
Abstract
Microbially mediated organohalide cycling in the ocean has profound implications for global biogeochemical cycles and climate, but the geographic distribution and diversity of the halogenation-dehalogenation cycling microorganisms remain unknown. Here, we constructed an organohalide-cycling gene database (HaloCycDB) to explore the global atlas of halogenation-dehalogenation cycling microorganisms and genes from 1473 marine metagenomes. Strikingly, 6204 out of 15,252 metagenome-assembled genomes (MAGs) carry organohalide-cycling genes, of which 84.30% are dehalogenating populations. Microorganisms of Pseudomonadota with even spatial distribution dominate both halogenation and dehalogenation potentials in the ocean, in contrast to lineages of Asgardarchaeota and Thermoproteota solely mediating dehalogenation in the Northern hemisphere. Notably, 80.91% of reductive dehalogenase (RDase) genes and 91.35% of RDase-containing prokaryotes represent uncharacterized lineages, substantially expanding known dehalogenation diversity. Further integration of microbial cultivation, protein structure prediction, and molecular docking revealed four unique “microorganism-RDase-organohalide” patterns for marine dehalogenation and its coupling with carbon/sulfur cycles, being distinctively different from their terrestrial patterns. These results advance our understanding of microbial organohalide cycling by providing insights into the halogenation-dehalogenation microbiomes in the ocean.
Similar content being viewed by others
Introduction
As the Earth’s largest reservoir of dissolved organic carbon, sulfur, and halogen species, the ocean is a hotspot for microbially-mediated biogeochemical element cycling, potentially influencing global food webs and climate1,2,3. In terms of the organic halogen species, over than 8000 organohalides have been identified to date from natural sources, of which the majority originate from marine environments4,5. These organohalides can serve as antibiotics and signaling molecules, profoundly affecting marine community structure and ecological competition5,6,7. For example, bromo-furanones produced by marine red alga change bacterial quorum sensing systems to inhibit biofilm formation6, and natural methoxylated polybrominated diphenyl ethers (MeO-PBDEs) disrupt endocrine and immune functions of higher trophic organisms through bioaccumulation in marine food webs8. Moreover, ocean-derived and short-lived organohalides (e.g., CH3Br) contribute up to 50% of ozone loss within the Antarctic springtime ozone hole7, suggesting their pivotal role in affecting global climate change. Nonetheless, in contrast to the extensively studied organic carbon and sulfur cycles9,10, investigation on the biogeochemical cycling of organohalides in the ocean is still in its infancy.
The biogeochemical cycling of organohalides is primarily driven by their biosynthesis (halogenation) and attenuation (dehalogenation) processes11, mediated by microorganisms that have evolved specialized enzyme systems4,12. For the halogenation process, accumulating biochemical evidence has revealed four groups of halogenases from phylogenetically diverse bacteria, algae, and fungi to catalyze this secondary metabolism process in marine and terrestrial ecosystems4,13, i.e., flavin-dependent halogenase (FlaHase), vanadium-dependent halogenase (VanHase), nonheme iron-dependent halogenase (NHFeHase), and S-adenosyl-L-methionine-dependent halogenase (SAMHase). By contrast, seven groups of dehalogenases, primarily from a small group of bacterial lineages of terrestrial sources, have been characterized to catalyze the dehalogenation of organohalides and mostly in energy-associated primary metabolic ways, including oxidative dehalogenase (OxDase)14, hydrolytic dehalogenase (HyDase)15, reductive dehalogenase (RDase)16,17, glutathione S-transferase (GSTDase)18, methyltransferase (MetDase)19, dehydrochlorinase (DehyDase)20, and halohydrin dehalogenase (HahyDase)21. These halogenases and dehalogenases, together with their host microorganisms and organohalide substrates, can form an extremely complex “microorganism-enzyme-organohalide” network, especially in the ocean with high salinity, oligotrophy, and extreme habitats22. The heterogeneity of microbial growth niches in the ocean23 can promote specialized and diverse “microorganism-enzyme-organohalide” patterns to drive the marine halogenation and dehalogenation cycling of organohalides. These patterns refer to acclimatization and interactions among niche-specific microorganisms, associated enzymatic machinery, and localized organohalide pools. Driven by redox potential and substrate availability, the “microorganism-enzyme-organohalide” patterns could shape the taxonomic and functional diversification for halogenation and dehalogenation of organohalides in ocean, which represent yet-to-be-explored microbial resources of novel organohalide-cycling microorganisms, enzymes, and active chemical species. Nonetheless, our current understanding of the halogenases/dehalogenases, as well as involved microorganisms and organohalides, is mainly based on cultivation and biochemical characterization studies, being largely hindered by the fact that both the medium-derived cultivation bias24,25 and unculturable microbial dark matter26,27 leave the majority of marine organohalide-cycling microorganisms and their functional genes uncharacterized. Moreover, previous studies mainly focused on the organohalide formation or dehalogenation potential of specific terrestrial regions (e.g., paddy soil28 and polluted urban rivers13) or microbial lineages of specific dehalogenation populations (e.g., organohalide-respiring bacteria of Dehalococcoides and Dehalobacter)29, lacking a comprehensive catalogue and characterization of organohalide-cycling microorganisms across the microbial kingdom and global ocean habitats. These limitations greatly impede our comprehension of the microbially-driven halogenation and dehalogenation cycling of organohalides in the ocean.
The culture-independent metagenomics offers opportunities to surmount these limitations30. Large-scale sampling and sequencing efforts (e.g., the Tara Oceans and Global Ocean Sampling expeditions) have generated a vast expanse of genetic resources to explore novel taxa and functions31. At the taxonomic level, novel microbial lineages continually emerge and expand marine biodiversity, including the recently identified uncultured bacterial phylum Candidatus Eudoremicrobiaceae to harbor an exceptionally high abundance of biosynthetic gene clusters for producing organohalides and other natural products32. At the functional level, metagenomic analyses have uncovered previously unrecognized metabolic capabilities of both known and novel microbial populations, exemplified by the identification of photosynthetic genes (e.g., pufLM) in the predatory Myxococcota33. Therefore, advances in metagenomics hold the potential to reveal a full picture of microbially-mediated halogenation and dehalogenation cycling of organohalides in the ocean, which requires orthology databases to support metagenomic analyses. Nonetheless, despite their unquestionable merits, currently available KEGG34 and similar comprehensive databases tend to be biased toward eukaryotes and the metabolism of model organisms for historical reasons35, and lack partial enzymatic routes for the microbially-mediated organohalide cycling, which often results in suboptimal functional assignment and false-positive outcomes35. Consequently, exploring organohalide-cycling microbiomes urges the development of a function-specific database.
To explore the global-scale microbially-mediated halogenation and dehalogenation cycling of organohalides in the ocean, we developed an organohalide-cycling gene database (HaloCycDB) to support organohalide-cycling metagenomic analyses, and analyzed 1473 metagenomes from 506 marine sampling sites to reveal the geographic distribution of organohalide-cycling microorganisms and functional genes at the global scale. Also, the microorganisms and associated functional genes for halogenation and dehalogenation cycling of organohalides were further catalogued to explore novel taxonomic and genetic resources. Additionally, the “microorganism-enzyme-organohalide” patterns for reductive dehalogenation were analyzed and confirmed with protein structure prediction, molecular docking, and laboratory cultivation experiments. To the best of our knowledge, this study provides insight into the halogenation and dehalogenation cycling of organohalides in the ocean, which provides a roadmap for exploring marine organohalide-associated bioresources.
Results
Database development to profile marine organohalide-cycling potential
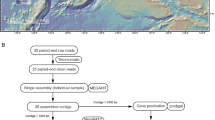
To facilitate the metagenome-based estimation of microbially mediated halogenation and dehalogenation potentials and to enable the robust projection of organohalide cycling in natural environments, an organohalide-cycling gene database (HaloCycDB) was manually curated to profile the halogenation-dehalogenation cycling genes (Fig. 1a). The HaloCycDB comprises a meticulously curated core database (coreDB) and a full database (fullDB) (Supplementary Fig. 1), containing sequences of 221 functionally-characterized genes and 187,289 representative homologous genes, respectively, and covering all of the four halogenation and seven dehalogenation processes (Fig. 1a). Compared to canonical KEGG, COG and their derivative databases, the annotation accuracy, coverage and sensitivity of the HaloCycDB were significantly improved in profiling organohalide-cycling genes (Fig. 1b), e.g., an accuracy of 99.97%, 79.20% and 70.89%, as well as a sensitivity of 100%, 58.76% and 42.28%, for the HaloCycDB, KEGG and COG databases, respectively. To ensure the gene annotation accuracy in data analyses, both sequence similarity and conserved regions/motifs (≥50% amino-acid/AA sequence similarity, or ≥30% AA sequence similarity plus conserved regions/motifs) were included as key screening parameters to prevent false annotation of halogenation and dehalogenation genes (Supplementary Fig. 1). Notably, with the strict screening criteria, the total abundance of confirmed halogenation and dehalogenation genes was decreased by 32.26–99.97% and 50.98–83.72%, respectively, relative to the widely employed less rigorous criteria (≥30% AA sequence similarity) (Fig. 1d).

