
Abstract
Elevating the charge cut-off voltage of the LiCoO2 positive electrode beyond 4.5 V has already been the focus to unlock its energy for portable electronics, whereas the severe phase transitions during the delithiation of LiCoO2 from 4.5 V to 4.7 V can ruin the electrode structure. Besides, the poor thermal and hydrolytic stabilities of traditional lithium salt (LiPF6) prevent batteries from working at high temperatures and increase production costs and environmental pollution. Here, we show that using covalent lithium nonafluoro-n-butanesulfonate as a fluorine-rich lithium salt to create a robust LiF-rich cathode electrolyte interphase, which effectively impedes the surface destruction, we successfully realize a stable LiCoO2 battery at a high voltage of 4.7 V and demonstrate a 2.14 Ah Li|| LiCoO2 pouch cell with a stack-level specific energy of 518 Wh kg−1 (without packaging). Furthermore, its satisfactory thermal stability empowers LiCoO2 to work at a harsh condition of 60 °C and 4.6 V. Even more, its antihydrolytic stability enables the LiCoO2 battery to work with the electrolyte with 200 or even 1000 ppm water contamination. Lithium nonafluoro-n-butanesulfonate presents itself as a potentially viable lithium salt for advanced lithium batteries, offering the prospect of high voltage, improved thermal stability and eco-friendliness.
Similar content being viewed by others

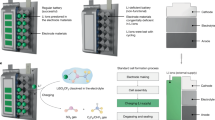
Introduction
Lithium-ion batteries (LIBs), as the most favorable energy storage devices, have achieved significant progress and are widely used. Among the various positive electrode materials in LIBs, lithium cobalt oxide (LiCoO2, LCO), which has the highest volumetric energy density, is employed as the prevailing positive electrode in batteries for portable electronics, such as cell phones, laptops, earphones, and smartwatches1,2.
Though the LCO has a high theoretical specific capacity of 274 mAh g−1, the commercial LCO electrode can only deliver a capacity of about 185 mAh g−1 at a charge voltage of 4.5 V1,2. In order to extend the working lifetime of batteries, improving the stability at high charge voltage of the positive electrode will release a much higher specific capacity and energy. Nowadays, much effort has been made to obtain a stable LCO electrode at 4.6 V, but realizing a decent 4.7 V LCO still remains a challenge due to the irreversible phase change at a highly delithiated state. During the charging process beyond 4.5 V, the LCO electrode will undergo a transition from the O3 hexagonal phase to the hybridized O1–O3 hexagonal phase (abbreviated as H1-3 phase), which has been regarded as a highly harmful phase with significant shrinkage of the c-axis and collapse of O–Co–O slabs3,4,5. When further elevating the charging voltage to extract the residual Li+, a more detrimental O1 phase will appear. Such H1-3 and O1 phases will lead to the collapse of the lattice structure with the formation of microcracks, leading to the oxidation and release of lattice oxygen3,4,5. Oxidized peroxide and highly reactive Co4+ on the positive electrode surface can easily attack the electrolyte to form a thick cathode electrolyte interphase (CEI) with high interfacial resistance, leading to a fast decay of the LCO electrode5,6,7.
To overcome the instability of LCO at high voltage, substantial efforts have been made. Such as surface coating8,9, bulk doping1,2,10,11, and electrolyte regulation for effective CEI5,12,13,14,15,16. Among these strategies, constructing a robust CEI using electrolytes has been seen as a simple yet effective tool, which can not only resist the lattice oxygen release and endless parasitic reactions between the positive electrode and electrolyte but also inhibit the phase change to some extent5,12,14,15. However, the traditional lithium salt, lithium hexafluorophosphate (LiPF6), cannot form an effective CEI on the positive electrode.
Besides, LiPF6, the dominant lithium salt in lithium batteries, is unstable at high temperatures or in the presence of water17,18,19,20. Though the salt anion PF6− is a stable coordination complex at room temperature, where the P is coordinated by six F atoms, the reaction equilibrium of LiPF6 ⇌ PF5 + LiF will shift to the right direction at a elevated temperature or in the presence of a trace amount of water (PF5 + H2O → POF3 + 2HF), thus leading to the severe decomposition of the salt anion. Furthermore, the strongly oxidative PF5 and POF3 are prone to catalyze the decomposition of the organic solvent, forming HF and O=PFxOyz-18,19,20,21,22,23. The HF formed from either organic solvent or water will further facilitate the degradation of the solvent and destroy the positive electrode with the dissolution of transition metal18, thus mutually leading to the early death of the battery. Unlike the coordination complex with a relatively fragile coordinate bond, a covalent salt anion that is only composed of stronger covalent bonds reasonably shows much higher stability24,25. Though lots of lithium salts have been utilized in lithium ion batteries, such as lithium bis(fluorosulfonyl)imide (LiFSI), lithium bis(trifluoromethylsulfonyl)imide (LiTFSI), lithium trifluoromethyl sulfonate (LiCF3SO3), lithium (difluoromethanesulfonyl)(trifluoromethanesulfonyl)imide (LiDFTFSI)18, none of them at a diluent concentration can solely enable the positive electrode to simultaneously tolerate the high voltage and high temperature, not to mention their stability in water. During the production and preservation process of LiPF6, the requirement of an ultra-dry environment (less than 10 ppm) substantially exacerbates the cost of chemical engineering. By contrast, using cost-effective and eco-friendly water to extract the residual lithium salts from electrodes and electrolytes will greatly promote the recycling of active components in electrolytes and reduce production costs and environmental pollution, thereby promoting the sustainable development of the battery industry.
In this work, lithium nonafluoro-n-butanesulfonate (LiNFBS), whose atoms only form covalent bonds in the salt anion, has been successfully employed to elevate the stability of the LCO battery at high voltage and high temperature. Owing to the fluorine-rich nature of LiNFBS, it can endow the CEI with abundant LiF components. Consequently, the dense LiF-rich CEI not only restrains the superfluous parasitic reaction between LCO electrode and electrolyte but also inhibits the oxygen release and indirectly decreases the ratio of harmful O1 phase, thus enabling the Li||LCO battery to achieve satisfactory cycling stability of 360 cycles at a high voltage of 4.7 V. And the anion NFBS− which is only composed of tightly covalent bonds, makes LiNFBS stable until about 400 °C before thermal decomposition and also empowers LiNFBS to coexist with water peacefully. As a result, the Li||LCO battery with LiNFBS-based electrolyte can stably operate at a high temperature of 60 °C along with high voltage, and the battery also shows negligible influence on the cycling performance when contaminating the electrolyte with 200 or even 1000 ppm water. Our electrochemical performances and experimental evidence highlight the potential of LiNFBS for the next-generation batteries to realize versatile requirements.
