
Abstract
Despite intensive development, P2X7 modulators have struggled in translation due to human genetic variability and species-dependent drug responses. Here, we identify PSFL1191, a portal-of-central-pocket (PCP)-site inhibitor selective for human and panda P2X7, but inactive against rodents. Cryo-EM structures revealed two distinct PCP sub-pockets: PCP1, a rigid base pocket demanding precise steric complementarity with PSFL1191, and PCP2, a conserved middle cavity targeted by JNJ-54175446, a clinical candidate unaffected by species differences. Species selectivity maps to a deep PCP1 motif (V312-Y295-M105-F103-P96). In P2rx7A312V/A312V mice, PSFL1191 markedly altered macrophage-mediated bacterial clearance and wound healing while preserving basal physiology, effects absent in wild-type animals. Our findings establish the structural basis for interspecies pharmacological divergence in P2X7 modulation and highlight transgenic models as powerful tools for predicting therapeutic efficacy, thereby enabling more precise and efficient drug discovery.
Similar content being viewed by others


Introduction
P2X receptors, comprising subtypes P2X1-7, play a crucial role in the pathogenesis of various diseases, including pain, tumors, inflammation, refractory chronic cough (RCC), rheumatoid arthritis, and depression1,2,3. Consequently, the P2X receptor family has emerged as a promising target in drug developments. Notably, antagonists like Gefapixant (AF-219)4, targeting the P2X3 receptor, have received clinical approval, while others such as BAY-1817080 (Bayer)5, BLU-5739 (Bellus)6, and S-600918 (Shionogi)7 are under clinical investigation, primarily for RCC. A few antagonists targeting P2X4 receptors, exemplified by NC-26008 for neuropathic pain and BAY23280659 for endometriosis, show promising therapeutic potential in Phase I or Phase II clinical assessments. Moreover, within the P2X family, the P2X7 receptor orchestrates a broad spectrum of physiological and pathological processes and emerges as the subtype harboring the highest tally of candidate drugs under clinical evaluation1. Illustrative examples encompass GSK-148216010 for nociceptive and inflammatory disorders, JNJ-5417544611 for depressive disorder, and JNJ-5530894212 for bipolar disorder, presently undergoing Phase I or Phase II clinical evaluations. Correspondingly, SGM-101913 for nonalcoholic steatohepatitis, CE-224,53514 for rheumatoid arthritis, AZD905615 for rheumatoid arthritis, and AK-178016 for chronic pain are also advancing through various stages of clinical development.
Despite quite a few candidate drugs targeting P2X receptors entering the main phases of clinical trial, few have been approved for market release17. The expedited progression of Gefapixant (AF-219) led to approval in Japan and the European Union, failing to obtain market authorization in major pharmaceutical markets such as the United States4. This highlights the sluggish progress and low success rate of clinical trials for P2X receptor-targeted drugs. Particularly striking is the case of P2X7 receptor antagonists, which have failed to advance beyond Phase II clinical trials18,19. One contributing factor to this challenge may be the presence of more non-synonymous polymorphism (nsSNP) sites in the P2rx7 gene compared to other family members20. The National Center for Biotechnology Information dbSNP database (https://www.ncbi.nlm.nih.gov/snp/) reveals over 1500 single SNPs in the P2rx7 gene, with several SNPs identified to be associated with human conditions such as depression and osteoporosis21. Clinical studies have also found that nsSNP can affect the binding of P2X7 with ligands in the human body22, thereby influencing drug efficacy. Moreover, differences in the P2rx7 gene between humans and model animals result in greater variations in ligand regulation of P2X7 receptors among different species23. Consequently, such interspecies differences, particularly in the drug-binding sites, present a formidable challenge in the rational design of P2X7 compounds24.
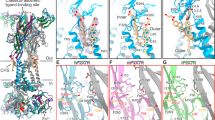
Currently, the clinical trial phase primarily encompasses P2X receptor allosteric modulators23. The P2X receptor is a trimer composed of three “dolphin-shaped” subunits25 (Fig. 1a). Three confirmed allosteric binding sites on P2X receptors have been identified (refer to Fig. 1a–c): one situated beneath the orthosteric binding site on the hP2X3 receptor, comprising the left flipper (LF) and adjacent subunits’ lower body (LB) and dorsal fin (DF) domains (known as the Gefapixant binding site)25; another recently confirmed within the head domain of the hP2X3 receptor26; and the third pocket positioned at the entrance of the central pocket of the P2X receptor, termed the portal of the central pocket (PCP, Fig. 1b, c). The residues forming the PCP are predominantly situated in the β-sheet of the upper body domains of these subunits and exhibit a degree of conservation across various P2X receptors (Fig. S1). As a result, PCP sites across different P2X subtypes are highly similar, exhibiting a characteristic dumbbell shape with bulkier ends and a narrower middle (Fig. 1c–f). Several high-affinity ligands have been verified to bind to the PCPs of various P2X subtypes, including the hP2X3 antagonist BLU-5937 (IC50 (half-maximal inhibitory concentration) = 0.025 μM)27, the P2X4 antagonist BX430 (IC50 = 0.54 μM)28, and the P2X7 antagonists A740003 (IC50 = 0.0437 μM), A804598, JNJ47965567, AZ10606120, and GW79134329 (Fig. S2 and Table S1). These antagonists not only show high affinity, with IC50 values in the nanomolar range, but also exhibit remarkable subtype selectivity, underscoring the advantage of allosteric modulation of PCP sites in P2X drug development28,29,30,31.

a Side view comparison of the apo/resting state (dark slate blue, PDB ID: 6U9V) and open state (medium violet red, PDB ID: 6U9V) structures of the rat P2X7 (rP2X7) receptor. b Representation of the orthosteric site and three allosteric sites of P2X receptors. Allosteric site 1 (upper right) resides within the Inner Pocket of the Head Domain (IP-HD), while allosteric site 2 (lower right) is situated beneath the orthosteric site, encompassing the left flipper and low body domain (LF-LBD). c Illustration of allosteric site 3 located at the portal of the central pocket (PCP), depicting its structure from both sides and vertical views. Composition of the PCP and three sub-pockets: PCP1 (green dotted circle), PCP2 (blue dotted circle), and PCP3 (brown dotted circle), shown from top (d) and side (e, f) views. The PCP primarily consists of rigid β-sheets, including β2, β3, β4, β7, and β8.
However, a significant limitation is species variability. For instance, A740003 exhibits a 37-fold difference in IC50 values between human P2X7 (hP2X7) and mouse P2X7 (mP2X7)32, BX-430 exhibits more than a 20-fold difference between human P2X4 (hP2X4) and mouse P2X4 (mP2X4)33. The precise mechanism underlying these interspecies differences induced by the PCP of P2X receptors remains unknown, and effective strategies to address this issue have yet to be proposed, limiting the progress of P2X receptor-targeted drug development.23,34 These species disparities pose significant challenges in evaluating high-affinity, highly specific human P2X inhibitors in animal models, hindering their advancement to clinical trials34. The PCP, particularly associated with the P2X7 receptor, is the only confirmed allosteric binding site for this subtype, suggesting a heightened likelihood of interspecies differences among allosteric modulators23. Therefore, understanding the specific mechanism by which the PCP causes interspecies differences and proposing viable solutions is crucial for advancing P2X receptors drug development.
In this work, we capitalize on the structural rigidity and sub-pocket architecture of the PCP to rationally design and synthesize berberine (P2X7 receptor inhibitor35) analogs, PSFL1191 and its stereoisomer PSFL1190, which exhibit substantially improved affinity for the human P2X7 receptor. Although PSFL1190 and PSFL1191 differ solely in chirality, they exhibit over a 50-fold difference in apparent affinity for hP2X7, highlighting the crucial role of sub-pocket shape complementarity in determining ligand affinity for the PCP. Through cryo-electron microscopy (cryo-EM), conformational sampling, and mutagenesis, we validate the interaction of PSFL1191, which displays interspecies diversity, and JNJ-54175446, which lacks interspecies differences, with human/panda P2X7 receptors. While both ligands bind to the PCP of human/panda P2X7, the interacting amino acid residues vary based on the depth of penetration into the pocket, explaining the interspecies differences in their interaction with human/panda and rodent receptors. This mechanism allows for the identification of inhibitors with (PSFL2780) or without (PSFL1633) interspecies differences as needed. Additionally, an examination of the distinct amino acid sequences of key PCP residues across species reveals that a single amino acid substitution in the mP2X7 (A312V) receptor is sufficient to eliminate interspecies differences in P2X7 inhibitors without altering the fundamental channel characteristics. The P2rx7A312V/A312V mice, engineered based on this principle, provide a solution for assessing the efficacy of interspecies-specific P2X7 inhibitors.
Results
PCP sites across P2X subtypes are high-affinity, subtype-specific, and druggable allosteric sites, showing species differences between human and murine receptors
Interspecies differences within the PCP likely represent a fundamental aspect of P2X receptor biology, with such variability being especially evident in the case of P2X7 receptor antagonists. Significant discrepancies have been found for P2X7 receptor inhibitors A74000332, JNJ4796556736, AZ11645373 and AZ1060612036 between hP2X7 and m/rP2X7 (Table S1). The apparent affinities (IC50) of AZ10606120 for hP2X7 are 500-fold and 1587-fold higher than those for mP2X7 and rP2X7, respectively (Table S1). A similar phenomenon is evident in the P2X4 receptor, where the IC50 value of BX430 for hP2X4 is more than 20-fold lower32 than for mP2X4 and rP2X4 (Fig. S2b and Table S1). However, not all drugs targeting the PCP exhibit pronounced species-specific differences. For instance, the clinical candidate BLU-5937, which targets P2X3-mediated chronic cough, and the P2X7 antagonist A438079, both act on the PCP with minimal variation in inhibitory effects between humans and rodents (Table S1)27,36. BLU-5937 binds primarily to the outer side of the PCP, referred to as the PCP3 sub-pocket37, whereas A740003 and A80459827 interact more deeply with the PCP1 sub-pocket (Fig. 1d-f). This suggests that the PCP’s volume accommodates small molecules in various binding modes. For example, our cryo-EM structure of the P2X7/JNJ-54175446 complex identified the PCP2 sub-pocket with partial overlap with PCP1 and PCP3 (Fig. 1f and see below). Despite these observations, the reason for the lack of species differences for JNJ-54175446 with hP2X7 and m/rP2X738, and for BLU-5937 with hP2X3 and mP2X327, in contrast to the significant differences seen with A740003 and A804598, remains unclear. Structural analysis reveals that β-sheets β2, β3, β7, β8, and β4 in the PCP are rigid (Fig. 1d), and the distinct sub-pockets—PCP1, PCP2, and PCP3—add to the complexity (Fig. 1e, f). These findings, combined with the discrepancies between human and animal models, underscore the need for further study to elucidate the mechanisms underlying these species-specific interactions.
PSFL1191 as a chemical tool for studying the species-specific nature of the druggable allosteric site PCP in P2X7 receptors
To elucidate the mechanism of ligand recognition at the rigid PCP site and explore species specificity, we employed various small-molecule tools (Figs. 2 and S3). Berberine, a natural isoquinoline alkaloid, is renowned for its diverse bioactivities35, yet its molecular targets remain elusive. Prior studies suggest that quaternary protoberberine alkaloids inhibit BzATP-induced hP2X7 activity35, though the role of berberine in modulating this receptor has not been thoroughly examined. Inspired by this, we posited that berberine could directly impact P2X7 receptor function. Indeed, our data affirm this hypothesis: berberine inhibits hP2X7 with an IC50 of 12.7 ± 0.9 μM. However, its inhibitory effects are substantially reduced in mouse and rat P2X7, with respective inhibition rates of 8.02 ± 2.70% and 19.2 ± 4.3% at 300 μM (Fig. 2a, b), underscoring significant interspecies pharmacodynamic variability. Furthermore, compared to the wild-type hP2X7 (h7WT), mutations within the PCP, including h7I68F, h7F88A, h7F103A, h7M140E, h7Y295F, h7E305F, h7V312A (P < 0.001) and h7E305C (P = 0.002), notably attenuated berberine’s inhibitory effect on hP2X7 (Fig. S3a, b). Subsequent study revealed a significant rightward shift in concentration-response curves for mutations h7I68F, h7Y295F, and h7E305F, with IC50 values of 39.9 ± 5.8, 46.6 ± 7.8, and 87.7 ± 4.3 μM, respectively. The solubility limits of berberine prevented the determination of the concentration-response curve for the h7V312A mutant; however, its IC50 was > 300 μM (Fig. S3c). These findings pinpoint the binding site of berberine within the PCP, a conclusion reinforced by synthesizing the nanomolar-level berberine analog PSFL1191 and determining the cryo-EM structure of the PSFL1191/pdP2X7 complex (see below).

