
Abstract
Protein design has become a critical method in advancing significant potential for various applications such as drug development and enzyme engineering. However, protein design methods utilizing large language models with solely pretraining and fine-tuning struggle to capture relationships in multi-modal protein data. To address this, we propose ProtDAT, a de novo fine-grained multi-modal data interaction framework capable of designing proteins from descriptive protein text input. ProtDAT builds upon the inherent characteristics of protein data to unify sequences and text as a cohesive whole rather than separate entities. It leverages a novel Multi-modal Cross-attention, integrating protein sequences and textual information for a foundational level and seamless integration. Evaluation metrics such as pLDDT, TM-score and RMSD are implemented to evaluate the structural plausibility, functionality, structural similarity, and validity of protein sequences. Experiments on 20,000 text-sequence pairs from Swiss-Prot within the ProtDAT framework demonstrate higher accuracy compared to the performance of the best method in the experiments, with a 23.34% increase in pLDDT, a 76.45% increase in TM-score, and a 24.41% reduction in RMSD.
Similar content being viewed by others
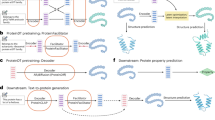
Introduction
Extensive research in the protein field has led to the accumulation of a large amount of multi-modal data, supporting advancements in protein design. In recent years, models for protein structure prediction based on protein sequences have been continuously emerging, such as ESMFold1, RoseTTAFold2, and AlphaFold3,4. They bridge the gap between protein sequence and three-dimensional protein structure information, enabling the rapid acquisition of one modality (e.g., protein structure) from the other (e.g., protein sequence)5. The unimodal learning tends to constrain the performance of protein language models6 since unimodal information can only partially capture the complexity of protein-related data, leading to an incomplete representation.
Protein design involves predicting or generating the sequences7, structures8, and functions9 of proteins to address specific scientific and engineering challenges. Thus, the integration of multi-modal data (e.g., text descriptions, amino-acid sequences, protein structures) represents a significant trend in the development of protein design models. While the connection between protein sequence and structure is well established, the relationship between protein textual information (e.g., protein function) and protein sequences has become a focal point. Consequently, a variety of Protein Language Models (PLMs) are emerging, offering new perspectives and methods that enrich the protein design process.
Existing protein language models based on large language models (LLMs) employ diverse methodologies, such as sequence-only autoregressive architectures, diffusion-based generative approaches, and multimodal frameworks. For instance, autoregressive models such as RITA10, ProtGPT211, and ProGen212 learn sequence generation patterns from large-scale protein data. Beyond autoregressive generation, models like EvoDiff13 leverage diffusion processes for de novo protein design, while ESM-314 integrates multimodal inputs for conditional generation. Other approaches include text-guided models such as ProteinDT15, NetGo3.016 and ProGen17 that use linguistic prompts to control protein generation. Retrieval-based systems like ProtST18 and ProTrek19 employ contrastive learning for functional protein search. Knowledge-graph enhanced methods including OntoProtein20 and PANNZER21 integrate heterogeneous biological data. Fine-tuning frameworks such as ProLLaMA22 and HelixProtX23 adapt pretrained models for multimodal generation. Also, broad-integration approaches like BioTranslator24 and BioT525 incorporate diverse biological contexts for functional protein design.
However, there are several restrictions on existing PLMs. Current text-guided protein generation methods demonstrate limited capacity for deep semantic understanding and integration of biological text, often relying on either single-modality sequence data or oversimplified text prompts. Multi-modal pretraining models for proteins present significant challenges, as they require high compatibility across various modalities, are susceptible to overfitting on training data, and encounter difficulties with modality alignment and expansion.
Recently, generative models have been widely applied in protein design, achieving notable results26. Previous work11,17 has demonstrated that generating protein sequences from texts is effective and has been validated to some extent. In Fig. 1a, the training paradigm of previous generative models captures the correlation between protein sequences and their descriptive texts, allowing for the expansion of knowledge, thereby enhancing the model’s compatibility. However, these methods are only built on original LLMs by pretraining or fine-tuning for different modalities, enabling only coarse-grained interactions among outputs from different modality-specific models. Inspired by the large language models, such as the GPT27 and LLaMA28 series models, we developed ProtDAT (Protein Design from Any Text Descriptions), a protein design framework that generates protein sequences based on any specified training dataset of textual descriptions. Our method designs Multi-modal Cross-attention Mechanism (MCM) for fine-grained protein sequence-text interactions of different modality information at the modality fusion decoder layer. By altering the framework structure, it directly integrates embeddings internally, improving the efficiency and accuracy of text-guided protein generation.

a Comparison between previous and our methods. Previous methods generally pretrain (P) or fine-tune (F) single or multi-modal data directly on Large Language Models (LLMs) without modifying internal structures like attention mechanisms, and they often treat modalities separately. In contrast, our method ProtDAT utilizes a Multi-modal Cross-attention Mechanism (MCM) to integrate multi-modal data within a single framework, with foundational structural adjustments for each modality, optimizing the framework for text-guided protein generation. b Components of the ProtDAT framework, including model input, pre-process model, decoder layers, model output and sequence generation module. c Preprocessing of the ProtDAT-Dataset training dataset, which includes three components: protein sequences, protein descriptive texts, and modality cross vectors. d Pretraining process of ProtDAT, where the model receives the output from (c) through a decoder-only architecture, generates entirely new protein sequences. e Flowchart of the MCM module, which applies self-attention and cross-attention mechanisms to the PTM, CIM, and PSM, respectively, ultimately producing the outputs of the current module.
To the best of our knowledge, ProtDAT is a de novo fine-grained trained framework that addresses the significant issue of inadequate guidance from protein description texts for protein sequence generation in previous PLMs. Additionally, ProtDAT removes the limitations of prompt-based input, enabling training on any standardized protein dataset composed of protein sequences and descriptions. It pushes forward for the boundary of the long-standing challenge in biological data by building effectively linking to vast multi-modal data, enabling organized natural language to be converted into new protein molecules with reasonable and novel sequence and structure. We first construct protein text-to-sequence datasets in a specific format and employ pretraining to develop a protein design model. ProtDAT does not require direct pretraining or fine-tuning on existing LLM architectures. Instead, it introduces improvements to the cross-attention mechanism within Transformer models based on an autoregressive generation framework. These enhancements broaden the model’s understanding of protein data and overcome the restrictions imposed by multi-modal pretrained models in previous protein design frameworks.
ProtDAT is pretrained on the constructed protein text-to-sequence dataset (ProtDAT-Dataset) without explicit protein structure information. It employs a Transformer decoder-only architecture to facilitate interactions between the two modalities. A novel cross-attention mechanism is proposed in ProtDAT, i.e., MCM, which is specifically designed for handling comprehensive information, providing an intuitive, human-logical pathway to protein design. Unlike other PLMs that handle interactions between individual modal models, ProtDAT integrates the interaction of the two modalities within a single model, establishing a robust connection between them from the ground up. During this unsupervised training process, ProtDAT acquires the relationship between protein sequences and descriptions, analogous to how natural language models learn semantic and syntactic rules. This framework combines information from both modalities, enabling multi-modal knowledge to interact throughout the entire training process, thereby allowing the model to generate protein sequences that satisfy specific goals.
In this work, the contributions of our research are as follows:
(1) We introduce ProtDAT, a de novo fine-grained trained framework capable of being trained on any specialized protein text-sequence dataset. By addressing the barrier of inadequate guidance from protein descriptive texts in previous methods, ProtDAT enables the generation of entirely new proteins with broad applicability and flexibility.
(2) A novel cross-attention mechanism, MCM, is designed for the interaction between protein sequences and textual modalities, pushing the boundaries of protein design. Unlike previous approaches that treat sequence and text as separate entities connected through conventional cross-attention mechanisms, MCM integrates both modalities at a foundational data level by establishing shared representations and alignment between protein sequences and text descriptions. This deep integration establishes effective linkages for multi-modal data transformation, providing a novel approach for paragraph-based text-guided protein sequence generation. It enables a unified approach that significantly enhances the model’s ability to generate proteins that align more closely with the given textual descriptions.
