
Abstract
The evolution of seeds transformed life on earth and is responsible for our most important food crops. Gymnosperms, the oldest living seed plants, are an untapped genomic reservoir for genes involved in seed evolution. To tap this resource, we assemble deep transcriptomes of 14 gymnosperms, four angiosperms, and two ferns and identified 22,429 phylogenetically informative ortholog groups. We observe that genes differentially expressed in ovules or leaves provide the majority of phylogenomic support for the evolutionary splits between 1) seed and non-seed plants; 2) gymnosperms and angiosperms; and/or 3) within gymnosperms (conifers vs. “ancient” gymnosperms). Our gymnosperm data identifies unreported candidate ovule regulated genes in Arabidopsis. Moreover, prior knowledge from Arabidopsis helps uncover 4,076 candidate ovule genes that influence these evolutionary splits. We validate the expression of candidate ovule genes in gymnosperm-specific ovule structures. Our work provides a resource for seed gene discovery, conservation, and crop improvement.
Similar content being viewed by others


Introduction
Seed-bearing plants make up the vast majority of plant species on Earth. Seeds constitute many of the staple foods for humans and livestock, including grains, legumes, and cooking oil. As of 2020, 38% of all global food production was derived from cereals and oil seeds, plus additional cropland dedicated to animal feed1. Thus, a deeper understanding of the genes that led to the evolution of seed structures may aid engineering or breeding for enhanced seed production and qualities.
The evolution of seeds from their ancestral spore-like morphologies enabled seed plants (gymnosperms and angiosperms) to thrive in diverse environments2,3. Non-seed plants, including ferns and lycophytes, develop a sporangium that releases single-celled spores that live independently from the sporophyte after dispersal. These spores develop into gametophytes, which in most species can produce both eggs and sperm (exceptions exist, such as Selaginella). By contrast, seed plants produce male microspores and female megaspores, with female gametophytes contained inside ovules attached to the sporophyte plant (Fig. 1A). After fertilization, the ovule develops into a seed4,5. The nucellus, the central part of the ovule, is analogous to the megasporangium and develops into an embryo with surrounding nutritive tissue, called endosperm in angiosperms. An integument surrounds the nucellus and develops into the seed coat. Whereas gymnosperms have one integument, most angiosperms contain two integuments surrounding the nucellus (Fig. 1A).

A Diagram illustrating homology between angiosperm ovule, gymnosperm ovule, and fern sporangium morphologies. i integument, ii inner integument, n nucellus, oi outer integument, s sporangium. B Species whose transcriptomes were assembled. C Female (seed plant) and homosporic (fern) reproductive structures of the twenty species in (B), in order from left to right, top to bottom. D Table showing Embryophyta_odb10 BUSCO scores (complete [C] + fragmented [F]) for each assembled transcriptome. E Table of final gene counts for each transcriptome.
Previous investigations into ovule development genes have predominantly focused on Arabidopsis and other model angiosperms6, rather than examining across seed plant groups. Ovule characteristics vary significantly across seed plants4,5,7 and some evolutionary splits between plant groups remain unresolved8,9. Supermatrices containing hundreds to thousands of genes have been employed to resolve the seed plant phylogeny9,10,11,12,13, though transcriptomes have only included data from single tissues, usually leaf12,13,14. Our prior study on genes driving seed plant evolution mainly examined genes under positive selection11. Gene expression has been integrated with species phylogenies before to obtain evolutionary insights into gene pathways14. However, the capacity of gene expression to inform phylogenies and relate genetic contributions to species divergence and structural evolution remains underutilized.
The relatively understudied clade of gymnosperms is a powerful model for seed evolution. Gymnosperms emerged during the lower Carboniferous period (~250 m.y.a.) and are likely the first extant group following the evolutionary split between seed and spore plants4. These species have relatively simple seed structures (gymnosperm means “naked seed”) borne directly on the plant without encasement in an ovary (Fig. 1A). Due to their large seed and ovule sizes15, gymnosperms have been instrumental in the discovery of seed development genes16,17. The low number of available genomic assemblies and annotations, however, hinders genetic mining of gymnosperms for insights into the evolutionary development of seeds.
To uncover genes that have driven the evolution of the ovule and its sub-structures, we mined deep transcriptomes of 14 gymnosperms in combination with angiosperms and ferns by integrating phylogenomics and gene expression. Of the 43,248 ortholog groups observed, we identified 4076 candidate ovule genes that influence the evolutionary splits between (1) seed and non-seed plants; (2) gymnosperms and angiosperms; and (3) within gymnosperms (conifers vs. cycads & Ginkgo). Our analysis and experimentation of gymnosperms reveal that these evolutionary splits are mainly supported by differentially expressed (DE) genes whose expression patterns and functions have been altered across plant groups.
Results
Phylogenomic reconstruction identifies 22,429 ortholog groups informing seed plant evolution
To identify genes that influenced seed evolution across plant groups, we collected leaf and reproductive (ovule or fern sporangia) tissues for deep RNA-seq analysis from 20 plant species, selected to span all major seed plant groups and the ferns (Fig. 1B, C, Supplementary Data 1). The seed plants include 14 gymnosperms (two Araucariales, five Cupressales, two Pinales, three Cycadales, Gnetum gnemon, and Ginkgo biloba) and four angiosperms (one monocot, Iris pseudoacorus; one eudicot, Arabidopsis thaliana, and two magnoliids, Liriodendron tulipifera and Piper nigrum) (Supplementary Data 2). For simplicity, genus names will signify the member species used in this study, unless otherwise indicated.
The twenty deep transcriptomes obtained for these species are highly complete, with combined BUSCO18 complete and fragmented scores ranging from 86.5% to 95.5% (Fig. 1D). For gymnosperms with published genomes, our transcriptome assemblies were close to or exceeded the BUSCO scores of their genome assembly counterparts (Supplementary Fig. 1). Gene sets obtained for each species ranged from 20,000 to 40,000 genes (Fig. 1E). We also assembled an expanded transcriptome of Taxus baccata, which is our gymnosperm experimental test system, using RNA extracted from leaf, ovule, green aril, and pollen tissues.
Most gymnosperm species in our dataset do not have published genome assemblies or annotations. Therefore, we used our revised PhyloGeneious pipeline19 (Supplementary Fig. 2) (see “Methods”) to identify the shared genes (orthologs) across these 20 species. Our analysis identified 43,248 ortholog groups in two or more species, with 1809 ortholog groups conserved across all 20 species, and 5934 groups shared between ferns and seed plants. Of the 37,314 seed plant-specific ortholog groups, 509 were conserved in all seed plants, 655 groups were specific to and conserved in all gymnosperms, and 2584 groups were specific to and conserved in all angiosperms (Supplementary Fig. 3). We also identified 1865 ortholog groups that were specific to the angiosperms and cycads, 470 of which were conserved in all angiosperms and 579 of which were only shared between cycads and Piper (Supplementary Fig. 4). Cycads had the most orthologs shared between angiosperms and any single gymnosperm order, followed by Gnetum which had 347 shared orthologs of which 169 were conserved in all angiosperms.
We then used ortholog groups present in four or more species to construct parsimony (Fig. 2A) and maximum likelihood20 (Supplementary Fig. 5) species trees for determining the genes driving phenotypic changes across plant groups. Ortholog groups consisted of one representative per species chosen from the species’ paralogs. This resulted in 23,525 ortholog groups used for phylogeny estimation, 22,429 of which were parsimony informative (i.e., containing at least one position in the ortholog protein alignment where a minimum of two different amino acid types were conserved in two or more species). The species relationships in our trees agree with previous literature findings10,11,21. The only conflict between our parsimony and likelihood trees is the placement of Gnetum as either basal to all gymnosperms or sister to the Pinales order, which also occurs in other studies9,10,11,12,13,21.

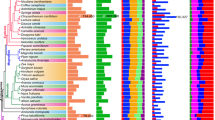
leaves of 20 species spanning ferns, gymnosperms, and angiosperms. A Parsimony total evidence phylogeny based on transcriptome assemblies of the 20 plant species, with 10,000 jackknife supports. Tree calculated from 22,429 ortholog groups. 1See Supplementary Data 7 for a list of ortholog groups. B Proportion of genes identified as leaf or OvS (ovule or sporangia) differentially expressed (DE) in each species (DESeq2, p-adj < 0.05, FC > 1.5). C Number of OvS DE genes identified in each species.