a Chemical structure of berberine and PSFL384-2. b Concentration-response curves for berberine on human P2X7 (hP2X7, solid lines are fits of the Hill 1 equation, n = 6 for 1 and 3 μM, and n = 5 for other concentrations), with ATP-evoked currents of panda P2X7 (pdP2X7, n = 4), mouse P2X7 (mP2X7, n = 7), and rat P2X7 (rP2X7, n = 4) tested in the presence of 300 μM berberine. Determination of the absolute configuration of PSFL1190 (c) and PSFL1191 (d) via X-ray single crystal diffraction (upper panel). Chemical structure (middle panel) and enantiomeric excess (ee) value (lower panel) of PSFL1190. e Representative current traces illustrating the inhibitory effects of PSFL1191 (1 μM) and PSFL1190 (30 μM) on the activation of human P2X7 (hP2X7) induced by ATP (1 mM). f Concentration–response curves of PSFL1191 (n = 3) and PSFL1190 (n = 4 for 0.3 and 10 μM, n = 5 for 1 and 100 μM, n = 3 for 3 μM, n = 8 for 30 μM) on hP2X7. Currents evoked by 1 µM ATP were normalized to those in the absence of PSFL1191. Each solid line represents a fit of the Hill 1 equation. g Representative current traces showing the inhibition by PSFL1191 on the activation of hP2X7, panda P2X7 (pdP2X7), mouse P2X7 (mP2X7), and rat P2X7 (rP2X7) induced by ATP (1 mM). PSFL1191 (1 or 30 μM) was applied as indicated. h Concentration-response curves of hP2X7 (n = 3) and pdP2X7 (n = 5 for 0.1, 1, 3 and 10 μM; n = 6 for 0.3 μM; n = 4 for 3 μM) receptors to PSFL1191. Currents evoked by 1 µM ATP were normalized to those in the absence of PSFL1191. Each solid line represents a fit of the Hill 1 equation. Additionally, ATP-evoked currents of mP2X7 (n = 6) and rP2X7 (n = 5) were tested in the presence of 30 µM PSFL1191. n = 3–5. (i) Concentration-response curves for PSFL1191 on hP2X7 (n = 3), with ATP-evoked currents of hP2X1 (n = 5), hP2X2 (n = 5), hP2X3 (n = 3), and hP2X4 (n = 5) tested in the presence of 30 μM PSFL1191. All summary data are expressed as mean ± SEM from independent cells.
To increase affinity and reveal interaction specifics at the PCP of P2X7, we developed the berberine-based compound PSFL384-2 (Fig. 2a, and see Methods). This compound, featuring two chiral centers, was separated into its enantiomers, PSFL1191 and PSFL1190 (Fig. 2c, d). Single-crystal diffraction established their absolute configurations and revealed significant conformational differences between the enantiomers (Fig. 2c, d). Consequently, PSFL1191 exhibited an affinity for hP2X7 (IC50 = 0.248 ± 0.017 μM) that was nearly 50-fold higher than that of PSFL1190 (12.6 ± 2.9 μM) (Fig. 2e, f). This result highlights the stringent conformational requirements of the PCP for recognizing P2X7 inhibitors.
PSFL1191 demonstrated similar inhibitory effects on hP2X7 and pdP2X7, with IC50 values of 0.248 ± 0.017 μM and 0.754 ± 0.018 μM, respectively. However, its efficacy was notably reduced against mP2X7 and rP2X7. At a concentration of 30 μM, PSFL1191 only inhibits mP2X7 and rP2X7 by 13.3 ± 3.04% and 4.94 ± 1.43%, respectively (Fig. 2g and h), and 30 μM PSFL1190 exhibits inhibition rates of 11.8 ± 6.1% and 12.1 ± 2.4% for mP2X7 and rP2X7, respectively (Fig. S3d). When tested against other P2X receptor subtypes, PSFL1191 showed inhibition rates of -8.35 ± 4.49% for hP2X1, 3.09 ± 7.46% for hP2X2, 20.6 ± 6.4% for hP2X3, and -17.6 ± 9.0% for hP2X4 at 30 μM (Fig. 2i). These findings highlight PSFL1191’s selectivity for P2X7 receptors, making it a valuable tool for studying PCP binding selectivity and efficacy differences between human and animal P2X7 receptors.
Cryo-EM structural determination of PSFL1191 and JNJ-54175446 bound to pdP2X7
Given the high similarity between human and panda P2X7 PCPs and their comparable inhibition profiles (Fig. 2i), along with the established binding of A8004598 to pdP2X7’s PCP29, we determined the cryo-EM structure of the pdP2X7/PSFL1191 complex. We also resolved the cryo-EM structures of pdP2X7 in complex with JNJ-54175446, a high-affinity inhibitor for human and rat P2X7 receptors (3.47 nM and 1.55 nM, respectively) and a clinical candidate for major depression therapy38. First, we expressed and purified the functional pdP2X7 construct previously used for structural studies39 and reconstituted the purified protein into lipid nanodiscs. Subsequently, the nanodisc-reconstituted protein was mixed with either PSFL1191 or JNJ-54175446 inhibitors and subjected to single-particle cryo-electron microscopy (Figs. S4 and S5). The resulting cryo-EM structures of the pdP2X7 complexes bound to PSFL1191 and JNJ-54175446 were resolved at resolutions of 4.0 Å and 3.3 Å, respectively (Table S2, and Figs. S4 and S5). The resolution of the pdP2X7/PSFL1191 complex structure is 4.0 Å, yet the cryo-EM density map for PSFL1191 and surrounding amino acids remains relatively clear (Fig. 3a, d), and it is possible to observe the binding of PSFL1191 to pdP2X7. In addition to the quality of the cryo-EM density map at the binding site, the characteristic atoms C12and C14 (Figs. 2d and 3b) facilitated the assignment of the binding pose of PSFL1191 to the cryo-EM density.

a Cryo-EM densities for PSFL1191 (gray mesh, left) and trimeric structures of PSFL1191-bound pdP2X7 (right), depicted in side-view and top-view orientations relative to the membrane. PSFL1191 is represented as spheres, while each subunit of the trimer is colored violet, yellow, and cyan, respectively. Electron density maps are contoured at 4.0 Å. b Close-up views of the PSFL1191 binding sites and their chiral carbons close to the side chain of V312. Both PSFL1191 (green) and key interacting residues are shown as stick models for emphasis. Residues from the three subunits are colored violet, yellow, and cyan, respectively. Yellow broken lines indicate hydrogen bonds between the ligand and receptor or interior contacts within the receptor. c The relative position of PSFL1191 and JNJ-54175446 in the PCP pocket. d Cryo-EM densities for binding residues (gray mesh) surrounding PSFL1191 (green sticks). e Cryo-EM densities for JNJ-54175446 (gray mesh, left) and trimeric structures of JNJ-54175446-bound pdP2X7 (right). JNJ-54175446 is represented as spheres. Electron density maps are contoured at 3.2 Å. f Zoomed-in view of JNJ-54175446 binding sites, with both the ligand and key residues shown as stick models for emphasis. All EM maps are contoured at X.0σ.
To validate the binding pose of PSFL1191 and the stability of the pdP2X7/PSFL1191 complex, we performed a 3.97-μs conventional molecular dynamics (CMD) simulation (Fig. S6). The simulation confirmed the stability of the PSFL1191/pdP2X7 complex, as evidenced by the RMSD values for P2X7 and PSFL1191 (Fig. S6a) and the analysis of bond rotation angles (Fig. S6d). Key residues involved in PSFL1191 binding, including Y298, D92, F108, Y295, and V312, remained stable throughout the simulation (Fig. S6b, c), and their roles were further supported by subsequent mutagenesis studies. Similarly, a 3.6-μs CMD simulation of the JNJ-54175446/pdP2X7 complex verified the stable binding of JNJ-54175446 to PCP2 (Fig. S7). In contrast, replacing PSFL1191 with its enantiomer PSFL1190 in the pdP2X7/PSFL1190 complex led to significant destabilization, as shown by a brief 1.5-μs CMD simulation (Fig. S8), with markedly increased RMSD values (Fig. S8a) and distinct bond rotation angle fluctuations (Fig. S8d). Interactions with residues such as M105, K297, Y298, and F293 decreased within the first 0.1 μs, while interactions with Y295, F103, and I310 increased (Fig. S8b, c), highlighting substantial differences in PCP interaction between PSFL1190 and PSFL1191. These results indicate that the PSFL1191/pdP2X7 and JNJ-54175446/pdP2X7 complexes represent stable conformations rather than transient states.
PSFL1191 and JNJ-54175446 both bind to the PCP (Fig. 3a, b, e, f), but they differ in the amino acids involved in their interactions and their binding sites. The binding site of PSFL191 is situated deeper within the PCP (Figs. 3b and 1h, identified as the PCP1 sub-site), where pivotal atoms C12 and C14 of PSFL1191 orient towards pdP2X7 V312, M105, and Y295, while the oxygen-containing heterocycles face inward the interior of PCP, in proximity to F103 and P96 (Fig. 3b, c). Additionally, its two methoxy groups align towards the outer aspect of PCP1, adjacent to F88, K297 and Y298 (Fig. 3b). These interactions are primarily hydrophobic or Van der Waals in nature, with PSFL1191’s O3 and N1 (Fig. 2d) establishing hydrogen bonds with the side chain of K297, whose conformation is influenced by the E305-K297 salt bridge, and the main chain oxygen of D92 (Fig. 3b), respectively.
The binding site of JNJ-54175446 is situated closer to the middle of PCP (Fig. 3c, f). Its nitrogen-containing heterocycle at the bottom is primarily accommodated by a sub-pocket (PCP2) formed by residues F103, F95, and Y298 within the middle region of the PCP (Fig. 3f), maintaining a certain distance from M105, V312, and Y295, distinguishing it from PSFL1191. Furthermore, the centrally methylated nitrogen-containing heterocycle interacts nonpolarly with Y298 and M105, while the chlorotrifluoromethyl group on the outer aspect of the PCP primarily interacts nonpolarly with F88. Unlike PSFL1191, JNJ-54175446 does not form hydrogen bonds with K297 and D92, instead forming hydrogen bonds with the carbonyl oxygen and the side chain of K110 (Fig. 3f).
Thus, the functional groups of PSFL1191 extend deeper into the PCP, beyond the F103-F95 sub-pocket, reaching V312, Y295, and P96 (PCP1). JNJ-54175446 is anchored within the middle region of the PCP by F103 and F95 (PCP2), interacting more prominently with F88.
Confirmation of PSFL1191 and JNJ-54175446 interaction with P2X7 via PCP1 and PCP2
For PSFL1191, the mutations pd7F88A, pd7D92F, pd7T94I, pd7P96G, pd7F103A, pd7F103W, pd7M105A, pd7M105F, pd7F108A, pd7Y295A, pd7K297R, pd7Y298A, pd7Y298F, pd7E305F, pd7V312F and pd7V312A significantly attenuated the inhibitory effect of 10 μM PSFL1191 (pd7Y298A, P = 0.004; other mutations, P < 0.001), while mutations pd7V71F, pd7Y93A, pd7F95A, pd7F95W, pd7F108W, pd7F293A, pd7F293W, pd7Y295W and pd7V312I did not (Fig. 4a, b). Even at the maximal soluble concentration of PSFL1191 (30 μM), the inhibition rate in pd7F88A, pd7F103W, pd7Y295A, pd7K297R, pd7Y298A and pd7V312A did not exceed 50%, indicating a more than 40-fold reduction in potency compared to pd7WT (IC50 = 0.754 ± 0.018 μM; Fig. 4c). Both pd7P96G (8.06 ± 0.00 μM), pd7E305F (1.57 ± 0.36 μM), and pd7M105F (2.06 ± 0.00 μM) displayed a rightward shift of concentration-response curves (Fig. 4c). These findings suggest that: 1. pd7F88, pd7D92, pd7D96, pd7F103, pd7V312, pd7Y295, pd7Y298 and pd7K297 play essential roles in mediating PSFL1191 binding, primarily through hydrophobic and other nonpolar interactions that stabilize the ligand within the binding pocket (like pd7F88A, Fig. 4 b, c); 2. pd7E305F may impair PSFL1191 binding by disrupting the salt bridge with K297 (Figs. 3b and 4b, c); 3. pd7F95 and pd7F293 are not essential residues in the formation of PCP1, as their side chain modifications (Ala or Trp) do not affect the inhibitory efficiency of PSFL1191 (Fig. 4b); 4. Maintenance of PSFL1191’s inhibitory efficacy requires that residue pd7V312 provides sufficient steric bulk to preserve the structural integrity of the binding pocket, as exemplified by the V312I substitution (Fig. 4b, c); and 5. pd7Y295 should possess adequate steric hindrance—conferred by bulky side chains such as tyrosine or tryptophan, but not alanine—to support ligand accommodation (Fig. 4b, c).

