
Abstract
Real-time in vivo monitoring of levodopa pharmacokinetics is essential to address its narrow therapeutic window in Parkinson’s disease (PD) therapy. However, current methods require excessive sample volumes, suffer low sampling frequencies, and fail to capture complete pharmacokinetic profiles. Here, we present an in vivo monitoring system for real-time tracking of levodopa levels in interstitial fluid (ISF) using a spindle-shaped carbon nanotube (CNT) fiber electrochemical sensor functionalized with a nanoscale molecularly imprinted polymer (nanoMIP) on single electroactive CNTs. The core-shell (CNT-nanoMIP) nanostructures provide sensitive, selective detection of levodopa, along with resistance to both biofouling and chemical fouling in continuous ISF detection. The system was validated for monitoring complete pharmacokinetic profiles and assessing temporal correlations between ISF/plasma levodopa pharmacokinetics in both PD rat models and healthy rats. We also demonstrated optimized and adjustable pharmacokinetic profiles through in vivo experiments with rationally designed levodopa dosing regimens, highlighting the system’s potential for personalized PD pharmacotherapy.
Similar content being viewed by others
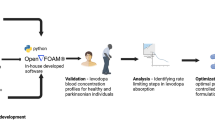


Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorder, associated with dysregulated motor and nonmotor symptoms, and currently affecting more than 8 million people worldwide1,2. It is characterized by the irreversible depletion of dopaminergic neurons in the substantia nigra and a subsequent progressive loss of the neurotransmitter dopamine3. Levodopa (L-Dopa), which has served as the gold-standard anti-parkinsonian medicine since its discovery in the 1960s4, can cross the blood-brain barrier and is metabolized into dopamine, thus helping PD patients restore and enhance dopamine levels in the brain and recover most motor function5,6. However, with long-term L-Dopa treatment, PD patients suffer from severe motor complications, such as dyskinesias and off-time episodes, due to the complex dose–response relationship related to L-Dopa’s short half-life and narrow therapeutic window7,8 (Supplementary Fig. S1). Real-time continuous monitoring of drug pharmacokinetics (PK) holds immense potential for enabling accurate and personalized drug treatment by providing clinicians with critical and quantitative pharmacological information9,10,11,12,13.
In therapeutic drug monitoring (TDM), gold-standard detection of L-Dopa levels is typically performed in laboratory settings using high-performance liquid chromatography (HPLC) or liquid chromatograph-mass spectrometer (LC-MS) analysis of plasma samples. The implementation of these approaches for at-home monitoring and timely medical intervention is hindered by challenges related to intensive labor requirements, complex instrumentation, and long turnaround times. Furthermore, issues such as rapid degradation of L-Dopa during sample transmission14, frequent sampling demands, and the absence of continuous dynamic information restrict the clinical adoption of L-Dopa PK monitoring. The development of electrochemical sensing technologies has facilitated the continuous monitoring of health-related and disease-specific biomarkers across various human biological fluids at the individual level15,16,17,18,19,20. However, current electrochemical sensors for L-Dopa are largely limited to point-of-care detection, relying on blood14,21 or sweat samples22,23,24,25 and requiring intermittent sampling. Recent advances in active iontophoresis stimulation have shown some promise for continuous sweat monitoring in PD patients26 but are unable to support long-term and uninterrupted sweat analysis. Alternatively, implantable sensors offer considerable promise for real-time, continuous, and on-site monitoring of L-Dopa27,28,29. Yet, their practical use in in vivo PK monitoring remains limited by challenges such as low target analyte concentration and severe electrode passivation caused by biofouling and L-Dopa’s self-polymerization30. What’s more, few of the existing L-Dopa sensors have been validated against the gold-standard method of plasma HPLC quantification. Therefore, the pharmacological relationship between L-Dopa levels in ISF and circulating blood remains unclear to date.
Here, we present a continuous L-Dopa sensing system for real-time and in vivo monitoring of L-Dopa PK in ISF (Fig. 1a, b). The system integrates a fiber-assembled implantable electrochemical sensor for sensitive and selective L-Dopa detection, along with a flexible circuit patch coupled with a Bluetooth module for wireless data transmission (Supplementary Figs. S2, S3). The key sensing fiber, referred to as the nanoMIP fiber sensor, is constructed by functionalizing nanoscale molecularly imprinted polymers (nanoMIP) onto individual carbon nanotubes (CNTs) from a spindle-shaped carbon nanotube fiber (SSCNTF)31. The CNT-core/nanoMIP-shell nanostructure imparts the nanoMIP fiber with L-Dopa-specific recognition via imprinted sites within the nanoMIP-shell and facilitates efficient electrochemical redox signal transduction through the electroactive CNT-core (Fig. 1c, d). The specific binding of L-Dopa results in an increased peak current in differential pulse voltammetry (DPV), while non-target analytes were excluded by the target-specific MIP shell due to their non-complementary structures. The design of core-shell nanostructures also offers multiple advantages for continuous L-Dopa monitoring in ISF, including resistance to both biofouling and chemical fouling, as well as good biocompatibility. Moreover, the nanostructures confer structural (Fig. 1e) and electrochemical (Fig. 1f) stability to the nanoMIP fiber sensor under strain conditions that mimic typical skin deformation32.

a Schematic of the in vivo L-Dopa sensing system, including a nanoMIP fiber sensor implanted subcutaneously for real-time ISF monitoring and a wearable flexible printed circuit board (FPCB) patch for DPV detection. The fiber sensor consists of a nanoMIP-coated spindle-shaped CNT fiber (SSCNTF), a CNT fiber (CNTF), and an Ag/AgCl-coated CNTF. b Schematic of PK optimization through timely dosage intervention guided by real-time L-Dopa monitoring. c Sensing mechanism: the nanoMIP shell specifically binds L-Dopa molecules, while the CNT core transduces the redox signal during L-Dopa oxidation. d TEM showing the core-shell nanostructure composed of a nanoMIP shell and CNT core. e Three-dimensional structure characterization of the nanoMIP fiber by X-ray tomography scanning (left). X-ray tomography of the nanoMIP fiber under 0% and 20% strain, with reconstructed images and surface area comparisons. f Electroactive surface area characterization before and after 20% stretching. g Photo of a rat model undergoing real-time L-Dopa PK monitoring using the sensing system. Inset: flexible printed circuit board (FPCB) patch. h Correlation between the nanoMIP fiber sensor and an electrochemical working station. Current signals were measured by differential pulse voltammetry (DPV) in simulated body fluid containing 20–80 μM L-Dopa. i DPV curves detected before (0 min) and after L-Dopa injection (40 min). Oxidation peak currents were used to calculate L-Dopa concentration. j L-Dopa PK profile following 0.12 mg·g⁻¹ dosing.
By addressing critical challenges in ISF-based L-Dopa monitoring through functional sensing electrodes, this system extends to in vivo PK monitoring in various rat trials (Fig. 1g). Prior to in vivo application, the sensing accuracy was validated using a commercial electrochemical workstation (Fig. 1h). When subcutaneously implanted in a rat model, the nanoMIP fiber sensor enables sensitive and selective detection of L-Dopa level changes (Fig. 1i), as well as continuous monitoring over a full PK cycle (Fig. 1j). In addition, we showcase the potential of the system for accurate PK profiling by examining correlations between dynamic L-Dopa concentrations and key PK parameters obtained from ISF (via the sensor) and blood (via HPLC). To further support the development of precise PD therapies, we characterized L-Dopa PK in both healthy and 6-hydroxydopamine (6-OHDA)-induced PD rat models and analyzed PK variations across different L-Dopa dosing regimens, including low-dose intermittent injection and variable-rate continuous infusion.
Results
Fabrication and characterization of the nanoMIP fiber electrode
The L-Dopa nanoMIP fiber was fabricated by co-electropolymerizing functional monomers with L-Dopa template molecules on the spindle-shaped CNTF (SSCNTF) electrode and then electrochemically removing the template L-Dopa molecules. In this work, we used an expanded SSCNTF as the electrode substrate that enables high electrochemical activity (Supplementary Fig. S4) with increased accessible CNT sites and enlarged network pores for mass diffusion31,33,34. The substantial individual CNT networks obtained in SSCNTF can provide sufficient surface area for MIP deposition on each electroactive CNT (Supplementary Fig. S5). The expanded SSCNTFs exhibited consistent redox capability toward the K3[Fe(CN)6]/K4[Fe(CN)6] redox couple (Supplementary Fig. S6). The fabrication process is illustrated in Fig. 2a. In detail, the nanoMIP layer was first deposited onto the CNT surface by electropolymerization in a mixed solution of pyrrole, resorcinol, and L-Dopa using cyclic voltammetry (CV, −0.2 V to +0.8 V)35. The L-Dopa template was subsequently eluted from the nanoMIP shell by applying a potential of 0.6 V in 0.1 M NaOH. These approaches could accelerate the electrochemical oxidation of the bound L-Dopa and disrupt its non-covalent interactions within the polymer matrix. The bound L-Dopa thus rapidly desorbs from the MIP layer, generating specific binding cavities with a complementary conformation to L-Dopa. This process effectively exposed the CNT core to the target drug molecule.