Basal seed plant divergences are primarily supported by differentially expressed ortholog groups
Conserved or changing expression patterns can inform how an ortholog group functions across species. As an initial step to classify candidate ovule development genes, we used differential expression to identify genes that were more highly expressed in seed plant ovule (Ov) or fern sporangia (S) tissues (from here referred to as OvS) relative to leaves (L) in each species. This yielded 53,248 OvS DE genes across all species, representing 19,250 ortholog groups (Fig. 2B, C). 7910 of these ortholog groups had OvS differential expression in two or more species. L differential expression included 58,560 genes across all species, representing 18,084 ortholog groups (Figs. 1E and 2B).
To determine whether ovule development genes were a major factor driving seed plant divergence, we compared our ortholog groups containing DE genes with the ortholog groups that were informative for our parsimony phylogeny (Fig. 3). We found that ortholog groups DE in any tissue (i.e., at least one DE member gene) were significantly more likely to be informative than random chance, with this being even more significant for groups DE in two or more species (Supplementary Fig. 7, Fig. 3B). Conversely, ortholog groups that contained no DE genes were significantly less likely to be informative (Fig. 3B, Supplementary Data 3). We identified a third category of ortholog groups, “Mixed DE”, where DE is found across multiple species but with regulatory divergence such that some species have L DE and other OvS DE. Ortholog groups DE in either tissue type (OvS, L, or Mixed) were also significantly more likely to have a higher number of informative positions and significantly more likely to have informative positions fall within predicted protein domains, suggesting functional relevance (Supplementary Fig. 8, Supplementary Data 3). Functional analysis of informative groups containing Arabidopsis orthologs found significant enrichment (p-adj < 0.05) for GO terms related to chloroplast and transcriptional regulation, while slightly less enriched terms (p-val < 0.01) included plant organ morphogenesis, flower and tissue development, and vesicle-mediated transport (Supplementary Data 4).

A Diagram of the three major evolutionary splits focused on in this study. i integument, ii inner integument, n nucellus, oi outer integument, s sporangium. B Numbers of all identified ortholog groups and ortholog groups informative to the species phylogeny, grouped by functional category. Significant p values of R pnorm test observation greater than random permutation (Methods): Mixed DE = 0, Conserved leaf DE = 1.03E-182, Conserved OvS DE = 2.86E-306. C Numbers of informative ortholog groups within the top 10% of absolute partition branch support values (|PBS|) for the component clades of each basal evolutionary split, grouped by functional category. Significant p values of R pnorm test observation greater than random permutation (Methods): Split 1 Conserved leaf DE = 9.05E-05, Split 1 Mixed DE = 4.84E-166, Split 2 Conserved leaf DE = 6.73E-06, Split 2 Mixed DE = 3.79E-211, Split 3 Conserved OvS DE = 2.31E-05, Split 3 Conserved leaf DE = 2.43E-04, Split 3 Mixed DE = 1.40E-167, All splits Conserved leaf DE = 0.000627, All splits Mixed DE = 4.59E-59. ***p value < 0.001; **p value < 0.005; *p value < 0.05. All p values are available in Supplementary Data 3.
We next examined how individual ortholog groups influenced species divergence at our three most basal evolutionary splits: (1) the split between seed and spore plants, (2) the split between gymnosperms and angiosperms, and (3) the split between cycad&Ginkgo and conifer gymnosperms (Fig. 3A). We achieved this by performing partition branch support22 (PBS) analysis with our parsimony tree, which measures the amount of support in amino acid changes each data partition (ortholog group) provides to a given evolutionary clade in the tree calculation. We found that several of the ortholog groups with the strongest PBS values for each split corresponded to flowering and reproductive development genes (Supplementary Data 5). A broader look also revealed that the majority of the ortholog groups highly supporting these evolutionary splits had Mixed DE supporting evolutionary splits at a much higher proportion than random chance (p-val <<< 0.01) (Fig. 3C, Supplementary Data 3). The particularly high enrichment of mixed DE genes as informative to clade divergences suggests that both sequence and regulatory divergence underlie evolutionary splits.
Integrating phylogenomic support and gene expression uncovers previously validated and candidate genes involved in seed development
To uncover the genes that have driven seed evolution across species, we integrated our phylogenomic and evolutionary findings with gene expression and compared our evolutionary informative ortholog groups against prior knowledge of genes involved in ovule development. Below, we describe three different strategies for integrating these two knowledge sources: (i) mapping differential expression patterns to orthology and phylogeny, (ii) utilizing known ovule genes to identify related orthologs, and (iii) characterizing ortholog groups with strong evolutionary influence and evidence of ovule-related function. We highlight some of our key discoveries obtained from each strategy.
Genes with ovule differential expression in many species strongly support seed divergence from spores
To test whether combining phylogenomics and expression could identify genes of importance to seed evolution, we started looking for ortholog groups implicated in ovule-related function in a majority of species. Most ortholog groups with conserved OvS differential expression were only conserved in a few species, with only 10 groups containing OvS DE genes from ten or more species (Fig. 4A). Four of the most frequently ovule DE ortholog groups included Arabidopsis genes: ACOS5/4CLL1, CYP704B1, DRL1/TKPR1, and CYP703A2 (Fig. 4A). These four genes all have documented roles in angiosperms in the formation of exine, the outer layer of the pollen wall, and prior experimental evidence suggests these proteins interact23,24. All four exine ortholog groups support the seed|spore evolutionary split (Fig. 4B) and slightly influence the split of cycad&Ginkgo in the conifers (Fig. 4B, C). The ACOS5 ortholog also strongly supports the separation of Gnetum from the other gymnosperms, with a PBS value of 11 (Supplementary Data 6). These pollen gene orthologs are highly DE in all fern sporangia and most gymnosperm ovules yet show little differential expression in angiosperms (with the exceptions of the Iris CYP704B1 ortholog in ovules and the Zamia CYP703A2 ortholog in leaves) (Fig. 4C). The Taxus orthologs of all four genes also show similar or higher expression levels in pollen relative to ovules in our expanded transcriptome. This gymnosperm-specific expression pattern matches prior morphological observations that the gymnosperm megaspore wall is similar to pollen exine15,25, while angiosperm megaspore walls are absent or highly reduced26,27 (Fig. 4D).

A Counts of ortholog groups with conserved ovule or sporangia (OvS) differential expression (DE), grouped by OvS DE frequency across species. Boxes highlight all ortholog groups containing Arabidopsis that were DE in nine or more species. Bolded names are Arabidopsis genes involved in pollen wall development. B Partition branch support (PBS) contributions of four exine development ortholog groups to the seed|spore (1), gymnosperm|angiosperm (2), and cycad&Ginkgo|conifer (3) evolutionary splits. 1 = seed plant clade, gym gymnosperm clade, ang angiosperm clade, cyc cycads&Ginkgo clade, con conifer clade. Percentage of partition supports for given clades with equal or greater magnitude: Split 1 CYP704B1 = 1.63%, Split 1 CYP703A2 = 0.91%. *p value ≤ 5%, **p value ≤ 2%, ***p value ≤ 1%. C OvS differential expression patterns across species of four exine development ortholog groups. D Diagram of spore wall conservation in gymnosperm and angiosperm megaspores. ms megaspore, msw megaspore wall, sp spore, sw spore wall.
The second ortholog group observed as ovule DE in thirteen species included Ginkgo GbMADS8, one of two gymnosperm-specific duplications of Arabidopsis AGL6 that is expressed throughout the developing ovule28 (Supplementary Fig. 9). GbMADS8 strongly supports the cycad&Ginkgo|conifer split. The PRX17 and RAD50 ortholog groups that were ovule DE in nine species are also known to have roles in reproductive development in Arabidopsis. Overall, these results demonstrate that integrating orthology with cross-species expression data yields key insights into conserved developmental pathways.
Broader conservation of known angiosperm ovule genes in gymnosperms than reported
To assess our method’s ability to recover established seed development genes, we searched our ortholog groups for 39 known ovule development genes6 and 480 validated embryo lethal (”seed”) genes29 from Arabidopsis (Fig. 5A) (Supplementary Data 8). Our pipeline found orthologs in other species for 26 of the known ovule genes and 405 of the known seed genes (Supplementary Data 9).

A Diagram of phylogenomic and gene expression integration with prior knowledge of seed genes. Reproductive GO annotations were required to have IMP (inferred from mutant phenotype) evidence. B Bar chart of ovule genes identified in gymnosperms using Arabidopsis annotations vs. annotation of reproductive genes in Arabidopsis based on ovule differential expression in gymnosperms.