a Two-dimensional (2D) interaction diagrams generated using LigPlot, highlighting the key residues involved in PSFL1191 binding. b Inhibitory effect of PSFL1191 (10 μM) on ATP (1 mM)-induced activation of pdP2X7 wild type (pd7WT) and indicated mutants. Each circle represents an independent cell. Sample sizes (n) are annotated in the plots. P value was compared to WT, one-way ANOVA followed by Dunnett’s multiple comparisons test, F (26, 107) = 48.02. c Concentration response curves to PSFL1191 of pd7WT and several mutants. Solid lines were fits of the Hill equation. n = 5 (0.1, 1, 3, and 10 μM), 6 (0.3 μM) or 4 (30 μM) for pdP2X7. n = 3 for P96G. n = 3 (0.3, 1 and 30 μM) or 4 (3 and 10 μM) for M105F. n = 5 (0.1, 0.3 and 30 μM), 7 (1 μM) or 6 (3 and 10 μM) for E305F. The ATP-evoked currents of pd7F88A (n = 3), pd7F103W (n = 3), pd7Y29A (n = 3), pd7K297R (n = 3), pd7Y298F (n = 5), and pd7V312A (n = 5), were tested in the presence of 30 µM PSFL1191. d 2D interaction diagrams generated using LigPlot, highlighting the key residues involved in JNJ-54175446 binding. e Inhibitory effect of JNJ-54175446 (100 nM) on ATP (1 mM)-induced activation of pd7WT and the indicated mutants. Each circle represents an independent cell. Sample sizes (n) are annotated in the plots. P value was compared to WT, one-way ANOVA followed by Dunnett’s multiple comparisons test, F (22, 81) = 29.78. f Concentration response curves to JNJ-54175446 of pd7WT and several of its mutants. Solid lines were fits of the Hill 1 equation. n = 5 for pdP2X7. n = 4 (1 and 3 nM), 3 (10 nM),6 (30, 1000, 3000 and 10000 nM) or 7 (100 and 300 nM) for F88A. n = 4 (3000 nM) or 5 for F95A. n = 3 for F103A and V312A. n = 4 (30 nM) or 5 for Y298F. g Confirmation of PCP as the action site of PSFL1191 through DTNB covalent occupancy. Residues (pink) close to bound PSFL1191 (green). h Pooled data illustrating the effects of DTNB (5,5’-Dithiobis-(2-nitrobenzoic acid)) on PSFL1191 inhibition in pdP2X7F88C, pdP2X7F108C, and pdP2X7I310C. Each line represents an independent cell. Sample sizes (n) are annotated under the circle. Statistical significance was assessed using a two-tailed paired t-test. Validation of PCP as the action site of JNJ-54175446 through DTNB covalent occupancy. Residues (green) close to JNJ-54175446 (pink) (i) and pooled data (j) showing the effects of DTNB on JNJ-54175446 inhibition in pdP2X7V84C and pdP2X7F88C. Each line represents an independent cell. Sample sizes (n) are annotated under the circle. Statistical significance was assessed using a two-tailed paired t-test. All summary data are expressed as mean ± SEM from independent cells84.
For JNJ-54175446, mutations pd7F88A, pd7F95A, pd7F103A, pd7F103W, pd7K110A, pd7Y298A, and pd7Y298F, significantly reduced the inhibitory effect of 0.1 μM JNJ-54175446 (pd7F103W, P = 0.002; other mutations, P < 0.001; Fig. 4d, e). Unlike PSFL1191, weakened JNJ-54175446 inhibition, while pd7V312F, pd7V312A, pd7M105A, pd7M105F, pd7Y295A, pd7Y295W, and pd7Y305F had a much lesser impact on JNJ-54175446 compared to PSFL1191 (Fig. 4e). The apparent affinities for pd7F88A (0.681 ± 0.109 μM), pd7F95A (0.269 ± 0.018 μM), pd7F103A (0.067 ± 0.047 μM), pd7Y298F (0.160 ± 0.020 μM) and pd7V312A (0.0547 ± 0.0107 μM) were 3-40-fold lower compared to pd7WT (0.0161 ± 0.0031 μM) (Fig. 4f).
To further validate the binding of JNJ-54175446 and PSFL1191 to pdP2X7, we conducted covalent occupancy experiments using Cys mutations of PCP (Fig. 4g, i). Treatment with 2 mM 5,5’-dithiobis (2-nitrobenzoic acid) (DTNB) significantly increased the inhibitory effect of PSFL1191 on pd7F88C (P = 0.008, before vs after DTNB modification), suggesting a critical, albeit relatively weak, interaction between F88 and PSFL1191 within the pdP2X7/PSFL1191 complex. Conversely, modification of pd7I310C decreased the inhibitory effect of PSFL1191 (P = 0.003). DTNB covalent occupancy did not significantly alter the effect on pd7F108C (P = 0.376, Fig. 4h), consistent with the finding in pd7F108W (Fig. 4b). Similarly, since JNJ-54175446 binds closer to the central-lateral side of PCP, covalent occupancy of pd7F88C and pd7V84C significantly reduced the inhibitory effect of JNJ-54175446 (P = 0.008 and < 0.001, after DTNB modification vs. before, Fig. 4j), further confirming the interaction between JNJ-54175446 and the PCP2 region of pdP2X7.
These findings collectively suggest that PSFL1191 and JNJ-54175446 interact with the PCP of the P2X7 receptor, albeit with varying residues. Notably, Y298 is a shared residue for PCP1 and PCP2, but JNJ-54175446’s binding is more sensitive to its mutations (Ala or Phe, Fig. 4a–d). While F88 plays a role in binding both compounds, its impact differs between PCP1 and PCP2, as seen with F88C covalent occupancy (Fig. 4f, h). Additionally, V312 significantly affected PSFL1191 but had minimal influence on JNJ-54175446 (Fig. 4a–d). Thus, although their binding sites partially overlap within PCP, PSFL1191 predominantly interacts with PCP1, while JNJ-54175446 binds at PCP2.
Determinants of affinity, selectivity, and species specificity in P2X7-PCP/inhibitor interactions
To elucidate the mechanisms underlying high affinity, subtype selectivity, and species-specific ligand recognition by P2X7 PCP1 and PCP2, we superimposed the structures of the PSFL1191/pdP2X7 and JNJ-54175446/pdP2X7 complexes with apo pdP2X7 (PDB ID: 5U1L). PSFL1191 binding induced more significant conformational changes in PCP and its surrounding amino acids (RMSD = 0.807 Å, with bound PSFL1191 vs. apo, Fig. 5a) compared to JNJ-54175446 (RMSD = 0.573 Å, Fig. 5b). Analysis of the fluctuation amplitudes of various rotatable bonds during CMD simulations revealed that JNJ-54175446 (Fig. S7d) exhibited fewer fluctuations than PSFL1191 (Fig. S6d). Consequently, the apparent affinity of PSFL1191 is approximately 10-20 times lower than that of JNJ-54175446 (Fig. 4b, d). This difference in affinity is likely due to the rigid β-sheet surrounding PCP, which requires substantial free energy to compensate for the strain energy between the protein and the small molecule upon induced fit binding. This may also explain why PSFL1190, despite having the same scaffold as PSFL1191, exhibits a more than 50-fold decrease in affinity due to its molecular shape not fitting well with PCP1 (Figs. S6 and S8). Additionally, the pd7K110L mutation disrupts the hydrogen bond between JNJ-54175446 and PCP1 but does not significantly affect its inhibitory activity (Fig. 4c). Therefore, to achieve high inhibitory activity with P2X7-PCP-targeted allosteric drugs, enhanced shape complementarity with PCP, whether PCP1 or PCP2, is required rather than polar contacts. Furthermore, due to the non-conserved nature of certain amino acids in the PCP region of P2X1-7 (Fig. S1a) and its inherent rigidity, inhibitors of various P2X-PCPs exhibit P2X subtype selectivity (Table S1).

Top view of superimposed structures of apo pdP2X7 (dark blue, PDB ID: 5U1L) and PSFL1191-bound pdP2X7 (green, a) or JNJ-54175446-bound pdP2X7 (light coral, b). Zoomed-in views of the PCP pocket are provided in the corresponding boxes. Root mean square deviation (RMSD) is calculated compared to apo pdP2X7. c Amino acid sequence alignment of human, panda, mouse, and rat P2X7. Amino acids at positions 108 and 312 are highlighted in red for emphasis. d Superimposed view of PSFL1191 (green) onto JNJ-54175446 (light coral) in the cryo-EM structure. Residues F108 and V312 are indicated with sticks for emphasis. e PSFL1191 (1 μM) effect on ATP (1 mM)-induced activation of hP2X7-WT (h7WT), mP2X7-WT (m7WT), and the indicated mutants. Each circle represents an independent cell. Sample sizes (n) are annotated in the plots. Statistical significance was assessed using one-way ANOVA followed by Tukey’s multiple comparisons test, F (4, 14) = 62.65. f Concentration-response curves to PSFL1191 of h7WT (n = 5) and m7A312V (n = 3 for 0.1 and 30 μM; 6 for 0.3, 1 and 3 μM and 5 for 10 μM). Solid lines were fits of the Hill 1 equation. Additionally, ATP-evoked currents of m7WT, were tested in the presence of 30 µM PSFL1191 (n = 6). g A804598 (0.1 μM) effect on ATP (1 mM)-induced activation of h7WT, m7WT, and its indicated mutants. Each circle represents an independent cell. Sample sizes (n) are annotated in the plots. Statistical significance was assessed using one-way ANOVA followed by Tukey’s multiple comparisons test, F (2, 9) = 40.91. All summary data are expressed as mean ± SEM from independent cells.
Similarly, the rigidity of PCP and its limited ability to adapt to ligands may underlie species-specific differences in PCP1 residue sequences. The core regions of PCP in hP2X7, pdP2X7, mP2X7, and rP2X7 exhibit notable differences at two residues: F108 and V312 (numbered according to hP2X7, Fig. 5c, d). We generated mutations substituting mouse and human PCP residues, specifically mP2X7Y108F, mP2X7A312v, and mP2X7Y108F/A312V. Compared to m7WT, 1 μM PSFL1191 showed no significant change in inhibition of mP2X7Y108F (P = 0.332), while inhibition was significantly increased by ~21- and 26-fold for mP2X7A312Vand mP2X7Y108F/A312V (P < 0.001) (Fig. 5e). There was no significant difference in PSFL1191 inhibition between mP2X7A312V and mP2X7Y108F/A312V (P = 0.202, Fig. 5e). Furthermore, compared to m7WT (30 μM inhibition rate of only 13.3 ± 3.0%), the IC50 of mP2X7A312V was 0.908 ± 0.026 μM (Fig. 5f). Although this value is 2-3 times higher than hP2X7 (0.248 ± 0.017 μM), it is reasonable to consider mP2X7A312Vas a gain-of-function mutation for PSFL1191. Thus, h7V312 is a critical amino acid responsible for the species-specific differences of PCP1-targeting ligands, forming a sub-pocket with V312-F103-Y295-M105-P96 and exhibiting hydrophobic interactions with PSFL1191 (Fig. S6c). The difference between h7F108 and m7Y108 is not a major factor, as evidenced by the unchanged inhibitory effect of PSFL1191 on pd7F108W (Fig. 4b) and h7F108C after DTNB modification (Fig. 4g, h). JNJ-54175446 is confined to the central PCP2 (formed by F103-Y298-F88, Fig. 3f), with no tight hydrophobic interaction with h7V312, thus not contributing to species differences.
We also examined the impact of m7A312V on other ligands showing species differences between mP2X7 and hP2X7. At 0.1 μM, A804598 exhibited significantly lower inhibition of m7WT compared to h7WT (P < 0.001). m7A312V significantly enhanced A804598 inhibition of mP2X7 (P < 0.001), with its inhibitory effect on m7A312V almost identical to that of h7WT (P = 0.986, Fig. 5g). These findings further validate that V312, positioned at the bottom of PCP1 within the rigid β8 (Fig. 5d), is a crucial amino acid contributing to species-specific differences in P2X7 receptor inhibition.
Discovery of P2X7 inhibitors with species-specific and non-specific properties through high-throughput virtual screening (HTVS) of PCP1 and PCP2
To support these observations, HTVS was conducted using the PCP1 and PCP2 conformations. The PCP2 conformation from the JNJ-54175446/pdP2X7 complex served as the template for virtual screening (Fig. 6a), which yielded a panel of P2X7 inhibitors (Fig. 6b). Promising compounds with significant inhibition rates were then revalidated (Fig. 6c). PSFL1633, which exhibited the highest inhibition rate (Fig. 7a), along with PSFL1632, were selected for further analysis of binding sites and species specificity. Notably, mutations. pd7F95A (specific to PCP2) and pd7Y298A significantly attenuated the inhibitory effect of 1 μM PSFL1633 (P = 0.038 and P = 0.002 compared with h7WT, Fig. 7a). Similarly, both pd7F95A and pd7Y298A mutations attenuated PSFL1632’s inhibitory efficacy (P = 0.042 and P < 0.001 compared with h7WT, Fig. 7a), mechanistically confirming its interaction with the PCP2 of pdP2X7 (Fig. 6a). Additionally, the apparent affinity of PSFL1633 in pd7Y298F (2.65 ± 0.76 μM) decreased sevenfold compared to pd7WT (0.417 ± 0.077 μM), indicating PSFL1633’s interaction with the PCP2 of pdP2X7 (Fig. 7b). Importantly, 50 μM PSFL1633 inhibited hP2X7, mP2X7, and rP2X7 by 0.930 ± 0.013, 0.773 ± 0.038, and 0.765 ± 0.032 times that of pdP2X7, respectively (Fig. 7c), demonstrating its detachment from significant species-specific differences. PSFL1632 also exhibited species-independent inhibition, with potencies of 1.00 ± 0.046 (hP2X7), 0.937 ± 0.022 (mP2X7), and 0.900 ± 0.035 (rP2X7) relative to pdP2X7 (Fig. 7c).

a–c Virtual screening and validation of species-independent P2X7 inhibitors. Initially, virtual screening for P2X7 receptor inhibitors was conducted based on the JNJ-54175446/pdP2X7 cryo-structure (a). Subsequently, the inhibitory effects of the compounds identified through virtual screening were preliminarily validated (b, n = 1), and compounds that exhibited significant inhibitory effects underwent repeat validation (50 μM, c). d–f Virtual screening and validation of species-specific differenced P2X7 inhibitors. Initially, virtual screening for P2X7 receptor inhibitors was conducted based on the PSFL1191/pdP2X7 cryo-structure (d). Subsequently, the inhibitory effects of the compounds identified through virtual screening were preliminarily validated (e, n = 1), and compounds that exhibited significant inhibitory effects in the preliminary validation underwent repeat validation (30 μM, f). Sample sizes (n) are annotated in the plots. Summary data are presented as mean ± SEM from independent cells.