Results
Physical properties
The molecular structures of LiPF6 and LiNFBS are shown in Fig. 1a. PF6- is a coordination complex with a coordinate bond (F→P) in which one of the F- atoms donates a lone pair electron to P, forming the equilibrium of LiPF6 \(\rightleftharpoons\) PF5+LiF. Since PF5 is a strong oxidant, the reactions between PF5 and organic solvents will become increasingly violent at elevated temperatures, leading to the equilibrium shifts to the right with the vanishing of PF6-. Besides, trace contaminated or residual water in the electrolyte will catalyze the decomposition of LiPF6 with the formation of aggressive PF5, POF3 and highly corrosive HF (LiPF6 → PF5 + LiF, PF5 + H2O → POF3 + 2HF)18,19,20,21,22, thus leading to the premature failure of the battery. The moisture-sensitive feature of LiPF6 also brings great difficulties to the production process and recycling, exacerbating environmental pollution and going against green chemical engineering for the modern battery industry (Fig. 1a). In contrast, NFBS− only consists of sturdy covalent bonds, so LiNFBS is anticipated to exhibit satisfactory thermal and hydrolytic stabilities, and we also place great hope on the fluorine-rich NFBS− for producing LiF-rich CEI to stabilize the LCO electrode (Fig. 1a). Therefore, LiNFBS may be a competitive candidate of future lithium salt for high-voltage (>4.6 V) and high-temperature lithium battery with environment-friendliness (Fig. 1b). Though LiNFBS has been used in previous reports26,27, its detailed physical and chemical properties have not been fully explored, and its advantage for stabilizing high-voltage battery (>4.6 V) at a diluent concentration (~1 M) has not been exploited.

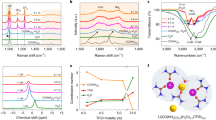
a Molecular structures of LiPF6 and LiNFBS. b Schematic of thermal/hydrolytic stabilities and the discrepancies in forming CEI of LiPF6 and LiNFBS. c Thermogravimetry (TG) curves of the two lithium salts. Photographs (d, e) of the electrolytes using the different lithium salts before and after storage at 60 °C for 4 days (d) or with the addition of 1000 ppm H2O before and after storage for 4 days (e), and the corresponding 19F nuclear magnetic resonance (19F-NMR) spectra (f, g).
Then, thermogravimetry-differential scanning calorimetry (TG-DSC) tests were first carried out to evaluate the thermal stabilities of the two lithium salts, LiNFBS and LiPF6. Unlike LiPF6 which suffers a severe mass loss above about 70 °C accompanied by a large amount of heat absorption, LiNFBS presents good stability until about 400 °C (Fig. 1c and Supplementary Fig. 1). Then the thermal and hydrolytic stabilities of the practical electrolytes with 1 M lithium salt (LiNFBS and LiPF6) in fluoroethylene carbonate (FEC) and ethyl methyl carbonate (EMC) were determined, and the electrolytes were sealed in vials under an Ar atmosphere. After resting the electrolytes in an incubator at 60 °C or storing the electrolytes with 1000 ppm H2O for 4 days, the LiPF6-based electrolytes turned dark brown while the LiNFBS-based electrolytes remained unchanged (Fig. 1d, e). Subsequently, 19F nuclear magnetic resonance (19F-NMR) spectra were employed to track the evolution of the lithium salts in electrolytes. From Fig. 1f, we can clearly see that the signals of PF6- were weakened and nearly disappeared after thermolysis and hydrolysis, respectively. Besides, the peaks of O=PFxOyz- were intensified, wherein the strong peaks at −73.6 ppm belonging to O=PF(OH)2 emerged, and some small peaks at −76~(−82) ppm can be assigned to the signals of decomposed solvents, such as O=PF(OC2H5)2 and OPF2(OC2H5)18,22. However, no difference can be observed in 19F-NMR spectra for the LiNFBS-based electrolyte, whether after thermolysis or hydrolysis (Fig. 1g), unambiguously proving the thermolytic and hydrolytic superiority of the LiNFBS.
Considering that the anodic stability of the electrolytes on the Al current collector is the premise to guarantee the battery operates stably at high voltage, Li||Al cells were assembled. Encouragingly, the LiNFBS and LiPF6-based electrolytes exhibit little anodic current without observable Al corrosion, in contrast to the phenomenon that LiFSI and LiCF3SO3 based electrolytes tend to severely corrode the Al foil (Supplementary Figs. 2 and 3). Moreover, the LiNFBS-based electrolyte manifests a better wettability on the Celgard membrane (Supplementary Fig. 4), and shows a pretty high Li+ transfer number (0.92) with a moderate ionic conductivity (2.4 mS cm−1) due to the relatively slow movement of the large-size anion (Supplementary Figs. 5 and 6).
LCO electrochemical performance
Based on the high oxidation stabilities of LiNFBS and LiPF6-based electrolytes beyond 5.0 V (Supplementary Fig. 2a), we checked the compatibility of the electrolytes for high-voltage (>4.6 V) LCO battery with a quasi-practical areal capacity of about 2.2 mAh cm−2 (i.e., 235 mAh g−1) at 0.1 C (1C = 200 mA g−1), and the cut-off voltage of the Li||LCO battery is challengingly fixed on 4.7 V to fully unlock the capacity of the LCO electrode. Pushing the charging voltage to 4.8 V can further release the capacity of LCO, but the tremendous O1 phase above 4.7 V can irreversibly ruin the electrode structure, which is still a great challenge even now. From Fig. 2a, a pair of plateaus at the final charge and initial discharge of the battery with LiPF6-based electrolyte can be observed, which can be assigned to the phase transition of LCO between H1-3 phase and O1 phase. It’s widely known that the O1 phase is regarded as the most destructive phase transition, with a huge shrinkage of c-axis and collapse of O–Co–O slabs, releasing O2 from the lattice and forming the irreversible spinel phase3,4,5. Then the formed oxidized oxygen and oxidant Co4+ are prone to attack the organic solvent, forming a thick CEI layer to impede the charge transfer (Supplementary Figs. 7 and 8). As a consequence, the collapse of the crystal structure and increased charge transfer resistance jointly contribute to the fast decay of the battery. As can be seen from Fig. 2a–c, the LiPF6-based battery with the emergence of the O1 phase attenuates quickly at a high voltage of 4.7 V, while the LiNFBS-based battery with a mild phase transition can cycle for 360 cycles with a capacity retention of 70%. Besides, the LiNFBS-based battery also shows a higher average Coulombic efficiency (CE), i.e., 99.88% for LiNFBS and 98.35% for LiPF6 (Fig. 2c), and low CE in the LiPF6-based electrolyte can be imputed to the continuous parasitic reaction between the LCO electrode and electrolyte (Supplementary Fig. 9). Encouragingly, our cycling performance at a high voltage of 4.7 V is competitive compared with the reported Li||LCO batteries (Supplementary Fig. 10 and Table S1)7,10,13,28,29,30,31,32.

