
Abstract
Migraine is a complex neurological disorder with substantial heritability, yet genome-wide association studies (GWAS) have explained only a fraction of its genetic component. We developed InsightGWAS, a Transformer-based model, to enhance genetic discovery for migraine by integrating functional annotations and leveraging transfer learning from GWAS datasets of major depressive disorder (MDD). Applying InsightGWAS to migraine GWAS datasets comprising 53,109 cases and 230,876 controls, we identified 293 previously unreported loci, influencing genes such as CACNA1D, HTR3C, and NLGN1, respectively. Furthermore, two loci rs4320030 (SCN11A) and rs5763529 (HORMAD2) were validated in independent sequencing studies, demonstrating the model’s precision in uncovering migraine-associated loci. Compared to traditional GWAS results, enrichment analyses of InsightGWAS-predicted loci uncovered new signaling pathways, including nitrogen compound metabolism and cation binding, offering novel insights into the metabolic and ionic mechanisms underlying migraine susceptibility. These findings demonstrate the impact of InsightGWAS in complementing conventional approaches and advancing our understanding of migraine genetics.
Similar content being viewed by others

Introduction
Migraine is a highly heritable neurological disorder, with estimates of heritability in the range of 30% to 50%1. Recent genome-wide association studies (GWAS) have made substantial progress in elucidating the genetic architecture of migraine by identifying numerous loci associated with the condition2,3,4. Complementary approaches combining GWAS with transcriptome-wide association studies (TWAS)5,6,7, expression quantitative trait loci (eQTL) and protein-level Mendelian randomization analyses have further prioritized causal genes and tissue-specific mechanisms in migraine pathogenesis8,9. Despite these advancements, current GWAS results explain only a fraction of the heritability. To comprehensively elucidate the genetic architecture underlying migraine, significantly larger sample sizes are needed to detect variants with smaller effect sizes that may contribute to disease susceptibility. However, expanding cohort sizes requires extensive efforts in both sample collection and data processing, as well as substantial financial investments in genotyping and sequencing. These challenges limit the feasibility and efficiency of traditional GWAS and may therefore hinder further elucidation of the genetic underpinnings of migraine.
Mental disorders, including depression, bipolar disorder, and schizophrenia, exhibit robust epidemiological and genetic associations with migraine10,11,12. Epidemiological studies consistently report elevated comorbidity rates between migraine and these mental health conditions, suggesting shared underlying mechanisms11. Furthermore, genetic correlation analyses support this hypothesis by revealing significant overlaps in genetic risk factors between migraine and various psychiatric disorders12,13,14. These findings indicate a complex interplay between neurological and psychological factors in the etiology of migraine. Building on these insights, researchers have employed novel statistical methods such as cross-trait analyses to successfully unravel the shared genetic basis of migraine and mental disorders12. By integrating genome-wide association data from both migraine and psychiatric conditions, these methods not only enhance the power to detect shared genetic factors but also provide a more comprehensive understanding of the biological mechanisms linking migraine and mental health disorders.
Recently, the application of deep learning techniques has opened new avenues for enhancing gene discovery based on GWAS results15,16,17,18. By integrating functional annotations from genomic, transcriptomic, and epigenomic evidence into GWAS analyses, these approaches prioritize genetic variants that are more likely to play a crucial role in disease biology. Such integration not only boosts the statistical power of GWAS but also provides deeper insights into the biological mechanisms driving complex traits. Furthermore, transfer learning—a technique where a model trained on one task is adapted to another—offers a promising strategy for applying insights from mental disorder genetics to the study of migraine.
In this study, we aimed to investigate the genetic architecture of migraine using both cross-trait GWAS analysis and deep learning techniques. By examining the genetic correlations between migraine and multiple mental disorders, we jointly analyzed the largest available summary statistics for migraine and Major Depressive Disorder (MDD), which was selected as the primary cross-trait phenotype due to its strong genetic correlation with migraine, large GWAS sample size, and extensive catalog of genome-wide significant loci. Leveraging this overlap, we aimed to uncover pleiotropic loci shared between the two disorders. In parallel, we applied transfer learning by training Transform-based deep learning models on MDD data to enhance the detection of migraine-associated loci. By integrating functional insights from multi-omics public data with these advanced methodologies, we uncovered multiple novel genetic loci, thereby deepening our understanding of the biological pathways underlying migraine pathophysiology.
Results Genetic correlations between migraine and mental disorders
To investigate the shared genetic basis between migraine and several mental disorders, we performed linkage disequilibrium score regression (LDSC) using GWAS summary statistics for migraine and six psychiatric conditions (Table S1), including major depressive disorder (MDD), bipolar disorder (BIP), schizophrenia (SCZ), attention-deficit/hyperactivity disorder (ADHD), and anxiety disorders (ANX). The genetic correlations (rg) between migraine and each disorder are presented in Table 1.
The most significant genetic correlation was observed between migraine and MDD (rg = 0.29, P = 1.78 × 10-33), indicating a substantial overlap in their genetic architectures. This strong association aligns with epidemiological evidence of high comorbidity and reinforces the hypothesis that migraine and depression share common biological pathways. Additionally, significant genetic correlations were identified between migraine and ADHD (rg = 0.2, P = 5.33 × 10-11) and ASD (rg = 0.12, P = 0.0029), although these associations were less pronounced compared to MDD. Collectively, these findings indicate the shared genetic underpinnings of migraine and psychiatric disorders, with the strongest overlap observed for MDD, suggesting convergent mechanisms that may contribute to their frequent co-occurrence.