a Halogenation and dehalogenation modules in the HaloCycDB. The modules were numbered with a pound sign (#). b The accuracy assessment (including sensitivity, false-negative rate, false-positive rate, accuracy rate, and coverage) for KEGG, eggNOG, COG, arCOG, and HaloCycDB. c 1473 marine microbial community genomes (metagenomes) were collected from 506 globally distributed sites. The map was generated using the R package maps124, in which the “world” data was derived from the Natural Earth (v2.0) (https://www.naturalearthdata.com/). d Impact of gene data filtering based on conserved domains/motifs and/or ≥50% amino-acid sequence identity on distribution of organohalide-cycling genes and taxa. See Supplementary Fig. 1 for the detailed strict criteria of gene data filtering. Heatmap illustrates gene abundances based on RPKM (Reads Per Kilobase per Million mapped reads) values. Source data are provided as a Source Data file.
To profile the cross-kingdom microbially-mediated cycling of organohalides in marine environments, we performed a large-scale metagenomic assembly on 1473 marine water and sediment metagenomes across the globe (Fig. 1c; Supplementary Fig. 2; Supplementary Data 1). There were a total of 145,897,437 contigs assembled from the 1473 metagenomes, which were employed to profile the halogenation and dehalogenation genes in marine water and sediment microbiomes by the HaloCycDB (Fig. 1d). In contrast to Ascomycota as the major eukaryotic player in dehalogenation, phylogenetically diverse Prokaryotes harbored 98.89% of all organohalide-cycling genes and were dominant players to mediate both the halogenation and dehalogenation processes of organohalides in the ocean (Fig. 1d; Supplementary Fig. 3a). These prokaryotes predominantly employed FlaHase/VanHase halogenases and HyDase/OxDase/RDase dehalogenases to mediate halogenation and dehalogenation, respectively (Fig. 1d; Supplementary Fig. 3a). Based on the HaloCycDB, we further reconstructed a total of 6204 non-redundant prokaryotic metagenome-assembled genomes (MAGs) with organohalide-cycling potential by harboring halogenation or dehalogenation genes (completeness ≥50% and contamination ≤10%; Supplementary Fig. 3b and 3c). Interestingly, the 6,204 organohalide-cycling MAGs accounted for 40.68% of all 15,252 reconstructed MAGs from global-scale marine metagenomes, of which 84.30% and 15.70% were identified to be dehalogenation and halogenation populations, respectively (Supplementary Fig. 3d). These results suggested the widespread presence and consequent potential critical roles of organohalide-cycling microorganisms in marine biogeochemical cycles, which were neglected by previous studies.
Prokaryote-mediated organohalide cycling and their geographic distribution in the ocean
To examine prokaryote-mediated organohalide cycling in the ocean, the 1473 ocean metagenomes were used to profile their halogenation and dehalogenation potentials, as well as their three-dimensional spatial distribution (Fig. 2). Results showed a core group of bacteria dominate the marine organohalide-cycling metabolisms. For example, Pseudomonadota being widespread in marine water and sediment environments mediated 65.73% and 65.14% potential halogenation and dehalogenation activities, respectively (Fig. 2a, b; Supplementary Fig. 4a). In addition, Chloroflexota mediated 5.98% of potential organohalide-cycling activities (Fig. 2a), and shared a similar geographic distribution pattern in the ocean with the organohalide-cycling Pseudomonadota (Fig. 2b; Supplementary Fig. 4b). Notably, Dehalococcoidia of the Chloroflexota mainly involved in FlaHase- and HyDase-mediated halogenation and dehalogenation, respectively, and the well characterized terrestrial obligate organohalide-respiring bacteria (OHRB) of this class (i.e., Dehalococcoides and Dehalogenimonas) for RDase-mediated reductive dehalogenation were absent in the marine water and sediment niches (Fig. 2a). In contrast, Anaerolineae instead of the Dehalococcoidia were major players of the phylum Chloroflexota to host RDase genes for reductive dehalogenation in the ocean29.

a Major taxonomic distribution of organohalide-cycling genes in prokaryotes. b Vertical distribution of organohalide-cycling microorganisms of Pseudomonadota, Chloroflexota, Cyanobacteriota, Asgardarchaeota, and Thermoproteota at different latitudes. The distribution of halogenation-dehalogenation cycling genes in different marine niches based on abundance without biomass correction (c) and with biomass correction (d). The thickness of the lines in the Sankey diagram (in a, c, d) represents the RPKM values. Sediments are divided into cold seep (CS), hydrothermal vent (HV), trench (Tre), and other sediments (OS). Source data are provided as a Source Data file.
There were phylogenetically diverse prokaryotic groups solely mediating dehalogenation in the ocean (Fig. 2a, b), including the sediment-inhabiting and HyDase/RDase-harboring archaea of Lokiarchaeia (Asgardarchaeota) and Bathyarchaeia (Thermoproteota) classes (Fig. 2a, b). Interestingly, compared to their even distribution along the longitude (Supplementary Fig. 4d), these HyDase/RDase gene-harboring Asgardarchaeota and Thermoproteota mainly (>80.73%) gathered in sediment of the Northern hemisphere (0–90°N) (Fig. 2b). In contrast to the sediment-inhabiting and HyDase/RDase-harboring archaea, microorganisms (PCC-6307 order) of Cyanobacteriota were the only halogenating prokaryotes without dehalogenation capability (Fig. 2a) and mainly colonized at the surface ocean (Fig. 2b; Supplementary Fig. 4c), particularly the tropical and temperate regions (45°S–45°N) with appropriate temperature and illumination conditions for their cell growth (Fig. 2b).
Of the organohalide-cycling gene distribution in the overall marine cross-sections, FlaHase and HyDase/OxDase/RDase were predominant halogenation and dehalogenation genes, respectively, accounting for 9.72% and 64.20%/12.61%/6.32% (totally 92.85%) of all potential organohalide-cycling activities (Fig. 2c; Supplementary Data 2). Specifically, in contrast to FlaHase as a dominant halogenase accounting for 76.20% of all halogenase genes, HyDase, OxDase, and RDase as major dehalogenases reached 73.59%, 14.45%, and 7.25% of total dehalogenase genes. The vertical distribution patterns of these organohalide-cycling genes were in accord with their increasing relative abundance along with marine water depth, i.e., 8.63%, 14.38%, 17.81% and 21.29% of the total organohalide-cycling genes in microbiomes of epipelagic (EPI), mesopelagic (MES), bathypelagic (BAT) and abyssopelagic (ABY) layers, respectively (Fig. 2c). In contrast, heterogeneous distribution patterns of these organohalide-cycling genes were observed in the cold seep, hydrothermal vent, trench and other sediment, e.g., 43.04% and 14.31% RDase genes, as well as 10.16% and 39.88% HyDase genes, were present in the cold seep and trench, respectively (Fig. 2c; Supplementary Data 2). Due to the very different biomass of microbiomes in the four marine water layers and the four sedimentary environments, the distribution of these organohalide-cycling genes varied distinctively from the above-described patterns when incorporating the biomass abundance (Fig. 2d). Particularly, in the epipelagic and mesopelagic layers, where micro-biomass abundance accounted for 70.03% of total marine microbial mass36,37, the HyDase and OxDase genes achieved 41.59% and 36.64% of their total abundance in the ocean, respectively (Fig. 2d; Supplementary Data 2). In contrast to the concentration of potential HyDase and OxDase activities in the surface ocean, 49.28% of total RDase genes were gathered in sedimentary environments (Fig. 2d; Supplementary Data 2), which might support the establishment of microbiomes in oligotrophic sediment by converting recalcitrant organohalides into comparatively labile organic matter for downstream microbially-mediated organic metabolisms.
Expanded catalog of organohalide-cycling genes and genomes from global marine water and sediment microbiomes
To uncover the halogenation/dehalogenation gene diversity, we clustered genes encoding major halogenases (FlaHase) and dehalogenases (HyDase and RDase) in marine water and sediment microbiomes. The FlaHase, HyDase, and RDase genes were clustered at 90%, 75%, and 90% amino acid identity, respectively, which were commonly used standards for grouping their functional-like genes38. Based on the functional gene annotations from the strict criteria of HaloCycDB, 32.73%, 53.01% and 80.91% were identified to be unknown FlaHase, HyDase and RDase genes, respectively, greatly expanding the current diversity of halogenation and dehalogenation genes (Fig. 3a; Supplementary Fig. 5). Notably, the ocean-derived FlaHase and HyDase genes were mainly clustered based on their substrate specificities, and showed high coverages of their phylogenetic tree lineages: (1) the ocean-sourced FlaHases evenly covered all major FlaHase groups, including group-2 (PltA/HrmQ/Mpy16-like), group-3/4 (PrnA/KtzQ/SpmH-like) and group-5 (BrvH-like) FlaHases to specifically catalyze halogenation of pyrrole-containing natural products, L-tryptophan and indole derivatives, respectively (Supplementary Fig. 5a); (2) the marine HyDases covered all major groups, but were mainly clustered into the group-6 (62.74%) HyDases that degraded haloalkanes (Supplementary Fig. 5b), which together with predominance of HyDases in the overall dehalogenation potential suggested the critical role of halogenated alkanes in sustaining microbiomes by conversion of persistent organohalides into labile organic matter in oligotrophic marine environments39. In contrast to the high coverage of ocean-sourced FlaHases and HyDases, marine RDases formed 6 new clusters (groups 6–11) that were separated from previously reported terrestrial RDases (groups 1–5; Supplementary Fig. 5c). This observation indicated the different evolutionary processes of the terrestrial and marine RDases, which could be partially due to the varied RDase-hosting organohalide-respiring microorganisms in terrestrial and marine environments. Moreover, 95.20% of the marine RDases, lacking a highly conserved twin-arginine motif (Tat) and a small associated membrane anchor protein (RdhB), were different from the terrestrial RDases, which mostly possess Tat and RdhB (Fig. 3a), suggesting that the RDase-based dehalogenation of organohalides proceeds in the cytoplasm of marine OHRB rather than in the periplasm of terrestrial OHRB.

