
Abstract
Introducing CO2-concentrating mechanisms (CCM) into C3 crops represents a major frontier in synthetic biology with potential to enhance photosynthetic efficiency and yields. Despite decades of progress in elucidating CCM components, mechanisms and genetics (including structures of native Rubisco-containing compartments), installing algal pyrenoids or cyanobacterial carboxysomes into plants remains a formidable challenge. This is due to the requirement for chloroplast engineering to facilitate sufficient expression, and specificity of condensate proteins that impedes use of heterologous Rubiscos without extensive genetic redesign. Here, we present a modular streamlined alternative, a synthetic system using encapsulin nanocompartments from Quasibacillus thermotolerans (QtEnc). By fusing a short cargo-loading peptide to diverse Rubisco isoforms, we achieve targeted encapsulation within QtEnc while retaining CO2-fixing activity. Our isoform-agnostic design establishes a foundation for constructing plant-compatible synthetic carboxysome mimics. While carbonic anhydrase remains to be incorporated, our system offers a simpler tractable path towards integrating a functional CCM in crops.
Similar content being viewed by others


Introduction
Synthetic biology is rapidly emerging as a transformative approach to address global food security by reimagining how we can boost crop productivity using less water and nitrogen1. While synthetic microbial engineering is creating novel and nutritious food ingredients2, a parallel frontier lies in redesigning plant metabolism to deliver more sustainable, higher productivity crops3. A major metabolic bottleneck in C3 crops like wheat, rice, soybean, and potato is the inefficiency of the CO2-fixation step in photosynthesis catalysed by ribulose 1,5-bisphosphate (RuBP) carboxylase/oxygenase (Rubisco; Fig. 1a). Rubisco’s relatively slow carboxylation rate (kcatc ~ 2 to 5 s−1 in plants4,5) and competing reactivity with O2 results in the formation of 2-phosphoglycolate (2-PG), a toxic byproduct that must be recycled via photorespiration5. This process not only consumes ATP but also leads to the re-emission of already fixed CO2 (Fig. 1a), decreasing overall photosynthetic efficiency by 25 to 50%6.

a The photosynthetic efficiency and productivity of C3 crops (examples shown) is encumbered by Rubisco’s slow carboxylation rate, sub-saturating CO2 affinity, and wasteful resource costs (ATP, NADPH consumption, CO2 loss) from high photorespiratory flux. Asterisk (*) indicates crops amenable to plastome transformation. b Introducing a chloroplast CCM is envisaged to circumvent these deficiencies by introducing inorganic carbon (Ci) pumping systems to elevate stromal HCO3− levels, while housing Rubisco with carbonic anhydrase (CA) in a localising structure to elevate CO2 supply. As seen in C4-plants, a saturating CO2 maximises Rubisco rate, limits oxygenic 2PG production and photorespiratory flux (indicated by thinner red dashed lines), all combining to improve the Calvin-Benson-Bassham (CBB) cycle flux and stimulate plant productivity (indicated by bolder black lines). Rubisco localising structures considered for integration into leaf chloroplasts include: (c) the multi-component pyrenoids from microalgae12,13, and (d) carboxysomes from α- or β-cyanobacteria, which comprise a porous shell composed of several Cso and Ccm proteins, respectively11. Rubisco in both systems is encased as phase-separated condensates by Rubisco-linking proteins. e This work examines the feasibility of developing Q. thermotolerans encapsulin as a genetically simple, Rubisco isoform-agnostic platform to develop CO2-fixing compartments for downstream use in plants as a synthetic CCM.
To mitigate the metabolic inefficiencies of Rubisco, many autotrophic organisms have evolved CO2-concentrating mechanisms (CCMs) through either biochemical or biophysical strategies. Biochemical CCMs—such as C2, C4, and CAM metabolism – achieve spatial or temporal carbon concentration via multicellular coordination7. In contrast, the biophysical CCMs of unicellular phototrophs rely on subcellular compartmentalisation and metabolic adaptations that locally elevate CO2 levels within phase-separated Rubisco condensates8,9,10. These CCM systems employ inorganic carbon membrane transporters to accumulate bicarbonate (HCO3−) and co-localise carbonic anhydrase with Rubisco to provide it with near saturating levels of CO2 supply11. Prominent Rubisco encasing components include the starch-edged pyrenoids of algae and the icosahedral protein-shelled carboxysomes of cyanobacteria9,10,12,13. These architectures are suited to housing Rubisco variants with higher carboxylation rates (kcatc) but lower CO2 affinity, with the elevated CO2 microenvironments around Rubisco suppressing unwanted reaction with O2 (Fig. 1b)5. In contrast, C3 plants lack a CCM and thus rely on Rubiscos with higher CO2 affinity but lower kcatc, necessitating its production in abundance – often 25-50% of soluble leaf protein – but at a significant metabolic and nitrogen cost3,4.
Due to their economic importance, there is significant interest in improving photosynthetic efficiency in C3 plants. Compared to C3 plants, C4 species benefit from enhanced photosynthetic efficiency and improved use of water, nitrogen and light14. These advantages have garnered synthetic biology efforts to engineer C4-like traits into C3 crops, such as rice15, as well as develop comparable synthetic metabolic pathways, such as photorespiratory bypass systems that have delivered gains in crop productivity16. Other efforts are focused on integrating biophysical CCMs from algae and cyanobacteria into C3-crop photosynthesis, which are projected to boost plant yields by up to 60%8,17. Between these endeavours, the algal CCMs are more genetically complex (Fig. 1c), with more progress made towards producing carboxysomes in plant chloroplasts18,19. A key challenge with carboxysome production is achieving the precise expression of the multiple shell proteins (Fig. 1d), as imbalances in stoichiometry lead to malformed structures18,19. Rubisco encapsulation also requires structural compatibility with its condensate-forming proteins20, limiting the suitability of heterologous Rubisco isoforms unless re-engineered, often at the cost of catalytic vigour21,22. Even packaging non-plant Rubiscos in leaf chloroplasts is problematic, as they fail to meet the assembly needs of algal Rubiscos23, while cyanobacterial Rubisco production in leaves is more than 10-fold below that used to simulate productivity gains4,17. These limitations have prompted interest in developing synthetic carboxysome-like systems with simpler protein shell architectures capable of housing diverse Rubiscos11.
Encapsulins, non-viral protein nanocompartments found in bacteria and archaea, are promising scaffolds for producing a synthetic CO2-fixing organelle mimic24. Encoded by a single gene, encapsulin proteins self-assemble to form robust and uniform icosahedral compartments, with well-defined sizes ranging from 18 to 42 nm in diameter depending on their species of origin. Encapsulins have been repurposed into synthetic vaccine scaffolds, delivery vehicles and enzyme nanoreactors25,26,27,28,29. Cargo loading is typically achieved via a 10 to 40 residue cargo-loading peptide (CLP), which binds to the interior surface of encapsulins during assembly, enabling modular multi-component encapsulation.