a Effects of PSFL1632 (3 μM) and PSFL1633 (1 μM) on ATP-induced activation in pd7WT and the indicated mutants. Each circle represents an independent cell. Statistical significance was assessed using one-way ANOVA followed by Dunnett’s multiple comparisons test, F (2, 8) = 62.02 and F (2, 9) = 11.38. b Concentration-response curves to PSFL1633 (the P2X7 inhibitor without species-specific differences) of pd7WT and its mutants (solid lines were fits of the Hill 1 equation). n = 6 (3 μM) or 5 (other concentrations) for pd7WT, and 4 for pd7Y298F. c The effects of PSFL1632 and PSFL1633 (50 μM) on ATP-induced activation of pdP2X7, hP2X7, mP2X7, and rP2X7 were examined. Each circle represents an independent cell. d Effects of PSFL2763, PSFL2780, PSFL2783, PSFL2784, and PSFL2787 (P2X7 inhibitors with species-specific differences, 30 μM) on ATP-induced activation of pd7WT and its mutants. Each circle represents an independent cell. Statistical significance was assessed using one-way ANOVA followed by Dunnett’s multiple comparisons test, F (2, 9) = 13.46, F (2, 12) = 101.7, F (2, 10) = 5.395, F (2, 11) = 13.44, and F (2, 14) = 16.30. e Concentration-response curves to PSFL2780 (the P2X7 inhibitor with species-specific differences) of pd7WT and m7A312V (solid lines were fits of the Hill 1 equation). n = 5 for pd7WT, and 3 (0.03 and 0.3 μM) or 4 (other concentrations) for pd7A312V. Additionally, ATP-evoked currents of pd7F88A (n = 4) and pd7V312A (n = 6) were tested in the presence of 30 µM PSFL2780. f Effect of PSFL2763, PSFL2780, PSFL2783, PSFL2784, and PSFL2787 (30 μM) on ATP-induced activation of pdP2X7, hP2X7, mP2X7, and rP2X7. Statistical significance was assessed using one-way ANOVA followed by Dunnett’s multiple comparisons test, F (3, 9) = 78.07, F (3, 10) = 82.10, F (3, 10) = 24.28, F (3, 10) = 24.28, and F (3, 10) = 22.42. Sample sizes (n) of a, c, d, and f are annotated in the plots. All summary data are expressed as mean ± SEM from independent cells.
Using the PCP1 conformation from the PSFL1191/pdP2X7 complex for HTVS, we identified various P2X7 inhibitors (Fig. 6d–f). Compounds with substantial inhibitory effects, such as PSFL2763, PSFL2780, PSFL2783, and PSFL2784, were validated for their binding sites. These inhibitors exhibited significantly reduced efficacy against pd7F88A and pd7V312A mutations compared to pd7WT (Fig. 7d). For instance, PSFL2780, even at the highest solubility concentration of 30 μM, did not inhibit pd7F88A and pd7V312A mutations by more than 50%, indicating an IC50 greater than 30 μM and suggesting a primary reliance on pdP2X7’s PCP1 (Fig. 7e). Similarly, these compounds demonstrated significant species-specific differences in their inhibitory effects on P2X7: 10 μM PSFL2763 inhibited ATP-mediated currents in hP2X7, mP2X7, and rP2X7 by 0.740 ± 0.010, 0.413 ± 0.024, and 0.0800 ± 0.0404 times that of pdP2X7, respectively; PSFL2780 showed proportions of 1.10 ± 0.01, 0.503 ± 0.067, and 0.107 ± 0.058; PSFL2783 exhibited proportions of 0.491 ± 0.066, 0.190 ± 0.035, and 0.230 ± 0.089; and PSFL2784 presented proportions of 0.767 ± 0.065, 0.0833 ± 0.0463, and 0.177 ± 0.126 (Fig. 7f).
Moreover, the mP2X7A312V mutation significantly enhanced the inhibitory effect of PSFL2780 on mP2X7, resulting in an IC50 of 0.306 ± 0.031 μM (Fig. 7e). These findings highlight the critical role of PCP1 and PCP2 in determining the species specificity of P2X7 receptor inhibitors.
Creation and functional validation of mice harboring the P2rx7 A312V/A312V mutation to study the species-specific P2X7 inhibitors
Understanding the mechanisms underlying the species-specific or non-specific binding of PCP1 and PCP2 to P2X7 receptor inhibitors is crucial for addressing inefficiencies in drug development caused by interspecies differences between humans and animal models. When certain inhibitors demonstrate efficacy against hP2X7 but not against mP2X7 or rP2X7, despite promising in vitro characteristics, evaluating their efficacy and safety in vivo requires an animal model. The m7A312V mutation enhances the affinity of species-specific P2X7 inhibitors for mP2X7 (Figs. 5f, 5g, and 7e). Importantly, the m7A312V does not significantly alter the fundamental functions of mP2X7: (1) The maximum current density of m7WT and m7A312V in comparable (P = 0.568, Fig. 8a); (2) The apparent affinity for ATP is approximately 1 mM (EC50 = 0.782 ± 0.071 and 1.42 ± 0.17 mM for m7WT and m7A312V, respectively, Fig. 8b); (3) P2X7 receptors exhibit current facilitation characteristics40, where initial ATP-induced currents are followed by a gradual increase to maximum second-phase currents, known as sensitization (Fig. 8c). The current facilitation characteristics of m7A312V on mP2X7 are unaffected (P = 0.397, Fig. 8d); (4) P2X7 receptors can permeate small molecules like NMDG41, and the m7A312V does not affect NMDG permeability (P = 0.725, Fig. 8e). Thus, mouse models with m7A312V gene editing are considered viable for further studies.

a–e The A312V mutation in mP2X7 does not alter its primary function. No significant difference in current density (pA/pF) was observed between m7WT and m7A312V (a). Concentration-response curves to ATP of m7WT and m7A312V indicate similar ATP affinities (b; solid lines were fits of the Hill 1 equation, n = 5). Representative currents demonstrate that current facilitation is inherent to mP2X7 (c), with no significant difference observed between m7WT and m7A312V (d). Compared to the sustained ATP stimulation-induced current (Imax), the initial ATP-induced current (I1) is typically smaller (c). Macropore permeability of P2X7 between m7WT and m7A312V remains comparable (e). Sample sizes (n) of a, d and e are annotated in the plots. Statistical significances were assessed using two-tailed unpaired t-test. f Flowchart depicting the full-thickness wound infection model. g Photographs of wounds treated with PSFL1191 (7.5 mg/ml, 50 μL, subcutaneous) and solutions (control) for 0, 1, 3, 5, and 7 days (scale bar = 2 mm). The green dashed circle outlines the wound’s edge. h Changes in wound size following various treatments. Significant difference between P2rx7A312V/A312V-PSFL1191 and P2rx7+/+-PSFL1191 treatments was assessed using two-way ANOVA followed by Tukey’s multiple comparisons test, F (21, 294) = 5.442. i Wound area of P2rx7A312V/A312V and P2rx7+/+ after continuous administration for 7 days (Statistical significances were assessed using two-way ANOVA followed by Tukey’s multiple comparisons test, F (21, 294) = 5.442). Sample sizes are annotated under the boxes. Boxes and lines in the boxes represent inter-quantile range (Q3–Q1) and median of relative area, respectively. Lower and upper whiskers show the minimum and maximum values of the relative area. All summary data are expressed as mean ± SEM from independent cells (a-e) or animals (h and i).
Subsequently, P2rx7A312V/A312V transgenic mice were generated to study the physiological functions and pharmacological activities. Comparative hematological and biochemical analyses revealed no significant differences between P2rx7A312V/A312V and P2rx7+/+ in white blood cell (WBC) levels or 28 other parameters (Fig. S9). The only notable difference was in total protein (TP) levels, which were 59.7 ± 1.5 and 65.3 ± 1.4 g/L for P2rx7+/+ and P2rx7A312V/A312V (Fig. S9). However, these values remained within normal physiological ranges42. A comprehensive battery of behavioral tests—including open field experiments (Fig. S10a–i), tail suspension tests (Fig. S10j), hot and mechanical pain assays (Fig. S10k and l), social dominance tests (Fig. S10m), and pole climbing tests (Fig. S10n)—showed no significant differences between the two genotypes. These results suggest that P2rx7A312V/A312V KI mice maintain normal physiological and behavioral functions, making them suitable for evaluating the efficacy and safety of P2X7 receptor inhibitors.
Different effects of PSFL1191 on wound healing in P2rx7 A312V/A312V and P2rx7 +/+ mice
The P2X7 receptor is implicated in a range of pathophysiological processes1,15,43,44. This study examines the role of P2X7 in the immune system, particularly in the phagocytic response of macrophages against bacterial pathogens. Specifically, inhibition of the P2X7 receptor in macrophages, which are critical for bacterial engulfment45, results in reduced bacterial clearance and impaired wound healing46. To explore this, we employed a mouse wound model47,48, introducing the Gram-positive bacterium Staphylococcus aureus (S. aureus) into the wound and administering PSFL1191 or a control solvent subcutaneously around the wound. By comparing wound healing times between P2rx7A312V/A312V and P2rx7+/+ mice treated with PSFL1191, we aimed to evaluate the in vivo effects of species-specific P2X7 receptor inhibitors (Fig. 8f).
First, the wound healing rates in P2rx7+/+ and P2rx7A312V/A312V mice treated with the control solution were comparable (Fig. 8g, h). No significant differences in wound area were observed between the two groups from days 1 to 7 post-treatment (P = 0.691, 0.978, 0.998, 0.999, 0.986, 0.760, and 0.750 for days 1-7, respectively; Fig. 8h, i and S11), indicating that the A312V mutation does not alter the wound healing rate.
Secondly, PSFL1191 significantly delayed wound healing in P2rx7A312V/A312V mice compared to those treated with the control solution (Fig. 8g, h). From day 2 post-treatment, the wound area in PSFL1191-treated P2rx7A312V/A312V mice was significantly larger than in the control group (days 2–7: P < 0.001, Fig. 8h, i and S11), demonstrating that PSFL1191 impedes wound healing in these mice.
Thirdly, a comparison between P2rx7+/+ and P2rx7A312V/A312V mice, both treated with PSFL1191, revealed that wound healing was significantly slower in the mutant mice (Fig. 8g, h). From day 3 post-treatment, the wound area in PSFL1191-treated P2rx7A312V/A312V mice was significantly larger than in P2rx7+/+ mice (day 3: P = 0.002, days 4-7: P < 0.001, Fig. 8h, i and S11), suggesting that the delayed wound healing effect of PSFL1191 is related to the P2X7 receptor.
Fourth, unlike its impact on P2rx7A312V/A312V mice, PSFL1191 did not influence the wound healing rate in P2rx7+/+ mice (Fig. 8g, h). No significant differences in wound area were observed between PSFL1191-treated and control solvent-treated P2rx7+/+ mice from days 1 to 7 post-treatment (P = 0.998, 0.604, 0.965, 0.999, 0.999, 0.987, and 0.953 for days 1-7, respectively; Fig. 8h, i, and S11), indicating that PSFL1191 effects are specific to P2rx7A312V/A312V mice.
To further investigate whether PSFL1191 decelerates wound healing by inhibiting P2X7 receptors in macrophages and thus impairing their phagocytic activity against bacteria, we assessed its direct effect on macrophage phagocytosis of S. aureus. Given that PSFL1191 inhibits human P2X7 but not mouse P2X7, we utilized human monocytic leukemia (THP-1) cells for this experiment. THP-1 cells were sequentially induced into M0 and M1 macrophages, and their phagocytic activity against bacteria was subsequently tested (Fig. 9a). We first excluded any direct bacteriostatic effect of PSFL1191, as 8 μM of the compound did not affect various concentrations of S. aureus (P = 0.229 and 0.880, PSFL1191 vs. control, Fig. 9b). THP-1 cells were differentiated into M0 macrophages using phorbol 12-myristate 13-acetate (PMA), and M0 cells were polarized into M1 macrophages using lipopolysaccharide (LPS) and IFN-γ, with polarization confirmed by cytokine analysis (Fig. 9c). Importantly, 8 μM of PSFL1191 significantly reduced the phagocytic activity of THP-1 M1 cells against S. aureus (P < 0.001, vs. control, Fig. 9d).

a Flowchart outlining the macrophage phagocytosis experiment. THP-1 cells were differentiated into M0 macrophages using Phorbol-Myristate-Acetate (PMA) and polarized to M1 macrophages with lipopolysaccharide (LPS) and interferon-γ (IFN-γ). Phagocytosis assays were performed on THP-1 M1 macrophages incubated with Staphylococcus aureus (S. aureus). THP-1 M1 macrophages were pretreated with PSFL1191 (8 μM) or vehicle before the phagocytosis assay, and bacterial concentration analysis was performed to assess the effect of PSFL1191 on macrophage phagocytosis. b PSFL1191 exhibits no bactericidal activity against S. aureus. c Relative mRNA levels of various factors in THP-1 M1 macrophages. RT-PCR was used to assess LPS and IFN-γ activation characteristics using selected M1 markers (TNF-α and IL-1β) or M2 markers (Arg-1 and CD206). d PSFL1191 inhibits S. aureus phagocytosis by THP-1 M1 macrophages. e THP-1 M1 macrophages exhibit high expression levels of the hP2rx7 gene. f Interference RNA targeting the P2rx7 gene reduces hP2rx7 gene expression in THP-1 M1 macrophages. In b, d and f, statistical significances were assessed using two-tailed unpaired t-test. g Reduced hP2rx7 gene expression attenuates the effect of PSFL1191 on macrophage phagocytosis (Statistical significance was assessed using one-way ANOVA followed by Tukey’s multiple comparisons test, F (3, 11) = 6.310). All summary data are expressed as mean ± SEM from independent experiments, and all ample sizes (n) are annotated in the plots. MTT Thiazolyl blue; SDS Sodium dodecyl sulfate, TNF-α tumor necrosis factor-α, IL-1β interleukin-1β, Arg-1 arginase 1.