(3) Experiments demonstrate that in the protein generation process, textual information plays a dominant role with MCM module. ProtDAT achieves the state-of-the-art results across various evaluation metrics for both sequence and structure on the Swiss-Prot29 dataset.
Results
ProtDAT training framework and data preparation
The ProtDAT framework in Fig. 1b consists of three primary input components: protein description text, protein sequence, and cross-modality tensor. These multi-modal inputs are first processed by a preprocessing module that vectorizes the information. The vectorized embeddings are then passed through decoder layers with the MCM module, which enables the model to generate modality-specific output vectors. Finally, these vectors are utilized in downstream protein generation tasks, facilitating the synthesis of novel protein sequences based on the integrated textual and sequence information.
The ProtDAT framework in Fig. 1d includes 12 decoder layers, each containing layer normalization, Rotary Position Embedding (RoPE)30, MCM, and a feedforward network. Embeddings of the multi-modal inputs in Fig. 1c are processed through decoder layers to generate corresponding output vectors. They are then transformed via a linear mapping and a Softmax function to produce probability vectors of protein sequences. Similar to previous autoregressive methods, ProtDAT is also trained with a cross-entropy loss function.
ProtDAT leverages MCM, specially designed to integrate protein sequences with their descriptive texts, as shown in Fig. 1e. Unlike conventional cross-attention approaches, MCM employs a Cross-Modality Interaction Module (CIM) to integrate information from the Protein Sequence Module (PSM) and the Protein Text Module (PTM), enhancing learning between modalities. Built upon Multi-Head Attention (MHA), MCM incorporates self-attention, cross-attention, Cross-Concat-Attention (CCA) mechanisms and corresponding custom masking strategies to ensure controlled interaction and prevent error accumulation in generation tasks. In autoregressive generation, if the initial protein sequences are generated with errors or inconsistencies, subsequent sequences will continue building upon these flawed fragments. This leads to accumulating deviations, causing the final output to progressively diverge from the target requirements. ProtDAT overcomes the limitations of previous models, which treated text annotations and sequences as separate modules, by integrating diverse modality data seamlessly within a unified framework.
We constructed the ProtDAT-Dataset shown in Supplementary Table 1 by obtaining a non-redundant set of 469,395 protein entries from the well-annotated Swiss-Prot29 database, with each entry consisting of a protein description paired with its corresponding protein sequence. The protein description texts include information on protein function, subcellular localization, and protein family, which are embedded utilizing PubMedBERT31, while the protein sequences are tokenized by the unique protein sequence tokenizer (see details in Supplementary Note. 6) of ESM series models (e.g., ESM-1b32, ESM-233, ESM-C34, ESM-314). ProtDAT-Dataset is partitioned into 402,395 entries for the training set, 47,000 entries for the validation set, and 20,000 entries for the test set. The dataset partitioning and statistical characteristics are detailed in Supplementary Table 2. Additionally, ProtDAT does not enforce rigid formatting requirements on the dataset, allowing for flexible construction based on task-specific description content.
ProtDAT generates protein sequences from multi-modal data
To evaluate the effectiveness of protein sequences generated by ProtDAT, we utilize the test set of ProtDAT-Dataset, containing 20,000 text-sequence pairs, as a reference dataset. ProtDAT employs a generation strategy that combines a temperature coefficient, Top-p decoding, and a repetition penalty to separately enhance diversity, reduce errors, and prevent repetitive sequences. This probabilistic sampling approach (see details in Supplementary Note. 8) draws multiple samples at the start of generation, further optimizing the diversity of the generated sequences. Therefore, we further develop five prompt methods for Progen212, ProtGPT211, ProLLaMA22, ESM-314 and ProtDAT, which perform the following protein generation tasks. The generation parameters for all models and the composition methodology of the Generated-Dataset are comprehensively documented in Supplementary Table 3.
Prompt method 1 (PM1). Only the protein description texts corresponding to the reference protein sequence are utilized as prompts during the generation process.
Prompt method 2 (PM2). Based on PM1, the first 10 amino acids of the corresponding sequence are added, with both the sequence and text modalities jointly guiding the generation process.
Prompt method 3 (PM3). Only the first 10 amino acids of the reference protein sequence are used as the prompts in the generation process.
Prompt method 4 (PM4). Building on PM3, the subcellular localization information from the protein annotations corresponding to the reference sequences is incorporated.
Prompt method 5 (PM5). Building on PM3, the corresponding InterPro35 keywords function information (as ESM-3 required) from the original protein annotations corresponding to the reference sequences is incorporated.
Each of Progen2 and ProtGPT2 generates 20,000 protein sequences by applying PM3, as they lack the ability to process coherent textual input. While ProLLaMA adopts a dual-modality approach using PM4, it only incorporates protein superfamily information. Therefore, 10,805 protein pairs are extracted from the ProtDAT-Dataset test dataset that match the protein superfamilies dataset of ProLLaMA. Given that ESM-3 is able to process protein functional annotation information from InterPro, we employ PM5 as a protein generation prompt strategy. By such a dual-modal prompt, a total of 20,000 protein sequences are successfully generated. We used these superfamily-sequence pairs as prompts to guide the protein generation process. Our framework, ProtDAT, employs PM1 and PM2 on the same test set, generating a total of 40,000 protein sequences—20,000 sequences for each method. We refer to the entities of all the generated protein sequences as Generated-Dataset, illustrated in Table 1.
We employ ESMFold to predict the structures for the protein sequences generated by each method. These predicted structures are then evaluated against reference structures (which serve as the ground truth) derived from the corresponding original sequences in the ProtDAT-Dataset test set, also leveraging ESMFold. The following metrics are leveraged to evaluate the generated proteins.
(1) Global sequence identity, which intuitively evaluates the similarity between the generated and original sequences through global protein sequence alignment.
(2) pLDDT3, which is used to represent the confidence level of the protein structure prediction model (e.g., ESMFold) of the structural prediction for each amino acid, mainly reflecting the reliability of individual residues.
(3) TM-score3, which measures the similarity between two protein structures and is more sensitive to overall shape changes, making it suitable for comparing proteins of different lengths.
(4) RMSD36, an indicator that compares the atomic coordinate differences between two structures, is used to quantify the discrepancy between the predicted structure and the reference structure.
Except for RMSD, where lower values indicate smaller differences, all other metrics are better when higher. These evaluation metrics for sequences in Generated-Dataset are shown in Table 2 and Fig. 2a–d. The pLDDT values are directly obtained from the structural Protein Data Bank (PDB)37 files, while TM-score and RMSD values are acquired through TM-align38.

a The average and maximum lengths of generated sequences by different methods (with a maximum length limit of 500). b–d Mean pLDDT, TM-score, and RMSD values of generated proteins by different methods, with sequence lengths ranging from 1 to 500. e–h The evaluation metric results for the subcellular localization dataset include each model’s accuracy, micro precision, micro recall, and F1 score across both Top-1 and Top-3 conditions. The Top-1 condition refers to the single most likely location, while Top-3 includes the top three most probable locations (or fewer if less than three are predicted). i–m Attention heat maps of MCM in ProtDAT. Assume \({{{{\mathcal{D}}}}}_{t},{{{{\mathcal{D}}}}}_{c},{{{{\mathcal{D}}}}}_{s}\) represent the lengths of the text, cross-modality tensor, and sequence, respectively. The attention weight diagrams are visualized using at most 100 tokens. (i) Self-attention mechanism weight diagram for PTM text embeddings. (j) Cross-attention weight diagram between CIM cross embeddings and PTM text embeddings. k Magnified views of the yellow specific regions in (l), while the CIM attention weight shaped \({{{{\mathcal{D}}}}}_{c}\) is condensed in one dimension. It provides a magnified view of the first 20 tokens of \({{{{\mathcal{D}}}}}_{s}\). l CCA weight diagrams between PSM sequence embeddings and CIM cross embeddings. m Magnified views of the red-specific regions in (l), a zoomed-in view of the first \({{{{\mathcal{D}}}}}_{c}\) columns, detailing CIM attention weights in sequence modality fusion. n The proportion of the overall attention weight contributed by CIM in protein sequence generation procedure, as shown in the CCA weights in (l). This is compared against the reference line (y = text_len / (text_len + current_seq_len)), which shows the ideal theoretical distribution: the influence of text tokens should decrease as the protein sequence grows longer during the autoregressive process. Source data are provided as a Source Data file.