Here, we present the construction of universally adaptable CO2-fixing nanocompartments by encapsulating Rubisco within the 42 nm encapsulin from Quasibacillus thermotolerans (QtEnc; Fig. 1e)30. We develop strategies to encapsulate three distinct Rubisco isoforms that are known to express well in Escherichia coli and plant chloroplasts. This includes the phylogenetically distinct Form I Rubiscos from tobacco (Nicotiana tabacum, Form IB)31 and the bacterium Rhodobacter sphaeroides (Form IC)32 that structurally comprise a catalytic core of eight RbcL subunits and two tetrads of the RbcS subunits (Fig. 2a). We also analyse the structurally simpler Form II Rubisco from Rhodospirillum rubrum, which only comprises a dimer of RbcL (Fig. 2a)33. We evaluate alternative CLP tagging strategies, assess their catalytic impacts, and compare co-induction versus staged induction for Rubisco-filled QtEnc assembly and function.
Results
Engineering Rubisco for encapsulin-mediated compartmentalisation
Enzymatic cargo is typically loaded into encapsulins via a C-terminal CLP tag that, for QtEnc, necessitates the addition of only 14 amino acids (Fig. 1e). For encapsulating Rubisco, the appropriate sites to append a CLP tag were determined by structure-function considerations. For the Form IB Rubisco from N. tabacum (NtRubisco) and Form IC Rubisco from R. sphaeroides (RsRubisco), their analogous hexadecameric L8S8 structures comprise a central core of four 50 kDa large subunit dimers (RbcL2) flanked at each end by tetrads of 15 kDa or 17 kDa small subunits (RbcS), respectively (Fig. 2a, Supplementary Fig. 1). Each RbcL2 houses two catalytic sites whose activation requires carbamylation via CO2 and Mg2+ binding5. This catalytic priming step enables the correct positioning of RuBP, triggering closure of the RbcL mobile loop 6 region over the active site. Loop closure is then stabilised by the flexible RbcL C-terminus via a ‘latching’ mechanism that serves to correctly orientate RuBP for reaction with CO2 or O232. Given the critical role of the RbcL C-terminus on catalytic fidelity34, modifications at the site, such as fusion of a CLP tag, may compromise enzymatic function.

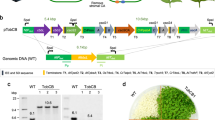
a RbcL and RbcS assembly in Form I L8S8 RsRubisco and NtRubisco and the Form II L2 Rr Rubisco. Structures depicted using ChimeraX with the PDB coordinates indicated. b Partial transparent surface renders of each Rubisco to view one RbcS as a ribbon structure in both Form I enzymes (their five C-terminal amino acids (C) shown in blue) and one RbcL in ribbon format in the Rr Rubisco dimer (the N-terminus (N) and C-terminus also in blue). An expanded view of the RbcS structures is provided in Supplementary Fig. 1. c Schematic of Rubisco expression plasmids with the amino acid sequence of the terminally appended Ser-Gly linker and cargo loading peptide (CLP) sequence shown. d Coomassie-stained standard native PAGE gels (8 µg protein/lane) and Rubisco antibody immunoblots (2 µg protein/lane) for analysis of cell soluble protein, enabling the quantity and mobility of each CLP-tagged (L8Sc8, cL2, Lc2) and untagged (L8S8, L2) Rubisco to be compared. EV = pET16 empty vector control. Data shown is representative of at least two independent experiments. Source data are provided as a Source Data file.
The RbcS subunits, although spatially distant from the active sites, contribute to L8S8 Rubisco conformational stability and mediate allosteric effects that catalytically prime the eight active sites within the RbcL8 core34,35. The C-terminal residues of RbcS are structurally disordered and extend from the distal surface of the holoenzyme (Fig. 2b, Supplementary Fig. 1b), making them attractive candidates for CLP fusion with reduced risk of perturbing catalytic function.
Based on these structural insights, we engineered CLP fusions by appending the QtEnc CLP, preceded by a flexible six-residue glycine-serine linker, to the C-terminus of RbcS in both NtRubisco and RsRubisco (Fig. 2c). These engineered complexes are hereafter referred to as NtL8Sc8 and RsL8Sc8, respectively. For comparison, the Form II Rubisco from R. rubrum (RrRubisco), which lacks RbcS and forms a homodimeric RbcL2 complex (Fig. 2a), presents structurally disordered N- and C-terminal regions that are solvent exposed (Fig. 2b). We therefore generated QtEnc CLP fusions at both termini, yielding two constructs: RrcL2 containing an N-terminal CLP fusion and RrLc2 containing a C-terminal CLP fusion (Fig. 2c).
Rubisco biogenesis is preserved following CLP tagging
To confirm that the appropriate oligomeric state of Rubisco was preserved upon CLP-tagging, all four CLP-tagged Rubiscos and their respective untagged wild-type controls were expressed in E. coli and analysed by native PAGE (Fig. 2d). Both CLP-tagged and untagged forms of RsRubisco and NtRubisco were expressed at comparable levels, as confirmed by immunoblotting with Rubisco-specific antibodies. The CLP-tagged L8Sc8 complexes had reduced electrophoretic mobility relative to their untagged counterparts, consistent with their larger molecular mass arising from fusion of the CLP tag (Fig. 2d). Aside from antibody detection of RbcL-associated chaperonin complexes (GroESL+RsRbcL or CPN60+NtRbcL, labelled with asterisks in Fig. 2d), no intermediary assembly states or degradation products were observed, indicating that the CLP tags on RbcS did not adversely affect the assembly or stability of either RsL8Sc8 or NtL8Sc8 Rubisco. Similarly, CLP tagging at the N- or C-terminus of RrRubisco did not impair assembly. Both RrcL2 and RrLc2 formed the expected dimeric complexes, with an increased molecular weight relative to the native RrcL2 due to the presence of the CLP tag (Fig. 2d).
To quantify the impact of CLP tagging on the biogenesis of active Rubisco, we measured their binding of radiolabelled 14C-carboxyarbinitol bisphosphate (14CABP) inhibitor, normalised to the cell soluble protein. CLP tagging to RbcL was found to have no measurable impact on RrRubisco formation. Meanwhile, the amount of CLP-tagged RsL8Sc8 enzyme that showed binding to 14CABP was reduced by 28% compared to the untagged RsRubisco control (Fig. 3a). CLP tagging on the RbcS of NtRubisco also appeared to reduce the amount of NtL8Sc8 enzyme the cells could produce, although the effect was not statistically significant. These findings suggest that despite the complex biogenesis requirements of NtRubisco in E. coli—which necessitates the co-expression of seven additional chloroplast chaperones to ensure proper RbcL folding and assembly (Supplementary Fig. 2)31—CLP-tagged RbcS subunits were successfully incorporated into functional NtL8Sc8 complexes.

Comparisons of (a) Normalised E. coli expression levels of Rubisco based on 14CABP binding measurements, and (b–f) catalytic properties for each Rubisco isoform as indicated. Small white squares denote the mean value, black vertical lines represent the median value, while the box plots show the 25th to 75th percentile range with whiskers showing the minimum and maximum values. Circles represent independent biological replicate measurements. T-test derived statistical differences between CLP tagged (RsL8Sc8, NtL8Sc8, RrcL2, RrLc2) and untagged wild-type (RsL8S8, NtL8S8, RrL2) values are denoted * (p < 0.05), ** (p < 0.01), and *** (p < 0.001). Kinetic values are listed in Supplementary Table 1. The response of Kc to oxygen for each Rubisco is compared in Supplementary Fig. 3. Source data are provided as a Source Data file.