Finally, to assess whether the reduction in phagocytic activity of THP-1 M1 cells by PSFL1191 is associated with P2X7, we first confirmed the high expression of the hP2rx7 gene in these cells (Fig. 9e). We then utilized siRNA targeting P2rx7 to downregulate hP2rx7 expression in THP-1 M1 cells (P = 0.006, siRNA-P2X7 vs. control, Fig. 9f). The knockdown of hP2rx7 effectively abolished the impact of PSFL1191 on the phagocytic capacity of THP-1 M1 cells (without siRNA-P2X7 treatment, P = 0.043; with siRNA-P2X7 treatment, P = 0.462, PSFL1191 vs. control, Fig. 9g). These results demonstrate that PSFL1191 reduces the phagocytic activity of THP-1 M1 cells by inhibiting P2X7 receptor activity, supporting the conclusion that PSFL1191 impairs wound healing in P2rx7A312V/A312V mice through P2X7 receptor inhibition in macrophages.
Thus, the P2rx7A312V/A312V mouse model proves to be an effective system for evaluating the in vivo effects of species-specific P2X7 receptor inhibitors (Fig. 10). Importantly, this model retains the functionality of mP2X7 while preserving the fundamental physiological processes in the mice.

Inhibitors binding to the P2X7 receptor exhibit varying depths of entry into the portal of the central pocket (PCP), leading to occupancy of different sub-pockets (PCP1-3). The deepest sub-pocket, PCP1, surrounded by rigid β-sheets (β4, β7, and β8), has a high probability of producing species-specific differences in its bound inhibitors, with the amino acid at position 312 of β8 in PCP1, the narrowest middle of PCP, being a key factor. P2rx7A312V/A312V mice can eliminate these species-specific differences between humans and rodents, facilitating the development of relevant mouse disease models and advancing these inhibitors toward clinical trials. hP2X7 human P2X7, mP2X7 mouse P2X7.
Discussion
Unveiling allosteric mechanisms and identification of allosteric sites has opened new avenues for drug development, transforming previously undruggable targets into viable ones49. Unlike highly conserved orthosteric sites, allosteric sites are more diverse, providing greater selectivity and reduced off-target effects49. This is exemplified by the allosteric site PCP on P2X receptors, where P2X7 inhibitors such as A74000332, A80459836, JNJ4796556736, AZ1060612036, and GW79134350 demonstrate excellent subtype selectivity and high affinity. Almost all identified subtype-specific allosteric inhibitors of the P2X7 receptor bind to the PCP site.23,29 However, sequence variations in PCP allosteric sites among species result in interspecies variability, leading to discrepancies or ineffectiveness in drug evaluations using rodent models compared to humans51. Since PCP is the only confirmed allosteric site on the P2X7 receptor23, understanding the mechanisms underlying species-specific differences at this site is critical. In this study, we identified PSFL1191 as a species-specific negative allosteric modulator and determined its cryo-EM structure in complex with the pdP2X7 receptor, alongside the species-independent negative allosteric modulator JNJ-54175446. The differences in their binding modes highlighted key factors for achieving high affinity, subtype selectivity, and species specificity for molecules targeting the P2X7 receptor’s PCP site. This insight enables the selective screening of P2X7 receptor inhibitors with tailored species specificity. To address the challenge of evaluating species-specific P2X7 receptor inhibitors’ efficacy and safety in rodent models, we developed a genetically modified mouse, P2rx7A312V/A312V, to assess the in vivo effects of human-specific P2X7 receptor inhibitors. Additionally, we confirmed that JNJ-54175446 binds to the PCP site of the P2X7 receptor (Fig. 3f). This drug is currently in phase II clinical trials for major depression (NCT04116606), providing a detailed atomic-level foundation for receptor-ligand interactions in depression therapy. Furthermore, the P2X3 receptor antagonist S-600918 from Shionogi30 and the P2X4 inhibitor BX43028 also targets this site, suggesting that PCP may be a crucial target for drug development across various P2X receptor subtypes.
The PCP site of the P2X receptor is notably large29. Despite various inhibitors binding to this site, their precise binding locations differ. We have categorized the PCP into three sub-pockets (Fig. 1d). BLU-5937 binds specifically to PCP3 of P2X3 receptors (Fig. 1d), situated at the outermost edge of the PCP27. The flexibility of the amino acid residues in PCP3 may explain why BLU-5937 exhibits high subunit selectivity without species specificity in P2X327. Furthermore, BLU-5937 demonstrates selectivity for both P2X3/2 heterotrimers and P2X3 homotrimers, suggesting that PCP is a valuable site for achieving both subtype and heterotrimer selectivity. This is critical for distinguishing the therapeutic benefits of RCC from the side effects related to taste disorders52. In contrast, P2X7 inhibitors primarily target the inner regions of PCP1 and PCP227. These sub-pockets are predominantly composed of rigid β-sheets (Figs. 1d and 5a, b), which may limit receptor-induced fit interactions with ligands. PCP1 is situated deeper within the PCP compared to PCP2, with the deepest point involving residues V312, Y295, M105, F103, and P96. PCP2’s deepest section (formed by residues F95, F103, and Y298) is more central. There is some overlap between these regions, including residues Y298 and F88. However, if a small molecule interacts primarily with the hydrophobic region encompassing V312, Y295, M105, and P96—mainly in rigid β4, 7, and 8—it may lead to species differences between human and animal P2X7 receptors, even if other parts of the molecule interact with outer residues like Y298 and F88. Minimizing interaction with this hydrophobic region may reduce species differences, as seen with JNJ-54175446. Although PSFL1191’s interaction with the PCP site does not involve F108, molecules binding to both V312 and F108 could induce species differences due to variations at this site between humans (Phe) and rodents (Tyr). F108 is located in the more exposed outer region of the PCP, while V312 is situated at the narrower bottom (Fig. 1d–f). Thus, species differences in PCP-targeted ligands are primarily influenced by V312. Variations in interactions between V/A-312 and the ligand may also be significant. A similar issue is observed with the P2X4-targeted BX430, where residue I315 is critical for zfP2X4’s sensitivity to BX430, whereas the corresponding mutation to W312 in rP2X4 reduces sensitivity53,54. Cryo-EM structures reveal that hydrophobic interactions between I315 and BX430 are responsible28, underscoring the impact of this site on species differences across P2X receptors. Species-selective ligand binding is a complex, multifactorial process, and residue V312 alone might be not fully account for the interspecies differences observed among PCP-targeting compounds. Notably, in addition to V312, residue F95 has been implicated as a critical determinant of the species-specific inhibitory activity of AZ11645373 on human versus rat P2X7 receptors55. However, since position 95 is conserved between human and mouse P2X7 (Fig. S1a), it is unlikely to explain the differential inhibition observed in these species. Furthermore, the combined Y108F/A312V mutation significantly enhances the potency of PSFL1191: while the A312V single substitution leads to a 21-fold increase in inhibitory activity, the double mutant achieves a 26-fold enhancement. Therefore, we do not exclude the potential translational value of an animal model carrying the Y108F/A312V double substitution.
Rodent models, such as mice and rats, are invaluable for drug development. Nevertheless, significant species differences in drug affinity between humans and these models can hinder preclinical studies34. Small molecules binding to the PCP of the P2X7 receptor often show pronounced species differences. To overcome this issue, the creation of genetically edited mice that address these disparities will support the evaluation of P2X7 receptor inhibitors and their safety profiles. We introduce the P2rx7A312V/A312V genetically edited mice, which maintain their essential physiological functions and respond to PSFL1191—unlike wild-type mice. Importantly, the characteristic features of the P2X7 receptor, such as low ATP affinity40, current facilitation40, and small molecule permeation,41,56 remain consistent. These features are thought to be associated with disease onset1. Disease models using P2rx7A312V/A312V mice are expected to reflect those created with wild-type mice, thereby facilitating the efficacy testing of drugs targeting the P2X7 receptor. Additionally, P2rx7A312V/A312V mice provide a platform for investigating the P2X7 receptor’s role in disease initiation. For instance, while the P2X7 receptor’s involvement in tumor development is established, its precise role in promoting or inhibiting tumor growth remains debated57,58,59. This uncertainty may be due to differing expressions of the P2X7 receptor in tumor and immune cells within the tumor microenvironment, each with unique functions43. The current challenge is distinguishing the receptor’s effects on various cell types in vivo. The use of P2rx7A312V/A312V mice and PSFL1191 could address this: by inoculating WT mouse-derived tumor cells into P2rx7A312V/A312V mice and administering PSFL1191, one can selectively inhibit P2X7A312V receptors in immune cells while sparing WT P2X7 receptors. This approach could partially elucidate the P2X7 receptor’s role in tumor development across different cell types. Further investigation is required to explore this hypothesis more thoroughly.
In summary, interspecies variation presents a significant challenge to protein allosteric regulation sites, as exemplified by the PCP of the P2X7 receptor, complicating preclinical studies and clinical trials of P2X7 receptor antagonists due to interspecies variations. We elucidated the specific binding mechanisms of ligands to the PCP using PSFL1191 and JNJ-54175446 as probes. Through cryo-electron microscopy, electrophysiology, and molecular dynamics, we identified differences in ligand binding associated with interspecies variations. We determined that shape complementarity is a crucial factor influencing these differences, rather than ligand interactions like hydrogen bonding. Building on these insights, we developed P2rx7A312V/A312V mice, which maintain the characteristics of mP2X7 and fundamental physiological functions, and show in vivo activity with interspecies variant compounds. This approach provides a valuable method for evaluating the efficacy and safety of P2X7 inhibitors across species variations (Fig. 10).
Methods
Drugs, mutagenesis, and cell cultures
Unless otherwise stated, all compounds were purchased from Sigma-Aldrich (USA). The pdP2X7 plasmid was synthesized by Convenience Biology, and the hP2X3 plasmid was purchased from Open Biosystems. cDNAs for hP2X1, hP2X2, hP2X7, and rP2X7 were synthesized by GKN and subcloned into the pcDNA3.1 vector. The pcDNA3-hP2X4 plasmid was generously provided by Dr. Alan North and Dr. Linhua Jiang, and the mP2X7 cDNA was generously provided by Dr. Ming Zhou and subcloned into the pcDNA3.1 vector. All mutations were generated using the KOD-Plus-Mutagenesis Kit (TOYOBO, catalog number KOD-401) and confirmed by DNA sequencing. HEK293 cells (Catalog number: GNHu 43) and THP-1 cells (Catalog number: TCHu 57) were purchased from the National Collection of Authenticated Cell Cultures (Shanghai Institutes for Biological Sciences, China). HEK293 cells were cultured in Dulbecco’s Modified Eagle Medium (Gibco) supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, and 1% GlutaMAX™ at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. THP-1 cells were cultured in Roswell Park Memorial Institute 1640 Medium (Gibco) supplemented with 10% FBS. Plasmids were transfected into cells using a calcium phosphate transfection reagent.
Chemical synthesis of PSFL1190 and PSFL1191
Materials and Methods
All the reagents for chemical synthesis were purchased from a commercial chemical reagent company and directly used without further purification. Analytical thin-layer chromatography (TLC) was performed on HSGF 254. All products were characterized by their 1H NMR, 13C NMR, LR-MS, and HR-MS. The purity of the target compounds was determined by HPLC (Gemini-C18, 5 μm, 4.6 mm × 150 mm, 35 °C, UV 214/254/280 nm, flow rate = 1.0 mL/min, MeOH (0.25% TEA)/H2O = 80/20(v/v)) for 30 min, and the peak areas were calculated at 280 nm. Chiral Separation of compound PSFL384-2 and ee value determination was conducted by Daicel Chiral Technologies (China) Co., Ltd (CHIRALPAK IG-3(IG30CD-WE016), 4.6 mm × 150 mm, 35 °C, UV 214 nm, flow rate = 1.0 mL/min, Hexane/EtOH = 98/2(v/v)) (Fig. S12).
Synthesis of 13-allyl-9,10-dimethoxy-5,8,13,13a-tetrahydro-6H-1,3dioxolo[4,5-g]isoquinolino[3,2-a]isoquinoline (PSFL384-2)
1-1 (berberine, 3.0 g, 8.0 mmol) and K2CO3 (3.33 g, 24.1 mmol) were dissolved in 100 mL of methanol, to which a solution of 5% NaOH (3 mL) containing NaBH4 (334 mg, 8.8 mmol) was added dropwise. The mixture was kept stirring for 2 h at room temperature. Then the precipitated product was filtered, washed with water (20 mL), 30% ethanol (20 mL), and absolute ethanol (50 mL) sequentially to provide the intermediate 1-2 as a yellow solid.
