
Abstract
Dipteran insects include numerous harmful species that cause significant agricultural damage. However, assembly of genomes for species in this order has been difficult due to their small body size, poor conservation of telomere and centromere structures, high levels of heterozygosity, and complex genetic backgrounds. In this study, we assemble a high-quality 596 Mb telomere-to-telomere genome for Bactrocera dorsalis, a fruit crop pest, using a strategy with a low-input HiFi CCS library from a male individual and an ONT sequence from pooled inbred individuals. The assembly includes complete structural organization information for centromeres and telomeres, providing insights into the evolution of chromosome structure in insects. Comparative genomic analysis reveals the polyphyletic origin of sex chromosomes across Diptera. Furthermore, we identify a homolog of ATPsynβ as a Y chromosome-specific gene that is highly expressed across multiple male tissues and may provide critical support for male-specific physiological activities. Additionally, we discover several tandem duplications of odorant receptor genes, including a triplet of the OR88a family, which was validated to be involved in the behavioral response to methyl eugenol. In summary, this complete reference genome provides a foundation for future genomic research in Diptera and offers genetic insights for the control of B. dorsalis.
Similar content being viewed by others

Introduction
Genomic approaches are becoming increasingly important in pest control research, aiding in the discovery of molecular targets, understanding resistance mechanisms, analyzing genetic diversity and environmental adaptation, and developing biological control technologies1,2,3,4,5. Obtaining a high-quality reference genome is fundamental to these approaches, and with the advent of third-generation sequencing technology, genome research has entered the telomere-to-telomere era, as demonstrated by the human and rice genome projects6,7,8. The completeness and accuracy of genome assembly have been greatly improved through the use of Oxford Nanopore Technology (ONT) ultralong reads and Pacific Biosciences (PacBio) high-fidelity sequencing technologies9. However, both of these technologies require relatively high DNA inputs (>5 μg). Therefore, assembling the genomes of small insects remains challenging due to the limited availability of high-molecular-weight (HMW) DNA and the complexity of their heterogeneous genetic backgrounds10,11,12,13,14,15.
To overcome the limitations of DNA input and genetic heterogeneity in insect genome research, researchers often pool individuals with high relatedness derived from inbreeding, which works well for organisms that are easy to breed, such as Drosophila species16. However, this approach is not possible for most pest insects, which are difficult to breed for multiple generations or develop high genetic heterogeneity, homozygous lethality, and the loss of certain wild-type phenotypes after frequent inbreeding. Additionally, pooling individuals inherently increases the number of haplotypes in the data, leading to inflated consensus error rates and lower assembly contiguities17. In Dipteran species, the low conservation of telomere and centromere sequences among members present additional complexities18,19. These limitations have hindered the application of T2T assembly in functional genomics research and pest control technologies for Dipteran pests.
Tephritid flies comprise approximately 4600 species12,20, many of which are major pests in fruit and vegetable production and cause both direct losses and indirect losses due to international trade restrictions, with an estimated annual impact exceeding $2 billion21. The oriental fruit fly, B. dorsalis, is a representative tephritid pest that affects more than 150 fruit crops12 and has spread to 75 countries across Asia, Africa, and Oceania22. Despite the availability of several genome assemblies for B. dorsalis, including assemblies compiled from pooled individuals23,24,25,26, a complete telomere-to-telomere (T2T) genome assembly is needed. The T2T genome is beneficial for the development of novel pest control technologies because it not only facilitates the assembly of typically challenging regions, such as telomeres, centromeres, and other highly repetitive sequences, but also enhances the detection of structural variations, gene duplications, and complex genomic features that might have been missed or misrepresented in previous assemblies. For example, accurately assembling genome information on X/Y chromosomes is crucial for the sterile insect technique (SIT), as it enables targeted manipulation of genes related to sex determination, improving the efficiency and effectiveness of generating sterile insect populations for pest control.
Inspired by the existing T2T projects, we leveraged advanced library construction techniques to construct a 596 Mb T2T genome of a male B. dorsalis individual, including verified chromosomal structure elements. This new genome will facilitate comparative genome research within Diptera and provide fundamental information for pest control strategies.
Results
Gapless T2T genome assembly of a male individual
The genetic background was assessed with 23.06 Gb Illumina sequencing data of pooled DNA from five individuals using GenomeScope, which revealed an average heterozygosity of 1.81% (Supplementary Fig. 1 and Supplementary Data 1). One female adult and one male adult (both raised with antibiotic feed) were used for PacBio HiFi low-input library construction (Fig. 1a). HiFi CCS reads were generated for each library, with a 29.83 Gb yield and an average read length of 11.54 kb for the female and a 19.55 Gb yield and an average read length of 12.34 kb for the male. Nanopore (94.79 Gb) and Hi-C sequencing (33.40 Gb) were also performed using pooled individuals from close inbreds (Supplementary Data 1 and Supplementary Fig. 2).

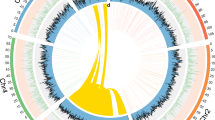
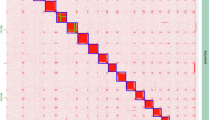
a Sample DNA content and sequencing scheme. b The genome assembly procedure (detailed in the method section). c Circos plot showing the T2T genome assembly of B. dorsalis. The tracks from A to M are as follows: [A] HiFi CCS from a male individual; [B] HiFi CCS from a female individual; [C] ONT reads coverage from mixed samples; [D] GC% content; [E] maximum FPKM value in a 500-bp bin from multiple RNA-seq data; [F] gene density; [G] ChIP-seq peak of histone H3K4me3 modification; [H] location of centromeres, telomeres and histone array; [I] LTR density; [J] DNA density; [K] nonLTR density; and [L] SSR density. d Hi-C contact map for assembly. Chromosome Y is enlarged for visualization. e Collinear regions between assemblies from the two assembling strategies.
HiFi CCS reads were first compared against bacterial and fungal RefSeq genomes to identify potential contaminants. Both Hifiasm and HiCanu were used to assemble contigs of bacterial and fungal origin as completely as possible, and the corresponding contaminant sequences were removed. As a result, 14.31% of reads from the female individual and 2.1% from the male individual were excluded (Supplementary Data 2). After removing contaminant HiFi CCS reads, 25.67 Gb ( ~43X coverage) and 19.06 Gb (~32X coverage) of clean data were retained from the female and male samples, respectively. Due to a high contamination ratio, genetic heterozygosity, and concerns about full spectral karyotyping, the male sample data were selected for the final reference assembly (Supplementary Data 3).
Due to coverage limitation and high heterogeneity, we adopted a multi-step assembly strategy, as shown in Fig. 1b and detailed in Method section. The final assembly of the male B. dorsalis was 596.16 Mb, consisting of 5 + XY chromosomes with no gaps (Fig. 1c, d), which is close to the 539 Mb estimated in the genome survey and the 521 Mb suggested by flow cytometry25. The error rate across the whole genome was estimated by Merqury to be less than 2 errors per 1 Mb (Phred Q42.64), with quality values (QVs) ranging from 37.47 to 49.17 for each chromosome, using a 21-mer set derived from HiFi CCS reads. Assembly continuity achieves a GCI score of 31.5 for the whole genome, and 51.4 and 57.0 for X and Y chromosomes separately. Genome completeness was estimated at 99.38%, as calculated by Merqury using a 21-mer set from Illumina paired-end reads obtained from five individuals across several generations.
The final assembly was more consistent than the Hifiasm+purged_dups+HiC assembly and other published genome assemblies of B. dorsalis (Fig. 1e and Supplementary Fig. 3). The level of genome completeness achieved was comparable to that of the T2T status, including the identified telomeres and centromeres. The inconsistencies and gaps are concentrated in the regions containing centromeres and sex chromosomes, which are notoriously difficult to sequence and assemble because of their intrinsic repetitive nature combined with high genetic diversity27,28.
The gene completeness of the assembly is comparable to that of the published fruit fly assemblies. Specifically, the assembly achieved 99.57% completeness for complete single-copy genes, 0.18% complete duplicated copies and 0.12% fragmented copies, as assessed using 3285 Benchmarking Universal Single-Copy Orthologs (BUSCO) genes (BUSCO v5 with diptera_odb10) (Supplementary Data 4). A comparative analysis between the telomere-to-telomere (T2T) assembly and the RefSeq reference genome (ASM2337382) revealed 199 uniquely identified genes in the T2T assembly and 8 exclusively annotated genes in the RefSeq genome (Supplementary Data 5).
Repeat annotation revealed approximately 337.94 Mb of repetitive sequence, accounting for 56.69% of the assembly (Supplementary Data 6). DNA transposable elements constituted a major portion of the repeats, accounting for 26.16% of the assembly.
By integrating ab initio prediction, homology prediction, and expressed transcript (RNA-seq and ISO-seq) evidence, we identified 15,574 protein-coding genes with high confidence and precision. A total of 15,139 genes (97.21%) were functionally annotated with reference to at least one public database (NR, KEGG, GO, or KOG) (Supplementary Data 7).
Characteristics of candidate centromeres and telomeres
Both centromeres and telomeres play key roles in maintaining genome stability and mediate the precise equal distribution of genetic material during cell division. Unlike other insects, species within the order Diptera exhibit high diversity in their telomeric and centromeric motifs19,29.
Our assembly presents 5 + X gapless centromeres characterized by high structural integrity and unique sequence features.
First, we identified three representative types of centromere satellite monomers (cenSats), BdoSat1, BdoSat2, and BdoSat3, with lengths of 180 bp, 166 bp, and 166 bp, respectively. These intact monomers occupy a total of 42.98 Mb across the genome. Each type of centromeric monomer is predominantly and distinctly localized within unitigs, forming organized structures within chromosomes (Fig. 2a, Supplementary Data 8 and Supplementary Data 9).

a Pericentromeric regions visualized by StainedGlass. The locations of the gene models, three types of centromeric satellites, and reverse transcriptase are indicated by purple bars. b Subtypes of centromeric satellite monomers. The upper plot shows the coefficient of variation of the sequence divergence rate for the BdoSat consensus, as annotated by RepeatMasker in hifiasm unitigs. The x-axis represents the average identity to the BdoSat monomer consensus for each unit. The lower plot shows the identity distribution of the BdoSat monomer consensus in the final T2T assembly. Annotations with lengths less than 95% of the consensus length were filtered from the RepeatMasker output. c Phylogenetic relationships of subtypes within BdoSat2 and BodSat3. d FISH analysis of the centromeric region of the chromosome, showing the most abundant subtype for each BdoSat. Images shown are representative of at least three independent experiments.