Cross-trait meta-analysis between migraine and mental disorders
To further investigate the shared genetic architecture between migraine and MDD, we performed cross-trait meta-analyses using two complementary approaches: multi-trait analysis of GWAS (MTAG)19 and conditional false discovery rate (condFDR)20. Both methods leverage pleiotropic effects to enhance statistical power for detecting shared loci associated with migraine and MDD. Using MTAG, we identified 31 pleiotropic loci associated with migraine, including two novel loci previously reported in GWAS of depression (Table S2)21,22. Loci with a linkage disequilibrium (LD) r2 less than 0.05 with previously reported loci associated with migraine were considered novel. The first novel locus, rs77960, is located within the NUDT12 gene, which has been implicated in multiple psychiatric disorders23,24. The second, rs7140660, resides within the PROX2 gene, which is involved in regulating mechanosensory neurons and has been linked to neurological processes, including esophageal motility25. In contrast, the condFDR method, applied with a cfdr12 threshold of <0.01, did not identify any additional novel loci for migraine (Table S3). To assess whether other psychiatric phenotypes could enhance migraine locus discovery through shared polygenic architecture, we additionally performed cross-trait analyses using ADHD, ASD, ANX, BIP and SCZ. However, neither MTAG nor condFDR identified any novel migraine-associated loci from these analyses (Tables S4-S13). These findings suggest that traditional methods may be approaching their detection ceiling. Uncovering new loci relevant to migraine may require either more extensive cohorts or advanced analytic techniques.
Developing the transformer-based deep learning model using MDD GWAS data
Inspired by the previously established DeepGWAS16, which models genetic associations using a multi-layer neural network, we developed InsightGWAS by integrating an expanded set of genetic features and incorporating targeted modifications to improve predictive accuracy. InsightGWAS models the probability of variant association with a phenotype of interest by utilizing a transformer-based neural network that processes a set of input features. Each genetic variant is treated as a distinct observation, represented by a carefully selected feature set designed to capture relevant genetic signals. These features include GWAS summary statistics, population-level genetic metrics, and functional annotations relevant to neurological traits (see Methods).
To address challenges in defining true binary labels for variant associations, we implemented a two-step labeling strategy leveraging GWAS datasets with differing statistical power. Association labels were derived from a large MDD GWAS dataset, while input features were obtained from a smaller, independent GWAS with lower power. Following training on the MDD dataset, InsightGWAS was adapted to migraine through transfer learning, enabling cross-trait insights from the high-powered MDD analysis to enhance variant detection in the smaller migraine dataset (Fig. 1). Unlike traditional GWAS methods that rely on statistical tests to generate association p-values, InsightGWAS does not perform hypothesis testing. Instead, it functions as a post-GWAS variant prioritization framework, using machine learning to re-rank SNPs based on their predicted relevance to the trait of interest. This approach allows InsightGWAS to operate effectively even when statistical power is limited and enables the identification of potentially meaningful loci that fall below genome-wide significance in conventional analyses. As such, InsightGWAS complements standard GWAS by providing a probabilistic prioritization of variants rather than replacing statistical association testing.

Training and Inference Pipeline (left panel): The pipeline includes training and inference stages. A baseline model is trained using MDD GWAS data and fine-tuned with migraine GWAS data through transfer learning. The fine-tuned model is then used to identify enhanced SNPs in the migraine GWAS dataset. Model Architecture (right panel): The model architecture is based on a Transformer encoder, designed to capture nonlinear relationships among GWAS features. The architecture includes input embedding, positional encoding, and multi-head attention layers, followed by a fully connected layer with a sigmoid activation function for binary classification of variant associations. Source: The MDD and migraine icons are from vecteezy.com, available under a Free License.
Compared to DeepGWAS, InsightGWAS introduces key architectural and conceptual innovations that improve variant prioritization in underpowered GWAS settings. While DeepGWAS integrates genetic features using a multi-layer perceptron, it is limited in capturing complex nonlinear dependencies and relies on undersampling to address class imbalance, which can lead to information loss and reduced model generalizability. In contrast, InsightGWAS employs a Transformer-based architecture with multi-head self-attention, which enables the model to learn context-dependent relationships among input features by attending to their pairwise interactions. This design allows the integration of complex nonlinear dependencies between statistical and functional annotations, rather than treating them as independent predictors. To empirically assess feature contributions, we implemented a permutation-based pairwise interaction test (Fig. S1). Specifically, we first evaluated the model’s baseline performance on the validation set, then measured how much predictive accuracy (area under the ROC curve (AUC) and average precision (AP)) dropped when shuffling the values of individual features, and finally repeated the procedure while shuffling two features simultaneously. Interaction effects were quantified by comparing the paired drop with the sum of the two individual drops, with a greater-than-expected decline interpreted as evidence of nonlinear, non-additive interactions. This analysis revealed that the strongest interactions occur among GWAS statistical features, while regulatory and QTL annotations contribute primarily marginal effects. This pattern is expected, as the summary statistics (Beta and P-values) are the only features directly reflecting migraine biology and therefore anchor the model to migraine-specific signals. Other annotations, such as eQTLs and sc-eQTLs, serve in a complementary role, helping to distinguish between borderline statistical signals that may arise by chance and those with greater functional plausibility, particularly when observed in brain tissues or neuronal cell types relevant to migraine. In line with the ablation experiments, removal of GWAS statistical features produced the largest performance decline (AUC = 0.9869 | Accuracy = 0.9634). Exclusion of position features (AUC = 0.9968 | Accuracy = 0.9765), molecular QTLs (AUC = 0.9987 | Accuracy = 0.9723), and regulatory/OCR annotations (AUC = 0.9986 | Accuracy = 0.9827) also reduced performance, indicating that the model does not rely solely on P-values. Rather, the Transformer flexibly integrates diverse biological signals, while the dominant predictive power remains concentrated in GWAS-derived features.
The choice of MDD as the source trait for transfer learning was grounded in both biological and statistical considerations. Among a range of psychiatric disorders evaluated, MDD exhibited the strongest genetic correlation with migraine. In addition, the MDD GWAS included 170,756 cases and 329,443 controls, yielding 102 genome-wide significant loci and approximately 4600 associated variants for training. In contrast, other psychiatric traits such as ADHD and ASD demonstrated weaker genetic correlations with migraine and were based on smaller GWAS sample sizes (e.g., ADHD: 38,691 cases; ASD: 18,381 cases), with fewer genome-wide significant loci and markedly fewer positive training variants (1428 for ADHD and 93 for ASD, compared to 1834 for migraine). These limitations rendered them suboptimal as source phenotypes for effective model pretraining. Furthermore, as confirmed in our supplementary analyses, cross-trait GWAS and deep learning using ADHD or ASD did not yield any additional migraine-associated loci. Together, these results support the selection of MDD as a statistically robust and biologically relevant source trait for enhancing migraine variant detection through transfer learning.