Our analysis identified some unexpected orthology within the type II MADS-box gene family, whose members are frequently reported to play roles in reproductive development. SEPALLATA (SEP) is well-known as one of the major ovule-identity determinators in angiosperms30, forming a complex with AGAMOUS, SHATTERPROOF 1&2, and SEEDSTICK31. SEPALLATA has also been documented as exclusively found in angiosperms32,33,34. While paralogs SEP1, 2, and 4 appear to be specific to Arabidopsis in our analysis, we found SEP3 orthologs in all four angiosperm species as well as in two gymnosperm cycads, Zamia and Stangeria (Supplementary Fig. 10). These cycad homologs are highly similar to the angiosperm genes in the sequence alignment, although the Stangeria homolog lacks a portion of the conserved MADS-box domain (Supplementary Fig. 11), and running BLAST searches of both cycad sequences in the NCBI nr database only yielded matches to SEP3 sequences from other plant species. The SEP ortholog group was informative to our species tree (though not a high supporter of the major evolutionary splits), and 10 of the 27 informative sites occurred within the conserved K-box domain. All angiosperm SEP3 orthologs were DE and highly expressed in ovules, while the cycad SEP3 orthologs were expressed at very low levels, with the Zamia ortholog observed exclusively in ovules. We did not observe a SEP3 ortholog in our Cycas transcriptome, and searching the Cycas panzhihuaensis genome35 also found no match. Among the other four genes that complex with SEP, most gymnosperm orthologs were distantly related, although Zamia had an additional ortholog that clustered directly with SEEDSTICK (Supplementary Fig. 12). There were also several clades of gymnosperm-specific MADS-box genes.
Our pipeline further identified gymnosperm orthologs in other ovule gene groups known for angiosperm-specific functions, such as HECATE (Supplementary Data 9). HECATE (HEC) is a group of bHLH transcription factors known to be involved in the development of the carpel and funiculus, structures that form the angiosperm ovary and connect the ovule to it36. Only HEC1 and HEC2 have been previously reported in gymnosperms37. We identified a gymnosperm-specific HECATE clade present in ten species, including cycads and conifers (Supplementary Fig. 13). These gymnosperm HECATEs were ovule DE in Ginkgo and three conifer species (Supplementary Fig. 13), and the group was informative to our species tree (Fig. 2A, Supplementary Data 9). MEME38 motif analysis also identified two unique protein motifs occurring in the conifer HECATE orthologs (Supplementary Fig. 14). RNA in situ hybridization of the Taxus HECATE homolog yielded no results due to low expression.
Other notable ovule gene findings included the discovery of 8 possible gymnosperm orthologs to INNER NO OUTER (INO), a gene required for the development of the angiosperm-specific outer integument39, along with orthologs of the ovule-expressed Ginkgo YAB1B28 in 11 additional gymnosperms and a Zamia ortholog of the angiosperm-only YABBY 528 (Supplementary Fig. 15). We also uncovered orthologs of WUSCHEL (WUS), a major regulator of the stem apical meristem and ovule development40, in seven gymnosperm species (Supplementary Fig. 16). Finally, we found six conifer orthologs of STIMPY/WOX9, a known regulator of INO and WUS in ovule patterning41. Both Sciadopitys WUS and STIMPY orthologs are also ovule DE (Supplementary Fig. 17).
Lastly, we further interrogated the power of our gymnosperm data to enhance the annotation of ovule-related genes in the model Arabidopsis. We identified 5437 Arabidopsis genes that had orthologs in multiple gymnosperms (Supplementary Data 10), then combined ovule differential expression with existing Arabidopsis reproductive annotations (Fig. 5A). Both Arabidopsis and gymnosperms were able to uniquely annotate genes from other species. Our gymnosperm expression data annotated the most Arabidopsis genes as ovule-related, the vast majority of which (1224) were genes not identified by existing Arabidopsis reproductive annotations (Fig. 5B, Supplementary Data 11) from a curated set of GO-term annotations. For these orthologs with known expression patterns in Arabidopsis, 77.7% were expressed in flowers, and more than 60% were expressed in other reproductive tissues (Supplementary Data 12). In addition, 96.9% were expressed during petal differentiation and expansion (enrichment p-val < 0.05), 91.0% were expressed during the embryo cotyledonary stage (enrichment p-adj < 0.05), and 88.8% were expressed in mature plant embryos (enrichment p-adj < 0.05) (Supplementary Data 13). GO term analysis found two of the 1224 ortholog groups were annotated for embryo and post-embryonic development (enrichment p-adj < 0.05), and one group was annotated for meiotic cell cycle (enrichment p-val < 0.01). There was also significant GO enrichment (p-adj < 0.05) for chloroplast and mild enrichment (p-val < 0.01) for GO terms related to plastids, organelle envelope, phosphorylation, ubiquitination, positive regulation, mRNA binding, DNA metabolism, anatomical morphogenesis, organophosphate and thiamine metabolism, hydrolase activity, and response to water deprivation (Supplementary Data 14). PFAM enrichment analysis showed mild enrichment for PPR and leucine-rich repeats among individual domains and mild enrichment for WD proteins and S33 serine aminopeptidases when examining domain combinations. There was also a general prevalence of protein kinases, DEAD/DEAH box helicases, F-box proteins, TPR repeats, and RNA recognition motifs (Supplementary Data 15 and 16).
4076 candidate ovule genes greatly influence major evolutionary splits
Our next goal was to leverage Arabidopsis prior knowledge in conjunction with our phylogenomic and expression data for the discovery of candidate ovule genes. We identified 15 ovule6 and 102 seed29 gene ortholog groups conserved in 15 or more species and used their differential expression pattern (log2 fold change) across species to search for additional ortholog groups with correlated expression (Fig. 5A) (Supplementary Fig. 18). This comparison approach also controls for potential differences in the ovule developmental stages collected across species. Our analysis identified 2396 highly correlated ortholog groups, with 42 groups correlated to known ovule and known seed genes, 29 groups correlated only to known ovule genes, and 2325 groups correlated only to known seed genes. In many cases, these correlated groups included genes with known ovule or reproductive functions; for example, the known ovule gene ADA2B/PRZ1, a histone acetyltransferase regulator required for proper integument development in angiosperms. This analysis also identified known reproductive development genes, including the mRNA processing gene RDM1642, the meiotic DNA repair gene RAD50, and multiple embryo-lethal mutants as correlated ortholog groups (Supplementary Fig. 19, Supplementary Data 17).
We now sought to fully integrate our evolutionary findings with functional evidence to identify genes with the largest impact on ovule evolution. To accomplish this, we specifically focused on ortholog groups that had high PBS scores for one of the major splits, as well as support for ovule-related function from expression and/or prior knowledge evidence (Fig. 6A). These criteria identified 4076 candidate ortholog groups associated with ovule evolution, of which 2267 support one basal evolutionary split, 1211 support two evolutionary splits, and 598 give high support for all three splits (Fig. 6B). These candidate ortholog groups included three of the known Arabidopsis ovule genes6 (HLL, ADA2B, and SEUSS) and 188 of the known seed genes29, as well as 397 other Arabidopsis genes annotated with reproductive roles (see “Methods”). Most of the candidate ortholog groups supported either (i) only the cycad&Ginkgo|conifer split, (ii) the seed|spore and gymnosperm|angiosperm splits, or (iii) only the gymnosperm|angiosperm split(Fig. 6B). Interestingly, very few orthologs only supported the seed|spore split, and none of those groups relate to known ovule genes (Fig. 6B). To further confirm the quality of our PBS scores for identifying valuable evolutionary candidates, we looked for ortholog groups with significant positive selection in seed plants relative to ferns and identified 557 orthologs, 315 of which were included in our candidate ortholog groups (Supplementary Data 18 and 19).

A Flowchart illustrating the phylogenomic and gene expression criteria used to identify candidate ovule development genes. B Upset plot of ortholog groups that support at least one major evolutionary plant split and have expression and/or phylogenetic evidence for influencing ovule development, with overlaps showing supporting evidence. The proportion of ortholog groups in each intersect with known Arabidopsis reproductive genes (Fig. 5A) is indicated in purple. Values on the left indicate the total number of candidate ortholog groups supporting the indicated combination of splits. On the far right, known ovule development genes1 are highlighted in purple, and candidate orthologs we further investigate here are highlighted in black. (1) = seed|spore split, (2) = gymnosperm|angiosperm split, (3) = cycad&Ginkgo|conifer split. 1Gasser and Skinner, 20196, 2Meinke, 202029, 3see Arabidopsis prior knowledge sources in Fig. 5A.