a Schematic illustration of the fabrication process of the nanoMIP fiber electrode and the corresponding structural changes in the CNT. b Scanning electron microscopy (SEM) image of MIP polymerized on CNTF (CNTF/MIP). Surface and internal structure of SSCNTF/MIP before (c) and after (d) template molecule extraction. Left: SEM image of SSCNTF/MIP; right: TEM image showing the nanoMIP structure. Raman spectra of SSCNTF, SSCNTF/MIP, and SSCNTF/NIP before and after template extraction, showing characteristic peaks of N 1s (e) and O 1s (f). a.u., arbitrary units. g DPV curves of SSCNTF/MIP and SSCNTF/NIP in response to 100 μM L-Dopa. h DPV curves of SSCNTF/MIP and CNTF/MIP in response to 10 μM L-Dopa.
The electrochemical polymerization of the nanoMIP fiber was first characterized and compared with non-imprinted polymer (NIP) (Supplementary Fig. S7), which was electropolymerized without the template molecule. In the polymerization CV curves, no oxidation signal of L-Dopa was observed in the NIP, while the MIP efficiently polymerized the L-Dopa template, showing a distinct oxidation peak in the first scan (Supplementary Fig. S8). Following polymerization, fourier transform infrared spectroscopy (FTIR) analysis was conducted to elucidate the interactions between the functional monomers and the template molecule. The results revealed that the binding was mainly governed by O–H···O–H and O–H···N–H hydrogen bonds, while additional N–H···O–H hydrogen bonding was also identified between the two functional monomers (Supplementary Fig. S9). These non-covalent interactions provide the selective recognition of L-Dopa within the monomer matrix. Moreover, the representative electroactivity of the MIP/NIP fiber was analyzed before and after template removal (Supplementary Fig. S10). Prior to L-Dopa elution, a larger double-layer current was observed in the MIP compared to the NIP. This difference is primarily attributed to the presence of L-Dopa, which leads to variations in the thickness and purity of the conductive polypyrrole (PPy) matrix. After elution, decreased double-layer current was identified in nanoMIP fiber, indicating the restoration of conductive pathways for redox electron transfer in MIP. These results confirm the successful incorporation of L-Dopa molecules into the nanoMIP layer before chronoamperometry (CA) elution and their effective removal afterward.
To further verify the extraction of the templates, the surface chemistry of the MIP and NIP was performed. As shown in SEM and TEM images (Fig. 2c), the MIP fiber exhibited a dense network of core-shell nanostructures, each comprising an individual CNT core and an encircling polymer shell. The elution process led to distinct morphological changes in the core-shell nanostructures. The MIP shell exhibited a significant reduction of ~20 nm in thickness after the detachment of L-Dopa from the polymeric matrix (Fig. 2d), whereas the NIP retained an intact shell with an almost unchanged thickness, showing only a slight decrease of ~4 nm (Supplementary Figs. S11, S12). XPS analysis of this shell revealed a decreased carbon content (Supplementary Fig. S13) and the emergence of nitrogen (N) signals characteristic of PPy (Fig. 2e) in the SSCNTF after MIP deposition. The MIP composition was further validated by Raman spectroscopy, confirming the presence of PPy with its characteristic peaks at 925, 981, and 1039 cm⁻¹ (Supplementary Fig. S14). In addition, the MIP shell contained a higher oxygen (O) content than the NIP, which was attributed to the incorporation of L-Dopa templates (Fig. 2f). After removing the templates, O content in NIP showed a significant increase, likely resulting from the introduction of oxidative functional groups under the overpotential of 0.6 V36. In contrast, the decreased O content observed in the MIP reflected a diminished level of bound L-Dopa. The FTIR spectra of the MIP showed decreases in the broad band at 3200–3600 cm⁻¹ after template removal, due to the loss of –OH and –NH₂ groups from L-Dopa, whereas no such change was observed for the NIP (Supplementary Fig. S15). In addition, at ~1600 cm⁻¹ (C=O and aromatic C=C stretching), the MIP exhibited a clear decrease after elution, indicating the elimination of C=O groups from L-Dopa. These observations indicated the selective extraction of template molecules, which may help to generate specific cavities embedded within non-specific polymer layers.
To illustrate the specific recognition of L-Dopa, we applied DPV to determine 10–100 μM L-Dopa in both MIP/SSCNTF and NIP/SSCNTF electrodes. No oxidation peak was found on the NIP even at a high concentration of 100 μM (Fig. 2g). However, detection with MIP/SSCNTF enables well-defined L-Dopa oxidation peaks with a flat baseline across all concentrations, indicating the successful fabrication of an L-Dopa-specific MIP electrode (Supplementary Fig. S16). Beyond specificity, high sensitivity is also important for drug sensing featuring a low in vivo concentration. CNTF-based MIP sensors (CNTF/MIP) have been previously reported for cortisol and lactate detection37,38, typically consisting of an electroactive CNTF as a redox core and a selective polymeric shell. Following a similar strategy, we fabricated a CNTF-based L-Dopa MIP electrode (CNTF/MIP) by replacing SSCNTF with CNTF. The CNTF/MIP exhibited a microscale MIP shell coating on the CNTF surface, referred to as microMIP (Fig. 2b). We evaluated its electrochemically active surface area and sensing performance towards L-Dopa with a low concentration (10 μM). This microMIP exhibited a smaller active surface area (~0.0099 cm2) than nanoMIP (~0.0496 cm2) (Supplementary Fig. S17) and limited sensitivity for L-Dopa sensing, with no detectable oxidation current (Fig. 2h). The comparison of the DPV responses of CNTF/MIP with SSCNTF/MIP electrodes demonstrates the benefits of the nanoscale core-shell sensing structures in enhancing molecular binding and signal transduction, which are crucial for in vivo monitoring of L-Dopa.
The sensitivity and selectivity of the nanoMIP electrode exhibit an inverse relationship, determined largely by the structure of the MIP shell, which is influenced by both electropolymerization and template extraction conditions (Supplementary Fig. S18). These parameters were evaluated by comparing the L-Dopa response between MIP and NIP electrodes. The response of NIP represents a non-specific electrochemical reaction on the CNT surface, which is exposed to the target sample due to an incomplete or degraded shell structure. An optimal condition should be characterized by a strong MIP response combined with little signal from the NIP. Here, we show that electrochemical template extraction using chronoamperometry (CA) at 0.6 V in 0.1 M NaOH enabled more efficient elution, yielding a greater signal difference between MIP and NIP compared to extraction using CV (0–0.6 V, Supplementary Fig. S19) or extraction in 0.1 M PBS (Supplementary Fig. S20), 0.1 M NH₃·H₂O, and 0.1 M KOH (Supplementary Fig. S21). Following these extraction conditions, the number of CV polymerization cycles (Supplementary Fig. S22), the duration of CA elution (Supplementary Fig. S23), the template concentration (Supplementary Fig. S24), and the incorporation of resorcinol in MIP polymerization (Supplementary Fig. S25) were further optimized. The results showed that the resorcinol-incorporated MIP prepared with 18 CV cycles, 30 s CA extraction, and 15 mM template concentration enabled an L-Dopa-specific current 12.2-fold greater than that of ascorbic acid39, a common bio-interferent analyte. In conclusion, a favorable trade-off between sensitivity and selectivity has been achieved in the nanoMIP fiber sensor, thereby improving the accuracy of L-Dopa quantification.
In vitro evaluation of the implantable nanoMIP fiber sensor
We tested the sensing performance of the nanoMIP fiber sensor in a series of in vitro and in vivo experiments. We first established an optimized DPV method for detecting L-Dopa, using a modulation time of 0.015 s and a step voltage of 0.005 V (Supplementary Fig. S26). In vitro DPV measurements of the nanoMIP fiber sensor in PBS showed a clear current gain in response to increasing L-Dopa concentrations (0.5–100 μM) (Supplementary Fig. S27). The physiological concentration range of L-Dopa in humans typically falls between 0 and 30 μM14. DPV measurements (Fig. 3a) and calibration curve (Fig. 3b) within this range demonstrate an experimentally determined detection limit of 0.5 μM and an ultra-high sensitivity of 6.45 μA·μM-1, which surpass those reported by most voltammetric methods for L-Dopa detection (Supplementary Table S1). We next evaluated the selectivity of the nanoMIP fiber sensor by monitoring the changes in current response to 100 μM L-Dopa upon the sequential addition of potential biological interferents, including glucose (Glu), urea (Urea), lactic acid (Lac), creatinine (Cre), cortisol (Cor), ascorbic acid (AA), uric acid (UA), the commonly used adjuvant drug carbidopa, and another interfering drug, acetaminophen (Fig. 3c), as well as various essential amino acids (Supplementary Fig. S28). These interferents produced negligible changes in the oxidation current of L-Dopa, demonstrating the excellent anti-interference capability of the nanoMIP sensor for in vivo monitoring. The reversibility of the MIP between high (30 μM) and low (0 μM) L-Dopa concentrations was also evaluated both in vitro (Fig. 3d and Supplementary Fig. S29) and under simulated in vivo conditions (Supplementary Fig. S30). The nanoMIP sensor exhibited reliable reversibility over 40 switching cycles, showing stable peak current differences (\(\Delta {{\rm{Current}}}\)) with relative standard deviations of 9.8% in vitro and 6.7% in vivo. This reversibility results from the electrochemical oxidation of L-Dopa and its subsequent desorption from the MIP within the DPV window (0–0.6 V)15. We also observed consistent sensing performance of the nanoMIP fiber sensor after 7 days of storage in deionized water at 4 °C, suggesting its excellent storage stability and convenience (Supplementary Fig. S31).