Rubisco catalytic activity remains largely intact after CLP-tagging
Comprehensive kinetic analyses revealed that the CLP tags did not significantly impact the catalytic performance of RsL8Sc8, RrcL2 or RrLc2. Specifically, the carboxylation rates (kcatc, Fig. 3b) and the Michaelis constants (Km) for CO2 (Kc, Fig. 3c), inhibitory O2 (Ko, Fig. 3d and Supplementary Fig. 3) and RuBP (KRuBP, Fig. 3e) were comparable to the untagged RsRubisco and RrRubisco controls. The only instance where CLP tagging had an impact was NtRubisco, resulting in a less than two-fold reduction in kcatc (Fig. 3b), but no significant impact across all other kinetic parameters measured. Across all Rubisco variants, the specificity for CO2 over O2 (Sc/o) also remained unchanged by CLP tagging (Fig. 3f). These findings are consistent with prior observations by Martin-Avila et al.36 where inclusion of a C-terminal polyhistidine tag on RbcS subunits impaired the kcatc of NtRubisco by 30% without altering Sc/o.
The differential impact of the CLP on kcatc likely reflects structural distinctions between Form IB and IC Rubisco. As illustrated in Fig. 2b and Supplementary Fig. 1, the RbcS in RsRubisco possesses an extended C-terminal β-hairpin that interacts with RbcL residues at the entrance to the central solvent channel, contributing to enzyme stability and function35. In contrast, the shorter C-terminus of Form IB plant RbcS engages in differing RbcL residue interactions that appear more catalytically sensitive, rendering it more susceptible to kcatc perturbations by C-terminal modifications36,37.
CLP tagging enables encapsulation of RsRubisco complexes
To evaluate whether CLP-tagged Form I Rubiscos could be successfully encapsulated within QtEnc compartments, we initially focused on RsRubisco as it can be abundantly produced in both E. coli38 and leaf chloroplasts32, the latter being our intended long-term site for CCM bioengineering. E. coli was co-transformed with pACYC-QtEnc, which encodes for arabinose-inducible QtEnc expression (Fig. 4a), along with either pCDF-RsLS (encoding wild-type RsL8S8) or pCDF-RsLSc (encoding CLP-tagged RsL8Sc8).

a Overview of producing Rs Rubisco (L8S8 control) and QtEnc-Rs Rubisco compartments in E. coli using co-induced (Co-I) or staged (ST) QtEnc expression strategies. Coloured circles indicate the genes being induced at each step. b Coomassie-stained blue native PAGE gel for analysis of E. coli total soluble protein and (c) corresponding Rs Rubisco immunoblot (following in-gel SDS denaturation prior to nitrocellulose transfer) enable comparison of RsL8S8 and RsL8Sc8 production and the formation of QtEnc-Rs Rubisco complexes following Co-I and ST QtEnc expression. Each experiment was repeated independently at least twice with similar results. d SDS-PAGE, 14CABP binding and kcatc of the purified QtEnc, Co-I and ST complexes, with the Rs Rubisco produced in tobRsLS leaves32 provided as a positive control for quantitative measurements and subunit stoichiometry. Ratio of measurements between samples is shown as bubble plots, with relative RbcL and RbcS content between samples determined by gel densitometry on Coomassie-stained bands, along with quantification of purified QtEnc concentration against a BSA standard. e Schematic representation drawn to scale, depicting the contents of purified QtEnc Co-I and ST compartments. The Co-I sample contains unassembled RsRbcSc and inactive RsL?Sc? subcomplexes of unknown oligomeric state, while the CO2-fixing ST sample contains functional RsL8Sc8, as well as a minor amount of unassembled RsRbcSc. Source data are provided as a Source Data file.
Blue-Native PAGE analysis revealed that co-induction of untagged RsRubisco and QtEnc resulted in a distinct band corresponding to free RsL8S8 as a ~ 540 kDa complex (Fig. 4b, lane 3), with only a small proportion of non-specific encapsulated RsRubisco detectable by immunoblotting (Fig. 4c). In contrast, no free complexes of CLP-tagged RsL8Sc8 were detected when co-induced with QtEnc. Instead, immunoblot analysis revealed that the RsRubisco was exclusively associated with QtEnc compartments ( ~ 7.7 MDa; Fig. 4c, lane 5), indicating encapsulation of the CLP-tagged cargo. Furthermore, the structural integrity of these QtEnc-Rubisco compartments was maintained following sonication (see also Supplementary Fig. 4), a process otherwise shown to trigger carboxysome rupture39.
Given that efficient encapsulation can require temporally coordinated expression of cargo enzyme and protein compartment40, we hypothesised that the complex biogenesis of Form I Rubiscos might necessitate a staged expression strategy. To test this hypothesis, CLP-tagged RsRubisco production was first induced with IPTG, followed by washing and transferral to fresh media containing arabinose to induce QtEnc expression (Fig. 4a). Native PAGE analysis confirmed that the CLP-tagged RsRubisco produced in the first stage of expression was quantitatively encapsulated after QtEnc production in the second stage (Fig. 4b, c, lane 6).
Staged expression is essential for encapsulating functional RsL8S8 Rubisco inside QtEnc
The QtEnc-RsRubisco complexes produced by staged expression and co-induction were purified alongside an empty QtEnc control (Fig. 4d), with Rubisco content and activity assessed by 14CABP binding and kcatc assays. Catalytic activity was only observed for the QtEnc-RsL8Sc8 complex produced via staged expression. The kcatc of the purified QtEnc-RsRubisco complexes from staged expression (2.3 s−1) was ~60% of that measured for the RsRubisco produced in tobRsLS leaves (Fig. 4d, lane 2) and the unencapsulated RsL8Sc8 produced in E. coli (Fig. 3b). These results indicate that the staged expression strategy was successful in encapsulating functional RsL8Sc8, and that the wild-type QtEnc compartment is sufficiently porous to permit the exchange of RuBP and its structural analogue 14CABP, supporting a reasonable rate of Rubisco catalysis even in the absence of any compartment engineering efforts.
No activity was detected in the purified co-induced QtEnc-RsRubisco sample, despite the presence of RsRubisco being detected by immunoblotting (Fig. 4c, lane 5) and both RsRbcL and RsRbcSc subunits observed by SDS-PAGE (Fig. 4d, lane 3). Comparing the stoichiometric ratio of subunits between the purified samples from co-induction, staged expression, and crude tobRsLS-derived leaf RsRubisco (Fig. 4d), the co-induced sample had an unbalanced ratio with excess RbcSc. Taken together, these observations suggest that the encapsulated material was composed of unassembled RbcSc as well as non-functional RbcL subcomplexes containing at least one RbcSc to mediate their CLP-dependent encapsulation by QtEnc (Fig. 4e).
