
Abstract
Determining the appropriate sample size (N) for bulk RNA sequencing experiments is critical for obtaining reliable results. We show in two N = 30 profiling studies, comparing wild-type mice and mice in which one copy of a gene has been deleted, the N required to minimize false positives and maximize true discoveries found in the N = 30 experiment. Results from experiments with N = 4 or less are shown to be highly misleading, given the high false positive rate and the lack of discovery of genes later found with higher N. For a cut-off of 2-fold expression differences, we find an N of 6-7 mice is required to consistently decrease the false positive rate to below 50%, and the detection sensitivity to above 50%. More is always better for both metrics – and an N of 8-12 is significantly better in recapitulating the full experiment.A common way to reduce the false discovery rate in underpowered experiments is to raise the fold cutoff. We show that this strategy is no substitute for increasing the N of the experiment: it results in consistently inflated effect sizes and causes a substantial drop in sensitivity of detection. These data should be helpful to scientists in choosing their Ns.
Similar content being viewed by others

Introduction
A typical RNA-expression study aims to find genes whose RNA levels differ significantly between two conditions. RNA sequencing (RNA-seq) measurements are subject to technical noise and biological variability, the impact of which is expected to diminish with increasing sample size, N, in each group. Too few samples result in differentially expressed genes being missed (type 2 errors, or false negatives), spurious findings (type 1 errors, or false positives), and inflated effect sizes (type M errors, or the “winner’s curse”1,2). Underpowered mouse studies are a major factor driving the lack of reproducibility in the scientific literature3,4.
The intuitive preference for more replicates in study design is balanced in practice by resource constraints, and by ethical concerns about using more animals than needed. Therefore, it is imperative to objectively assess effects of differing sample sizes and determine guidelines for future experiments, reflecting an optimal tradeoff between the errors incurred at low N and the resource constraints associated with always preferring a high N.
Analytical power calculations for RNA-seq studies are challenged by the observed long-tailed dispersed distribution of sequence count data, often modeled as a negative binomial distribution. Previous studies have employed parametric statistical tools that model power as a function of expected effect sizes, dispersion of the data, sequencing depth, and other factors5,6,7,8,9,10. Though potentially useful when accompanied by user-friendly tools, researchers seldom know the values of the parameters on which these models depend upon. More recent tools10,11 estimate these parameters from existing studies that the researcher believes will be comparable to their planned one. Even so, different tools give discrepant results and perform poorly for low fold changes12.
These theoretical approaches are complemented by studies that infer appropriate sample sizes directly from empirical data. Baccarella, et al.13 compare a human monocyte data set to a gold standard obtained from additional studies, finding that sample size has a much larger impact than read depth on precision and recall, with performance dropping notably below seven replicates. A similar analysis of six public data sets14 highlights the importance of sample size as well as gene expression variability (dispersion), and notes the challenge of accurately estimating the latter. Finally, Schurch et al.15 sub-sample from a large cohort of yeast to assess the impact of sample size on accurately calling differentially expressed genes, using their full cohort as the gold standard.
While these empirical studies highlight key principles of study design, none of them include the most-studied model organism used in biology – the mouse. Mice are often studied due to their status as mammals combined with the availability of inbred strains, which is hoped to decrease variability between study subjects; also, techniques for genetic manipulation – creation of knockouts, conditional knockouts, and transgenics – are well-established in mice. Since they are mammals, mice are more closely related to humans evolutionarily than other well-established genetic models such as yeast, C. elegans worms, Drosophila, and zebrafish.
In this study, we transcriptionally profile large (N = 30) cohorts of genetically modified and wild-type pure strain C57BL/6 mice and report comparisons between subsets of this cohort. We find that N = 5 and lower Ns fail to recapitulate the full signature, and systematically overstate effect sizes. The adequately sized subsets are found to be a larger N than is often encountered in the literature, in the range of 8 or greater mic per group. In contrast, it seems that group sizes of 3 to 6 are frequently found in published papers – casting doubt on the reported claims of differentially expressed genes, especially ones with low expression. We find that “more is always better” when it came to discovery rates, at least within our maximum sets of N = 30. Our results suggest an N of 6–7 as a minimum, and 8–12 if possible, setting a new suggested guideline for future bulk RNA-seq experiments.