a Rooted maximum likelihood phylogenetic trees of FlaHase, HyDase, and RDase genes identified from marine samples and HaloCycDB. b Rooted maximum likelihood phylogenetic tree of organohalide-cycling MAGs. The color of the internal branches indicates whether the organohalide-cycling MAGs were reported for the first time (functionally novel; red) or reported previously (known; light gray). c The percentages of functionally novel organohalide-cycling MAGs at different taxonomic levels. Bootstrap of >70% in the phylogenetic trees are indicated as gray circles. Source data are provided as a Source Data file.
To reveal new microbial populations with halogenation/dehalogenation potential, we clustered the 4430 non-redundant prokaryotic MAGs with a quality score (completeness − 5 × contamination) of ≥50 containing halogenation/dehalogenation genes at multiple taxonomic levels (from genus to phylum; Fig. 3b; Supplementary Fig. 6). The 4430 prokaryotic MAGs were assigned to 60 bacterial and 5 archaeal phyla, with Pseudomonadota (n = 1784), Chloroflexota (n = 422), Actinomycetota (n = 408), Bacteroidota (n = 395) and Planctomycetota (n = 230) as predominant bacterial phyla, and Asgardarchaeota (n = 37) and Thermoproteota (n = 29) as dominant archaeal phyla (Fig. 3b). Notably, at the genus level, 48.73%, 82.01% and 91.35% of FlaHase-, HyDase- and RDase-hosting prokaryotes, respectively, were unknown halogenation/dehalogenation populations (functionally novel organohalide-cycling microorganisms) identified as mediating organohalide cycling (Fig. 3c). Especially, the largely expanded diversity of RDase-containing microorganisms represented a vast yet-to-be-explored resource in ocean for potential bioremediation applications. Compared to the HyDase- and RDase-containing dehalogenation prokaryotes, FlaHase-based halogenation microorganisms had larger genome sizes, lower protein-coding density, and a higher proportion of fast growers (Supplementary Fig. 7), which suggested their distinctively different niche-colonizing capabilities and ecological roles40. Interestingly, in contrast to Pseudomonadota as predominant host microorganisms of FlaHase and HyDase with a comparatively small size range (<1000 amino acids; Fig. 3a), both RDases and their host microorganisms were phylogenetically diverse, and the marine RDases were clustered based on the taxonomy and redox-potential-related metabolisms of their host microorganisms: (1) obligate anaerobes, these anaerobic OHRB were capable of sulfate reduction and/or fermentation, and contained small-size RDases (<500 amino acids) that accounted for 78.01% of all anaerobic-OHRB RDases; (2) facultative anaerobes and aerobes, these OHRB were major hosts of the medium- and large-size RDases accounting for 51.14% and 42.72% of all facultative-anaerobic and aerobic OHRB RDases, respectively (Supplementary Data 3).
Structure-based phylogeny of RDases and associated “microorganism-enzyme-organohalide” patterns
Due to the high percentages of novel RDases and functionally novel RDase-hosting microorganisms in ocean, the RDase was selected to investigate the phylogeny of AlphaFold2-predicted structures of marine RDases and associated “microorganism-enzyme-organohalide” patterns (Fig. 4). Notably, marine RDase structures can be clustered into 7 distinct evolutionary groups (groups 1–7, or Gstr 1–7) with varied preferences of host microorganisms and associated growth niches, of which phylogeny is aligned with RDase sequence-based phylogenetic clustering (Gseq), i.e., Gstr 1, 2, 3, 4, 5, 6 and 7 mainly derived from Gseq 11, 5, 10, 4, 1/2/3/6, 7/8 and 9, respectively (Fig. 4a; Supplementary Data 4). For example, group-1 and group-2 RDases are preferentially hosted by aerobic and seawater-originated populations of α/γ-Proteobacteria (Fig. 4b). Moreover, the group-1 and group-2 RDases exhibit larger channel volumes (Fig. 4c; ANOVA, p < 0.05) and greater accessible vertices (Fig. 4d; ANOVA, p < 0.05) relative to other RDase groups. In contrast, group-3 and group-4 RDases are mainly employed by anaerobic microorganisms (e.g., sulfate-reducing bacteria, SRB) from low-redox sedimentary environments, including the cold seep (Fig. 4b). The groups 5–7 RDases have a comparatively more diverse range of host OHRB relative to the groups 1–4 RDases, and consequently present in all marine habitats (Fig. 4b).

a Structure-based phylogenetic tree of 100 representative marine RDases based on AlphaFold2-predicted structures. b Distribution of the 7 RDase groups across the different marine water and sediment habitats. The predicted volume of cavity (c) and max accessible vertex area (d) of the 7 groups of marine RDases. The number of RDases contained in each group is shown at the top of the graphs. Central line and whiskers in each box represent the median and 1.5 times the interquartile range, respectively. Boxes indicate the interquartile range between 25th and 75th percentiles. Data are shown as circular symbols, and mean values are shown as squares. The same definition is applicable to the boxplots shown in (e). e The RDase-organohalide binding energies for halogenated pyrrole, PCE, and halomethane. The number of RDase-organohalide pairs analyzed for binding energies in each RDase group is shown at the top of the graphs. Refer to Supplementary Fig. 8 for the RDase-organohalide binding energies for halogenated alkaloid, benzene, diphenyl ether, and phenol compounds. f Correlation between the RDase-organohalide binding energy and max accessible vertex area for PCE, tetrachloride, and hexabromo-2,2’-bipyrrole (n = 100). Linear regression model with two-sided test was used for the statistical analysis. The gray area around the smooth line indicates the 95% confidence interval. g Four model patterns of “microorganism-enzyme-organohalide” for reductive dehalogenation of organohalides in the ocean. Refer to Supplementary Fig. 9 for their detailed metabolic potential in the four models. Source data are provided as a Source Data file.
Based on a ligand library comprising 66 representative ocean-sourced organohalides (Supplementary Data 5), the binding energies for 6600 protein-ligand (RDase-organohalide) pairs were calculated to range from 0.59 to −8.51 kcal/mol (Fig. 4e; Supplementary Fig. 8; Supplementary Data 6). The ocean-sourced RDases generally have high affinity for prevalent marine organohalides, including halogenated pyrroles, phenols, and benzenes (Fig. 4e; Supplementary Fig. 8). Moreover, tetrachloroethylene (PCE), as a common dechlorination substrate of diverse RDases has a mean RDase-PCE binding energy of −3.24 kcal/mol, within the dechlorination-active range (Fig. 4e). In contrast, the lower binding affinity of RDases to halomethanes (Fig. 4e) suggests that the dehalogenation of halomethanes in the ocean is probably mediated by other processes rather than RDase-based reductive dehalogenation. Further exploration of the substrate specificity shows an intriguing observation that the molecular size of organohalides significantly affects the RDase-organohalide binding specificity. Specifically, small molecular organohalides (e.g., PCE and halomethane) exhibit RDase-independent binding energies (Fig. 4e; ANOVA, p > 0.05). Conversely, the RDase-organohalide binding energy of large molecular organohalides (e.g., halogenated pyrroles) is RDase-specific (ANOVA, p < 0.05), e.g., groups 1, 2, and 5 RDases exhibit higher affinities for the large molecular organohalides than other RDase groups (Fig. 4e). To elucidate the underlying mechanism, structural features of RDases have been examined to show that binding energy difference may arise from variations in max accessible vertices of these RDases. Specifically, binding energies of small molecular organohalides (PCE and halomethanes) show no significant correlation with protein max accessible vertices (p > 0.05), whereas the binding energies of large molecular organohalides (hexabromo-2,2’-bipyrrole) exhibit a significant positive correlation with the protein max accessible vertices (p < 0.001) (Fig. 4f). These observations indicate that the binding energy of large molecules is more easily affected by the enzyme’s channel structure and active site accessibility relative to small molecules.