Charge-discharge profiles of the LCO batteries with LiPF6 (a) and LiNFBS (b) based electrolytes at 0.5 C after 0.1 C activation for two cycles, 1C = 200 mA g−1. c Cycling stability of the batteries with LiPF6 and LiNFBS based electrolyte at 3.0–4.7 V. Charge-discharge profiles (d) and cycling stability (e) of the 2.14 Ah Li||LCO pouch cell at 4.7 V and 0.2 C after two formation cycles at 0.1 C, stack-level specific energy (without packaging) is calculated to be 518 Wh kg−1.
Then an ampere-hour-level Li||LCO pouch cell with thin Li foil (50 μm Li, N/P = 1.9), high LCO mass loading (~23.5 mg cm−2) and lean electrolyte (2.0 g Ah−1) was assembled to verify the practicality of LiNFBS-based electrolyte for realizing high specific energy (Fig. 2d, e and Supplementary Fig. 11). As can be seen from Fig. 2d, the 4.7 V Li||LCO pouch cell delivers a capacity of 2.14 Ah during the activation process at 0.1 C, which the specific capacity of LCO electrode and the specifc energy of the pouch cell can be calculated to be 232 mAh g−1 and 518 Wh kg−1 (Supplementary Table S2), respectively, and the pouch cell can cycle about 50 cycles at 0.2 C (Fig. 2d, e). Though LiNFBS can stabilize the Li metal to some extent compared with LiPF6 (see detailed discussion in Supplementary Figs. 12–20), insufficiently high Li plating/stripping CE restricts the cycling lifetime of the practical pouch cell in such harsh conditions, and we believe that using advanced solvents or extra additives to stabilize the Li negative electrode will prolong the cycling lifetime of the pouch cell.
LCO structure evolution
To unravel the phase evolution during the charging process, in-situ X-ray diffraction (XRD) measurements were conducted on the LCO positive electrodes (Fig. 3a–d and Supplementary Fig. 21). Since the (003) peak related to the layer distance of the c-axis is prone to change violently during the delithiation, the (003) peak is usually chosen to track the structure evolution of LCO2,3,32, and the intensities of pristine (003) peak were normalized to the same value. For the LCO electrode charged in LiPF6-based electrolyte, the (003) peak shifts to a much larger angle with a high proportion of harmful H1-3/O1 phase (67.2%) (Fig. 3a, b), intergrowth of H1-3 and O1 phases4,33. However, the (003) peak of the LiNFBS-based LCO electrode shows a small shift with a weak H1-3/O1 phase (41.5%) (Fig. 3c, d). Besides, a stronger peak of H2a intermediate belonging to the H2 phase at the initial charging process in LiNFBS can be noted (Fig. 3c, d)34, which may contribute to the delay of phase transition.

In situ XRD evolution of (003) peak of LCO during 0.2 C charging in LiPF6 (a, b) and LiNFBS (c, d) based electrolytes. SEM images of the LCO electrodes after 50 cycles in LiPF6 (e) and LiNFBS (f) based electrolytes. Differential electrochemical mass spectrometry (DEMS) profiles of the LCO batteries during the charging process at 0.2 C with LiPF6 (g) and LiNFBS (h) based electrolytes. i Electron paramagnetic resonance (EPR) signals of the LCO electrode at different states. j O K-edge soft X-ray absorption spectroscopy (sXAS) spectra of the LCO electrode after different cycles.
To explore the detrimental effects of H1-3 and O1 phase transitions, we use a scanning electron microscope (SEM) to visualize the morphology changes. From Fig. 3e, f, and Supplementary Fig. 22, we can observe obvious cracks on the LiPF6-based LCO electrode due to the internal stress-induced lattice oxygen release, while the LiNFBS-based LCO electrode remains intact. Then, in-situ differential electrochemical mass spectrometry (DEMS) tests were carried out to check the gas evolution during the charging process (Fig. 3g, h). For the LiNFBS-based LCO electrode, no obvious gas evolution can be detected (Fig. 3h). In contrast for the LiPF6-based LCO electrode, O2 from the oxidized lattice oxygen evolves accompanied by the generation of CO2 from the oxidative decomposition of electrolyte (Fig. 3g). The release of lattice oxygen will produce oxygen vacancies, and the electron trapped in oxygen vacancy or the unpaired electron in oxidized oxygen (Oα-2, 0<α<2) can be detected by electron paramagnetic resonance (EPR) spectra, which shows sharp absorption with a g-factor of about 2.0035,36. Due to the partial overlapping of O 2p and Co 3d orbitals, lattice oxygen will be partially oxidized at high voltage37. When charging the LCO electrode in LiPF6-based electrolyte at 4.7 V, the LCO electrode exhibits a distinct EPR signal of oxygen vacancy or oxidized oxygen, and the signal still remains at a high intensity after discharge, which means the irreversible oxygen oxidation or the release of lattice oxygen forms permanent oxygen vacancies/radicals. In contrast, though the LCO electrode charged in LiNFBS-based electrolyte shows some EPR signal, it nearly disappears after discharge (Fig. 3i and Supplementary Fig. 23), indicating the reversible oxygen redox enabled by LiNFBS.
Then, soft X-ray absorption spectroscopy (sXAS) tests were carried out to investigate the transition of electronic structure at the surficial layer of LCO. As shown in Fig. 3j, the peaks associated with the excitation of photoelectrons from the O 1s orbital to Co 3d-O 2p hybridized orbitals can be observed. More specifically, the highest peaks ~530.6 eV can be assigned to the O 2p hybridized with the eg of Co3+, while the peaks before 530 eV can be assigned to the O 2p hybridized with the eg and t2g of Co4+ 9,38. The presence of massive Co4+ in the LiPF6-based LCO electrode can be attributed to the formation of spinel phase from the collapse of LixCoO2, which can block the diffusion channel of Li+39, thus impeding the reversible redox of Co4+/Co3+ due to the lack of charge compensation. The structure deterioration of the LiPF6-based LCO electrode has been confirmed by the high-resolution transmission electron microscope (HRTEM). As can be seen in Supplementary Fig. 24, the original layer structure of the LCO electrode was transformed into a spinel phase in LiPF6-based electrolyte, while that is well preserved in LiNFBS-based electrolyte, further demonstrating the superiority of our LiNFBS-based electrolyte for stabilizing the high voltage LCO electrode.
