
Abstract
Germavinylidenes (R2C=Ge), the heavier congeners of vinylidenes, have long remained elusive, both as free species and in Lewis base-stabilized forms. Herein, we report the isolation of a base-stabilized germavinylidene employing an imine-functionalized cyclic (alkyl)(amino)carbene (imino-CAAC) ligand. Comprehensive structural characterization, supported by theoretical calculations, confirms the presence of a Ge=C double bond, with the divalent germanium center coordinated by the imine donor. Preliminary reactivity studies reveal a rich chemical profile: the complex undergoes selective methylation at the germanium center, forms a π-dominated coordination complex with iron carbonyl, and acts as a germanium atom transfer reagent to generate a rare tetraazagermylene. Furthermore, a [2 + 2] cycloaddition with isocyanate provides compelling evidence for the Ge=C double bond character. These results underscore the previously unexplored potential of imino-CAAC ligands in stabilizing reactive and uncharted p-block main group species.
Similar content being viewed by others
Introduction
For decades, the synthesis of heavier element-element multiple bonds was regarded as an elusive goal, thereby solidifying the foundational ‘double-bond rule’1. This stipulated that p-block elements with a principal quantum number greater than two, starting with aluminum, were intrinsically unable to form multiple bonds, either among themselves or with other elements. This long-standing paradigm was decisively challenged around the 1980s through a series of landmark discoveries: Lappert’s pioneering synthesis of a Sn = Sn bond2, West’s isolation of a Si = Si species3, Yoshifuji’s structurally authenticated P = P bond4, and Brook’s report on a Si = C linkage5. These breakthroughs ignited a renaissance in the study of heavier multiple bonds, enabling the generation of heavier congeners of classical organic molecules. In contrast to their lighter counterparts, these heavier analogs manifest fundamentally distinct features in terms of stability, bonding, and reactivity6,7,8,9. As a result, they have evolved into powerful platforms for the synthesis of main group species and the development of unconventional catalytic transformations10,11,12.
Vinylidenes (R2C = C) are compounds featuring a terminal divalent carbon atom with six electrons in its valence shell, establishing them as pivotal reactive intermediates in various organic transformations (Fig. 1a)13. Difluorovinylidene, F2C = C, stands as the sole vinylidene variant capable of spectroscopic analysis, sustainable at temperatures up to 15 K14. Despite this extreme instability, coordination with transition metals has proven to be a viable method for stabilizing these elusive vinylidene species15. While acetylene is well-known for its stability as a bench chemical, its vinylidene isomer, H2C = C, remains elusive, existing only transiently with an extremely brief lifetime of approximately 10−10 s under severe conditions16,17,18,19. Substituting the carbon atoms in H2C = C with heavier group 14 elements facilitates access to their corresponding heavier analogs, H2E = E (E = Si, Ge, Sn, Pb). Theoretical investigations indicate that H2E = E achieve their most stable configurations compared to their heavier acetylene counterparts, H–E ≡ E–H20,21,22,23,24. However, the incorporation of sterically demanding substituents in R2E = E compromises the stability of vinylidene, predisposing the molecular structure towards acetylene forms due to intense steric repulsion among adjacent bulky groups25,26,27,28. This has been experimentally validated through the isolation of heavier acetylenes, R–E ≡ E–R, which exhibit a formal E ≡ E triple bond and a trans-bent geometry29,30,31,32. The pioneering efforts in isolating these compounds have opened new avenues for further exploration33,34,35,36. Despite these advances, the stabilization of heavier vinylidenes remains a rare achievement and a formidable challenge, predominantly due to the lack of sufficient steric protection around the terminal E atom in its low oxidation state and the absence of efficient synthetic methodologies37,38.

a Representation of acetylene, vinylidene, and heavier analogs with germanium. b Selected examples of germanium-containing heavier vinylidenes. c Imino-NHC Ge(0) adduct/mesoionic germylene. d This work: an isolable monomeric germavinylidene. Tip = 2,4,6-iPr3C6H2, Ar* = 2,6-Dipp2C6H3, Dipp = 2,6-iPr2C6H3.
Specifically, in the context of germanium-containing vinylidenes, Scheschkewitz and colleagues introduced silagermenylidene I with bulky substituents and N-heterocyclic carbene (NHC) coordination (Fig. 1b)39,40. Wesemann et al. synthesized a phosphine-stabilized digermavinylidene II41, and Aldridge’s group reported a free digermavinylidene III42. Mo and co-workers described the isomerization of stannagermenylidene IV to germastannenylidene V via double 1,2-migration43. Computational insights indicate that the simplest 1-germavinylidene H2C = Ge is thermodynamically more stable than its germyne counterpart (H–C ≡ Ge–H) by approximately 160 kJ/mol44,45, a conclusion supported by its detection through laser spectroscopy46,47. Leung and co-workers synthesized the stable bis(germavinylidene) VI48, which serves as a precursor for reactive monomeric base-ligated germavinylidene38. In 2013, this group reported an oligomeric germanvinylidene consisting of the germavinylidene moiety [(Ph)2P = NSiMe3)(Ph2P)C = Ge], where the Ge atom is coordinated with two PPh2 groups49. Apeloig et al. synthesized genuine germenyl anions featuring a Ge = C double bond via the germavinylidene intermediate50. More recently, the group of Dong reported a dicationic monomeric 1-germavinylidene51, where the incorporation of positive charges suppresses oligomerization of the highly reactive C = Ge moiety via Coulombic repulsion. In a related study, Kinjo and co-workers disclosed a unique hybrid species exhibiting dual resonance character between an imino-NHC-stabilized germylone and a mesoionic germylene (Fig. 1c)52, characterized by π-electron delocalization across the GeC2N2 five-membered ring. Despite these, the isolation of a neutral monomeric germavinylidene species remains a formidable challenge.