Subunit stoichiometry for the purified staged expression sample was more balanced, more closely matching the ratio found in the tobRsLS control, and consistent with the amount of Rubisco as quantified by 14CABP binding (i.e., matching bubble plot diameters in Fig. 4d). These data suggest that staged expression resulted in encapsulation of fully assembled, functional RsL8Sc8 holoenzymes. Based on 14CABP binding and QtEnc densitometry, we estimated that the QtEnc compartments encapsulate an average of ~8 RsL8Sc8 complexes upon staged expression (Fig. 4c), assuming stoichiometric and near irreversible binding of one 14CABP per active site (i.e., 8 per L8S8 complex41) and QtEnc compartments composed of 240 copies of the 32.2 kDa protein29. Of note, however, the encapsulated sample still shows a slight disproportionality towards a higher amount of RsRbcSc relative to tobRsLS (Fig. 4d). This indicates that in addition to encapsulating complete RsL8Sc8 complexes, a minor amount of unassembled RsRbcSc protomers were also encapsulated (Fig. 4e). This observation aligns with prior findings that RbcL folding and assembly primarily limit L8S8 Rubisco biogenesis in E. coli, thus often leaving an excess of unassembled soluble RbcS42. Indeed, the presence of excess RbcSc in both the staged and co-induced samples suggests that premature binding of unassembled RbcSc to QtEnc may limit their availability for L8Sc8 enzyme incorporation, especially during co-induction when early QtEnc expression may sequester some RbcSc before it is able to assemble with RsRbcL (Fig. 4e).
Functional NtRubisco and RrRubisco can also be encapsulated within QtEnc
The staged expression approach also proved successful in encapsulating tobacco NtRubisco (Supplementary Fig. 4). SDS PAGE densitometry and 14CABP binding analyses of purified QtEnc-NtRubisco compartments confirmed that NtRbcL abundance was a reliable proxy for the amount of encapsulated holoenzyme (Fig. 5a). Similar to RsRubisco, the QtEnc compartments contained a mixture of fully assembled NtL8Sc8 and some unassembled NtRbcSc (Fig. 5a). Stoichiometry estimates from duplicate preparations of purified QtEnc-NtL8Sc8 after staged induction indicated an average of ~7 NtL8Sc8 complexes were encapsulated per QtEnc compartment (Fig. 5a).

a SDS PAGE and 14CABP binding of purified QtEnc-Rubisco compartments following staged (ST, blue shading) E. coli expression in biological duplicate experiments for tobacco NtL8Sc8 Rubisco and (b) co-induced (Co-I, yellow shading) and ST samples for R. rubrum RrcL2 or RrLc2 Rubisco. Protein samples from tobacco and tobRr leaves33 are provided as controls for quantitative measurements and subunit stoichiometry for NtL8S8 and RrL2, respectively. Measurements between samples are shown as bubble plots, with the relative RbcL, RbcS and QtEnc contents quantified by gel densitometry on Coomassie-stained bands. Supplementary Fig. 4 provides a native PAGE analysis of each Rubisco before and after encapsulation. Each experiment was repeated independently at least twice with similar results. c Negative stain transmission electron micrographs of each purified ST QtEnc-Rubisco sample show that compartment morphology is preserved upon Rubisco encapsulation. Representative images shown are from a set of at least nine different images taken for each sample. Source data are provided as a Source Data file.
Compared to the heterooligomeric complexity of Form I Rubiscos, the structurally simple homodimeric architecture of RrRubisco facilitated efficient assembly of the RbcL2 enzyme, resulting in its abundant (Fig. 3a) and almost fully soluble expression in E. coli43. Consistent with this efficient assembly, the levels of encapsulated N-terminally tagged RrcL2 and C-terminally tagged RrLc2 versions of RrRubisco were comparable, regardless of whether QtEnc compartments were co-induced or staged (Fig. 5b). SDS-PAGE gel densitometry and 14CABP binding analyses on purified samples revealed that approximately 110 RrLc2 complexes were encapsulated per QtEnc compartment in both co-induced and staged samples, compared to ~60 RrcL2 complexes in corresponding preparations (Fig. 5b). While the reason for lower packaging efficiency in the case of N-terminal tagging remains unclear, one possibility is that the omission of the flexible Ser-Gly linker in the RrcL2 construct (Fig. 1c) impaired the efficacy of CLP-mediated binding within the QtEnc compartment. Additionally, structural hindrance arising from a greater limitation in the mobility of the RrRbcL N-terminus and/or limitations in the ability of assembling QtEnc compartments to recognise the non-canonical N-terminal CLP fusion may have further impaired RrcL2 encapsulation.
QtEnc ultrastructure and stability are preserved following Rubisco encapsulation
Transmission electron microscopy (TEM) confirmed that the morphology and size of each QtEnc-Rubisco compartment produced by staged expression closely matched those of empty wild-type QtEnc compartments produced in E. coli (Fig. 5c). Dynamic light scattering (DLS) measurements showed that the hydrodynamic radii of all samples were comparable to that of empty QtEnc (Supplementary Fig. 5a), indicating that Rubisco encapsulation does not alter the overall structure of QtEnc. Thermal stability, assessed via differential scanning fluorimetry (DSF), revealed that the melting temperatures of Rubisco-loaded QtEnc shells were similar to those of empty QtEnc (Supplementary Fig. 5b). Collectively, these data demonstrate that Rubisco encapsulation does not significantly compromise the structural integrity, size, or thermal stability of the QtEnc compartment.
Rubisco kinetic estimates may reflect changes in substrate accessibility
Initial CO2-fixation assays confirmed that all encapsulated Rubisco isoforms retained catalytic activity. However, their kcatc rates were reduced up to two-fold relative to their unencapsulated, CLP-tagged counterparts (see Figs. 3b, 6). This reduction may have resulted from a combination of two possible factors. Firstly, structural constraints imposed by anchoring one or more Rubisco subunits to the inner surface of the QtEnc shell may have impacted the mobility of active-site-associated regions such as RbcL loop 6 and the C-terminus, thereby impairing catalytic fitness. Secondly, the decrease in activity could reflect a limitation in substrate availability44,45—specifically the diffusional supply of RuBP, CO2 and/or bicarbonate through the pores of the QtEnc compartment. To explore the latter possibility, we compared the apparent Km values for RuBP and CO2 between each purified QtEnc-Rubisco complex and its corresponding unencapsulated control (i.e., L8Sc8, RrcL2, and RrLc2).