CEI characterization and analysis
To disclose the lithium salt-induced difference in LCO cycling performance, X-ray photoelectron spectroscopy (XPS) was used to analyze the components of CEI on the LCO surface. From Fig. 4a, Supplementary Fig. 25 and Supplementary Tables S3–S6, we can see the distinct decomposed products of lithium salts. The F 1s spectra display the LiPF6-derived CEI shows the dominant proportion of LixPOyFz with little LiF (Fig. 4a and Supplementary Table S3)40,41, but the LiNFBS-derived CEI layer is composed of a large amount of LiF along with the partially decomposed -CFx (Fig. 4a and Supplementary Fig. 25a)41,42. The decomposition of lithium salts can also be validated by the Li 1s, P 2p and S 2p spectra (Supplementary Fig. 25b–d) because the corresponding lithium salt anion is the unique source of P or S element in the positive electrode and electrolyte. The CEI of LCO in contact with LiPF6 electrolyte with an ignorable amount of LiF cannot inhibit the decomposition of organic solvent, thus leading to the much stronger C–O signal in C 1s spectra (Supplementary Fig. 25a and Supplementary Table S4). The roughnesses and mechanical properties of the CEI on both LCO electrodes have been assessed by atomic force microscope (AFM), which indicates that the CEI on the LCO in LiPF6 electrolyte is uneven with significant variations in height (Supplementary Fig. 26a), and the organic-rich CEI shows a much lower Young’s modulus (Supplementary Fig. 26b). In contrast, LiNFBS-derived LiF-rich CEI exhibits a much higher Young’s modulus, enabling it to stabilize the LCO surface with a smoother morphology (Supplementary Fig. 26c,d). Therefore, the LiF-rich CEI enabled by the LiNFBS naturally endows the LCO electrode with prominent cycling stability at high voltage. Though no report claims the CEI can directly inhibit the positive electrode phase transition, lots of scientists report robust CEI or coating layer on the positive electrode surface can not only impede the continuous side reactions between the positive electrode and electrolyte but also prevent the lattice oxygen release and cobalt dissolution43,44,45,46, thereby avoiding the irreversible formation of spinel phase to retain the integrity of positive electrode and restrain the phase transition to some extent. Additionally, we provide a different viewpoint on the CEI-induced inhibition of phase transition as follows. Assuming the conversion of the O3 phase to H1-3 and O1 phases (O3\(\rightleftharpoons \,\)H1-3 \(\rightleftharpoons\) O1) is reversible, once the lattice oxygen in metastable H1-3 or O1 phase releases and subsequently reacts with electrolyte (H1-3/O1+ electrolyte→ spinel + byproducts), the H1-3 or O1 phase will irreversible form spinel phase and promote the conversion of O3 phase to H1-3 or even O1 phase. Based on the above analysis, we can believe that our LiF-rich CEI can indirectly restrain the phase transition to a certain extent by stabilizing the LCO electrode surface, thus endowing the LCO electrode with a sterling cycling lifetime at a high voltage of 4.7 V.

a F 1s X-ray photoelectron spectroscopy (XPS) spectra of the LCO electrodes after 50 cycles. Time-of-flight secondary-ion mass spectrometry (TOF-SIMS) depth profiles (b) and corresponding 3D mapping images (c) of the LiNFBS-based LCO surface after 50 cycles, acquisition area was 50 μm × 50 μm. d Electrochemical potentials of redox couples. e Gibbs free energy profiles of the decomposition pathway of NFBS−.
Then, time-of-flight secondary-ion mass spectrometry (TOF-SIMS) was utilized as a complementary technique to XPS to further characterize the chemical composition of CEI and visualize its distribution in a 3D viewer. Then, we can deduce the CEI components from the mass spectra and visualize the relative content of CEI from their intensity. As presented in Fig. 4b, c, we can clearly observe the signal of F- fragments dominating from the surface to the bulk of CEI, which mainly originates from LiF. However, the signals of organic components such as CHO2- and C2HO- are sparsely distributed in the CEI layer. Furthermore, we can observe that the signal of S- from NFBS− is much stronger than that of Li2CO3- and LiCO3- from organic solvent, demonstrating that the LiF-rich CEI endowed by the LiNFBS inhibits the decomposition of organic carbonate solvent. Though the -SO3--derived CEI may also contribute to protection of the positive electrode43,44,45,46, its low content compared to LiF diminishes the actual effectiveness in CEI.
Though the Highest Occupied Molecular Orbital energy level obtained from density functional theory (DFT) calculation is highly associated with the oxidation potential of the molecule, their difference cannot be ignored47,48. Accurate redox potential can be obtained from the Nernst equation (ΔG = –nFEabs), where Eabs is the potential versus absolute vacuum scale and can be converted into Li+/Li scale by a conversion factor of 1.4 V47,48,49. The optimized molecular structures are presented in Supplementary Data 1–4. As the cycling voltage range of the Li||LCO batteries is restricted to 3.0–4.7 V, the Fermi level of the LCO electrode can be 3.0–4.7 V vs. Li+/Li. As shown in Fig. 4d and Supplementary Tables S7, and 8, NFBS− delivers the lowest oxidation potential (5.08 V vs. Li+/Li), which conforms to the Linear sweep voltammetry (LSV) results in Supplementary Fig. 2a, indicating the preferential decomposition of NFBS−. Based on the Gibbs free energy changes from DFT calculations, the energetically favorable decomposition pathways of NFBS− to CEI can be illustrated in Fig. 4e. With the 4.7 V voltage applied on the Li||LCO battery, the energy for oxidizing NFBS− (C4F9SO3- - e- = C4F9 + SO3) on the LCO electrode can decrease from 6.48 eV to 0.38 eV, which makes the energy gap easy to be surpassed. Then the Ox- from lattice oxygen or decomposed electrolyte can react with SO3 and C4F9 to form Li2SO4, C3F7, LiF, Li2CO3, CO2, etc. Additionally, C3F7 can be further degraded into a lower homologue (CxFy) or completely decomposed in a manner similar to C4F9. The downtrend of Gibbs free energy during the decomposition means the decomposition reactions are thermodynamically feasible, and the decomposition products were evidenced by XPS spectra and TOF-SIMS results (Fig. 4a–c and Supplementary Fig. 25). Therefore, we can firmly believe that LiNFBS-derived LiF-rich CEI endows the high LCO electrode with satisfactory stability.