Results
In order to determine an ideal range of N for an RNA-seq study, we first performed a large scale set of comparative expression studies, with a maximum N = 30, across four organs (heart, kidney, liver, and lung) from wild-type and heterozygous mice - in which one copy of a gene was deleted. This choice of maximum N, close to an order of magnitude larger than typically reported in published studies, was defined to be the gold-standard, capturing the true underlying biological effects as accurately as possible, and serving as a benchmark for comparison against subsets with smaller N. To mitigate the possible concern that a particular gene deletion may be atypically variable in gene expression changes across all tissues, we separately studied two distinct gene heterozygotes, resulting in a total of 360 RNA-seq samples. We sequenced 30 mice heterozygous for Dachsous Cadherin-Related 1 (Dchs1), 30 mice heterozygous for Fat Atypical Cadherin 4 (Fat4), together with 30 wild type (WT) mice, each group derived from the same litters; these heterozygous lines were picked as representative comparators versus wild-type animals. DCHS1 and FAT4 are large cell adhesion molecules that act as a tethered ligand–receptor pair on adjacent cells to mediate planar cell polarity. Homozygous null mutations of either gene in mice are lethal at neonatal stages and have similar phenotypes affecting many organs. These include postnatal lethality, decrease in body weight, small cystic kidneys, abnormal skeletal morphology, curly tails, small lungs and cardiovascular abnormalities. Heterozygous Dchs1 and Fat4 mice exhibit less severe phenotypes16. Every effort was made to control for confounding factors and reduce variation between individuals, including use of a highly inbred pure strain C57BL/6NTac line, identical diet and housing, IVF derivation from the same male, same day tissue harvesting and same day sequencing. In this text, we focus our observations around mice heterozygous for the Dchs1 allele. Results for the kidney and liver of Fat4, included in the supplement, showed analogous patterns, while heart and lung yielded too few gene changes (4 and 7 genes, respectively, were perturbed by at least 50%, see Supplementary Table 1) to examine meaningfully.
Dchs1 heterozygous (Het) mice showed strong gene expression changes relative to WT mice in all four tissues assayed. The liver and kidney showed the most perturbations, with key tissue markers and functions strongly affected. Gene signatures derived using the full 60 (30 versus 30 comparison) mouse cohort are designated the gold standard for differentially expressed genes (DEGs). Note that a separate gold standard set is calculated for each combination of P-value, fold change, and absolute expression thresholds considered.
We assessed the impact of replicate number on the sensitivity and false discovery rate (FDR) using a down-sampling strategy (Fig. 1). For a given sample size N, we randomly sampled N Het and N WT samples without replacement (N ranges from 3 to 29), performed DEG analysis, and compared the resulting signature to the gold standard (N = 30). We define sensitivity as the percent of gold standard genes detected in the sub-sampled signature. Conversely, the percent of sub-sampled signature genes missing from the gold standard is the false discovery rate (FDR- not to be conflated with the multiple hypothesis testing term). These definitions rely on both statistical significance (P-value) and absolute fold change (ratio of perturbation in either direction) thresholds being met in both signatures, and differ from analogous studies15 that define agreement based solely on shared statistical significance. We justify the former approach in the discussion, but also provide results using the latter definition in Supplementary Figs. 9 and 10. Figure 2A shows the results of these virtual experiments (40 Monte Carlo trials for each N), using an absolute fold change cutoff of 1.5 (50% up- or down-regulation) and adjusted P-value of less than 0.05. As expected, FDR drops towards zero while sensitivity rises towards 100% as N increases and the experiments more closely resemble the gold standard one. For a sample size of 3, over a third (38%) of genes found to be perturbed in the heart represent false discoveries, either because they didn’t meet statistical significance in the gold standard, or were perturbed by less than 50%. The kidney and lung have similar FDR values at this low N, while liver is slightly lower (28%). Though there is no clear inflection point, the FDR appears to taper around N = 8 to 10, depending on tissue, indicating diminishing returns at higher N values. Sensitivity increases more smoothly, after a marked jump from N = 5 to 6. For heart, kidney, and liver, a median sensitivity of 50% is attained by N = 8, while the lung required a sample size of 11. The liver signature of Fat4 hetrozygous mice showed analogous results, with FDR showing smaller decreases beyond N = 8 (Fig. S11). Sensitivity increased smoothly, though without the jump. FDR for the Fat4 kidney signature did not have an obvious change in slope, while sensitivity jumped at a higher N ~ 9. Overall, false discovery rates were higher, and sensitivity lower, in the Fat4 Hets than in Dchs1, reflecting the general tendency that a stronger overall effect (as reflected by the number of genes perturbed) leads to better agreement.

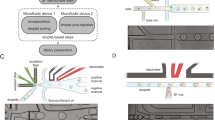
We define the ‘gold standard’ signature as the set of DEGs in the full cohort of mice (30 Dchs1 Het vs 30 WT). From this full cohort, we generated a smaller “mini-experiment” (trial) by randomly selecting N Het and N WT mice (Down-sampling strategy, top middle). The DEG from this trial were compared to the gold standard signature to assess sensitivity, specificity, and effect size bias. The number of mice selected (N) range from 3 to 29, and 40 trials were performed for each N. A separate approach (disjoint sets) was used to assess consistency between pairs of mini-experiments. Here, two mini-experiments of size N are created in each trial, and the resulting two DEG sets are compared. Higher consistency between DEGs is interpreted as each DEG capturing more biological signal.