a DPV curves for L-Dopa detection using the nanoMIP fiber. b Corresponding calibration plot with linear fitting. (n = 3 independent samples, mean ± SD). c Selectivity of the sensor for 30 μM L-Dopa against 1 mM glucose (Glu), 1 mM urea, 1 mM lactic acid (Lac), 100 μM creatine (Cre), 1 μM cortisol (Cor), 100 μM ascorbic acid (AA), 100 μM uric acid (UA), 30 μM carbidopa (C-Dopa), and 100 μM acetaminophen (Ace). IL-Dopa and Iinterference represent the L-Dopa response and signal change with interferents, respectively. (n = 3 independent samples, mean ± SD). d Representative reversible sensing behavior of the nanoMIP sensor. The nanoMIP was alternately tested in 30 μM L-Dopa and 0 μM L-Dopa in 0.1 M PBS for 40 switching cycles. e Schematic of chemical fouling during the electrochemical oxidation of L-Dopa to dopaquinone. Oligomerization of oxidation byproducts, 5,6-dihydroxyindole-2-carboxylic acid (DHICA) and 5,6-dihydroxyindole (DHI), causes a fouling layer on the electrode surface. f Change in oxidation current over 1–50 DPV scans in 100 μM L-Dopa. Ip: the peak current at each cycle; I1st: initial peak current. (n = 3 independent samples, mean ± SD). g SEM images of nanoMIP fiber and SSCNTF electrodes after 50 DPV scans. Calibration plots of SSCNTF (h) and nanoMIP fiber (i) in PBS before and after subcutaneous implantation in rats for 2 h. (n = 3 independent samples, mean ± SD). The insets were created with BioRender.com/fifwmcd and BioRender.com/cshw283. j Fluorescence images of NIH/3T3 cells cultured for 5 days in extract solutions from 1, 4, or 7 nanoMIP fibers. Live cells (green: calcein acetoxymethyl ester, Calcein-AM), dead cells (red: propidium iodide, PI). k Cell viability quantified by Calcein-AM/PI ratio. (n = 4 independent samples, mean ± SD). l Representative hematoxylin and eosin (H&E) staining of rat dorsal skin after 1 and 4 days of subcutaneous nanoMIP fiber implantation.
Chemical fouling of the electrode surface poses a major challenge in the long-term electrochemical detection of phenolic analytes such as dopamine, serotonin, and L-Dopa, resulting from the excessive accumulation of their oxidation by-products40,41,42. In the case of L-Dopa oxidation (Fig. 3e), not only is L-dopaquinone produced, but undesirable derivatives, including 5,6-dihydroxyindole-2-carboxylic acid (DHICA) and 5,6-dihydroxyindole (DHI), are also generated30. These species can spontaneously polymerize into oligomers and adsorb onto the electrode surface, forming a chemical fouling layer. This fouling layer can significantly reduce the sensitivity, response rate, and detection stability of the L-Dopa sensor, thereby limiting its applicability in continuous monitoring. We next assessed the changes in current response during continuous L-Dopa detection (100 μM) over 50 successive DPV scans on both nanoMIP fiber and SSCNTF electrodes without MIP modification. As a result, the current response from the nanoMIP fiber retained 99% of its initial current after 50 consecutive scans. In contrast, the bare SSCNTF electrode retained only 26% due to the severe chemical fouling (Fig. 3f and Supplementary Fig. S32). To verify this, we characterized the surface morphology of the tested electrodes using SEM. As observed in Fig. 3g, clusters of polymeric by-products were adsorbed on the CNT surface within the SSCNTF electrode, whereas no such aggregation was found on the nanoMIP fiber. Furthermore, Raman spectra before and after 50 consecutive L-Dopa detections were examined (Supplementary Fig. S33). The SSCNTF electrode exhibited an H–C–H wagging deformation band at 1400 cm⁻¹, indicative of the formed L-Dopa polymerization products, and its ID/IG ratio—a parameter reflecting surface defects and structural disorder—increased from 0.32 to 0.65 (Supplementary Fig. S33a)41. In contrast, the nanoMIP showed nearly unchanged ID/IG values, confirming its resistance to chemical fouling (Supplementary Fig. S33b). These findings suggest that the nanoMIP shell structure effectively mitigates the self-polymerization and adsorption of the by-products on the electroactive CNT surface. This effect may be attributed to the spatially confined oxidation of individual L-Dopa molecules within the imprinted cavities, which suppresses subsequent polymerization reactions of the oxidation products (Supplementary Fig. S34). With its high resistance to chemical fouling, the nanoMIP fiber sensor is capable of achieving stable and precise continuous monitoring.
Implantable sensors are exposed to dynamic biological fluids with complex components and are susceptible to rapid biofouling or degradation in such in vivo environments43. On the basis of anti-chemical fouling capability, we anticipated that the nanoMIP fiber sensor would also provide protection against biofouling during in vivo monitoring, owing to the sequestration of reactive sites within the nanoMIP shell. For this purpose, we subcutaneously implanted the nanoMIP fiber and SSCNTF electrodes into a live rat and measured their in vitro sensing signals before and after implantation (Supplementary Fig. S35). After a 2-h incubation, the SSCNTF electrodes exhibited a drastic loss in sensitivity in subcutaneous ISF, dropping from 24.38 μA·μM-1 to 0.24 μA·μM-1 (Fig. 3h), while the nanoMIP fiber sensor retained most of its electrochemical sensing capability (2.67 μA·μM-1 from an initial 4.02 μA·μM-1) (Fig. 3i). Furthermore, the sensitivity of the nanoMIP sensor was assessed after 1, 2, 3, and 4 h of in vitro incubation in simulated body fluid, yielding values of 0.93–1.07 μA·μM-1 with no statistically significant differences (p = 0.8389) (Supplementary Fig. S36). These comparative results suggest that the nanoMIP fiber sensor possesses biofouling resistance to a certain extent owing to its protective nanoMIP shell, demonstrating suitability for ISF-based in vivo monitoring.
We also investigated the biocompatibility of the nanoMIP fiber sensor. Cytocompatibility was first evaluated by culturing NIH/3T3 mouse fibroblast cells in the extract media incubated with 1, 4, and 7 nanoMIP fibers. After five days of culture, cells were stained with Calcein-AM and propidium iodide (PI) to visualize live/dead cell distribution (Fig. 3j). Quantitative analysis of cell viability, based on the ratio of Calcein-AM– to PI–positive areas, demonstrated consistently high viability (>99%) in both the experimental and control groups (cells cultured in untreated medium), indicating good cytocompatibility of the sensor (Fig. 3k). To further evaluate tissue compatibility, we subcutaneously implanted the sensor on the dorsal skin of rats for 1 and 4 days before collecting the skin tissues for histological analysis. Representative hematoxylin and eosin (H&E) staining (Fig. 3l) and Masson’s trichrome staining (Supplementary Fig. S37) presented no apparent inflammatory response at the implantation site, compared to control skin without implantation. Collectively, these results confirm the suitability of the nanoMIP fiber sensor for safe, stable, continuous in vivo monitoring of low-concentration L-Dopa in ISF.
In vivo validation of the L-Dopa sensor for PK monitoring
We assessed the real-time, continuous detection capabilities of the nanoMIP fiber sensor in living rats. We first assembled the nanoMIP fibers, together with counter and reference electrode fibers, into a three-electrode electrochemical sensor and tested their current response to gradient concentrations of L-Dopa in simulated body fluid (SBF) for calibration curve fitting. These curves were used for the calibration of L-Dopa concentration from the DPV data in all in vivo measurements described throughout the manuscript. We then subcutaneously implanted the developed nanoMIP fiber sensor on the backs of two healthy rats to continuously monitor changes in L-Dopa concentration in ISF following intraperitoneal injection of L-Dopa at doses of 0.06 mg·g−1 and 0.12 mg·g−1, respectively44,45 (Fig. 4a). The sensors responded to the L-Dopa dosing in real time and exhibited similar kinetic profiles—characterized by a rapid increase followed by a gradual decline—although the detected concentrations differed between the two administered doses (Fig. 4b).