Higher-order repeat (HOR) structures of cenSats in chromosomes are surrounded by repetitive elements, lacking H3K4me3 modifications or gene annotations (Supplementary Fig. 4). Both BdoSat1 and BdoSat2 clusters were uniquely present in autosomes, whereas the BdoSat3 cluster was predominantly found on the sex chromosome and was rarely observed in the centromere region of chromosome 3. This distribution of BdoSat3 is consistent with the results from fluorescence in situ hybridization (FISH) (Fig. 2d). The BdoSat2 and BdoSat3 monomers presented a high degree of similarity, with 71.2% sequence identity, both characterized by a relatively low GC content (29.51% and 30.12%, respectively), whereas BdoSat1 presented a relatively high GC content of 38.89%, slightly exceeding the genome-wide average of 36.34%.
The completeness and accuracy of the assembly allowed us to achieve high-resolution differentiation of HORs and obtain their chromosome-specific distributions (Fig. 2b). The three satellites were further divided into seven subtypes according to their diversity and location, represented by BdoSat1A, BdoSat1B, BdoSat2A, BdoSat2B, BdoSat2C, BdoSat3A, and BdoSat3B, each exhibiting chromosome-specific sequences and structural variants. Notably, chr5 and chr6 contained sequences of BdoSat2A, whereas chr2, chr3, and chr4 contained different types of BdoSat2B and BdoSat2C. The different compositions of cenSats across chromosomes imply distinct evolutionary origins of the chromosomes, leading to the unique structural characteristics observed in their centromeres.
In contrast to the complex telomeric structures found in Drosophila30,31, we identified a 528 bp repeat unit at the extreme terminal ends of the autosomes that was absent in the sex chromosomes. In sex chromosomes, many tandemly repeated rDNA sequences are concentrated at the terminal end and mixed with reverse transcriptase genes, particularly on the short arm of the X chromosome, which is consistent with the FISH results of 18S rDNA probes exclusively on the X and Y chromosomes in the closely related Bactrocera oxeae32. Like those in Drosophila and Bombyx mori33, rDNA insertions are prevalent in both the X and Y chromosomes of B. dorsalis, accounting for approximately 48% of the ONT read estimation (Supplementary Fig. 5a). A 2.4 kb DNA fragment inserted exclusively in 28S ribosomal RNA genes, which contain an non-LTR retrotransposon R1, which identified in Bactrocera tryoni, and a partial segment of a Gypsy-type LTR, was annotated as a nonlong terminal repeat (Supplementary Fig. 5b). In D. melanogaster, approximately 44% of the rDNA units have R1 retrotransposons inserted at specific locations within their 28S regions34. These insertions prevent the production of functional 28S rRNA35, although experiments have shown that R1 expression increases through prestalling of Pol I during heat shock36.
Cross-species comparisons in Drosophila obscura flies support the ancestral configuration of the rDNA cluster in sex chromosomes37. In D. melanogaster, the rDNA cluster is located in the pericentromeric heterochromatin of the X chromosome long arm and the base of the short arm of the Y chromosome38. The heterochromatic state suppresses recombination between the two chromosomes, helping to maintain the structural integrity of both the X and Y chromosomes. Recently, studies have shown that rDNA loci located on sex chromosomes act as cis-elements for nonrandom sister chromatid segregation39. Unequal sister chromatid exchange is suspected to increase rDNA copy number, helping maintain germline immortality by countering the spontaneous loss of rDNA over generations. This characteristic of rDNA, which helps preserve chromosome stability and continuity across generations, is somewhat analogous to the role of telomeres in protecting chromosome ends and preventing genomic degradation.
Characteristics of nuclear–mitochondrial DNA segment (NUMT) insertions
Mitochondrial DNA has been shown to be continuously inserted into the nuclear genome in both humans and insects, displaying high diversity across human populations40. Leveraging the completeness of our assembly, we identified 105 loci containing sequence relics of NUMT insertions, each exceeding 100 bp in length (Supplementary Data 10). Notably, these loci can be further clustered into 11 mitochondrial synteny regions, which are distributed across the genome and are located mainly on chromosome 2 and the X chromosome, with microsyntenic regions representing more than 75% of the mitochondrial genome (Fig. 3a). Consistent with observations in human cancer cells, the breakpoints of the inserted mitochondrial sequences are not random but are concentrated near the D-loop region, which is the replication origin for mitochondria41.

a Relationships between NUMTs and the mitochondrial genome. We randomly selected representative cases from the three phylogenetically distinct NUMT clusters to demonstrate the progressive erosion of homologous syntenic architecture over evolutionary timescales. Circos plot illustrating the locations of NUMTs in the nucleus and their syntenic regions in mtDNA genomes. The tracks from outer to inner represent annotation features, variants between NUMTs and mtDNA, repeats between NUMTs, and maximum FPKM values in 500-bp bins from multiple RNA-seq datasets, and coordinates with color bars indicate annotation features. b Phylogenetic relationships and multiple attributes among the 11 large blocks of NUMTs and the mtDNA genome. Ti/Tv, GC/AT transitions, and the proportions of NUMT fragments and repeats in large blocks are shown in pie charts. Sequence identity, GC content in NUMT fragments, and GC content in syntenic regions of the mtDNA genome are shown in bar plots. The final bar shows the ratio of variant types attributed to GC/AT transitions.
The expression levels of mitochondrial gene regions within the 11 NUMTs are extremely low. When we examined the sequence composition, we observed that with increasing time since insertion, the inserted mitochondrial sequences presented a reduced transition-to-transversion (Ti/Tv) ratio, along with increasing GC content, gradually increasing from the average level of 28% in the mitochondria to 40% in the nuclear genome (Fig. 3b). Older NUMT copies are expected to exhibit a stronger AT-to-GC mutation bias compared to the mitochondrial reference genome, as highlighted by the base composition differences observed between chr2MT5 and chr5MT1. This turnover may reflect a change in evolutionary pressure or a difference in the error proneness of DNA repair mechanisms42. This process is also accompanied by an increase in repeat insertions, reflecting the common characteristics of exogenous sequences integrated into the genome. Additionally, the insertion of transposable elements may inactivate these foreign sequences, and the overall composition tends to align with the average composition of nuclear genes.
Characteristics of sex chromosomes and related gene expression
We successfully assembled both sex chromosomes, with the X chromosome spanning 67.73 Mb and the Y chromosome spanning 7.17 Mb (Fig. 4a). A total of 578 genes were annotated on the X chromosome, and 52 genes were annotated on the Y chromosome.

a Diagram of sex chromosomes. Gene expression levels are calculated as maximum FPKM values in 500-bp bins for biological replicates of peripheral tissues. Expression levels above the baseline represent male tissues (red), whereas expression levels below the baseline represent female tissues (blue). For Y chromosomes, mixed samples of males and females were used for illustration with the label “M&F”. The gene model structures are depicted as solid purple boxes above the chromosome coordinates. LSU rRNA, SSU rRNA, reverse transcriptase, and BdoSat are represented by purple boxes below the chromosome coordinates. Assembly continuity is represented with HiFi CCS and ONT reads, which are filtered according to the read length and mapping status (see methods). b Heatmap showing the DEGs shown significant calling between male and female samples in any of the peripheral tissues. Genes with significant differential expression in female relative to male tissues were visualized using “log2FoldChange” value. DEGs identified by DESeq2 (two-sided Wald test; BH-FDR adjusted; significance at padj <0.05). c Statistics for sex-related differentially expressed genes (DEGs). d Comparison of GO enrichment profiles between male and female sex-related DEGs using compareCluster. e Phylogenic relationships among the ATPsynβ family in species from Diptera and B. mori. The green and blue clades represent two distinct subfamilies. Members from D. melanogaster and B. dorsalis are highlighted in purple and red, respectively. f TPM values of the three homologs of ATPsynβ across different tissues of B. dorsalis. Chromosome Y contains BdoYG000019. g Experimental validation of BdoYG000019 at the genome and transcript levels. Gel images are representative of at least two independent experiments each for gDNA and cDNA. Unprocessed gel images were deposited in Source Data file.
Dosage compensation is particularly evident on the X chromosome. In six peripheral tissues, the genes on the long arm of the X chromosome exhibited active gene expression, whereas those on the short arm and near the centromere were not actively expressed. In Drosophila, dosage compensation is achieved by hyperactivating the single male X chromosome. At the male X:A ratio (0.5), autosomal Dpn repressors outweigh X-encoded Sis activators, silencing the X-encoded Sxl gene. The male-specific MSL2 protein (which suppressed by sxl) assembles the MSL/DCC complex to bind the X and acetylates histone H4K16 to loosen chromatin dosage compensation. However, in Bactrocera species, sex determination mechanism does not involve Sxl43. Whether a similar dosage compensation mechanism exists remains unclear and warrants further investigation.
Across these tissues, 5523 sex-related DEGs were identified, with the largest differences in expression observed in the external genitalia (Fig. 4b and Supplementary Data 11). Among these DEGs, 2246 were female-specific genes, and 2712 were male-specific genes, including 80 and 102 genes located on the X chromosome, respectively. In addition, 151 female-specific and 174 male-specific genes contained more than six transmembrane domains (TMRs) and were predicted to be involved in signal transduction (Fig. 4c). The minimal overlap of sex-biased genes between male and female tissues suggests that sex-differentially expressed genes form distinct, specialized networks during development. The sex-specific expression patterns of these genes reflect their potential involvement in the development of sex-specific phenotypes. Comparative gene set enrichment analysis revealed that male-specific genes were significantly enriched in pathways related to energy production (Fig. 4d). The high energy requirements of male flies might be a result of their lekking behavior and male display activities44, which is thought to be energetically expensive, similar to observations in birds45. In addition to lekking behavior, male B. dorsalis are likely to face increased energy demands because of the exclusive costs associated with long-distance travel to locate male attractants46 and the subsequent metabolic conversion of these compounds into sex pheromones47.
A previous study reported that the Y chromosome-specific gene MoY is involved in sex determination during early development43. Our assembly results indicate that this gene is present in three copies, all of which exhibit the conserved sequence “KHNSRT” in the N region, as previously reported, and we confirmed the authenticity of this sequence through PCR validation (Supplementary Fig. 6). Moreover, we discovered a previously unreported Y-specific gene, BdoYG000019, which was annotated as ATP synthase β subunit (ATPsynβ) and was highly expressed in all examined peripheral tissues (Fig. 4a, g). This gene is located in the central region of the Y chromosome and maintains high expression across all male peripheral tissues. Two homologous copies are located on the X chromosome, one of which is highly expressed in peripheral tissues, supposing to be involving in regular energy production within the mitochondria (Fig. 4f). Evolutionary analysis suggested that this Y-specific gene originated from one of the X chromosome homologs, BdoXG000104 (Fig. 4e). Notably, rDNA and retrotransposon-related repeat elements are clustered near BdoYG000019, suggesting that it may have originated through recombination.