To learn the types of candidate genes that drove ovule evolution, we performed functional enrichment of PFAM (protein family) domains for the candidates supporting each combination of the three evolutionary splits. Enrichment was performed both for single domains (Supplementary Data 20) and for co-occurring domain groups (Supplementary Data 21). Candidate ortholog groups supporting the seed|spore and gymnosperm|angiosperm splits were highly enriched for WD proteins, DEAD/DEAH box helicases, cytochrome P450s, and Rubisco, indicating roles in the early divergence of the seed plants (Supplementary Data 22). DEAD/DEAH box genes were also enriched in candidates only supporting the gymnosperm|angiosperm and cycad&Ginkgo|conifer splits, while cytochromes were additionally enriched in candidates only supporting the cycad&Ginkgo|conifer split. PPR proteins were also frequently enriched among candidates supporting all three splits, solely the gymnosperm|angiosperm and cycad&Ginkgo|conifer splits, or only the cycad&Ginkgo|conifer split, possibly reflecting their expansion within the seed plants43. Single domain enrichment further identified TPR and SET domains among seed|spore and gymnosperm|angiosperm split supporters, F-box domains among groups supporting all splits or only the cycad&Ginkgo|conifer split, and ankyrin repeats among groups supporting all splits or solely the gymnosperm|angiosperm and cycad&Ginkgo|conifer splits. These candidate orthologs (Fig. 6, Supplementary Data 23) are a treasure trove for discovering uncharacterized ovule genes and dissecting the evolutionary history of previously identified genes.
Validation of candidate ovule genes reveals changing regulatory programming in gymnosperms
Having identified several genes as candidate drivers of ovule evolution, we next sought to confirm whether these genes’ support for evolutionary splits corresponded to changing roles in ovule development in non-model species. To test this, we chose a gymnosperm-specific BELL ortholog group containing the Gnetum gene MELBEL1, which strongly supported our cycad&Ginkgo|conifer split (Fig. 6B: Split 3) (Supplementary Fig. 20). Gnetum MELBEL1 is expressed in ovules throughout the developing nucellus44, while the Ginkgo homolog GibiBEL1-2 has expression more restricted to the megaspore mother cell and ovule base28,45. However, MELBEL1 has not yet been characterized in conifers. Our MELBEL1 ortholog group was ovule DE in five of fourteen gymnosperms, all conifers (Supplementary Fig. 20), and leaf DE in Gnetum. We also examined a neighboring uncharacterized gymnosperm-specific BELL group, here dubbed GYMNOSPERM BELL 2 (GBEL2), which was ovule DE in seven of thirteen gymnosperms and not observed in Gnetum.
In order to validate the role of these gymnosperm-specific BELLs in conifer ovules, we performed in situ hybridization of MELBEL1 and GBEL2 orthologs in the conifer Taxus baccata in three stages of developing ovules and young vegetative tissue (Fig. 7). Ovules of Taxus species are notable for the fleshy fruit-like aril that surrounds the seed (Fig. 7B); fused arils are also observed in Cephalotaxus species while this structure is absent in all other gymnosperm groups46 (Fig. 7A).

A Representation of Taxus and Cephalotaxus aril morphology relative to other seed plants. B Photographs of aril development in Taxus baccata, the species used to validate the pipeline. C–M In situ hybridization. Imaged sections of Taxus baccata ovules. Purple color indicates the presence of RNA. TbMELBEL1 expression in shoot apical meristem (C), Stage 1 ovule (D), Stage 3 ovule (E), Stage 4 ovule (F), and Stage 5 ovule (G), from six technical replicates. Arrows indicate the megaspore mother cell. TbGBEL2 expression in Stage 1 ovule (H), Stage 3 ovule (I), Stage 4 ovule (J), and Stage 5 ovule (K), from six technical replicates. Expression of control sense probes in young shoot and ovule sections for TbMELBEL1 (L) and TbGBEL2 (M), from three technical replicates. a aril, b bract, i integument, ms megaspore, n nucellus, sam shoot apical meristem. Scale bars (C–M) = 200 µm.
Our in situ experiments showed that TbMELBEL1 was expressed throughout the shoot apical meristem and adjacent leaf primordia in young vegetative tissue (Fig. 7C). In Stage 1 ovules, TbMELBEL1 was strongly expressed throughout the nucellus (Fig. 7D). In Stage 3 ovules, TbMELBEL1 was expressed throughout the nucellus and in the interior of the integuments, as well as throughout the developing aril (Fig. 7E). TbMELBEL1 was also very highly expressed in the megaspore mother cell (Fig. 7F). In Stage 5 ovules, TbMELBEL1 was expressed throughout the developing megaspore and the cone bracts, but no longer observed in the arils (Fig. 7G). To confirm the tissue-specific expression of TbMELBEL1, we observed this gene’s quantified expression in the expanded transcriptome of leaf, ovule, green aril, and pollen tissues. The expanded Taxus transcriptome confirmed that TbMELBEL1 is highly expressed in ovules and green arils, with lower expression in pollen and leaves.
The previously uncharacterized TbGBEL2 was highly expressed throughout the ovule primordia in Stage 1 ovules based on in situ hybridization (Fig. 7H). In Stage 3 and 4 ovules, TbGBEL2 was expressed in the nucellus, with expression in the developing aril observed in some but not all sections (Fig. 7I, J, Supplementary Fig. 21). No further expression was observed in the integument. In Stage 5 ovules, TbGBEL2 was expressed throughout the nucellus and developing megaspore (Fig. 7K). Our expanded Taxus transcriptome also showed TbGBEL2 as highly expressed in ovules and green arils, with much lower expression in leaves and pollen. No expression was observed in the sense probe controls for either TbMELBEL1 or TbGBEL2 (Fig. 7L, M).
These localization results highlight the role of TbMELBEL1 and TbGBEL2 in Taxus ovule development, and support our evolutionary prediction that MELBEL1 function changed in conifers relative to cycads&Ginkgo.
Discussion
Our study assembled what is, to our knowledge, the largest collection of gymnosperm ovule transcriptomes to date. This informational resource spanning all major seed plant clades enabled us to identify a large number of previously uncharacterized orthologs involved in ovule development, including gymnosperm orthologs of gene groups previously reported to only be found in the angiosperms, and to analyze their contribution to the evolution of these species. Our identification of unreported candidate ovule genes in Arabidopsis via our gymnosperm expression data, and the validation that many of these genes are reported as expressed in Arabidopsis reproductive tissues, both highlight the ability of our dataset to generate key insights and demonstrate the limitations of relying on annotations from a single species for understanding broad species groups.
The division between ferns and seed plants involves not only the development of the ovule but also the transitions to heterospory and endospory. While there are some heterosporous model non-seed plants such as Selaginella moellendorfii, the heterospory trait has evolved independently in multiple plant lineages4, and the evolutionary drivers behind this trait in other lineages may not be the same as those in seed plants. Further transcriptomic resources within the ferns and basal seed plants may provide insights into the molecular mechanisms underlying the independent emergence of heterospory.
Due to difficulties in sampling and long life cycles, cycad species have been frequently excluded from evolutionary comparisons of the land plants, particularly when comparing gymnosperms and angiosperms. This can be seen in the multiple prior studies that erroneously documented the SEPALLATA clade as exclusive to angiosperms32,33,34. However, we identified hundreds of ortholog groups that were exclusively expressed in the cycads and angiosperms, more than any other gymnosperm order, especially among Zamia and Stangeria (Supplementary Fig. 4). This finding cements the status of the cycads as one of the oldest gymnosperm clades, and highlights the importance of sampling all gymnosperm clades when performing comparative analyses across seed plants. Our ortholog groups also suggest there may have been ancestral gene losses within the Cycas genus, indicating a need to sequence additional cycad genomes. Our discovery of broader conservation of ovule development genes between gymnosperms and angiosperms, particularly among MADS-box genes, corroborates previous observations of shared genetic programming between flower and ovulate cone structures32,47.
Our maximum likelihood tree supports the placement of Gnetum as sister to the Pinales (“gnepine”); this placement is a frequently supported hypothesis among recent phylogeny studies9,10,12,13. However, our parsimony analysis placed Gnetum as sister to the other gymnosperms. This gymnosperm-sister hypothesis is also supported by other total evidence and gene group studies11,21. Even studies that favor the gnepine hypothesis have noted a large number of conflicting sites supporting alternate placements9,10,48. We do not believe the exclusion of Gnetum from our cycad&Ginkgo|conifer split branch support analysis greatly affects our identification of orthologs broadly changing between the two plant groups. Gnetum also shared a fair number of ortholog groups solely with angiosperms, which could suggest ancient ancestry, convergent evolution9, or horizontal gene transfer49.