a Experimental scheme for real-time monitoring of L-Dopa level in ISF in a healthy rat model, with simultaneous blood sampling for HPLC analysis. L-Dopa was administered via intraperitoneal injection. Created with BioRender.com/41h623j. b Variation of L-Dopa levels from the sensor (ISF) and HPLC (blood) following L-Dopa injection at doses of 0.06 mg·g⁻¹ (i) and 0.12 mg·g⁻¹ (ii). c Correlation of L-Dopa levels between ISF and blood at (i) 0.06 mg·g⁻¹ and (ii) 0.12 mg·g⁻¹, considering data within 0–240 min after injection. d Schematic of a two-compartment pharmacokinetic model and representative PK curves of the central (blood) and peripheral compartment (ISF), including key PK parameters: \({{\rm{AUC}}}\) (area under curve), \({C}_{\max }\) (maximum concentration), \({T}_{\max }\) (time to \({C}_{\max }\)), and \({T}_{1/2}\) (elimination half-life). Created with BioRender.com/tiym1by. Comparisons of \({{\rm{AUC}}}\) (e) and \({C}_{\max }\) (f) at two doses. g Schematic of targeted injection of 6-hydroxydopamine (6-OHDA) into the medial forebrain bundle (MFB) to induce a PD rat model. Created with BioRender.com/7bbvnmy. h Confocal fluorescence images of brain sections from saline-injected (control) and 6-OHDA-injected (PD) rats, stained with 4′,6-diamidino-2-phenylindole (DAPI, blue FL) and cyanine 3 (Cy3, red FL). i Quantification of mean red fluorescence intensity of dopaminergic neurons (n = 3 independent rats, mean ± SD). j Experimental scheme of PK monitoring in healthy/normal and PD rat models. Created with BioRender.com/d0vupnd. L-Dopa PK profiles from normal rats (k) and PD rat models (l).
In parallel, we collected blood samples from the retro-orbital venous plexus at intermittent time points during the on-site sensor monitoring. L-Dopa concentrations in these blood samples were subsequently quantified using standard HPLC analysis conducted in an analytical laboratory. During the long-distance transport of blood samples, L-Dopa is susceptible to degradation and oxidation, which can lead to inaccurate HPLC readouts14. To alleviate these problems, we implemented pretreatment of blood samples involving stabilization in an acidic solution and removal of proteins, as detailed in Supplementary Fig. S38. As a result, we observed typical L-Dopa kinetics in the HPLC readouts, aligned with those obtained from the sensor signals, with clear absorption and elimination phases present in both measurements. Moreover, both methods consistently showed higher L-Dopa concentrations following the 0.12 mg·g−1 dose compared to the 0.06 mg·g−1 dose. We noted that ISF L-Dopa exhibited delayed (average delay of ~13.5 min) and attenuated kinetics (average retention of ~14%) relative to blood. These differences may be attributed to the heterogeneous distribution and transport of L-Dopa molecules from blood to ISF13. To validate the physiological relevance of L-Dopa in ISF, sensor signals were correlated with blood readouts from the two dosing experiments. Strong positive correlations were observed with Pearson’s r ranging from 0.95 to 0.96 (Fig. 4c). Hence, the nanoMIP fiber sensor that enables real-time in vivo L-Dopa profiling may serve as a viable tool for PK study.
Next, we extended this study to investigate the PK characteristics of L-Dopa in relation to the administered doses using a compartment PK model. In this model, blood was considered the central compartment, with ISF serving as the peripheral compartment (Fig. 4d). We derived the key PK parameters (\({{\rm{AUC}}}\) and \({C}_{\max }\)) for both blood (central) and ISF (peripheral) by fitting their concentration-time profiles to the model (Supplementary Fig. S39). In pharmacokinetics (PK) studies, \({{\rm{AUC}}}\) reflects the total drug exposure, and \({C}_{\max }\) describes the maximum drug concentration; both are meaningfully associated with therapeutic efficacy and safety. As shown in Fig. 4e, the \({{\rm{AUC}}}\) values in ISF were higher in the high-dose PK compared to the low-dose PK, showing a trend consistent with the \({{\rm{AUC}}}\) results from blood samples. Likewise, higher \({C}_{\max }\) values were observed at the high dose condition in both ISF and blood PK profiles (Fig. 4f). Despite differences in absolute values, ISF and blood PK parameters exhibited comparable dose-dependent trends. Notably, \({{\rm{AUC}}}\) and \({C}_{\max }\) values were not directly proportional to the administered dose, showing only 1.38-fold and 1.51-fold increases from the low to high dose, respectively, suggesting a nonlinear dose–response relationship that may complicate L-Dopa therapy.
To investigate L-Dopa PK under pathological conditions, we established a Parkinson’s disease (PD) rat model by unilaterally injecting the neurotoxin 6-hydroxydopamine (6-OHDA) into the medial forebrain bundle (MFB) (Fig. 4g and Supplementary Fig. S40). The neurotoxin 6-OHDA can selectively induce dopaminergic neurodegeneration in the lesioned hemisphere, thereby modeling the pathological features of primary Parkinson’s disease46. Two weeks post-surgery, we observed a significant reduction in the number of dopaminergic neurons—immunostained for tyrosine hydroxylase (TH; red fluorescence)—in the substantia nigra (Fig. 4h) and striatum (Supplementary Fig. S41) of 6-OHDA-lesioned rats compared to saline-injected controls. Quantification of TH fluorescence intensity revealed a significant difference between the two groups (p < 0.05), confirming the successful establishment of the PD rat model (Fig. 4i).
We then demonstrated the capability of our L-Dopa sensing system to monitor complete PK profiles in both PD rat model and healthy rats. In the in vivo experiments (Fig. 4j), all rats were implanted with the nanoMIP fiber sensor for continuous monitoring over a 4-h period to enable full capture of the PK profiles, including both absorption and elimination phases. L-Dopa was administered intravenously at a dose of 0.12 mg·g⁻¹. The recorded ISF PK profiles for the PD rat models and healthy/normal rats are displayed in Fig. 4k and l, respectively. We derived the PK parameters using the same fitting method described above (Supplementary Figs. S42, S43), including \({{\rm{AUC}}}\), \({C}_{\max }\), \({T}_{\max }\) (time to reach \({C}_{\max }\)), \({T}_{1/2}\) (apparent half-life), \({T}_{1/2{{\rm{\alpha }}}}\) (distribution half-life), and \({T}_{1/2{{\rm{\beta }}}}\) (elimination half-life) (Table 1). In normal rats, both \({C}_{\max }\) and \({{\rm{AUC}}}\) showed considerable inter-individual variability, with \({C}_{\max }\) ranging from 1.97 to 7.81 mg·L−1 and AUC ranging from 188.32 to 716.49 mg·min·L−1. Similar variability was observed in PD rat models, with \({C}_{\max }\) ranging from 1.85 to 10.4 mg·L−1 and AUC from 119.6 to 895.37 mg·min·L−1. The comparison between normal and PD rat models revealed no significant differences in \({{\rm{AUC}}}\), \({C}_{\max }\), \({T}_{\max }\), \({T}_{1/2}\), and \({T}_{1/2{{\rm{\alpha }}}}\), indicating that the PD pathology does not markedly affect the overall absorption quantity and early distribution of L-Dopa in vivo. However, a significant difference was observed in \({T}_{1/2{{\rm{\beta }}}}\) between PD and normal rats. The reduced values in the PD group suggest an accelerated elimination of L-Dopa, potentially induced by disease-related physiological changes. Nonetheless, nearly all PK parameters appeared to be more strongly influenced by individual physiological variability than by PD pathology itself, underscoring the importance of real-time, individualized monitoring of L-Dopa dynamics. Furthermore, we confirmed the short half-life of L-Dopa, with mean \({T}_{1/2}\) values of 30.98 min in normal rats and 35.29 min in PD rats. This short half-life restricts sustained therapeutic efficacy, suggesting the need for more frequent dosing to maintain consistent drug levels.
PK optimization using adaptive dosing regimens
L-Dopa administration with frequent and low-dose regimens is preferred in clinical practice to maintain a stable and effective therapeutic concentration, avoid excessively high or low plasma levels, and thereby reduce dyskinesia-related adverse effects (due to supratherapeutic levels) and the risk of off-time episodes (due to subtherapeutic levels) in PD therapy47,48. On the basis of the L-Dopa-sensing system, we demonstrated the capability to manage and optimize in vivo L-Dopa PK using intermittent dosing regimens. We monitored the PK profiles of L-Dopa in a single healthy rat following two or four intermittent injections at doses of 0.06 mg·g⁻¹ or 0.03 mg·g⁻¹ per injection, administered at ~30-min intervals based on the drug’s half-life (Fig. 5a). The two regimens were separated by a 24-h washout period to ensure complete clearance and excretion of residual L-Dopa. In the two-dose intermittent injection regimen, two absorption peaks were observed at 50 min and 110 min, responding to the first and second injections, respectively (Fig. 5b). In contrast, under the four-dose intermittent regimen, a single absorption peak appeared at 20 min in response to the initial injection, followed by a gradual increase in L-Dopa levels until a relatively steady state was reached at 110 min (Fig. 5c). These results indicate that multiple doses can lead to a more stable L-Dopa PK, achieving a steady state that is desirable for maintaining therapeutic efficacy.