Based on above-mentioned findings, four representative models are summarized to show the “microorganism-enzyme-organohalide” patterns for RDase-catalyzing dehalogenation of organohalides in ocean (Fig. 4g; Supplementary Fig. 9; Supplementary Data 7): Model-I for reductive dehalogenation and aerobic degradation of organohalides, aerobic bacteria of α/γ-Proteobacteria initially employ group-1 and group-2 RDases to remove halogens from halogenated pyrroles and similar large molecular organohalides, and then degrade the dehalogenation products to achieve complete mineralization (Fig. 4g; Supplementary Fig. 9a); Model-II for facultative dehalogenation and sulfate reduction, anaerobic SRB originated from cold seeps employ group-3 or group-4 RDases to dehalogenate organohalides, or utilize Dsr to mediate sulfate reduction with the sulfate and organohalides as competitive electron acceptors (Fig. 4g; Supplementary Fig. 9b); Model-III for reductive dehalogenation and anaerobic degradation of organohalides, anaerobic SRB initially dechlorinate chlorophenols or similar organohalides, and then couple degradation of dechlorination products with sulfate reduction; this process is generally proceeded under oligotrophic marine water and sediment conditions in shortage of carbon sources (Fig. 4g; Supplementary Fig. 9c); Model-IV for dechlorination and fermentation, the deep-sea sediment-colonized fermenting OHRB initially dehalogenate organohalides and then ferment dehalogenation products to generate acetate and H2, which can further support growth of methanogens and other microorganisms (Fig. 4g; Supplementary Fig. 9d). These “microorganism-enzyme-organohalide” patterns, together with the FlaHase-based halogenation process, can drive the assembly of a FlaHase-RDase-host microbiome for the halogenation and dehalogenation of organohalides in the ocean (Fig. 4h).
Cultivation evidence for the RDase-based “microorganism-enzyme-organohalide” patterns
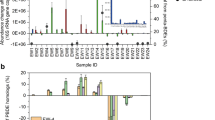
To experimentally confirm the above-mentioned OHRB-mediated metabolic networks in RDase-based dehalogenation microbiomes, 32 marine sediment samples were collected from varied geographic sites (Supplementary Data 8) to set up dehalogenation cultures with PCE or tetrachloride as an electron acceptor to support organohalide respiration of OHRB (Fig. 5a). PCE was selected as a model substrate for its broad reactivity, optimal energy yield, and dual natural-anthropogenic sources. In contrast to observing no tetrachloride dechlorination activity after three months of incubation, 12 out of the 32 cultures were shown to dechlorinate PCE to cis-dichloroethene (cis-DCE) via trichloroethene (TCE), notably, without generation of trans-DCE that was generally present as a PCE/TCE dechlorination product of obligate OHRB, i.e., Dehalococcoides, Dehalogenimonas, and Dehalobium29 (Fig. 5a). 16S rRNA gene-based high-throughput sequencing analyses confirmed the presence of facultative OHRB and the near absence of obligate OHRB (Fig. 5a), further indicating the different OHRB to mediate reductive dehalogenation of organohalides in the marine and terrestrial environments.

a PCE dechlorination and sulfate reduction in marine sediment cultures and presence of known OHRB. b RDase-host MAGs retrieved from metagenomics of marine sediment cultures and characterization of their RDase genes (rdhA). metabolic potentials of LS2-bin13 of phylum Desulfobacterota (c) and LS2-bin5 of phylum Chloroflexota (d). e cold seep culture-inferred interaction network among fermenting and sulfate-reducing OHRB (Ferment-OHRB and SR-OHRB) and associated microorganisms. ANME, anaerobic methanotrophic archaea. Refer to Supplementary Data 9 for the detailed annotation of functional genes with the key metabolic pathways shown in Fig. 5c–e. Source data are provided as a Source Data file.
To identify the OHRB and their metabolic networks in the PCE-dechlorinating cultures, 34 RDase-hosting MAGs were retrieved from metagenomic data of these cultures (Fig. 5b). MAGs of Halodesulfovibrio, Desulfoluna, Carboxylicivirga and Marinifilum genera were identified as dominant OHRB in the PCE-dechlorinating cultures based on 16S rRNA gene sequencing analysis (Fig. 5a). In addition, the 34 MAGs were assigned to 8 bacterial phyla, all of which were facultative OHRB with metabolisms of dissimilatory sulfate reduction, fermentation, and/or nitrate reduction (Fig. 5b–d). Particularly, instead of well-characterized terrestrial sources of obligate OHRB Dehalococcoidia from the same phylum of Chloroflexota, Anaerolineae was identified as the facultative OHRB in culture LS2 (Fig. 5d), confirming the RDase-host Anaerolineae as a major reductive dehalogenation population of the Chloroflexota in the ocean. In addition, RDases with length over 1000 amino acids were identified in the facultative anaerobes of Sulfitobacter that were capable of aerobic respiration and nitrate reduction (Fig. 5b). Based on the MAGs-inferred metabolic potentials, similar “microorganism-enzyme-organohalide” models of the facultative OHRB-mediated RDase-based dehalogenation were observed in the PCE-dechlorinating cultures (Fig. 5b–d; Supplementary Data 9) with these in global marine environments (Fig. 4g). These facultative OHRB including fermenting OHRB and sulfate-reducing OHRB formed a metabolic interaction network with dissimilatory sulfate-reducing bacteria, methanogenic archaea and other microorganisms (e.g., methane-oxidizing archaea in PCE-dechlorinating culture SY4), based on the carbon/electron sources and organohalides/sulfate as electron acceptors (Fig. 5e; Supplementary Data 9). Consequently, these cultivation experiments further corroborated the metagenomics-derived dehalogenation patterns in the ocean.
Discussion
This study provides the insight into the global-scale microbially mediated organohalide cycling in the ocean, which greatly expands the diversity of organohalide-cycling genes and microorganisms. The microbial-driven cycling of organohalides in marine environments significantly impacts the global biogeochemical cycles and climate systems, which may generate far-reaching ecological consequences. For example, marine microorganisms employ halogenases to synthesize a diverse range of recalcitrant organohalides for varied biological functions (e.g., signal transduction and chemical defense5,6,7), playing an important role in marine cycling of organohalides and organic carbon. Subsequently, these recalcitrant organohalides can be converted into labile organic carbon through dehalogease-catalyzed carbon-halogen bond cleavage, providing both energy and carbon sources for marine microbiomes. In oligotrophic oceans, organohalides have comparable concentrations to other organic compounds41, enabling a variety of marine microorganisms to function as facultative populations that employ organohalide-cycling activities to complement other metabolic processes (e.g., fermentation and sulfate reduction) and to enhance competitiveness17. Take the marine microorganism - Desulfoluna as an example, this facultative dehalogenator can couple bromobenzene-to-phenol conversion with sulfate reduction, linking organohalide respiration with sulfur cycling17. These dehalogenators can also act as metabolic hubs and provide carbon sources and reducing equivalents for methanogens and other microorganisms, which is particularly important to sustain marine microbiomes in oligotrophic oceans. Notably, scenarios are different for the organohalide cycling in terrestrial and marine environments. Terrestrial organohalides either originate from natural sources at relatively low concentrations compared to abundant organic matter in surrounding environments or exist as anthropogenic pollutants in high concentrations13. Therefore, organohalide-cycling microorganisms may play a minor role in organic matter cycling in pristine terrestrial environments, or evolve into obligate organohalide-cycling microorganisms under the selective pressure of organohalide pollution13. Take the obligate organohalide-respiring bacterium Dehalococcoides as an example, it is generally present in organohalide-polluted terrestrial environments and almost absent in the ocean42, which was a nitrogen-fixation microorganism and evolved to solely use organohalides as electron acceptors for energy metabolism43. Therefore, the above-mentioned different organohalide-derived selection pressures in the ocean and in terrestrial environments could play a central role in governing the community assembly and associated “microorganism-enzyme-organohalide” patterns in organohalide-cycling microbiomes.
The global-scale profiling of marine organohalide-cycling microbiomes is enabled by developing the HaloCycDB database that has advantages over KEGG and other databases in terms of accuracy and sensitivity, highlighting the importance of devising a function-specific database and integrating sequence similarity and conserved regions/motifs in homologous gene screening. The sequence similarity alone may bring in false organohalide-cycling genes with high sequence similarity but without key motifs (e.g., hydrogenases share high sequence similarity with reductive dehalogenases44), and underestimate novel organohalide-cycling genes that have low sequence similarity with current known genes, e.g., novel RDase groups 6–11 in Supplementary Fig. 5c. Therefore, both sequence similarity and conserved regions/motifs should be included as key screening parameters in developing functional gene databases, especially in exploring novel functional genes. In this study, the greatly expanded diversity of organohalide-cycling genes and microorganisms provides valuable resources for future biomedical and bioremediation applications. For example, through synthetic biology approaches, the novel RDases identified in this study could be further integrated into engineered strains such as VCOD-1545, broadening their substrate range and enhancing degradation efficacy. To further explore these bioresources, following studies are warranted: (1) metagenomics analyses can only predict the potential of gene functions and microbial activities, of which validation requires multi-omics data (e.g., transcriptomics and proteomics)46 and biogeochemical assays; nonetheless, current acquisition of marine meta-transcriptomics and proteomics data is challenged by sample collection, transportation and treatment during ocean expeditions47; (2) exploring the prodigious resources of novel organohalide-cycling genes and taxa urge the development of high throughput approaches for varied biophysiochemical tests, including the high throughput heterogeneous expression of organohalide-cycling genes; (3) mechanistic understanding of the intricate “microorganism-enzyme-organohalide” patterns for marine organohalide cycling necessitate the employment of artificial intelligence (AI)-driven data mining, which are largely depended on the development of biological AI models48. With these advancements, in-depth understanding and comprehensive exploration of the organohalide cycling in the ocean can be expected in the near future.