While the imine-functionalized cyclic (alkyl)(amino)carbene ligand 1 (imino-CAAC)53 has emerged as a superior ligand framework54,55,56—exhibiting enhanced σ-donor strength and π-accepting capability relative to NHCs—its utility in the chemistry of p-block main group elements has remained largely unexplored. Herein, we report the isolation of a crystalline, monomeric, base-stabilized germavinylidene species supported by ligand 1. Comprehensive experimental and theoretical investigations confirm the presence of a bona fide Ge = C double bond. Moreover, the coordination behavior of this germavinylidene toward iron carbonyl complexes, its reactivity with electrophiles and isocyanates, and its potential utility as a germanium transfer reagent are elucidated.
Results and discussion
Aiming to investigate its potential, we treated 1 with two equivalents of GeCl2·dioxane, resulting in the formation of 2 with a 64% yield (Fig. 2). In the proton-decoupled 13C NMR spectrum of 2, the resonance attributed to the germanium-bound carbon is observed at 233.1 ppm, which is significantly low-frequency-shifted compared to that of 1 (307.5 ppm). The resonance of the imine carbon manifests at 197.3 ppm, exhibiting a high-frequency shift relative to that of 1 (179.4 ppm).

Condition (i) reagent: 1.0 equiv. of 1; solvent: toluene; reaction temperature (time), –40 °C to room temperature (12 h). (ii) reagent: 1.0 equiv. of 2, solvent: THF; reaction temperature (time), room temperature (12 h).
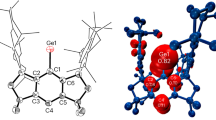
In the solid state of 2, Ge(1) manifests a three-coordinate configuration, adopting a distorted trigonal pyramidal geometry, while C(1) resides in a nearly planar environment with the sum of bond angles amounting to 359.2° (Fig. 3a). The Ge(1)–C(1) bond length (2.0398(16) Å) is consistent with those observed in carbene-ligated germyliumylidene ions, which range from 2.011(5) to 2.0588(19) Å52,57,58. The Ge(1)–N(2) bond length at 2.1231(14) Å is longer than that recorded for Kinjo’s germyliumylidene cation supported by an imino-NHC ligand (2.0560(16) Å)52. Additionally, the C(1)–N(1) bond length at 1.286(2) Å closely parallels the imine N(2)–C(5) double bond length of 1.289(2) Å. These observations collectively suggest that 2 can be aptly described as a hybrid entity, integrating characteristics of both a base-stabilized germyliumylidene ion and an imine-stabilized germylene (Fig. 2).

a Thermal ellipsoid drawing of 2 at 30% probability. b Thermal ellipsoid drawing of 3 at 30% probability. For clarity, all hydrogen atoms and the anionic fragment in 2 are omitted, and Dipp substituents are shown in wireframe style.
A THF solution of 2 was treated with four equivalents of potassium graphite (KC8) and stirred at room temperature overnight (Fig. 2). Following workup, compound 3 was isolated as a dark blue solid, yielding 82%. This species demonstrated exceptional thermal stability; no decomposition was observed even when its THF solution was boiled for an hour. In the proton-decoupled 13C NMR spectrum, the resonance of the germanium-bound carbon in 3 shifted to a lower frequency at 223.2 ppm from 233.1 ppm in 2, while the imine carbon showed a decrease to 179.5 ppm from 197.3 ppm. Additionally, the UV-visible spectroscopy of 3 revealed absorption bands at 352 nm and 649 nm, which are attributable to the π-π* transitions of the Ge–C bond (Supplementary Fig. 38).
Compound 3 crystallizes in the monoclinic space group P21/c (Fig. 3b), with Ge(1) adopting a di-coordinated configuration, bonding with both a carbon atom and an imine nitrogen atom. The N(1)–Ge(1)–C(1) bond angle of 78.9(3)° is slightly smaller than the 80.59(6)° observed in the imino-NHC coordinated Ge(0) complex52. The Ge(1)–C(1) bond distance at 1.835(6) Å is considerably shorter than those in dimeric (VI) (1.905(8) and 1.908(7) Å)48 and oligomeric (VII) germanvinylidene forms (1.975(5) Å)49 and the dicationic germanvinylidene (1.977(5) Å)51, yet it is similar to the authenticated Ge = C double bond (1.825(2) Å) in CAAC-derived bis(digermene)s59. Additionally, the Ge(1)–N(2) distance of 2.2150(49) Å is much longer than those in imino-NHC and diimino-NHC coordinated Ge(0) complexes52,58, but it aligns closely with dative N→Ge bonds in diimino-phenyl germylenes60. This length is also greater than that in 2 (2.1231(14) Å), contrasting with the reductions observed in germylone complexes, where Ge–N distances typically decrease52,58. Collectively, these observations indicate a pronounced localized Ge = C double bond character in 3, designating it as a base-stabilized germavinylidene.