Comparative assessment of transformer and conventional models
To assess the performance of the Transformer-based model (InsightGWAS) against traditional machine learning approaches in GWAS analysis, we conducted a series of comparative experiments with classical models, including neural networks (DeepGWAS), logistic regression, XGBoost, and ridge regression, using the MDD dataset. Each model was trained with consistent input features to ensure comparability across approaches (see Methods). For the neural network model, we used a multi-layer perceptron (MLP) architecture similar to DeepGWAS, comprising 14 fully connected layers with ReLU activation to capture nonlinear feature interactions effectively.
Experimental results showed that the Transformer model (InsightGWAS) outperformed traditional approaches in managing data noise and class imbalance, as demonstrated by ROC and DET curve analyses (Fig. 2). Specifically, ROC analysis revealed superior performance of InsightGWAS over comparator models across both sensitivity and specificity metrics. To further validate model robustness and prevent overfitting, we implemented bootstrap resampling, which consistently confirmed the enhanced performance of InsightGWAS (Fig. S2). The model’s attention mechanism efficiently minimized noise, facilitating accurate identification of disease-associated SNPs. DET curves further underscored its robustness, particularly in low false positive rate (FPR) regions where conventional models exhibited limitations.

a Zoomed-in ROC Curves: the Receiver Operating Characteristic (ROC) curves demonstrate the true positive rate (TPR) versus the false positive rate (FPR) for various models, including Ridge Regression, Transformer (InsightGWAS), Neural Networks (DeepGWAS), Logistic Regression, and XGBoost. The Transformer model shows superior performance with a sharper curve indicating higher sensitivity and specificity. b Zoomed-in DET Curves: the Detection Error Tradeoff (DET) curves illustrate the tradeoff between false negative rate (FNR) and false positive rate (FPR) for the same set of models. The Transformer model exhibits the lowest error rates, emphasizing its robustness for SNP prioritization. c t-SNE projections of prediction results from different models. Compared to traditional models, the Transformer model demonstrates a clearer separation between positive (Label 1) and negative samples (Label 0).
Transfer learning enhances migraine GWAS via a transformer-based framework
We next applied transfer learning to adapt the trained model for migraine association analysis. Transfer learning is particularly well-suited for scenarios where the target phenotype has limited statistical power but shares partial structure with a better-powered source phenotype. In our framework, the Transformer-based InsightGWAS model was first pretrained on the MDD GWAS, which offers a large sample size and extensive association signals, to learn generalizable patterns linking variant-level features with association likelihood. To adapt the model to migraine, we then conducted a fine-tuning step using two large-scale migraine GWAS datasets2,3. During this stage, model parameters were updated with a reduced learning rate (0.0001), allowing gradual adjustment while preserving relevant information learned from MDD. This two-phase design enables the model to transfer useful statistical and biological representations while remaining flexible to migraine-specific signals.
The migraine dataset was split into training (80%) and validation (20%) sets. To assess the benefit of transfer learning, we compared two models trained under identical conditions: a baseline Transformer trained from scratch on migraine data only, and a transfer model initialized with pretrained weights from MDD GWAS. Both models were trained for 20 epochs with the same learning rate and training-validation split. The transfer model achieved a validation accuracy of 99.37%, consistently outperforming the baseline model (Fig. S3). These results highlight the advantage of MDD-based pretraining in accelerating convergence and stabilizing model performance. To evaluate the model’s performance on the full set of ~7.5 million variants, we applied a stringent decision threshold to prioritize specificity and reduce false positives: variants were predicted as associated only if assigned a probability ≥ 99%. This resulted in 682 true positives and only 30 false positives, yielding a low false positive rate of ~0.0004% (30/7,500,000). These results align with expectations and demonstrate the model’s robustness and high specificity in identifying migraine-associated loci through cross-trait transfer learning.
Enhancements were conducted on the largest available migraine GWAS dataset, using summary statistics from Hautakangas et al.2, excluding the 23andMe data due to policy restrictions. As shown in Fig. 3a, there is substantial overlap between significant SNPs (P < 5 × 10⁻⁸) from the no-23andMe Hautakangas dataset and those predicted by InsightGWAS. Of the 13,585 SNPs predicted by InsightGWAS, 1820 significant SNPs were successfully identified, with only 14 missed. These results demonstrate InsightGWAS’s capacity to detect new associations while maintaining high sensitivity for known loci, resulting in an approximate eightfold increase in SNP coverage through transfer learning.

(a) Overlap Before Clumping: Venn diagram showing the overlap of migraine-associated SNPs from the full GWAS of Hautakangas et al. (“ALL”), the dataset excluding 23andMe (“no23andme”), and SNPs predicted by InsightGWAS. (b) overlap after clumping: Venn diagram showing the overlap of independent signals (SNPs after clumping to remove linkage disequilibrium).