A For each sample size N (x-axis), N Dchs1 Hets and N WTs were randomly chosen, Het-vs-WT DEGs calculated, and compared to the gold standard (full cohort) signature. Left panel shows FDR, calculated as the fraction of DEGs from the sub-sampled trial that was absent from the gold standard signature. Right panel shows sensitivity, defined as the fraction of gold standard DEGs also detected in the sub-sampled trial. Signature genes were those perturbed by 50% or more, with a multiple hypothesis adjusted P < 0.05, as calculated using the DeSeq2 package’s negative binomial model. The three horizontal lines of each boxplot correspond to the 25th, 50th, and 75th percentile of the distribution, while whiskers extend to the most extreme observation that is still within 1.5 times the interquartile range (IQR) of the box. Points beyond 1.5*IQR are plotted individually. B Plots are as in (A), except showing a single tissue, liver, with varying absolute fold change (2^abs(log2FC), perturbation in either direction) thresholds along each row. C Plots are as in (A), except showing a single tissue, liver, with varying P-value thresholds along each row. D Plots are as in (A), except showing a single tissue, liver, with varying minimum absolute expression (in TPM) along each row.
The variability in false discovery rates across trials is particularly high at low sample sizes. In the lung, the FDR ranges between 10 and 100% depending on which N = 3 mice are selected for each genotype. In all tissues, this variability drops markedly by N = 6. Because the size of the overlap between sampled subsets increase with sample size, we expect more consistency between trials at high N, though considerable variability persists even with double-digit sample sizes, particularly in the Fat4 experiment. Notably, this variability is lower in kidney and liver, the tissues most affected by the genetic modification. The variation in sensitivity across trials falls more graduallywith higher Nand this relationship is less apparent in lung, or in the Fat4 Hets.
We next examined the impact of varying fold change thresholds. Can researchers salvage an underpowered study by limiting their purview to highly perturbed genes? As shown in Fig. 2B, narrowing the focus to large effect sizes actually increases the false discovery rate, as compared to a gold standard derived using the same absolute fold change cutoff (in either up or down direction) for a cut-off of 2-fold expression differences, we find an N of 6-7 mice is required to consistently decrease the false positive rate to below 50%, and the detection sensitivity to above 50%. Intuitively, this happens because achieving a statistically significant P-value in an underpowered experiment requires an extreme, and so likely inflated, effect size that was not observed in the gold standard signature. Note that for a given N, high-fold-change genes are more likely than low-fold-change ones to be perturbed at all, or above a fixed threshold. For example, a gene perturbed by 20–50% (1.2 to 1.5-fold) in an N = 4 sub-sampling of heart samples has less than a 5% chance of also being found in a gold standard heart signature that was derived with absolute fold change of 1.5 (Fig. S2A). Genes perturbed by 50–100% (1.5 to 2-fold) in the same sub-sampling have about an even chance of being found in that same gold standard signature, while genes perturbed by 3 to 5-fold are almost certain to be found. Thus, true positives (using this alternative definition of a gold standard gene set, whose fold change threshold need not match that in the sub-sampled trial), are enriched more in the higher fold change bins (Fig. S2A). However, the observed fold change at lower N, often inflated due to type M error (an error of Magnitude), should not be taken at face value. In addition, limiting our analysis to a subset of genes above a high fold change threshold greatly impacts sensitivity to detect true changes, as one might expect (Fig. S2B).
Increasing the stringency of the P-value (alpha), rather than fold change filter, reduces FDR, while also reducing sensitivity to detect (Fig. 2C). This strategy is therefore more conservative than a fold change filter, though its utility is limited: the FDR decrease between an alpha of 0.05 vs 0.01 is modest (Fig. S1B). By contrast, applying a minimum abundance threshold leaves the FDR largely unaffected, while greatly increasing sensitivity to detect gold standard genes also passing this expression filter (Fig. 2D). The impacts of these various filters are consistent across all tissues assayed (Figs. S1A, S1B, S1C), and in the Fat4 experiment (Figs. S12A,S12B, S12C), though here imposing an abundance threshold yielded minimal improvement in sensitivity
To evaluate the robustness of our sampling approach, we also performed comparisons between groups of wild type samples randomly selected from the Dchs1 cohort. Since the mice being compared have the same genotype, we interpret all discovered genes as false positives. Few such false positives are seen for N = 5 or greater, though a few rare trials have many false positives (Fig. S3). These outlier trials tend to have lower N, but we do see a few even at N greater than 10.
A limitation of the down-sampling approach for assessing an “optimal” sample size is that the same mice are used to derive the gold standard and the random trial gene signatures. Our second approach avoids this circularity by randomly sampling mice to create two disjoint experiments of size N in each trial, and comparing their DEGs (Fig. 1, bottom). As N increases, signatures from the two sub-sampled, independent experiments should both better capture the underlying common biology and hence resemble each other more. Fig. 3 shows that this holds for all tissues and fold change thresholds tested. Agreement between DEGs asymptotes to an N value between 8 and 10 for heart, kidney, and liver, consistent with the tapering of FDR values observed with the down-sampling approach (Fig. 2A). Lung DEGs showed lower agreement than other tissues, and the asymptote was less clear, particularly for smaller absolute fold change cutoffs. The Fat4(+/−) vs Fat4(+/+) liver comparison showed a similar pattern, though with lower overall concordance (Fig. S12). Interestingly, signatures from Fat4 kidney samples showed little concordance even with large N, suggesting that the initial gold standard signature may itself not be robust. Limiting to genes with a minimum expression level does not appreciably change these results (Fig. S4).