Previous studies have reported that ATPsynβ can ectopically localize on the surface of vascular endothelial cells as a receptor for apolipoprotein A-I, which is the major component of HDL48. ATPsynβ mediates the uptake and resecretion of apolipoprotein A-I and is further involved in cholesterol homeostasis49. In the Alliance genome database, ATPsynβ was validated to interact with various genes (22 genes validated by yeast two-hybrid and others 54 supported by expression correlation), implying its involvement in multiple cellular processes, such as cytoskeletal organization (Act87E), lipid metabolism (Agpat4), synaptic function (Brp), calcium signaling (CanB), and the oxidative stress response (Sod3), as well as developmental processes (Dsx, Tll) (Supplementary Data 12). These interactions highlight the central role of genes in metabolic and regulatory pathways.
Phylogenetics and X chromosome evolution
To explore the genome evolution of B. dorsalis, we compared our assembly with 22 chromosome-level assemblies of Dipteran insects (Supplementary Data 13). The assembly of the Lepidopteran B. mori was used as the outgroup in the analysis. With 1914 concatenated single-copy orthologous genes, we inferred the phylogenomic tree and indicated that species within the genus Bactrocera represent a relatively late-diverging lineage within the Tephritidae family of the broader Brachycera clade (Fig. 5a). Their recent adaptive radiation has been collectively driven by global geographic expansion and rapid host-associated adaptive coevolution50.

a Total 1914 single-copy shared orthologous genes were used for phylogenetic tree construction. b Synteny comparison of genomic data within the sub Brachycera, highlighting the evolutionary origin trajectory of the X chromosome in orange. Chromosomes are highlighted by color according to the B. dorsalis reference genome.
Comparative genomic analyses revealed that within the suborder Brachycera, the evolutionary origin of the X chromosome varies significantly across species (Fig. 5b). Teleopsis dalmanni, D. melanogaster, and B. dorsalis each possess X chromosomes derived from distinct evolutionary origins, reflecting divergent evolutionary trajectories. This observation aligns with the paradigm-shifting findings of Vicoso and Bachtrog51, who demonstrated that Diptera have undergone an independent sex-chromosome transition, challenging the traditional view of stable XY systems in this order. Specifically, the syntenic conservation between the B. dorsalis X chromosome and D. melanogaster chromosome 4 (Muller F) suggests ancestral Diptera pattern, where the dot chromosome typically served as the proto-sex chromosome. In certain Drosophila strains, the nucleolus organizer region (NOR), which contains rDNA, is located on chromosome 4. This suggests that during the early evolutionary process, rDNA may have played a crucial role in maintaining the structural integrity of sex chromosomes52,53. Moreover, these species have experienced different chromosomal fusion and fission events, highlighting the dynamic structural evolution of their genomes.
T2T genome data facilitate functional genomics to develop molecular-target-based pest control techniques
The insect chemical receptor superfamily comprises gustatory receptors and odorant receptors, which are among the largest families of functionally diverse genes in multicellular organisms54. The odorant receptor (OR) families have evolved dynamically, with frequent sequence and structural variations, often requiring manual annotation with an accurate genome ref. 55. With the improved T2T B. dorsalis genome, we identified 110 odorant receptors (BdoORs) by considering conserved domains (Supplementary Data 14) and homologous information (Fig. 6a). Compared with the limited number of odorant receptors in D. melanogaster and mosquitoes (approximately 60–70), there are a greater number of ORs in B. dorsalis, which has evolved over the past few million years. This confers a significant advantage in adapting to diverse environments and a broad host range, facilitating ecological adaptation. The BdoOR families we identified are located on autosomes, with more than 50% (65 of 110) existing as tandem repeats with 2 to 10 members. Among them, BdorOR33s and BdorOR7s in particular have been subjected to robust tandem duplication, containing as many as 6 to 10 copies (Fig. 6b). A total of 22 tandem duplication clusters were detected, along with a few diversities in exon–intron structures and expression patterns (Fig. 6c).

a Maximum-likelihood phylogenetic tree based on amino acid sequences for OR subfamily members in Diptera. The blue clade represents GRs, while the red clade represents ORs (with purple stars indicating B. dorsalis ORs). b Distribution of B. dorsalis ORs across chromosomes. c Clusters, gene structures, and expression patterns of tandemly duplicated B. dorsalis ORs. The corresponding abbreviations for tissues are as follows: A—Antennae, M—Mouthpart, F-L—Foreleg, M-L—Midleg, H-L—Hindleg, G-L—Genitalia. d Analysis of junction events in BdorOR88a and its chimeric transcripts via RT‒PCR and RNA-seq. e Schematic design for the establishment of BdorOR88 knockout strains and subsequent experiments. f Quantification of EAG responses (mean ± SEM) to different concentrations of ME in WT and BdorOR88a12−/− males, with 11 and 15 recordings per group, respectively. Statistical test was performed by two-side unpaired t test (normally distributed data) or Mann–Whitney U test (non-normally distributed data). ns denotes P > 0.05. g Behavioral responses (mean ± SEM) of WT and BdorOR88a12−/− males to ME, with n = 5 biological replicates, each containing 30 individuals. Statistical test was performed by two-side unpaired t test (normally distributed data) or Mann–Whitney U test (non-normally distributed data). Significance: ns denotes P > 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 via statistical test, the exact P-values are provided in Supplementary Data 1. Source data are provided as Source Data files.
Among these genes, BdorOR88a has been reported as a methyl eugenol (ME) receptor56 that plays a key role in mediating the well-known male annihilation technique (MAT) for tephritid control57. In our assembly, the BdorOR88a gene cluster, consisting of three members, spans approximately 14 kb in the genome and exhibits a high degree of nucleotide sequence identity (over 92%), except in exon 4 (80.56%) and intron 2 (54.70%) (Supplementary Fig. 7a). Notably, RNA-seq analysis and PCR verification detected multiple chimeric transcripts across the three copies. These transcripts are formed by the splicing of exons between adjacent BdorOR88a members (Fig. 6d and Supplementary Fig. 7b), potentially increasing isoform diversity and enhancing functional resilience by allowing connected exons to maintain gene function despite genomic sequence alterations. Chimeric transcripts, which can be generated through either trans-splicing or cis-splicing, are widely observed across eukaryotes58. However, the understanding of the functions of these transcripts is relatively limited, with most studies focusing on their pathogenetic roles, particularly related to human cancer59.
We subsequently used CRISPR/Cas9 to simultaneously knock out the highly expressed BdorOR88a1 and BdorOR88a2 to verify their impact on the electrophysiology and behavior of B. dorsalis in response to methyl eugenol (Fig. 6f, Supplementary Fig. 7b–f, Supplementary Data 15 and Supplementary Data 16). Although the electroantennogram (EAG) response did not significantly differ between wild-type adults and BdorOR88a12−/− adults, the chemotaxis behavior toward ME was significantly attenuated (Fig. 6g, Supplementary Data 17 and Supplementary Data 18). To ensure that the attenuation of ME-directed chemotaxis behavior was not affected by potential functional compensation from the typically low-expressed BdorOR88a3, we conducted transcriptomic analysis of the mutants. The results revealed that the transcriptional expression pattern of BdorOR88a3 remained unchanged after the knockout of BdorOR88a1 and BdorOR88a2. This finding rules out the possibility of functional compensation from BdorOR88a3, confirming that the observed behavioral changes are primarily attributable to the loss of function of the two highly expressed gene copies (Supplementary Fig. 7g–i). Since the behavior of the mutants was not completely abolished and the EAG response remained intact, the detection of MEs in B. dorsalis might not fully depend on BdorOR88as, and the olfactory receptor that plays an essential role in mediating ME-directed attractive behavior still needs to be further elucidated. BdorOR88a may indirectly affect other olfactory genes, leading to the observed behavioral changes. For example, different chemosensory genes can influence each other’s expression, thereby affecting neural perception60. Future research could further explore the functions of the BdorOR88a gene copies to better understand how genetic diversity from gene duplication contributes to behavior and ecological adaptation.
Discussion
Despite the advances in genome assembly that have enabled T2T assemblies for most species, obtaining sufficient DNA for T2T genome assembly remains challenging for small insects. Unlike plant and animal genomes, which can be sequenced from a single individual, thereby minimizing data heterogeneity and allowing haplotype-resolved assembly through coverage estimates and parental Hi-C data, the genomes of small insects such as B. dorsalis have typically required HiFi-CCS, ONT, and Hi-C data from different pooled individuals for assembly. Additionally, laboratory insect populations typically require regular supplementation with wild individuals to mitigate inbreeding depression, including lethal homozygosity caused by laboratory propagation. Due to the resulting high internal heterozygosity, and the requirement for samples from multiple individuals in library preparation, ONT and Hi-C data cannot be effectively utilized to resolve highly repetitive regions to achieve a T2T status using current assemblers with a standard pipeline.
In this study, we successfully assembled the T2T genome of a male B. dorsalis using sequencing data from a single individual, establishing a framework for assembling the genomes of small insects with limited sequencing resources. Our assembly includes integrated sex chromosome, genome content and genome structure information. Our genome provides a valuable example of a complete genome assembly within Diptera species. The continuous generation of new high-quality genomes not only advances comparative genomics research but also enhances our understanding of genome evolution in pest species that are in constant conflict with humans.
Notably, Dipteran species have evolved telomerase-independent mechanisms for chromosomal end maintenance, with two distinct telomeric architectures identified to date61. The telomeric structure of D.melanogaster can extend through several kilobases and consists of a mixed repetitive array of HetA/TART/TAHRE elements, along with telomere-associated sequences30,31. The other type, found in lower Diptera, consists of long tandemly repeated sequences61. The telomeric element sequence of B.dorsalis differs substantially from those of the other seven species of the suborder Brachycera, all of which have the long repetitive element at the end of the chromosome.
We identified rDNA arrays on both the X and Y chromosomes in species of Dipteran species other than Drosophila. Interestingly, despite the presence of rDNA arrays on both sex chromosomes, the origins of the sex chromosomes appear to be distinct. This finding aligns with previous observations in the D. nobura species group, where rDNA clusters are found on both the X chromosome and the Y chromosome, suggesting a conserved ancestral state37. Our discovery in this non-Drosophila species highlights the potential variability in the evolutionary pathways of sex chromosomes involving the establishment of rDNA arrays. These results suggest that while the presence of rDNA on both sex chromosomes may be a shared feature across Diptera, the mechanisms and evolutionary history governing their distribution and retention on X and Y chromosomes could vary significantly among taxa.
Furthermore, in species such as lepidopterans and other insects, ancestral chromosomal synteny is often retained even after millions of years of divergence, suggesting that gene content conservation plays a crucial role in maintaining functional stability62. Synteny analysis revealed that B. dorsalis retains relatively conserved gene content within chromosomes in the suborder Brachycera, although chromosomal structures can be more dynamic (Supplementary Fig. 8). A previous study highlighted the high frequency of horizontal transfer of DNA transposons rather than retrotransposons in the insect genome63. Except Lepidoptera, most insect genomes harbor an abundance of DNA transposons64, which may contribute to disrupting the synteny of chromosomes. The cut-and-paste mechanism of DNA transposons increases the likelihood of rearrangements by producing double-stranded DNA breaks.