We observed through integrating phylogenetic support and expression analysis that genes DE between ovules and leaves have a disproportionately strong influence on our total evidence species phylogeny. Remarkably, ~37% of the conserved ortholog space drives more than 85% of the basal evolutionary changes distinguishing major plant groups (Fig. 8). This finding strongly suggests that it is not just evolving gene sequences, but also changes in expression of these functional genes, that are driving the phenotypic diversity across seed plants. The strength of this phenomenon may in part be due to the broad evolutionary scale we examine; many of the species in our phylogeny are separated by millions of years of evolution. Moreover, even “closely related” genera in our phylogeny, such as Taxus and Cephalotaxus, are distinguished by many morphological differences (Fig. 1C). We also found that the vast majority of parsimony informative positions identified in our ortholog groups fell within predicted protein domains (Supplementary Fig. 8), potentially indicating that conserved protein domains are undergoing more positive selection across the species under study compared to non-conserved protein regions. However, this observation is likely biased by the conserved protein domains aligning the best across the different orthologs, and therefore most likely to meet the informative requirements. It is also possible that at these broad time scales, neutrally evolving amino acid sites are more likely to appear as randomly switching in the protein alignments and therefore less likely to impart phylogenetic information. The enrichment of regulatory and plant organ morphogenesis GO terms among genes informative to our phylogeny was not surprising, given the large number of morphological differences between the species in our study.

Result: ~37% of the identified ortholog space drives more than 80% of phylogenomic support, particularly for orthologs with different tissue expression in leaves vs. ovules/sporangia across species.
The current study also identified pollen exine genes supporting the split between seed and spore plants, which were highly up-regulated across gymnosperm ovules (Fig. 4). This evolutionary split corresponds not just to the development of the ovule but also to the transition from a single spore type to separate male and female spores. These genes support the existence of common developmental pathways between the megaspore wall and pollen exine, and indicate that gene pathways involved in ovule and pollen development may be less distinct in gymnosperms than angiosperms. Orthologs of the pollen gene CYP703A2 have been shown to impact silique development in Arabidopsis24. The other four exine genes do not have prior reported roles in female reproductive development. While in most angiosperms pollination and fertilization occur in rapid succession (<48 h in A. thaliana36), in gymnosperms these events are separated by a time gap ranging from a few months up to 2 years15. It is possible that some of the young gymnosperm ovules we collected RNA may have been pollinated, but not yet fertilized, leading to pollen contamination in our ovule transcriptomes. However, we find it unlikely that we would observe such a consistent differential expression trend across species if this were the sole cause of these transcripts, especially when so few ortholog groups had consistent expression patterns. Other reproductive genes are also expressed in both male and female gymnosperm cones47,50. Angiosperm-specific tissue subfunctionalization is already reported for the ovule gene WUS40,51, which we identified, supporting the cycad&Ginkgo|conifer split. WUS-like duplications have also been reported in multiple conifer species, although the paralog function is not well characterized51.
The PBS support patterns among our candidate ovule genes indicate that orthologs that drove ancient evolutionary splits, such as between seed and spore plants, are more likely to contribute to later evolutionary splits (Fig. 6). By contrast, more recent evolutionary splits are more likely to include exclusive supporters; for the cycad&Ginkgo|conifer split, this seems primarily due to the evolution of lineage-specific genes within gymnosperms. The large proportion of ortholog groups with mixed differential expression supporting our basal evolutionary splits suggests that many of the morphological differences observed between plant groups are due to gene regulation. Thus, there may be additional genetic changes within noncoding regulatory regions that would not be captured by RNA sequencing. These observations indicate there is yet a wealth of information on the evolution of seed structures contained within the extant gymnosperms that remains to be tapped.
We believe our study provides a wealth of uncovered candidate genes for hypothesis testing of their roles in seed development. One example of this validation is our study of the in situ expression of the TbMELBEL1 and TbGBEL2 genes in ovule development in Taxus (Fig. 7). The ovule expression of TbMELBEL1 we detected in Taxus largely coincided with the expression patterns reported in Gnetum44. Specifically, the high expression of TbMELBEL1 in shoot apical meristems indicates it may play other developmental roles outside of reproduction, while TbGBEL2 expression appears to be important throughout Taxus ovule development (Supplementary Fig. 21). The expression of both orthologs in the early aril (Fig. 7, Supplementary Fig. 21) supports that the regulation and/or functions of these two transcription factors were modified for the development of this unique ovule structure in Taxus. This observation suggests that additional ovule organ structures can develop via adaptation of existing ovule development pathways. In general, BELL genes have been shown to be critical for proper egg development across land plants, including in mosses and bryophytes45, and BEL1 regulates ovule genes WUS and INO in Arabidopsis52. Our study indicates that further ovule developmental roles are yet to be uncovered.
Our unique approach of combining phylogenomic and gene expression data unveiled genetic trends associated with the evolution of the ovule, both at the broad scale (seed vs. spore plants) and within individual plant groups (gymnosperms). This approach can also be used with other species to elucidate genes driving major evolutionary adaptations. Our combined results provide a vast community resource for candidate ovule gene discovery, as well as expanding knowledge for known reproductive genes.
Methods
RNA collection and de novo transcriptome assembly
Young leaf and young ovule or fertile leaf tissues were collected for Iris pseudacorus, Piper nigrum, Liriodendron tulipifera, Agathis macrophylla, Podocarpus matudae, Callitropsis nootkatensis, Metasequoia glyptostroboides, Cephalotaxus sinensis, Taxus baccata, Sciadopitys verticillata, Pseudotsuga menziesii, Tsuga canadensis, Gnetum gnemon, Ginkgo biloba, Cycas rumphii, Zamia furfuracea, Stangeria eriopus, Adiantum capillus-veneris, and Ceratopteris richardii. Collection details and accession IDs are in (Supplementary Data 1 and 25). We did not have access to comparable tissue samples from any basal angiosperms to include in this work. Additionally, our review of published RNA-seq datasets for these species failed to find samples compatible with our young leaf and ovule tissue comparison. RNA extractions were completed according to the protocols listed in Supplementary Data 25. The Qiagen RNEasy Plant Extraction kit was used with the following modifications: (1) adding 100 μL of 10% PEG 4000 per 1 mL of either RLT or RLC Buffer; (2) adding 0.025 g PVP 40 to the RLT/RLC-PEG solution (CAT ID:74104). RNA Quality was checked using Qubit and Tapestation assessments. For all species except Ceratopteris richardii, RNA libraries were processed using either the Kapa mRNA Hyper Prep kit or the Epicenter Scriptseq v2 RNA seq Library Kit with Ribozero (Supplementary Data 25). Libraries were sequenced at Cold Spring Harbor Laboratory with Illumina NextSeq 500 paired-end 150 sequencing (Mid Output). Ceratopteris richardii RNA libraries were processed and sequenced by Novogene Co., Ltd with Illumina paired-end 150 sequencing. The majority of species have three samples from leaf and ovule or fertile leaf, with the exception of Ginkgo (one leaf). Only two Stangeria leaf samples were used for expression analyses, as the expression profile of leaf sample L1 (93814-P) clustered separately from the other samples (Supplementary Fig. 22).
For the expanded tissue Taxus baccata RNA-seq, young leaves, ovules, pollen cones, and dissected young (green) arils were collected from Taxus baccata (accessions 1168/41*C and accession 1194/41*A) at the New York Botanical Garden. Taxus RNA was extracted from ovules with modification of the QIAGEN RNeasy mini-kit (Qiagen, Hilden, Germany) with β-mercaptoethanol53. Four different tissues were processed for a total of 12 samples for sequencing. The total RNA was extracted as previously described, and the quality of the total RNA was assessed using Qubit2.0 (Thermo Fisher Scientific) and Agilent Technologies 2100 Bioanalyzer. For the preparation of the sequencing libraries, only good quality and undegraded total RNA was used(Ratio A260/A280 ≈ 2 and RIN ≥ 8). RNA-seq libraries were prepared using NEBNext Poly(A) mRNA Magnetic Isolation Module Library Prep Kit (New England Biolabs). The resulting libraries were paired-end (PE) sequenced (2 × 150 bp) using an Illumina HiSeq2000. The average sequencing depth for each sample was 40 million reads.
Read quality was assessed with FastQC v0.11.9 [http://www.bioinformatics.babraham.ac.uk/projects/fastqc/] and reads were trimmed using a custom trimming script trimAdapterAndQualityPE_082416.py (see supp files), which removes Illumina adapter sequences, low quality 3′ bases (if the mean of a 5 bp sliding window drops below Phred score of 20), and trimmed reads smaller than 25 bp. Contigs were assembled using Trinity54 v2.15.0 using default parameters, and protein predictions were made with Transdecoder55 v5.5.0 using --retain_pfam_hits for PFAM domains identified by hmmscan. CD-Hit-EST56,57 v4.8.1 was run with parameters -T 0 -c 0.98 -G 0 -aS 1 -n 8 -r 1 -g 1 to cluster and remove redundant sequences. Non-plant sequences were identified and removed by running BLASTX58 searches against the NCBI nr database with the PhyloGeneious pipeline script qsblastg.pl. Protein sequences were finally filtered to only keep the longest isoform for each gene.