a Intermittent L-Dopa dosing scheme in a normal rat: two doses (0.06 mg·g⁻¹ each) or four doses (0.03 mg·g⁻¹ each) at 30 min intervals, total 0.12 mg·g⁻¹. Created with BioRender.com/qrwgwmo. L-Dopa PKs under two-dose (b) and four-dose (c) injection regimens. Box and distribution plots show fluctuations of L-Dopa concentration (d) and L-Dopa variation rate (e) monitored under two-dose and four-dose regimens. The variation rate, calculated as the difference quotient of concentration, reflects the extent of change in L-Dopa levels. f Continuous drug administration scheme using an infusion pump. Created with BioRender.com/cuxc20n. g Continuous drug infusion scheme in a normal rat: four rates, total 0.12 mg·g⁻¹. Created with BioRender.com/5va7dto. h L-Dopa PK profiles under four infusion rates: ultra-high-speed (4 μg·g⁻¹·s⁻¹), high-speed (4 μg·g⁻¹·min⁻¹), medium-speed (2 μg·g⁻¹·min⁻¹), and low-speed (1 μg·g⁻¹·min⁻¹) continuous infusion. i Comparison of L-Dopa PK parameters under the four infusion rates. j Box and distribution plots show fluctuations of L-Dopa concentration monitored under four infusion rates. k Compilation of data on L-Dopa concentration and its variation rate in response to progressively increasing infusion rates within a single PK monitoring session. The mean variation rate was calculated at 30, 60, 90, 120, 180, 210, and 240 min. All box plots: mean (center), box = 25th–75th percentiles, whiskers = 1.5 × IQR.
We also analyzed the distribution characteristics of L-Dopa concentration data under the two dosing regimens (Fig. 5d). The four-dose regimen showed a narrower distribution and a lower mean concentration (0.31 mg·L−1) compared to the two-dose regimen (mean concentration of 0.62 mg·L−1), demonstrating that high-frequency and low-dose administration can reduce PK fluctuations. To verify this, we calculated the first-order derivative of the L-Dopa concentration over time, which we defined as the L-Dopa variation rate. As shown in Fig. 5e, the four-dose regimen yielded a more concentrated distribution of L-Dopa variation rate, whereas the two-dose regimen exhibited greater variability. These comparisons suggest that L-Dopa dosing with higher frequency can maintain a relatively stable L-Dopa level. We noted that the two regimens achieved comparable average steady-state concentrations between 140 and 240 min (0.41 mg·L−1 for the two-dose and 0.406 mg·L−1 for the four-dose regimen). These results strongly support that the high-frequency intermittent L-Dopa administration is capable of enhancing therapeutic efficiency and offer valuable insights into the practical and clinical application of in vivo L-Dopa PK in the management of PD.
Multiple daily intakes pose practical challenges for PD patients due to considerable inconvenience and poor compliance. Continuous L-Dopa infusion pumps have been widely proposed to address these challenges; however, their clinical application remains limited9. To investigate its potential for optimizing PK, we conducted in vivo L-Dopa monitoring in a rat receiving continuous intraperitoneal infusion at different rates via a syringe pump (Fig. 5f, g)48. The sensor implantation methods and drug washout periods followed the same procedures as those in the above experiments. Four continuous PK profiles were obtained at ultra-high-speed (4 μg·g⁻¹·s⁻¹), high-speed (4 μg·g⁻¹·min⁻¹), medium-speed (2 μg·g⁻¹·min⁻¹), and low-speed (1 μg·g⁻¹·min⁻¹) infusion rates (Fig. 5h). From these profiles, we calculated three PK indicators—\({{\rm{AUC}}}\), \({C}_{\max }\), and \({T}_{\max }\)—which meaningfully represent the medication’s bioequivalence (Fig. 5i). We demonstrated that these indicators vary significantly in relation to the infusion rate. Specifically, both \({{\rm{AUC}}}\) and \({T}_{\max }\) increased as the infusion rate decreased, displaying a positive PK-rate relationship. Conversely, \({C}_{\max }\) exhibited a negative relationship with infusion rates, as rapid infusion led to supratherapeutic peak concentrations that may result in unsafe L-Dopa therapy. Of these parameters, \({{\rm{AUC}}}\), \({C}_{\max }\), and \({T}_{\max }\), which are known to affect the drug bioavailability in vivo, could be quantitatively measured from continuous PK data using our L-Dopa sensing system.
Next, we set out to assess the fluctuation degree of drug concentration in relation to different infusion rates. To this end, we calculated the L-Dopa variation rate from concentration profiles (Supplementary Fig. S44). Box plots of L-Dopa concentration (Fig. 5j) and variation rate (Supplementary Fig. S45) were used to visualize the impact of infusion rate on concentration fluctuation. Ultra-speed infusion (within 30 s) resulted in the greatest fluctuation, while slower infusion rates produced tighter distributions in both L-Dopa concentration and variation rate47. To further verify this, we analyzed the coefficient of variation (CV) and degree of fluctuation (DF) for L-Dopa concentration data (Supplementary Fig. S46). Compared with the ultra-high-speed infusion, medium- and low-speed infusion markedly reduced L-Dopa level variability, with CV and DF decreasing by an average of 55.9% and 68.0%, respectively (from 88.7% to 39.1% for CV and from 232.5% to 74.3% for DF). This analysis demonstrated the feasibility of continuous L-Dopa administration at low or medium infusion rates for maintaining stable L-Dopa levels and may help alleviate motor fluctuations.
Finally, we performed a continuous L-Dopa delivery with a progressively increasing infusion rate over 90 min in the same rat. Changes in L-Dopa concentration were accurately detected using the implanted nanoMIP fiber sensor (Fig. 5k). The L-Dopa concentration exhibited a markedly slow increase during the first 60 min (corresponding to infusion rates of 1 and 2 μg·g⁻¹·min⁻¹), followed by a more rapid rise under the higher infusion rate of 4 μg·g⁻¹·min⁻¹, reaching its peak at 130 min (\({T}_{\max }=\,130{\mathrm{min}}\)), eventually stabilizing during the final 120 min. In the L-Dopa variation rate profile, three distinct peaks were observed during the infusion period, each corresponding to a different infusion rate. Meanwhile, the average L-Dopa variation rate also increased with higher infusion rates. To further assess the influence of infusion rate, we also conducted the reverse experiment in the same rat, in which the rate was decreased from high to low (Supplementary Fig. S47). In this case, the L-Dopa concentration showed a rapid and sustained rise, reaching a maximum at 90 min (\({T}_{\max }\,=\,90{\mathrm{min}}\)). The average L-Dopa variation rate was highest at 4 μg·g⁻¹·min⁻¹ and declined progressively as the infusion rate decreased. Notably, in both the low-to-high and high-to-low infusion models, the high-speed infusion produced a larger variation rate compared to the low- or medium-speed phases. This rate-adjustable continuous delivery demonstrates the ability to modulate in vivo L-Dopa PK, which can be monitored in real time to guide feedback-based dosing, and offers valuable insights into the preclinical application of PK-guided therapy.
Discussion
Continuous L-Dopa monitoring provides insights into pharmacokinetic profiles, facilitating data-driven dosage individualization. However, current ISF-based sensors often lack sufficient in vivo validation and face biofouling issues. These limitations hinder their clinical translation, particularly for real-time dose adjustment to mitigate motor complications in Parkinson’s disease.
To address these challenges, we developed a nanoMIP fiber sensor featuring a rationally designed core-shell nanostructure in which a functional MIP shell is coated on a single CNT. This unique core-shell nanostructure enables the specific and sensitive detection of target L-Dopa, along with resistance to both biofouling and chemical fouling, thereby achieving continuous and accurate monitoring of a complete PK cycle in subcutaneous ISF. Chemical fouling resistance is pivotal for longitudinal and accurate monitoring, yet this critical phenomenon remains understudied in L-Dopa sensors. A key challenge arises from the polymerization of catechol groups and quinone intermediates during the detection/oxidation of L-Dopa, which leads to severe electrode passivation40,42. Resistance to the chemical fouling has been well established in our nanoMIP fiber sensor. Nevertheless, the chemical fouling mechanism needs to be further investigated at the molecular dynamics level. In addition, cytotoxicity and tissue inflammation tests confirmed its favorable biocompatibility.
We validated the accuracy of the nanoMIP fiber sensor by comparing its output with HPLC, the gold standard for L-Dopa quantification9. In vivo experiments using the nanoMIP fiber sensor validated a strong correlation between L-Dopa levels in ISF and blood and revealed a lag time of 12–15 min between the two compartments. This delay is closely matching the 10-min delay previously reported between plasma L-Dopa levels and motor symptom performance in PD patients14, suggesting the pharmacodynamic relevance of ISF monitoring. When fitted with a compartmental model, the PK characteristics of ISF and blood also exhibited consistent trends in relation to drug dosage, further confirming the capability of the nanoMIP fiber sensor for rapid and accurate PK analysis.
We applied the nanoMIP sensor to investigate pharmacokinetic characteristics in both healthy and Parkinson’s disease (PD) model rats. The nanoMIP fiber sensor provided accurate PK profiles in both healthy and PD rat models, capturing rapid absorption and short half-life characteristics of L-Dopa. In PD rat models, statistical analysis of PK parameters indicated delayed absorption and accelerated elimination, likely attributable to disease-induced physiological alterations. In addition, substantial inter-individual variability was observed within each group, in line with clinical observations49. These results emphasize the need for personalized L-Dopa therapy, particularly through low-dose, high-frequency dosing regimens to maintain stable therapeutic effects.