The conservation of ancestral chromosomal synteny demonstrates extraordinary evolutionary persistence across insect lineages. In species such as lepidopterans and other insects, ancestral chromosomal synteny is often retained even after millions of years of divergence, suggesting that gene content conservation plays a crucial role in maintaining functional stability62. Our analysis revealed that gene content of Muller elements remains stable across the species we examined. In suborder Brachycera, and this conservation can even be traced back to the Anopheles gambiae genome65. The mechanistic basis of this evolutionary stasis appears multifaceted. In Drosophila, comparative genomic study and phylogenetic reconstruction demonstrates that large-scale intra-arm inversions dominate chromosomal rearrangement66. At the nucleotide sequence level, comparison between D. simulans and D. yakuba shows that most inversion breakpoints are consistent with the staggered-breakpoint model, while only a few are associated with repetitive sequences66. Comparison among D.melanogaster and two closely related species, Drosophila simulans and Drosophila yakuba, suggests the mechanism of staggered breaks, forming inverted duplication sequence, primarily drives the generation of inversions67.
Gene duplication can occur through various mechanisms, such as whole-genome duplication (WGD), segmental duplication, tandem duplication (TD), and transposon element-mediated duplication68,69,70. In plants, genes encoding interacting proteins tend to be retained after WGD, whereas genes associated with stress resistance tend to be retained after TD71,72. Unlike plants, insects cannot utilize WGD73 but generally adopt TD to gain functional diversity in genes. For example, in the clonal raider ant, at least 93% of ORs originate from duplications, forming 41 tandem arrays with 2 to 89 gene copies each. Duplications of Diptera ORs have been systematically studied in drosophilids, with an average of 1 to 18 duplications. Drosophila grimshawi, with 27 duplications, represents an extreme example that is likely adapted to the complex environmental conditions of Hawaii74. Compared with reported OR duplications in Diptera genomes, B. dorsalis shows relatively extensive duplication (69% of ORs, 27 duplications, including 22 tandem events), potentially supporting its broad host range and contributing to its high invasive and adaptive capabilities. Moreover, tandem repeat ORs, such as BdorOR88a (with over 90% sequence identity), facilitate the production of chimeric transcripts, suggesting that tandem repeats increase genetic stability and reliability, thereby underscoring the role of gene duplications in adaptation and evolution. Here, our current study has certain limitations. Specifically, we were unable to distinguish the functions of individual transcript isoforms, as the high sequence similarity (>90%) among the tandemly duplicated copies of BdorOR88a presents a significant challenge for accurate expression quantification. To overcome these limitations, future studies could adopt more sensitive techniques, such as RNAscope, and integrate ligand identification, mutant analyses, and electrophysiological approaches to uncover how BdorOR88a contributes to behavioral adaptation and environmental responses in B. dorsalis.
In conclusion, we present a model pipeline that successfully achieved T2T assembly from a low-input library of a single dipteran insect using PacBio HiFi CCS sequencing. This assembly enabled the identification of novel centromeric and telomeric satellite sequences. The newly resolved structure of the sex chromosomes, along with identified sex-related and olfaction-related genes, provides a valuable foundation for future research on pest control, population genetics, and comparative genomics within Diptera. This work lays a solid foundation for advancing our understanding of dipteran insects.
Methods
Sample collection, DNA and RNA extraction, and sequencing
The experimental population was obtained from the Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences. The lines were kept under a 14-hour light/10-hour dark photoperiod at a temperature of 26 ± 1 °C and a relative humidity of 60 ± 5%. The larvae were provided with an artificial diet composed of banana, corn flour, yeast, sugar, and cellulose paper. Mature larvae were transferred to moist sand to pupate. The pupae were placed in a small cage (18 cm × 12.5 cm × 14 cm) for emergence. A single pair of adults (F1 parents) was mated, and their offspring were maintained. The F1 generation was further bred and maintained (experimental population) for use in all subsequent experiments.
For general DNA extraction, tissue was ground into a fine powder with liquid nitrogen and lysed in SDS lysis buffer at the appropriate temperature. After centrifugation, the supernatant was subjected to phenol-chloroform extraction, and nucleic acids were precipitated with isopropanol. The precipitates were washed twice with ethanol, air-dried, and dissolved in TE buffer overnight to obtain high-quality DNA. For RNA extraction, total RNA was isolated from tissues using the TRIzol reagent (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s instructions. RNA integrity was assessed using the Fragment Analyzer 5400 (Agilent Technologies, CA, USA).
For the genome survey, five samples were randomly selected and mixed for DNA extraction. A 150 bp paired-end sequencing library was prepared using the Next® UltraTM DNA Library Prep Kit for Illumina (E7645L, NEB, USA) and sequenced via the Illumina NovaSeq 6000 platform by Novogene Biotechnologies, Inc. (Beijing, China).
For PacBio HiFi low-input library construction, a single 12-day-old male adult and a female adult were selected from the F1 generation. The digestive tracts were removed, and the samples were frozen in liquid nitrogen. The HiFi library was prepared using the SMRTbell® Express Template Prep Kit 2.0. Starting with 1.7 μg of high molecular weight (HMW) genomic DNA (most fragments >30 kb), and the DNA was sheared and an average size of ~15 kb was retained. Sequencing was performed using the PacBio Sequel II platform by Novogene Biotechnologies, Inc. (Beijing, China).
The Hi-C Library construction was carried out following the standard protocol described by Belton et al., with some modifications. The sample was ground in liquid nitrogen and crosslinked with 4% formaldehyde under vacuum at room temperature for 30 min. The reaction was quenched with 2.5 M glycine. After centrifugation and washing with buffer, the sample pellet was resuspended in lysis buffer for nuclei isolation. The nuclei were solubilized with diluted SDS, incubated at 65 °C for 10 min, and neutralized with Triton X-100. DNA was digested with the 4-cutter restriction enzyme MboI, followed by biotin labeling and blunt-end ligation. Crosslinks were reversed through proteinase K treatment, and DNA was purified using phenol-chloroform extraction. Unligated biotin was removed using T4 DNA polymerase. DNA fragments were then sheared by sonication, repaired, and enriched using streptavidin-coated magnetic beads to isolate biotin-labeled fragments. Finally, A-tails were added to the DNA ends, and Illumina paired-end sequencing adapters were ligated. The Hi-C library was amplified by PCR and sequenced using the Illumina NovaSeq 6000 platform with 150 bp paired-end reads. Raw data was filtered to remove adapter sequences, reads with no base information, and low-quality reads, resulting in clean reads. Clean reads were further processed to remove duplicates. The quality control criteria included Q30 > 80%, an error rate of less than 1%, and a GC content distribution without significant separation for individual reads.
For the Oxford Nanopore ultralong library, HMW DNA extraction included DNA fragmentation with diluted FRA, rapid adapter ligation, and DNA precipitation with PPT buffer. All steps were performed with care to preserve long DNA fragments, utilizing wide-bore tips to avoid mechanical shearing. For the Oxford Nanopore ultra-long library preparation, 78.3 μg of long DNA, extracted from a mixture of five larvae and exhibiting a prominent band around 100 kb, was size-selected and processed using the Ligation Sequencing Kit (SQK-ULK001; Oxford Nanopore Technologies, Oxford, UK) following the manufacturer’s instructions. Sequencing was subsequently performed on the PromethION sequencer with an R9.4 chip (Oxford Nanopore Technologies, Oxford, UK).
For genome annotation, samples from different organ parts were collected for RNA extraction and RNA-seq. Adult samples included the head, thorax, abdomen, wings, and reproductive organs. To cover genes related to olfactory behavior, the antennae, mouthparts, legs, and genitalia from both sexes were collected. Pooled samples of larvae, pupae, and adults were collected for iso-seq. All the samples were stored at −80 °C until use. Details of sample collection are listed in (Supplementary Data 19). -All the samples were submitted to Novogene for further processing. The library was prepared by using NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, USA). Briefly, mRNA was purified using poly-T oligo-attached magnetic beads and fragmented using divalent cations under elevated temperature. First-strand cDNA was synthesized with random hexamer primers and M-MuLV Reverse Transcriptase, followed by second-strand cDNA synthesis using DNA Polymerase I and RNase H. Overhangs were converted to blunt ends, and 3′ adenylation was performed before ligating NEBNext Adaptor with hairpin loop structure. Size selection of ~250–300 bp cDNA fragments was performed using AMPure XP beads, and USER Enzyme was applied to process adaptor-ligated fragments. PCR amplification was carried out with Phusion High-Fidelity DNA Polymerase and indexed primers. The library was purified and assessed on an Agilent Bioanalyzer 2100 system. Finally, clustering was performed on an Illumina cBot system using the TruSeq PE Cluster Kit v3, and sequencing was conducted on the Illumina NovaSeq 6000 platform to generate 150 bp paired-end reads.
Adapter sequences, reads with uncertain bases (> 10%), and low-quality reads were removed from raw data, resulting in clean reads with Q30 > 90% and an error rate of less than 1%. All quality control steps were performed using Fastp v0.19.7. The details of the statistical description of QC were added to Supplementary Data 20 and 21.
Raw data preprocessing
For the HiFi CCS reads, 28 bp were removed from both ends, and only reads with a QV of 20 were retained for further analysis. The ONT reads were filtered using NanoFilt75 (--headcrop 50 --tailcrop 50), and only those longer than 80 kb were retained for assembly. Iso-seq reads were preprocessed to cluster transcript sequences using the IsoSeq v3 pipeline (v3.8.0, https://github.com/PacificBiosciences/IsoSeq). The Illumina paired-end reads used for the genome survey and RNA-seq were quality filtered with fastp76 (v0.23.4).
Genome size and heterozygosity estimation
One library with an insert length of 300 bp was constructed with a sample of 5 individual mixed siblings and further sequenced on the Illumina platform. Jellyfish77 (v2.1.3) and GenomeScope78 (v2.0) were used to estimate genome size and heterozygosity.
Contamination remove for HiFi CCS reads
Due to the small size of the individual insects and the potential for bacterial contamination, the midgut was removed, and the sample was washed multiple times before HiFi CCS library preparation. However, residual internal and external microorganisms remained, resulting in a high level of contamination in the raw data. To assemble contigs of bacterial and fungal origin as completely as possible, we first assembled the contaminant-containing HiFi CCS reads using Hifiasm79,80 (r603) with default parameters and HiCanu81 (v2.2) with parameters (useGrid=0, genomeSize = 540 m, minReadLength = 6000, utgOverlapper=minimap, minOverlapLength = 3000, contigFilter = “2 0 0.8 0.5 2”, -pacbio-hifi).
Bacterial and fungal genomes were downloaded from RefSeq (release205). Minimap282 was used to align the QC-filtered HiFi CCS reads to the bacterial and fungal reference genomes with default parameters.
