection, and STI-free vs mono-infection) was assessed by χ2 test.
Logistic regression models were applied to assess the associations between different STI phenotypes and vaginal microbiota taxa at species level, including 68 core species with RA > 0.1% in at least 1% samples. The relative abundance was transformed using the centred additive log-ratio (CLR) transformation in association analysis. NG was not included in all analyses, and TV, MG, HSV-2 was not included in the mono-infection analysis, due to limited case number (Supplementary Data 1). And for pairwise co-infection model, we only focused on the co-infections with enough cases in the downstream analysis, including HPV-UU (N = 82), HPV-MH (N = 92), HPV-CT (N = 48), HPV-CA (N = 43), and UU-MH (N = 41).
In total three models were tested in this study, which were similar to the analysis of alpha diversity:
Model1: STI (STI-free vs infection) ~ taxa abundance + age + BMI + centre
Model2: STI (STI-free vs mono-infection) ~ taxa abundance + age + BMI + centre
Model3: STI (STI-free vs co-infection) ~ taxa abundance + age + BMI + centre
We also applied MaAsLin262 to investigate the association between STIs and vaginal microbiome, and this analysis was also performed using CLR transformation method and adjusting for age, BMI and study centre. Results were considered as significant at false discovery rate (FDR) < 0.05 by Benjamini–Hochberg method.
Menstrual cycle was reported to be associated with vaginal microbiome. We further performed sensitivity analysis in subjects with information on menstrual cycle (N = 4091), to confirm if the association between STI and vaginal microbiota were confounded by the different menstrual cycle phase. The sensitivity analysis was performed by additionally adjusting for menstrual cycle phase in the above models, as well as by stratifying samples by menstrual cycle phase and assessing the association again using the same models above. The stratified analyses were performed in women in follicular phase (N = 1891) and luteal phase (N = 2016) respectively. The ovulatory phase was not tested due to the limited sample size (N = 184) and case number for each STI in this group.
The association between STI and TCT was also assessed by logistic regression model: TCT (normal vs abnormal) ~ STI (STI-free vs infection) + age + BMI + centre, and the analysis was performed in all samples, HPV-positive samples and HPV-negative samples respectively. Logistic regression model was applied to assess the associations between TCT and vaginal microbiota taxa at species level: TCT (normal vs abnormal) ~ taxa abundance/Shannon diversity + age + BMI + centre. In addition, we also did stratified analysis in subgroup of samples, including HPV-positive samples, HPV-negative samples, STI-free subjects, and STI positive subjects (positive in any STI measured in this analysis), using the same model mentioned above. Sub-group analysis of different grade of TCT was performed for ACS-US (N = 257) and LSIL (N = 100) that had enough case number.
To assess the contribution of each STI to vaginal microbiome conditional upon the others, the full model was fit first: Shannon/Taxa ~ age + BMI + centre + HPV + MH + UU + CT + TV + MG + NG + HSV-2 + CA. Subsequently several reduced models were fit where one STI was missing. The R² for this full model was then compared against the model with the missing level. The difference between the reduced model and the full model (R²_full − R²_reduced) was taken as a measure of the variance explained by that STI when accounting for the effects of the other STIs. Similarly, we also assessed the contribution of each STI on TCT, where the pseudo-R² was used instead of R2.
Cox regression was applied to assess the association between baseline features and progress of STIs (MH and HPV) and TCT. The progress of STIs (MH and HPV) and TCT was defined according to baseline and follow-up status. Samples were further stratified based on the baseline status to define the outcome of persistency (P, baseline positive follow-up positive vs baseline positive follow-up negative) and new-onset (N, baseline negative follow-up positive vs baseline negative follow-up negative) respectively. The following associations were tested:
Analysis1: HPV/MH (persistent negative vs new-onset/persistent infection vs transient) ~ baseline Shannon diversity + age + BMI + centre
Analysis2: TCT (persistent negative vs new-onset/persistent positive vs transient) ~ baseline Shannon diversity/HPV/MH + age + BMI + centre (in all samples)
Analysis3: TCT (persistent negative vs new-onset/persistent positive vs transient) ~ baseline Shannon diversity/MH + age + BMI + centre (in baseline HPV positive and negative samples, respectively).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The full summary statistics to support the findings of this study, and data used to generate main figures are included within the Supplementary Data file. The microbiome sequencing data and limited metadata generated in this study have been deposited in the Genome Sequence Archive under study accession ID: HRA010936 (https://ngdc.cncb.ac.cn/gsa-human/). Requests for the deidentified individual participant data and the data dictionary that support the findings of this study can be directed to hzhou@smu.edu.cn with a response expected within four weeks.