In Fig. 2a, the maximum length of generated sequences for each method is set to 500 tokens. Given that the average length of sequences in the ProtDAT-Dataset test set is 387 tokens across 20,000 sequences, results indicate that while most methods can reach this maximum length, sequences generated by ProLLaMA are noticeably shorter and do not achieve the full 500 tokens. It is noteworthy that ESM-3 generates sequences with a mask generation method. Therefore, we directly use the sequence lengths from the ProtDAT-Dataset test set as generation prompts.
As the length of generated sequences varies in Fig. 2a, the evaluation metrics of protein sequences and structures remain stable across various lengths, and there is no decline in confidence as the sequence length of ProtDAT increases. Leveraging the complete and coherent protein description text as the sole prompt allows the framework to generate sequences that more closely resemble natural proteins. With the additional content of protein sequence prompts, it can further improve the quality of generated sequences and reduce error accumulation.
In Table 2, ProtDAT achieves the best performance across all metrics under both prompt methods, with average values of the global sequence identity at 0.334, pLDDT at 64.41, TM-score at 0.607, and RMSD at 3.477. In Fig. 2b, pLDDT shows an average improvement of 18.65, while in Fig. 2c and Fig. 2d, it is evident that the structural similarity of ProtDAT shows a significant improvement. The confidence in structural prediction shows a steady improvement, especially with the structural similarity metric TM-score, which increased by ~0.26 on average, while RMSD decreased by more than 1.2 Å. These metrics indicate that the protein sequences generated by ProtDAT align reasonably well with the detailed descriptions from the text, even when the global sequence identity is low. The evaluation metrics distributions can be found in Supplementary Note. 5.
The subcellular localization and function analysis of model generated sequences
To evaluate whether the protein sequences generated by each model reflect the characteristics present in their respective textual information, a study is conducted to assess the subcellular localization of the generated protein sequences. The sequences corresponding to the ten subcellular localizations defined in DeepLoc2.139 are selected to create the subcellular localization dataset. Since the text prompts for protein sequences generated by ProtDAT in PM1 and PM2 within the Generated-Dataset contained subcellular localization information, it is necessary to regenerate this subset of sequences. This study extracts 10,705 text entries with localization annotations from the test dataset of ProtDAT-Dataset, removes the localization information, and subsequently generates corresponding protein sequences under both modes. This process yields 21,410 sequences in total, which are incorporated into the subcellular localization dataset. This regeneration approach eliminates potential biases from localization cues in the generated sequences, thereby enhancing the fairness and transparency of the experiments. The subcellular localization dataset additionally incorporates 10,705 sequences each from Progen2, ProtGPT2 and ESM-3, along with 6249 protein sequences from ProLLaMA—all derived by filtering the corresponding model-generated sequences containing localization information from the Generated-Dataset.
Furthermore, each subcellular region can be treated as a class, and subcellular localization could be considered as a multiclass classification problem. Due to the diversity and imbalance of subcellular localization regions, the micro evaluation method is more appropriate than the macro strategy. The micro method aggregates class contributions before calculating the metric, thus addressing class imbalances by giving more weight to frequent classes. This ensures a more accurate assessment of overall model performance, particularly when certain localization regions are underrepresented. The evaluation metrics for subcellular localization are computed as detailed in Methods section.
Subsequently, we utilize DeepLoc2.1 to obtain the subcellular localization information of the protein sequences in the subcellular localization dataset. The results are then compared with the actual localization information from the corresponding texts. Based on the evaluation metrics, the results are presented in Fig. 2e-h.
In this experiment, we evaluate the performance of ProtDAT (in both Mode I and Mode II), Progen2, ProLLaMA, ESM-3 and ProtGPT2. The evaluation metrics included accuracy, micro precision, micro recall, and F1 score under both Top-1 and Top-3 subcellular region conditions.
In terms of Top-1 accuracy shown in Fig. 2e, ProtDAT outperforms all other models with a value close to 0.6. Even under the Top-3 condition, ProtDAT maintains the highest accuracy across all models. This result indicates that ProtDAT has a significant advantage in accurately predicting subcellular localization, successfully assigning most proteins to the correct subcellular regions. For micro precision in Fig. 2f, ProtDAT exhibits the best performance, with precision exceeding 0.5 in the Top-1 scenario, indicating a high level of classification accuracy. In contrast, the precision of the other models is significantly lower, highlighting the superior ability of ProtDAT to correctly classify target categories. Regarding micro recall in Fig. 2g, ProtDAT’s performance mirrors that of micro precision. Whether under Top-1 or Top-3 conditions, its recall rates significantly outperform those of the other models, with an improvement of ~0.2. This indicates that ProtDAT is particularly robust in identifying the correct subcellular regions. In the evaluation of F1 score in Fig. 2h, ProtDAT again demonstrates its superiority by balancing both precision and recall. In the Top-1 condition, its F1 score approaches the highest value of 0.592, underscoring the model’s robust ability to maintain a balance between accuracy and recall.
To validate the functional annotation similarity between the generated protein sequences and their corresponding source sequences, a new study employs Gene Ontology (GO) term prediction to assess functional consistency. Leveraging the MMseqs240 tool for sequence similarity searches, we construct two target databases, generated_db and origin_db, from the generated sequences in the Generated-Dataset and their source sequences from ProtDAT-Dataset, respectively. Additionally, we integrate 449,395 non-redundant sequences from the training and validation sets of the ProtDAT-Dataset to form the query database query_db, ensuring no overlap with the Generated-Dataset test sets. From this query database, the filtered 439,486 protein sequences with GO annotations are used to establish a functional reference set.
The experiment performs double-blind searches using MMseqs2 with the following parameters: sensitivity 3, E-value threshold 1e-4, minimum coverage 0.5, 32 CPU threads for parallel computation, and a limit of one top hit per query. For each pair of generated and source sequences, we extract the GO term sets of their closest matches in the query database and quantified functional annotation overlap using the Jaccard similarity coefficient, defined as \(J(A,B)={|A}\cap {B|}/{|A}\cup {B|}\), where \(A\) and \(B\) represent the GO term sets of the generated and source sequences, respectively. Finally, we statistically analyze the distribution of GO term relevant indicators, with detailed results of each model presented in Table 3.
As shown in Table 3, ProtDAT-generated protein sequences under functional text prompts demonstrate superior performance across all metrics, particularly with a significantly higher proportion in Filter Seq compared to baseline methods. This indicates that ProtDAT effectively integrates functional text annotations from both PM1 and PM2, encoding them into protein sequences as reflected by GO term consistency.
MCM’s role in ProtDAT sequence generation
In Fig. 1b-e, MCM proficiently integrates PSM, PTM, and CIM, enabling ProtDAT to generate protein sequences guided by description texts. To further illustrate the role of different attention mechanisms in facilitating the sequence generation within MCM, we visualize the attention weights across different modules during the protein sequence generation process. We applied to the 20,000 generated sequences using PM1 prompts of ProtDAT as described in Table 1. All attention weight maps in Fig. 2i-n represent the averaged weights from the sequence generation process across both the 12 decoder layers and 12 attention heads.