Methods
Development and assessment of organohalide-cycling gene database HaloCycDB
An organohalide-cycling gene database (HaloCycDB) was developed to support meta-omics analyses, which had both a core database and a full database. The core database included 221 functionally characterized organohalide-cycling genes, as well as host organisms and viruses (i.e., 194, 26 and 1 from prokaryotes, eukaryotes and virus, respectively), catalyzing reactions and substrates/products (Supplementary Data 10). Notably, the core database included eukaryotes- and virus-derived organohalide-cycling genes due to that these genes can potentially be transferred horizontally from eukaryotes and viruses to prokaryotes49,50. For the construction of the core database, we first conducted literature searches on PubMed using two sets of keywords “halogenase/dehalogenase” and “halo/dehalo + enzyme”. All functionally characterized halogenases and dehalogenases based on cultivation, physiochemical experiments, and/or multi-omics analyses were included in the core database, along with the conserved regions, functional domains, and motifs of these halogenase/dehalogenase-encoding genes. Moreover, information on the taxonomy of host organisms and viruses, as well as halogenase/dehalogenase-catalyzing reactions and substrates/products was collected and included in the core database. Subsequently, protein sequences of these characterized halogenase/dehalogenase genes were extracted from the Swiss-Prot51 and TrEMBL52 databases, of which the accuracy was manually checked based on their annotation and conserved-regions with InterPro53 and Pfam files using hmmsearch from HMMER54 (v3.1) with e-value ≤ 1e-5. For the motif filtering, a custom Python script (motif_search_ident.py) was created to perform a one-to-one search of the motif sequences with the target dehalogenase/halogenase gene sequences.
The full database contained 187,289 homologous genes with taxonomic information of their host organisms and viruses. The pre-full database sequences were mainly obtained from five public databases (i.e., NCBI nr55, COG56, arCOG57, eggNOG58, and KEGG34): (1) homologous genes containing keywords of “halogenase” and “dehalogenase” were retrieved and collected from the five public databases; (2) the five public databases were searched against the core database using USEARCH59 (v.11) to retrieve organohalide-cycling homologous genes with a global identity >30%, which could fully capture the distantly related but functionally similar sequences60,61,62. To prevent false positive results, the pre-full database was further filtered with the conserved regions and motifs of halogenase/dehalogenase genes, using the same filtering methods as described for the core database construction. Subsequently, the pre-full database was searched against the NCBI RefSeq databases59,63 of bacteria, archaea, and eukaryotes using USEARCH (v.11) with an e-value ≤ 1e-6 and identity >30% to determine the taxonomy of these gene-host organisms and viruses. All gene sequences from the pre-full database were further clustered by CD-HIT64 (v4.8.1) at 100% identity with parameters ‘-c 1.0 aS 1.0 -aL 1.0’ to remove completely identical gene sequences and avoid the database sequence redundancy. Finally, the accuracy of the taxonomy identifier (TaxIds) was checked with TaxonKit63 (v.0.20.0) for the representative sequences annotated from NCBI Refseq database, which was further employed to transform and normalize TaxIds to seven taxonomic levels (i.e., kingdom/domain, phylum, class, order, family, genus, and species). These analyses enabled accurate and standardized host taxonomic identification of all representative sequences for full database construction.
To evaluate the accuracy of HaloCycDB, an artificial dataset including 10,000 organohalide-cycling gene sequences and 10,000 non-organohalide-cycling gene sequences (that were highly similar to the organohalide-cycling gene sequences) was constructed to compare the accuracy, false-negative rate, false-positive rate, sensitivity, and coverage of HaloCycDB with eggNOG, KEGG, COG, and arCOG. The dataset was searched against HaloCycDB, KEGG, COG, and arCOG using USEARCH with an identity >30%, and against the eggNOG database using eggNOG-mapper with an e-value of ≤1e-4 to obtain the accuracy of these databases for functional annotation. Organohalide-cycling gene sequences annotated to incorrect genes were identified as false-positive annotations, and the failed annotated organohalide-cycling genes were considered as false-negative annotations. The following formulas were used to calculate the accuracy (1), false-negative rate (2), false-positive rate (3), sensitivity (4), and coverage (5) for evaluating the accuracy of HaloCycDB:
HaloCycDB and Python scripts for identifying organohalide-cycling genes and microorganisms were set to be available on GitHub (https://github.com/metabiolab-wang/HaloCycDB).
Global-scale marine metagenomic datasets collection, assembly, and binning
Metagenomes of the Tara Oceans31, BioGEOTRACES65, Hawaiian Ocean Time-series66, Bermuda-Atlantic Time-series Study67, and Malaspina68 expeditions were downloaded from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (NCBI-SRA). To further complement these ocean expedition metagenomic datasets, keywords including “seawater”, “ocean sediment”, “cold seep sediment”, “hydrothermal vent sediment”, and “trench sediment” were employed to retrieve relevant literatures from PubMed (https://pubmed.ncbi.nlm.nih.gov/), and associated metagenomic data were collected from the NCBI-SRA database. The water and sediment samples were collected from 506 sites of four different depth-based water layers (epipelagic, mesopelagic, bathypelagic and abyssopelagic layers with a depth of 0–10,905 m) and four typical sedimentary environments (cold seep, hydrothermal vent, trench, and other sediment) in the ocean (Supplementary Fig. 2a, b). To catch the organohalide-cycling potential of cross-kingdom microorganisms, in contrast to unfiltered sediment, water samples were selected based on three groups of filtering size, i.e., 0–3 μm, prokaryote-rich; 0–20 μm, particle-rich; and unfiltered (Supplementary Fig. 2c; Supplementary Data 1). These metagenomic datasets-associated sampling and environmental information, including ecosystem classification, latitude, longitude, and water depth, were manually curated in the corresponding literature (Supplementary Data 1).
Metagenomic raw reads data were filtered to remove low-quality bases/reads using trim_galore69 (v0.6.10) with default parameters. Clean reads from each sample were assembled individually into contigs using MEGAHIT v1.2.9 (k-mer: 21,29,39,59,79,99,119,141)70 with subsequent quality assessment using QUAST71 (v5.0.2). Contigs were taxonomically assigned to taxa using both CAT72 (v5.2.3) and Kaiju73 (v1.9.2) to improve the accuracy and breadth of annotations. The contigs were annotated with CAT by predicting open reading frames (ORFs) with Prodigal74 (v2.6.3; parameter: -meta) and by comparing them with DIAMOND blastp75 (v2.1.7) to the non-redundant set of proteins in GTDB (GTDB taxonomy release_214)76. In addition, the contigs annotation with Kaiju was performed utilizing the NCBI nr database that included bacteria, archaea, viruses, fungi, and other eukaryotic microorganisms for annotating contigs with default parameters.
For metagenomic binning, MAGs were constructed from the contigs of over 1000 bp using three different binning methods (i.e., --metabat277 --maxbin278 –concoct79) in the metaWRAP80. MAGs were further refined using the bin_refinement module of metaWRAP. To obtain optimal genome quality, metagenomic sequencing reads were further mapped to each MAG and then reassembled with metaSPAdes81 via the reassemble_bins module of metaWRAP. CheckM82 (v.1.0.12) with lineage-specific marker sets was used to assess the completeness and contamination of each MAG. dRep83 (v3.4.3) was used to dereplicate high- and medium-quality MAGs (completeness ≥50% and contamination ≤10%) at 95% average nucleotide identity. The dereplicated MAGs with a quality score (completeness − 5 × contamination) ≥50 were retained for downstream analysis. MAGs were taxonomically assigned using GTDB-tk v2.1.0 with reference to GTDB taxonomy release_21484. The phylogenetic tree of MAGs was constructed based on the multiple sequence alignment of 40 specific marker genes retrieved from MAGs using fetchMGs85 (v1.1). The phylogenetic tree was inferred by FastTree86 (v2.1.11) under the model WAG + GAMMA and visualized in iTOL87 v6. To determine the relative abundance of MAGs in each sample, clean reads were mapped to dereplicated MAGs using (v 0.6.0, https://github.com/wwood/CoverM/) with parameters ‘-genome’ and ‘-m rpkm’ to calculate RPKM values. The ORFs were predicted from each MAG by Prodigal (v2.6.3; parameter: -meta). The predicted ORFs were functionally annotated using eggnogmapper58 (v 2.1.12) with an e-value ≤ 1e-5. Metabolic pathways of these MAGs were predicted using the KEGG server (BlastKOALA)88 and METABOLIC89 (v4.0). The genome size and GC content were calculated using CheckM82 (v1.0.12). Protein coding density was calculated as the number of predicted proteins per kilobase of the genome. Based on codon usage bias, the maximum growth rate of bacteria was predicted using the R package gRodon90 (v2.3.0). The minimum doubling time (MDT) was further calculated based on the tight relationship between codon usage bias and bacterial maximum growth rate using the ‘predictGrowth’ function in the gRodon package. In case of incomplete genomes, the function parameter was set to ‘partial’. Only bacteria with a predicted MDT < 5 h were considered fast growers in this study.