Comparing how the encapsulation of each CLP-tagged Rubisco (open circles) impacts their apparent Michaelis-Menten constants (Km) for (a) RuBP (KRuBP) and (b) CO2 in the absence of oxygen (Kc) relative to unencapsulated enzyme (filled circles). Individual data points (N = 3 to 6 biological replicates) are shown alongside the mean values for KRuBP and Kc (± standard deviation). These parameters were fitted to the Michaelis-Menten equation (see Supplementary Method 4) to generate the fitted curves. Bold lines encapsulated CLP-tagged Rubisco, and thin lines unencapsulated enzyme. Carboxylase activity (Vc) was normalised to the extrapolated Vcmax for each sample to allow comparison across each sample. Values of kcatc derived from the Kc assays for the encapsulated enzymes were 2.3 ± 0.1 s−1 (RsRubisco), 1.5 ± 0.1 s−1 (NtRubisco), 5.7 ± 0.4 s−1 (cL2-Rr Rubisco) and 7.0 ± 0.5 s−1 (Lc2-Rr Rubisco). Statistical significance was assessed using paired two-sided t-tests, with p-values indicated as follows: p < 0.05 (*) and p < 0.01 (**). A summary of the kinetic parameters and sampling size (N) is provided in Supplementary Table 2. KRuBP assays were conducted under saturating CO2 (30 mM NaH14CO3 for Rr Rubisco, 20 mM NaH14CO3 for RsRubisco, 10 mM NaH14CO3 for NtRubisco) and KC assays were performed with 0.5–1 mM RuBP (Supplementary Table 3). Source data are provided as a Source Data file.
Encapsulation of NtL8Sc8, RrcL2 and RrLc2 Rubisco variants led to substantial increases in KRuBP, with no significant change observed only in the case of RsL8Sc8 encapsulation (Fig. 6a). While the effects of anchoring Rubisco to the QtEnc shell may play a role, the general increase in KRuBP could be due in part to diffusional limitations in RuBP supply, especially under sub-saturating concentrations highlighted with yellow shading on the kinetic plots in Fig. 6a. The lack of detectable supply limitations for encapsulated RsL8Sc8 may potentially be explained by the intrinsically higher KRuBP of RsRubisco, which is more than four-fold higher than the other Rubisco variants. This elevated KRuBP likely leads to slower rates of RuBP consumption under lower substrate concentrations, thereby reducing the impact of diffusional constraints.
In contrast to the KRuBP values, no significant differences were observed in the Kc values of encapsulated versus unencapsulated NtL8Sc8, RrcL2, and RrLc2 enzymes, with RsL8Sc8 again as the only exception, suggesting that CO2 diffusion through QtEnc pores is generally not limiting (Fig. 6b). In a functional QtEnc-based CCM where carbonic anhydrase (CA) is co-encapsulated with Rubisco, the primary concern is not the permeability of the QtEnc protein cages to CO2, but rather to bicarbonate (HCO3⁻). If the diffusion of HCO3⁻ into the cage is not rate-limiting, then the apparent CO2 affinity of Rubisco within QtEnc-CA compartments should match that observed for non-encapsulated Rubisco under saturating CA conditions. However, such experiments have yet to be conducted, pending the development of a fully functional QtEnc-based CCM.
Taken together, these findings suggest that while the native QtEnc architecture may allow sufficient CO2 permeability, further protein engineering may be necessary to enhance RuBP accessibility and potentially enhance HCO3⁻ transport.
Discussion
Engineering a CCM into C3 crops is a long-standing objective in synthetic biology, with the potential to significantly enhance photosynthetic efficiency and productivity3,8,17. Despite major advances in understanding the genetic and structural basis of native CCMs10,12,13, efforts to reconstitute these systems in plants have been constrained by the complexity of chloroplast genome engineering and the structural specificity of native condensate proteins, which limit their compatibility with heterologous Rubisco isoforms9,46.
To address these challenges, this study describes the successful development of a modular and genetically streamlined compartment engineering platform using Q. thermotolerans encapsulin. By fusing its CLP to structurally permissive termini of Rubisco subunits, we achieved encapsulation of three different Rubisco isoforms within QtEnc (Fig. 4b, c, Supplementary Fig. 4), while preserving the uniform structure, size, and high thermal stability of the compartments (Fig. 5, Supplementary Fig. 5). The simple genetic encoding of QtEnc encapsulation systems offers advantages over more complex encapsulation systems like pyrenoids and carboxysomes (Fig. 1c, d) by enabling robust expression in heterologous hosts and simplifying stoichiometric control.
Rubisco activity was generally retained upon CLP tagging and subsequent encapsulation. Enzyme assembly and catalytic parameters were not impacted by CLP tagging, except for the reduced kcatc of NtRubisco, whose structurally sensitive RbcS C-terminus appears to pervasively impact catalysis (Fig. 3b)36,37. While encapsulated Rubisco retained CO2-fixation activity, the purified compartments showed some reduction in kcatc. This effect may be in part due to potential diffusional restrictions for RuBP but not CO2 under sub-saturating concentrations, as indicated by increases in KRuBP for encapsulated Rubiscos with stronger RuBP affinity (Fig. 6). Fortuitously, the structural simplicity of encapsulins makes them highly amenable to tuning porosity without significantly impacting compartment assembly and stability44,47. For example, encapsulin variants with engineered pores of different size and charge have been reported to improve catalytic performance of encapsulated enzymes (LigM demethylase, NanoLuc, nitroreductase) when compared to encapsulation in wild-type compartments, an effect attributed to increased flux of substrates and products25,48,49.
The staged expression experiments highlight the complexity of encapsulating functional Form I Rubiscos, given their complex biogenesis requirements and assembly into heterooligomeric L8S8 hexadecamers. Sequential expression of Rubisco followed by QtEnc was essential for encapsulating fully assembled RsL8Sc8, as simultaneous co-induction led to the inadvertent encapsulation of unassembled RsRbcSc subunits and inactive RbcL-containing subcomplexes (Fig. 4d, e). Consistent with these observations, the simpler assembly requirements of Form II L2 RrRubisco enabled its efficient encapsulation without the need for temporal control of expression (Fig. 5b). The ability of QtEnc to encapsulate all three Rubisco isoforms highlights their generic protein packaging capability and the versatility of our modular encapsulation approach, enabled by temporal control of protein expression.
Our findings underscore the potential of QtEnc as a versatile synthetic biology platform for constructing plant-compatible CCMs. In addition to future pore engineering efforts to optimise RuBP flux, other changes are likely needed to enable operation within a photosynthetic host. While the modularity of QtEnc enables packaging compatibility with any form of Rubisco, including faster variants engineered by directed evolution4,38, enhancing carbon fixation efficiency will require co-packaging with carbonic anhydrase to boost localised CO2 supply11. For Form I enzymes, encapsulation with their cognate Rubisco activase is also likely required, possibly necessitating additional pore engineering to maintain an adequate supply of ATP to drive its removal of sugar phosphate inhibitors from Rubisco3,5. To support these future endeavours, we have recently developed an in vitro platform for QtEnc assembly50 that enables rapid prototyping of multiple enzyme cargoes in different ratios. Together with staged and tunable expression strategies, this in vitro platform will enable the encapsulation of Rubisco, carbonic anhydrase, and Rubisco activase to be rapidly optimised, ready for downstream in vivo implementation.