Quantum chemical calculations were performed to elucidate the electronic structure of 3. Intrinsic bond orbital (IBO)61,62 analysis further delineates that 3 possesses two σ-bonding orbitals at Ge(1): the Ge(1)–C(1) and Ge(1)–N(2) bonds (Fig. 4a,b). Notably, the latter exhibits substantial polarization towards N(2), with an electron density distribution of 80.3% at N(2) and 15.4% at Ge(1), indicative of significant lone pair character at N(2) and a pronounced dative bond character between Ge(1) and N(2). Moreover, an out-of-plane π-bonding orbital exists between C(1) (46.4%) and Ge(1) (47.7%) (Fig. 4c), alongside a dominant nonbonding lone pair orbital at Ge(1) (98.6%) (Fig. 4d). These orbital features correspond with electron localization function (ELF)63,64 calculations, highlighting pronounced π-type electron density in the valence region between Ge(1) and C(1), and localized electron density around Ge(1) (Supplementary Fig. 42). In addition, conceptual DFT calculations revealed that the dual descriptor (DD)65 value of Ge(1) (−0.35) is most negative, indicating its predominant nucleophilic behavior (Fig. 4h).

a Ge(1) − C(1) σ-bonding orbital. b Ge(1) − N(2) σ-bonding orbital. c Ge(1) − C(1) π-bonding orbital. d Ge(1) lone-pair orbital. e HOMO. f LUMO + 5. g HOMO − 1. h DD value.
The highest occupied molecular orbital (HOMO) at −4.65 eV primarily features the Ge(1)–C(1) π-bonding orbital, exhibiting antibonding interactions with the lone pair on N(1) (Fig. 4e). The lowest unoccupied molecular orbital (LUMO) at −0.06 eV corresponds to the π*-antibonding orbital of the C(5)–N(2) bond in the imine group (Supplementary Fig. 39), while the LUMO + 5 at 1.19 eV corresponds to the π*-antibonding orbital of the Ge(1)–C(1) bond (Fig. 4f). The σ-type lone pair on Ge(1) significantly contributes to the HOMO − 1 at −6.95 eV (Fig. 4g). Additionally, the high ellipticity (ϵ = 0.34) of the Ge(1)–C(1) bond, calculated using the quantum theory of atoms in molecules (QTAIM)66,67, underscores the double bond character of this interaction. Taken as a whole, these computational insights strongly support the characterization of 3 as a base-stabilized germanvinylidene.
Delving into the orbital interactions of the Ge(1)–C(1) and Ge(1)–N(2) bonds, principal interacting orbital (PIO)68,69 analysis provided deep insights into the bonding between Ge(1) and the imino-CAAC fragment (Fig. 5). Two PIO pairs characterize the Ge(1)–C(1) bond: the unique, a π-bond, emerges from the interaction of 4p orbital of Ge(1) with 2p orbital of C(1) (or the π*-antibonding orbital of the C(1)–N(2) double bond), contributing 0.90e and 1.10e respectively, yielding a PIO-based bond index (PBI) of 0.99. The second, a σ-bond, involves the sp2 hybrid orbital of C(1) (1.56e) interacting with the 4p orbital of Ge(1) (0.44e), resulting in a slightly lower PBI of 0.69. An overall PBI of 1.68 distinctly indicates the formal double bond character of Ge(1) = C(1), corroborated by a Wiberg bond index of 1.33 for the Ge(1)–C(1) bond based on natural bond orbital analysis, which is comparable with that of 1.35 for the Ge–C bond in CAAC = Ge(Tip)SiMe2(2-NMe2-Ph) (Tip = 2,4,6-iPr3-C6H2)54. In contrast, the interaction between Ge(1) and N(2), involving a donation of 0.18e from the lone pair electrons of N(2) to the 4p orbital of Ge(1), is considerably weaker, reflected in a modest PBI of 0.32. Notably, a similar bonding scenario between Ge(1)–C(1) and Ge(1)–N(2) was observed in an additional model species where the nitrogen in the CAAC is replaced by a quaternary carbon (Supplementary Figs. 46, 47). These findings underscore the general representation of the electronic structure of 3 as R2C = Ge stabilized by an imine ligand.

a Ge(1) − C(1) π-bonding mode. b Ge(1) − C(1) σ-bonding mode. c Ge(1) − N(2) σ-bonding mode. d Fragment segmentation of 3 for PIO analysis.
Aligned with computational results, 3 displays nucleophilicity. The treatment of 3 with Me-OTf leads to the formation of 4 in an 85% yield (Fig. 6), regardless of the amount of Me-OTf used. This reaction contrasts with prior observations on the imino-NHC chelated germylone, where increasing the amount of Me-OTf induce a double methylation at Ge52. In the solid state of 4 (Fig. 7a), both the N(1)–C(1) and N(2)–C(5) bonds measure 1.292(5) Å, indicative of typical N = C double bonds, reminiscent of the bonding scenario observed in 2 (Fig. 6).