Annotation, phylogeny, and abundance of organohalide-cycling genes and host microorganisms
To identify organohalide cycling genes, ORFs of contigs or MAGs were predicted using Prodigal74 (v2.6.3; parameter: -meta), with which organohalide-cycling genes were identified based on both protein sequence similarity and conserved regions/motifs against HaloCycDB. In brief, protein sequences were searched against HaloCycDB using DIAMOND blastp75 (v2.1.7) with identity ≥30% and e-value ≤ 1e-4. Hmmsearch (e-value ≤ 1e-4) from hmmer54 (v3.1) was also applied to identify homologs of organohalide-cycling genes based on the conserved regions. Protein sequences being annotated as organohalide-cycling genes were further classified into each gene family and filtered using functionally conserved motifs. The organohalide-cycling genes without motifs were filtered with criteria of sequence similarity of ≥50% with confirmed organohalide-cycling gene sequences. The taxa of medium-high quality MAGs (completeness ≥50% and contamination ≤10%) and contigs with length over than 5 kb containing organohalide-cycling genes were considered as organohalide-cycling microorganisms. To determine the relative abundance of organohalide-cycling genes in contigs, clean reads were mapped to the contigs using CoverM (v 0.6.0, https://github.com/wwood/CoverM/) with parameter ‘-contig’ and cut-off values of 75% identity and 75% alignment coverage for mapped reads, which generated coverage profiles of each contig and normalized as RPKM. The RPKM were calculated using the equation:
Where numReads is the number of reads mapped to a sequence; seqLength is the length of the sequence; totalNumReads is the total number of mapped reads of a sample. Then, the relative abundance of organohalide-cycling genes was calculated by dividing RPKM values of individual genes by the sum of RPKM values of all genes. Phylogenetic trees of halogenases (FlaHase) and dehalogenases (HyDase and RDase) were used to construct phylogenetic clades of organohalide-cycling genes. Briefly, the protein sequences of contigs-retrieved FlaHases, HyDases, and RDases, together with the corresponding reference sequences from HaloCycDB, were first clustered at 90%, 75%, and 90% identity, respectively, using CD-HIT64 (v4.8.1). Then, representative protein sequences of FlaHase, HyDase, and RDase were further aligned using MUSCLE91 (v3.8.1) and trimmed using TrimAL92 (v1.4) with default options. Maximum-likelihood trees of FlaHase, HyDase, and RDase were constructed using FastTree (v2.1.11)86 under the model WAG + GAMMA. All phylogenetic trees were visualized using iTOL (v6)87.
Biomass variations in microbiomes in the four water depth layers and four sedimentary environments were considered in analyzing the distribution of organohalide-cycling genes in marine regions/habitats (Supplementary Data 11), specifically through the following steps: (1) volume estimation, the total volumes of global seawater and seafloor sediment were determined as described37,93. The seawater and sediment volumes of specific regions/habitats were allocated proportionally according to water depth and areal percentage, respectively36,94,95; (2) total microbial cell abundance estimation, cell concentration data for different regions were obtained from previous studies96,97,98,99,100,101,102,103,104,105; the regional total microbial cell abundances were calculated by multiplying region-specific microbial cell concentrations by their corresponding volumes; then, the estimated regional microbial cell abundances were refined using the total microbial cell abundance derived from seawater and seafloor sediments (as calculated in Step 1). Subsequently, the abundances of organohalide-cycling genes in each region were proportionally adjusted according to the corrected microbial cell abundance ratios. Finally, to minimize unequal-sampling-size derived bias, the total abundance of organohalide cycling genes in each habitat was normalized by dividing the sample numbers in corresponding habitats.
Protein structure prediction and molecular docking
Based on the phylogeny of the above-described RDase genes (Supplementary Fig. 5), 100 RDases were selected from all clustered groups of the marine RDase gene phylogenetic tree to further predict their protein structures using AlphaFold2106 (v2.3) (Supplementary Data 4). The AlphaFold2 prediction generated five models for each RDase, and the top-ranked model (ranked_0) with the highest average pLDDT score was selected for subsequent analyses. Protein structural visualizations were generated using PyMOL107 (v2.6). The structural pair alignment was performed using 3Di and amino acid-based alignment, implemented with Foldseek108 (v10.941cd33). A phylogenetic tree based on the RDase protein structures was constructed using Foldtree109 (https://github.com/DessimozLab/fold_tree) and visualized using iTOL87 (v6).
To investigate the distribution of different structure-based RDase groups in marine habitats, we considered the potential bias caused by limited sampling of the structure tree. Therefore, we opted to use the distribution of marine habitats corresponding to sequence-based RDase groups that aligned with the phylogenetic clustering of the structure-based RDase groups, as a proxy for the distribution and information in the structure-based RDase tree, thereby minimizing bias introduced by the limited sampling of the structure tree.
Molecules in sdf files were retrieved from PubChem110 and further converted to mol2 files using Open Babel111 (v2.3.1) for molecular docking. LeDock (v1.0) was used to predict the binding poses of 66 natural organohalides in different RDases (RMSD: 1.0 Å; number of binding poses: 20). The properties of protein clefts/pockets, including the total volume and accessible vertices of the largest surface cleft, were analyzed using the PDBsum server112 to provide insight into ligand molecule binding mechanisms113.
Cultivation of marine organohalide-respiring microorganisms
A total of 32 marine sediment samples were collected from 8 ocean regions and shipped to the laboratory at an ambient temperature (Supplementary Data 8). Microcosm setup was conducted in an anaerobic chamber soon after arrival of these samples as described16,114. Briefly, 90 mL of bicarbonate-buffered mineral salt medium amended with 10 mM lactate, 450 mM NaCl, and 20 mM Na2SO4 was dispensed into 160 mL serum bottles containing around 2 g of sediment samples. The serum bottles were sealed with black butyl rubber septa and secured with aluminum crimp caps. To maintain low redox potential, 0.2 mM L-cysteine and 0.2 mM Na2S·9H2O were added as reductants, and 0.02 mM resazurin was added as a redox indicator. Microcosms were spiked with 0.2 mM perchloroethene (PCE) or tetrachloride as an electron acceptor. All cultures were set up in triplicate and incubated in the dark at 30 °C without shaking. Duplicate abiotic controls (without microbial inocula) and no-organohalide controls (without PCE and tetrachloride injection) were set up for each experiment.
Analytical methods
Chloroethenes13 and tetrachloride115 were analyzed as described. Headspace samples of chloroethenes, ethene and tetrachloride were injected manually with a gastight, luer lock syringe (Hamilton, Reno, NV, USA) into a gas chromatograph (GC) with a flame ionization detector (Agilent 7890B, Wilmington, DE, USA) and a Gas-Pro column (30 m × 0.32 mm; Agilent J&W Scientific, Folsom, CA, USA). Nitrogen, hydrogen, and air were used as the carrier, fuel, and oxidant gases, respectively. Sulfide was measured using a UV-Vis spectrophotometer (UV-2100, Shimadzu, Kyoto, Japan) at a wavelength of 675 nm as described in the methylene blue method114,116,117.
DNA extraction, sequencing, and analyses
Samples for genomic DNA (gDNA) extraction were collected from PCE-dechlorinating cultures, of which gDNA was extracted using the FastDNA Spin Kit DNA extraction kit (MP Biomedicals, Carlsbad, CA, USA) according to the manufacturer’s instructions114. For the 16S rRNA gene amplicon sequencing, V4-V5 regions of the 16S rRNA genes were amplified using the primer set 515 F/909 R with unique 8-mer barcodes for multiplex PCR amplicons, and amplicons were purified as described previously118. Then, the purified PCR products were pooled and sequenced using the Illumina NovaSeq 6000 platform (PE250; Illumina; San Diego, CA, USA) by MAGIGENE (Shenzhen, China). Paired-end reads (2 × 250 bp) were processed to generate amplicon sequence variants (ASVs) using the DADA2119 (v1.6) package in R (v4.3.2), including quality filtering, dereplication, merging, and chimera removal. Taxonomic classification was conducted using the RDP naive Bayesian classifier in conjunction with the GTDB (r214) download from the DADA2 website118. To ensure unbiased microbial community analysis, ASV abundance tables were normalized to a uniform sequencing depth using the vegan package120 (v.2.6.4). For metagenomic analysis, DNA library preparation and Illumina HiSeq sequencing services were provided by BGI (Shenzhen, China). The metagenomic data analyses followed the same analytical procedures as the above-mentioned global marine metagenomic data analyses.