Arabidopsis thaliana transcriptome assembly
Arabidopsis transcripts for ovule, young leaf, and young petiole were used from Klepikova et al.59. Accession files SRR3581388, SRR3581838, SRR3581383, SRR3581837, SRR3581727, and SRR3581889 were downloaded from the NCBI Sequence Read Archive (SRA) database [https://www.ncbi.nlm.nih.gov/sra/] with sra-tools v2.10.9. The Arabidopsis thaliana reference genome Araport11_20210660 was downloaded from TAIR61, and reads were mapped to the reference with HISAT262 v2.2.1 and samtools63 v1.12. Read counts were obtained with stringtie64 v2.1.6.
Gene annotation
BUSCO18 v5.0.0 was run with Embryophyta_odb10 with predicted protein sequences to get completeness estimates for each transcriptome.
InterProScan65 (v5.65-97.0) was run individually for each species to obtain predicted protein domains and annotations, using the following parameters:
interproscan.sh -i species.faa --seqtype p -T assemblies/interpro -f TSV,JSON,GFF3 --goterms --iprlookup --pathways.
Our list of known Arabidopsis ovule development genes was obtained from Gasser & Skinner 20196, and our list of known Arabidopsis embryo lethal (“seed”) genes was obtained from Meinke 202029 (Supplementary Data 8). To identify additional Arabidopsis genes with known reproductive roles, gene ontology (GO) terms, GO slim terms, evidence codes, gene symbols, and gene descriptions were obtained using TAIR61 Bulk Data Retrieval tools [https://www.arabidopsis.org/tools/bulk/index.jsp]. Genes were filtered for IMP (Inferred from Mutant Phenotype) evidence codes and GO or GO slim annotations containing at least one of the following strings: “reprod,” “ovule,” “flower,” “pollen,” “pollination,” “gamet,” “sporangi,” “megaspor,” “sepal,” “polar nucl,” and “generative cell.” For known ovule and seed genes without TAIR gene symbols, gene symbols and descriptions were annotated manually.
Additional BLAST searches of individual protein sequences to identify specific known genes or confirm annotations were conducted using BLAST+ v2.15.058.
Orthology and maximum parsimony tree
We used a modified version of our PhyloGeneious pipeline19 for our evolutionary inferences across species (Supplementary Fig. 2). OrthologID (Supplementary Fig. 2, top) was used to identify shared ortholog groups across the different species. To summarize, protein sequences from all species were pooled and then organized into unsupervised “gene family” clusters based on BLAST similarity scores. Gene trees were then calculated for each family cluster, and ortholog groups were defined from the branches. Ortholog groups were then reduced to 1:1 representations by randomly selecting one representative paralog per species present for each group. These 1:1 ortholog groups were then filtered for parsimony informative positions, which were concatenated into a partitioned sequence matrix, with each partition corresponding to a different group. The parsimony species tree was calculated using this matrix along with jackknife support from 10,000 replicates. An orthology matrix was generated using the PhyloGeneious script generate_pres_abs_matrix.py, and the data was imported into R and plotted with the heatmap.2 function from gplots (see supp files).
Maximum likelihood tree
A maximum likelihood tree was calculated with RAxML20 v8.2.12 with 20 ML searches and 100 bootstrap trees, using a partitioned sequence matrix on the 23,412 ortholog groups present in four or more species. Ortholog groups were filtered with the PhyloGeneious script orth2matrix.pl with parameter -m 4. Partitioned ML searches were run with parameters:
raxmlHPC-PTHREADS-AVX2 -T 30 -m PROTGAMMAJTTF -p 12345 -# 20 -o Adicap,Cerric -j –silent
and bootstrap search was run with parameters:
raxmlHPC-PTHREADS-AVX2 -T 30 -m PROTGAMMAJTTF -p 12345 -x 12345 -# 100 -o Adicap,Cerric –silent
The top ML tree and the bootstraps results were combined using:
raxmlHPC-PTHREADS-AVX2 -f b -m PROTGAMMAJTTF -t RAxML_bestTree.RAXML_MLfull1.tre -z RAxML_bootstrap.RAXML_fullbootstrap.tre -o Adicap,Cerric --silent.
Differential expression
Differential expression analysis was performed for each species individually. The Trinity script align_and_estimate_abundance.pl54 was run with bowtie266 (v2.3.2) and RSEM67 (v1.3.0) to estimate read counts per gene. RSEM counts were imported into R [https://www.R-project.org] using tximport68, except for Arabidopsis counts, which were imported using stringtie64. Data was pre-filtered to remove genes with low expression (read count <10 across all samples). DESeq269 was used in R to calculate gene differential expression between leaf and reproductive (ovule, developing spore, or fertile leaf) tissues with a p-adj cutoff of 0.01. For Arabidopsis, young leaf and petiole samples were both labeled as leaf. PCA plots were made from DESeq2 read counts to confirm organ type as the primary source of variation in each species. DESeq2 results were further filtered to remove NAs and add cutoff |log2FoldChange| > 0.58.
Information from paralogs was included in determining whether ortholog groups had conserved differential expression in ovules, conserved differential expression in leaves, mixed differential expression in both tissues, non-conserved differential expression across species, or were non-DE. If some member species contained paralogs DE in both ovules and leaves, then only the species with differential expression in one tissue were used to classify that ortholog group; otherwise, if all DE member species had mixed expression patterns among paralogs, the ortholog group was classified as mixed. Ortholog groups were classified as non-conserved when differential expression was only observed in one species.
Permutation testing for overlap statistical significance
To test the significance of overlaps of DE ortholog group classifications with evolutionary analyses, permutation tests were run in R with 10,000 rounds of random sampling without replacement, and results were compared to actual observed values using the R pnorm function. All code is available in the script informative_analysis.R.
For example, to test the significance of the overlap of the 13,911 ovule DE ortholog groups with parsimony-informative ortholog groups (Fig. 3), 13,911 ortholog groups were randomly sampled from the full set of ortholog groups 10,000 times, and the number of parsimony-informative groups was counted in each sample.
For the significance of the percentage of each ortholog group that was parsimony-informative, the observed informative percentage of each group was calculated by dividing the number of informative sites by the alignment length. During permutation, the mean percentages of the samples were compared to the mean percentage of all informative groups.
For the significance of informative site overlap with annotated protein domains, overlap percentages of samples were compared to the average percent overlap during permutation.
For the significance of influence (strong PBS) on major parsimony tree splits, the number of ortholog groups among the top 10% influencers was counted for each split. During permutation, that number was sampled from all informative groups, and the number overlapping with the given DE classification was counted.
For the significance of evolutionary split supporters identified as candidates, random samples were taken from the parsimony-informative groups with evidence of ovule function (gene family, correlation, expression), and the number of supporters was counted.
For the significance of groups with evidence of ovule function identified as candidates, random samples were taken from the parsimony-informative groups with support for one of the three major evolutionary splits, and the number of groups with evidence was counted.
Partition branch support analysis
The PhyloGeneious output MatrixRecording.log was used to identify the parsimony-informative ortholog groups. The PBS value is determined by comparing the number of amino acid changes within an ortholog group within the current tree to the number of changes in the best tree that does not have the chosen evolutionary clade22. PBSs were calculated from the maximum parsimony species tree and 1:1 ortholog sequence matrix with TNT via the PhyloGeneious script pbs_split_bowery_gil.pl. GO enrichment for each branch split was conducted using the PhyloGeneious R script PBS_GO_enrich.R, based on the GO annotations of Arabidopsis orthologs.
Positive selection analysis
Positive selection dN/dS analysis was performed on the 5934 ortholog groups shared between ferns and seed plants using HyPhy v2.5.24 [https://hyphy.org/] with the seed plants as foreground branches and the two fern species as background. We used the aBSREL and BUSTED methods to identify ortholog groups under positive selection and filtered the results with the following criteria:
aBSREL: At least one foreground branch with dN/dS > 1 and corr-p_val <0.05, and no background branch with dN/dS > 1.
BUSTED: Foreground unconstrained dN/dS > 1, with portion of sites > 0, and corr-p_val <0.05; background either unconstrained dN/dS not > 1, or > 1 but the portion of sites = 0; and no background branches have dN/dS > 1 in the aBSREL results.
We then used the MEME method to identify the sites under analysis and further filtered for ortholog groups with significant sites.
Correlated expression
Corresponding ortholog groups and gene families for known Arabidopsis seed and ovule development genes were identified. The full list of orthologs (parse_partitionMembers_paralogs.txt), a list of ortholog groups DE in multiple species, and related information for the known seed and ovule genes were imported into R. Numbers of seed and ovule gene orthologs identified were obtained for each species. Correlation analysis was only run with ortholog groups that were present in at least ten of the 20 species to ensure reliable pattern resolution. Ortholog groups and sequence IDs were converted into matrix format, and a matrix was generated with the representative log2FC value of each ortholog group in each species. For species with multiple paralogs, the log2FC of the paralog most DE in the reproductive tissue was used for that ortholog group. For species with no observed ortholog, the log2FC was set to 0. Correlation was calculated with the rcorr function from the Hmisc R package [https://hbiostat.org/r/hmisc/].