InsightGWAS identifies novel migraine risk loci with functional relevance
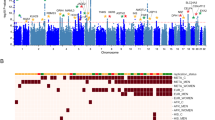
To further refine the results and isolate independent GWAS signals, we applied PLINK’s clumping procedure to remove SNPs in LD with r² greater than 0.05. This analysis yielded 367 independent GWAS signals, including all 41 significant signals in the no-23andMe Hautakangas GWAS (48,975 cases and 540,381 controls). We then compared our results to the 157 independent GWAS signals from the full Hautakangas GWAS (102,084 cases and 771,257 controls)2, which includes the 23andMe data. Beyond the 41 signals from the no-23andMe subset, InsightGWAS successfully predicted additional 53 signals reported by Hautakangas et al. demonstrating the model’s robustness in identifying novel migraine-associated loci (Figs. 3b and 4). We identified 293 loci in low linkage disequilibrium (r² <0.05) with previously reported loci in migraine, suggesting they represent novel signals. Notably, many of the newly identified variants by InsightGWAS are located near gene regions potentially linked to migraine pathophysiology including rs9311507 (CACNA1D), rs12629252 (HTR3C) and rs35198830 (NLGN1), which are associated with calcium channel signaling, serotonergic pathways, and synaptic plasticity, respectively (Fig. 4b). A comprehensive list of additional candidate genes is provided in Supplementary Data 1 and visualized in Fig. S4, with further discussion in the main text (see Discussion). Furthermore, a recent large-scale sequencing study on migraine identified 13 novel loci26. Of these, four were located within established GWAS signal regions, suggesting that certain genomic regions may contain both common and rare variants contributing to migraine susceptibility. Notably, among the remaining nine loci, rs4320030 (SCN11A) and rs5763529 (HORMAD2) fell within genomic regions newly predicted by InsightGWAS. Interestingly, variant rs4320030 is in strong LD (R2 = 0.97) with the top SNP rs33985936 predicted by InsightGWAS near SCN11A, further supporting the involvement of the SCN11A locus in migraine genetics. Similarly, rs5763529, located upstream of HORMAD2 shows moderate LD (R2 = 0.97) with InsightGWAS-predicted rs5753008, suggesting that the genomic region surrounding HORMAD2 may contribute to migraine susceptibility.

a Manhattan plot for the migraine GWAS from Hautakangas et al. (excluding 23andMe data), highlighting variants predicted by InsightGWAS in orange. b Manhattan plot for independent signals (clumped SNPs) predicted by InsightGWAS, with key loci highlighted in orange. Genes associated with these key loci are functionally annotated and categorized into Neurotransmitter Signaling and Synaptic Regulation (green), Neurodevelopment and Synaptic Connectivity (purple), and Ion Transport and Cellular Signaling (blue). Note: The association p-values displayed are from the original GWAS; no new statistical tests were applied for this visualization.
To further validate the loci identified by InsightGWAS, we leveraged a recently released and independent large-scale migraine GWAS from the VA Million Veteran Program (MVP)27. This resource includes summary statistics from two migraine phenotypes: (i) self-reported migraine headache (GCST90475546; 28,635 cases and 315,668 controls), and (ii) ICD-based migraine diagnosis (PheCode 340; GCST90475837; 31,836 cases and 437,667 controls). For each of the 293 novel loci prioritized by InsightGWAS, we examined the lead variant or a proxy in high linkage disequilibrium and assessed replication using a nominal significance threshold (P < 0.05). Despite differences in phenotype definitions and ascertainment across datasets, 117 loci showed replication in at least one of the two MVP migraine datasets, and 23 loci replicated in both (Supplementary Data 2). Additionally, four InsightGWAS-prioritized loci (rs6883741, rs61865730, rs5763529, and rs9607782) were also identified in another large-scale European migraine GWAS meta-analysis (comprising UKB, IHGC (Gormley), and GERA)4, including rs5763529 near HORMAD2, which additionally showed consistent effects in the MVP dataset, further supporting its role as a robust migraine risk locus. Although this meta-analysis included overlapping samples from UKB and IHGC used in our primary analysis (Hautakangas GWAS without 23andMe), the GERA cohort was independent, providing a partially independent replication and complementary supporting evidence. Taken together, these results provide additional support for the validity of many of the loci prioritized by InsightGWAS, further reinforcing the method’s ability to uncover robust migraine-associated genetic signals that generalize across diverse populations and study designs.
Gene set enrichment analysis
To gain biological insight into the newly identified loci, we next sought to explore their functional relevance at the gene and pathway levels. Using MAGMA, we prioritized candidate genes associated with risk loci by examining regions within 500 kb upstream and downstream of each locus. Genes with p-values below 0.01 were selected for subsequent pathway enrichment analysis to identify biological processes potentially implicated in migraine pathophysiology.
In our analysis of the 41 significant loci identified by Hautakangas et al.2, excluding 23andMe data, pathway enrichment was observed only in AMP deaminase activity. In contrast, analysis of the full set of 138 loci, which included 23andMe data, revealed a broader range of pathways potentially linked to migraine, including negative regulation of response to stimulus, regulation of neurogenesis, and steroid hydroxylase activity (Fig. 5a), aligning with current understanding of migraine’s neurobiological and hormonal underpinnings. Notably, analysis of the 387 loci predicted by InsightGWAS not only recapitulated these pathways but also uncovered additional pathways, including regulation of nitrogen compound metabolic processes and cation binding (Fig. 5b).

a Enrichment results from the significant loci identified in the migraine GWAS by Hautakangas et al., including 23andMe data. b Enrichment results from loci predicted by InsightGWAS. GO terms are color-coded to represent their functional categories: Biological Process (blue), Cellular Component (orange), and Molecular Function (green). Migraine-related terms are highlighted with striped bars.
Discussion
Our study demonstrates that advanced deep learning approaches can extend the reach of genetic discovery in migraine when conventional methods approach their limits. Through comparisons with traditional and cross-trait GWAS techniques, we show that transformer-based approaches effectively capture additional genetic signals. These findings suggest that deep learning methods may be crucial for fully harnessing existing datasets and achieving a more comprehensive understanding of migraine genetics.
The latest migraine GWAS demonstrate that increasing statistical power through larger sample sizes can accelerate the discovery of novel risk loci2,3. However, evidence from other complex diseases suggests that as GWAS approaches near peak efficiency, the discovery of additional loci demands exponentially larger datasets compared to earlier efforts28,29. For migraine, doubling or even tripling sample sizes may enhance statistical power, but poses logistical and financial challenges that are increasingly difficult to overcome. Cross-trait analysis offers an alternative approach by leveraging genetic correlations between migraine and other complex disorders, particularly MDD. Prior studies using cross-trait methods have successfully identified new loci by focusing on shared genetic signals between MDD and migraine, even with smaller datasets12,30. However, our application of cross-trait methods to the latest migraine GWAS yielded limited additional loci, suggesting that both conventional GWAS and cross-trait approaches may be approaching a plateau in locus discovery for migraine.