The sample size N is shown along the x-axis, while the y-axis shows Cohen’s unweighted kappa (κ) measure, calculated between expression signatures from two independently constructed experiments of size N. Kappa values range from zero (no agreement) to one (perfect agreement, identical DEGs).
In addition to the loss of sensitivity and specificity, underpowered studies lead to inflated effect size estimates. To study this further, we compared the fold changes of DEGs from down-sampled trials with those in the gold standard. Focusing on a representative gebe example, Trex1 in liver, we observe fold changes exceeding that seen in the gold standard (horizontal black line in Fig. 4B)—but only among trials yielding a statistically significant P-value for this gene. In trials where the Trex1 expression change did not reach significance, the log2 fold change (log2FC) is accurately estimated, though with considerable variation around this estimate. The P-value filter thus acts as an “effect overestimate generator”, since only large effect sizes attain significance in underpowered trials. With increasing N and adequate power, the observations reverse: estimated log2FCs for significant trials approach the gold standard estimate, while non-significant trials, which at higher N must be underestimating the true effect size (else they would reach significance) deviate from the gold standard estimate, and trend towards zero log2FC. With high enough N, non-significant trials disappear altogether. Fig. S5 shows several randomly chosen gold standard genes for each tissue, each reflecting the same trend of significant (P < 0.05) trials converging on the fold change observed in the 30-vs-30 signature. Though genes vary in how quickly they converge, and at what N significant trials start to appear, the trend clearly demonstrates the reproducibility of these observations, at least in two different experimental models.

A Percentage of DEGs in each trial whose effect size (2^abs(log2FC), perturbation in either direction) in the sub-sampled trial exceeds the effect size in the 30-vs-30 comparison Sample size N is shown along the x-axis. The dotted line at 50% indicates no bias, where a gene is equally likely to over- or under-estimate the effect. B Effect size estimates for a representative gene (Trex1) in liver. For each N (x-axis), the Het-vs-WT log2 fold change estimate of Trex1 is shown separately for statistically significant (cyan), or not significant (red) trials. Statistical significance was defined as having amultiple hypothesis adjusted P-value of 0.05 or lower, as calculated using the DeSeq2 package’s negative binomial model. With small N, fold change estimates from significant trials overestimate that of the gold standard (solid horizontal line), but approach it as N increases. Non-significant trials by contrast show no bias at small N.
Assessing further the generality of fold change inflation, we find that for small sample size trials, the overwhelming majority of DEGs in all tissues overestimate the true fold change, regardless of fold change cutoff (Fig. 4A). Even at very high N, most DEGs show perturbations more extreme than those observed in the gold standard signature. This pattern holds even after excluding low-expression genes (Fig. S6A). Fat4 kidney and liver also recapitulate this pattern (Fig. S14). Interestingly, shrinking the observed fold changes using the ashr algorithm almost eliminates this bias, though some over-estimation persists at low N, particularly in the lung (Fig. S6B).
For comparison with prior work, we applied an alternative definition for matching DEGs, based solely on shared statistical significance, regardless of effect size. Here, genes are considered true positives if they have an adjusted P-value below 0.05 in both the full data set, and the sub-sampled trial. False negatives achieve significance only in the former, false positives only in the latter. Relative to results derived using our definition (Fig. 2A), this approach reduces the false discovery rate (Fig. S9), since significantly perturbed genes with incorrect effect sizes now count as true positives. Sensitivity to detect true changes is also reduced, owing to a large number of low-fold change gold standard genes going undetected in underpowered small-N trials.
Though the alternative definition of true and false positives is fold change agnostic, we can still ask how focusing on genes with larger effect sizes impacts sensitivity and FDR. Following Schurch, et al.15, we calculate these metrics only for the subset of genes whose fold change exceeds a certain threshold in the 30-vs-30 signature, regardless of whether those genes’ perturbations are statistically significant in either the full or sub-sampled experiment. Consistent with their work, and with Fig. S2C, the sensitivity (true positive rate) is higher (Fig. S10A), and the FDR lower (Fig. S10B), among more-perturbed genes. Applying a basic abundance filter further increases the true positive rate. Also consistent with their results, and with our WT-vs-WT comparisons, the false positive rate is well controlled across all sample sizes.
Since perturbations of individual genes are not faithfully captured at low N, we briefly explored whether the overall pattern of gene changes is consistent with that in the gold standard comparison. Fig. S7A shows correlations between log2 fold changes (log2FC) in the sub-sampled experiment, and the log2FC in the gold standard (N = 30) experiment. Even at N = 3, these correlations (Pearson’s ⍴) range around 0.5–0.6 depending on tissue, increasing to 0.7–0.75 at N = 10. Analogous correlations using the Wald statistic calculated by DESeq2, rather than the log2FC, show even higher agreement, with ⍴ between 0.5 and 0.75 at N = 3 (Fig. S7B). Correlations between sub-sampled trial pairs (disjoint strategy), yielded overall lower values (Fig. S8), but these associations are still strongly significant, and outside the range of correlations observed between WT subsets and the gold standard signature (Fig. S7, blue boxes).