Condition (i) reagent: 1.0 equiv. of 3; solvent: toluene; reaction temperature (time), room temperature (5 h). (ii) reagent: 1.0 equiv. of 3, solvent: THF; reaction temperature (time), room temperature (12 h). (iii) reagent: 1.0 equiv. of 3, solvent: toluene; reaction temperature (time), room temperature (12 h). (iv) reagent: 1.0 equiv. of 3, solvent: toluene; reaction temperature (time), −30 °C to room temperature (12 h).

a Thermal ellipsoid drawing of 4 at 30% probability. b Thermal ellipsoid drawing of 5 at 30% probability. c Thermal ellipsoid drawing of 7 at 30% probability. For clarity, all hydrogen atoms and the anionic fragment in 4 are omitted, and Dipp substituents are shown in wireframe style.
The coordination of 3 with a half equivalent of Fe2(CO)9 in THF yielded complex 5 in a 54% yield, accompanied by the release of CO (Fig. 6). Unlike previously reported germylone complexes, which typically provide two lone pairs for coordination70, only the monoiron species 5 was obtained, even in the presence of excess Fe2(CO)9, underscoring the distinct property of this complex.
The molecular structure of 5 was confirmed via single crystal X-ray diffraction (Fig. 7b). Notably, the structure reveals the coordination of the Fe(CO)4 unit to the germavinylidene moiety, oriented out of the N(1)–C(1)–Ge(1) plane, in stark contrast to the trigonal planar geometry characteristic of imino-NHC stabilized Ge–M(CO)5 adducts (M = Cr, Mo, W)71. Moreover, the Ge atom in 5 adopts a distorted pyramidal geometry, diverging from the trigonal planar geometry observed in similar germavinylidene coordinated metal carbonyls72,73. The Ge(1)–C(1) bond of 1.917(10) Å is elongated relative to that in 3 (1.835(6) Å). The Ge(1)–Fe(1) distance is 2.3988(18) Å, comparable with that of the silagermenylidene Fe(CO)4 complex (2.3780(6) Å)37, and the C(1)–Ge(1)–Fe(1) bond angle is 119.2(3)°. These structural features in 5 suggest a predominant π-coordination interaction between the germavinylidene and the Fe(CO)4 unit. This specific orientation results from the concerted coordination of the Ge(1)–C(1) π-bond (HOMO) and the Ge(1) lone pair (HOMO-9) to the Fe(1) center (Supplementary Fig. 49). IBO analysis further delineates a π-type C(1)–Ge(1)–Fe(1) three-center-two-electron bonding orbital, with contributions of 17.1% from C(1), 45.4% from Ge(1), and 23% from Fe(1), while the lone pair is primarily localized at the Ge(1) center (Ge(1), 84.7%; C(1), 5.6%; Fe(1), 4.0%) (Supplementary Fig. 50). Indeed, the observed reaction selectivity aligns perfectly with the orbital-weighted dual descriptor calculations74 for 3, indicating a stronger nucleophilicity for the π-orbital of the Ge(1) = C(1) bond compared to the lone pair orbital of Ge(1) towards electrophiles (Supplementary Fig. 43).
In a striking reaction, the treatment of 3 with two equivalents of sterically bulky terphenyl azide (MesTerN3, where MesTer = 2,6-Mes2-C6H3 and Mes = 2,4,6-Me3C6H2) resulted in the immediate liberation of N2. Subsequent workup yielded light yellow crystals of 6, which X-ray diffraction identified as a tetraazagermylene with a divalent Ge atom (Fig. 6). An in-situ NMR study verified the quantitative synthesis of 6, alongside the concurrent liberation of the free ligand 1 (Supplementary Fig. 30).
Within the structure of 6, a coplanar five-membered GeN4 ring manifests, with the sum of its inner angles reaching 540° (Supplementary Fig. 36). The N–Ge–N angle is determined at 85.247(86)°. Notably, the Ge–N and Ge–N bond lengths, measuring 1.7856(18) Å and 1.7092(19) Å respectively, are substantially shorter than those observed in tetraazagermoles, which range from 1.8399(16) to 1.889(11) Å75. This reduction in bond length is attributed to the robust π-donation from the nitrogen atoms to the Ge(II) center. Additionally, the N(1)–N(2), N(3)–N(4), and N(2)–N(3) bonds at 1.4810(39) Å, 1.5474(70) Å, and 1.3281(80) Å, respectively, are markedly longer than the bond lengths typically recorded in tetraazagermoles (N–N, avg. 1.39 Å; N = N, avg. 1.27 Å)75. Remarkably, compound 6 is the solely documented example of a tetraazagermylene, underscoring the synthetic versatility of 3 in generating unique germanium species. The NICS(1)-zz values calculated for the N4Ge ring are –14.3 and –17.3 ppm, reflecting significantly weaker aromaticity relative to benzene (–30.3 ppm, –30.3 ppm), consistent with suboptimal π-overlap between Ge and N.