A foreseeable application of our generic QtEnc-Rubisco encapsulation platform includes deployment in the chloroplasts of photoautotrophic organisms. Their application in plant systems may be advantaged by the use of nuclear transformation strategies to append a CLP tag to one or more of the endogenous RbcS gene copies, or to introduce an additional RbcS-CLP transgene. Upon import into the chloroplast and assembly with RbcL and untagged endogenous RbcS subunits into the holoenzyme, the resulting partly-CLP-tagged Rubiscos would be compatible with QtEnc encapsulation5,36,51. A key technical challenge is the exceptionally high concentration of Rubisco in chloroplasts (typically 100 to 200 mg/mL), which would necessitate the production of a large number of QtEnc compartments. Achieving this level will likely require stable integration of the QtEnc gene into the chloroplast genome, a genetic approach primarily employed to express cyanobacterial CCM components in adequate abundance8,18,19. Looking forward, the successful implementation of a chloroplast-located QtEnc-based CO2-fixing compartment will also require the coordinated expression of chloroplast envelope bicarbonate transporters and suppression of stromal carbonic anhydrase activity (Fig. 1b)10,11. These modifications are essential to establish a functional CO2 gradient and enable efficient carbon fixation within the encapsulated microenvironment.
Ultimately, the strength of our QtEnc-Rubisco platform lies in its engineering simplicity and adaptability. QtEnc compartments offer a genetically compact, engineerable platform for constructing synthetic carboxysome-like organelles. With further development, this system holds promise for introducing an artificial CCM into C3 crops and advancing the broader goal of enhancing photosynthetic performance through synthetic biology.
Methods
Molecular cloning
All genes were synthesised and supplied as gene blocks (gBlocks Integrated DNA Technologies; gene sequences supplied in Supplementary Data 1). Cloning and construct design was simulated using Geneious Prime 2024.0.7 using Golden Gate cloning, modified components from the EcoFlex Kit52 using BsaI and BsmBI cloning. Prior to use in Golden Gate cloning reactions, PCR fragments were purified by agarose gel electrophoresis, followed with gel extraction using Wizard SV Gel and PCR Clean-Up System (Promega). Chemically competent XL1-Blue E. coli cells (Agilent) were transformed with cloned plasmids via heat shock, and single colony cultures were grown overnight, and their plasmid DNA purified using Wizard Plus SV Minipreps DNA Purification System (Promega). Plasmids were screened by restriction mapping before either short-read Sanger sequencing (Macrogen) or whole-plasmid sequencing (Plasmidsaurus).
As outlined in Fig. 2c, rbcL and rbcS genes encoding RsRubisco were cloned into pCDF (spectinomycin resistance) under independent T7-promoter control, while those encoding RrRubisco and NtRubisco were cloned into pET16 (ampicillin resistance). A QtEnc gene encoding the 32.2 kDa QtEnc was cloned into pACYC (chloramphenicol resistance) within an arabinose-inducible pBAD promoter system. QtEnc CLP sequences were appended onto the N-terminus or C-terminus of RsRbcS, NtRbcS or RrRbcL by PCR (Fig. 2c). Full plasmid and amino acid sequences are listed in Supplementary Data 1.
Recombinant protein expression
Electrocompetent BL21 Star (DE3) E. coli (Invitrogen) were transformed with cloned plasmids. For N. tabacum Rubisco biogenesis, the pET16-NtL-NtS(–/+QtCLP) plasmid was co-transformed with the pCDF-NtAssembl plasmid, which expresses the 7 auxiliary chloroplast assembly/folding chaperones (cpn60α, cpn60β, cpn20, raf1, raf2, rbcX, and bsd2) under IPTG induction (Supplementary Fig. 2)31. Protein expression was undertaken in small (100 mL for Rubisco content and kinetics) or large volumes (400 mL for QtEnc-Rubisco purification) of antibiotic-containing LB medium (50 µg/mL ampicillin, 50 µg/mL spectinomycin and/or 30 µg/mL chloramphenicol). Cultures were grown at 37 °C to an OD600 of ~0.7 before inducing Rubisco expression with 1 mM IPTG at 23 °C for 8–16 h (or 24 h for cultures expressing NtL8S8) before harvesting the cells by centrifugation (6000 g, 10 min, 4 °C), flash freezing in liquid N2 and storing at –80 °C. The same process was used to co-induce Rubisco and QtEnc with 0.2% w/v arabinose added along with 1 mM IPTG. For staged Rubisco-QtEnc expression, the cells were harvested following IPTG induction by centrifuging (4000 g, 4 °C, 10 min) under sterile conditions, and the cells were resuspended in LB with 0.2% w/v arabinose, and incubated for 22–24 h at 20 °C before harvesting as above. The protein induction processes are summarised in Fig. 4a.
Protein extraction, enzyme activity measurements, PAGE, and immunoblotting
The following methods pertain to generating clarified cell lysates for conducting Rubisco assays and PAGE analyses, with additional detail provided in Supplementary Methods 1–5. E. coli cell pellets were resuspended in 1 mL of ice-cold extraction buffer (50 mM EPPS-NaOH, pH 8.0, 15 mM MgCl2, 0.5 mM EDTA, 5 mM DTT, 1 mM PMSF), sonicated on ice for 30 s at 60% amplitude (Qsonica Q125, 20 kHz) and centrifuged at 13000 g, 4 °C for 2–10 min to obtain the total cytoplasmic protein fraction. Soluble E. coli protein concentration was determined with Coomassie Plus Protein Assay Reagent (Pierce, Thermo Scientific) against bovine serum albumin standards, and the Rubisco content was quantified by 14C-carboxyarabinitol bisphosphate binding53. Enzyme kinetics were measured using 14CO2-fixation assays for kcatC and KC, while the [1-3H]-RuBP consumption assay was used to measure SC/O51. The same soluble protein fraction was also analysed by non-denaturing native PAGE (4-12%, Tris-Glycine Gels; Invitrogen), blue native PAGE (NativePAGE 3-12% Bis-Tris Gel, Invitrogen) and by SDS-PAGE (4–12% Bis-Tris Gels; Invitrogen)54. Coomassie-stained protein gels were scanned using Perfection V700 Photo Flatbed Scanner (Epson), while immunoblots were visualised using Clarity™ Western ECL (Bio-Rad) with ChemiDoc MP Imaging system (Bio-Rad).
Purification of QtEnc compartments
Frozen cell pellets were resuspended in 25 mL SEC buffer (50 mM EPPS pH 8, 200 mM NaCl, 20 mM MgCl2, 1 mM EDTA, 1 mM DTT) with DNase I (10 µg/mL), lysozyme (100 µg/mL), 1× protease inhibitor (cOmplete Protease Inhibitor Cocktail, Roche), and kept on ice for 30 min before lysis using a Sonoplus HD 4050 probe sonicator with TS-106 probe (Bandelin; 55% amplitude and pulsing on ice for 11 min at 8 s on and 10 s off). Following centrifugation (17,000 g, 1 h, 4 °C) the supernatant was collected and solid ammonium sulphate was added to 20% w/v. After 15 min rocking at 4 °C, the sample was centrifuged (10,000 g, 15 min, 4 °C), the supernatant collected, and ammonium sulphate added to 50% w/v, then after 15 min centrifuged again. The protein pellet was resuspended in 5 mL of SEC buffer then successively dialysed against 1 L volumes of fresh SEC buffer for 2 h then 16 h at 4 °C using Snakeskin Dialysis Tubing (3.5 kDa MWCO, ThermoFisher). The sample was then subjected to anion exchange chromatography on a HiPrep Q XL 16/10 column at 5 mL/min. QtEnc was collected in the unretained fraction and further purified on a Sephacryl S-500 HR SEC column at 1 mL/min. QtEnc-containing fractions were concentrated using Amicon Ultra-15 centrifugal filters (100 kDa MWCO, Merck) and applied to a Superose 6 Increase 10/300 GL column at 0.5 mL/min. The purified QtEnc complexes that eluted at ~8 mL were stored at 4 °C until further analysis.