To overcome these challenges, we developed InsightGWAS, a Transformer-based deep learning model, to expand traditional GWAS methodologies for complex traits like migraine. By incorporating a broad range of genetic features and utilizing transfer learning from large, well-powered datasets such as those for MDD, InsightGWAS successfully identified both known and novel loci associated with migraine. In contrast to earlier neural network models, like DeepGWAS, which struggled with nonlinear dependencies and data imbalance, InsightGWAS leverages the Transformer’s attention mechanism to dynamically focus on relevant features, enhancing its ability to detect genetic signals within diverse genomic landscapes. The model’s performance is further elevated by the careful selection of an extensive set of features, including mQTLs and single-cell eQTLs (sc-eQTLs) that capture cell-type-specific regulatory activity in brain tissues, pathogenic annotations that integrate multiple predictive scores for variant functionality, and transcription factor binding sites (TFBS), ensuring robust detection of both coding and non-coding variant effects.
By leveraging InsightGWAS to enhance the latest migraine GWAS excluding 23andMe data (48,975 cases and 540,381 controls), we successfully predicted over half of the novel loci identified in the full GWAS (102,084 cases and 771,257 controls). This demonstrates the model’s ability to enhance the detection of genetic loci, achieving a sensitivity comparable to the statistical power gained from substantially larger GWAS sample sizes. InsightGWAS also identified several previously unreported gene loci potentially associated with migraine, including those related to ion channels and neurotransmitter pathways. Importantly, two novel signals identified by InsightGWAS, rs33985936 (SCN11A) and rs5753008 (upstream of HORMAD2), have been independently validated by a recent large-scale sequencing study on migraine26, further supporting the robustness and reliability of the model in detecting genetic factors underlying migraine susceptibility.
Among the novel ion channel genes identified by InsightGWAS, SCN11A plays a critical role in nociception by modulating neuronal excitability to regulate pain thresholds26. SCN9A was also previously linked to inherited pain disorders and migraine31, implying the shared contribution of sodium channel dysfunction to the hyperexcitability underlying migraine pathophysiology. Similarly, calcium channel genes CACNA1D and CACNA2D3, identified here, expand on the well-established role of CACNA1A in familial hemiplegic migraine2,32, suggesting broader involvement of calcium signaling pathways in synaptic transmission and neuronal plasticity. TRPM3, a member of the transient receptor potential channel family, contributes to sensory signal transduction and nociception, with supporting evidence linking it to migraine susceptibility33. SLC9B1, which encodes a sodium/hydrogen exchanger and has been repeatedly implicated in migraine through multiple cross-trait GWAS meta-analyses12,34, further demonstrating the role of ion channel dysregulation in migraine susceptibility and pain processing. Moreover, InsightGWAS detected genes implicated in neurotransmitter pathways, including GABBR2 and GABRR3, which encode components of the GABAergic system. Dysregulation of these genes may impair inhibitory signaling, exacerbating neuronal hyperexcitability—a hallmark of migraine35. Additionally, the identification of SLC1A2, a glutamate transporter gene, implies possible disruptions in excitatory neurotransmission, consistent with previous research linking glutamatergic dysfunction to migraine susceptibility and severity36. Furthermore, the discovery of HTR3C, encoding a subunit of the serotonin (a monoamine neurotransmitter) receptor, suggests the role of the serotonergic system in pain perception and central sensitization37. This aligns with prior evidence implicating HTR1A in migraine2. Collectively, these results point to a coordinated interplay of GABAergic, glutamatergic, and serotonergic pathways in migraine susceptibility.
This study also identifies loci associated with neurodevelopment and synaptic connectivity, suggesting the neuronal basis of migraine. For example, NLGN1 is critical for synapse formation and function38, while ROBO1 and SLIT3 regulate axon guidance and neuronal connectivity. These findings align with emerging evidence that migraine is not solely a vascular disorder but also involves intricate neuronal and synaptic mechanisms. In consistent with this, the identification of AUTS2 and LINGO2 further supports the hypothesis that neurodevelopmental pathways may influence susceptibility, potentially by modulating the sensitivity of pain-processing networks.
Gene enrichment analysis further reveals the biological significance of the loci identified by InsightGWAS. Compared to the latest GWAS findings, the expanded set of loci predicted by InsightGWAS uncovered additional pathways, including nitrogen compound metabolic processes and cation binding, both of which are intricately linked to migraine pathophysiology. Nitrogen compound metabolic processes are central to neurotransmitter synthesis and signaling, with nitrogenous molecules such as nitric oxide (NO) implicated in vasodilation and neurogenic inflammation39. Cation binding, involving the regulation of ions such as calcium, sodium, and potassium, is critical for neuronal excitability, synaptic function, and the cortical spreading depression associated with migraine attacks40. These findings demonstrate that InsightGWAS not only increases the number of detectable loci but also provides a more comprehensive and biologically informed perspective on the genetic architecture of migraine.
This study has limitations. Although the current version of InsightGWAS is developed and optimized in the context of migraine, we acknowledge that in the present study the performance gains from transfer learning over a baseline Transformer were modest. In fact, once functional features were carefully curated and expanded, the incremental benefit of adopting a Transformer architecture compared with deep neural networks or other classifiers was limited. The primary contribution of this work lies in demonstrating that feature integration substantially improves the precision of variant prioritization, and in showing that these findings are reliable through replication in the independent migraine GWAS from MVP. More broadly, the benefit of transfer learning may not generalize across all diseases, as its value depends on whether suitable genetically correlated traits exist and whether sufficiently large GWAS datasets are available. In some scenarios transfer learning may offer advantages, while in others a baseline Transformer may perform equally well or better. For this reason, we have provided both models, and future work will extend evaluations across phenotypes with diverse genetic architectures to better delineate when transfer learning is most beneficial.