The germavinylidene character of 3 was further corroborated by its reactivity toward 4-tolyl isocyanate, which undergoes a formal [2 + 2] cycloaddition to furnish carboxamido germylene 7 (Fig. 6)76. Compound 7 exhibits excellent thermal stability, with no detectable changes observed when a C6D6 solution of 7 was refluxed for 12 h (Supplementary Fig. 31). The molecular structure of 7 features the nitrogen atom of the NCO moiety coordinated to the Ge center (Fig. 7c), while the carbonyl group is connected to the former carbene carbon via a C–C bond measuring 1.558(4) Å. The Ge atom adopts a pyramidal coordination geometry. Notably, the Ge(1)–C(1) bond elongates to 2.020(3) Å, substantially longer than that in 3 (1.835(6) Å), indicative of a classical Ge–C single bond. In contrast, the Ge(1)–N(3) bond measures 1.956(2) Å—significantly shorter than the dative Ge(1)–N(2) interaction (2.294(2) Å)—supporting the formation of a covalent Ge–N linkage.
To gain mechanistic insight into this transformation, theoretical studies were performed (Fig. 8). The proposed pathway begins with a nucleophilic attack of the Ge = C π-bond on the electrophilic carbon of the NCO moiety, proceeding via transition state TS1 and surmounting an energy barrier of 22.8 kcal/mol. This step yields a zwitterionic intermediate (Int1), which lies 13.4 kcal/mol above the energy of the separated reactants. A subsequent C-migration event, accompanied by [2 + 2] cycloaddition, results in the concerted formation of Ge–N and C–C bonds to generate the final product 7. This step is both thermodynamically favorable (ΔG = –29.4 kcal/mol) and kinetically accessible, with a relatively low activation barrier of 6.5 kcal/mol. Collectively, these findings underscore the unique germavinylidene character of 3 and its propensity for nucleophilic cycloaddition reactivity.

DFT-computed free energy profile (in kcal/mol) at the SMD(toluene)-M06-2X-D3/def2-TZVP//M06-2X/def2-SVP level.
More than two decades since the initial reports of dimeric 1-germavinylidene structures48, this work establishes that monomeric germavinylidene can be synthetically realized through ligation with a bulky imino-CAAC ligand that provides crucial kinetic stabilization to the low-valent germanium center. This germavinylidene is characterized by a Ge = C double bond, with the germanium atom further stabilized by the proximal imine ligand. This Ge = C double bond exhibits [2 + 2] cycloaddition reactivity with an isocyanate. Notably, the higher-energy Ge = C π bond takes precedence over the Ge lone pair during coordination processes, leading predominantly to the formation of an iron π complex rather than the typical σ complex. Additionally, our work delineates the capacity of germavinylidene to drive the synthesis of previously unexplored tetraazagermylenes. Given the successful use of imino-NHC ligands in isolating diverse, unique main group species, it is anticipated that the introduction of the imino-CAAC ligand into main group chemistry will inspire further interest and exploration in this domain.
Methods
All manipulations were carried out under a dry argon atmosphere using Schlenk line and glovebox techniques. Toluene, benzene, THF and n-hexane were dried by stirring with sodium. Deuterated benzene and deuterated THF were dried by refluxing with sodium/potassium under argon prior to use, and deuterated chloroform was dried by stirring with CaH2. The NMR (1H, 13C, 19F and 2D) spectra were recorded on Bruker Avance II 500 MHz and 700 MHz spectrometers. High-resolution mass spectrometry (HRMS) was conducted using a Thermo Fisher Scientific Q-Exactive MS System. Commercial reagents were purchased and used as received.
Synthetic method of 3 is as follows. More experimental and calculational details can be found in the Supplementary Information file.
3: To the mixture of 2 (150 mg, 0.19 mmol) and KC8 (103 mg, 0.76 mmol) was added THF (50 mL) at room temperature. After stirring for 30 min, a dark blue solution formed. Then the mixture was kept stirring overnight. After filtration, the filtrate was evaporated under vacuum, and the residue was washed with n-hexane (3 mL) to yield dark green solid in 82% yield (90 mg). Crystals suitable for X-ray diffraction were obtained from the toluene solution at room temperature.
Data availability
Additional synthetic, spectroscopic, crystallographic, and computational data generated in this study are provided in the Supplementary Information file. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Center, under deposition numbers CCDC 2402153 (2), 2402154 (3), 2402156 (4), 2402155 (5), 2402158 (6) and 2425191 (7). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Source data are provided in this article. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Dasent, W. E. Non-existent compounds. J. Chem. Edu. 40, 130 (1963).
Goldberg, D. E., Harris, D. H., Lappert, M. F. & Thomas, K. M. A new synthesis of divalent group 4B alkyls M[CH(SiMe3)2]2 (M = Ge or Sn), and the crystal and molecular and molecular strcuture of the tin compound. J. Chem. Soc., Chem. Commun. 261-262 (1976).
West, R., Fink, M. J. & Michl, J. Tetramesityldisilene, a stable compound containing a silicon-silicon double bond. Science 214, 1343–1344 (1981).
Yoshifuji, M., Shima, I., Inamoto, N., Hirotsu, K. & Higuchi, T. Synthesis and structure of bis(2,4,6-tri-tert-butylphenyl)diphosphene: isolation of a true phosphobenzene. J. Am. Chem. Soc. 103, 4587–4589 (1981).
Brook, A. G., Abdesaken, F., Gutekunst, B., Gutekunst, G. & Kallury, R. K. A solid silaethene: isolation and characterization. J. Chem. Soc., Chem. Commun. 191-192 (1981).