Transmission electron microscopy
Purified QtEnc samples were diluted to 150 µg/mL in 50 mM EPPS pH 8, 100 mM NaCl. Copper grids (300-mesh coated with Formvar-carbon film, EMS) were made hydrophilic via glow discharge at 25 mA for 30 s before floating on 10 μL of sample for 2 min, blotting, and staining with 20 μL of 2% (w/v) uranyl acetate for 10 s. Grids were then blotted and air-dried. Negative stain TEM images were captured using a Tecnai T12 microscope at 120 keV using a Gatan Rio CMOS camera at Sydney Microscopy and Microanalysis.
Dynamic light scattering and thermal stability measurements
Encapsulin samples were diluted to ~1 mg/mL in SEC buffer (without DTT) and loaded onto Prometheus Panta (NanoTemper). Isothermal DLS measurements were taken using the Size Analysis function (10 acquisitions, 5 s each, 100% laser intensity, 25 °C). For differential scanning fluorimetry, the ~1 mg/mL samples were loaded into standard capillaries (NanoTemper) and sealed at the ends (Capillary Sealing Paste, NanoTemper). Unfolding was determined by nanoDSF on a Prometheus Panta (NanoTemper) using a heating ramp from 25 to 110 °C using an increment of 1 °C and a 20% sensitivity setting. The melting temperature was calculated by the Prometheus Panta software as the point where the curve’s first derivative is at a maximum and its second derivative is zero.
Statistical analysis
Statistical comparisons between paired conditions were performed using a two-tailed paired t-test and p-value < 0.05 considered statistically significant. Normality of differences was assessed using the Shapiro-Wilk test using OriginPro 2024 (OriginLab Corp, MA, USA).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the findings of this work are available within the paper and its Supplementary Information files. A reporting summary for this Article is available as a Supplementary Information file. Source data are provided with this paper.
References
Sargent, D., Conaty, W. C., Tissue, D. T. & Sharwood, R. E. Synthetic biology and opportunities within agricultural crops. J. Sustain. Agric. Environ. 1, 89–107 (2022).
Chen, R. et al. Synthetic biology for the food industry: advances and challenges. Crit. Rev. Biotechnol. 45, 23–47 (2025).
Bailey-Serres, J., Parker, J. E., Ainsworth, E. A., Oldroyd, G. E. D. & Schroeder, J. I. Genetic strategies for improving crop yields. Nature 575, 109–118 (2019).
Gionfriddo, M., Rhodes, T. & Whitney, S. M. Perspectives on improving crop Rubisco by directed evolution. Semin. Cell Dev, Biol. 155, 37–47 (2024).
Sharwood, R. E. Engineering chloroplasts to improve Rubisco catalysis: prospects for translating improvements into food and fiber crops. N. Phytol. 213, 494–510 (2017).
South, P. F., Cavanagh, A. P., Lopez-Calcagno, P. E., Raines, C. A. & Ort, D. R. Optimizing photorespiration for improved crop productivity. J. Integr. Plant Biol. 60, 1217–1230 (2018).
Sage, R. F., Sage, T. L. & Kocacinar, F. Photorespiration and the evolution of C4 photosynthesis. Annu Rev. Plant Biol. 63, 19–47 (2012).
Hennacy, J. H. & Jonikas, M. C. Prospects for engineering biophysical CO(2) concentrating mechanisms into land plants to enhance yields. Annu Rev. Plant Biol. 71, 461–485 (2020).
Barrett, J. et al. A promiscuous mechanism to phase separate eukaryotic carbon fixation in the green lineage. Nat. Plants 10, 1801–1813 (2024).
Fei, C., Wilson, A. T., Mangan, N. M., Wingreen, N. S. & Jonikas, M. C. Modelling the pyrenoid-based CO2-concentrating mechanism provides insights into its operating principles and a roadmap for its engineering into crops. Nat. Plants 8, 583–595 (2022).
Nguyen, N. D. et al. A carboxysome-based CO concentrating mechanism for C crop chloroplasts: advances and the road ahead. Plant J. 118, 940–952 (2024).
Fauser, F. et al. Systematic characterization of gene function in the photosynthetic alga Chlamydomonas reinhardtii. Nat. Genet. 54, 705–714 (2022).
Li, X. et al. A genome-wide algal mutant library and functional screen identifies genes required for eukaryotic photosynthesis. Nat. Genet. 51, 627–635 (2019).
von Caemmerer, S. & Furbank, R. T. Strategies for improving C4 photosynthesis. Curr. Opin. Plant Biol. 31, 125–134 (2016).
Furbank, R., Kelly, S. & von Caemmerer, S. Photosynthesis and food security: the evolving story of C4 rice. Photosynth. Res. 158, 121–130 (2023).
Chen, G. et al. Synthetic photorespiratory bypass improves rice productivity by enhancing photosynthesis and nitrogen uptake. Plant Cell 37, koaf015 (2025).
McGrath, J. M. & Long, S. P. Can the cyanobacterial carbon-concentrating mechanism increase photosynthesis in crop species? A theoretical analysis. Plant Physiol. 164, 2247–2261 (2014).
Chen, T. et al. Engineering α-carboxysomes into plant chloroplasts to support autotrophic photosynthesis. Nat. Commun. 14, 2118 (2023).
Long, B. M. et al. Carboxysome encapsulation of the CO2-fixing enzyme Rubisco in tobacco chloroplasts. Nat. Commun. 9, 3570 (2018).
Adler, L. et al. New horizons for building pyrenoid-based CO2-concentrating mechanisms in plants to improve yields. Plant Physiol. 190, 1609–1627 (2022).
Atkinson, N. et al. Rubisco small subunits from the unicellular green alga Chlamydomonas complement Rubisco-deficient mutants of Arabidopsis. N. Phytol. 214, 655–667 (2017).
Atkinson, N. et al. The pyrenoidal linker protein EPYC1 phase separates with hybrid Arabidopsis–Chlamydomonas Rubisco through interactions with the algal Rubisco small subunit. J. Exp. Bot. 70, 5271–5285 (2019).
Whitney, S. M., Baldet, P., Hudson, G. S. & Andrews, T. J. Form I Rubiscos from non-green algae are expressed abundantly but not assembled in tobacco chloroplasts. Plant J. 26, 535–547 (2001).
Giessen, T. W. & Silver, P. A. Engineering carbon fixation with artificial protein organelles. Curr. Opin. Biotechnol. 46, 42–50 (2017).
Jenkins, M. C. & Lutz, S. Encapsulin nanocontainers as versatile scaffolds for the development of artificial metabolons. ACS Synth. Biol. 10, 857–869 (2021).
