
Abstract
Colorectal cancer (CRC) is the fourth most common cancer and the third leading cause of cancer-related mortality worldwide, with incidence rising among younger populations. The significant clinical and economic burden highlights the need for minimally invasive technologies capable of detecting pre-malignant and early-stage disease. Although liquid biopsy approaches have advanced, they have not achieved sufficient performance for clinical adoption when compared with colonoscopy, the current diagnostic gold standard. CRC is a mucosal pathology, yet current diagnostic methods have not leveraged mucosal biology. Here we demonstrate the clinical utility of rectal mucus specimens, collected using a minimally invasive device in an outpatient setting, without bowel preparation. Through a hologenomic approach integrating host and microbial genomics, we identify genetic and epigenetic aberrations and perturbations in microbial communities that drive the detection of adenomatous polyps and CRC in rectal mucus. Hologenomic integration enables superior stratification of CRC by disease site and stage compared with single-omics methods. In summary, we demonstrate the clinical utility of rectal mucus sampling combined with hologenomic analysis as a translatable prospective tool for diagnostic application.
Similar content being viewed by others

Introduction
Colorectal cancer (CRC) is the fourth most common cancer and third most common cause of cancer-related mortality worldwide1. Higher incidence, particularly in younger populations2, is driving an increase in disease burden together with factors such as population aging and increasing obesity3. While overall CRC mortality rates continue to fall, higher incidence underlies increased mortality among people under 504.
Five-year survival rates for CRC are strongly correlated with stage at diagnosis, with 85–90% of patients diagnosed with Stages I–II surviving for five years or more, compared with 10–65% for Stages III–IV5. Additionally, decreased incidence and mortality among populations receiving preventative screening6,7 have prompted the expansion of screening programmes to younger groups8,9. This has engendered higher demand for screening, increasing the burden on healthcare systems10,11 that have traditionally relied upon primarily endoscopic and imaging-based techniques12, highlighting a need for a minimally invasive triage method. Population-scale deployment of quantitative faecal immunochemical testing (qFiT)13 in symptomatic cohorts has shown utility as a non-invasive colonoscopy qualification tool14, although debate remains about optimal thresholding in symptomatic and asymptomatic cohorts. Advanced imaging techniques, such as capsule endoscopy, have been trialled15,16 and adopted on a very limited scale17.
Recent advances have focused on developing methods that utilise blood-based specimens, collectively referred to as liquid biopsies. These assays involve various analytes, including circulating ensembles of tumour-associated cells (C-ETACs)18,19,20, circulating tumour cells (CTC)21, protein biomarkers22,23, cell-free DNA (cfDNA)24,25, and RNA26,27. Liquid biopsy targeting mutation and DNA methylation profiles in circulating tumour DNA (ctDNA) in blood has clinical utility in detecting, monitoring and guiding treatment of several cancers28,29,30,31. However, this technology is limited by the small quantities of pathology-derived material, hindering biomarker detection32,33 and leading to poor sensitivity in pre-malignant and early-stage disease34,35,36. Various mitigation measures have been employed to overcome this, including increased sample volumes and plasmapheresis. In parallel with improvements in detection technology, sample handling and processing play important roles ensuring target biomarkers are sampled and preserved in detectable quantities, allowing for robust and reproducible analyses37,38. Immune profiles that correlate with disease39,40 and treatment response41,42 represent another area with diagnostic and therapeutic potential.
Sample matrices with anatomical proximity to the tumour site (e.g., stool, urine, and peritoneal fluid), which are likely to contain higher concentrations of target analytes and biomarkers, have the potential to complement or supplant blood-based techniques for specific indications43,44,45. Tumour-proximal sampling enables analysis of biomarkers that are not readily detectable in a blood-based matrix. For instance, increasing evidence implicates the microbiome in colorectal disease46,47 and highlights its potential utility in cancer diagnosis48,49.