Fischer, R. C. & Power, P. P. π-Bonding and the lone pair effect in multiple bonds involving heavier main group elements: developments in the new millennium. Chem. Rev. 110, 3877–3923 (2010).
Power, P. P. An update on multiple bonding between heavier main group elements: the importance of Pauli repulsion, charge-shift character, and London dispersion force effects. Organometallics 39, 4127–4138 (2020).
Zhao, L., Pan, S., Holzmann, N., Schwerdtfeger, P. & Frenking, G. Chemical bonding and bonding models of main-group compounds. Chem. Rev. 119, 8781–8845 (2019).
Melen, R. L. Frontiers in molecular p-block chemistry: from structure to reactivity. Science 363, 479–484 (2019).
Power, P. P. Main-group elements as transition metals. Nature 463, 171–177 (2010).
Präsang, C. & Scheschkewitz, D. Reactivity in the periphery of functionalised multiple bonds of heavier group 14 elements. Chem. Soc. Rev. 45, 900–921 (2016).
Weetman, C. Main group multiple bonds for bond activations and catalysis. Chem. Eur. J. 27, 1941–1954 (2021).
Bruce, M. I. Organometallic chemistry of vinylidene and related unsaturated carbenes. Chem. Rev. 91, 197–257 (1991).
Breidung, J. et al. Difluorovinylidene, F2C = C. Angew. Chem., Int. Ed. Engl. 36, 1983–1985 (1997).
Roh, S. W., Choi, K. & Lee, C. Transition metal vinylidene and allenylidene mediated catalysis in organic synthesis. Chem. Rev. 119, 4293–4356 (2019).
Skell, P. S. & Plonka, J. H. Chemistry of the singlet and triplet C2 molecules. Mechanism of acetylene formation from reaction with acetone and acetaldehyde. J. Am. Chem. Soc. 92, 5620–5624 (1970).
Hayes, R. L., Fattal, E., Govind, N. & Carter, E. A. Long live vinylidene! A new view of the H2C = C: → HC≡CH rearrangement from ab initio molecular dynamics. J. Am. Chem. Soc. 123, 641–657 (2001).
Schork, R. & Koppel, H. Barrier recrossing in the vinylidene-acetylene isomerization reaction: a five-dimensional ab initio quantum dynamical investigation. J. Chem. Phys. 115, 7907–7923 (2001).
Joseph, S. & Varandas, A. J. C. Accurate MRCI and CC study of the most relevant stationary points and other topographical attributes for the ground-state C2H2 potential energy surface. J. Phys. Chem. A 114, 13277–13287 (2010).
Shimizu, T. & Frenking, G. Structures and bonding situation of Pb2X2 (X = H, F, Cl, Br and I). Theor. Chem. Acc. 130, 269–277 (2011).
Momeni, M. R. & Shakib, F. A. Theoretical description of triplet silylenes evolved from H2Si = Si. Organometallics 30, 5027–5032 (2011).
Schaefer, H. F. III. The silicon-carbon double bond: a healthy rivalry between theory and experiment. Acc. Chem. Res. 15, 283–290 (1982).
Stogner, S. M. & Grev, R. S. Germyne, H–C≡Ge–H, and the excited states of 1-germavinylidene, H2C = Ge. J. Chem. Phys. 108, 5458–5464 (1998).
Brody, H. K., Magers, D. H. & Leszczyński, J. Ab initio studies of methylenecarbene and isoelectronic species. Struct. Chem. 6, 293–300 (1995).
Apeloig, Y. & Karni, M. Triple bonds to silicon. Substituent effects on the thermodynamic and kinetic stabilities of silynes relative to their isomeric silylidenes and silavinylidenes. Organometallics 16, 310–312 (1997).
Danovich, D. et al. Silynes (RC≡SiR′) and disilynes (RSi≡SiR′): Why are less bonds worth energetically more?. Angew. Chem., Int. Ed. 40, 4023–4026 (2001).
Bibal, C., Mazières, S., Gornitzka, H. & Couret, C. A route to a germanium–carbon triple bond: first chemical evidence for a germyne. Angew. Chem., Int. Ed. 40, 952–954 (2001).
Wu, P.-C. & Su, M.-D. Triply bonded stannaacetylene (RC≡SnR): theoretical designs and characterization. Inorg. Chem. 50, 6814–6822 (2011).
Ding, Y., Li, Y., Zhang, J. & Cui, C. Synthesis of an N-heterocylic boryl-stabilized disilyne and its application to the activation of dihydrogen and C−H bonds. Angew. Chem., Int. Ed. 61, e202205785 (2022).
Sugiyama, Y. et al. Synthesis and properties of a new kinetically stabilized digermyne: new insights for a germanium analogue of an alkyne. J. Am. Chem. Soc. 128, 1023–1031 (2006).
Sekiguchi, A. Disilyne with a silicon-silicon triple bond: A new entry to multiple bond chemistry. Pure Appl. Chem. 80, 447–457 (2008).
Power, P. P. Silicon, germanium, tin and lead analogues of acetylenes. Chem. Commun. 2091-2101 (2003).
Stender, M., Phillips, A. D., Wright, R. J. & Power, P. P. Synthesis and characterization of a digermanium analogue of an alkyne. Angew. Chem., Int. Ed. 41, 1785–1787 (2002).