Discussion
Over the last several years, there has been an increased focus on “rigor and reproducibility” when it comes to biological data. This is a result of several studies indicating that quite a few high profile papers were found to be non-reproducible upon attempts to repeat those studies17,18. What followed were many discussions as to what might be improved – and suggestions ranged from focus on journal policies19, education20,21,22, and policies of funding organizations23.
Of course, a fundamental component of addressing reproducibility is to perform studies that directly query the ability to reproduce the data from a particular type of experiment. Bulk tissue RNA-seq studies have become an indispensable and widespread technology for transcriptome wide analysis of DEGs over the past 15 years, and remain a staple of current comparative functional genomics. Given this ubiquity, it is important to understand what range of N is required to better assure that the conclusions made will be found to be predictive of subsequent analyses of the same sort of comparison.
One particularly common bulk RNA-seq experiment involves comparing knockout or transgenic animals with wild-type controls. When a gene is deleted, it is of interest to learn how this change perturbs mRNA expression, because this gives indications as to the molecular mechanisms which are normally affected by the gene of interest. We chose heterozygotes in this study to model a system where a particular gene was still in place but potentially reduced in effect, since a phenotype was observable, as in the case of the animals used here - because many biological settings where RNAseq is performed profile this sort of setting. As a comparator, wild-type animals were used – animals in which the gene is left unperturbed.
A particularly common mouse strain used for such experiments are C57BL/6 mice. Since this is an inbred mouse strain, one might expect that genetic variation would be minimized in comparison to outbred animals; indeed, the ability to use fewer animals is a common rationale to study inbred strains of mice. Even though this inbred mouse line is relatively genetically homogenous in comparison to outbred, or truly “wild” mice, this study makes it clear that considerable variety in gene expression across mice still persists, with implications for study design and interpretation.
Our results demonstrate how underpowered RNA-Seq experiments result in type I, type II, and type M (magnitude) errors, and offer guidance about adequate sample sizes to mitigate them. Commonly reported sample sizes of 5 or less are to be avoided, since for our studies seeking to identify genes perturbed by at least 1.5-fold, one finds over 25% false discoveries, less than 50% power to detect true changes of equal or greater magnitude, and inflated fold changes a large majority of the time. Moreover, our WT-vs-WT comparisons detect at least some false positives for samples sizes less than 6.
Considerable discrepancies persist between the gold standard N = 30 signature and those from sub-sampled experiments, which diminish with increasing N. Therefore, one simple conclusion is that “more is always better” in the case of sample size – there is no point at which adding samples doesn’t help reduce error – at least within our sample sets, where the maximum N was 30. Given realistic constraints such as budget and colony sizes, we looked for a point of diminishing returns. Both the down-sampling and disjoint approach suggest that this is reached around an N of 8–11. Formally of course, we can only say these were our findings in these two mouse models. But the broader guidance is that scientists should be more aware of the need to understand fidelity of gene expression differences in their models, as part of a “system validation” study, before going forward with an RNAseq experiment at lower Ns. We offer this experiment for some guidance in case it simply isn’t economically feasible for researchers to do their own high N experiment.
In keeping with the guidelines outlined by the Microarray Quality Control (MAQC) project24, this paper defines DEGs based on both statistical significance and effect size. A gene that is statistically significant in both sub-sampled and gold standard signatures, but only exceeds the fold change threshold in the latter, we classify as a false negative. This is a stringent criterion: others may classify it as a true positive, reasoning that the effect is “real” if the gene perturbation is significant in both conditions. Our view is that, if the absolute fold change threshold is meaningful, failing to exceed it means the observation did not reproduce. The same logic designates as false positives genes exceeding the fold change threshold in only the sub-sampled experiment. Our conclusions (e.g., that FDR increases with fold change stringency) therefore differ from studies using the alternative, P-value alone definition. Such studies mirror our results in Fig. S2, which show the opposite FDR trend as ours (Fig. 2B), provided that false positives are defined as genes absent from a fixed gold standard. However, this increase in specificity is offset by a substantial drop in sensitivity, our ability to detect true gene changes (Fig. S2B), and in the high likelihood that observed effect sizes are over-estimated. The widespread practice of focusing on highly perturbed genes therefore fails to fully capture the biology of the studied model; higher sample sizes are needed.
Nearly all observed fold changes overestimate the true effect for N less than 6–7, though considerable over-estimation persists even at higher N. The reason for this is that smaller, underpowered studies require a larger effect size to achieve significance. This observation also explains why false discovery rates increase as we raise the fold change cutoff for both the sub-sampled and gold standard (N = 30) experiments: the latter tend to show more modest changes, so genes will often fall below the fold change threshold in N = 30, but exceed it in sub-sampled trials. Such genes will be counted as false positives. The intuitive solution is to apply a stringent fold change cutoff to only the low-N trial, as in Fig. S2A, while keeping in mind that the true changes are almost certainly more muted than those observed.