Figure 2i-n visualizes the average attention weights of the three attention mechanism modules in MCM during the generation process. In Fig. 2i and Fig. 2j, the absence of a masked attention mechanism results in no typical diagonal weight boundary, whereas Fig. 2l and its zoomed-in subfigure exhibit distinct diagonal patterns due to the application of a causal mask.
The self-attention mechanism originating from the Transformer41 in Fig. 2i appropriately integrates single modality data. The attention weights of the protein description text tokens gradually decrease as the text length increases, indicating that textual tokens in the beginning positions have a more substantial influence on guiding the generation method. In Fig. 2j, the weight differences for each token in PTM are not significant, indicating that the cross-attention mechanism effectively integrates the textual information into the cross-embedding (with \({{{{\mathcal{D}}}}}_{c}\) is 50), laying the foundation for subsequent fusion with protein sequence information.
In Fig. 2l, the CCA module is performed under a causal mask shaped \({{{{\mathcal{D}}}}}_{s}\times {{{{\mathcal{D}}}}}_{s}\). It only applies to the last \({{{{\mathcal{D}}}}}_{s}\) columns of CCA attention weights, where it can be observed that the selection of each amino acid is closely related to the weights of CIM and the preceding few amino acid tokens of PSM. As illustrated in Fig. 2k, the compact attention weights of CIM play a significant role in protein sequence generation, holding a substantial weight and serving as a critical part of the procedure. Furthermore, the weights of a few amino acid tokens just before the current generation time step are relatively high. The results indicate that error accumulation is a persistent phenomenon when using autoregressive models for designing proteins. The inclusion of CIM in the generation procedure addresses this issue to some extent, stabilizing the whole sequence generation process.
Figure 2m is another detailed view of Fig. 2l. The attention weights decrease to some extent as the dimension of \({{{{\mathcal{D}}}}}_{s}\) (i.e., the number of rows) increases, indicating that the guidance of the protein description text on sequence generation diminishes as the protein sequence lengthens, while still maintaining a certain level of influence.
Thus, Fig. 2n provides a more intuitive display of average attention weights in Fig. 2l in facilitating the generation process as sequence length (maximum to 500) grows. Assuming using \({{{{\mathcal{D}}}}}_{c}\) amino acid tokens to assist ProtDAT in generation, a function is derived as \({{{{\mathcal{D}}}}}_{c}*1/({{{{\mathcal{D}}}}}_{c}+m)\) to demonstrate the weights of the prompt tokens, where \(m\) denotes the length of already generated sequence tokens in the procedure. This function is presented as a curve named ‘Reference Value’ in Fig. 2n. It is observed that as the protein sequence length rises, the contribution of the protein sequence portion used as a prompt to subsequent sequence generation diminishes. However, in our method, when the sequence length reaches 100, the contribution of the protein descriptive text to the generation stabilizes at around 20%, regardless of the sequence length. This further demonstrates that the textual description ensures the accurate instruction of essential amino acid tokens during the early stages of generation and continues to provide directional support as the protein sequence grows. This effectively addresses the issue of instability in generation as the protein sequence lengthens.
In conclusion, MCM plays a critical role in guiding protein generation. As a bridge connecting protein description texts and protein sequences, CIM significantly contributes to both the integration of information from protein texts and the guidance of sequence generation. MCM tackles the problem of insufficient guidance from protein descriptive texts in existing methods, facilitating robust multi-modal integration and offering a cohesive framework to handle both modalities. By leveraging MCM, ProtDAT greatly enhances the precision of generated protein sequences, ensuring better alignment to the provided textual descriptions.
ProtDAT designs remote homologs protein sequences
To further investigate whether the protein sequences generated by ProtDAT possess the characteristics described in the text, we compared 40,000 sequences generated from ProtDAT with different prompt methods in Generated-Dataset with the test set of ProtDAT-Dataset. ESM series models have extensively learned the representation patterns of protein sequences, it produces protein representation vectors that encode diverse structural and functional properties. These vectors can be further analyzed and compared in the vector space, enabling a deeper understanding of the relationships among proteins through the dimensionality reduction technique t-SNE42 and clustering algorithm K-means43. The dimensionality reduction specifically revealed that text-conditioned sequences generated by ProtDAT formed distinct clusters aligned with their semantic descriptions in the embedding space of ESM series models, demonstrating ProtDAT’s effective text-to-sequence translation capability. This approach facilitates not only protein classification but also the prediction of novel functional insights by exploring the vector space for hidden patterns.
In Fig. 3a, it is evident that the distribution of the generated and reference protein sequences in the vector space exhibits a significant similarity, indicating that the generated sequences share similar underlying properties with the test set. Notably, the clustering of sequences is predominantly concentrated at the bottom of Fig. 3a, suggesting a high density of points in this region. This pattern of distribution further emphasizes the model’s ability to capture and replicate the spatial characteristics of sequences in the embedding space, reinforcing its effectiveness in generating biologically relevant protein representations.

a The point cloud distribution of protein vectors visualizes three groups: ‘ProtDAT(PM1)’ and ‘ProtDAT(PM2)’ (sequences generated using two distinct prompt methods), and ‘Test’ (the reference protein sequences from the ProtDAT-Dataset test set). The structures of the reference and generated sequences are depicted separately in yellow and blue, with UniProt IDs and results of global sequence identity and TM-score. b A case of ProtDAT design process with PM1, comprises four components: prompt input, protein sequence generation, protein sequence alignment, and protein evaluation. c The similarities between the protein sequences generated by ProtDAT under different generation parameters and natural protein sequences, calculated by KL divergence. d, e The amino acid residue distribution of protein sequences generated (‘Gen Seqs’) separately in PM1 and PM2 under the conditions of Top-p = 0.85 and T (temperature coefficient) =1.0, compared to the corresponding natural sequences (‘Test Seqs’) from ProtDAT-Dataset test set.
To further assess the level of similarities between the generated sequences and their corresponding descriptive texts, we selected six protein sequences from different regions in the mapped space with UniProt44 IDs. These generated protein sequences exhibit an average structural similarity of over 90% while maintaining a sequence similarity of less than 25%. Such characteristics align with the definition of remote homologs, which are proteins with low global sequence similarity but high structural resemblance. This result highlights the capability of the generation process to explore and replicate the principles of remote homolog evolution, producing novel sequences that deviate at the sequence level while significantly preserving structural conservation.
The protein generation and evaluation process of ProtDAT is outlined as follows, with Fig. 3b illustrating one case. First, ProtDAT is informed of the generation task through the input of different prompt methods, followed by the selection of generation parameters to produce protein sequences. Subsequently, the generated sequences are subjected to a similarity analysis against the references to obtain global sequence similarity. Additionally, the natural and generated sequences can be input into protein structure models such as ESMFold to derive their structures. These structures are then compared using TM-align to assess TM-score, pLDDT, and RMSD, which measure structural quality and similarity.
ProtDAT generates sequences with natural protein characteristics
In the ProtDAT generation module, whether the generated protein sequences closely resemble the natural arrangement of amino acids is a crucial evaluation criterion. Therefore, designing novel sequences is of paramount importance.
To determine the values of the generation parameters, Optuna45 hyperparameter optimization framework is deployed, randomly selecting 3000 protein sequences of the ProtDAT-Dataset test set. The similarities between generated sequences and natural sequences are calculated by KL divergence46, thereby determining the range of generation parameters, as shown in Fig. 3c. Subsequently, the model generates sequences via protein description texts under different Top-p and temperature coefficient parameters, while Top-p filters out tokens with low probabilities and temperature coefficient controls the diversity in the sequence generation process. Since the global sequence identity47 cannot directly determine whether protein sequences have similar functions, therefore TM-score is implemented to evaluate the generation results from the 3D structures’ perspective. Experiments are conducted with the repetition penalty set to 1.2 according to our local tests, which prevents excessive repetition of tokens, ensuring sequence variability.