To conclude, these results demonstrate the capability of InsightGWAS to uncover novel genetic loci and pathways relevant to migraine, expanding beyond the limits of traditional GWAS. By integrating advanced deep learning approaches and biologically informed features, InsightGWAS provides a powerful framework for deciphering the complex genetic architecture of migraine and offers valuable insights into its pathophysiology.
Methods
GWAS data
Summary statistics for two migraine GWAS datasets were obtained through application to the International Hedache Genetics Consortium. The first dataset, reported by Hautakangas et al. (2022)2, comprises 102,084 migraine cases and 771,257 controls from five large-scale studies: the International Headache Genetics Consortium (IHGC2016), 23andMe, UK Biobank, GeneRISK, and HUNT. The second dataset, described by Gormley et al. (2016)3, includes 59,674 cases and 316,078 controls from 22 non-overlapping case-control cohorts. Due to policy restrictions from 23andMe, their data were excluded from both GWAS datasets. Following this adjustment, the no-23andMe Hautakangas dataset consisted of 53,109 cases and 230,876 controls, while the Gormley dataset included 29,209 cases and 172,931 controls.
Summary statistics for several psychiatric disorders were obtained from the Psychiatric Genomics Consortium (PGC). For Major Depressive Disorder (MDD), two datasets were utilized: the first included 170,756 cases and 329,443 controls from a meta-analysis of UK Biobank and PGC cohorts21, while the second comprised 59,851 cases and 113,154 controls from PGC, deCODE, UK Biobank, and iPSYCH cohorts41. Bipolar Disorder (BIP) data included 41,917 cases and 371,549 controls42, and Attention-Deficit/Hyperactivity Disorder (ADHD) data included 38,691 cases and 186,843 controls43. Autism Spectrum Disorder (ASD) data consisted of 18,381 cases and 27,969 controls, integrating family-based and iPSYCH cohorts44. For Anxiety Disorders (ANX), summary statistics included 7016 cases and 14,745 controls45, while Schizophrenia (SCZ) data comprised 76,755 cases and 243,649 controls, representing the largest GWAS meta-analysis for this disorder to date28. Details of all datasets are summarized in Table S1.
Genetic correlation analysis
Genetic correlation (rg) between migraine and psychiatric traits was estimated using linkage disequilibrium score regression (LDSC) with GWAS summary statistics restricted to HapMap3 variants, as recommended46. SNP-based heritability of the analyzed traits was also calculated using LDSC. Precomputed linkage disequilibrium scores for HapMap3 SNPs, based on European ancestry individuals from the 1000 Genomes Project, were used in the analysis. SNP markers with an imputation INFO score below 0.9 were excluded to ensure data quality.
Genome-wide cross-trait analysis
To identify shared genetic loci between migraine and psychiatric traits, we employed two complementary approaches: Multi-Trait Analysis of GWAS (MTAG)19 and Conditional False Discovery Rate (CondFDR)20.
MTAG is a generalized meta-analysis approach that leverages genetic correlations across traits to increase power for detecting shared loci19. Variants were filtered to exclude non-common SNPs, strand-ambiguous SNPs, and duplicates. Pairwise genetic correlations were estimated using LD Score Regression (LDSC) to calibrate the variance-covariance matrix of the random effect component. MTAG then conducted a random-effects meta-analysis to compute SNP-level summary statistics specific to each trait. In this analysis, migraine was designated as the primary trait and MDD as secondary traits.
The conditional false discovery rate (condFDR) extends the standard FDR framework by leveraging genetic associations from a secondary phenotype (MDD) to re-rank the test statistics of a primary phenotype (migraine)20. This re-ranking process incorporates information from the secondary trait, allowing for more refined prioritization of genetic variants associated with the primary phenotype. By conditioning on the secondary phenotype, condFDR adjusts the significance threshold dynamically, enhancing the power to detect shared genetic loci while maintaining stringent control over false discoveries.
Transformer-based model
The InsightGWAS model employs a Transformer encoder architecture to unravel complex, nonlinear relationships between genetic features. This approach is particularly well-suited for genomic data, where interactions between features like sQTLs, eQTLs, and positional information play a pivotal role in understanding disease-associated loci. The encoder consists of two layers, each with four multi-head attention mechanisms and a feed-forward neural network. Input features are embedded and positionally encoded, enabling the model to consider both feature interactions and spatial relationships within the genome. The self-attention mechanism dynamically assigns importance to input features, allowing the model to focus on key genetic elements while accounting for their dependencies. Multi-head attention further enhances this capability by capturing diverse perspectives on feature relationships. The output of the encoder is passed through a fully connected layer and sigmoid activation to predict disease associations, bypassing the need for a decoder. This architecture enables efficient and robust analysis of genetic data, making it an ideal choice for GWAS applications. The model was optimized for genomic data by tuning the hidden dimensions (64 in this case) and leveraging layer normalization and residual connections to stabilize training. The implementation is publicly available on GitHub (https://github.com/ziangmeng/MA-MDD-Transformer-based-model-/tree/main). Further technical details, including model architecture, training parameters, and evaluation metrics, are provided in the Supplementary Methods.
Input features
The InsightGWAS model integrates a comprehensive set of features to prioritize migraine-associated loci effectively. Key predictors include GWAS summary statistics (effect sizes, p-values, sample sizes, and minor allele frequencies) and population genetics metrics, such as linkage disequilibrium (LD) scores derived from European ancestry samples in the 1000 Genomes Project. Functional annotations enrich the genetic interpretation, incorporating QTL data (e.g., brain-specific eQTLs and sQTLs from BrainMeta and GTEx datasets, mQTLs from large-scale meta-analyses, and single-cell eQTLs for brain cell types, Table S14) and epigenomic marks. These include open chromatin regions, transcription factor binding sites, and regulatory annotations from ENCODE, such as active enhancers, promoters, and repressive regions.