Although adjusting the fold change threshold proved to have drawbacks, our data suggest that filtering out low-abundance genes substantially increases our likelihood of accurately detecting changes, without major changes to the false discovery rate. We also show that fold change shrinkage may be desirable to mitigate the effect size over-estimation seen most acutely at low N. Finally, despite the high rate of type I, type II, and type M errors encountered when comparing sub-sampled and gold standard DEGs, overall concordance of changes, as measured by correlation of log2FCs or Wald statistics, was strongly significant. This lends support to the idea of favoring gene-aggregation approaches rather than focusing on individual gene changes. Pathway analysis may be fairly robust for sample sizes of 6 or greater25, though these results vary by algorithm, pathway size, and other factors26.
We had similar findings with two separate sets of heterozygous animals – analyzing two distinct genes. Fat4 heterozygosity showed a less pronounced phenotype than did Dchs1, leading us to exclude the largely unaffected heart and lung from the Fat4 analysis. The Fat4 (+/−) kidney also showed a weaker signature than other tissues, and didn’t reach an asymptote by N = 14 in the disjoint method. Notwithstanding, both kidney and liver recapitulated the relationships between sample size and FDR/sensitivity we observed for Dchs1, as well as the impact of varying fold change, P-value, or absolute expression filters. Given that these relatively high Ns were found for an inbred strain, it should also be acknowledged that it’s highly likely that even higher Ns would be needed for outbred strains, as well as human studies. One might ask whether there is some idiosyncracy in the two heterozygotic lines studied that would make the data adduced in this study more variable than that found in other settings. Of course, the only way to answer this would be to do similar studies in still more genetically modified lines vs wild-type controls. The expression and function of Dchs1 and Fat4 are not known to be circadian, or feeding-dependent, for example, or dependent on other highly variable factors. Therefore, there is no particular reason to believe that the two heterozygous strains highlighted in this paper are unusually variable.
From both an ethical and financial perspective, one might like to minimize the number of animals used in an experiment to the degree possible. One should of course use as few mice as necessary, but no fewer. Sacrificing mice in the service of an underpowered experiment yielding misleading or irreproducible results is also a major concern, the correction of which will likely involve the use of many more animals. We hope this study will be generally useful for the determination of N in future RNA-seq studies, and in evaluating the utility of prior-published work. This study should also help contextualize studies done with comparatively low Ns.
Methods
The research in this study comport with all relevant ethical regulations. Mouse protocols were approved by the company’s internal IACUC committee.
Transgenic mice
C57BL/6NTac mice were purchased from Taconic Biosciences (USA) and maintained at Regeneron animal holding facilities under specific pathogen free (SPF) conditions. Mice were housed in groups of 4–5 per cage with controlled temperature and light (22 °C, 12-h light/12-h dark cycle: lights on at 0600 h/lights off at 1800h) and with ad libitum access to food (PicoLab Rodent Diet 20, Lab Supply) and water. Samples were obtained from 11–12 week old C57BL/6NTac male mice after overnight fasting. As Fat4 (−/−) and Dcsh1(−/−) are lethal as homozygotes, comparisons were done between heterozygous (+/−) and WT mice taken from the same litters. Transgenic mice were generated using Regeneron’s VelociGene technology27,28. Both the Fat4 and Dchs1 deletions removed cExon1, starting at the ATG. LacZ was used as a reporter. All animal procedures were conducted in compliance with protocols approved by the Regeneron Pharmaceuticals Institutional Animal Care and Use Committee.
RNASeq processing
RNA was prepped from tissues stored in RNAlater using MagMAX Nucleic Isolation Kits on KingFisher Instruments (ThermoFisher). Strand-specific RNA-seq libraries were prepared from 500 ng RNA using KAPA Stranded RNA-Seq Kit for Illumina Platforms (Roche). Twelve-cycle PCR was performed to amplify libraries. Sequencing of single-end, 33 base pair reads was performed on Illumina HiSeq®2500 (Illumina) by multiplexed sequencing with 33 cycles. Sequencing depth across samples had a mean of 23.8 million reads, standard deviation of 3.6 million.