LCO stability at high temperatures and with water
To meet the demand for high-temperature batteries in various applications, such as electronics used in summer or confined spaces with poor heat dissipation, improving the thermal stability of the electrolyte is a prerequisite for ensuring the battery performs well at high temperatures. However, the parasitic reactions between the positive electrode and electrolyte can be accelerated at elevated temperatures, exhausting the electrolyte and forming a thick passive layer on the positive electrode surface. Besides, LiPF6 is prone to decomposing into PF5 at elevated temperatures, which will further promote the decomposition of electrolytes and facilitate the side reactions at the positive electrode surface18,19,20,21. Consequently, realizing a stable cycling performance of the battery at high temperatures is very tricky, to say nothing of enabling a battery to stably work under both high temperature and high voltage simultaneously.
After evaluating the thermal stability of the electrolytes (Fig. 1c–g), the two electrolytes were used to check their feasibility in practical batteries under high voltage and high temperature (Supplementary Fig. 27). When we increase the temperature to 60 °C and elevate the cut-off voltage to 4.6 V (Fig. 5a–c), the LCO battery with LiPF6-based electrolyte quickly attenuates within 39 cycles in this harsh environment. Besides, when we rest the charged LCO batteries at 60 °C, the open circuit voltage of the LiPF6-based battery decreases quickly (Supplementary Fig. 28). Thanks to the satisfactory thermal stability of LiNFBS and its ability to form LiF-rich components to stabilize the positive electrode and negative electrode surfaces (Supplementary Fig. 29 and Supplementary Table S9), our LiNFBS-based electrolyte enables the battery to cycle 200 cycles with a capacity retention of 70% and a high Coulombic efficiency of 99.75% (Fig. 5b, c).

Charge-discharge profiles (a, b) and corresponding cycling performance (c) of the LCO batteries at 60 °C with 0.5C charge and 1C discharge after two formation cycles at 0.1 C. d–f Charge-discharge profiles of the LCO batteries with different electrolytes and water contents at 0.5C (0.1C for the initial two cycles), and the corresponding cycling performance (f).
In the commercial production and storage of the commonly used lithium salt LiPF6, the humidity of the environment should be strictly controlled, and the water content of the prepared electrolyte should be less than 10 ppm, which notably raises the commercialization costs. Besides, LiPF6 can also react with organic solvents to produce water when the battery is exposed to hot weather or undergoes thermal runaway. The water will catalyze the decomposition of LiPF6 with the formation of PF5 and HF (LiPF6 → PF5 + LiF, PF5 + H2O → POF3 + 2HF), and the aggressive PF5, POF3 and HF are apt to attack the electrolyte and corrosive the positive electrode with the formation of additional water18,19,20,21,22, leading to vicious circle and the premature death of the battery. After checking the hydrolytic stability of the electrolytes in Fig. 1e–g, we further examined the cycling stabilities of the LCO batteries under a high cut-off voltage of 4.7 V with the two electrolytes in the presence of water. As shown in Fig. 5d–f, after adding 200 ppm water to the LiPF6-based electrolyte, the charging curves of the LCO battery fluctuate along with the emergence of the overcharging behavior (Fig. 5d), making the battery quickly expire with a low average CE of 61.23% (Fig. 5f). In sharp contrast, owing to the satisfactory antihydrolytic stability of LiNFBS and its originated LiF-rich CEI, despite adding 200 ppm water to the LiNFBS-based electrolyte, the LCO battery shows comparable cycling performance with the battery using the pristine LiNFBS-based electrolyte (Fig. 2c, and Supplementary Fig. 30). Even if exposing the LCO battery to the LiNFBS-based electrolyte with 1000 ppm water, the cycling lifetime of the LCO battery still shows negligible inferiority comparing with the battery using dry electrolyte (Figs. 2c, and 5e, f). It should be noted that we just intend to show the superiority of our lithium salt under harsh environments in this paragraph, and adding extra water to the real battery is not advisable because water can react with either Li metal electrode or lithiated graphite electrode50,51. Pretreating the electrolyte with molecular sieve can also lower the water content50,51, whereas our lithium salt (LiNFBS), with significantly higher thermal and hydrolysis stabilities, can lower production and preservation costs, as well as ensure the electrolyte withstands the hydrolysis of water originating from high temperature and parasitic reactions.
Discussion
In summary, LiNFBS, a covalent and fluorine-rich lithium salt with high anodic stability (>5 V vs. Li+/Li), and high thermal and antihydrolytic stability, has been reported for the high-voltage and high-temperature Li||LCO battery. The LiF-rich CEI derived from the salt anion makes the LCO equipped with a sturdy surface to prevent the oxidization and release of lattice oxygen, thus preventing the reactive Co4+ and oxidized oxygen from attacking the electrolyte and enabling the Li||LCO battery to operate at 4.7 V for 360 cycles with a capacity retention of 70%. The superiority of the 4.7 V LCO battery has been embodied in a 2.14 Ah Li||LCO pouch cell, which shows a high stack-level specific energy of 518 Wh kg−1. Besides, the high thermal stability of LiNFBS coupled with its derived LiF-rich CEI empowers the Li||LCO battery to operate 200 cycles simultaneously at high voltage (4.6 V) and high temperature (60 °C). Furthermore, when we add 200 or even 1000 ppm water into the LiNFBS-based electrolyte, the batteries can still run for 360 and 340 cycles with a capacity retention of 70%, respectively, which means LiNFBS’s antihydrolytic ability renders the Li||LCO battery to show negligible decay on the cycling performance. We firmly believe LiNFBS can behave as a promising lithium salt for the next-generation lithium batteries to achieve high voltage, high thermal stability, low cost, and environmental friendliness.