Using this real-time PK monitoring system, we explored various L-Dopa delivery regimens, including intermittent injection and continuous infusion. Our results showed that increasing the dosing frequency in intermittent injection reduced L-Dopa fluctuations and prolonged stable drug levels, while continuous infusion allowed fine-tuned control over \({C}_{\max }\) and therapeutic duration by adjusting infusion rates. Moderate-speed infusion achieved higher AUCs than fast infusion, likely due to reduced clearance and prolonged systemic exposure. These findings offer insight into optimizing L-Dopa PK while minimizing risks of supratherapeutic peaks or off-time episodes. Clinically, controlled-release formulations and adjunct therapies—such as oral tablets and intestinal gels—are used to maintain stable L-Dopa levels50,51,52,53, while inhalable formulations like CVT-301 offer rapid symptom relief during off periods54. Our sensing system combined with L-Dopa continuous infusion could support both strategies by enabling real-time, personalized adjustment of dosing.
Despite these promising outcomes, the study has several limitations. All experiments were conducted in rat models, which may not fully represent the pharmacokinetics or symptom dynamics of human PD patients. Long-term variation of PK dynamics over days or weeks should be further evaluated. Moreover, the clinical translation of ISF-based L-Dopa sensors will require further validation of therapeutic thresholds and dynamic correlations with motor symptom changes.
Our findings support the feasibility of combining real-time PK monitoring with adaptive drug delivery systems to further implement a closed-loop strategy for L-Dopa therapy. Future work should focus on the development of responsive dosing algorithms, expansion to multi-drug treatment regimens that enhance L-Dopa bioavailability, and clinical validation in human patient populations. Beyond Parkinson’s disease, this sensing system may also be applied to neurological and psychiatric research, where L-Dopa is also used to probe dopaminergic function. Overall, our L-Dopa sensing system provides a foundation for in vivo therapeutic drug monitoring and enables personalized, feedback-driven pharmacotherapy, potentially improving treatment precision and quality of life.
Methods
Fabrication of the nanoMIP fiber electrode
Spindle-shaped carbon nanotube fibers (SSCNTFs) were fabricated from carbon nanotube fibers (CNTF, Suzhou Tanfeng Graphene Technology, China) via electrochemical expansion31. Briefly, half of the CNTF was immersed in 1 M H₂SO₄, while the other half was kept in air. In a two-electrode configuration with CNTF as the cathode and a Pt plate as the anode, a voltage of −6 V was applied to the CNTF until a spindle-shaped expansion formed at the liquid–air interface. The MIP layer was electropolymerized onto SSCNTFs by cyclic voltammetry (CV, −0.2 V to 0.8 V) in a mixed solution containing 15 mM pyrrole (Aladdin Biochemical Technology, China), 15 mM L-Dopa, and 5 mM resorcinol (Sinopharm Chemical Reagent, China). L-Dopa templates were extracted by chronoamperometry at 0.6 V for 30 s in 0.1 M NaOH. After extraction, the electrodes were thoroughly rinsed with deionized water (DI water) for ~1 min to obtain the MIP-modified electrode, referred to as SSCNTF/MIP. All the chemicals were purchased from Sigma-Aldrich (USA) unless otherwise specified.
Optimization of the fabrication of nanoMIP
To achieve both high sensitivity and selectivity of the SSCNTF/MIP electrode toward L-Dopa, several key parameters were optimized, including the electrochemical protocol and solution used for template extraction, the number of electropolymerization cycles, the duration of template extraction, and the concentration of the template molecule. The L-Dopa response (100 µM) of SSCNTF/MIP and SSCNTF/NIP electrodes was measured to evaluate electrode sensitivity. In addition, the response of both electrodes to ascorbic acid (AA) was assessed to determine selectivity. In this way, a SSCNTF/MIP electrode with simultaneous high sensitivity and selectivity could be acquired.
Characterization of nanoMIP fiber electrode
Electrochemical characterization
Electrochemical activity was evaluated by CV (0 V to 0.6 V) in a solution containing 1 mM K₃[Fe(CN)₆] and 1 mM K₄[Fe(CN)₆] as the redox probe. Electrochemical impedance spectroscopy (EIS) was also performed in the same solution over a frequency range of 10 kHz to 0.1 Hz. Sensing performance was assessed by differential pulse voltammetry (DPV) using the following parameters: 5 mV step potential, 0.1 V modulation amplitude, 0.01 s modulation time, and 0.5 s interval. All electrochemical measurements were carried out using a µAutolab III electrochemical workstation (Metrohm, Switzerland) with a three-electrode system: a platinum wire as the counter electrode (CE) and a standard Ag/AgCl electrode as the reference (RE).
Chemical and physical characterization
X-ray photoelectron spectroscopy (XPS, Escalab 250Xi, Thermo Fisher Scientific, U.K.) was used to analyze the elemental composition (C, N, O) of SSCNTF, SSCNTF/MIP, and SSCNTF/NIP electrodes. Raman spectroscopy (LabRAM HR Evolution, HORIBA, France) was employed to investigate the chemical structure of bare SSCNTF and SSCNTF/MIP before and after template extraction. Fourier-transform infrared spectroscopy (Nicolet iS50, Thermo Fisher) was used to analyze the compositional and bonding changes of SSCNTF/MIP before and after extraction, as well as the interactions between monomers and between monomers and the template.
Surface morphology of SSCNTF/MIP and SSCNTF/NIP was observed using scanning electron microscopy (SEM, Nova NanoSEM 450, FEI, Eindhoven, Netherlands) and transmission electron microscopy (TEM, HT-7700, Hitachi, Japan). The three-dimensional morphology of SSCNTF/MIP was characterized by X-ray computed tomography (ZEISS Xradia 610 Versa). To enhance imaging contrast, SSCNTF/MIPs were stained with 5% potassium iodide (KI) for 1 h. Reconstructed tomograms were visualized using Avizo software (Thermo Scientific).
Sensing performance of nanoMIP fiber electrode
The sensing performance of the nanoMIP fiber sensor was evaluated in 0.1 M PBS (pH 6.7, Beyotime Biotechnology, China) containing 1–100\μM L-Dopa. Selectivity was assessed by measuring the response of the sensor to 100 µM L-Dopa with sequential additions of potential interfering species: 1 mM glucose (Glu), 1 mM urea, 1 mM lactic acid (Lac), 100 μM creatine (Cre), 1 μM cortisol (Cor), 100 μM ascorbic acid (AA), 100 μM uric acid (UA), 30 μM carbidopa (C-Dopa), 100 μM acetaminophen (Ace), 50 μM valine (Val), 50 μM leucine (Leu), 50 μM l-isoleucine (Ile), 50 μM lysine (Lys), 50 μM methionine (Met), 50 μM phenylalanine (Phe), 50 μM threonine (Thr), and 50 μM histidine (His). C-Dopa was purchased from Shanghai Bepharm Science & Technology.
Anti-fouling performance was evaluated under two conditions. For chemical fouling, 50 consecutive differential pulse voltammetry (DPV) scans were performed in 100 μM L-Dopa. For biofouling, the sensor was implanted subcutaneously in a live Sprague–Dawley rat for 2 h, and DPV measurements were recorded before and after implantation.
Sensing stability was assessed over 7 days of storage at 4 °C in deionized water. Cytocompatibility was evaluated using NIH/3T3 mouse embryonic fibroblast cells (Mus musculus, male embryo; adherent morphology), which were authenticated by short tandem repeat (STR) profiling (CL-0171, Wuhan Procell Life Science & Technology, China). Culture medium was replaced with nanoMIP fiber soaking solutions, prepared by incubating 1, 4, and 7 fibers in complete medium for 5 days, with daily medium refreshment. After 72 h of incubation, cell viability was evaluated using the Calcein acetoxymethyl ester/propidium iodide (Calcein-AM/PI) Live/Dead Viability/Cytotoxicity Kit (Dojindo, Japan). Tissue biocompatibility was assessed via histological analysis. Skin tissues containing implanted sensors (1 and 4 days post-implantation) were excised (~1.5 cm × 1.5 cm × 0.5 cm) after euthanasia of SD rats. Samples were rinsed with 0.9% NaCl, fixed in 10% paraformaldehyde (v/v) in PBS at 25 °C for 24 h, and sectioned into ~3 μm slices. Sections were stained with hematoxylin and eosin (Pinocell Biotechnology, China) and Masson’s trichrome kits (Pinocell Biotechnology, China) to assess tissue morphology. Stained slices were mounted with neutral resin and imaged using a light microscope (ECLIPSE Ci, Nikon, Japan).
Design of the flexible DPV circuit
The flexible DPV circuit integrates an STM32F412RET6 MCU (ARM Cortex-M4, 100 MHz, 64-pin LQFP, 10 mm × 10 mm) for system control. An AD5941 analog front-end (AFE; Analog Devices) performs signal excitation, acquisition, and processing for DPV detection. Data are wirelessly transmitted to a PC via a dual-mode Bluetooth SoC (BK3432) interfaced through UART. Power is regulated to 3.3 V by an RT9013 LDO, supporting stable operation despite a 250 mV drop from a 3.7 V lithium battery.