Enrichment analysis
Enrichment analysis was performed with the script annotation_enrichment_general.py, which uses the scipy fisher_exact function.
Parent GO terms were included in the GO enrichment analyses. Ortholog group sets were compared against all ortholog groups with GO-annotated Arabidopsis genes.
To obtain PFAM annotations for ortholog groups, InterProScan65 PFAM predictions were obtained for all member orthologs and filtered down to those present in at least two species. Enrichment analyses were performed for single PFAM domains and combined PFAM annotations, using all ortholog groups with PFAM annotations as background. For combined PFAM enrichment, PFAM annotations were sorted and concatenated for each ortholog group (e.g., an ortholog group with “domain A;domain B” would have a distinct annotation from an ortholog group with “domain A”).
RNA extraction for in situ hybridization
Young leaves and ovulate cones were collected from Taxus baccata (accessions 1168/41*C and accession 1194/41*A) at the New York Botanical Garden. See collection dates and stages in Supplementary Fig. 23. A subset of these were frozen and ground in liquid nitrogen for RNA extraction. Taxus RNA was extracted from ovules with modification of the QIAGEN RNeasy mini-kit (Qiagen, Hilden, Germany).
Tissue sectioning
Tissue samples were fixed overnight in formaldehyde–acetic acid–ethanol (FAA; 3.7% formaldehyde, 5% glacial acetic acid, 50% ethanol, 35% DI water) under vacuum (20 in Hg). Samples were dehydrated in a standard ethanol series, embedded in paraffin wax with a Leica TP 1020 tissue processor, and stored at 4 °C until use. Samples were sectioned 10 µm thick with a Microm HM 315 rotary microtome (FisherScientific, Pittsburgh, PA, USA).
In situ hybridization
cDNA was synthesized from total RNA from ovulate cones using SuperScript III First-Strand Synthesis System (Invitrogen, Grand Island, NY, USA) with oligodT20 primers, following the manufacturer’s instructions. PCR was conducted using EconoTaq PLUS GREEN 2x with the following ratios: 12.5 µL EconoTaq, 2.5 µL forward primer, 2.5 µL reverse primer, 2 µL DNA template, 5.5 µL nuclease-free water (Lucigen, Middleton, WA, USA). Primers were designed to avoid conserved domains and tested against the NCBI nt database and our full set of transcriptome sequences using Primer-BLAST to ensure specificity. Primer sequences, fragment sizes, and amplification temperatures used for PCR are in Supplementary Data 26. The fragments for probe synthesis were cleaned using the QIAquick PCR purification kit (Qiagen, Valencia, CA, USA). Digoxigenin-labeled RNA probes were prepared using T7 RNA polymerase (Roche, Switzerland), a murine RNAse inhibitor (New England Biolabs, Ipswich, MA, USA), and RNA labeling mix (Roche, Switzerland) according to each manufacturer’s protocol.
Probe synthesis and RNA in situ hybridization for TbMELBEL1 and TbGBEL2 were performed following Ambrose et al.70. Probes were used at 1:50 dilutions. Cover slips were mounted with Permount (Thermo Fisher Scientific, Waltham, MA, USA). Slides were viewed and photographed with a Zeiss Axioplan compound microscope equipped with an Axiocam 712 color digital camera using ZEN 3.6 software. Taxus ovule developmental stages were defined following previous descriptions46,71 (Supplementary Data 27). In situ hybridization was also performed using sense probes of TbMELBEL1 and TbGBEL2 to ensure the specificity of the observed signal.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw RNA reads and transcriptome assemblies generated in this study have been deposited in the NCBI database under BioProject PRJNA1180065, accessions SRR31173681-SRR31173792 and SRR35806086-SRR35806097 for RNA reads and accessions GKZZ00000000, GLAA00000000, GKZX00000000, GKZY00000000, GKZW00000000, GKZV00000000, GLAE00000000, GKZN00000000, GKZO00000000, GKZP00000000, GKZQ00000000, GKZR00000000, GKZS00000000, GKZT00000000, GKZU00000000, GLAB00000000, GLAC00000000, GLAD00000000, GLJD00000000 for transcriptomes. The Arabidopsis RNA reads used in this study are available in the NCBI database under BioProject PRJNA314076, accessions SRR3581388, SRR3581838, SRR3581383, SRR3581837, SRR3581727, and SRR3581889. Enrichment results, partition branch supports, significance values, selection results, ADA2B correlated expression results, and candidate ortholog group lists generated in this study are provided in the Supplementary Data file. Raw outputs from analyses conducted in this study (including annotation, orthology, expression, enrichment, PBS, selection, etc.) have been deposited in an OSF project [https://osf.io/c34h9/].
Code availability
Our updated version of the PhyloGeneious pipeline used in this study is available on GitHub with release tag v2.2024 [https://github.com/coruzzilab/PhyloGeneious/tree/2024_update]. All other scripts used in this study have been deposited in an OSF project [https://osf.io/c34h9/].
References
FAO. World Food and Agriculture—Statistical Yearbook 2022. https://doi.org/10.4060/cc2211en (2022).
Rudall, P. J. Evolution and patterning of the ovule in seed plants. Biol. Rev. https://doi.org/10.1111/brv.12684 (2021).
Smith, D. L. The evolution of the ovule. Biol. Rev. 39, 137–159 (1964).
Andrews, H. N. Early seed plants. Science 142, 925–931 (1963).
Singh, H. Embryology of Gymnosperms (Gerbrüder Borntraeger, 1978).
Gasser, C. S. & Skinner, D. J. Development and evolution of the unique ovules of flowering plants. Curr. Top. Dev. Biol. 131, 373–399 (2019).
Davis, G. L. Systematic Embryology of the Angiosperms. Madroño; a West Am. J. Bot. 19, 95 (1967).
Doyle, J. A. Phylogenetic analyses and morphological innovations in land plants. in Annual Plant Reviews Online, Vol. 45 (eds Ambrose, B. A. & Purugganan, M.) 1–50 (John Wiley & Sons, Ltd, 2017).
Ran, J. H., Shen, T. T., Wang, M. M. & Wang, X. Q. Phylogenomics resolves the deep phylogeny of seed plants and indicates partial convergent or homoplastic evolution between Gnetales and angiosperms. Proc. R. Soc. B Biol. Sci. 285, 20181012 (2018).
Wickett, N. J. et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA 111, E4859–E4868 (2014).
Lee, E. K. et al. A functional phylogenomic view of the seed plants. PLoS Genet. 7, e1002411 (2011).
Leebens-Mack, J. H. et al. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685 (2019).
Stull, G. W. et al. Gene duplications and phylogenomic conflict underlie major pulses of phenotypic evolution in gymnosperms. Nat. Plants 7, 1015–1025 (2021).
Boachon, B. et al. Phylogenomic mining of the mints reveals multiple mechanisms contributing to the evolution of chemical diversity in Lamiaceae. Mol. Plant 11, 1084–1096 (2018).
Bhatnagar, S. P. & Moitra, A. Gymnosperms (New Age International (P) Ltd., 1996).
Becker, A. et al. A novel MADS-box gene subfamily with a sister-group relationship to class B floral homeotic genes. Mol. Genet. Genom. 266, 942–950 (2002).
Lovisetto, A., Guzzo, F., Busatto, N. & Casadoro, G. Gymnosperm B-sister genes may be involved in ovule/seed development and, in some species, in the growth of fleshy fruit-like structures. Ann. Bot. 112, 535–544 (2013).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: assessing genomic data quality and beyond. Curr. Protoc. 1, e323 (2021).
Eshel, G. et al. Plant ecological genomics at the limits of life in the Atacama Desert. Proc. Natl. Acad. Sci. USA 118, e2101177118 (2021).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Schmidt, M. & Schneider-Poetsch, H. A. W. The evolution of gymnosperms redrawn by phytochrome genes: the Gnetatae appear at the base of the gymnosperms. J. Mol. Evol. 54, 715–724 (2002).
Baker, R. H. & Desalle, R. Multiple sources of character information and the phylogeny of Hawaiian Drosophilids. Syst. Biol. 46, 654–673 (1997).
Lallemand, B., Erhardt, M., Heitz, T. & Legrand, M. Sporopollenin biosynthetic enzymes interact and constitute a metabolon localized to the endoplasmic reticulum of tapetum cells. Plant Physiol. 162, 616 (2013).