By analyzing GFA (Graphical Fragment Assembly) information, we identify bacterial homologous sequences within the assembled fragments (unitigs, contigs, and super-contigs). If a significant portion of a contig shows homology to a bacterial genome, we consider the entire contig to be contaminated. In our approach, a cutoff value of 0.4 is applied, validated by comparing the entire fragment with the NT database. If a contig is identified as contamination, all reads comprising that contig are excluded. Additionally, reads with more than 70% overlap with contaminated reads were excluded from further assembly.
Notably, some low-coverage assembled regions showed HiFi CCS reads containing short microbial DNA fragments embedded within host genomic sequences when aligned to the NCBI NT database. This suggests possible chimeric reads generated during library preparation due to erroneous ligation events.
The filtered, clean HiFi CCS reads were used for the following genome assembly.
Genome assembly
General assembly procedure with Hifiasm, purged_dups and Juicer pipeline
Initial assembly was assembled with HiFi CCS reads by using hifiasm (r603) with parameters (--hg-size 540 m--primary -r 5 -D 6 -m 1000000 --b-cov 2 -l 1 -O 2). The primary supers-scaffold (p_utg) produced by Hifiasm underwent haplotypic duplication removal using Purge_Dups (v1.2.6). The Juicer83 (v1.6) and 3D-DNA pipelines84 (v2.20) were used to anchor contigs to pseudochromosomes with Hi-C data. The results were further manually refined in Juicebox85 (v2.20).
This finalized assembly was employed to facilitate the identification of candidate regions corresponding to X chromosome fragments, telomeric sequences, and centromeric sequences.
Unitigs validation
ONT reads were mapped to hifiasm unitig contigs with minimap2 (v2.26), retaining only those reads with a mapping length of at least 30 kb and a mapping length coverage above 95%. For each 200 bp genomic bin, the relative distance between the mapped reads and genome loci (calculated as (read_center-genome_loci)/read_length) was computed. This value is expected to follow a uniform distribution within the range of [−0.5, 0.5]. The Anderson‒Darling and Shapiro‒Wilk normality tests were applied to evaluate this distribution, excluding terminal 8 kb regions of contigs from calculation. The 200 loci with the lowest p-values were manually inspected, and the 12 suspicious loci with disproportionate read distributions were resolved by breaking the corresponding contigs (Examples shown in Supplementary Fig. 10). Additionally, approximately 10 kb were trimmed from the newly generated contig ends to avoid potential erroneous extensions caused by individual HiFi CCS reads.
Sex chromosome assembling
We first aimed to assemble the Y chromosome, considering its small size, abundant repetitive sequences, and weak Hi-C interactions.
HiFi CCS reads from male and female samples were aligned to initial hifiasm unitigs and contigs using Winnowmap286 (v2.03), chosen for its superior performance in repetitive regions. Sex chromosome candidate fragments were identified based on differences in read coverage, assuming that the coverage of autosomes should follow a binomial distribution. Differential fragment coverage was analyzed using DEGseq87, applying a stringent cutoff of p-value < 0.001 to select candidate sex chromosomes contigs. Candidate contigs rich in repetitive elements, such as histone or rDNA sequences, were excluded. Several validated Y chromosome markers, including the GIGYF family protein (MT978097) and Moy protein, previously identified as transcribed Y-specific markers of B.dorsalis, were used to confirm contigs belonging to the Y chromosome.
For the X chromosome, the primary assembly relied on the p_utg contigs. Structural continuity was manually confirmed by assessing the alignment consistency of unitigs and HiFi CCS reads.
The mapping result of Nanopore long reads (>80 kb) was extracted and converted into a connected graph of unitigs. Relationships among contigs were manually verified and extended based on maximal read overlaps and minimal mismatches.”
Identification of genome structural features
The terminal ends of the original hifiasm-derived unitigs and contigs were extended using ONT long reads to characterize repetitive centromeric and telomeric elements. The “Hifiasm+purged_dups+Hi-C” based genome assembly was generated to facilitate the candidate selection. Given that no centromeric repeat units had been previously reported for B.dorsalis, we hypothesized that they might be located near the ends of the assembled super-contigs.
For monomer sequences of centromere and telomere, the detailed analysis is shown in the following corresponding sections.
Scaffolding
Telomeric, centromeric, histone, and rDNA arrays in assembled unitigs (r_utg) were annotated using RepeatMasker and visualized as “yarn balls” in Bandage88 (v0.8.1). Based on repetitive element composition, unitig graph was divided into low-complexity, repeat-rich regions (primarily derived from heterochromatic and scattered regions) and high-complexity, gene-rich regions.
For low-complexity regions (high repetitive in sequence), contigs along with their neighboring regions were isolated from assemble graph. Contig path surrounding the centromere and telomere was resolved by using the primary unitig graph and was further simplified through graph alignment and gap closing with ONT reads:
ONT reads were mapped to hifiasm r_utg.gfa using GraphAligner89 (v1.0.17) was used to map ONT reads to hifiasm “r_utg.gfa”. Only reads meeting the following criteria were retained: (1) 95% of the length aligned to the genome (to simplify the assembly graph and construct the longest contig connection), and (2) at least one terminal alignment longer than 20 kb and an NM value/mapping length <0.05. If the ends covered multiple contigs, the alignment had to support the existing r_utg contig connection. New connection relationships were established using mutual best alignments inferred from Minimap2, Winnowmap2, and GraphAligner.
For high-complexity regions, hifiasm outputs were verified and redundancies removed based on Hi-C interactions (Supplementary Fig. 11) and ONT alignments to remove additional alternate contigs in the diploid bubble structure.
Genome polish
Due to the high heterozygosity in the population, short-read or ONT reads-based polishing would introduce more structural diversity. Thus, we limited our approach to using unitigs and super contigs to replace shorter paths within bubble regions, effectively creating a hifiasm primary-like assembly. We first called SNVs with ONT reads (>40 kb) using SVision90. Homozygous deletions or insertions > 3 kb, supported by more than 10 reads, were manually verified at 8 loci by aligning ONT/HiFi CCS reads. Suspected errors at 5 loci were corrected using assembled contig sequences.
Gap filling for histone region
The only remaining gap in the assembly was a histone cluster located in the pericentromeric region of chromosome 2, which could not be directly closed using long ONT reads or string graphs. The copy number of the histone gene cluster was estimated to be approximately 170 copies based on ONT reads and approximately 150 copies based on HiFi CCS reads. Due to high sequence conservation (Supplementary Fig. 9), the final assembly filled this gap with 150 copies of the histone gene, comprising approximately 802 kb. The heterogeneous contigs and scaffolding in the Hifiasm results were manually validated using Hi-C information. Finally, the assembly was polished with primary unitigs and contigs.
Candidate centromere identification
We first aligned the long ONT reads (>80 kb) to the hifiasm assembly with winnowmap2 and extracted overhanging sequences. After extending the terminal regions of the hifiasm assembly with ONT reads longer than 80 kb via winnowmap2, we extracted the overhanging sequences and performed tandem repeat identification with TRF91 (v4.09.1) (parameters: 2 7 7 80 10 50 500 -f -d -m). The identified repeat units were used as library seed files for whole-genome identification with RepeatMasker. All repeat regions were further characterized using TRF for repeat unit identification.
The results were then aligned using MARS92 and manually curated, followed by sequence alignment via MAFFT93 (v7.505). Clustering was performed according to the similarity distribution of the sequences, and consensus sequences were generated using cons in EMBOSS94 (version 6.6.0) with the parameters (-identity 100 -plurality 50). The structure and location of centromeres within the genome were illustrated using the R packages karyoploteR95 and StainedGlass96 (v0.6).
Telomere identification
An additional sequence of 528 bp repeats was identified in the most terminal region of the primary assembly. This structure was further confirmed through analysis with srf97 (https://github.com/lh3/srf) on >80 kb ONT ultralong reads. Among these, 214 ultralong ONT reads were found to contain this specific repeat unit, which was localized exclusively either at the end of a read or extending across the full length of a read. TRF (v4.09) and MAFFT (v7.505) were employed to extract and construct the final consensus for these telomere-associated sequences.
Assembly assessment
To evaluate the diploid assembly, we employed Merqury98, a reference-free k-mer-based tool. Compleasm99 (v0.2.5) was used to assess assembly completeness using the diptera_odb10 gene set (n = 3285) from BUSCO v5. GCI100 (v1.0) was used for continuity estimation. To verify the accuracy of sequence assembly, minimap2 (v2.26) and winnowmap2 (v2.03) were used for Hifi CCS and ONT read mapping. Mapping reads were filtered with the following criteria: (1) for HiFi CCS reads: length>9 kb, NM tag/mapping length<0.01, primary or supplementary alignment, and mapping length >90% of read length; (2) for ONT reads: length >40 kb, NM tag/mapping length <0.05, and mapping length >90% of read length. A Circos plot was generated using TBtools101 (v2.096), and a profile plot along the sex chromosomes was generated via the karyoploteR package (v1.28.0).
Gene annotation
Gene structure annotation was performed with a combination of three approaches: transcriptome-based prediction, homology-based prediction, and ab initio annotation.
All RNA-seq data were spliced-aligned against the genome assembly with HISAT2102 (v2.2.1). The transcripts were constructed with StringTie103 (v2.1.7) and further merged with TACO104. Iso-seq data were processed with the Iso-seq3 workflow (https://github.com/PacificBiosciences/pbbioconda). The CDS region was refined with TransDecoder (v5.5.0).
The protein sequences of Bactrocera cucurbitae (GCF_000806345.2), Bactrocera oleae (GCF_001188975.3), Bactrocera tryoni (GCF_016617805.1), and Bactrocera latifrons (GCF_001853355.1) were obtained from NCBI RefSeq. Homolog-based gene prediction was performed using the gth tool from GenomeThreader105 (v1.7.1).
Ab initio gene structure annotation was performed using Augustus106 (v3.4.0).
The CDS annotation files generated with the three approaches were manually integrated. First, extract the CDS information from the annotation files of the three methods separately and merge them using GffRead107 (v0.12.7). If different prediction methods generate conflicting CDS structures at the same gene locus, use the following priority to determine the final structure: transcript-based evidence > homology-based prediction > ab initio annotation. For CDS structures obtained from ab initio annotation, if multiple short CDS structures are integrated into the same gene locus, this indicates that the gene may be a fragmented gene. For Augustus genes containing introns longer than 5000 bp, prioritize retaining transcript structures supported by ISO-Seq data. If no ISO-Seq data is available, retain only structures with a complete Open Reading Frame (ORF).
Gene functions were assigned according to the best match by aligning the protein sequences using BLASTP (v2.12.0, E value < 10−3) to the NCBI nr database. KEGG pathways were assigned with the KofamKOALA web server108 (KEGG release v110). Functional classification (GO categories) was performed via InterProScan109 (v 5.55-88.0). Functional enrichment analysis and plotting were performed with the “compareCluster” function in the clusterProfiler110 package (v4.10.1). Transmembrane regions were predicted via DeepTMHMM111 (v1.0.8). Differentially expressed genes between males and females were analyzed for multiple tissues with DESeq2112.