Efforts to develop a minimally-invasive, population-scale CRC screening tool have sought to address these issues and to improve performance via multiomics approaches, targeting genetic, epigenetic and biochemical markers50,51. Molecular biomarkers are long-established indicators of dysregulation, with many studies implicating genetic mutations and epigenetic marks in CRC. Moreover, the use of molecular markers has been shown to be cost-effective, deployable and have clinical utility in the field of diagnostics. Here we leverage the unique features of rectal mucus to assess host genomics and non-host prospective biomarkers from the microbiome, which are either underrepresented or not present in other biospecimens. In this study of 800 patients referred with lower gastrointestinal symptoms through the UK National Health Service (NHS) Suspected Colorectal Cancer (Two-Week Wait) pathway and patients with newly diagnosed colorectal adenocarcinoma (from the Colorectal Multidisciplinary team meeting) assessed in the outpatient setting, we exploit multiple omics to investigate the utility of rectal mucus as a novel clinical specimen for detecting genetic, epigenetic and microbial markers of pre-cancerous lesions (advanced adenomas defined as polyps > 10 mm in diameter or with high-grade dysplasia) and CRC.
Results
Study design and participant demographics
The OriCol™ sampling device (Supplementary Fig. 1) was used for rectal mucus retrieval from patients referred with suspected colorectal cancer in four NHS colorectal surgical outpatient clinics. Sampling was performed without any bowel preparation and provided the patient could tolerate a digital rectal examination (Supplementary Fig. 2). Data on the safe use of this device in the target population and the identification of exfoliated tumour material in the collected sample have been published52,53,54.
Eight hundred participants were enroled in the ORI-EGI-02 study (Supplementary Fig. 3). Figure 1b–i describe the demographics of 419 participants, the combined total of all participants analysed by each genomic assessment. Trial demographics for all 800 participants are detailed in Supplementary Fig. 4.

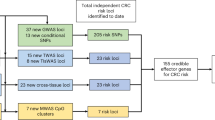
a PRISMA flow diagrams illustrating three sample-processing pathways (ecNGS, EM-seq, and whole-genome shotgun metagenomic sequencing) and the inclusion/exclusion process. Summary of study demographics, including biological sex (b), smoking status (c), BMI (d), age (e), Rockwood frailty score (f), and clinical category (g). CRC cases were subdivided by stage (h) and pathology site (i). E-Polyps + , Early-stage polyps; L-Polyps + , Late-stage polyps; C+ Other, Small bowel cancer; No CRC, Not diagnosed with CRC and not assessed by gold-standard examinations.
From this pool, three sub-cohorts were selected (Fig. 1a), weighted for CRC to increase analytical power. Sample aliquots were processed using three laboratory pipelines: error-corrected next-generation sequencing (ecNGS), enzymatic methyl sequencing (EM-seq), and whole-genome shotgun metagenomic sequencing.
Cohort sizes were determined by participant and sample inclusion criteria, where participants who had a complete clinical assessment, an OriCol™ sample successfully collected, and a complete colonoscopy were available for analysis. Additionally, samples were not included if biological material was not available, or where sample pre-processing quality controls (QC), such as DNA extraction QC criteria, were not met.
Once selected for processing and analysis, samples were further excluded if library preparation or sequencing QC criteria were not met (Methods).
Final sample inclusion yielded the following cohorts: ecNGS = 161, EM-seq = 208, shotgun metagenomics = 379. Eighty samples were common to the ecNGS and EM-seq pipelines, and 68 were common to all three pipelines. Demographics of analytical sub-cohorts (ecNGS, EM-seq and shotgun metagenomics) and intersecting analytical methods are illustrated in Supplementary Figs. 5–8 and Supplementary Data 1.
Clinical labels reported throughout this study are defined as follows. Early-stage polyps (E-Polyps + ) refers to polyps ≤10 mm in diameter and with low-grade dysplasia, hyperplasia or sessile serrated lesions. Late-stage polyps (L-Polyps + ) are >10 mm in diameter or with high-grade dysplasia. C+ Other refers to a diagnosis of small bowel cancer. No CRC refers to participants who were referred through the pathway but not assessed by gold-standard examinations and not diagnosed with CRC by the 12-month follow-up appointment.