Methods
Materials
Lithium perfluorobutanesulfonate (LiNFBS, 99.6%) was purchased from Tokyo Chemical Industry, battery-grade LiPF6 (99.99%), fluoroethylene carbonate (FEC, >99%), ethyl methyl carbonate (EMC, 99.9%) and N-methylpyrrolidone (NMP, >99.5%, anhydrous) were purchased from Aldrich. Lithium bis(fluorosulfonyl)imide (LiFSI, 99.9%) and lithium trifluoromethyl sulfonate (LiCF3SO3, 99.9%) were purchased from DoDoChem. Polyvinylidene difluoride (PVDF) and Super P carbon black were purchased from Kejing Materials Technology Co., Ltd. The solvents were dried with molecular sieves for at least one week, and the electrolytes were prepared by dissolving the lithium salts into FEC/EMC (v/v = 3:7) to obtain 1 M solution. LCO was purchased from Ronbay Technology. The LCO slurries were prepared with NMP and blended by an automatic mixer, and the weight ratio of the LCO powder, PVDF and Super P in the LCO electrodes is 96.4:1.8:1.8. The mass loadings of the LCO electrodes in the coin cell (single-side coated) and pouch cell (double-side coated) are about 9.6 and 23.5 mg cm−2, respectively, and the tap densities of the LCO electrodes are 3.5 g cm−3. Fresh Li chips (99.9%; diameter: 15.6 mm, thickness: 450 μm) and Li-Cu composite foils (99.9% Li and 99.99% Cu; 50 μm Li on both sides of 6 μm Cu) were purchased from China Energy Lithium Co., Ltd. All the negative/positive electrodes and electrolyte solutions were sealed and used in an Argon-filled glove box (H2O or O2 purity: <0.1 ppm). Al2O3-coated polyethylene membrane (12 μm PE and 2 μm Al2O3; 60 mm width) and polypropylene membrane (Celgard 2500, 25 μm) were purchased from Guangdong Canrd New Energy Technology Co., Ltd, and the Al2O3-coated side was in contact with the negative electrode. In the Li||LCO pouch cell, 9 pieces of Li@Cu electrode (45 × 56 mm2) were stacked with 8 pieces of LCO electrodes (45 × 56 mm2), separated by the Al2O3-coated membrane. The storage of cell components and the assembly of cells were conducted at 25 ± 1 °C.
Characterizations
LSV, chronoamperometry (CA), and electrochemical impedance spectroscopy (EIS) tests were performed on CHI760 electrochemical workstation. TG-DSC tests were carried out on HITACHI STA200 under N2 atmosphere, and the temperatures were increased from room temperature to 450 °C at a heating rate of 5 °C min−1. 19F-NMR spectra were recorded on Bruker 600 MHz, and fluorobenzene was used as the internal standard. In-situ XRD measurements were performed on D2 PHASER XE-T X-ray Diffractometer System with a scan speed of 2° (2θ) min−1, then the evolutions of (003) peak of the LCO electrodes can be tracked during the charging process of Li||LCO batteries at 0.2 C rate. In-situ DEMS tests were performed on QMG220 (Linglu Instrument Co. Ltd) to record the gas evolution during the charging process, and the charge current was 0.25 C, and the flow rate was 0.8 mL/min. Only XRD and DEMS tests were performed with in-situ methods, whereas the other characterizations were carried out with ex-situ methods. For ex-situ characterizations, the cycled coin-type Li||LCO and Li||Li cells were disassembled in an Argon-filled glove box to avoid the parasitic reactions with moisture and oxygen in air, then the cycled LCO electrodes and Li negative electrodes were washed with anhydrous dimethyl carbonate to remove the residual electrolytes. After drying the electrodes under vacuum, they were transferred to the specimen chamber of the characterization equipment with vacuum transfer chambers to prevent electrode contamination. The morphologies of the cycled LCO positive electrode and Li negative electrode were conducted on Quattro S SEM and an atomic force microscope (AFM, Bruker Dimension Icon). X-ray photoelectron spectra (XPS) tests were performed on Thermo Scientific Nexas with Al K-Alpha X-ray source, and C 1s spectrum at 284.8 eV was adopted to calibrate the spectra. The X-ray spot size was set to 400 µm, and the working voltage and current were 12 kV and 6 mA, respectively. High-resolution XPS was collected 5 times to improve the signal-to-noise ratio. HRTEM tests were conducted on FEI Helios 5. TOF-SIMS analysis of the detached positive electrode was carried out on ToF.SIMS 5–100 instrument (IONTOF GmbH) with high mass resolution mode, 30 keV Bi+ ion beam was used for depth profiling and 1 keV Cs+ ion beam was used for sputtering the cycled positive electrodes. The total sputtering time and area were 1200 s and 200 μm × 200 μm, respectively, and the acquisition area was 50 μm × 50 μm. Bruker EMXplus-6/1 was utilized for EPR spectra tests to detect the unpaired electron and defect in the LCO electrode. Soft X-ray absorption spectra tests were performed at Beamlines MCD-A and MCD-B (Soochow Beamline for Energy Materials) of the National Synchrotron Radiation Laboratory (NSRL) in the University of Science and Technology of China. The spectra data were acquired in total electron yield mode under vacuum and calibrated with NiO.
Electrochemical measurements
All the coin cells were CR2032-type, made with a 304 stainless steel case and spring, and assembled in an argon-filled glove box (H2O or O2 purity: <0.1 ppm). The storage, assembly and electrochemical measurements of cells were conducted at 25 ± 1 °C. The Coulombic efficiencies (CEs) for the Li||Cu cell are determined by the ratio of discharge capacity to charge capacity. In contrast, for the Li||LCO cell, the calculation is reversed. Potentiostatic polarization at 5.0 V was conducted by Li||Al cells to evaluate the electrochemical stability of the electrolyte on the Al foil at high voltage, and the potentiostatic polarization at 4.7 V was conducted by Li||LCO cells to determine the electrochemical stability of the electrolyte on the highly delithiated LCO electrode. Here, Li metals were used as negative electrodes and reference electrodes for LSV and CA tests, thus, the potentials can be referenced to Li+/Li. The ionic conductivities of the electrolytes were measured using SS||SS coin cells (where SS denotes stainless steel disc with a diameter of 15.6 mm) by potentiostatic EIS with a voltage amplitude of 5 mV, calibrated with 0.1 M KCl in water52. Galvanostatic intermittent titration technique tests were conducted on LAND CT2001A with a current pulse at 0.2 C for 20 min and a relaxation time of 2 h, one data point was acquired every second during relaxation. Coulombic efficiencies (CEs) were evaluated by Li||Cu cells with Aurbach’s methods at 0.5 mA cm−2 53. External pressure of 1 MPa was applied on the Li||LCO pouch cell during test, and the current densities during activation and cycling process were 0.1 C and 0.2 C, respectively.