H3K4me3 and H3K27me3 ChIP-seq data from the thorax muscles of the Hainan population of B. dorsalis were downloaded from NCBI BioProject PRJNA911513. The raw data were filtered with fastp, and the T2T assembly was aligned with bwa-mem2113 (v2.2.1). The ChIP-seq signal peaks were called using MACS2 (v2.2.9.1) with the parameters (--broad -f BAMPE).
Repeat annotation
We used Repbase114 (v26.11), specifically, the Arthropoda, angrep, and invrep datasets, as a reference set of known repeats. Repeat annotation was performed using EDTA115 (v2.0.1) and RepeatModeler2116 (v2.0.2a), and the repeat seeds were dereplicated via CD-HIT117 (v4.8.1) with the parameters (-aS 0.8 -c 0.8 -g 1 -G 0 -A 80 -M 10000). Unknown repeat seeds were further annotated using PASTEClassifier118 and DeepTE119. Finally, the repeat seed library was unified with naming according to Wicker’s code, and the assembly was annotated with RepeatMasker (v 4.1.1).
NUMT detection and analysis in nuclear DNA sequences
Mitochondrial DNA sequences in the nuclear genome were identified via nucmer120 (v4.0.0rc1) with the options --maxmatch -c 100 -d 0.2 -g 1000. The putative concatenated NUMTs were identified by clustering the adjacent mitochondrial-like DNA blocks using the following criteria: (1) same chromosome, (2) same direction (plus or minus strand), (3) continuous coordinates, and (4) more than 10% of the mitochondrial genome length. Using the above criteria, 29 concatenated NUMTs were detected in the nuclear genome (Supplementary Data 10). Among these NUMTs, mitochondrial genome sequence represented more than 75% of the total sequence in 11 cases. These 11 NUMTs were aligned to the mitochondrial genome sequence via MAFFT (v7.487) and used to construct the evolutionary tree via PhyML121 (v3.3.20190909). To identify variants between these NUMTs and the mitochondrial genome sequence, we first built the chain file using crossmap workflow (https://github.com/soybase/crossmap-workflow) on the basis of the delta file generated by nucmer and then identified the variants via the transanno (v0.4.0) chain-to-bed-vcf command based on the chain file. We also annotated gene models for these NUMTs by transferring the gene annotations to the mitochondrial genome sequence using CrossMap (v0.6.5) based on the chain file. The relationships between the NUMTs and the mitochondrial genome were visualized via TBtools-II (v2.096).
Additional 8 NUMTs were shown in Supplementary Fig. 12.
DEGs for RNA-seq data
The RNA-Seq reads obtained from B. dorsalis peripheral nervous tissue were quality-filtered as described in “Raw data preprocessing”. The filtered reads were aligned to the genome using HISAT2102 (v2.2.1) with default parameters. Based on the alignment results, featureCounts (v2.0.1) was employed to quantify annotated genes in the genome, compiling a raw gene expression matrix and a TPM (Transcripts Per Million) matrix for the peripheral nervous tissue. The DESeq2112 was applied to analyze sex-specific differentially expressed genes (DEGs) in the peripheral nervous tissue, with thresholds set at fold change > 1.5 and adjusted p-value (p.adjust) <0.05. DEGs between sexes across different tissues were subsequently extracted for further analysis. Heatmaps were visualized using the pheatmap package in R.
Refine the structure of OR genes
Protein sequences of olfactory receptor (OR) genes from dipteran insects published in NCBI were collected, and incomplete sequences were filtered out. Using the hmmbuild function in HMMER, a Hidden Markov Model (HMM) was constructed based on these protein sequences. This model, along with the 7tm_6 odorant receptor HMM profile (PF02949) from the Pfam database, was applied via the hmmsearch function to screen all B. dorsalis protein sequences, retaining hits with e-value < 1e‒3 as candidate OR genes supported by domain evidence. Separately, OR gene protein sequences of D.melanogaster from the Swiss-Prot database were collected and aligned against all B. dorsalis protein sequences using PSI-BLAST. Hits with e-value < 1e−3 were selected as candidate OR genes supported by homology evidence. The intersection of the two candidate sets was taken to obtain the final candidate OR gene list. The protein sequences of the final candidate genes were analyzed with InterProScan (v5.55–88.0) against the Pfam, TMHMM, Phobius, and PANTHER databases to validate domains and perform functional predictions. Genes predicted as ORs or containing 7-transmembrane domains (7tm_6) were retained as the final candidate OR genes.
Publicly available OR and gustatory receptor (GR) protein sequences of D.melanogaster, B.correcta, B.cucurbitae, B.tau, B.minax, as well as OR protein sequences of E.balteatus and E.corollae, were searched and downloaded online. These sequences, along with the amino acid sequences of OR genes annotated in this study from B. dorsalis, were subjected to multiple sequence alignment using MAFFT (v7.515). A phylogenetic tree was then constructed by the maximum likelihood method with IQ-TREE (v2.2.0.3) using parameters: -bb 1000 -nt 20 -m MFP -bnni.
OR-related expression data were extracted from the peripheral nervous tissue expression profiles and differential expression analysis results mentioned in the previous sections.
Phylogenetics and synteny analysis
Nonredundant protein sequences from 22 species of Diptera and one outgroup species (B. mori) were prepared for ortholog analyses (Supplementary Data 13). Orthologs and orthogroups were inferred using OrthoFinder122 (v2.5.4) with the default settings and ‘-M msa’ activation. A phylogenetic tree was constructed with Gblocks, MAFFT (v 7.490) and IQ-TREE123 (v2.3.3) by using single-copy orthologs. Syntenic blocks were detected with MMseqs2124 (v13.45111) and WGDI125 (v0.6.5), and syntenic relationships among the selected species were visualized with plot_riparian in the GENESPACE126 package (v1.3.1). The orthogroup composition of OrthoFinder result and the genomic coordinate information used for the synteny analysis are provided in Supplementary Data 22 and Supplementary Data 23, respectively.
Fluorescence in situ hybridization (FISH)
The brains of 3rd instar (or younger) larvae were dissected in PBS and immediately transferred to 1% hypotonic sodium citrate solution and incubated for 15 min. These samples were subsequently fixed in a methanol:acetic acid solution (3:1) and incubated for 5 min at room temperature. After fixation, the brains were treated with 60% acetic acid until they became transparent. The brains were placed on a slide, and a drop of 60% acetic acid was added. A coverslip was placed on top, and the sample was lightly tapped with a silicone hammer for approximately 10 s. The slide was then frozen in liquid nitrogen, after which the coverslip was quickly removed with a razor blade. These slides were subsequently placed in ethanol at −20 °C for 10 min before use. To prepare chromosomes for hybridization, the slides were washed in PBS with 0.1% Tween for 5 min and then transferred through a series of prehybridization solutions, as follows: 5 min in 2×SSC plus 0.1% Tween, 5 min in 2× SSC plus 0.1% Tween and 50% formamide, 2.5 min at 92 °C in 2× SSC plus 0.1% Tween and 50% formamide, 5 min 2× SSC plus 0.1% Tween and 50% formamide, and then 20 min at 60 °C in 2× SSC plus 0.1% Tween and 50% formamide.
The DNA probes were designed according to the target sequences (Supplementary Data 24). The 5′ FAM- or Cy3-conjugated probes were provided by GENEWIZ® and were purified by HPLC. The probes were diluted in hybridization buffer, denatured for 2.5 min at 95 °C and immediately chilled on ice. Hybridization was performed at 37 °C overnight, and unbound probes were removed by washing for 10 min in 2× SSC at 60 °C. Finally, the slides were washed three times for 10 min each in PBS at room temperature. A drop of mounting medium (SlowFade Gold antifade reagent with DAPI) was applied before placing the coverslip. The hybridized chromosomes were analyzed under a Nikon Ni microscope at 1000× magnification using appropriate filter sets.
Establishment of the CRISPR/Cas9-based tandem repeat OR knockout strain
The tandem repeat odorant receptor (OR) knockout strain was generated following the methods described by previous study127. This included the synthesis of sgRNAs, sgRNA injection into embryos, and mutant screening.
Genomic DNA was extracted from the mid-legs of adult flies, and the target regions corresponding to the aforementioned genes were amplified via PCR with specific primers (details of the primers and PCR conditions are provided in Supplementary Data 15). To verify the BdorOR88a1, BdorOR88a2, and BdorOR88a3 sequences, the PCR products for each gene were subsequently cloned and inserted into blunt-end vectors (TransGen Biotech, Beijing, China). Subsequently, 20 individual bacterial colonies were screened for each gene product to identify conserved regions within the genes for the purpose of designing targets for gene editing. A total of 4 sgRNAs were designed across the first exons in the CDS of BdorOR88as, and only sgRNA2 was found to target both BdorOR88a1and BdorOR88a2 (Supplementary Data 25). All of the sgRNAs were designed via gRNA-Cas9-AI software128 and possessed targeting sites of 20 base pairs in length that included an adjacent protospacer adjacent motif (PAM) of NGG or CCN. The sgRNAs were synthesized using commercial kits (GeneArt gRNA Kit, Thermo Fisher Scientific).
Adult insects aged 9–12 days were kept in transparent cages and provided with sufficient food and water. The insects were allowed to mate for three days, after which an embryo collection apparatus containing orange juice was introduced. Embryos collected within a 10-minute window were used for injections. Due to the high sequence similarity among BdorOR88a gene copies (90%), achieving specific knockout mutants was challenging, and the likelihood of generating mutations in multiple similar locations with a single sgRNA was low. Therefore, a mixture of sgRNAs (sgRNA1 + 2 + 3 + 4) was injected to increase the likelihood of targeting different gene copies. For example, if sgRNA1 induced a mutation in 88a1, another sgRNAs might induce a mutation in 88a2, resulting in a BdorOR88a12 double mutant. The working concentrations were set to 300 ng/µL for the sgRNA and 150 ng/µL for the Cas9 protein. After the mixture was prepared, embryo injections were performed using the FemtoJet and InjectMan 4 systems (Eppendorf, Hamburg, Germany). The injected embryos were incubated at 26.5 °C with 60% relative humidity. Hatched larvae were transferred with a soft brush to an artificial diet and reared under laboratory conditions.