Participant demographics (Fig. 1b–i) showed a balance of biological sex, with 50.2% male and 49.8% female. Among all participants, 56.6% had never smoked, 33.4% were ex-smokers and 10.0% were current smokers. Body mass index (BMI) skewed towards overweight categories, with 30.8% overweight (BMI 25–29.9 kg/m2). The distribution of CRC sites was 24.1% right-colon, 22.8% mid-colon (hepatic flexure to splenic flexure), 17.9% left-colon (descending and sigmoid), and 34.5% rectal.
Somatic mutation profiling reveals site-responsive disease signals in CRC tumour suppressors and oncogenes
Initial technology assessments were undertaken to determine the most appropriate methodologies for mutation profiling analysis. Methods employing error-corrected NGS with dual unique molecular identifiers were assessed, as these approaches use molecule-specific read consensus sequences such that low-frequency somatic mutations detected on both strands can be distinguished from sequencing errors and PCR errors associated with traditional NGS. Libraries prepared using the Duplex Sequencing Universal Kit (TwinStrand Biosciences, Seattle, USA) exhibited the highest proportion of sequencing reads usable for consensus sequence assembly and subsequent variant calling (Supplementary Fig. 9).
Fifty cancer-associated genes were assessed by ecNGS55 (Duplex Sequencing) and variant pathogenicity was annotated using the National Library of Medicine ClinVar database (release 20241223). Additionally, loss-of-function predictions were used to supplement ClinVar labelling for tumour suppressor genes (APC, TP53, FBWX7 and SMAD4). The dataset was filtered to include only variants with evidence of a pathogenic impact on cancer development. Twenty-five genes remained after filtering across all clinical groups.
Among the OriCol™ samples collected from CRC participants, the most frequently mutated genes were TP53 (mutated in 79% of CRC cases), FBXW7 (65%), KRAS (63%), ERBB2 (54%), APC (49%), BRAF (33%), PIK3CA (18%) and SMAD4 (15%). Thirty genes were not mutated in CRC cases. The median number of mutated genes per CRC was 4 (range = 0–10) (Fig. 2a).

a OncoPrint of CRC cases (n = 78) in the ecNGS cohort across the 25 genes which remained after filtering. The top horizontal bar is per-participant mutation burden and the right vertical bar is per-gene mutation count. b Hierarchically clustered heatmap of per-gene, per-participant maximum VAFs across the 161-participant ecNGS cohort for 25 genes that remained after filtering (control n = 47; CRC n = 78; early-stage polyps n = 19; late-stage polyps n = 7; small bowel cancer n = 1; no CRC n = 9). The bars along the bottom of the heatmap denote clinical category, pathology site, and cancer stage. E-Polyps + , Early-stage polyps; L-Polyps + , Late-stage polyps; C+ Other, Small bowel cancer; No CRC, Not diagnosed with CRC and not assessed by gold-standard examinations.
To compare observed and expected mutation frequencies, we used cBioPortal to curate a multistudy cohort (n = 1605) of CRC cases with tumour mutation data. In this cohort, the expected mutation frequencies for the genes reported above were: TP53 (60%), FBXW7 (15%), KRAS (35%), ERBB2 (5%), APC (61%), BRAF (14%), PIK3CA (22%) and SMAD4 (13%). For the 30 unmutated genes, the median expected frequency was 2% (range = 0–8%) (Supplementary Fig. 10; Supplementary Data 2). Where genes were mutated in both cohorts, a strong correlation (r2 = 0.91) was seen between expected and observed frequencies of mutated genes (Supplementary Fig. 10).