Density functional theory calculations and molecular dynamics simulations
All DFT calculations were carried out by Gaussian 16A software54. Structure optimizations were performed at the level of B3LYP-D3(BJ)/6-311g(d,p) under a solvation environment, and the solvation effects were quantified by SMD (solvation model based on density) solvation model with methyl ethanoate as the solvent and 22.7 as the dielectric constant55,56. High-accuracy electronic energies were calculated at the level of B2PLYP/ma-def2-TZVP, then we can get the redox potential from the Nernst equation (ΔG = –nFEabs), where Eabs is the potential versus absolute vacuum scale and can be converted into Li+/Li scale by a conversion factor of 1.4 V47,48,49. Classic molecular dynamics simulations were conducted on Gromacs package, version 2018.857. For the LiNFBS-based electrolyte, it contains 100 NFBS−, 100 Li+, 359 FEC, and 592 EMC, and the LiPF6-based electrolyte includes 100 PF6-, 100 Li+, 383 FEC and 629 EMC (See Supplementary Table S10 for details). The RESP20.5 charges of the optimized molecules were calculated by Gaussian at the level of B3LYP-D3(BJ)/def2-TZVP with the help of Multiwfn program58. FEC, EMC, NFBS− and PF6- were modeled by the general Amber force field, and Li+ was modeled by the Universal force field. After initial energy minimization at 298.15 K and 1 atm, 1000 ps with a time step of 1 fs was used to equilibrate the simulation system, and 500 ps with a time step of 1 fs was used for production runs. The particle-mesh Ewald method with a Coulomb cutoff of 1.0 nm was employed to account for the long-range electrostatic interactions, and Verlet cut-off scheme was used as the default buffer size. The radial distribution functions were calculated by Visual Molecular Dynamics software with a step size of 0.02 nm. The initial and final configuration of simulation results are shown in Supplementary Data 6–8.
Data availability
The data supporting the findings of this study are available within the paper and its Supplementary Information, and from the corresponding author upon request. Source data are provided with this paper.
References
Liu, Q. et al. Approaching the capacity limit of lithium cobalt oxide in lithium ion batteries via lanthanum and aluminium doping. Nat. Energy 3, 936–943 (2018).
Zhang, J.-N. et al. Trace doping of multiple elements enables stable battery cycling of LiCoO2 at 4.6V. Nat. Energy 4, 594–603 (2019).
Ohnishi, T., Mitsuishi, K. & Takada, K. In situ X-ray diffraction of LiCoO2 in thin-film batteries under high-voltage charging. ACS Appl. Energy Mater. 4, 14372–14379 (2021).
Wu, Z. et al. Unveiling the evolution of LiCoO2 beyond 4.6 V. ACS Energy Lett. 8, 4806–4817 (2023).
Zhang, J. et al. Interfacial design for a 4.6 V high-voltage single-crystalline LiCoO2 cathode. Adv. Mater. 34, e2108353 (2022).
Yang, X. et al. Enabling stable high-voltage LiCoO2 operation by using synergetic interfacial modification strategy. Adv. Funct. Mater. 30, 2004664 (2020).
Yang, X. et al. Pushing lithium cobalt oxides to 4.7 V by lattice-matched interfacial engineering. Adv. Energy Mater. 12, 2200197 (2022).
Qian, J. et al. Electrochemical surface passivation of LiCoO2 particles at ultrahigh voltage and its applications in lithium-based batteries. Nat. Commun. 9, 4918 (2018).
Wang, Y. et al. An in situ formed surface coating layer enabling LiCoO2 with stable 4.6 V high-voltage cycle performances. Adv. Energy Mater. 10, 2001413 (2020).
Yan, Y. et al. One-step surface-to-bulk modification of high-voltage and long-life LiCoO2 cathode with concentration gradient architecture. Adv. Mater. 36, e2308656 (2024).
Huang, Y. et al. Mg-pillared LiCoO2: towards stable cycling at 4.6 V. Angew. Chem. Int. Ed. 60, 4682–4688 (2021).
Zhang, F. C. et al. Phytate lithium as a multifunctional additive stabilizes LiCoO2 to 4.6 V. Energy Environ. Sci. 16, 4345–4355 (2023).
Wu, D. et al. Mechanically and thermally stable cathode electrolyte interphase enables high-temperature, high-voltage Li||LiCoO2 batteries. Angew. Chem. Int. Ed. 63, e202315608 (2024).
Yang, C. et al. Phosphate-rich interface for a highly stable and safe 4.6 V LiCoO2 cathode. Adv. Mater. 35, e2210966 (2023).
Yan, Y. et al. Tailoring electrolyte dehydrogenation with trace additives: stabilizing the LiCoO2 cathode beyond 4.6 V. ACS Energy Lett. 7, 2677–2684 (2022).
Yuhang, L. et al. Present and future of functionalized Cu current collectors for stabilizing lithium metal anodes. Nano Res. Energy 2, e9120048 (2023).
Yang, S., Meng, T., Wang, Z. & Hu, X. Unveiling decaying mechanism of non-flammable all-fluorinated carbonate electrolytes in lithium metal batteries with 4.6-V LiCoO2 cathodes at elevated temperatures. Energy Storage Mater. 65, 103177 (2024).
Qiao, L. et al. Stable non-corrosive sulfonimide salt for 4-V-class lithium metal batteries. Nat. Mater. 21, 455–462 (2022).
Terborg, L. et al. Investigation of thermal aging and hydrolysis mechanisms in commercial lithium ion battery electrolyte. J. Power Sources 242, 832–837 (2013).
Liu, M. et al. Hydrolysis of LiPF6-containing electrolyte at high voltage. ACS Energy Lett. 6, 2096–2102 (2021).
Spotte-Smith, E. W. C., Petrocelli, T. B., Patel, H. D., Blau, S. M. & Persson, K. A. Elementary decomposition mechanisms of lithium hexafluorophosphate in battery electrolytes and interphases. ACS Energy Lett. 8, 347–355 (2022).
Campion, C. L., Li, W. & Lucht, B. L. Thermal decomposition of LiPF6-based electrolytes for lithium-ion batteries. J. Electrochem. Soc. 152, A2327 (2005).
Wang, Y.-Y., Zhang, X.-Q., Zhou, M.-Y. & Huang, J.-Q. Mechanism, quantitative characterization, and inhibition of corrosion in lithium batteries. Nano Res. Energy 2, e9120046 (2023).
Lu, Z., Yang, L. & Guo, Y. Thermal behavior and decomposition kinetics of six electrolyte salts by thermal analysis. J. Power Sources 156, 555–559 (2006).
Ping, P., Wang, Q., Sun, J., Xiang, H. & Chen, C. Thermal stabilities of some lithium salts and their electrolyte solutions with and without contact to a LiFePO4 electrode. J. Electrochem. Soc. 157, A1170 (2010).
He, J., Qi, S., Wang, H., Ma, J. & Artrith, N. Highly antioxidative lithium salt enables high-voltage ether electrolyte for lithium metal battery. ACS Appl. Energy Mater. 8, 343–354 (2025).
Wang, D. et al. A long-lasting dual-function electrolyte additive for stable lithium metal batteries. Nano Energy 75, 104889 (2020).
Zhou, A. et al. Al2O3 surface coating on LiCoO2 through a facile and scalable wet-chemical method towards high-energy cathode materials withstanding high cutoff voltages. J. Mater. Chem. A 5, 24361–24370 (2017).