Genomic DNA was extracted from the mid-legs of mutant adult flies for genotyping via the specific primers and PCR conditions described above. PCR-amplified fragments were sequenced via the Sanger platform, and individuals with overlapping peaks at the target site were identified as carriers of edited genes. The PCR products were subsequently cloned and inserted into blunt-end vectors for precise genotype determination. Only strains with simultaneous knockouts of both BdorOR88a1 and BdorOR88a2 were retained. Adult G0 flies were backcrossed with wild-type flies, and G1 offspring were genotyped to isolate heterozygous mutants. The desired G1 heterozygotes were selected and crossed with wild-type flies to expand the population and generate G2 heterozygotes, which were then intercrossed to yield G3 homozygotes. G3 homozygous lines were established and maintained for further experiments. Additional information regarding knockout mutant screening is provided in Supplementary Data 16.
Electroantennogram (EAG) recordings
EAG recordings were performed using a modified version of the procedure described by Xu et al.129. Twelve-day-old male adults were prepared and decapitated. The heads of the males were subsequently injected with glass electrodes filled with 0.1 M KCl solution. One antenna was cut at the tip and attached to another glass electrode, which was also filled with 0.1 M KCl solution. Recordings were taken from 11 to 15 adults of each genotype, specifically from unmated males that were 12 days old. EAG recordings were conducted using a BX 51 microscope (Olympus).
The tested chemicals were prepared at concentrations of 0.1–100 μg/μl in paraffin oil, and 10 μl was added to a Pasteur pipette. Airflow stimulation was performed using a CS-55 controller (Syntech, Kirchzarten, Germany) at a rate of 1.4 ml/min for 300 milliseconds. The signals were collected using a universal probe preamplifier and converted with an IDAC-4-USB digital-to-analog converter (Syntech, Netherlands). The data were analyzed using EAGpro 2.0 software (Syntech, Netherlands). Relative EAG responses were calculated by subtracting the baseline signal obtained with paraffin oil alone from the signal produced by the test compound.
Four-quadrant olfactometer assay for behavioral observations
Behavioral experiments were conducted in a four-quadrant olfactometer and recorded using an automated camera system. The olfactometer was cleaned with 75% ethanol and dried for 5 min (repeated three times) to ensure cleanliness. The gas flow stability was tested to confirm consistent flow rates in all four quadrants (0.4 L/min). Adult males were placed in the ventilated olfactometer and allowed to acclimate to airflow for even distribution. Methyl eugenol (ME) at a concentration of 1 μg/μL in paraffin oil was added to the glass odor chambers, and the odor source was linked to two quadrants, while the controls were linked to the opposite quadrants. Behavioral responses were observed for 10 min. Experiments were conducted between 8:00 and 9:00 AM under 280–300 lux light intensity, 26 ± 1 °C temperature, and 60 ± 5% humidity. The videos of the responses were analyzed by manually counting the number of adults in each quadrant every minute. The attraction index was calculated as follows: (number of adults in test quadrants − number of adults in control quadrants)/(number of adults in test quadrants + number of adults in control quadrants). The experiments were replicated five times with both wild-type and mutant flies, with 30 adults per replicate.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the Article and its Supplementary Data files. The raw sequence data generated in this study have been deposited in the Genome Sequence Archive (GSA) database under accession id CRA027090, CRA027091, CRA027092, CRA027178 and CRA027308 [[https://ngdc.cncb.ac.cn/gsa/browse/CRA027308]. The whole genome assembly reported in this paper have been deposited in the Genome Warehouse (GWH) database under accession GWHFHGK00000000.1 [https://ngdc.cncb.ac.cn/gwh/Assembly/86355/show]. All data was cataloged under the umbrella project accession number PRJCA031569. Source Data is provided as a Source Data file. Source data are provided with this paper.
References
Papanicolaou, A. et al. The whole genome sequence of the Mediterranean fruit fly, Ceratitis capitata (Wiedemann), reveals insights into the biology and adaptive evolution of a highly invasive pest species. Genome Biol. 17, 192 (2016.
McKenna, D. D. et al. Genome of the Asian longhorned beetle (Anoplophora glabripennis), a globally significant invasive species, reveals key functional and evolutionary innovations at the beetle-plant interface. Genome Biol. 17, 227 (2016).
Matthews, B. J. et al. Improved reference genome of Aedes aegypti informs arbovirus vector control. Nature 563, 501–507 (2018).
Li, S. et al. The genomic and functional landscapes of developmental plasticity in the American cockroach. Nat. Commun. 9, 1008 (2018).
Wan, F. et al. A chromosome-level genome assembly of Cydia pomonella provides insights into chemical ecology and insecticide resistance. Nat. Commun. 10, 4237 (2019).
Aganezov, S. et al. A complete reference genome improves analysis of human genetic variation. Science 376, eabl3533 (2022).
Mao, Y. & Zhang, G. A complete, telomere-to-telomere human genome sequence presents new opportunities for evolutionary genomics. Nat. Methods 19, 635–638 (2022).
Shang, L. et al. A complete assembly of the rice Nipponbare reference genome. Mol. Plant 16, 1232–1236 (2023).
Li, H. & Durbin, R. Genome assembly in the telomere-to-telomere era. Nat. Rev. Genet. 25, 658–670 (2024).
You, M. et al. A heterozygous moth genome provides insights into herbivory and detoxification. Nat. Genet. 45, 220–225 (2013).
Richards, S. & Murali, S. C. Best practices in insect genome sequencing: what works and what doesn’t. Curr. Opin. Insect Sci. 7, 1–7 (2015).
Christenson, L. D. & Foote, R. H. Biology of Fruit Flies. Annu. Rev. Entomol. 5, 171–192 (1960).
Li, F. et al. Insect genomes: progress and challenges. Insect Mol. Biol. 28, 739–758 (2019).
Deng, Y. et al. A high heterozygosity genome assembly of Aedes albopictus enables the discovery of the association of PGANT3 with blood-feeding behavior. BMC Genomics 25, 336 (2024).
Jia, H. et al. Low-input PacBio sequencing generates high-quality individual fly genomes and characterizes mutational processes. Nat. Commun. 15, 5644 (2024).
Drosophila 12 Genomes, C. et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450, 203–218 (2007).
Adams, M. et al. One fly-one genome: chromosome-scale genome assembly of a single outbred Drosophila melanogaster. Nucleic Acids Res. 48, e75 (2020).
Danilevskaya, O. N., Lowenhaupt, K. & Pardue, M. L. Conserved subfamilies of the Drosophila HeT-A telomere-specific retrotransposon. Genetics 148, 233–242 (1998).
Kuznetsova, V., Grozeva, S. & Gokhman, V. Telomere structure in insects: a review. J. Zool. Syst. Evol. Res. 58, 127–158 (2020).
White, I. M. & Elson-Harris, M. M. Fruit flies of economic significance: their identification and bionomics. Environ. Entomol. 22, 1408–1408 (1992).
Shelly T, Epsky N, Jang EB, Reyes-Flores J, Vargas R (eds). Trapping and the Detection, Control, and Regulation of Tephritid Fruit Flies: Lures, Area-Wide Programs, and Trade Implications (Springer, 2014).
Zeng, Y. et al. Global distribution and invasion pattern of oriental fruit fly, Bactrocera dorsalis (Diptera: Tephritidae). J. Appl. Entomol. 143, 165–176 (2019).
Jiang, F., Liang, L., Wang, J. & Zhu, S. Chromosome-level genome assembly of Bactrocera dorsalis reveals its adaptation and invasion mechanisms. Commun. Biol. 5, 25 (2022).
Yang, Y. et al. Chromosome-level genome assembly reveals potential epigenetic mechanisms of the thermal tolerance in the oriental fruit fly, Bactrocera dorsalis. Int. J. Biol. Macromol. 225, 430–441 (2023).
Wang, Y. et al. Behavioral and genomic divergence between a generalist and a specialist fly. Cell Rep. 41, 111654 (2022).
Zhang, Y. et al. Genomes of the cosmopolitan fruit pest Bactrocera dorsalis (Diptera: Tephritidae) reveal its global invasion history and thermal adaptation. J. Adv. Res. 53, 61–74 (2023).
Logsdon, G. A. et al. The variation and evolution of complete human centromeres. Nature 629, 136–145 (2024).
Rabanal, F. A. et al. Pushing the limits of HiFi assemblies reveals centromere diversity between two genomes. Nucleic Acids Res. 50, 12309–12327 (2022).
Palomeque, T. & Lorite, P. Satellite DNA in insects: a review. Heredity (Edinb.) 100, 564–573 (2008).
de Lima, L. G., Svartman, M. & Kuhn, G. C. S. Dissecting the satellite DNA landscape in three cactophilic sequenced genomes. G3-Genes Genom. Genet. 7, 2831–2843 (2017).
Mason, J. M., Frydrychova, R. C. & Biessmann, H. Drosophila telomeres: an exception providing new insights. Bioessays 30, 25–37 (2008).
Drosopoulou, E. et al. Sex chromosomes and associated rDNA form a heterochromatic network in the polytene nuclei of Bactrocera oleae (Diptera: Tephritidae). Genetica 140, 169–180 (2012).
Wilkinson, M. E., Frangieh, C. J., Macrae, R. K. & Zhang, F. Structure of the R2 non-LTR retrotransposon initiating target-primed reverse transcription. Science 380, 301–308 (2023).
Ye, J. & Eickbush, T. H. Chromatin structure and transcription of the R1- and R2-inserted rRNA genes of Drosophila melanogaster. Mol. Cell Biol. 26, 8781–8790 (2006).
Stage, D. E. & Eickbush, T. H. Origin of nascent lineages and the mechanisms used to prime second-strand DNA synthesis in the R1 and R2 retrotransposons of Drosophila. Genome Biol. 10, R49 (2009).
Raje, H. S., Lieux, M. E. & DiMario, P. J. R1 retrotransposons in the nucleolar organizers of Drosophila melanogaster are transcribed by RNA polymerase I upon heat shock. Transcription 9, 273–285 (2018).
Bracewell, R. & Bachtrog, D. Complex Evolutionary History of the Y Chromosome in Flies of the Drosophila obscura Species Group. Genome Biol. Evol. 12, 494–505 (2020).
Bianciardi, A., Boschi, M., Swanson, E. E., Belloni, M. & Robbins, L. G. Ribosomal DNA organization before and after magnification in Drosophila melanogaster. Genetics 191, 703–723 (2012).
Watase, G. J., Nelson, J. O. & Yamashita, Y. M. Nonrandom sister chromatid segregation mediates rDNA copy number maintenance in Drosophila. Sci. Adv. 8, eabo4443 (2022).
Wei, W. et al. Nuclear-embedded mitochondrial DNA sequences in 66,083 human genomes. Nature 611, 105–114 (2022).
Ju, Y. S. et al. Frequent somatic transfer of mitochondrial DNA into the nuclear genome of human cancer cells. Genome Res. 25, 814–824 (2015).
Kiktev, D. A., Sheng, Z., Lobachev, K. S. & Petes, T. D. GC content elevates mutation and recombination rates in the yeast Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA 115, E7109–E7118 (2018).
Meccariello, A. et al. Maleness-on-the-Y (MoY) orchestrates male sex determination in major agricultural fruit fly pests. Science 365, 1457–1460 (2019).