Statistical analysis
Statistical analyses were carried out in R v4.2.3. One-way ANOVA with post hoc LSD test was performed to assess the statistically significant differences between the compared groups using the R package agricolae121. The linear regression model in the R package stats122 was used to analyze the correlation between the RDase-organohalide binding energy and max accessible vertex. p values less than 0.05 were considered significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequencing reads of the 16S rRNA gene amplicons and metagenomic data generated in this study have been deposited into the European Nucleotide Archive database with accession numbers PRJEB88795 (https://www.ebi.ac.uk/ena/browser/view/PRJEB88795) and PRJEB88842 (https://www.ebi.ac.uk/ena/browser/view/PRJEB88842), respectively. Protein structures of RDases are available at https://doi.org/10.6084/m9.figshare.30305311. HaloCycDB for identifying organohalide-cycling genes and microorganisms were set to be available on GitHub (https://github.com/metabiolab-wang/HaloCycDB). Source data are provided with this paper.
Code availability
Python scripts and codes related to the identification of genes involving in organohalide cycling are publicly available at Github (https://github.com/metabiolab-wang/HaloCycDB) and Zenodo123 (https://doi.org/10.5281/zenodo.17299792).
References
York, A. Marine biogeochemical cycles in a changing world. Nat. Rev. Microbiol. 16, 259–259 (2018).
Danovaro, R., Levin, L. A., Fanelli, G., Scenna, L. & Corinaldesi, C. Microbes as marine habitat formers and ecosystem engineers. Nat. Ecol. Evol. 8, 1407–1419 (2024).
Heneghan, R. F. et al. The global distribution and climate resilience of marine heterotrophic prokaryotes. Nat. Commun. 15, 6943 (2024).
Agarwal, V. et al. Enzymatic halogenation and dehalogenation reactions: pervasive and mechanistically diverse. Chem. Rev. 117, 5619–5674 (2017).
Gribble, G. W. Naturally occurring organohalogen compounds—a comprehensive review. Prog. Chem. Org. Nat. Prod. 121, 1–546 (2023).
Borchardt, S. et al. Reaction of acylated homoserine lactone bacterial signaling molecules with oxidized halogen antimicrobials. Appl. Environ. Microbiol. 67, 3174–3179 (2001).
Hossaini, R. et al. Efficiency of short-lived halogens at influencing climate through depletion of stratospheric ozone. Nat. Geosci. 8, 186–190 (2015).
Teuten, E. L., Xu, L. & Reddy, C. M. Two abundant bioaccumulated halogenated compounds are natural products. Science 307, 917–920 (2005).
Fakhraee, M. et al. The history of Earth’s sulfur cycle. Nat. Rev. Earth Environ. 6, 106–125 (2024).
Regnier, P., Resplandy, L., Najjar, R. G. & Ciais, P. The land-to-ocean loops of the global carbon cycle. Nature 603, 401–410 (2022).
Atashgahi, S., Häggblom, M. M. & Smidt, H. Organohalide respiration in pristine environments: implications for the natural halogen cycle. Environ. Microbiol. 20, 934–948 (2018).
Butler, A. & Sandy, M. Mechanistic considerations of halogenating enzymes. Nature 460, 848–854 (2009).
Qiu, L. et al. Organohalide-respiring bacteria in polluted urban rivers employ novel bifunctional reductive dehalogenases to dechlorinate polychlorinated biphenyls and tetrachloroethene. Environ. Sci. Technol. 54, 8791–8800 (2020).
Pimviriyakul, P., Jaruwat, A., Chitnumsub, P. & Chaiyen, P. Structural insights into a flavin-dependent dehalogenase HadA explain catalysis and substrate inhibition via quadruple π-stacking. J. Biol. Chem. 297, 100952 (2021).
Chan, P. W. et al. Defluorination capability of l-2-haloacid dehalogenases in the HAD-like hydrolase superfamily correlates with active site compactness. ChemBioChem 23, e202100414 (2022).
Wang, S. et al. Genomic characterization of three unique Dehalococcoides that respire on persistent polychlorinated biphenyls. Proc. Natl. Acad. Sci. USA 111, 12103–12108 (2014).
Peng, P. et al. Organohalide-respiring Desulfoluna species isolated from marine environments. ISME J. 14, 815–827 (2020).
Raes, B. et al. Aminobacter sp. MSH1 mineralizes the groundwater micropollutant 2, 6-dichlorobenzamide through a unique chlorobenzoate catabolic pathway. Environ. Sci. Technol. 53, 10146–10156 (2019).
Murdoch, R. W. et al. Identification and widespread environmental distribution of a gene cassette implicated in anaerobic dichloromethane degradation. Glob. Change Biol. 28, 2396–2412 (2022).
Shrivastava, N., Prokop, Z. & Kumar, A. Novel LinA type 3 δ-hexachlorocyclohexane dehydrochlorinase. Appl. Environ. Microbiol. 81, 7553–7559 (2015).
van Hylckama Vlieg, J. E. et al. Halohydrin dehalogenases are structurally and mechanistically related to short-chain dehydrogenases/reductases. J. Bacteriol. 183, 5058–5066 (2001).
Giordano, N. et al. Genome-scale community modelling reveals conserved metabolic cross-feedings in epipelagic bacterioplankton communities. Nat. Commun. 15, 2721 (2024).
Gralka, M., Szabo, R., Stocker, R. & Cordero, O. X. Trophic interactions and the drivers of microbial community assembly. Curr. Biol. 30, R1176–R1188 (2020).
Callaway, E. These are the 20 most-studied bacteria—the majority have been ignored. Nature 637, 770–771 (2025).
Kapinusova, G., Lopez Marin, M. A. & Uhlik, O. Reaching unreachables: obstacles and successes of microbial cultivation and their reasons. Front. Microbiol. 14, 1089630 (2023).
Rappé, M. S. & Giovannoni, S. J. The uncultured microbial majority. Annu. Rev. Microbiol. 57, 369–394 (2003).
Schloss, P. D. & Handelsman, J. Status of the microbial census. Microbiol. Mol. Biol. Rev. 68, 686–691 (2004).
Chen, C. et al. Influence of redox conditions on the microbial degradation of polychlorinated biphenyls in different niches of rice paddy fields. Soil Biol. Biochem. 78, 307–315 (2014).
Adrian, L. & Löffler, F. E. Organohalide-respiring bacteria (Springer, 2016).
Daniel, R. The metagenomics of soil. Nat. Rev. Microbiol. 3, 470–478 (2005).
Sunagawa, S. et al. Tara Oceans: towards global ocean ecosystems biology. Nat. Rev. Microbiol. 18, 428–445 (2020).
Paoli, L. et al. Biosynthetic potential of the global ocean microbiome. Nature 607, 111–118 (2022).
Li, L. et al. Globally distributed Myxococcota with photosynthesis gene clusters illuminate the origin and evolution of a potentially chimeric lifestyle. Nat. Commun. 14, 6450 (2023).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Darzi, Y., Falony, G., Vieira-Silva, S. & Raes, J. Towards biome-specific analysis of meta-omics data. ISME J. 10, 1025–1028 (2016).
Nunoura, T. et al. Hadal biosphere: insight into the microbial ecosystem in the deepest ocean on Earth. Proc. Natl. Acad. Sci. USA 112, E1230–E1236 (2015).
Whitman, W. B., Coleman, D. C. & Wiebe, W. J. Prokaryotes: the unseen majority. Proc. Natl.Acad. Sci. USA 95, 6578–6583 (1998).
Hug, L. A. et al. Overview of organohalide-respiring bacteria and a proposal for a classification system for reductive dehalogenases. Philos. Trans. R. Soc., B 368, 20120322 (2013).
Kunka, A., Damborsky, J. & Prokop, Z. Haloalkane dehalogenases from marine organisms. Methods Enzymol. 605, 203–251 (2018).
Rodríguez-Gijón, A. et al. Linking prokaryotic genome size variation to metabolic potential and environment. ISME Commun. 3, 25 (2023).
Gribble, G. W. Naturally occurring organohalogen compounds. Acc. Chem. Res. 31, 141–152 (1998).
Xu, G., Zhao, X., Zhao, S., Rogers, M. J. & He, J. Salinity determines performance, functional populations, and microbial ecology in consortia attenuating organohalide pollutants. ISME J. 17, 660–670 (2023).
Lee, P. K., He, J., Zinder, S. H. & Alvarez-Cohen, L. Evidence for nitrogen fixation by “Dehalococcoides ethenogenes” strain 195. Appl. Environ. Microbiol. 75, 7551–7555 (2009).
Molenda, O. et al. Insights into origins and function of the unexplored majority of the reductive dehalogenase gene family as a result of genome assembly and ortholog group classification. Environ. Sci. Process. Impacts 22, 663–678 (2020).
Su, C. et al. Bioremediation of complex organic pollutants by engineered Vibrio natriegens. Nature 642, 1024–1033 (2025).
Tarazona, S., Arzalluz-Luque, A. & Conesa, A. Undisclosed, unmet and neglected challenges in multi-omics studies. Nat. Comput. Sci. 1, 395–402 (2021).
Pinto, Y. & Bhatt, A. S. Sequencing-based analysis of microbiomes. Nat. Rev. Genet. 25, 829–845 (2024).
Brixi, G. et al. Genome modeling and design across all domains of life with Evo 2. Preprint at https://doi.org/10.1101/2025.02.18.638918 (2025).
Keeling, P. J. & Palmer, J. D. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 9, 605–618 (2008).