To examine the impact of background somatic mutations, we evaluated mutation burden after pathogenic filtering compared to controls. No clustering was observed when visualised by heatmap (Supplementary Fig. 11). We then calculated maximum variant allele frequency (mVAF) to give a measure of signal strength and visualised the resulting matrix of per-gene, per-participant mVAFs as a heatmap with hierarchical clustering (Fig. 2b). High mVAFs (particularly in APC, TP53, KRAS, and SMAD4) resulted in clustering of CRC and late-stage polyps cases towards each end of the heatmap. Control participants with low mVAFs generally clustered towards the centre, and early-stage polyps cases were distributed throughout. mVAFs for APC (Mann–Whitney–Wilcoxon test Benjamini–Hochberg-adjusted P = 0.0055) and TP53 (adjusted P = 0.0055) were significantly higher in CRC cases than controls (Supplementary Data 3), while no significant differences were found between polyps cases and controls (Supplementary Data 8 and 9).
Right-sided (proximal) CRC is more difficult to detect in stool-based liquid biopsies50,56. We therefore expected pathology sites nearer to sample collection to have stronger signal. To explore this, we selected the eight most frequently mutated genes (Fig. 2a). Per-gene, per-participant mVAFs for pathogenic variants detected in OriCol™ samples collected from CRC cases were ordered by site of cancer, followed by those for controls and plotted as a heatmap (Fig. 3a). mVAFs ≥0.03 are most frequently seen in rectal cancers and least among controls. The distribution of mVAFs by pathology site was plotted for each gene for CRC cases, polyps cases and controls (Fig. 3b–i). APC and TP53 showed the clearest gradients of high rectal mVAFs to low right-sided mVAFs. By contrast, ERBB2 and FBXW7 did not show a clear gradient, where mVAFs were comparable between clinical categories and pathology sites. Consistent with these trends, mVAFs for APC (Mann–Whitney–Wilcoxon test Benjamini–Hochberg-adjusted P = 0.00003) and TP53 (adjusted P = 0.001) were significantly higher in rectal CRC cases than controls (Supplementary Data 4). mVAFs for APC (adjusted P = 0.0431) were also higher in left-colon CRC cases than controls (Supplementary Data 5), whereas per-gene mVAFs were not significantly different in mid-colon and right-colon cases relative to controls after correcting for multiple testing (Supplementary Data 6 and 7). Together, these findings indicate that more pathology-derived material was collected when the pathology was closer to the biospecimen collection site (the rectum).

a Heatmap of per-gene, per-participant maximum VAFs for the eight most frequently mutated genes across CRC cases and controls, ordered by site of pathology (none (control) n = 47; unknown site CRC n = 1; right-colon CRC n = 14; mid-colon CRC n = 14; left-colon CRC n = 18; rectal CRC n = 31). b–i Per-gene boxplots of maximum VAFs for CRC cases, polyps cases and controls ordered by site of pathology (control n = 47; CRC n = 78; early-stage polyps n = 19; late-stage polyps n = 7; small bowel cancer n = 1; no CRC n = 9). E-Polyps + , Early-stage polyps; L-Polyps + , Late-stage polyps; C+ Other, Small bowel cancer; No CRC, Not diagnosed with CRC and not assessed by gold-standard examinations.
DNA hypermethylation in CRC associates with gene promoters, 5′ ends and CpG islands marked by transcriptionally active or bivalent chromatin in a rectal mucosa reference epigenome
We performed targeted enzymatic methyl sequencing (New England Biolabs, Ipswich, USA) using DNA extracted from OriCol™ samples. This technology uses enzymatic conversion of unmethylated cytosines to uracils, which are then converted to thymines during PCR, resulting in reduced DNA degradation and higher quality sequencing libraries compared to bisulfite sequencing57,58,59,60. In a preliminary technical assessment, each EM-seq library had an enzymatic conversion rate >99%, and libraries derived from the same genomic DNA sample clustered together when applying hierarchical clustering to a distance matrix derived from pairwise Spearman rank (rs) correlation coefficients calculated for DNA methylation levels across target CpG sites (Supplementary Fig. 12). These findings show consistent performance of EM-seq for DNA methylation profiling across different sample types, providing the basis for applying this technology to investigate the extent to which colorectal pathology-associated DNA methylation patterns are detectable in rectal mucus samples.