To ensure biological relevance, additional annotations highlight variants disrupting microRNAs, snoRNAs, and transcription factor binding sites, as well as those associated with neurological or psychiatric traits based on data from the GWAS Catalog and Open Targets. Pathogenicity metrics, including conservation scores (e.g., GERP + +), functional predictors (e.g., PolyPhen-2, SIFT, CADD), and variant impact assessments, provide further insights into potential deleterious effects. Together, these features enable InsightGWAS to systematically evaluate genetic variants, facilitating robust identification and interpretation of biologically significant loci. More details are provided in the Supplementary Methods.
Comparison with alternative methods
To assess the predictive performance of the Transformer-based model, we compared it with several established methods, including a neural network, XGBoost, logistic regression, and Ridge regression. The neural network, implemented using Keras, followed an architecture similar to that employed in DeepGWAS16, with densely connected layers and ReLU activation functions. The output layer utilized a sigmoid activation to estimate the probability of each variant being significantly associated with the trait. Training was conducted over 30 epochs with binary cross-entropy as the loss function, incorporating batch normalization to enhance convergence and model stability.
The XGBoost model, a widely used decision-tree-based ensemble algorithm, was configured to perform logistic regression within a gradient boosting framework. The model employed root mean square log error (RMSLE) as the evaluation metric, with early stopping applied to mitigate overfitting. Logistic regression, implemented with scikit-learn, served as a baseline classifier, modeling the probability of variant-trait associations. Ridge regression, also implemented via scikit-learn, incorporated L2 regularization to control overfitting, with optimal regularization strength determined through cross-validation.
Model performance was evaluated comprehensively using true positive rate (TPR), F1 score, and receiver operating characteristic (ROC) curves. These metrics allowed for a balanced assessment of sensitivity, precision, and overall classification efficacy, providing a robust framework for comparing the Transformer-based model with traditional and machine-learning approaches.
Transfer learning
The Transformer model employed a transfer learning approach to enhance prediction performance for migraine-associated loci. Initially, the model was pre-trained on a major depressive disorder (MDD) dataset, enabling it to learn complex feature patterns relevant to genetic associations. This pre-training phase provided a foundation for understanding generalizable genetic relationships. Subsequently, the model was fine-tuned on the migraine dataset to adapt to disease-specific features. During fine-tuning, the learning rate was set to 0.0001, and an early stopping strategy was implemented to prevent overfitting and ensure effective convergence. Input features in the migraine dataset were standardized, and the data was split into training and validation sets to monitor performance. Model parameters were updated iteratively through backpropagation to minimize the loss function, while validation loss and accuracy were recorded to track convergence and stability.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All datasets used in this study are publicly available. The GWAS summary statistics for migraine can be obtained upon application from the International Headache Genetics Consortium (IHGC). Summary statistics for mental disorders are available for direct download from the Psychiatric Genomics Consortium (PGC) website at: https://pgc.unc.edu/for-researchers/download-results/. Source data are provided with this paper.
Code availability
All code used in this study is publicly available at our GitHub repository (https://github.com/ziangmeng/MA-MDD-Transformer-based-model-)47. To support flexibility and reproducibility, we provide configurable training pipelines, pretrained model checkpoints, and documentation. Both the baseline (non-transfer) and transfer learning versions of the model are included to facilitate comparative analysis and transparent reporting.
References
Olofsson, I. A. Migraine heritability and beyond: A scoping review of twin studies. Headache 64, 1049–1058 (2024).
Hautakangas, H. et al. Genome-wide analysis of 102,084 migraine cases identifies 123 risk loci and subtype-specific risk alleles. Nat. Genet 54, 152–160 (2022).
Gormley, P. et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet 48, 856–866 (2016).
Choquet, H. et al. New and sex-specific migraine susceptibility loci identified from a multiethnic genome-wide meta-analysis. Commun. Biol. 4, 864 (2021).
Meyers, T. J. et al. Transcriptome-wide association study identifies novel candidate susceptibility genes for migraine. HGG Adv. 4, 100211 (2023).
Gui, J. et al. A cross-tissue transcriptome-wide association study reveals novel susceptibility genes for migraine. J. Headache Pain. 25, 94 (2024).
Ghaffar, A. & Nyholt, D. R. Integrating eQTL and GWAS data characterises established and identifies novel migraine risk loci. Hum. Genet 142, 1113–1137 (2023).
Xiong, Z. et al. Proteome-wide Mendelian randomization identified potential drug targets for migraine. J. Headache Pain. 25, 148 (2024).
Sun, X. et al. Multi-omics Mendelian randomization integrating GWAS, eQTL and pQTL data revealed GSTM4 as a potential drug target for migraine. J. Headache Pain. 25, 117 (2024).
Minen, M. T. et al. Migraine and its psychiatric comorbidities. J. Neurol. Neurosurg. Psychiatry 87, 741–749 (2016).
Antonaci, F. et al. Migraine and psychiatric comorbidity: a review of clinical findings. J. Headache Pain. 12, 115–125 (2011).
Bahrami, S. et al. Dissecting the shared genetic basis of migraine and mental disorders using novel statistical tools. Brain 145, 142–153 (2022).
Yang, Y. et al. Shared Genetic Factors Underlie Migraine and Depression. Twin Res Hum. Genet 19, 341–350 (2016).
Viudez-Martinez, A., Torregrosa, A.B., Navarrete, F. & Garcia-Gutierrez, M.S. Understanding the Biological Relationship between Migraine and Depression. Biomolecules 14 (2024).
Nicholls, H. L. et al. Reaching the End-Game for GWAS: Machine Learning Approaches for the Prioritization of Complex Disease Loci. Front Genet 11, 350 (2020).
Li, Y. et al. DeepGWAS: Enhance GWAS Signals for Neuropsychiatric Disorders via Deep Neural Network. Res Sq, (2023).
Lakiotaki, K. et al. Automated machine learning for genome wide association studies. Bioinformatics 39 (2023).