Yuan, W., Yu, J. & Li, Z. Rapid functional activation of horizontally transferred eukaryotic intron-containing genes in the bacterial recipient. Nucleic Acids Res. 52, 8344–8355 (2024).
Bairoch, A. & Boeckmann, B. The SWISS-PROT protein sequence data bank. Nucleic Acids Res. 20, 2019 (1992).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504 (2005).
Galperin, M. Y., Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269 (2015).
Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Archaeal clusters of orthologous genes (arCOGs): an update and application for analysis of shared features between Thermococcales, Methanococcales, and Methanobacteriales. Life 5, 818–840 (2015).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Tu, Q., Lin, L., Cheng, L., Deng, Y. & He, Z. NCycDB: a curated integrative database for fast and accurate metagenomic profiling of nitrogen cycling genes. Bioinformatics 35, 1040–1048 (2019).
Ji, M. et al. Biodiversity of mudflat intertidal viromes along the Chinese coasts. Nat. Commun. 15, 8611 (2024).
Song, X. et al. Rhizosphere-triggered viral lysogeny mediates microbial metabolic reprogramming to enhance arsenic oxidation. Nat. Commun. 16, 4048 (2025).
Shen, W. & Ren, H. TaxonKit: A practical and efficient NCBI taxonomy toolkit. J. Genet. Genom. 48, 844–850 (2021).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Anderson, R. F. GEOTRACES: Accelerating research on the marine biogeochemical cycles of trace elements and their isotopes. Ann. Rev. Mar. Sci. 12, 49–85 (2020).
Karl, D. M. & Church, M. J. Microbial oceanography and the Hawaii Ocean Time-series programme. Nat. Rev. Microbiol. 12, 699–713 (2014).
Michaels, A. F. & Knap, A. H. Overview of the US JGOFS Bermuda Atlantic Time-series Study and the Hydrostation S program. Deep Sea Res. Part II 43, 157–198 (1996).
Duarte, C. M. Seafaring in the 21st century: the Malaspina 2010 circumnavigation expedition. Limnol. Oceanogr. Bull. 24, 11–14 (2015).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Von Meijenfeldt, F. B., Arkhipova, K., Cambuy, D. D., Coutinho, F. H. & Dutilh, B. E. Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol. 20, 217 (2019).
Menzel, P., Ng, K. L. & Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7, 11257 (2016).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119 (2010).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794 (2022).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Wu, Y.-W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158 (2018).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 11, 2864–2868 (2017).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2020).
Mende, D. R., Sunagawa, S., Zeller, G. & Bork, P. Accurate and universal delineation of prokaryotic species. Nat. Methods 10, 881–884 (2013).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650 (2009).
Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82 (2024).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731 (2016).
Zhou, Z. et al. METABOLIC: high-throughput profiling of microbial genomes for functional traits, metabolism, biogeochemistry, and community-scale functional networks. Microbiome 10, 33 (2022).
Weissman, J. L., Hou, S. & Fuhrman, J. A. Estimating maximal microbial growth rates from cultures, metagenomes, and single cells via codon usage patterns. Proc. Natl. Acad. Sci. USA 118, e2016810118 (2021).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Kallmeyer, J., Pockalny, R., Adhikari, R. R., Smith, D. C. & D’Hondt, S. Global distribution of microbial abundance and biomass in subseafloor sediment. Proc. Natl. Acad. Sci. USA 109, 16213–16216 (2012).
LaRowe, D. E., Burwicz, E., Arndt, S., Dale, A. W. & Amend, J. P. Temperature and volume of global marine sediments. Geology 45, 275–278 (2017).
D’Hondt, S., Pockalny, R., Fulfer, V. M. & Spivack, A. J. Subseafloor life and its biogeochemical impacts. Nat. Commun. 10, 3519 (2019).
Glud, R. N. et al. High rates of microbial carbon turnover in sediments in the deepest oceanic trench on Earth. Nat. Geosci. 6, 284–288 (2013).
Jamieson, A. J., Fujii, T., Mayor, D. J., Solan, M. & Priede, I. G. Hadal trenches: the ecology of the deepest places on Earth. Trends Ecol. Evol. 25, 190–197 (2010).
Liu, R., Wang, L., Wei, Y. & Fang, J. The hadal biosphere: recent insights and new directions. Deep Sea Res. Part II 155, 11–18 (2018).
Hiraoka, S. et al. Microbial community and geochemical analyses of trans-trench sediments for understanding the roles of hadal environments. ISME J. 14, 740–756 (2020).
Hoehler, T. M. & Jørgensen, B. B. Microbial life under extreme energy limitation. Nat. Rev. Microbiol. 11, 83–94 (2013).
Johnson, H. P. & Pruis, M. J. Fluxes of fluid and heat from the oceanic crustal reservoir. Earth Planet. Sci. Lett. 216, 565–574 (2003).
Edwards, K. J., Wheat, C. G. & Sylvan, J. B. Under the sea: microbial life in volcanic oceanic crust. Nat. Rev. Microbiol. 9, 703–712 (2011).
Hu, S. K. et al. Microbial eukaryotic predation pressure and biomass at deep-sea hydrothermal vents. ISME J. 18, wrae004 (2024).
Briggs, B. et al. Macroscopic biofilms in fracture-dominated sediment that anaerobically oxidize methane. Appl. Environ. Microbiol. 77, 6780–6787 (2011).
Jørgensen, B. B. & Boetius, A. Feast and famine—microbial life in the deep-sea bed. Nat. Rev. Microbiol. 5, 770–781 (2007).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Schrodinger, L. L. C. The PyMOL molecular graphics system, version 1.8 (New York, NY, USA, 2015).
Van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–246 (2024).
Moi, D. et al. Structural phylogenetics unravels the evolutionary diversification of communication systems in gram-positive bacteria and their viruses. Nat. Struct. Mol. Biol. 1–11 https://doi.org/10.1038/s41594-025-01649-8 (2025).
Kim, S. et al. PubChem 2023 update. Nucleic Acids Res. 51, D1373–D1380 (2023).
O’Boyle, N. M. et al. Open Babel: An open chemical toolbox. J. Cheminform. 3, 1–14 (2011).
Laskowski, R. A. PDBsum 1: a standalone program for generating PDBsum analyses. Protein Sci. 31, e4473 (2022).
Lee, D., Redfern, O. & Orengo, C. Predicting protein function from sequence and structure. Nat. Rev. Mol. Cell Biol. 8, 995–1005 (2007).
Wang, S. et al. Generation of zero-valent sulfur from dissimilatory sulfate reduction in sulfate-reducing microorganisms. Proc. Natl. Acad. Sci. USA 120, e2220725120 (2023).
Ding, C., Zhao, S. & He, J. AD esulfitobacterium sp. strain PR reductively dechlorinates both 1, 1, 1-trichloroethane and chloroform. Environ. Microbiol. 16, 3387–3397 (2014).
Mousavi, M. & Sarlack, N. Spectrophotometric determination of trace amounts of sulfide ion based on its catalytic reduction reaction with methylene blue in the presence of Te (IV). Anal. Lett. 30, 1567–1578 (1997).
Lawrence, N. S., Davis, J. & Compton, R. G. Analytical strategies for the detection of sulfide: a review. Talanta 52, 771–784 (2000).
Liang, Z. et al. Mechanistic insights into organic carbon-driven water blackening and odorization of urban rivers. J. Hazard. Mater. 405, 124663 (2021).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Oksanen, J., Blanchet, F. G., Kindt, R. & Legendre, P. R Package ‘vegan’: Community Ecology Package. https://cran.r-project.org/web/packages/vegan/vegan.pdf. (2014).
de Mendiburu, F. & de Mendiburu, M. F. Package ‘agricolae’. R Package, version 1 (2019).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2020).
Wang, S. et al. Microbially-mediated halogenation and dehalogenation cycling of organohalides in the ocean. Zenodo. https://doi.org/10.5281/zenodo.17299792 (2025).
Becker, R. A. & Wilks, A. R. maps: Draw geographical maps. R package version 3.4.2. https://doi.org/10.32614/CRAN.package.maps (2023).
Acknowledgements
This study was financially supported by the Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai) (SML2021SP317 to S.W. and SML2024SP022 to Z.H.) and National Natural Science Foundation of China (42161160306 to S.W., 42107129 to Z.L. and 42430707 to Z.H.). We acknowledge the computational resources provided by the SongShan Lake HPC Center at Great Bay University.
Author information
Authors and Affiliations
Contributions
S.W. designed the study. Z.L. and N.Z. established the HaloCycDB. N.Z. conducted cultivation experiments. N.Z., Q.L., Z.L., H.W., Z.H., and S.W. analyzed and visualized the data. N.Z., Q.L., P.L., and K.Y. predicted the protein’s tertiary structure. K.Y. and C.Z. provided marine sediment samples. S.W., N.Z., and Q.L. drafted the manuscript. All authors reviewed the results and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Joeselle Serrana and Mirna Vazquez for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, N., Li, Q., Liang, Z. et al. Microbially-mediated halogenation and dehalogenation cycling of organohalides in the ocean. Nat Commun 16, 10670 (2025). https://doi.org/10.1038/s41467-025-65696-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65696-x