Genome and transcriptome reference
To NCBI’s GRCm38/mm10 genome assembly, we added 22 short sequences corresponding to markers of interest (including the LacZ reporter), whose sequences are shown in Supplementary Data File 1). All transcripts’ exon regions, which include 3’ and 5’ untranslated regions (UTRs), are listed in the GTF file (Supplementary Data File 2). The transcripts used in the analysis are derived from 20,670 protein-coding genes, 3292 non-coding RNAs, 326 pseudogenes, 119 snoRNAs, 22 tRNAs, 10 rRNAs, 5 snRNAs, as well as the 22 marker genes. The genes are further annotated in Supplementary Data File 3. Supplementary Data Files 1–3 are also hosted on our Github page, DOI:files were converted to Fastq format via Illumina Casava 1.8.2. Reads were decoded based on their barcodes, mapped to the mouse transcriptome (described above) using Omicsoft’s OSA aligner, with the following parameters: Read Trim Quality = 2 (Bases are trimmed from the 3’ end until the first base with quality score > 2 is encountered. Reads with fewer than 17 bp remaining are excluded from analysis.); Maximum penalty = 2 (If a read cannot be perfectly aligned, up to 2 mismatches or indels are permitted); Report Cutoff = 10. (Reads with more than 10 non-unique mappings are excluded). Reads that failed to align with the transcriptome were mapped to the full genome reference. For each RNA sample, the fraction of intronic, intergenic, exonic, duplicated, and other mapping quality statistics are available in Supplementary Data File 4 and 5 (Dchs1and Fat4, respectively). Further details of the alignment algorithm are described in Hu et al.29, and here: https://resources.omicsoft.com/downloads/whitepaper/OmicsoftAligner.pdf. Exon mapped reads were summed at the gene level using Omicsoft Studio software (Summarize Gene/Transcript Count Module). Briefly, this module quantifies reads mapped to the transcriptome by counting individual reads. An Expectation-Maximization (EM) algorithm statistically allocates reads mapping to multiple transcripts based on their likelihood of originating from each transcsript. From the allocated reads, Omicsoft calculates transcript-level expression values in TPM (Transcripts Per Million), dividing the raw count of reads mapped to a transcript by its length, and then scaling these values by the total across all transcripts. To obtain gene-level expression, Omicsoft sums the transcript-level TPM values for all transcripts belonging to a specific gene. The samples from one Dchs1 WT mouse (5153265) were excluded from the analysis, as these clustered with Dchs1 heterozygotes in all four tissues assayed. Plots corresponding to these observations are included in the Supplementary Information file.
P-values and fold changes (effect sizes) for determining differentially expressed genes were obtained using DeSeq230 (1.34.0), with default parameters, except for alpha, which was set to 0.05. For a subset of the analysis, fold changes were shrunk using the ashr algorithm31.
Comparison between gene signatures
Down-sampling (Figs. 2B–D and 3) was performed by selecting N WT and N Het mice without replacement from the initial 30 + 30 cohort. DEGs were then derived for all four tissues assayed using the selected mice and compared to the gold standard (N = 30) signature.
For the alternative, “disjoint” approach (Fig. 2), two non-overlapping sets of N WT and N Het mice were selected, and the DEGs from these two sets were compared using Cohen’s unweighted kappa (κ) measure, which quantifies to what extent two judges agree in categorizing elements. The four inputs are: number of genes found in both signatures, number of genes found in neither signature, number of genes unique to signature 1, number of genes unique to signature 2. The first two numbers represent agreement, the next two, disagreement. The measure was calculated with the Kappa function from the vcd R package. As an alternative to Cohen’s κ, the intra-class correlation coefficient (ICC) was also calculated, using the icc function of the irr package. The input here is a Gx2 matrix, where G is the number of genes assayed. The two columns represent the two signatures being compared, with one and zero values indicating presence or absence in the signature, respectively. Though Cohen’s κis typically used for categorical variables, and the ICC for continuous ones, the two approaches yielded strikingly similar results.
For showing effect size differences, representative genes were chosen randomly from each tissue’s gold standard DEG list, with adjusted P-value threshold of 0.05. For each tissue, 20 genes were selected, evenly distributed among: DEGs with absolute fold change between 1–1.2 (up to 20% up- or down-regulation); between 1.2–1.5; between 1.5–2; and greater than 2. All plots were generated using the ggplot2 package32.
Data visualization
For all boxplots, the center line denotes the median of the distribution; the lower and upper hinges of the box denote the 25th and 75th percentile, respectively, and the distance between these two is the interquartile range (IQR); “whiskers” extend from both upper and lower hinges to the farthest data point within 1.5 IQR from the hinge; points more than 1.5 IQR from a hinge are plotted individually.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Summarized counts and TPM data, raw fastq files, as well as sample metadata, have been deposited to the Gene Expression Omnibus (GSE272152 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE272152], Bioproject PRJNA1135223).
Code availability
All code and data necessary to reproduce these results are available on Github at https://github.com/regeneron-mpds/mouse_RNA-Seq_sample_size. We have also used Zenodo to assign a DOI to the repository: https://doi.org/10.5281/zenodo.17137282.
References
Gelman, A. & Carlin, J. Beyond power calculations: assessing type S (sign) and type M (magnitude) errors. Perspect. Psychol. Sci. 9, 641–651 (2014).
Button, K. S. et al. Power failure: why small sample size undermines the reliability of neuroscience. Nat. Rev. Neurosci. 14, 365–376 (2013).
Ioannidis, J. P. Why most published research findings are false. PLoS Med. 2, e124 (2005).
Ioannidis, J. P. Why most discovered true associations are inflated. Epidemiology 19, 640–648 (2008).
Bi, R. & Liu, P. Sample size calculation while controlling false discovery rate for differential expression analysis with RNA-sequencing experiments. BMC Bioinforma. 17, 146 (2016).
Guo, Y., Zhao, S., Li, C. I., Sheng, Q. & Shyr, Y. RNAseqPS: a web tool for estimating sample size and power for RNAseq experiment. Cancer Inf. 13, 1–5 (2014).
Li, C. I., Su, P. F. & Shyr, Y. Sample size calculation based on exact test for assessing differential expression analysis in RNA-seq data. BMC Bioinforma. 14, 357 (2013).