In Fig. 3c, the optimal settings are identified as Top-p = 0.95 and T = 1.0. Therefore, we select Top-p values ranging from 0.55 to 1.0 with a step of 0.15 and temperature coefficients from 0.4 to 1.4 with a step of 0.2 for further generation experiments. The TM-Vec model48 is employed to rapidly assess structural similarity from the generated protein sequences. In Table 4, the TM-score reaches the highest value when Top-p is 0.85 and the temperature coefficients are 1.0 and 1.2. To introduce randomness in sequence generation, the temperature coefficient in the subsequent model generation processes is set to 1.0.
With generation parameters of Top-p = 0.85 and T = 1.0, and a repetition penalty of 1.2 applied consistently across all tasks, ProtDAT achieves randomness in protein sequence generation, reducing excessive consecutive repetitions of amino acids and minimizing low-probability tokens, thereby enhancing the generation accuracy. As shown in Fig. 3d and Fig. 3e, the generated protein sequences from ProtDAT exhibit a high degree of similarity with the amino acid sequence distribution of natural proteins, indicating that the model has effectively learned the specific patterns of text-guiding protein generation process.
Ablation study
To further validate the effectiveness of the ProtDAT framework and the Multi-modal Cross-attention Mechanism (MCM), we conduct corresponding ablation experiments. Specifically, while maintaining the overall architecture of ProtDAT, MCM is replaced with the standard widely used cross-attention from Transformers41. The model is trained and validated on the same training/validation dataset and subsequently evaluated on the same test set (see Supplementary Table 2 for detailed data). Similarly, all parameters of ProtDAT are kept unchanged, and a new model, denoted as ProtDAT (without MCM), is trained for 200 epochs. Subsequently, this model is used to generate protein sequences under the PM2 condition on the test set of ProtDAT-Dataset. These sequences are then structurally predicted using ESMFold, followed by the subsequent experimental analyses.
For clarity, we designate the model trained with MCM as ProtDAT. This model is developed under the PM2 framework as ProtDAT, and its experimental results have been comprehensively presented in the Results section. The comprehensive ablation studies are conducted by comparing ProtDAT with ProtDAT (without MCM) across three critical dimensions:
(1) Sequence generation quality. We evaluate 20,000 protein sequences generated by each model using standard protein assessment metrics, with quantitative results presented in Table 2.
(2) Conditional generation capability. Specifically examining the models’ performance on 10,705 protein sequences generated without subcellular localization prompts, measured through localization classification experiments as shown in Table 5.
(3) Functional preservation analysis. Systematically comparing the functional similarity scores of 20,000 generated sequences from each model, with detailed metrics documented in Table 3.
The experimental results demonstrate that our proposed ProtDAT achieves significant improvements over ProtDAT (without MCM) across multiple evaluation dimensions. Specifically: (1) For protein sequence and structure assessment, MCM exhibits comprehensive superiority in all key metrics; (2) In subcellular localization tasks, the performance gains are particularly substantial across all evaluation indicators; (3) Notably, for functional similarity retrieval, ProtDAT not only returns more qualified protein sequences but also shows markedly better retrieval metrics. These systematic advantages conclusively validate the critical value of MCM in the ProtDAT framework, representing a substantial advancement over conventional attention mechanisms.
Discussion
Proteins can be described through various modalities, including natural language text, sequences, and structures, making the integration of different modalities crucial for a comprehensive understanding. ProtDAT stands as a de novo fine-grained trained framework that can be trained on any specialized protein text-sequence dataset. By overcoming the persistent challenge of insufficient guidance from protein descriptive texts in previous methods, ProtDAT enables the generation of entirely new proteins with wide-ranging applicability and flexibility. We introduce MCM, a novel cross-attention mechanism specifically designed to facilitate interactions between protein sequences and textual data. Unlike existing methods that handle sequence and text independently, MCM integrates both modalities from the ground up, establishing robust connections for multi-modal data transformation. Experimental results demonstrate that the protein sequences generated by ProtDAT effectively incorporate text information, achieving promising performance in terms of structural plausibility, functionality and structural similarity with an average 18.65 improvement in pLDDT, a 0.26 increase in TM-score, and a 1.2 Å reduction in RMSD. Relevant resources can be found at (https://github.com/GXY0116/ProtDAT).
In the future, we plan to pretrain ProtDAT by expanding its linguistic capacity with a wider range of annotated protein datasets for more nuanced text-guided protein generation. Additionally, we aim to broaden the MCM by incorporating structural attention mechanisms, extending the current protocol to cover more modalities, creating a more efficient tool for the field of protein design. Furthermore, we intend to extend ProtDAT beyond protein sequences to other biological languages, such as RNA, drug design and single-cell data, by training the framework on datasets from different domains with advanced mechanisms.
Methods
Construction and preprocessing of ProtDAT-Dataset
To train the ProtDAT, we obtained a non-redundant set of 469,395 protein entries from Swiss-Prot29 database, including all entries available before 2023-02-22. We extracted protein text-based information and protein sequence from Swiss-Prot. Each entry in the dataset is a pair consisting of a protein description and its corresponding protein sequence. The protein description texts consist of three components: protein functions, subcellular localization, and protein families. These characteristics are described using expressive biomedical texts49. To identify the correlations between sequence fragments and description sentences, the model preprocesses the two modalities using different tokenizers.
The texts are processed using pre-trained PubMedBERT31 to obtain description embeddings. The tokenizer of PubMedBERT has a vocabulary size of 30,522, which covers most biomedical terms, enabling effective text embedding with a dimension of 768. For protein sequences, the unique protein sequence tokenizer of ESM series models (see details in Supplementary Note. 6) is applied for tokenization, as the ESM series models are proficient in capturing the relationships between sequences. Protein sequences are made up of combinations of amino acids, which makes it difficult to identify whether specific groups of amino acids have particular biological functions. To address this challenge, each amino acid is tokenized individually, which maximizes the model’s ability to learn the relationship between the entire protein sequence and the description text. This approach enhances the likelihood of identifying protein sequence fragments with specific functions during the protein generation. To ensure uniformity in batch processing, the length of the text is limited to 512 tokens, and the length of the protein sequence is limited to 1024 tokens. The masking pretraining process for the model is outlined in MCM module.
Additionally, ProtDAT does not impose a strict format requirement on the dataset, as long as it is constructed based on specific description content tailored to different tasks. Here, we provide a method for extracting protein texts from Swiss-Prot to construct the dataset. During the training of ProtDAT, the format of the input ProtDAT-Dataset in Fig. 1c is shown in Supplementary Table 1. The protein description texts primarily include protein function, subcellular localization, and protein family information, while the protein sequence is represented by its amino acid tokens. Each sequence has at least one of the aforementioned textual annotations. The specific distribution of all data pairs is shown in Supplementary Table 2, which indicates that these three types of description texts have a relatively high proportion in the overall dataset.
Decoder-only masking pretraining framework of ProtDAT
The ProtDAT pretraining framework is based on the decoder-only architecture and trained using the autoregressive generation loss function50 (denoted as \({{{\mathcal{L}}}}\)). It is constructed through 12 decoder layers in Fig. 1d, where each layer utilizes different attention mechanisms of different types of data. The aim is to embed multi-modal information to guide the generation of protein sequences.