Lagoutte, P. et al. Simultaneous surface display and cargo loading of encapsulin nanocompartments and their use for rational vaccine design. Vaccine 36, 3622–3628 (2018).
Lau, Y. H., Giessen, T. W., Altenburg, W. J. & Silver, P. A. Prokaryotic nanocompartments form synthetic organelles in a eukaryote. Nat. Commun. 9, 1311 (2018).
Moon, H., Lee, J., Min, J. & Kang, S. Developing genetically engineered encapsulin protein cage nanoparticles as a targeted delivery nanoplatform. Biomacromolecules 15, 3794–3801 (2014).
Szyszka, T. N., Adamson, L. S. R. & Lau, Y. H. in Microbial production of high-value products. (eds. B.H.A. Rehm & D. Wibowo) 309–333 (Springer International Publishing, Cham; 2022).
Giessen, T. W. et al. Large protein organelles form a new iron sequestration system with high storage capacity. eLife 8, e46070 (2019).
Buck, S. et al. Escherichia coli expressing chloroplast chaperones as a proxy to test heterologous Rubisco production in leaves. J. Exp. Bot. 74, 664–676 (2023).
Zhou, Y., Gunn, L. H., Birch, R., Andersson, I. & Whitney, S. M. Grafting Rhodobacter sphaeroides with red algae Rubisco to accelerate catalysis and plant growth. Nat. Plants 9, 978–986 (2023).
Whitney, S. M. & Sharwood, R. E. Construction of a tobacco master line to improve Rubisco engineering in chloroplasts. J. Exp. Bot. 59, 1909–1921 (2008).
Andersson, I. & Backlund, A. Structure and function of Rubisco. Plant Physiol. Biochem 46, 275–291 (2008).
Joshi, J., Mueller-Cajar, O., Tsai, Y. C., Hartl, F. U. & Hayer-Hartl, M. Role of small subunit in mediating assembly of red-type Form I Rubisco. J. Biol. Chem. 290, 1066–1074 (2015).
Martin-Avila, E. et al. Modifying Plant photosynthesis and growth via simultaneous chloroplast transformation of Rubisco large and small subunits. Plant Cell 32, 2898–2916 (2020).
Lin, M. T., Stone, W. D., Chaudhari, V. & Hanson, M. R. Small subunits can determine enzyme kinetics of tobacco Rubisco expressed in Escherichia coli. Nat. Plants 6, 1289–1299 (2020).
Zhou, Y. & Whitney, S. Directed evolution of an improved Rubisco; in vitro analyses to decipher fact from fiction. Int J. Mol. Sci. 20, 5019 (2019).
Holthuijzen, Y. A., Kuenen, J. G. & Konings, W. N. Activity of ribulose-1,5-bisphosphate carboxylase in intact and disrupted carboxysomes of Thiobacillus neapolitanus. FEMS Microbiol. Lett. 42, 121–124 (1987).
Patterson, D. et al. Encapsulation of Pseudomonas aeruginosa elastase inside the P22 virus-like particle for controlling enzyme–substrate interactions. Biotechnol. J. 17, 2200015 (2022).
Blayney, M., Whitney, S. & Beck, J. NanoESI mass spectrometry of Rubisco and Rubisco activase structures and their Interactions with nucleotides and sugar phosphates. J. Am. Soc. Mass Spectrom. 22, 1588–1601 (2011).
Mueller-Cajar, O. & Whitney, S. M. Evolving improved Synechococcus Rubisco functional expression in Escherichia coli. Biochem. J. 414, 205–214 (2008).
Mueller-Cajar, O., Morell, M. & Whitney, S. M. Directed evolution of Rubisco in Escherichia coli reveals a specificity-determining hydrogen bond in the form II enzyme. Biochem 46, 14067–14074 (2007).
Adamson, L. S. R. et al. Pore structure controls stability and molecular flux in engineered protein cages. Sci. Adv. 8, eabl7346 (2022).
Williams, E. M., Jung, S. M., Coffman, J. L. & Lutz, S. Pore engineering for enhanced mass transport in encapsulin nanocompartments. ACS Synth. Biol. 7, 2514–2517 (2018).
Nam, O. et al. A protein blueprint of the diatom CO(2)-fixing organelle. Cell 187, 5935–5950.e5918 (2024).
Tasneem, N., Szyszka, T. N., Jenner, E. N. & Lau, Y. H. How pore architecture regulates the function of nanoscale protein compartments. ACS Nano 16, 8540–8556 (2022).
Kwon, S., Andreas, M. P. & Giessen, T. W. Pore engineering as a general strategy to improve protein-based enzyme nanoreactor performance. ACS Nano 18, 25740–25753 (2024).
Zmyslia, M. et al. A nanoengineered tandem nitroreductase: designing a robust prodrug-activating nanoreactor. RSC Chem. Biol. 6, 21–35 (2025).
Szyszka, T. N. et al. High-fidelity in vitro packaging of diverse synthetic cargo into encapsulin protein cages. Angew. Chem. Int. Ed. Engl. 64, e202422459 (2025).
Wilson, R. H., Martin-Avila, E., Conlan, C. & Whitney, S. M. An improved Escherichia coli screen for Rubisco identifies a protein–protein interface that can enhance CO2-fixation kinetics. J. Biol. Chem. 293, 18–27 (2018).
Moore, S. J. et al. EcoFlex: a multifunctional MoClo kit for E. coli synthetic biology. ACS Synth. Biol. 5, 1059–1069 (2016).
Whitney, S. M. & Sharwood, R. E. Rubisco engineering by plastid transformation and protocols for assessing expression. Methods Mol. Biol. 2317, 195–214 (2021).
Whitney, S. M. & Sharwood, R. E. Linked Rubisco subunits can assemble into functional oligomers without impeding catalytic performance. J. Biol. Chem. 282, 3809–3818 (2007).
Acknowledgements
This research was supported by an Australian Research Council grant DP230101045. We thank students Riley Furbank and Daniel Bronitt for undergraduate experimental assistance on the project and the support of Dr Errin Johnson at Sydney Microscopy and Microanalysis. We further acknowledge the core facilities from Sydney Analytical for infrastructure support.
Author information
Authors and Affiliations
Contributions
T.N.S., S.M.W., and Y.H.L. conceptualised the study. T.N.S., D.S.W., S.M.W., and Y.H.L. designed the experiments. T.N.S. and D.S.W. performed the cloning and protein expression studies, with T.R. and N.P. assisting. T.N.S. performed the encapsulin purification experiments. D.S.W. performed the gel electrophoresis experiments, with T.R. and N.P. assisting. D.S.W. and S.M.W. performed the kinetic analyses. T.N.S., R.S., and A.L. performed DLS/DSF and TEM experiments for encapsulin characterisation. T.N.S., D.S.W., S.M.W. and Y.H.L. wrote the manuscript with input from R.S. and A.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ismael Bustos-Jaimes and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Szyszka, T.N., Wijaya, D.S., Siddiquee, R. et al. Reprogramming encapsulins into modular carbon-fixing nanocompartments. Nat Commun 16, 9493 (2025). https://doi.org/10.1038/s41467-025-65307-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65307-9