Wu, H., Wang, C. & Wu, Z. PROPER: comprehensive power evaluation for differential expression using RNA-seq. Bioinformatics 31, 233–241 (2015).
Yu, L., Fernandez, S. & Brock, G. Power analysis for RNA-Seq differential expression studies. BMC Bioinforma. 18, 234 (2017).
Zhao, S., Li, C. I., Guo, Y., Sheng, Q. & Shyr, Y. RnaSeqSampleSize: real data based sample size estimation for RNA sequencing. BMC Bioinforma. 19, 191 (2018).
Li, F. et al. SSizer: determining the sample sufficiency for comparative biological study. J. Mol. Biol. 432, 3411–3421 (2020).
Poplawski, A. & Binder, H. Feasibility of sample size calculation for RNA-seq studies. Brief. Bioinform. 19, 713–720 (2018).
Baccarella, A., Williams, C. R., Parrish, J. Z. & Kim, C. C. Empirical assessment of the impact of sample number and read depth on RNA-Seq analysis workflow performance. BMC Bioinforma. 19, 423 (2018).
Ching, T., Huang, S. & Garmire, L. X. Power analysis and sample size estimation for RNA-Seq differential expression. RNA 20, 1684–1696 (2014).
Schurch, N. J. et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use?. RNA 22, 839–851 (2016).
Mao, Y. et al. Characterization of a Dchs1 mutant mouse reveals requirements for Dchs1-Fat4 signaling during mammalian development. Development 138, 947–957 (2011).
Begley, C. G. & Ellis, L. M. Drug development: Raise standards for preclinical cancer research. Nature 483, 531–533 (2012).
Rodgers, P. & Collings, A. What have we learned? Elife 10 e75830 (2021).
Stodden, V., Seiler, J. & Ma, Z. An empirical analysis of journal policy effectiveness for computational reproducibility. Proc. Natl. Acad. Sci. USA 115, 2584–2589 (2018).
Broman, K. Recommendations to Funding Agencies for Supporting Reproducible mlResearch. https://www.amstat.org/asa/files/pdfs/POL-ReproducibleResearchRecommendations.pdf (2017).
Glass, D. J. A critique of the hypothesis, and a defense of the question, as a framework for experimentation. Clin. Chem. 56, 1080–1085 (2010).
Glass, D. J. NIH grants: focus on questions, not hypotheses. Nature 507, 306 (2014).
Collins, F. S. & Tabak, L. A. Policy: NIH plans to enhance reproducibility. Nature 505, 612–613 (2014).
Consortium, M. et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 24, 1151–1161 (2006).
Maleki, F., Ovens, K., McQuillan, I. & Kusalik, A. J. Size matters: how sample size affects the reproducibility and specificity of gene set analysis. Hum. Genom. 13, 42 (2019).
Mubeen, S., Tom Kodamullil, A., Hofmann-Apitius, M. & Domingo-Fernandez, D. On the influence of several factors on pathway enrichment analysis. Brief Bioinform. 23, bbac143 (2022).
Poueymirou, W. T. et al. F0 generation mice fully derived from gene-targeted embryonic stem cells allowing immediate phenotypic analyses. Nat. Biotechnol. 25, 91–99 (2007).
Valenzuela, D. M. et al. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat. Biotechnol. 21, 652–659 (2003).
Hu, J., Ge, H., Newman, M. & Liu, K. OSA: a fast and accurate alignment tool for RNA-Seq. Bioinformatics 28, 1933–1934 (2012).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Stephens, M. False discovery rates: a new deal. Biostatistics 18, 275–294 (2017).
Wickham, H. in Use R!,, Edn. 2nd 1 Online Resource (XVI, 260 pages 232 illustrations, 140 illustrations in color (Springer International Publishing: Imprint: Springer, Cham, 2016).
Acknowledgements
We thank George Yancopoulos, Andrew Murphy, Len Schleifer, and the entire Regeneron research community for their support of this work. We would also like to thank the Regeneron DNA Core; the Molecular Profiling Lab, including Joseph Song, Shuo Li, and Nicole Negron for their work on the RNA-Seq assay; and the staff of “Velocigene,” our group at Regeneron who designs and produces our knockout and trangenic rodent models. This work was funded entirely by Regeneron. No grants were received from outside sources.
Author information
Authors and Affiliations
Contributions
G.H.: experimental design and data analysis, and writing. JS: Mouse breeding, and tissue harvesting. NG: RNA prep. MN: RNA prep. WKL: Data analysis. GSA: Experimental design and data analysis. YB: Data analysis and advice. DJG: Experiment conception, data analysis, writing and editing.
Corresponding author
Ethics declarations
Competing interests
All authors were employees of Regeneron when their contributions to this manuscript were performed. Many of the authors own stock and/or stock options in the company.
Peer review
Peer review information
Nature Communications thanks Nicholas Schurch, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Halasz, G., Schmahl, J., Negron, N. et al. Optimized murine sample sizes for RNA sequencing studies revealed from large scale comparative analysis. Nat Commun 16, 10173 (2025). https://doi.org/10.1038/s41467-025-65022-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65022-5