Phillips, A. D., Wright, R. J., Olmstead, M. M. & Power, P. P. Synthesis and characterization of 2,6-Dipp2-H3C6SnSnC6H3-2,6-Dipp2 (Dipp = C6H3-2,6-Pri2): a tin analogue of an alkyne. J. Am. Chem. Soc. 124, 5930–5931 (2002).
Pu, L., Twamley, B. & Power, P. P. Synthesis and characterization of 2,6-Trip2H3C6PbPbC6H3-2,6-Trip2 (Trip = C6H2-2,4,6-i-Pr3): a stable heavier group 14 element analogue of an alkyne. J. Am. Chem. Soc. 122, 3524–3525 (2000).
Sekiguchi, A., Kinjo, R. & Ichinohe, M. A stable compound containing a silicon-silicon triple bond. Science 305, 1755–1757 (2004).
Rivard, E. Group 14 inorganic hydrocarbon analogues. Chem. Soc. Rev. 45, 989–1003 (2016).
Leung, W.-P., Chan, Y.-C. & So, C.-W. Chemistry of heavier group 14 base-stabilized heterovinylidenes. Organometallics 34, 2067–2085 (2015).
Jana, A., Huch, V. & Scheschkewitz, D. NHC-stabilized silagermenylidene: a heavier analogue of vinylidene. Angew. Chem., Int. Ed. 52, 12179–12182 (2013).
Jana, A., Majumdar, M., Huch, V., Zimmer, M. & Scheschkewitz, D. NHC-coordinated silagermenylidene functionalized in allylic position and its behaviour as a ligand. Dalton. Trans. 43, 5175–5181 (2014).
Krebs, K. M. et al. Phosphine-stabilized digermavinylidene. J. Am. Chem. Soc. 141, 3424–3429 (2019).
Rit, A., Campos, J., Niu, H. & Aldridge, S. A stable heavier group 14 analogue of vinylidene. Nat. Chem. 8, 1022–1026 (2016).
Qiao, Z., Li, X., Chen, M., Cao, F. & Mo, Z. Double 1,2-carbon migration at mixed heavier Sn = Ge vinylidenes. Angew. Chem., Int. Ed. 63, e202401570 (2024).
Yang, Z. et al. Gas-phase preparation of 1-germavinylidene (H2CGe; X1A1), the isovalent counterpart of vinylidene (H2CC; X1A1), via non-adiabatic dynamics through the elementary reaction of ground state atomic carbon (C; 3P) with germane (GeH4; X1A1). J. Phys. Chem. Lett. 14, 430–436 (2023).
Hostutler, D. A., Clouthier, D. J. & Pauls, S. W. The ground state of germylidene (H2C = Ge). J. Chem. Phys. 116, 1417–1423 (2002).
Harper, W. W. et al. Laser spectroscopic detection of the simplest unsaturated silylene and germylene. J. Am. Chem. Soc. 119, 8361–8362 (1997).
Hostutler, D. A., Smith, T. C., Li, H. & Clouthier, D. J. The electronic spectrum, molecular structure, and oscillatory fluorescence decay of jet-cooled germylidene (H2C = 74Ge), the simplest unsaturated germylene. J. Chem. Phys. 111, 950–958 (1999).
Leung, W.-P., Wang, Z.-X., Li, H.-W. & Mak, T. C. W. Bis(germavinylidene) [(Me3SiN = PPh2)2C = Ge → Ge =C(Ph2P = NSiMe3)] and 1,3-dimetallacyclobutanes [M{μ2-C(Ph2P = NSiMe3)2}]2 (M = Sn, Pb). Angew. Chem., Int. Ed. 40, 2501–2503 (2001).
Leung, W.-P., Chiu, W.-K. & Mak, T. C. W. Synthesis and structural characterization of base-stabilized oligomeric heterovinylidenes. Inorg. Chem. 52, 9479–9486 (2013).
Goldshtein, Y. et al. Synthesis of genuine germenyl lithiums and the first persistent germenyl radicals. Angew. Chem., Int. Ed. 61, e202202452 (2022).
Li, Y. et al. Dicationic 1-germa and 1-stannavinylidenes: synthesis, structure, and reactivity. JACS Au 5, 1289–1298 (2025).
Su, B., Ganguly, R., Li, Y. & Kinjo, R. Isolation of an imino-N-heterocyclic carbene/germanium(0) adduct: a mesoionic germylene equivalent. Angew. Chem., Int. Ed. 53, 13106–13109 (2014).
Chu, J., Munz, D., Jazzar, R., Melaimi, M. & Bertrand, G. Synthesis of hemilabile cyclic (alkyl)(amino)carbenes (CAACs) and applications in organometallic chemistry. J. Am. Chem. Soc. 138, 7884–7887 (2016).
Grünwald, A. et al. An isolable terminal imido complex of palladium and catalytic implications. Angew. Chem., Int. Ed. 57, 16228–16232 (2018).
Grünwald, A., Heinemann, F. W. & Munz, D. Oxidative addition of water, alcohols, and amines in palladium catalysis. Angew. Chem., Int. Ed. 59, 21088–21095 (2020).
Zhao, L. et al. Cyclic (alkyl)(amino)carbene ligand-promoted nitro deoxygenative hydroboration with chromium catalysis: scope, mechanism, and applications. J. Am. Chem. Soc. 143, 1618–1629 (2021).