To a solution of 1-2 (1.8 g, 5.3 mmol) and NaI (2.4 g, 16.0 mmol) in CH3CN (20 mL) was added allyl bromide (4.61 mL, 53.4 mmol). The mixture was refluxed under Ar for 16 h before cooling down. The solvent was evaporated under vacuum, and 100 mL of water was added. After ultrasonic oscillation for 20 min, the unstable iminium salt (1-3) was obtained by filtration as an orange solid. After drying on the oil pump, 1-3 (2.0 g, 5.3 mmol) was dispersed in 50 mL of methanol, to which NaBH4 (1.0 g, 26.4 mmol) was added slowly at 0 °C. The resulting mixture was stirred for 0.5 h at room temperature. Then another 1.0 g of NaBH4 was added at 0 °C, and the suspension was kept stirring for an additional hour at room temperature. The reaction was quenched by saturated NH4Cl. The solution was extracted with ethyl acetate (EA, 30 mL × 3), and the organic phase was combined and washed with saturated NaCl (50 mL × 2). After drying by Na2SO4, the solvent was evaporated under vacuum, and the residue was purified by column chromatography (Hexane/EA: 10/1) to provide the target product as a light-yellow solid (800 mg, yield: 26.2% for three steps). HPLC purity: 97.51%. M.p. 96-98 oC. 1H NMR (600 MHz, DMSO-d6) δ 6.89 (s, 1H), 6.86 (d, J = 8.5 Hz, 1H), 6.79 (d, J = 8.4 Hz, 1H), 6.68 (s, 1H), 5.96 (dd, J = 17.7, 1.1 Hz, 2H), 5.72–5.64 (m, 1H), 4.80–4.76 (m, 1H), 4.67 – 4.62 (m, 1H), 4.07 (d, J = 16.0 Hz, 1H), 3.77 (s, 3H), 3.73 (s, 3H), 3.59 (s, 1H), 3.39 (d, J = 15.9 Hz, 1H), 3.21–3.17 (m, 1H), 3.11–3.05 (m, 1H), 2.92–2.85 (m, 1H), 2.58–2.53 (m, 1H), 2.45–2.39 (m, 1H), 2.07–2.01 (m, 1H), 1.99–1.94 (m, 1H). 13C NMR (151 MHz, DMSO-d6) δ 149.88, 145.96, 145.31, 144.31, 138.16, 131.93, 129.16, 129.14, 127.78, 124.79, 115.13, 110.34, 108.04, 105.66, 100.55, 62.92, 59.44, 55.59, 53.83, 50.56, 42.87, 36.61, 29.15. ESI-MS m/z 380.0 [M + H]+. HR-MS: (ESI, m/z) calcd for C23H26NO4+ [M + H]+: 380.1856, found 380.1863 (Fig. S13).
Compounds PSFL1190 and PSFL1191 were obtained by chiral separation of PSFL384-2
(13S,13aR)-13-allyl-9,10-dimethoxy-5,8,13,13a-tetrahydro-6H-1,3dioxolo[4,5-g] isoquinolino[3,2-a]isoquinoline (PSFL1190)
The absolute configuration of compound PSFL1190 was unequivocally confirmed by X-ray crystallography (Fig. S14 and Table S3) (CCDC 2361922, PSFL1190). HPLC purity: 97.84%. ee: 99.73%. M.p. 99–100 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.89 (s, 1H), 6.86 (d, J = 8.5 Hz, 1H), 6.79 (d, J = 8.5 Hz, 1H), 6.68 (s, 1H), 5.96 (dd, J = 11.9, 1.1 Hz, 2H), 5.75–5.61 (m, 1H), 4.82–4.61 (m, 2H), 4.07 (d, J = 16.0 Hz, 1H), 3.77 (s, 3H), 3.72 (s, 3H), 3.59 (s, 1H), 3.39 (d, J = 16.0 Hz, 1H), 3.23–3.14 (m, 1H), 3.12–3.04 (m, 1H), 2.94–2.82 (m, 1H), 2.55 (d, J = 15.6 Hz, 1H), 2.46–2.36 (m, 1H), 2.09–1.90 (m, 2H). 13C NMR (126 MHz, DMSO) δ 149.85, 145.93, 145.29, 144.31, 138.13, 131.93, 129.14, 129.13, 127.76, 124.75, 115.08, 110.35, 108.01, 105.63, 100.53, 62.91, 59.42, 55.59, 53.82, 50.55, 42.88, 36.60, 29.14. ESI-MS m/z 380.0 [M + H]+. HR-MS: (ESI, m/z) calcd for C23H26NO4+ [M + H]+: 380.1856, found 380.1859 (Fig. S15).
(13R,13aS)-13-allyl-9,10-dimethoxy-5,8,13,13a-tetrahydro-6H-1,3dioxolo[4,5-g]isoquinolino[3,2-a]isoquinoline (PSFL1191)
The absolute configuration of compound PSFL1191 was unequivocally confirmed by X-ray crystallography (Fig. S16 and Table S4) (CCDC 2361725, PSFL1191). HPLC purity: 96.16%. ee: 99.93%. M.p. 98–100 °C. 1H NMR (400 MHz, DMSO-d6) δ 6.89 (s, 1H), 6.86 (d, J = 8.5 Hz, 1H), 6.79 (d, J = 8.4 Hz, 1H), 6.68 (s, 1H), 5.96 (dd, J = 12.0, 1.0 Hz, 2H), 5.75–5.61 (m, 1H), 4.82–4.58 (m, 2H), 4.07 (d, J = 16.0 Hz, 1H), 3.77 (s, 3H), 3.72 (s, 3H), 3.59 (s, 1H), 3.39 (d, J = 15.9 Hz, 1H), 3.22–3.16 (m, 1H), 3.11–3.04 (m, 1H), 2.94–2.82 (m, 1H), 2.55 (d, J = 15.7 Hz, 1H), 2.47–2.37 (m, 1H), 2.09–1.91 (m, 2H). 13C NMR (126 MHz, DMSO) δ 149.85, 145.93, 145.29, 144.31, 138.13, 131.93, 129.13, 129.12, 127.75, 124.75, 115.08, 110.34, 108.01, 105.63, 100.52, 62.90, 59.42, 55.58, 53.82, 50.55, 42.87, 36.60, 29.14. ESI-MS m/z 380.0 [M + H]+. HR-MS: (ESI, m/z) calcd for C23H26NO4+ [M + H]+: 380.1856, found 380.1863 (Fig. S17).
Conventional and nystatin-perforated whole-cell recordings
Recordings of hP2X1, hP2X2, hP2X7, mP2X7, rP2X7, and pdP2X7 receptor currents were performed using a conventional whole-cell patch configuration, as previously described60,61. For conventional whole-cell recordings, the pipette solutions comprised (in mM) 120 KCl, 30 NaCl, 0.5 CaCl2, 1 MgCl2, 10 HEPES, and 5 EGTA (pH 7.4, adjusted with Tris-base). To prevent rundown, hP2X3, mP2X3, and hP2X4 currents were recorded using nystatin (Sangon Biotech) perforated recordings. The nystatin (0.15 mg/mL) perforated intracellular solutions contained (in mM) 5 MgSO4, 75 K2SO4, 55 KCl, and 10 HEPES (pH 7.4). HEK293 cells were recorded after 24-48 hours of transfection at room temperature (25 ± 2 °C) using an Axopatch 200B amplifier (Molecular Devices, USA) with a holding potential of -60 mV. Current data were sampled at 10 kHz, filtered at 2 kHz, and analyzed using PCLAMP 10 (Molecular Devices, USA). HEK293 cells were bathed in standard extracellular solution containing (in mM) 2 CaCl2, 1 MgCl2, 10 HEPES, 150 NaCl, 5 KCl, and 10 glucose (pH 7.4, adjusted with Tris-base). ATP and other drugs were dissolved in a standard solution (SS) and applied via Y-tubes. 5,5’-dithiobis-(2-nitrobenzoic acid) (DTNB) was diluted to appropriate concentrations and perfused into the cell membrane immediately during the recording period.
Cryo-EM structures of PSFL1191-bound and JNJ-54175446-bound pdP2X7 receptors
Expression and purification of pdP2X7
The previously reported expression construct of giant panda (Ailuropoda melanoleuca) P2X7 (residues 22-359, N241S/N284S/V35A/R125A/E174K, XP_002913164.1)29 was synthesized (Genewiz, China) and subcloned into a modified version of the pFastBac vector (Invitrogen, USA) containing an octahistidine tag, Twin-Strep-tag, mEGFP, and human rhinovirus (HRV) 3 C protease cleavage site at N-terminus. Using the Bac-to-Bac system, the mEGFP-fusion pdP2X7 protein was expressed in baculovirus-infected sf9 cells. The cells were collected by centrifugation (5,400 × g, 10 min) and disrupted usingan ultrasonic homogenizer in TBS buffer (20 mM Tris pH 8.0, 150 mM NaCl) containing 1 mM phenylmethylsulfonyl fluoride (PMSF), 5.2 μg/mL aprotinin, 1.4 μg/mL pepstatin, and 1.4 μg/mL leupeptin. The supernatant was harvested by centrifugation (7600 × g, 20 min). Then the membrane fraction was isolated by ultracentrifugation (200,000 × g, 1 h), and solubilized with buffer A (50 mM Tris pH 7.5, 150 mM NaCl) containing 2% (w/v) n-dodecyl-beta-D-maltopyranoside (DDM) at 4 °C for 1 hour. The solubilized supernatant was collected by ultracentrifugation (200,000 × g, 1 h) again, and applied to a Strep-Tactin resin (Qiagen, USA) column equilibrated with buffer A containing 0.025% (w/v) DDM. After incubating the resin for 1 h, the column was eluted with buffer B (100 mM Tris pH 8.0, 150 mM NaCl, and 2.5 mM desthiobiotin, 0.025% (w/v) DDM). The eluted protein was concentrated to 2 mg/ml for nanodisc reconstitution.
Nanodisc reconstitution
Soybean polar lipid extract (Avanti Polar Lipids, USA) was dissolved in chloroform, dried under a nitrogen stream, and resuspended in reconstitution buffer (20 mg/ml soybean polar lipid, 20 mM HEPES pH7.,0 and 150 mM NaCl). After incubating at room temperature for 1 hour, the lipid suspension was sonicated for 5 minutes until the lipids became nearly transparent. Then DDM (Anatrace, USA) was added at a final concentration of 0.4%, and incubated at 4 °C for 2 hours. The mEGFP-fusion pdP2X7, MSP2N2 protein, and soybean polar lipid were mixed at the molar ratio of 1:3:180. The mixture was incubated at 4 °C for 1 h, then incubated with bio-beads (Bio-rad, USA) for 4 hours. After incubation, bio-beads were removed through filtration, and the mEGFP-fusion pdP2X7-containing nanodisc fractions were bound to Ni-NTA (Qiagen, USA) resin preequilibrated with wash buffer (20 mM HEPES pH7.5, 150 mM NaCl, 30 mM Imidazole) and then eluted with elution buffer (20 mM HEPES pH7.5, 150 mM NaCl, 300 mM imidazole). To cleave the N-terminal EGFP, the elution was mixed with HRV3C proteases at room temperature for 1 hour and then at 4 °C overnight. Nanodisc-reconstituted pdP2X7 protein was separated by size-exclusion chromatography using Superdex 200 Increase 10/300 column (Cytiva, USA) preequilibrated with SEC buffer (20 mM HEPES pH 7.5, and 150 mM NaCl), and concentrated to 1.2 mg/ml. P2X7 antagonists, either JNJ-54175446 or PSFL1191, were incubated with nanodisc-reconstituted pdP2X7 at a final concentration of 0.5 mM for 1 hour on ice. Following this incubation, samples were immediately prepared for cryo-EM by vitrification on glow-discharged grids.
EM data acquisition and analysis
For both JNJ-54175446-bound PSFL1191-bound pdP2X7 samples, a total of 2.5 μl of the nanodisc-reconstituted pdP2X7 was applied to the glow-discharged holey carbon-film grid (Quantifoil, Au 1.2/1.3, 300 mesh, USA), blotted with a Vitrobot (FEI, USA) system using a 3.0-s blotting time with 100% humidity at 9 °C and plunge-frozen in liquid ethane. Cryo-EM data collection was performed using a 300 kV Titan Krios microscope (FEI, USA) equipped with K3 direct electron detector (Gatan Inc., USA). The specimen stage temperature was maintained at 80 K. Images were recorded by beam-image shift data collection methods62 under the superresolution mode with a pixel size of 0.41 Å (a physical pixel size of 0.83 Å) with a magnification of 29,000 and a defocus ranging from −−1.5 µm to -2.3 µm. The dose rate was 20 e- s–1, and each movie was 1.758 s long, dose-fractioned into 40 frames with an exposure of 1.3 e- Å–2 for each frame. The cryo-EM data are summarized in Table S1.