Mieth, B. et al. DeepCOMBI: explainable artificial intelligence for the analysis and discovery in genome-wide association studies. NAR Genom. Bioinform 3, lqab065 (2021).
Turley, P. et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet 50, 229–237 (2018).
Smeland, O. B. et al. Discovery of shared genomic loci using the conditional false discovery rate approach. Hum. Genet 139, 85–94 (2020).
Howard, D. M. et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352 (2019).
Thorp, J. G. et al. Symptom-level modelling unravels the shared genetic architecture of anxiety and depression. Nat. Hum. Behav. 5, 1432–1442 (2021).
Cross-Disorder Group of the Psychiatric Genomics Consortium. Electronic address, p.m.h.e. & Cross-Disorder Group of the Psychiatric Genomics, C Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 179, 1469–1482.e11 (2019).
Coleman, J. R. I. et al. The Genetics of the Mood Disorder Spectrum: Genome-wide Association Analyses of More Than 185,000 Cases and 439,000 Controls. Biol. Psychiatry 88, 169–184 (2020).
Lowenstein, E. D. et al. Prox2 and Runx3 vagal sensory neurons regulate esophageal motility. Neuron 111, 2184–2200.e7 (2023).
Bjornsdottir, G. et al. Rare variants with large effects provide functional insights into the pathology of migraine subtypes, with and without aura. Nat. Genet 55, 1843–1853 (2023).
Verma, A. et al. Diversity and scale: Genetic architecture of 2068 traits in the VA Million Veteran Program. Science 385, eadj1182 (2024).
Trubetskoy, V. et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022).
Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Yang, Y. et al. Molecular genetic overlap between migraine and major depressive disorder. Eur. J. Hum. Genet 26, 1202–1216 (2018).
Nyholt, D. R. et al. A high-density association screen of 155 ion transport genes for involvement with common migraine. Hum. Mol. Genet 17, 3318–3331 (2008).
de Boer, I., Hansen, J. M. & Terwindt, G. M. Hemiplegic migraine. Handb. Clin. Neurol. 199, 353–365 (2024).
Krivoshein, G., Tolner, E. A., Maagdenberg, A. V. D. & Giniatullin, R. A. Migraine-relevant sex-dependent activation of mouse meningeal afferents by TRPM3 agonists. J. Headache Pain. 23, 4 (2022).
Tasnim, S., Wilson, S. G., Walsh, J. P., Nyholt, D. R. & International Headache Genetics, C. Shared genetics and causal relationships between migraine and thyroid function traits. Cephalalgia 43, 3331024221139253 (2023).
Bell, T. et al. GABA and glutamate in pediatric migraine. Pain 162, 300–308 (2021).
Jeong, H. et al. Gene Network Dysregulation in the Trigeminal Ganglia and Nucleus Accumbens of a Model of Chronic Migraine-Associated Hyperalgesia. Front Syst. Neurosci. 12, 63 (2018).
Panconesi, A. Serotonin and migraine: a reconsideration of the central theory. J. Headache Pain. 9, 267–276 (2008).
Zhao, J. Y. et al. Activity-dependent Synaptic Recruitment of Neuroligin 1 in Spinal Dorsal Horn Contributed to Inflammatory Pain. Neuroscience 388, 1–10 (2018).
Akerman, S., Williamson, D. J., Kaube, H. & Goadsby, P. J. Nitric oxide synthase inhibitors can antagonize neurogenic and calcitonin gene-related peptide induced dilation of dural meningeal vessels. Br. J. Pharm. 137, 62–68 (2002).
Yan, J. & Dussor, G. Ion channels and migraine. Headache 54, 619–639 (2014).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet 50, 668–681 (2018).
Mullins, N. et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet 53, 817–829 (2021).
Demontis, D. et al. Genome-wide analyses of ADHD identify 27 risk loci, refine the genetic architecture and implicate several cognitive domains. Nat. Genet 55, 198–208 (2023).
Grove, J. et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet 51, 431–444 (2019).
Otowa, T. et al. Meta-analysis of genome-wide association studies of anxiety disorders. Mol. Psychiatry 21, 1391–1399 (2016).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015).
Meng, Z. et al. Transformer-Based Deep Learning Enhances Discovery in Migraine GWAS. MA-MDD-Transformer-based-model-. https://doi.org/10.5281/zenodo.17338730.
Acknowledgements
The study was supported by grants from the Special Funds of Taishan Scholar Project, China (tsqn202211224, X.C.), the National Natural Science Foundation of China (32270661, X.C.), Excellent Youth Science Fund Project (Overseas) of Shandong China (2023HWYQ-082, X.C.), the China Postdoctoral Science Foundation (2024M761870, Y.S.), Shandong Postdoctoral Science Foundation (SDCX-ZG-202400042, Y.S.) and Shandong Province Higher Education Institution Youth Innovation and Technology Support Program (2023KJ179, Y.S.). The Institutional Development Funds from the Children’s Hospital of Philadelphia to the Center for Applied Genomics (H.H). We thank the International Headache Genetics Consortium (IHGC) for providing access to migraine GWAS data.
Author information
Authors and Affiliations
Contributions
Conceptualization: X.C., H.H. Methodology: Z.M., Y.S., Y.J., X.W., Y.Z., X.L. Investigation: Z.M., Y.S., Y.J., X.W., Y.Z., X.L. Visualization: Z.M., Y.S. Formal analysis: Z.M., Y.S., Y.J., X.W. Data curation: Z.M., Y.S. Funding acquisition: X.C., H.H. Project administration: X.C., H.H. Supervision: X.C., H.H. Writing—original draft: Z.M., Y.S. Writing—review & editing: X.C., H.H. Z.M., and Y.S. contributed equally to this work. X.C., and H.H. jointly supervised this study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wonil Chung, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Meng, Z., Song, Y., Jiang, Y. et al. Transformer-based deep learning enhances discovery in migraine GWAS. Nat Commun 16, 11023 (2025). https://doi.org/10.1038/s41467-025-65991-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65991-7