Code availability
The R code used for the data analysis can be found on GitHub (https://github.com/ZJJY-Bioinformatics/CAL2004_VM_STI) and Zenodo (https://doi.org/10.5281/zenodo.17059937)63.
References
Haggerty, C. L. et al. Identification of novel microbes associated with pelvic inflammatory disease and infertility. Sex. Transm. Infect. 92, 441–446 (2016).
Moreno, I. et al. Evidence that the endometrial microbiota has an effect on implantation success or failure. Am. J. Obstet. Gynecol. 215, 684–703 (2016).
Fettweis, J. M. et al. The vaginal microbiome and preterm birth. Nat. Med. 25, 1012–1021 (2019).
Edwards, V. L. et al. The cervicovaginal microbiota-host interaction modulates Chlamydia trachomatis infection. mBio 10, e01548-19 (2019).
Chen, Y. et al. Human papillomavirus infection and cervical intraepithelial neoplasia progression are associated with increased vaginal microbiome diversity in a Chinese cohort. BMC Infect. Dis. 20, 629 (2020).
Ravel, J. et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 108, 4680–4687 (2011).
Newman, L. et al. Global estimates of the prevalence and incidence of four curable sexually transmitted infections in 2012 based on systematic review and global reporting. PLoS One 10, e0143304 (2015).
Zhang, J., Ma, B., Han, X., Ding, S. & Li, Y. Global, regional, and national burdens of HIV and other sexually transmitted infections in adolescents and young adults aged 10-24 years from 1990 to 2019: a trend analysis based on the Global Burden of Disease Study 2019. Lancet Child Adolesc. Health 6, 763–776 (2022).
Dai, L., Wilson, L. G., Nakagawa, M. & Qin, Z. Coinfections with additional oncoviruses in HPV+ individuals: Status, function and potential clinical implications. J. Med. Virol. 96, e29363 (2024).
Li, Y. et al. The estimated lifetime quality-adjusted life-years lost Due to Chlamydia, Gonorrhea, and Trichomoniasis in the United States in 2018. J. Infect. Dis. 227, 1007–1018 (2023).
Zhong, C., Li, X., Teng, Y. & Tian, J. Co-infection with human papillomavirus and sexually transmitted infections among Chinese individuals. Micro Pathog. 185, 106395 (2023).
Pella-Saavedra, P. et al. Prevalence of coinfections in a cross-sectional cohort of women screened for multiple pathogens in Peru. Heliyon 9, e14257 (2023).
Ghasemian, E., Harding-Esch, E., Mabey, D. & Holland, M. J. When bacteria and viruses collide: a tale of Chlamydia trachomatis and sexually transmitted viruses. Viruses 15, 1954 (2023).
Usyk, M. et al. Cervicovaginal microbiome and natural history of HPV in a longitudinal study. PLoS Pathog. 16, e1008376 (2020).
Cheng, L. et al. Vaginal microbiota and human papillomavirus infection among young Swedish women. NPJ Biofilms Microbiomes 6, 39 (2020).
Andrade Pessoa Morales, J. et al. Vaginal microbiome components as correlates of cervical human papillomavirus infection. J. Infect. Dis. 226, 1084–1097 (2022).
Chiu, S.-F. et al. Vaginal microbiota of the sexually transmitted infections caused by Chlamydia trachomatis and Trichomonas vaginalis in women with vaginitis in Taiwan. Microorganisms 9, 1864 (2021).
Ardizzone, C. M. et al. Association of Chlamydia trachomatis burden with the vaginal microbiota, bacterial vaginosis, and metronidazole treatment. Front. Cell Infect. Microbiol. 13, 1289449 (2023).
Zhu, B., Tao, Z., Edupuganti, L., Serrano, M. G. & Buck, G. A. Roles of the microbiota of the female reproductive tract in gynecological and reproductive health. Microbiol. Mol. Biol. Rev. 86, e0018121 (2022).
Reimers, L. L. et al. The cervicovaginal microbiota and its associations with human papillomavirus detection in HIV-infected and HIV-uninfected women. J. Infect. Dis. 214, 1361–1369 (2016).