In vivo validation of nanoMIP fiber sensor for PK monitoring
Ethics statements
All animal experiments were conducted in compliance with institutional guidelines, and all animal procedures were approved by the Institutional Animal Care and Use Committee of Zhejiang University (ZJU20250146).
In vivo experiment protocol
Male Sprague–Dawley rats (wild-type, 8–10 weeks old, 300–400 g, Experimental Animal Center of Zhejiang University) were anesthetized with isoflurane and positioned on a surgical table (RWD Life Science, China). The dorsal area was shaved, cleaned, and disinfected with 75% ethanol. A 0.2 cm incision was made for subcutaneous implantation of the nanoMIP fiber electrode. A 30-min incubation was selected to allow baseline stabilization of the nanoMIP sensor before L-Dopa injection. L-Dopa (0.12 or 0.06 mg·g⁻¹) was administered via intraperitoneal injection. DPV measurements (−0.2 V to 0.4 V) were recorded every 10 min over 240 min. Prior to implantation, electrode performance was evaluated in simulated body fluid (SBF, Yuanye Bio-Technology, China) for calibration. Blood samples (0.3–0.5 mL) were collected from the retro-orbital vein at predefined time points.
High-performance liquid chromatography analysis
L-Dopa concentrations in blood were quantified using high-performance liquid chromatography with fluorescence detection (HPLC-FLD; Agilent 1100). Blood samples were centrifuged at ~720 × g for 10 min to obtain serum. For protein precipitation, 50 μL of 6% perchloric acid (Sinopharm Chemical Reagent, China) was mixed with 50 μL of serum, vortexed, and centrifuged again for 10 min. Approximately 60 μL of the supernatant was collected for analysis.
Pharmacokinetics analysis
The pharmacokinetic profile of L-Dopa was obtained by fitting its blood and ISF concentration data to a classic extravascular compartmental model (Eq. (1)) using the DAS 2.0 software.
Here, \(A\), \(B\), and \(G\) are weighting coefficients related to the volume of distribution and the administered dose; \(\alpha\) and \(\beta\) represent the rate constants for the distribution and elimination phases, respectively, corresponding to the initial rapid decline and the subsequent slower decline in drug concentration; \({K}_{{{\rm{a}}}}\) is the absorption rate constant; and \(C\) denotes the drug concentration.
The area under the concentration–time curve (\({{\rm{AUC}}}\)) from time 0 to \(t\) was calculated using the trapezoidal rule (Eq. (2)).
The maximum plasma concentration (\({C}_{\max }\)), the time to reach \({C}_{\max }\) (\({T}_{\max }\)), and the apparent elimination half-life (\({T}_{1/2}\)) were determined directly from the concentration–time data. The distribution half-life (\({T}_{1/2{{\rm{\alpha }}}}\)) and elimination half-life (\({T}_{1/2{{\rm{\beta }}}}\)) were calculated using the following Eqs. (3) and (4).
The fluctuation degree (\({{\rm{DF}}}\)) of L-Dopa concentration at steady state was calculated using Eq. (5).
Where \({C}_{{{\rm{ss}}}}\_{{{\rm{avg}}}}\), \({C}_{{{\rm{ss}}}\_\max }\), and \({C}_{{{\rm{ss}}}\_\min }\) represent the average, maximum, and minimum steady-state concentrations, respectively. Steady state was considered to be reached at 120 min post-administration.
L-Dopa variation rate (\({{\rm{d}}}C/{{\rm{d}}}t\)), representing the rate of change in drug concentration, was calculated using Eq. (6).
Studies of L-Dopa PKs on PD rat models
Establishment of PD rat model
A unilateral Parkinson’s disease (PD) rat model was established using male Sprague–Dawley rats (wild-type, 8–10 weeks old, 300–400 g, Experimental Animal Center of Zhejiang University). The animals received a stereotaxic injection of 6-hydroxydopamine (6-OHDA, 20 µg; MedChemExpress, USA) into the right medial forebrain bundle (MFB). Control rats received equal volumes of saline. All procedures were performed under anesthesia.
Validation of PD rat model (Immunohistochemistry)
Two weeks after 6-OHDA lesioning, rats were anesthetized and decapitated for brain extraction. Brain tissues were fixed, sectioned, and processed for immunohistochemistry. Sections were permeabilized with 0.5% Triton X-100 in PBS (pH 7.4, Yuanye Bio-Technology, China) for 10 min at room temperature, then blocked with 5% bovine serum albumin (BSA, Yuanye Bio-Technology, China) at 37 °C for 30 min. Slices were incubated overnight with rabbit anti-tyrosine hydroxylase (TH) antibody (AF6113, 1:200, Affinity Biosciences Technology, China), washed with PBS, and incubated with Cyanine 3-conjugated goat anti-rabbit immunoglobulin G (AS007, 1:200, ABclonal Technology, China) at 37 °C for 45 min. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Beyotime Biotechnology, China) for 3 min. Fluorescence images were captured using a confocal microscope (BX53, Olympus, Japan) and analyzed with ImageJ 1.54p software (NIH, USA).
Studies of L-Dopa PK based on different drug administration
Two dosing strategies were tested in healthy rats. For intermittent dosing, one rat received either two doses of 0.06 mg·g⁻¹ or four doses of 0.03 mg·g⁻¹, with 30 min intervals between injections. For continuous dosing, another rat was administered L-Dopa intraperitoneally via infusion pump at four rates: low (1 μg·g⁻¹·min⁻¹), medium (2 μg·g⁻¹·min⁻¹), high (4 μg·g⁻¹·min⁻¹), and ultra-high (4 μg·g⁻¹·s⁻¹). A 24-h washout period was implemented between in vivo trials to ensure complete drug clearance.
Statistics and reproducibility
All data visualization was performed using Origin 2021. Statistical comparisons between two groups were conducted using a Student’s t test (two-tailed, unpaired). Statistical comparisons between multiple groups were conducted using one-way ANOVA. For all analyses, a p-value < 0.05 was considered statistically significant. All statistical calculations were performed using GraphPad Prism v10. For the representative TEM image of the nanoMIP core-shell nanostructure shown in Fig. 1 and the SEM and TEM images of CNTF/MIP and SSCNTF/MIP before and after template extraction shown in Fig. 2, all experiments were repeated three times with similar results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the article and its supplementary files. Any additional requests for information can be directed to, and will be fulfilled by, the corresponding authors. Source data are provided with this paper.
References
Zhu, J. et al. Temporal trends in the prevalence of Parkinson’s disease from 1980 to 2023: a systematic review and meta-analysis. Lancet Healthy Longev. 5, e464–e479 (2024).
Dorsey, E. R. et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 17, 939–953 (2018).
Armstrong, M. J. & Okun, M. S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 323, 548–560 (2020).
Birkmayer, W. & Hornykiewicz, O. Der L-Dioxyphenylalanin (=L-DOPA)-Effekt beim Parkinson-Syndrom des Menschen: Zur Pathogenese und Behandlung der Parkinson-Akinese. Arch. Psychiatr. Neurologie 203, 560–574 (1962).
Dong-Chen, X., Yong, C., Yang, X., Chen-Yu, S. & Li-Hua, P. Signaling pathways in Parkinson’s disease: molecular mechanisms and therapeutic interventions. Sig Transduct. Target Ther. 8, 1–18 (2023).
Connolly, B. S. & Lang, A. E. Pharmacological Treatment of Parkinson Disease: A Review. JAMA 311, 1670–1683 (2014).
Yahr, M. D. Drugs Five Years Later: Levodopa. Ann. Intern Med. 83, 677 (1975).
Fox, S. H. Revisiting levodopa for advanced Parkinson’s disease. Lancet Neurol. 21, 1063–1065 (2022).
Teymourian, H. et al. Closing the loop for patients with Parkinson disease: where are we?. Nat. Rev. Neurol. 18, 497–507 (2022).
Teymourian, H. et al. Wearable Electrochemical Sensors for the Monitoring and Screening of Drugs. ACS Sens. 5, 2679–2700 (2020).
Probst, D., Batchu, K., Younce, J. R. & Sode, K. Levodopa: From Biological Significance to Continuous Monitoring. ACS Sens. 9, 3828–3839 (2024).
Seo, J.-W. et al. Real-time monitoring of drug pharmacokinetics within tumor tissue in live animals. Sci. Adv. 8, eabk2901 (2022).
Lin, S. et al. Wearable microneedle-based electrochemical aptamer biosensing for precision dosing of drugs with narrow therapeutic windows. Sci. Adv. 8, eabq4539 (2022).
Mahato, K. et al. Biosensor Strip for Rapid On-site Assessment of Levodopa Pharmacokinetics along with Motor Performance in Parkinson’s Disease. Angew. Chem. Int Ed. 63, e202403583 (2024).
Wang, M. et al. A wearable electrochemical biosensor for the monitoring of metabolites and nutrients. Nat. Biomed. Eng. 6, 1225–1235 (2022).
Sempionatto, J. R. et al. An epidermal patch for the simultaneous monitoring of haemodynamic and metabolic biomarkers. Nat. Biomed. Eng. 5, 737–748 (2021).