(1) Input embeddings of ProtDAT. In Fig. 1c, assume \({{{\bf{s}}}}=[{s}_{1},{s}_{2}\ldots,{s}_{n}]\) represents the protein sequence tokens obtained after the ESM unique sequence tokenization (see details in Supplementary Note. 6), and \({{{\bf{t}}}}=[{t}_{1},{t}_{2}\ldots,{t}_{m}]\) represents the corresponding protein text annotation tokens obtained from PubMedBERT. Since the framework is designed to generate protein sequences with the aid of text, the output of the final layer of the PubMedBERT model \({{{{\bf{E}}}}}_{{{{\bf{t}}}}\_{{{\bf{input}}}}}=[{E}_{{t}_{1}},{E}_{{t}_{2}}\ldots,{E}_{{t}_{m}}]\) is the protein text embedding. Due to the use of a causal mask during sequence training, ProtDAT does not have access to the full information of the protein sequence. Therefore, it is not suitable to directly input the entire sequence into the ESM series models to obtain embedding vectors. In ProtDAT, after mapping through a randomly initialized embedding layer, the sequence embedding is denoted as \({{{{\bf{E}}}}}_{{{{\bf{s}}}}\_{{{\bf{input}}}}}=\,[{E}_{{s}_{1}},{E}_{{s}_{2}}\ldots,{E}_{{s}_{n}}]\). Additionally, at the training step of each batch, a cross-modality tensor \({{{\bf{c}}}}=\,[{c}_{1},{c}_{2}\ldots,{c}_{{c\_size}}]\) is initialized (where \({c\_size}\) represents the length of \({{{\bf{c}}}}\) in ProtDAT). Upon transformation via the randomly initialized embedding layer, this vector is denoted as \({{{{\bf{E}}}}}_{{{{\bf{c}}}}\_{{{\bf{input}}}}}=[{E}_{{c}_{1}},{E}_{{c}_{2}}\ldots {,E}_{{c}_{{c\_size}}}]\). Specifically, the embedding layer of \({{{{\bf{E}}}}}_{{{{\bf{t}}}}\_{{{\bf{input}}}}}\) remains fixed as the pre-trained PubMedBERT weights without update, whereas both the embedding layers of \({{{{\bf{E}}}}}_{{{{\bf{s}}}}\_{{{\bf{input}}}}}\) and \({{{{\bf{E}}}}}_{{{{\bf{c}}}}\_{{{\bf{input}}}}}\) undergo trainable initialization with their weights iteratively optimized. The above three components serve as the protein sequence embedding inputs for the subsequent training process.
(2) Output embeddings of each decoder layer. In Fig. 1d, input embeddings are first passed through layer normalization before being fed into the attention module. Compared to absolute position encoding, the relationship between the sequence and the text is more closely relevant to relative position and neighborhood information. Therefore, during the attention matrix multiplication process, RoPE30 is applied. After the embeddings pass through the Multi-modal Cross-attention Mechanism (MCM) module, they are then passed through a feedforward network followed by another layer normalization. The output of the i-th decoder layer is given by.
where DL represents the decoder layer, \({{{{\bf{E}}}}}_{{{{\bf{s}}}}}^{{{{\bf{i}}}}},\,{{{{{\bf{E}}}}}_{{{{\bf{c}}}}}^{{{{\bf{i}}}}},{{{\bf{E}}}}}_{{{{\bf{t}}}}}^{{{{\bf{i}}}}}\) are also the input of the (i + 1)-th layer. The final output of the 12-layer decoder consists of the vectors denoted as: \({{{{\bf{E}}}}}_{{{{\bf{s}}}}\_{{{\bf{output}}}}}\),\(\,{{{{\bf{E}}}}}_{{{{\bf{t}}}}\_{{{\bf{output}}}}}\),\(\,{{{{\bf{E}}}}}_{{{{\bf{c}}}}\_{{{\bf{output}}}}}\), which respectively represent the embeddings of the protein sequences, the protein annotations, and the modality cross vectors.
(3) Loss function design. The loss function is the commonly used next-token prediction loss in autoregressive generative models, which is well-suited for sequential data generation tasks. Since ProtDAT is designed specifically for text-guided protein sequence generation, this loss function ensures that each token prediction aligns with the context provided by preceding tokens, enhancing sequence coherence. Therefore, the loss function is computed only for \({{{{\bf{E}}}}}_{{{{\bf{s}}}}\_{{{\bf{output}}}}}\).
where \({{{\mathcal{L}}}}\) represents the loss for generating the sequence at time step \(t\) based on the sequence from time steps 0 to \(t-1\), where it is implemented using the cross-entropy function.
Under this pretraining paradigm, the loss function does not directly involve \({{{{\bf{E}}}}}_{{{{\bf{t}}}}\_{{{\bf{output}}}}}\) and\(\,{{{{\bf{E}}}}}_{{{{\bf{c}}}}\_{{{\bf{output}}}}}\). The MCM module of each layer enables the fusion of the two modalities during training, ensuring that the sequence embedding incorporates information from its corresponding protein description text.
To handle variable-length sequences, ProtDAT imposes length limits using padding tokens to standardize the length within each batch. The protein generation model is trained for 200 epochs on 4 A100 GPUs for 5 days. The loss curves for the training and validation sets are shown in Supplementary Fig. 2.
ProtDAT is trained on 469,395 non-redundant protein text-to-sequence description pairs, with a parameter count of 279 M. The sequence data represent the arrangement of amino acids within protein molecules, while the textual data consists of information on protein functions, subcellular localization, and protein families. Details of training settings can be found in Supplementary Note. 3.
ProtDAT multi-modal cross-attention mechanism
To integrate information of diverse modalities, ProtDAT incorporates a Multi-modal Cross-attention Mechanism (MCM) specifically designed for protein sequences and their corresponding descriptive texts, as illustrated in Fig. 1e. MCM builds effective links for the transformation of multi-modal data. Unlike the traditional cross-attention mechanism41 in Transformer, which directly utilizes the query from one modality (e.g., protein sequence) to interact with the key and value from another modality (e.g., protein description text), MCM implements a different strategy. In the Transformer’s cross-attention module, each token of protein sequences represents an amino acid, and simply aligning medical text vocabulary with individual amino acid token makes it challenging to learn the relationships between them. Therefore, merely crossing the two modalities is unlikely to yield optimal results, especially in protein generation tasks where error accumulation during the generation process may cause serious consequences. To address this issue, we introduced the Cross-Modality Interaction Module (CIM) in MCM module to facilitate the nuanced transfer of information from natural language into the protein sequences.
MCM is built upon Multi-Head Attention (MHA) and incorporates the CIM. However, the masking strategy differs in its implementation (see Supplementary Note. 7 for details and examples). Additionally, we modified the attention masking mechanism specifically for the CIM, embedding it into the framework to ensure that the interaction between the sequence and textual information. This approach addresses the challenge faced by previous models, which requires pretraining textual annotations and sequences as separate modules. The MCM module includes three components: Protein Sequence Module (PSM), Protein Text Module (PTM), and the CIM. During training, the presence of CIM necessitates masking both the descriptive annotations and the protein sequences. For PSM, the token at time step \(t\) should only be able to attend to sequences from time steps 0 to \(t-1\), so a causal mask should be deployed. For PTM and CIM, since they utilize self-attention and a basic cross-attention mechanism, only a padding mask is required.