Wang, X. et al. Lithium-aluminum-phosphate coating enables stable 4.6 V cycling performance of LiCoO2 at room temperature and beyond. Energy Storage Mater. 37, 67–76 (2021).
Fu, A. et al. Highly stable operation of LiCoO2 at cut-off ≥ 4.6 V enabled by synergistic structural and interfacial manipulation. Energy Storage Mater. 46, 406–416 (2022).
Fan, T. et al. Highly stable 4.6 V LiCoO2 cathodes for rechargeable Li batteries by Rubidium-based surface modifications. Adv. Sci. 9, e2202627 (2022).
Zhuang, Z. et al. Ultrahigh-voltage LiCoO2 at 4.7 V by interface stabilization and band structure modification. Adv. Mater. 35, 2212059 (2023).
Ren, H. et al. Densification of cathode/electrolyte interphase to enhance reversibility of LiCoO2 at 4.65 V. Adv. Mater. 36, 2408875 (2024).
Lin, C. et al. Structural understanding for high-voltage stabilization of lithium cobalt oxide. Adv. Mater. 36, 2307404 (2024).
Hu, B. et al. A multifunctional manipulation to stabilize oxygen redox and phase transition in 4.6 V high-voltage LiCoO2 with sXAS and EPR studies. J. Power Sources 516, 230661 (2021).
Zhang, S.-D. et al. Surface engineering of LiCoO2 by a multifunctional nanoshell for stable 4.6V electrochemical performance. Energy Storage Mater. 57, 289–298 (2023).
Kong, W. et al. Tailoring Co3d and O2p band centers to inhibit oxygen escape for stable 4.6 V LiCoO2 cathodes. Angew. Chem. Int. Ed. 60, 27102–27112 (2021).
Mizokawa, T. et al. Role of oxygen holes in LixCoO2 revealed by soft X-ray spectroscopy. Phys. Rev. Lett. 111, 056404 (2013).
Huang, Y. et al. Lithium manganese spinel cathodes for lithium-ion batteries. Adv. Energy Mater. 11, 2000997 (2021).
Younesi, R., Hahlin, M., Björefors, F., Johansson, P. & Edström, K. Li-O2 battery degradation by lithium peroxide (Li2O2): A model study. Chem. Mater. 25, 77–84 (2013).
Xiong, Q. et al. Soluble and perfluorinated polyelectrolyte for safe and high-performance Li-O2 batteries. Angew. Chem. Int. Ed. 61, e202116635 (2022).
Yu, Y. et al. In situ designing a gradient Li+ capture and quasi-spontaneous diffusion anode protection layer toward long-life Li-O2 batteries. Adv. Mater. 32, e2004157 (2020).
Audren-Paul, M. et al. Stability of LiF deposited by ALD on high-voltage spinel/polyimide composite electrodes. ACS Appl. Energy Mater. 8, 3392–3403 (2025).
Liang, Z. et al. Novel insights into enhanced stability of Li-rich layered and high-voltage olivine phosphate cathodes for advanced batteries through surface modification and electron structure design. Adv. Sci. 12, 2413054 (2025).
Wu, F. et al. Dual-anion ionic liquid electrolyte enables stable Ni-rich cathodes in lithium-metal batteries. Joule 5, 2177–2194 (2021).
Huang, H. et al. Dextran sulfate lithium as versatile binder to stabilize high-voltage LiCoO2 to 4.6 V. Adv. Energy Mater. 11, 2101864 (2021).
Peljo, P. & Girault, H. H. Electrochemical potential window of battery electrolytes: the HOMO-LUMO misconception. Energy Environ. Sci. 11, 2306–2309 (2018).
Borodin, O. Challenges with prediction of battery electrolyte electrochemical stability window and guiding the electrode-electrolyte stabilization. Curr. Opin. Electrochem. 13, 86–93 (2019).
Trasatti, S. The absolute electrode potential: an explanatory note (Recommendations 1986). IUPAC 58, 955–966 (1986).
Sacci, R. L., Gill, L. W., Hagaman, E. W. & Dudney, N. J. Operando NMR and XRD study of chemically synthesized LiCx oxidation in a dry room environment. J. Power Sources 287, 253–260 (2015).
Chang, Z. et al. Sustainable lithium-metal battery achieved by a safe electrolyte based on recyclable and low-cost molecular sieve. Angew. Chem. Int. Ed. 60, 15572–15581 (2021).
Xue, W. et al. Ultra-high-voltage Ni-rich layered cathodes in practical Li metal batteries enabled by a sulfonamide-based electrolyte. Nat. Energy 6, 495–505 (2021).
Aurbach, D., Gofer, Y. & Langzam, J. The correlation between surface chemistry, surface morphology, and cycling efficiency of lithium electrodes in a few polar aprotic systems. J. Electrochem. Soc. 136, 3198 (1989).
Frisch, M. J. et al. Gaussian 16A, Gaussian, Inc. (Wallingford CT, 2016).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
Hall, D. S., Self, J. & Dahn, J. R. Dielectric constants for quantum chemistry and Li-ion batteries: Solvent blends of ethylene carbonate and ethyl methyl carbonate. J. Phys. Chem. C. 119, 22322–22330 (2015).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Acknowledgements
This research was supported by the National Key R&D Program of China under Project 2019YFA0705104 (C.Y.Z.). This work was supported in part by the InnoHK Project [Project 1.4—Flexible and stretchable technologies (FAST) for monitoring of CVD risk factors: Soft Battery and self-powered, flexible medical devices] at the Hong Kong Centre for Cerebro-cardiovascular Health Engineering (C.Y.Z.). We thank the Beamlines MCD-A and MCD-B (Soochow Beamline for Energy Materials) at the National Synchrotron Radiation Laboratory (NSRL) in the University of Science and Technology of China for measuring sXAS of O K-edge.
Author information
Authors and Affiliations
Contributions
Q.X. and C.Y.Z. developed the concept and designed the experiments; Q.X. performed the experimental works and DFT/MD simulations; R.J.W. collected the sXAS data; Q.X. wrote the manuscript; L.S., X.L.F. and Q.L. participated in the deep discussion of the results and provided suggestions; C.Y.Z. supervised the whole project; All authors participated in the discussion of the results.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Qifeng Zheng and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. [A peer review file is available.]
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiong, Q., Wang, R., Li, D. et al. Covalent and polyfluorinated lithium salt for stable LiCoO2 batteries at high temperature and high voltage. Nat Commun 16, 10325 (2025). https://doi.org/10.1038/s41467-025-65256-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65256-3