Target CpG sites were defined based on GRCh38 coordinates for loci in the Human Methylome Panel (Twist Bioscience, San Francisco, USA) and CpG islands (CGIs) in the UCSC Genome Browser database61 that overlap annotations for 17 genes whose DNA methylation levels have been previously implicated in CRC. To identify potential confounders, we evaluated clinical labels and DNA methylation levels for associations with participant demographic variables and EM-seq technical variables (Supplementary Figs. 13 and 14). Associated variables were included as covariates in the model matrix created for each contrast to evaluate differential methylation between controls and CRC cases.
Across all target loci, we identified 905 hypermethylated CpG sites (hyper-mCpGs) and 41 hypomethylated CpG sites (hypo-mCpGs) in CRC using DMRcate62 (FDR < 0.05) (Fig. 4a). To explore the functional context in which differentially methylated cytosines (DMCs) are located, we evaluated DMC overlap with (epi)genomic annotations. We accounted for annotation biases accompanying CGI-centric target enrichment by comparing observed overlaps with the overlap distribution from permuted sets of eligible CpG sites (within the target regions and evaluated for differential methylation). In permutation tests analysing DMC overlap with gene features in the Matched Annotation from NCBI and EMBL-EBI (MANE)63, hyper-mCpGs show significant overlap with 1-kb promoters, 5′ UTRs, first exons, all exons (each Benjamini–Hochberg-adjusted P = 0.0001), and regions 1–5 kb upstream of transcription start sites (TSSs; adjusted P = 0.0219) (Fig. 4a; Supplementary Data 10). Hyper-mCpGs overlap introns and 3′ UTRs significantly less than expected (each adjusted P = 0.0001) (Fig. 4a; Supplementary Data 10). Inverted trends were observed for hypo-mCpGs, which are largely in introns (adjusted P = 0.0062) and absent from features near gene TSSs (Fig. 4a; Supplementary Data 10). Hypermethylation near TSSs and in transcribed regions in CRC cells may be associated with altered transcription factor binding affinity and exon splicing64.

a Volcano plot of target CpG sites evaluated for differential DNA methylation in CRC cases (n = 77) relative to controls (n = 88) (FDR < 0.05), with sites colour-coded by their overlapping gene (top). Permutation tests analysing overlap of CRC differentially methylated CpG sites (DMCs) with MANE representative gene annotations (bottom; regions 1–5 kb upstream of the TSS, 1-kb promoters, 5′ UTRs, exons, introns, and 3′ UTRs)63. For each test, 10,000 sets of randomly selected eligible CpG sites (within the target regions and evaluated for differential methylation) of the same number as those within the given DMC set were defined. The overlap distribution from these 10,000 random sets (grey violins) was used to calculate the expected number of overlaps (black bars; mean permuted-set overlaps) and the number of overlaps at the significance level (orange bars; α = 0.05). Permuted overlaps were compared with the observed number of DMCs overlapping the annotation category of interest (blue bars) to calculate an empirical P-value (Supplementary Data 10). P-values were adjusted for multiple testing with the Benjamini–Hochberg method. b As in a, but showing overlap with CpG island (CGI) features (islands, shores (2-kb flanks of islands), shelves (2-kb flanks of shores), and inter-CGI regions (“open sea”)) in the UCSC Genome Browser database61. c As in a, but showing overlap with chromatin state segmentations for rectal mucosa reference epigenome E10165. 01 TssA, Active TSS; 02 TssAFlnk, Flanking active TSS; 03 TxFlnk, Transcribed at gene 5′ and 3′ ends; 04 Tx, Strong transcription; 05 TxWk, Weak transcription; 06 EnhG, Genic enhancers; 07 Enh, Enhancers; 08 ZNF/Rpts, ZNF genes and repeats; 09 Het, Heterochromatin; 10 TssBiv, Bivalent/poised TSS; 11 BivFlnk, Flanking bivalent TSS/enhancers; 12 EnhBiv, Bivalent enhancers; 13 ReprPC, Polycomb repressed; 14 ReprPCWk, Weakly Polycomb repressed; 15 Quies, Quiescent. d As in a, b, but showing target CpG sites evaluated for differential methylation in rectal CRC cases (n = 29) relative to controls (n = 88) (FDR < 0.05; Supplementary Data 11). e As in a, b, but showing target CpG sites evaluated for differential methylation in left-colon CRC cases (n = 16) relative to controls (n = 88) (FDR < 0.05; Supplementary Data 12). f As in a, b, but showing target CpG sites evaluated for differential methylation in mid-colon CRC cases (n = 18) relative to controls (n = 88) (FDR < 0.1; Supplementary Data 13). g As in a, b, but showing target CpG sites evaluated for differential methylation in right-colon CRC cases (n = 14) relative to controls (n = 88) (FDR < 0.5; Supplementary Data 14).