Xiong, Y., Yao, S., Tan, G., Inoue, S. & Driess, M. A cyclic germadicarbene (“germylone”) from germyliumylidene. J. Am. Chem. Soc. 135, 5004–5007 (2013).
Nguyen, M. T. et al. Ge(0) compound stabilized by a diimino-carbene ligand: synthesis and ambiphilic reactivity. J. Am. Chem. Soc. 142, 5852–5861 (2020).
Thömmes, A.-L., Morgenstern, B., Zimmer, M., Andrada, D. M. & Scheschkewitz, D. -Conjugated bis(germylene) adducts with NHC and CAACs. Chem. Eur. J. 29, e202301273 (2023).
Khan, S. et al. Monomeric Sn(ii) and Ge(ii) hydrides supported by a tridentate pincer-based ligand. Chem. Commun. 48, 4890–4892 (2012).
Knizia, G. & Klein, J. E. M. N. Electron flow in reaction mechanisms—revealed from first principles. Angew. Chem., Int. Ed. 54, 5518–5522 (2015).
Klein, J. E. M. N. & Knizia, G. cPCET versus HAT: a direct theoretical method for distinguishing X–H bond-activation mechanisms. Angew. Chem., Int. Ed. 57, 11913–11917 (2018).
Savin, A., Nesper, R., Wengert, S. & Fässler, T. F. ELF: The electron localization function. Angew. Chem. Int. Ed. Engl. 36, 1808–1832 (1997).
Lu, T. A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn. J. Chem. Phys. 161, 082503 (2024).
Morell, C., Grand, A. & Toro-Labbé, A. New dual descriptor for chemical reactivity. J. Phys. Chem. A 109, 205–212 (2005).
Bader, R. F. W. A quantum theory of molecular structure and its applications. Chem. Rev. 91, 893–928 (1991).
Bader, R. F. W. The quantum mechanical basis of conceptual chemistry. Monatsh. Chem. 136, 819–854 (2005).
Zhang, J.-X., Sheong, F. K. & Lin, Z. Unravelling chemical interactions with principal interacting orbital analysis. Chem. Eur. J. 24, 9639–9650 (2018).
Zhang, J.-X., Sheong, F. K. & Lin, Z. Principal interacting orbital: a chemically intuitive method for deciphering bonding interaction. WIREs Comput. Mol. Sci. 10, e1469 (2020).
Su, B., Ota, K., Li, Y. & Kinjo, R. Germylone-bridged bimetallic Ir and Rh complexes. Dalton. Trans. 48, 3555–3559 (2019).
Su, B., Ganguly, R., Li, Y. & Kinjo, R. Synthesis, characterization, and electronic structures of a methyl germyliumylidene ion and germylone-group VI metal complexes. Chem. Commun. 52, 613–616 (2016).
Leung, W.-P., So, C.-W., Kan, K.-W., Chan, H.-S. & Mak, T. C. W. Synthesis of a manganese mermavinylidene complex from bis(germavinylidene). Inorg. Chem. 44, 7286–7288 (2005).
Leung, W.-P., So, C.-W., Wang, J.-Z. & Mak, T. C. W. A novel synthesis of metallogermacyclopropane and molybdenum bis(iminophosphorano)carbene complexes from bisgermavinylidene. Chem. Commun. 21, 248–249 (2003).
Pino-Rios, R., Inostroza, D., Cárdenas-Jirón, G. & Tiznado, W. Orbital-weighted dual descriptor for the study of local reactivity of systems with (quasi-) degenerate states. J. Phys. Chem. A 123, 10556–10562 (2019).
Li, T. et al. A germanimidoyl chloride: synthesis, characterization and reactivity. Chem. Commun. 59, 1533–1536 (2023).
Tho Nguyen, M., Gusev, D. G., Dmitrienko, A., Pilkington, M. & Nikonov, G. I. Reversible coupling of germylone with isocyanates. Chem. Eur. J. 30, e202400613 (2024).
Acknowledgments
We thank the financial support from the Ministry of Education of the People’s Republic of China and Hunan Normal University. The National Natural Science Foundation of China [22501078 (B.L.), 22301122 (J.L.) and 22350004 (L.L.L.)], Natural Science Foundation of Hunan Province [2024JJ6306 (B.L.)], the Key Program of Hunan Education Committee [23A0089 (B.L.)] and Guangdong Basic and Applied Basic Research Foundation [2024A1515010842 (J.L.)] are acknowledged. We acknowledge Dr. Lei Guo at Hunan University for assistance with X-ray structure measurement. The theoretical work was supported by the Center for Computational Science and Engineering at SUSTech.
Author information
Authors and Affiliations
Contributions
B.L. conceptualized the project. B.L. and L.L.L. supervised the project. B.L., Y.Z., J.W. and Y.W. performed the experimental work. J.L. performed the computational work. B.L. and M.Z. performed the X-ray crystallographic analyses. B.L., J.L. and L.L.L. wrote the paper with input from all authors. All authors discussed the results in detail and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Henry F. Schaefer III and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, B., Li, J., Zhu, Y. et al. A base-stabilized germavinylidene. Nat Commun 16, 10015 (2025). https://doi.org/10.1038/s41467-025-65001-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65001-w