Kim, J. et al. Overexpression of the Panax ginseng CYP703 alters cutin composition of reproductive tissues in Arabidopsis. Plants 11, 383 (2022).
Pettitt, J. M. The megaspore wall in gymnosperms: ultrastructure in some zooidogamous forms. Proc. R. Soc. Lond. Ser. B Biol. Sci. 195, 497–515 (1977).
Brown, W. H. The nature of the embryo sac of Peperomia. Bot. Gaz. 46, 445–460 (1908).
Soltis, D. et al. Chapter 1. Relationships of angiosperms to other seed plants. in Phylogeny and Evolution of the Angiosperms (eds Soltis, D. et al.). https://doi.org/10.7208/chicago/9780226441757.003.0001 (The University of Chicago Press, 2018).
D’Apice, G. et al. Identification of key regulatory genes involved in the sporophyte and gametophyte development in Ginkgo biloba ovules revealed by in situ expression analyses. Am. J. Bot. 109, 887–898 (2022).
Meinke, D. W. Genome-wide identification of EMBRYO-DEFECTIVE (EMB) genes required for growth and development in Arabidopsis. New Phytol. 226, 306–325 (2020).
Biewers, S. M. Sepallata Genes and Their Role During Floral Organ Formation. University of Leeds (2014).
Favaro, R. et al. MADS-box protein complexes control carpel and ovule development in Arabidopsis. Plant Cell 15, 2603 (2003).
Shen, G., Yang, C.-H., Shen, C.-Y. & Huang, K.-S. Origination and selection of ABCDE and AGL6 subfamily MADS-box genes in gymnosperms and angiosperms. Biol. Res. 52, 1–15 (2019).
Zahn, L. M. et al. The evolution of the SEPALLATA subfamily of MADS-box genes: a preangiosperm origin with multiple duplications throughout angiosperm history. Genetics 169, 2209–2223 (2005).
Rijpkema, A. S., Zethof, J., Gerats, T. & Vandenbussche, M. The petunia AGL6 gene has a SEPALLATA-like function in floral patterning. Plant J. 60, 1–9 (2009).
Liu, Y. et al. The Cycas genome and the early evolution of seed plants. Nat. Plants 8, 389–401 (2022).
Gremski, K., Ditta, G. & Yanofsky, M. F. The HECATE genes regulate female reproductive tract development in Arabidopsis thaliana. Development 134, 3593–3601 (2007).
Pfannebecker, K. C., Lange, M., Rupp, O., Becker, A. & Purugganan, M. Seed plant-specific gene lineages involved in carpel development. Mol. Biol. Evol. 34, 925–942 (2017).
Bailey, T. L., Johnson, J., Grant, C. E. & Noble, W. S. The MEME Suite. Nucleic Acids Res. 43, W39–W49 (2015).
Skinner, D. J., Brown, R. H., Kuzoff, R. K. & Gasser, C. S. Conservation of the role of INNER NO OUTER in development of unitegmic ovules of the Solanaceae despite a divergence in protein function. BMC Plant Biol. 16, 1–12 (2016).
Groß-Hardt, R., Lenhard, M. & Laux, T. WUSCHEL signaling functions in interregional communication during Arabidopsis ovule development. Genes Dev. 16, 1129–1138 (2002).
Petrella, R. et al. Pivotal role of STIP in ovule pattern formation and female germline development in Arabidopsis thaliana. Development 149, dev201184 (2022).
Sharma, R. D., Bogaerts, B. & Goyal, N. RDM16 and STA1 regulate differential usage of exon/intron in RNA directed DNA methylation pathway. Gene 609, 62–67 (2017).
Manna, S. An overview of pentatricopeptide repeat proteins and their applications. Biochimie 113, 93–99 (2015).
Zumajo-Cardona, C. & Ambrose, B. A. Deciphering the evolution of the ovule genetic network through expression analyses in Gnetum gnemon. Ann. Bot. https://doi.org/10.1093/aob/mcab059 (2021).
Zumajo-Cardona, C., Little, D. P., Stevenson, D. & Ambrose, B. A. Expression analyses in Ginkgo biloba provide new insights into the evolution and development of the seed. Sci. Rep. 11, 21995 (2021).
Mundry, I. Morphologische und morphogenetische Untersuchungen zur Evolution der Gymnospermen. Bibl. Bot. 152, 1–90 (2000).
Chanderbali, A. S. et al. Conservation and canalization of gene expression during angiosperm diversification accompany the origin and evolution of the flower. Proc. Natl. Acad. Sci. USA 107, 22570–22575 (2010).
Pease, J. B., Brown, J. W., Walker, J. F., Hinchliff, C. E. & Smith, S. A. Quartet Sampling distinguishes lack of support from conflicting support in the green plant tree of life. Am. J. Bot. 105, 385–403 (2018).
Wu, C. S., Wang, R. J. & Chaw, S. M. Integration of large and diverse angiosperm DNA fragments into Asian Gnetum mitogenomes. BMC Biol. 22, 1–13 (2024).
Carlsbecker, A. et al. The DAL10 gene from Norway spruce (Picea abies) belongs to a potentially gymnosperm-specific subclass of MADS-box genes and is specifically active in seed cones and pollen cones. Evol. Dev. 5, 551–561 (2003).
Alvarez, J. M. et al. Analysis of the WUSCHEL-RELATED HOMEOBOX gene family in Pinus pinaster: new insights into the gene family evolution. Plant Physiol. Biochem. 123, 304–318 (2018).
Barro-Trastoy, D., Dolores Gomez, M., Tornero, P. & Perez-Amador, M. A. On the way to ovules: the hormonal regulation of ovule development. Crit. Rev. Plant Sci. 39, 431–456 (2020).
Wang, T., Zhang, N. & Du, L. Isolation of RNA of high quality and yield from Ginkgo biloba leaves. Biotechnol. Lett. 27, 629–633 (2005).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Hass, B. J. TransDecoder. https://github.com/TransDecoder/TransDecoder/wiki.
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Klepikova, A. V., Kasianov, A. S., Gerasimov, E. S., Logacheva, M. D. & Penin, A. A. A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. 88, 1058–1070 (2016).
Cheng, C. Y. et al. Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 89, 789–804 (2017).
Reiser, L. et al. The Arabidopsis Information Resource in 2024. Genetics 227, iyae027 (2024).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, 1–4 (2021).
Shumate, A., Wong, B., Pertea, G. & Pertea, M. Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLOS Comput. Biol. 18, e1009730 (2022).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, B. & Dewey, C. N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12, 1–16 (2011).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research 4, 1521 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Ambrose, B. A. et al. Molecular and genetic analyses of the Silky1 gene reveal conservation in floral organ specification between eudicots and monocots. Mol. Cell 5, 569–579 (2000).
Stützel, T. & Röwekamp, I. Female reproductive structures in Taxales. Flora 194, 145–157 (1999).
Acknowledgements
This project has received funding from the U.S. National Science Foundation’s Plant Genome Research Program (Grant numbers NSF-PGRP:IOS-0922738 to G.M.C., D.W.S., W.R.M., and NSF-PGRP:IOS-1758800 to W.R.M., D.P.L., G.M.C., D.W.S.) and the European Union’s Horizon 2020 RISE program (Marie Skłodowska-Curie grant agreement number 101007738 to S.N.). C.Z.-C. received funding from the STARS@UNIPD program through the project “SeedDive.” W.R.M. is the Davis Family Professor of Human Genetics at CSHL. Additional support and training to V.M.S. from the U.S. National Institutes of Health Quantitative Biological Systems Training (QBIST) Program (T32 GM132037). This work was supported in part through the NYU IT High Performance Computing resources, services, and staff expertise.
Author information
Authors and Affiliations
Contributions
V.M.S., G.E., K.V., W.R.M., D.P.L., B.A., D.W.S., G.M.C. designed research; V.M.S., S.F., C.Z.-C., S.N., L.D., T.S., B.A. performed research; V.M.S., G.E., W.R.M. analyzed data; V.M.S., G.E., K.V., D.P.L. wrote scripts; K.V., M.S.K., T.L.J., D.P.L., B.A., D.W.S., G.M.C. mentored and supported the research; and V.M.S., S.F., M.S.K., T.L.J., C.Z.-C., S.N., B.A., D.W.S., G.M.C. wrote the paper. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
W.R.M. is a founder, shareholder, and board member of Orion Genomics, which specializes in plant genomics. Orion Genomics was not involved in this research. Other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Stefan de Folter, Verónica Di Stilio, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sondervan, V.M., Eshel, G., Varala, K. et al. Developmentally regulated genes drive phylogenomic splits in ovule evolution. Nat Commun 16, 9589 (2025). https://doi.org/10.1038/s41467-025-65399-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65399-3