Wang, M. et al. Printable molecule-selective core–shell nanoparticles for wearable and implantable sensing. Nat. Mater. 24, 589–598 (2025).
Mishra, R. K. et al. Continuous Opioid Monitoring along with Nerve Agents on a Wearable Microneedle Sensor Array. J. Am. Chem. Soc. 142, 5991–5995 (2020).
Vora, L. K. et al. Microneedle-based biosensing. Nat. Rev. Bioeng. 2, 64–81 (2024).
Wu, J., Liu, H., Chen, W., Ma, B. & Ju, H. Device integration of electrochemical biosensors. Nat. Rev. Bioeng. 1, 346–360 (2023).
Brunetti, B., Valdés-Ramírez, G., Litvan, I. & Wang, J. A disposable electrochemical biosensor for L-DOPA determination in undiluted human serum. Electrochem. Commun. 48, 28–31 (2014).
Moon, J.-M. et al. Non-Invasive Sweat-Based Tracking of L-Dopa Pharmacokinetic Profiles Following an Oral Tablet Administration. Angew. Chem. Int. Ed. 60, 19074–19078 (2021).
Peng, H.-L., Zhang, Y., Liu, H. & Gao, C. Flexible Wearable Electrochemical Sensors Based on AuNR/PEDOT:PSS for Simultaneous Monitoring of Levodopa and Uric Acid in Sweat. ACS Sens. 9, 3296–3306 (2024).
Zhou, D.-W. et al. LDHzyme-assisted high-performance on-site tracking of levodopa pharmacokinetics for Parkinson’s disease management. Biosens. Bioelectron. 268, 116926 (2025).
Tai, L.-C. et al. Wearable Sweat Band for Noninvasive Levodopa Monitoring. Nano Lett. 19, 6346–6351 (2019).
Ding, S. et al. A fingertip-wearable microgrid system for autonomous energy management and metabolic monitoring. Nat. Electron 7, 788–799 (2024).
Friedel, M. et al. Opportunities and challenges in the diagnostic utility of dermal interstitial fluid. Nat. Biomed. Eng. 7, 1541–1555 (2023).
Goud, K. Y. et al. Wearable Electrochemical Microneedle Sensor for Continuous Monitoring of Levodopa: Toward Parkinson Management. ACS Sens. 4, 2196–2204 (2019).
Park, S. et al. Highly-sensitive single-step sensing of levodopa by swellable microneedle-mounted nanogap sensors. Biosens. Bioelectron. 220, 114912 (2023).
Yang, S.-J. et al. Codeposition of Levodopa and Polyethyleneimine: Reaction Mechanism and Coating Construction. ACS Appl. Mater. Interfaces 12, 54094–54103 (2020).
Zhou, Y. et al. Expanded Carbon Nanotube Fiber at the Liquid–Air Interface for High-Performance Fiber-Based Supercapacitors and Electrochemical Sensors. ACS Appl. Mater. Interfaces 15, 41839–41849 (2023).
Zhao, C., Park, J., Root, S. E. & Bao, Z. Skin-inspired soft bioelectronic materials, devices and systems. Nat. Rev. Bioeng. 2, 671–690 (2024).
Sun, H. et al. Hierarchical 3D electrodes for electrochemical energy storage. Nat. Rev. Mater. 4, 45–60 (2019).
Zhang, X., Lu, W., Zhou, G. & Li, Q. Understanding the Mechanical and Conductive Properties of Carbon Nanotube Fibers for Smart Electronics. Adv. Mater. 32, 1902028 (2020).
Pourhajghanbar, M., Arvand, M. & Habibi, M. F. Surface imprinting by using bi-functional monomers on spherical template magnetite for selective detection of levodopa in biological fluids. Talanta 254, 124136 (2023).
Tsai, T.-C. et al. Modification of platinum microelectrode with molecularly imprinted over-oxidized polypyrrole for dopamine measurement in rat striatum. Sens. Actuators B Chem. 171–172, 93–101 (2012).
Chen, Y. et al. Large-Scale Flexible Fabric Biosensor for Long-Term Monitoring of Sweat Lactate. Adv. Funct. Mater. 34, 2401270 (2024).
Hu, X. et al. Wearable and Regenerable Electrochemical Fabric Sensing System Based on Molecularly Imprinted Polymers for Real-Time Stress Management. Adv. Funct. Mater. 34, 2312897 (2024).
Zhang, L. et al. Engineering Carbon Nanotube Fiber for Real-Time Quantification of Ascorbic Acid Levels in a Live Rat Model of Alzheimer’s Disease. Anal. Chem. 89, 1831–1837 (2017).
Zhou, L. et al. COF-Coated Microelectrode for Space-Confined Electrochemical Sensing of Dopamine in Parkinson’s Disease Model Mouse Brain. J. Am. Chem. Soc. 145, 23727–23738 (2023).
Zhu, F. et al. Galvanic Redox Potentiometry for Fouling-Free and Stable Serotonin Sensing in a Living Animal Brain. Angew. Chem. Int. Ed. 62, e202212458 (2023).
Feng, T. et al. Low-Fouling Nanoporous Conductive Polymer-Coated Microelectrode for In Vivo Monitoring of Dopamine in the Rat Brain. Anal. Chem. 91, 10786–10791 (2019).
Chan, D. et al. Combinatorial Polyacrylamide Hydrogels for Preventing Biofouling on Implantable Biosensors. Adv. Mater. 34, 2109764 (2022).
Picconi, B. et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA–induced dyskinesia. Nat. Neurosci. 6, 501–506 (2003).
Winkler, C., Kirik, D., Björklund, A. & Cenci, M. A. L-DOPA-Induced Dyskinesia in the Intrastriatal 6-Hydroxydopamine Model of Parkinson’s Disease: Relation to Motor and Cellular Parameters of Nigrostriatal Function. Neurobiol. Dis. 10, 165–186 (2002).
Shin, W. et al. Nanoscale Magneto-mechanical-genetics of Deep Brain Neurons Reversing Motor Deficits in Parkinsonian Mice. Nano Lett. 24, 270–278 (2024).
Warren Olanow, C. et al. Continuous versus intermittent oral administration of levodopa in Parkinson’s disease patients with motor fluctuations: A pharmacokinetics, safety, and efficacy study. Mov. Disord. 34, 425–429 (2019).
Olanow, C. W., Obeso, J. A. & Stocchi, F. Drug Insight: continuous dopaminergic stimulation in the treatment of Parkinson’s disease. Nat. Rev. Neurol. 2, 382–392 (2006).
Khor, S.-P. & Hsu, A. The Pharmacokinetics and Pharmacodynamics of Levodopa in the Treatment of Parkinsons Disease. Curr. Clin. Pharmacol. 2, 234–243 (2007).
Woods, A. nneC., Glaubiger, G. eorgeA. & Chase, T. homasN. Sustained-release levodopa. Lancet 301, 1391 (1973).
MacMahon, D. G. et al. A comparison of the effects of controlled-release levodopa (Madopar CR) with conventional levodopa in late Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 53, 220–223 (1990).
Fernandez, H. H. et al. Long-term safety and efficacy of levodopa-carbidopa intestinal gel in advanced Parkinson’s disease. Mov. Disord. 33, 928–936 (2018).
Politis, M. et al. Sustained striatal dopamine levels following intestinal levodopa infusions in Parkinson’s disease patients. Mov. Disord. 32, 235–240 (2016).
Lipp, M. M., Batycky, R., Moore, J., Leinonen, M. & Freed, M. I. Preclinical and clinical assessment of inhaled levodopa for OFF episodes in Parkinson’s disease. Sci. Transl. Med. 8, 360ra136 (2016).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 82272120 to B.L.); the National Key Research and Development Program of China (Grant No. 2024YFA0920400 to B.L.); and the Natural Science Foundation of Zhejiang Province, China (Grant No. LTGG23H180001 to L.F. and Grant No. ZCLZ24F0101 to B.L.). The authors thank Dr. Yangfan Lu (School of Materials Science and Engineering) for assistance with XPS measurements and analysis. We are also grateful to Dr. Liang Lei (Westlake University) for support with X-ray characterization. We thank Dr. Yang Zhu (Zhejiang University) for valuable assistance with the animal experiments. We also thank Dr. Tingting Tu (Zhejiang University) for her valuable help in revising the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: B.L., Y.Z., and X.S.Y.; Methodology: Y.Z. and B.L.; Investigation: Y.Z., J.H.L., Z.Y.X., Y.X.Z, S.S.Z, T.L., Y.L.Y., L.F., and Y.C.; Visualization: Y.Z., Z.Y.X., Y.X.Z., and T.L.; Supervision: B.L. and X.S.Y.; Writing—original draft: Y.Z.; Writing—review & editing: B.L., Y.Z., S.S.Z, and X.S.Y.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kotagiri Yugender Goud, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, Y., Li, J., Xu, Z. et al. A nanoMIP sensor for real-time in vivo monitoring of levodopa pharmacokinetics in precision Parkinson’s therapy. Nat Commun 16, 10796 (2025). https://doi.org/10.1038/s41467-025-65853-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65853-2