Guo, C. et al. Cervicovaginal microbiota significantly changed for HPV-positive women with high-grade squamous intraepithelial lesion. Front. Cell Infect. Microbiol. 12, 973875 (2022).
Katki, H. A. et al. Cervical cancer risk for women undergoing concurrent testing for human papillomavirus and cervical cytology: a population-based study in routine clinical practice. Lancet Oncol. 12, 663–672 (2011).
Mitra, A. et al. The vaginal microbiota, human papillomavirus infection and cervical intraepithelial neoplasia: what do we know and where are we going next?. Microbiome 4, 58 (2016).
Gardella, B. et al. The complex interplay between vaginal microbiota, HPV infection, and immunological microenvironment in cervical intraepithelial neoplasia: a literature review. Int. J. Mol. Sci. 23, 7174 (2022).
France, M. T. et al. VALENCIA: a nearest centroid classification method for vaginal microbial communities based on composition. Microbiome 8, 166 (2020).
Salliss, M. E., Maarsingh, J. D., Garza, C., Łaniewski, P. & Herbst-Kralovetz, M. M. Veillonellaceae family members uniquely alter the cervical metabolic microenvironment in a human three-dimensional epithelial model. NPJ Biofilms Microbiomes 7, 57 (2021).
Mitra, A. et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci. Rep. 5, 16865 (2015).
Randis, T. M. & Ratner, A. J. Gardnerella and prevotella: co-conspirators in the pathogenesis of bacterial vaginosis. J. Infect. Dis. 220, 1085–1088 (2019).
Cox, C., Watt, A. P., McKenna, J. P. & Coyle, P. V. Mycoplasma hominis and Gardnerella vaginalis display a significant synergistic relationship in bacterial vaginosis. Eur. J. Clin. Microbiol. Infect. Dis. 35, 481–487 (2016).
Abavisani, M. & Keikha, M. Global analysis on the mutations associated with multidrug-resistant urogenital mycoplasmas and ureaplasmas infection: a systematic review and meta-analysis. Ann. Clin. Microbiol. Antimicrob. 22, 70 (2023).
Yang, T. et al. Antimicrobial Resistance in Clinical Ureaplasma spp. and Mycoplasma hominis and Structural Mechanisms Underlying Quinolone Resistance. Antimicrob. Agents Chemother. 64, e02560-19 (2020).
Rosário, A. et al. Impact of cervicovaginal microbiome on the risk of cervical abnormalities development. J. Med Virol. 95, e28762 (2023).
Chao, X.-P. et al. Correlation between the diversity of vaginal microbiota and the risk of high-risk human papillomavirus infection. Int J. Gynecol. Cancer 29, 28–34 (2019).
Moscicki, A.-B., Shi, B., Huang, H., Barnard, E. & Li, H. Cervical-vaginal microbiome and associated cytokine profiles in a prospective study of HPV 16 acquisition, persistence, and clearance. Front. Cell Infect. Microbiol. 10, 569022 (2020).
Torcia, M. G. Interplay among vaginal microbiome, immune response and sexually transmitted viral infections. Int. J. Mol. Sci. 20, 266 (2019).
Dabee, S., Passmore, J.-A. S., Heffron, R. & Jaspan, H. B. The complex link between the female genital microbiota, genital infections, and inflammation. Infect. Immun. 89, e00487–20 (2021).
Waltmann, A., Thomas, C. & Duncan, J. A. The role of the genital microbiota in the acquisition and pathogenesis of sexually transmitted infections. Curr. Opin. Infect. Dis. 36, 35–48 (2023).
Volesky-Avellaneda, K. D., Laurie, C., Tsyruk-Romano, O., El-Zein, M. & Franco, E. L. Human papillomavirus detectability and cervical cancer prognosis: a systematic review and meta-analysis. Obstet. Gynecol. 142, 1055–1067 (2023).
Liu, Y. et al. The functions of lncRNAs in the HPV-negative cervical cancer compared with HPV-positive cervical cancer. Apoptosis 27, 685–696 (2022).
Petry, K. U. et al. Evaluating HPV-negative CIN2+ in the ATHENA trial. Int J. Cancer 138, 2932–2939 (2016).