Image processing
A total of 4860 and 4980 movies for the PSFL1191-bound and JNJ-54175446-bound pdP2X7 samples were motion-corrected, respectively, and binned with MotionCor263 with 5 × 5 patches, producing summed and dose-weighted micrographs with a pixel size of 0.83 Å. Contrast transfer function (CTF) parameters were estimated by CTFFIND 4.164. Particle picking and further image processing were performed using RELION 3.165. For both JNJ-54175446-bound and PSFL1191-bound pdP2X7 samples, a total of 1,569,253 and 981,777 particles were auto-picked, respectively, and extracted with a box size of 256 × 256 pixels. After 2D classification, we performed 3D classification with C1 and C3 symmetry using RELION 3.1. Then, 135,394 particles for the JNJ-54175446-bound sample and 162,345 particles for the PSFL1191-bound sample were selected for non-uniform refinement with C3 symmetry by cryoSPARCv4.2.166. The final map resolution reached 4.0 Å for the PSFL1191-bound pdP2X7 and 3.3 Å for the JNJ-54175446-bound pdP2X7, respectively. The final resolution was estimated using the Fourier shell correlation (FSC) = 0.143 criterion on the corrected FSC curves, in which the influence of the mask was removed. The local resolution was estimated using cryoSPARCv4.2.1. The workflow for image processing and for the 3D reconstruction is shown in Figs. S4 andS5. The EM density maps without C3 symmetry imposed remained similarly symmetric but exhibited lower overall resolutions (Figs. S18,S19).
Model building
The initial models of pdP2X7 were manually built starting from the previously reported pdP2X7 structure (PDB ID: 5U1L). Manual model building was performed using Coot67. Real-space refinement was performed using PHENIX68. All structure figures were generated using PyMol (https://pymol.org/).
Conventional Molecular Dynamics (CMD) simulations and high-throughput virtual screening of pdP2X7
The simulation systems were built using Maestro’s System Builder, and CMD simulations were performed using DESMOND (V6.2)69, as implemented in Maestro molecular modeling suites, using NPT (constant number (N), pressure (P), and temperature (T)) ensemble configurations, as we previously described60,61,70. The DESMOND suite provides a comprehensive pipeline tailored to membrane protein preparation, benefiting from its integrated OPLS all-atom force field71, which supports hydrogen addition, charge assignment for both ligands and receptors, sidechain reconstruction in cases of partial residue loss, and localized energy minimization. A large 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer (300 K) was selected as the membrane system, comprising 252, 255, and 268 POPC molecules for the PSFL1190-, PSFL1191-, and JNJ-54175446-pdP2X7 complex systems, respectively. The position of the membrane was determined based on the pdP2X7 cryo-EM structures (PDB IDs: 8Z0Z and 8Z1D) in the Orientations of Proteins in Membranes database (https://opm.phar.umich.edu), and the automatic mode was selected to add the membrane. The ligand/pdP2X7 complex was dissolved in simple point charge (SPC) water molecules. The solution conditions were neutral, and counterions were added to compensate for the net negative charge of the system. In addition, the concentration of NaCl was chosen to be 150 mM. We did not employ enhanced (biased) conformational sampling, as CMD simulations (unbiased) more faithfully recapitulate physiological processes. Furthermore, in examining the interactions of PSFL1190 and PSFL1191 with P2X7, CMD revealed pronounced microsecond-scale differences, obviating the need for enhanced approaches. In CMD simulations, the RMSD values of both protein–ligand complexes (PSFL1191/P2X7 and JNJ-54175446/P2X7) rapidly converged to stable plateaus, indicating that the simulated systems had attained structural equilibrium.
Prior to initiating the production phase of the simulation, the system underwent a structured relaxation process. This default DESMOND relaxation protocol encompasses multiple stages of restrained or no constraints energy minimization to ensure system stability and accuracy in subsequent productions. Briefly, (1) simulations were performed for 100 ps in the NVT (constant number (N), volume (V), and temperature (T)) ensemble with Brownian dynamics at 10 K and solute heavy atoms constrained; (2) simulations were performed for 12 ps in the NVT ensemble using a Berendsen thermostat at 10 K and solute heavy atoms constrained. (3) 12 ps simulations in the NPT ensemble using a Berendsen thermostat and Barostat at 10 K and 1 atm with solute heavy atoms constrained. (4) 12 ps simulations using a Berendsen thermostat and Barostat at 300 K and 1 atm, with solute heavy atoms constrained; (5) 24 ps simulations using a Berendsen thermostat and Barostat at 300 K and 1 atm, with no constraints. After equilibration, the MD simulations were carried out for about 0.6–12µs. The long-range electrostatic interactions were calculated using the smooth particle grid Ewald method. The trajectory recording interval was set to 200-ps, and the other default parameters of DESMOND were used in the MD simulation runs. All simulations used the all-atom OPLS_2005 force field72,73, which is used for proteins, ions, lipids, and SPC waters. The Simulation Interaction Diagram (SID) module in DESMOND was used to explore the interaction analysis between PSFL1191 (or JNJ-54175446) and P2X7. Three replicate simulations were performed, each yielding consistent results. All simulations were performed on a DELL T7920 with NVIDIA TESLTA K40C or CAOWEI 4028GR with NVIDIA TESLTA K80. The simulation system was prepared, trajectory analyzed and visualized on a CORE DELL T7920 graphic workstation with 92 CPU cores.
High-throughput virtual screening (HTVS) of the CHEMDIV compound library (https://www.chemdiv.com; 1,535,478 molecules yielding 3,228,069 conformations) was conducted using the Virtual Screening Workflow (VSW) module within the Schrödinger suite, under the default parameters as previously described74. Compound selection followed a tiered Glide docking protocol, retaining the top 2% from HTVS, the top 10% from Standard Precision (SP), and the top 10% from Extra Precision (XP) docking stages. In consideration of the receptor-ligand induced-fit effect, we did not use the apoP2X7 structures of PCP1 and PCP2 as the starting points for our VSW. Instead, we employed the cryo-EM complex structures of PSFL1191/P2X7 (PCP2) and JNJ54175446/P2X7 (PCP1) as the starting templates. Prior to screening, the structures of these two complexes were carefully prepared. The generation of the PCP2 and PCP1 GRID files was centered around PSFL1191 or JNJ54175446, with a grid box extending 20 Å. From the resulting 200-300 compounds with higher XP scores, we performed manual filtering to remove obvious false positives, such as glycoside-containing or highly hydroxylated compounds, and those with excessive polarity or high-water solubility, given the relatively hydrophobic nature of the PCP binding sites. Finally, we selected 30-50 corresponding hits from commercial libraries for electrophysiological screening. In total, we purchased 79 hits corresponding to the two starting structures.
Animals
Using CRISPR/Cas9 technology, we engineered mice harboring a homozygous A312V substitution in the P2rx7 gene (P2rx7A312V/A312V). The targeted modification replaced the codon for alanine at position 312 with one encoding valine. These gene-edited animals were subsequently backcrossed onto a C57BL/6 background for over ten generations to ensure genomic stability and consistency. WT P2X7 (P2rx7+/+, C57BL/6J, 8–10 weeks) and P2rX7A312V/A312V mice (aged 8–10 weeks, 18–25 g) were used in all animal experiments. Animals were housed in a controlled environment with a temperature of 23 ± 2 °C, humidity of 50 ± 10%, and a 12-hour light/dark cycle, with ad libitum access to standard rodent chow and water. Blood routine and biochemistry analyses were conducted on both P2rx7+/+ and P2rX7A312V/A312V mice, following established protocols75. Behavioral tests, including the open field test76, tail suspension test77, hot plate test78, mechanical pain test79, tube-test ranking for social hierarchy80, and pole test81 were performed on P2rx7+/+ and P2rX7A312V/A312V mice, as described in previous studies.
Murine full-thickness wound infection model
The murine wound infection model was established following previously protocols47,48. Briefly, Staphylococcus aureus (S. aureus) was resuspended in saline and washed thrice with saline. Anesthetized mice had their fur removed from their backs, and the skin was disinfected with an iodine solution. A 6 mm diameter biopsy punch was used to create a skin wound, onto which a 20 μL suspension of S. aureus (1×107 CFU/ml) was applied. After a 24-hour pre-infection period, P2rx7+/+ and P2rX7A312V/A312V mice were randomly allocated into two groups and administered different treatments: 20% Sulfobutylether-β-Cyclodextrin in normal saline (solvent control, 50 μL) and PSFL1191 (7.5 mg/ml, 50 μL), via subcutaneous injection twice daily for 7 consecutive days. Wound healing progress was monitored daily, and the wound area was quantified using ImageJ analysis software.
Phagocytosis activity of S. aureus by THP-1
Phagocytosis activity of S. aureus by THP-1 macrophages was assessed following established procedures45,82. S. aureus suspensions were added to monolayer cultures of THP-1 macrophages and incubated at 37 °C for 30 minutes. During the experiment, cells were treated with either 8 μM PSFL1191 or 0.5% dimethyl sulfoxide (DMSO) as a negative control. Extracellular bacteria were eliminated by two consecutive washes with RPMI 1640 medium. The combined washes were subjected to the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) colorimetric assay to assess phagocytic efficiency. The assay was performed according to standard protocols82,83, where 100 μL of bacteria washes and 10 μL of MTT reagent were added to wells in 96-well plates. After incubation at 37 °C and 200 rpm for 30 minutes, 100-μL aliquots of 5% SDS-buffered DMSO solution were added to each well. Plates were maintained on a shaker at 200 rpm for 4 hours, after which absorbance values at 570 nm were measured using a microplate reader. The real-time quantitative PCR (qPCR) experiment was performed in accordance with the instructions of the PrimeScript™ RT reagent Kit (RR047A) from TAKARA (Beijing, China). All operations were conducted on ice throughout the process. The sequences of the qPCR primers are provided in Table S5.
Data analysis
Data were presented as mean ± standard error of the mean (SEM). Statistical analyses were performed using one-way analysis of variance (ANOVA) followed by Dunnett’s test or Tukey’s test for multiple independent groups comparison. Paired or unpaired t-tests were employed for comparisons between two groups. Concentration-effect curves were fitted using the Hill equation: I/Imax = 1/(1 + (IC50/[inhibitor])n), here I represents the normalized current at a given antagonist concentration, Imax is the maximum normalized current induced by the agonist, IC50 is the concentration of the antagonist exhibiting the half-maximum effect, [inhibitor] is the concentration of drugs, and n is the Hill 1 coefficient.
Ethical approval
The study was approved by the ethics committee of the China Pharmaceutical University. All experimental procedures adhered to the guidelines outlined in the Guide for the Care and Use of Laboratory Animals and were approved by the Ethics Committee of the China Pharmaceutical University (approval number: 2023-10-015).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-39716 (JNJ-54175446) and EMD-39723 (PSFL1191). The atomic coordinates of the pdP2X7 structure have been deposited in the Protein Data Bank under accession codes 8Z0Z (JNJ-54175446) and 8Z1D (PSFL1191). Previously resolved P2X7 structures were retrieved from the PDB under accession codes 6U9W, 6U9V, 5U1L, 5U1U, 5U1V, 5U1X, 5U1W, 5U1Y, 9BPC, and 8JV5. The MD simulation data have been deposited to figshare [https://doi.org/10.6084/m9.figshare.27132306]. All other relevant data are included in the paper or its supplementary information. Source data are provided with this paper.
References
Bartlett, R., Stokes, L. & Sluyter, R. The P2X7 receptor channel: recent developments and the use of P2X7 antagonists in models of disease,. Pharmacol. Rev. 66, 638–675 (2014).
Illes, P. et al. Update of P2X receptor properties and their pharmacology: IUPHAR Review 30,. Br. J. Pharmacol. 178, 489–514 (2021).
Schmid, R. & Evans, R. J. ATP-Gated P2X Receptor channels: molecular insights into functional roles. Annu. Rev. Physiol. 81, 43–62 (2019).
McGarvey, L. P. et al. Efficacy and safety of gefapixant, a P2X3 receptor antagonist, in refractory chronic cough and unexplained chronic cough (COUGH-1 and COUGH-2): results from two double-blind, randomised, parallel-group, placebo-controlled, phase 3 trials. Lancet 399, 909–923 (2022).
Friedrich, C. et al. Safety, Pharmacodynamics, and Pharmacokinetics of P2X3 Receptor Antagonist Eliapixant (BAY 1817080) in Healthy Subjects: Double-Blind Randomized Study. Clin. Pharmacokinet. 61, 1143–1156 (2022).
Smith, J. A. et al. Safety and Efficacy of BLU-5937 In the Treatment of Refractory Chronic Cough from the Phase 2b Soothe Trial. Am. J. Respir. Crit. Care Med. 205, A5778 (2022).
McGarvey, L. et al. A Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Phase 2b Trial of P2X3 Receptor Antagonist Sivopixant for Refractory or Unexplained Chronic Cough. Lung 201, 25–35 (2023).
Inoue, K. Nociceptive signaling of P2X receptors in chronic pain states. Purinergic Signal 17, 41–47 (2021).
Stokes, L. et al. P2X4 Receptor Function in the Nervous System and Current Breakthroughs in Pharmacology. Front. Pharmacol. 8, 291–305 (2017).
Ali, Z. et al. Pharmacokinetic and pharmacodynamic profiling of a P2X7 receptor allosteric modulator GSK1482160 in healthy human subjects. Br. J. Clin. Pharmacol. 75, 197–207 (2013).
Recourt, K. et al. Characterisation of the pharmacodynamic effects of the P2X7 receptor antagonist JNJ-54175446 using an oral dexamphetamine challenge model in healthy males in a randomised, double-blind, placebo-controlled, multiple ascending dose trial. J. Psychopharmacol. 34, 1030–1042 (2020).
Bhattacharya, A. et al. Neuropsychopharmacology of JNJ-55308942: evaluation of a clinical candidate targeting P2X7 ion channels in animal models of neuroinflammation and anhedonia. Neuropsychopharmacology 43, 2586–2596 (2018).