In Fig. 1e, \({{{{\bf{E}}}}}_{{{{\bf{s}}}}}^{{{{\bf{i}}}}-{{{\bf{1}}}}}\),\(\,{{{{\bf{E}}}}}_{{{{\bf{t}}}}}^{{{{\bf{i}}}}-{{{\bf{1}}}}}\) and \({{{{\bf{E}}}}}_{{{{\bf{c}}}}}^{{{{\bf{i}}}}-{{{\bf{1}}}}}\) are the inputs to the i-th decoder layer, while \({{{{\bf{E}}}}}_{{{{\bf{s}}}}\_{{{\bf{input}}}}},{{{{\bf{E}}}}}_{{{{\bf{t}}}}_{{{\bf{input}}}}},{{{{\bf{E}}}}}_{{{{\bf{c}}}}\_{{{\bf{input}}}}}={{{{\bf{E}}}}}_{{{{\bf{s}}}}}^{{{{\bf{0}}}}},{{{{\bf{E}}}}}_{{{{\bf{t}}}}}^{{{{\bf{0}}}}},{{{{\bf{E}}}}}_{{{{\bf{c}}}}}^{{{{\bf{0}}}}}\). After applying layer normalization and RoPE, linear transformation is used to obtain the Query \({{{{\mathcal{Q}}}}}_{t}^{i}\), Key \({{{{\mathcal{K}}}}}_{t}^{i}\), and Value \({{{{\mathcal{V}}}}}_{t}^{i}\) for PTM, and the Query \({{{{\mathcal{Q}}}}}_{c}^{i}\) for CIM.
where \({{{{\bf{E}}}}}_{{{{\bf{t}}}}}^{{{{\bf{i}}}}}\) represents the output of PTM from the self-attention mechanism at the current layer. \({{{{\bf{E}}}}}_{{{{\bf{c}}}}}^{{{{\bf{i}}}}}\) is the output of CIM from the cross-attention mechanism at the i-th decoder layer, which contains the compressed and unified text information, preparing for subsequent interaction with the protein sequence modality. Then MCM combines \({{{{\bf{E}}}}}_{{{{\bf{c}}}}}^{{{{\bf{i}}}}}\) to obtain \({{{{\mathcal{K}}}}}_{c}^{i}\) and \({{{{\mathcal{V}}}}}_{c}^{i}\), the updated representations of the sequence and annotation for the next layer are:
where \({{Linear}}_{{k\_c}}^{i}\) and \({{Linear}}_{{v\_c}}^{i}\) represent the linear layers that projected from \({{{{\bf{E}}}}}_{{{{\bf{c}}}}}^{{{{\bf{i}}}}}\) to the key and value for CIM.
Subsequently, the MCM module combines the text and sequence information, where \({{{{\mathcal{Q}}}}}_{s}^{i}\), \({{{{\mathcal{K}}}}}_{s}^{i}\) and \({{{{\mathcal{V}}}}}_{s}^{i}\) are the query, key, and value for PSM:
where \({{{{\bf{E}}}}}_{{{{\bf{s}}}}}^{{{{\bf{i}}}}}\) serves as the output of PSM within the CCA mechanism. It appropriately integrates the data of different modalities, making it a crucial factor for calculating the loss function and designing new protein sequences. Finally, \({{{{\bf{E}}}}}_{{{{\bf{s}}}}}^{{{{\bf{i}}}}}\), \({{{{\bf{E}}}}}_{{{{\bf{c}}}}}^{{{{\bf{i}}}}}\) and \({{{{\bf{E}}}}}_{{{{\bf{t}}}}}^{{{{\bf{i}}}}}\) are passed through residual connections, layer normalization, and a feedforward layer, formed as the inputs for the (i + 1)-th decoder layer.
The MCM module is built upon the traditional attention mechanism, centering around CIM to link PSM and PTM. This design successfully incorporates multi-modal information, enabling the model to identify associations between biomedical annotations and protein sequences. It also addresses the challenges faced by traditional cross-attention mechanisms, which result in a lack of comprehensive guidance from protein annotations throughout the generation process, thereby limiting the model’s ability to accurately capture functional and structural nuances. Notably, to prevent CIM from becoming overly complex and negatively impacting the training process, the vector needs to be reinitialized for each batch during the training process.
Protein sequence generation module of ProtDAT
To assess the ability to generate protein sequences and design novel proteins, the generation module is specifically constructed for an autoregressive approach: it generates protein sequences one token at a time step, from left to right, with each subsequent token being conditioned on all previously generated tokens. Assume\(\,[{x}_{1},{x}_{2}\ldots {x}_{t-1}]\) is the tokenized sequence at time step \(t-1\) and text information is \({{{\mathcal{T}}}}\), the calculation process of the probability of generating the protein sequence token at time step \(t-1\) is:
Due to the relatively small vocabulary size of protein sequences, Top-k parameter is unnecessary for decoding. Instead, a temperature coefficient is applied to control the diversity of the sequence generation process. Additionally, a Top-p decoding strategy is employed to eliminate tokens with very low probabilities, thereby reducing error accumulation. Finally, a repetition penalty parameter is applied to prevent the model from generating a large number of consecutive repetitive tokens during the generation process.
In the case of greedy search51, the generated sequences tend to have higher repetition and lower similarity to natural sequences. Beam search52, to some extent, can lead to error accumulation, as the autoregressive generation mechanism relies on previously generated sequences to guide subsequent token selection. Therefore, the model opts for a generation method based on probabilistic sampling, where multiple samples are drawn at the start of the procedure to further improve the diversity and orderliness of the generated sequences. In summary, ProtDAT employs a generation strategy53 that combines Top-p, temperature coefficient, and repetition penalty.
ProtDAT is able to design protein sequences either individually or in batches, the required input prompt can be provided in the following two modes.
Mode Ⅰ. Text-Only Input. ProtDAT only receives the protein descriptive text as input, which can be organized by selecting one or more aspects in protein function, subcellular localization, and protein family information. ProtDAT will generate a completely new protein sequence from scratch with the characteristics described in the given input text.
Mode Ⅱ. Text and Sequence Prompt Input. In addition to the input described in Mode Ⅰ, if there are specific requirements for the generated protein sequence, a prompt fragment can be given for a desired protein sequence at the N-terminal. ProtDAT will then generate the sequence based on the provided protein description text and the input protein sequence prompt. Examples are provided in Table 6, while pseudocodes are in Supplementary Note. 4.
Subcellular localization evaluation metrics
The evaluation metrics are as follows, where \({{TP}}_{i}\), \({{TN}}_{i}\), \({{FP}}_{i}\), \({{FN}}_{i}\) respectively represent the true positives, true negatives, false positives, and false negatives, for subcellular region \(i\):
(1) Accuracy, a metric used to evaluate the overall performance of a classification model, representing the proportion of correctly predicted samples out of the total samples, which is as follow:
(2) Micro Precision54, an evaluation metric for multi-class classification models that measures the overall accuracy by aggregating the true positives and false positives across all classes, which is suitable for imbalanced class distributions. Micro precision (MP) is represented as:
(3) Micro Recall54, a metric that measures the ability of a multi-class classification model to correctly identify positive samples across all classes. Micro recall (MR) is calculated by aggregating the true positives and false negatives from all classes:
(4) F1 Score54, the harmonic mean of precision and recall, used to provide a balanced evaluation of a classification model’s performance, especially in cases of class imbalance, which is as follow:
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Swiss-Prot dataset (https://www.uniprot.org/uniprotkb?query=reviewed:true) from the UniProt database (https://www.uniprot.org/) is deduplicated, shuffled and divided into distinct datasets for training, validating and testing the ProtDAT framework. The overall datasets (including training, validation and test) are publicly available at: (https://zenodo.org/records/14967237). The model weights are publicly available at: https://zenodo.org/records/14264096. Source data are provided with this paper.
Code availability
The code in this study is publicly available and has been deposited in Github at: (https://github.com/GXY0116/ProtDAT/tree/v1.0.0)55, under CC BY-NC 4.0 license. The specific version of the code associated with this publication is archived in Zenodo and is accessible via (https://doi.org/10.5281/zenodo.17218015). The online website can be found at: (http://www.csbio.sjtu.edu.cn/bioinf2/ProtDAT/).
References
Lin, Z. et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. BioRxiv 2022, 500902 (2022).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 630, 493–500 (2024).
Orengo, C. A., Todd, A. E. & Thornton, J. M. From protein structure to function. Curr. Opin. Struct. Biol. 9, 374–382 (1999).
Garagić, D. et al. Machine Learning Multi-Modality Fusion Approaches Outperform Single-Modality & Traditional Approaches. IEEE Aerospace Conference (pp. 1–9), 2021.
Samish, I., MacDermaid, C. M., Perez-Aguilar, J. M. & Saven, J. G. Theoretical and computational protein design. Annu. Rev. Phys. Chem. 62, 129–149 (2011).