Horner, P. et al. Should we be testing for urogenital Mycoplasma hominis, Ureaplasma parvum and Ureaplasma urealyticum in men and women? - a position statement from the European STI Guidelines Editorial Board. J. Eur. Acad. Dermatol Venereol. 32, 1845–1851 (2018).
Workowski, K. A. et al. Sexually transmitted infections treatment guidelines, 2021. MMWR Recommendations Rep. 70, 1 (2021).
Klein, C. et al. Mycoplasma co-infection is associated with cervical cancer risk. Cancers 12, 1093 (2020).
Adebamowo, S. N. et al. and ACCME Research Group Mycoplasma hominis and Mycoplasma genitalium in the vaginal microbiota and persistent high-risk human papillomavirus infection. Front. Public Health 5, 140 (2017).
Bautista, C. T. et al. Bacterial vaginosis: a synthesis of the literature on etiology, prevalence, risk factors, and relationship with chlamydia and gonorrhea infections. Mil. Med. Res. 3, 4 (2016).
Pekmezovic, M., Mogavero, S., Naglik, J. R. & Hube, B. Host-pathogen interactions during female genital tract infections. Trends Microbiol. 27, 982–996 (2019).
Gump, D. W., Gibson, M. & Ashikaga, T. Lack of association between genital mycoplasmas and infertility. N. Engl. J. Med. 310, 937–941 (1984).
Noda-Nicolau, N. M. et al. Genital mycoplasmas and biomarkers of inflammation and their association with spontaneous preterm birth and preterm prelabor rupture of membranes: a systematic review and meta-analysis. Front. Microbiol. 13, 859732 (2022).
Berman, S. M. et al. Low birth weight, prematurity, and postpartum endometritis. Association with prenatal cervical Mycoplasma hominis and Chlamydia trachomatis infections. JAMA 257, 1189–1194 (1987).
Plummer, E. L. et al. Are mycoplasma hominis, ureaplasma urealyticum and ureaplasma parvum associated with specific genital symptoms and clinical signs in nonpregnant women?. Clin. Infect. Dis. 73, 659–668 (2021).
Dessì, D. et al. Trichomonas vaginalis and Mycoplasma hominis: new tales of two old friends. Parasitology 146, 1150–1155 (2019).
Zhou, Z. et al. The association of HPV infection and vaginal microbiota of reproductive women in China: a multicenter cohort study protocol. Med. Microecol. 15, 100072 (2023).
Qing, W. et al. Species-level resolution for the vaginal microbiota with short amplicons. mSystems 9, e0103923 (2024).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
McDonald, D. et al. Greengenes2 enables a shared data universe for microbiome studies (Bioinformatics) https://doi.org/10.1101/2022.12.19.520774 (2022).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–596 (2013).
Cole, J. R. et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–642 (2014).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226 (2018).
Callahan, B. J. et al. Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women. Proc. Natl. Acad. Sci. USA 114, 9966–9971 (2017).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 (2014).
Mallick, H. et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput Biol. 17, e1009442 (2021).
Chen, M. et al. CALM2004 Consortium. Vaginal microbiome and sexually-transmitted pathogens in Chinese reproductive-age women: a multicentre cross-sectional and longitudinal cohort study. CAL2004_VM_STI, https://doi.org/10.5281/zenodo.17059937 (2025).
Acknowledgements
This study was supported by the Natural Science Foundation of China (NSFC 81925026 (H.Z.), 82002201 (M.C.), 82302610 (C.Q.), 82205157 (Y.Z.)) and Guangdong Basic and Applied Basic Research Foundation (2021B1515230007, H.Z.).
Author information
Authors and Affiliations
Consortia
Contributions
H.Z. and Y.H. designed and conceptualized the study. M.C., Y.H., and H.Z. supervised the study. C.Q. and W.Q. performed the statistical analyses and contributed to data management. Y.Z., Z.Z., Y.H., and J.O. processed biological samples and performed laboratory analyses. C.Q. and M.C. drafted the manuscript. Y.Z., Z.Z., and R.C. provided critical revision for important intellectual content. M.C., Y.H., and H.Z. were responsible for the decision to submit the manuscript. All authors reviewed the manuscript and approved the submitted final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, M., Qi, C., Qing, W. et al. Vaginal microbiome and sexually-transmitted pathogens in Chinese reproductive-age women: a multicentre cross-sectional and longitudinal cohort study. Nat Commun 16, 10002 (2025). https://doi.org/10.1038/s41467-025-64917-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64917-7