ClinicalTrials. Study of SGM-1019 in Patients with Nonalcoholic Steatohepatitis (NASH) https://clinicaltrials.gov/study/NCT03676231?term=SGM-1019&rank=1.
Stock, T. C. et al. Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J. Rheumatol. 39, 720–727 (2012).
Keystone, E. C. et al. Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann. Rheum. Dis. 71, 1630–1635 (2012).
Asahi-Kasei. Lilly and Asahi Kasei Pharma Announce License Agreement for Chronic Pain Drug Candidatehttps://www.asahi-kasei.com/news/2020/e210129_2.html
Iqbal. J. et al. A patent review of P2X7 receptor antagonists to treat inflammatory diseases (2018 - present), Expert Opin. Ther. Patents, 2024.
Tang H., et al. P2X7 receptors: a bibliometric review from 2002 to 2023, Purinergic Signal, 2024.
Genetzakis, E. et al. Development and clinical translation of P2X7 receptor antagonists: A potential therapeutic target in coronary artery disease?. Pharmacol. Ther. 237, 108228–108248 (2022).
Schäfer, W. et al. Origin, distribution, and function of three frequent coding polymorphisms in the gene for the human P2X7 ion channel. Front. Pharmacol. 13, 1033135–1033147 (2022).
De Marchi, E. et al. P2X7 receptor as a therapeutic target. Adv. Protein Chem. Struct. Biol. 104, 39–79 (2016).
Van Weehaeghe, D. et al. 11C]JNJ54173717, a novel P2X7 receptor radioligand as marker for neuroinflammation: human biodistribution, dosimetry, brain kinetic modelling and quantification of brain P2X7 receptors in patients with Parkinson’s disease and healthy volunteers. Eur. J. Nucl. Med. Mol. Imaging 46, 2051–2064 (2019).
Nicke, A. The P2X7 Receptor Methods and Protocols. Humana Press.
Cichero, E. & Tonelli, M. Targeting species-specific trace amine-associated receptor 1 ligands: to date perspective of the rational drug design process. Future Med. Chem. 9, 1507–1527 (2017).
Sheng, D. & Hattori, M. Recent progress in the structural biology of P2X receptors. Proteins 90, 1779–1785 (2022).
Guo, C. et al. Chronic cough relief by allosteric modulation of P2X3 without taste disturbance. Nat. Commun. 14, 5844–5862 (2023).
Denis, G. & Nathalie, C. BLU-5937: A selective P2X3 antagonist with potent anti-tussive effect and no taste alteration. Pulm. Pharmacol. Ther. Vol. 56, 56–62 (2019).
Shen, C. et al. Structural insights into the allosteric inhibition of P2X4 receptors. Nat. Commun. 14, 6437–6452 (2023).
Karasawa, A. & Kawate, T. Structural basis for subtype-specific inhibition of the P2X7 receptor. ELife 5, e22153 (2016).
Wang, D. P. et al. Druggable site near the upper vestibule determines the high affinity and P2X3 homotrimer selectivity of sivopixant/S-600918 and its analogue DDTPA. Br. J. Pharmacol. 181, 1203–1220 (2024).
Honore, P. et al. A-740003 [N-(1-{[(cyanoimino)(5-quinolinylamino) methyl]amino}-2,2-dimethylpropyl)-2-(3,4-dimethoxyphenyl)acetamide], a novel and selective P2X7 receptor antagonist, dose-dependently reduces neuropathic pain in the rat. J. Pharmacol. Exp. Ther. 319, 1376–1385 (2006).
Donnelly-Roberts, D. L. et al. Mammalian P2X7 receptor pharmacology: comparison of recombinant mouse, rat and human P2X7 receptors. Br. J. Pharmacol. 157, 1203–1214 (2009).
Ase, A. R. et al. Identification and Characterization of a Selective Allosteric Antagonist of Human P2X4 Receptor Channels. Mol. Pharmacol. 87, 606–616 (2015).
Sluyter, R. et al. Animal Models for the Investigation of P2X7 Receptors. Int. J. Mol. Sci. 24, 8225–8255 (2023).
Ga, L. et al. Synthesis and structure–activity relationships of novel, substituted 5,6-dihydrodibenzo [a,g] quinolizinium P2X7 antagonists. Bioorg. Med. Chem. Lett. 19, 954–958 (2009).
Bhattacharya, A. et al. Pharmacological characterization of a novel centrally permeable P2X7 receptor antagonist: JNJ-47965567. Br. J. Pharmacol. 170, 624–640 (2013).
Thach, T. et al. Mechanistic insights into the selective targeting of P2X3 receptor by camlipixant antagonist. J. Biol. Chem. 301, 108109 (2015).
Letavic, M. A. et al. 4-Methyl-6,7-dihydro-4H -triazolo [4,5-c] pyridine-Based P2X7 Receptor Antagonists: Optimization of Pharmacokinetic Properties Leading to the Identification of a Clinical Candidate. J. Med. Chem. 60, 4559–4572 (2017).
Sheng, D. et al. Structural insights into the orthosteric inhibition of P2X receptors by non-ATP analog antagonists. ELife 12, RP92829 (2024).
McCarthy, A. E., Yoshioka, C. & Mansoor, S. E. Full-Length P2X7 Structures Reveal How Palmitoylation Prevents Channel Desensitization. Cell 179, 659–670 (2019).
Harkat, M. et al. On the permeation of large organic cations through the pore of ATP-gated P2X receptors. Proc. Natl. Acad. Sci. USA 114, E3786–E3795 (2017).
Boehm, O. et al. Clinical chemistry reference database for Wistar rats and C57/BL6 mice. Biol. Chem. 388, 547–554 (2007).
Di Virgilio, F. et al. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 18, 601–618 (2018).
Dabbagh, K. et al. Preclinical and first-in human development of SGM-1019, a first-in-class novel small molecule modulator of inflammasome activity for the treatment of nonalcoholic steatohepatitis (NASH). J. Hepatol. 68, S60 (2018).
Zumerle, S. et al. Intercellular Calcium Signaling Induced by ATP Potentiates Macrophage Phagocytosis. Cell Rep. 27, 1–10 (2019).
Li, S. et al. Diagnostics for wound infections. Adv. Wound Care 10, 317–327 (2021).
Xi, Y. et al. Biomimetic elastomeric polypeptide-based nanofibrous matrix for overcoming multidrug-resistant bacteria and enhancing full-thickness wound healing/skin regeneration. ACS Nano 12, 10772–10784 (2018).
Zhang, H. et al. Switching from membrane disrupting to membrane crossing, an effective strategy in designing antibacterial polypeptide. Sci. Adv. 9, n771 (2023).
Lu, S. et al. Allosteric modulator discovery: from serendipity to structure-based design. J. Med. Chem. 62, 6405–6421 (2019).
Michel, A. D., Chambers, L. J. & Walter, D. S. Negative and positive allosteric modulators of the P2X(7) receptor. Br. J. Pharmacol. 153, 737–750 (2008).
Smith, N. J. & Milligan, G. Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol. Rev. 62, 701–725 (2010).
Garceau, D. & Chauret, N. BLU-5937: A selective P2X3 antagonist with potent anti-tussive effect and no taste alteration. Pulm. Pharmacol. Ther. 56, 56–62 (2019).
Pasqualetto, G. et al. Identification of the molecular determinants of antagonist potency in the allosteric binding pocket of human P2X4. Front. Pharmacol. 14, 1101023–1101132 (2023).
Ase, A. R., Therrien, E. & Seguela, P. An allosteric inhibitory site conserved in the Ectodomain of P2X receptor channels. Front. Cell. Neurosci. 13, 121–131 (2019).
Michel, D. et al. Mechanism of action of species-selective P2X7 receptor antagonists. Br. J. Pharmacol. 156, 1312–1325 (2009).
Di Virgilio, F., Schmalzing, G. & Markwardt, F. The Elusive P2X7 Macropore. Trends Cell Biol. 28, 392–404 (2018).
Lara, R. et al. P2X7 in cancer: from molecular mechanisms to therapeutics. Front. Pharmacol. 11, 793–822 (2020).
Sarti, A. C., Vultaggio-Poma, V. & Di Virgilio, F. P2X7: a receptor with a split personality that raises new hopes for anti-cancer therapy. Purinergic Signal 17, 175–178 (2021).
Ghiringhelli, F. et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 15, 1170–1178 (2009).
Wang, J. et al. Druggable negative allosteric site of P2X3 receptors. Proc. Natl. Acad. Sci. USA 115, 4939–4944 (2018).
Ma, X. et al. The long β2,3-sheets encoded by redundant sequences play an integral role in the channel function of P2X7 receptors. J. Biol. Chem. 298, 102002–102014 (2022).
Wu, C. et al. High-quality, high-throughput cryo-electron microscopy data collection via beam tilt and astigmatism-free beam-image shift. J. Struct. Biol. 208, 107396–107401 (2019).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. ELife 7, e42166 (2018).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Emsley, P. et al. Features and development of Coot. Acta Crystallogr. Sect. D. -Biol. Crystallogr. 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. D. -Struct. Biol. 75, 861–877 (2019).
Schrödinger L. Schrödinger Release 2018-1 Desmond. 2018.
Cui, W. et al. P2X3-selective mechanism of Gefapixant, a drug candidate for the treatment of refractory chronic cough. Comput. Struct. Biotechnol. J. 20, 1642–1653 (2022).
Jorgensen, W. L. et al. Development and testing of the OPL Sall-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996).
Jorgensen, W. L. & Maxwell, D. S. TiradoRives J. Development and testingoftheOPLSall-atomforcefield on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996). 12.
Banks, J. L. et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 26, 1752–1780 (2005).
Sun, M. et al. Vanilloid agonist-mediated activation of TRPV1 channels requires coordinated movement of the S1–S4 bundle rather than a quiescent state. Sci. Bull. 67, 1062–1076 (2022).
Stahl, F. R. et al. Laboratory diagnostics of murine blood for detection of mouse cytomegalovirus (MCMV)-induced hepatitis. Sci. Rep. 8, 14823 (2018).
Knight, P. et al. Sex differences in the elevated plus-maze test and large open field test in adult Wistar rats. Pharmacol. Biochem. Behav. 204, 173168 (2021).
Ueno, H. et al. Effect of simultaneous testing of two mice in the tail suspension test and forced swim test. Sci. Rep. 12, 9224 (2022).
Yamamoto, T., Nozaki-Taguchi, N. & Chiba, T. Analgesic effect of intrathecally administered orexin-A in the rat formalin test and in the rat hot plate test. Br. J. Pharmacol. 137, 170–176 (2002).
Modi, A. D., Parekh, A. & Pancholi, Y. N. Evaluating pain behaviours: Widely used mechanical and thermal methods in rodents. Behav. Brain Res. 446, 114417 (2023).
Fan, Z. et al. Using the tube test to measure social hierarchy in mice. Nat. Protoc. 14, 819–831 (2019).
Matsuura, K. et al. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J. Neurosci. Methods 73, 45–48 (1997).
Ordway, D. et al. Intracellular activity of clinical concentrations of phenothiazines including thioridiazine against phagocytosed Staphylococcus aureus. Int. J. Antimicrob. Agents 20, 34–43 (2002).
Benov, L. Improved formazan dissolution for bacterial MTT assay. Microbiol. Spectr. 9, e01637-21 (2021).
Laskowski, R. A. & Swindells, M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 (2011).
Acknowledgements
This study was supported with funds from National Natural Science Foundation of China (No. 32371289 to Y. Y., No. U23A20524 and 82474171 to C. G., No. 82130105, 22337003, and 82121005 to H. L., and No. 32271244, 32471247, 32411540020 to M. H.), Innovation and Entrepreneurship (Shuangchuang) Program of Jiangsu Province (JSSCTD202350 to Y. Y.), National Key R&D Program of China (2022YFA1302900 to H. L.), Changsha Jie Bang Gua Shuai Major Science and Technology Programs (KQ2301004, Y. Y.), the CAMS Innovation Fund for Medical Sciences (2019-I2M-5-074, Y. Y.), the Medical Innovation and Development Project of Lanzhou University (lzuyxcx-2022-156, Y. Y.), and the Open Research Fund of State Key Laboratory of Genetics and Development of Complex Phenotypes (No. SKLGDP2502, M. H.). We thank the staff scientists at the Center for Biological Imaging, Institute of Biophysics (Chinese Academy of Sciences), for technical assistance with cryo-EM data collection (project numbers: CBIapp202007004).
Author information
Authors and Affiliations
Contributions
Y.Y., H.L., and M.H. designed the project; C.G., T.L., J.Y., Y-Y. Z., D.W.,J.C,. and Y.H. performed cell culture and patch-clamp recording; C.G. and J.Y. performed animal experiments; D.S.,C.S., F.J., and M.H. performed Cryo-EM experiments; Y.Y., C.G., and P.C. performed MD simulations; J-Y. L., R.Z., J.L., J.W,. and Y.Z. performed chemical synthesis experiments; Y. Y. and C.G. analyzed data; and Y.Y., M.H., H.L., and C.G. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks David Thal, who co-reviewed with Felix Bennetts, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, CR., Sheng, D., Li, JY. et al. Understanding interspecies drug response variations between human and rodent P2X7 receptors. Nat Commun 16, 10827 (2025). https://doi.org/10.1038/s41467-025-65847-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65847-0