Shelly, T. E. & Kaneshiro, K. Y. Lek behavior of the oriental fruit fly,Dacus dorsalis, in Hawaii (Diptera: Tephritidae). J. Insect Behav. 4, 235–241 (1991).
Barske, J. et al. Energetics of the acrobatic courtship in male golden-collared manakins (Manacus vitellinus). Proc. Biol. Sci. 281, 20132482 (2014).
Steiner, L. F. Methyl eugenol as an attractant for oriental fruit fly. J. Economic Entomol. 45, 241–248 (1952).
Nishida, R. et al. Accumulation of phenylpropanoids in the rectal glands of males of the Oriental fruit fly,Dacus dorsalis. Experientia 44, 534–536 (1988).
Martinez, L. O. et al. Ectopic beta-chain of ATP synthase is an apolipoprotein A-I receptor in hepatic HDL endocytosis. Nature 421, 75–79 (2003).
Howard, A. D., Verghese, P. B., Arrese, E. L. & Soulages, J. L. The beta-subunit of ATP synthase is involved in cellular uptake and resecretion of apoA-I but does not control apoA-I-induced lipid efflux in adipocytes. Mol. Cell Biochem. 348, 155–164 (2011).
Zhao, Z., Carey, J. R. & Li, Z. The global epidemic of Bactrocera pests: mixed-species invasions and risk assessment. Annu. Rev. Entomol. 69, 219–237 (2024).
Vicoso, B. & Bachtrog, D. Numerous transitions of sex chromosomes in Diptera. PLoS Biol. 13, e1002078 (2015).
Kotov, A. A., Bazylev, S. S., Adashev, V. E., Shatskikh, A. S. & Olenina, L. V. Drosophila as a model system for studying of the evolution and functional specialization of the Y chromosome. Int. J. Mol. Sci. 23, 4184 (2022).
Roy, V. et al. Evolution of the chromosomal location of rDNA genes in two Drosophila species subgroups: ananassae and melanogaster. Heredity (Edinb.) 94, 388–395 (2005).
Benton, R. Multigene family evolution: perspectives from insect chemoreceptors. Trends Ecol. Evol. 30, 590–600 (2015).
Morillon, A. & Gautheret, D. Bridging the gap between reference and real transcriptomes. Genome Biol. 20, 112 (2019).
Liu, H., Chen, Z. S., Zhang, D. J. & Lu, Y. Y. BdorOR88a modulates the responsiveness to methyl eugenol in mature males of Bactrocera dorsalis (Hendel). Front Physiol. 9, 987 (2018).
Allwood, A., Vueti, E., Leblanc, L. & Bull, R. Eradication of introduced bactrocera species (Diptera: Tephritidae) in nauru using male annihilation and protein bait application techniques. In: Turning the Tide: The Eradication of Invasive Species 19–25 (IUCN Species Specialist Group, 2002).
Frenkel-Morgenstern, M. et al. ChiTaRS: a database of human, mouse and fruit fly chimeric transcripts and RNA-sequencing data. Nucleic Acids Res. 41, D142–D151 (2013).
Zhang, Y. et al. Chimeric transcript generated by cis-splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov. 2, 598–607 (2012).
Mika, K. et al. Olfactory receptor-dependent receptor repression in Drosophila. Sci. Adv. 7, eabe3745 (2021).
Lukhtanov, V. A. & Pazhenkova, E. A. Diversity and evolution of telomeric motifs and telomere DNA organization in insects. Biol. J. Linn. Soc. 140, 536–555 (2023).
Wright, C. J., Stevens, L., Mackintosh, A., Lawniczak, M. & Blaxter, M. Comparative genomics reveals the dynamics of chromosome evolution in Lepidoptera. Nat. Ecol. Evol. 8, 777–790 (2024).
Peccoud, J., Loiseau, V., Cordaux, R. & Gilbert, C. Massive horizontal transfer of transposable elements in insects. Proc. Natl Acad. Sci. USA 114, 4721–4726 (2017).
Sproul, J. S. et al. Analyses of 600+insect genomes reveal repetitive element dynamics and highlight biodiversity-scale repeat annotation challenges. Genome Res. 33, 1708–1717 (2023).
Coluzzi, M., Sabatini, A., della Torre, A., Di Deco, M. A. & Petrarca, V. A polytene chromosome analysis of the Anopheles gambiae species complex. Science 298, 1415–1418 (2002).
Schaeffer, S. W. Muller “Elements” in Drosophila: how the search for the genetic basis for speciation led to the birth of comparative genomics. Genetics 210, 3–13 (2018).
Ranz, J. M. et al. Principles of genome evolution in the Drosophila melanogaster species group. PLoS Biol. 5, e152 (2007).
Freeling, M. Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60, 433–453 (2009).
Panchy, N., Lehti-Shiu, M. & Shiu, S. H. Evolution of gene duplication in plants. Plant Physiol. 171, 2294–2316 (2016).
Kuzmin, E. et al. Exploring whole-genome duplicate gene retention with complex genetic interaction analysis. Science 368, eaaz5667 (2020).
Maere, S. et al. Modeling gene and genome duplications in eukaryotes. Proc. Natl Acad. Sci. USA 102, 5454–5459 (2005).
Cheng, F. et al. Gene retention, fractionation and subgenome differences in polyploid plants. Nat. Plants 4, 258–268 (2018).
Li, Z., Tiley, G. P., Rundell, R. J. & Barker, M. S. Reply to Nakatani and McLysaght: analyzing deep duplication events. Proc. Natl Acad. Sci. USA 116, 1819–1820 (2019).
Guo, S. & Kim, J. Molecular evolution of Drosophila odorant receptor genes. Mol. Biol. Evol. 24, 1198–1207 (2007).
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669 (2018).
Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. Imeta 2, e107 (2023).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Cheng, H. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 40, 1332–1335 (2022).
Nurk, S. et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305 (2020).
Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37, 4572–4574 (2021).
Durand, N. C. et al. Juicer Provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Jain, C., Rhie, A., Hansen, N. F., Koren, S. & Phillippy, A. M. Long-read mapping to repetitive reference sequences using Winnowmap2. Nat. Methods 19, 705–710 (2022).
Wang, L., Feng, Z., Wang, X., Wang, X. & Zhang, X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010).
Wick, R. R., Schultz, M. B., Zobel, J. & Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352 (2015).
Rautiainen, M. & Marschall, T. GraphAligner: rapid and versatile sequence-to-graph alignment. Genome Biol. 21, 253 (2020).
Lin, J. et al. SVision: a deep learning approach to resolve complex structural variants. Nat. Methods 19, 1230–1233 (2022).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Ayad, L. A. & Pissis, S. P. MARS: improving multiple circular sequence alignment using refined sequences. BMC Genomics 18, 86 (2017).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European molecular biology open software suite. Trends Genet 16, 276–277 (2000).
Gel, B. & Serra, E. karyoploteR: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 33, 3088–3090 (2017).
Vollger, M. R., Kerpedjiev, P., Phillippy, A. M. & Eichler, E. E. StainedGlass: interactive visualization of massive tandem repeat structures with identity heatmaps. Bioinformatics 38, 2049–2051 (2022).
Zhang, Y., Chu, J., Cheng, H. & Li, H. De novo reconstruction of satellite repeat units from sequence data. Genome Res. 33, 1994–200 (2023).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Huang, N. & Li, H. compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics 39, btad595 (2023).
Chen, Q., Yang, C., Zhang, G. & Wu, D. GCI: a continuity inspector for complete genome assembly. Bioinformatics 40, btae633 (2024).
Chen, C. et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Niknafs, Y. S., Pandian, B., Iyer, H. K., Chinnaiyan, A. M. & Iyer, M. K. TACO produces robust multisample transcriptome assemblies from RNA-seq. Nat. Methods 14, 68–70 (2017).
Gremme, G., Brendel, V., Sparks, M. E. & Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Inf. Softw. Technol. 47, 965–978 (2005).
Hoff, K. J. & Stanke, M. Predicting Genes in Single Genomes with AUGUSTUS. Curr. Protoc. Bioinform. 65, e57 (2019).
Pertea, G. & Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Res 9 https://doi.org/10.12688/f1000research.23297.2 (2020).
Aramaki, T. et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252 (2020).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Hallgren, J. et al. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv, Preprint at https://doi.org/10.1101/2022.04.08.487609 (2022).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Vasimuddin, M., Misra, S., Li, H. & Aluru, S. In: 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS) 314–324 (IEEE).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 (2019).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl Acad. Sci. USA 117, 9451–9457 (2020).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Hoede, C. et al. PASTEC: an automatic transposable element classification tool. PLoS ONE 9, e91929 (2014).
Yan, H., Bombarely, A. & Li, S. DeepTE: a computational method for de novo classification of transposons with convolutional neural network. Bioinformatics 36, 4269–4275 (2020).
Marcais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944 (2018).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Steinegger, M. & Soding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Sun, P. et al. WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol. Plant 15, 1841–1851 (2022).
Lovell, J. T. et al. GENESPACE tracks regions of interest and gene copy number variation across multiple genomes. Elife 11, e78526 (2022).
Yuan, J. et al. Protocols for CRISPR/Cas9 Mutagenesis of the Oriental Fruit Fly Bactrocera dorsalis. J. Vis. Exp. https://doi.org/10.3791/64195 (2022).
Xie, S., Shen, B., Zhang, C., Huang, X. & Zhang, Y. sgRNAcas9: a software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLoS ONE 9, e100448 (2014).
Xu, J. et al. Regulation of olfactory-based sex behaviors in the silkworm by genes in the sex-determination cascade. PLoS Genet. 16, e1008622 (2020).
Acknowledgements
This work was funded by the Shenzhen Science and Technology Program (Grant No. KCXFZ20240903093859009, KQTD20180411143628272), special funds for science technology innovation and industrial development of Shenzhen Dapeng New District (Grant No. PT202101-02), the National Key Research and Development Program of China (Grant No. 2022YFD1700200), and the IAEA Coordinated Research Project D41031. We thank Professor Quan Wang of the Agricultural Genomics Institute, Chinese Academy of Agricultural Sciences, for his generous guidance and assistance throughout the karyotype FISH experiment.
Author information
Authors and Affiliations
Contributions
G.R.W. and P.C. supervised and funded the project; W.L. and Q.L. designed the project. W.L., Q.W., J.Z., L.Y., and Y.Y.L. performed all experiments. Q.L., W.F.L., J.J. assembled the genome. Q.L., W.F.L., and Q.W., analyzed the data. Q.L. and W.L. wrote the manuscript with intellectual input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, W., Lin, Q., Wang, Q. et al. Telomere-to-telomere genome assembly of the Dipteran Bactrocera dorsalis from a single individual. Nat Commun 16, 10861 (2025). https://doi.org/10.1038/s41467-025-65870-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65870-1