Mizuguchi, K., Deane, C. M., Blundell, T. L., Johnson, M. S. & Overington, J. P. JOY: protein sequence-structure representation and analysis. Bioinformatics (Oxford, England) 14, 617–623 (1998).
Sadowski, M. & Jones, D. The sequence–structure relationship and protein function prediction. Curr. Opin. Struct. Biol. 19, 357–362 (2009).
Hesslow, D., Zanichelli, N., Notin, P., Poli, I. & Marks, D. Rita: a study on scaling up generative protein sequence models. arXiv Preprint arXiv:2205.05789 (2022).
Ferruz, N., Schmidt, S. & Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 13, 4348 (2022).
Nijkamp, E., Ruffolo, J. A., Weinstein, E. N., Naik, N. & Madani, A. Progen2: exploring the boundaries of protein language models. Cell Syst. 14, 968–978. e963 (2023).
Alamdari, S. et al. Protein generation with evolutionary diffusion: sequence is all you need. Preprint at BioRxiv, 2023.09.11.556673 (2023).
Hayes, T. et al. Simulating 500 million years of evolution with a language model. Science, 387, eads0018 (2025).
Liu, S. et al. A text-guided protein design framework. Nature Machine Intelligence, 7, 580–591 (2025).
Wang, S., You, R., Liu, Y., Xiong, Y. & Zhu, S. NetGO 3.0: protein language model improves large-scale functional annotations. Genomics, Proteomics & Bioinformatics, 2, 349–358. (2023).
Madani, A. et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 41, 1099–1106 (2023).
Xu, M. et al. Protst: Multi-modality learning of protein sequences and biomedical texts. International Conference on Machine Learning. pp. 38749–38767 (2023).
Su, J., He, Y., You, S. et al. A trimodal protein language model enables advanced protein searches. Nat. Biotechnol. https://doi.org/10.1038/s41587-025-02836-0 (2025).
Zhang, N. et al. Ontoprotein: protein pretraining with gene ontology embedding. International Conference on Machine Learning. 2022: 1786–1803.
Törönen, P. & Holm, L. PANNZER—a practical tool for protein function prediction. Protein Sci. 31, 118–128 (2022).
Lv, L. et al. Prollama: A protein large language model for multi-task protein language processing. IEEE Transactions on Artificial Intelligence, 1–12, https://doi.org/10.1109/TAI.2025.3564914 (2025).
Chen, Z. et al. Unifying sequences, structures, and descriptions for any-to-any protein generation with the large multimodal model HelixProtX. arXiv Preprint arXiv:2407.09274 (2024).
Xu, H., Woicik, A., Poon, H., Altman, R. B. & Wang, S. Multilingual translation for zero-shot biomedical classification using BioTranslator. Nat. Commun. 14, 738 (2023).
Pei, Q. et al. BioT5: Enriching Cross-modal Integration in Biology with Chemical Knowledge and Natural Language Associations. Proceedings of the 2023 Conference on Empirical Methods in Natural Language Processing (pp. 1102–1123). Association for Computational Linguistics. (2023).
Harshvardhan, G., Gourisaria, M. K., Pandey, M. & Rautaray, S. S. A comprehensive survey and analysis of generative models in machine learning. Computer Sci. Rev. 38, 100285 (2020).
Liu, X. et al. GPT understands, too. AI Open, 208–215 (2023).
Touvron, H. et al. Llama 2: Open foundation and fine-tuned chat models. arXiv Preprint arXiv:2307.09288 (2023).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic acids Res. 31, 365–370 (2003).
Su, J. et al. Roformer: Enhanced transformer with rotary position embedding. Neurocomputing 568, 127063 (2024).
Gu, Y. et al. Domain-specific language model pretraining for biomedical natural language processing. ACM Trans. Comput. Healthc. (HEALTH) 3, 1–23 (2021).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. 118, e2016239118 (2021).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Team, E. ESM Cambrian: Revealing the mysteries of proteins with unsupervised learning. EvolutionaryScale Website (2024).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2008).
Sapundzhi, F., Popstoilov, M. & Lazarova, M. RMSD Calculations for Comparing Protein Three-Dimensional Structures. International Conference on Numerical Methods and Applications. 279–288. Cham: Springer Nature Switzerland, 2022.
Berman, H. M. et al. The protein data bank. Acta Crystallogr. Sect. D: Biol. Crystallogr. 58, 899–907 (2002).
Zhang, Y. & Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic acids Res. 33, 2302–2309 (2005).
Ødum, M. T. et al. DeepLoc 2.1: multi-label membrane protein type prediction using protein language models. Nucleic Acids Research, gkae237 (2024).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Vaswani, A. et al. Attention is all you need. Adv. Neural Inf. Process. Systems 30, 5998–6008 (2017).
Van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. learning Res. 9, 2579−2605 (2008).
Macqueen, J. et al. Some methods for classification and analysis of multivariate observations. Proceedings of 5-th Berkeley Symposium on Mathematical Statistics and Probability/University of California Press. 281–297. (1967)
Consortium, U. UniProt: a worldwide hub of protein knowledge. Nucleic acids Res. 47, D506–D515 (2019).
Akiba, T., Sano, S., Yanase, T., Ohta, T. & Koyama, M. Optuna: A next-generation hyperparameter optimization framework. Proceedings of the 25th ACM SIGKDD international conference on knowledge discovery & data mining. 2623–2631. (2019)
Van Erven, T. & Harremos, P. Rényi divergence and Kullback-Leibler divergence. IEEE Trans. Inf. Theory 60, 3797–3820 (2014).
May, A. C. Percent sequence identity: the need to be explicit. Structure 12, 737–738 (2004).
Hamamsy, T. et al. Protein remote homology detection and structural alignment using deep learning. Nat. Biotechnol. 42, 975–985 (2024).
Tanabe, L. & Wilbur, W. J. Tagging gene and protein names in biomedical text. Bioinformatics 18, 1124–1132 (2002).
Radford, A. et al. Language models are unsupervised multitask learners. OpenAI blog 1, 9 (2019).
Chickering, D. M. Optimal structure identification with greedy search. J. Mach. Learn. Res. 3, 507–554 (2002).
Freitag, M. & Al-Onaizan, Y. Beam search strategies for neural machine translation. Proceedings of the First Workshop on Neural Machine Translation (pp. 56–60). Association for Computational Linguistics. https://doi.org/10.18653/v1/W17-3207 (2017).
Holtzman, A., Buys, J., Du, L., Forbes, M. & Choi, Y. The curious case of neural text degeneration. arXiv Preprint arXiv:1904.09751 (2019).
Yacouby, R. & Axman, D. Probabilistic extension of precision, recall, and f1 score for more thorough evaluation of classification models. Proceedings of the first workshop on evaluation and comparison of NLP systems. 79–91. (2020)
Guo, X. et al. Ab-initio amino acid sequence design from any protein text description with ProtDAT Zenodo https://doi.org/10.5281/zenodo.17218015 (2025).
Acknowledgements
This work is supported by the Science and Technology Commission of Shanghai Municipality (No.24510714300 to H.B.S.).
Author information
Authors and Affiliations
Contributions
X.Y.G., Y.F.L. and H.B.S. conceived the study, and H.B.S. supervised it. X.Y.G. proposed the computational model and implemented downstream tasks. X.Y.G. developed the model and conducted data analysis. H.B.S., X.P., Y.L. and Y.F.L. provided advice on data analysis. X.Y.G. drafted the manuscript. H.B.S., X.P., Y.L. and Y.F.L. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Muhammad Arif, Amy Lu, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, XY., Li, YF., Liu, Y. et al. Ab-initio amino acid sequence design from protein text description with ProtDAT. Nat Commun 16, 10544 (2025). https://doi.org/10.1038/s41467-025-65562-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65562-w