To examine the epigenomic landscape in which these changes may be occurring, we analysed DMC overlap with CGI features in the UCSC database61, and with chromatin state segmentations for a rectal mucosa reference epigenome65. Consistent with gene promoter overlap, hyper-mCpGs significantly overlap CGIs, chromatin state 01 (H3K4me3-marked “Active TSS”) and state 10 (H3K4me3- and H3K27me3-marked “Bivalent/Poised TSS”) (each adjusted P = 0.0001) (Fig. 4b, c; Supplementary Data 10). Elevated DNA methylation in these regions may be associated with silencing of tumour suppressors or dysregulation of genes involved in CRC tumourigenesis. Significant overlaps with chromatin state 13 (“Polycomb repressed”; adjusted P = 0.0156) and state 12 (“Bivalent enhancers”; adjusted P = 0.0199) were also observed (Fig. 4c; Supplementary Data 10). Associations with bivalent chromatin and Polycomb repression may reflect disruptions to tightly regulated expression programmes, with aberrant transcription potentially leading to cell cycle perturbations and cell proliferation66.
In contrast, hypo-mCpGs significantly overlap chromatin state 15 (“Quiescent”, characterised by condensed, transcriptionally inactive chromatin, with low levels of all histone marks and high DNA methylation; adjusted P = 0.0007) (Fig. 4c; Supplementary Data 10). Overlap with quiescent chromatin may reflect an association between decreased DNA methylation in cancer cells and a transition to more open chromatin in these regions, with possible implications for transcriptional regulation and genome stability.
We also identified DMCs in contrasts between controls and CRC cases grouped by pathology site. Consistent with stronger signals at disease markers in cases with pathology closer to the sample collection site, DMCs could be called in rectal (hyper-mCpGs = 1493, hypo-mCpGs = 299) and left-colon (hyper-mCpGs=35) cases with a significance threshold of FDR < 0.05, whereas DMCs could be called in mid-colon (hyper-mCpGs = 30) and right-colon (hyper-mCpGs = 1) cases only with more permissive thresholds (FDR < 0.1 and FDR < 0.5) (Fig. 4d–g). DMC overlap analyses for rectal cases revealed similar trends to those observed for CRC cases, while hyper-mCpGs in left-colon cases are more promoter-localised (Fig. 4d, e; Supplementary Data 11–14).
Similar to findings for right-sided CRC cases, weak signals of pathology at DNA methylation marker genes were also detectable in late-stage polyps (hyper-mCpGs = 7, hypo-mCpGs = 3) and early-stage polyps (hyper-mCpGs=32, hypo-mCpGs = 2) cases relative to controls, with a relaxed significance threshold applied (FDR < 0.5) (Supplementary Figs. 15 and 16; Supplementary Data 15 and 16). Hyper-mCpGs in late-stage polyps significantly overlap chromatin state 01 (H3K4me3-marked “Active TSS”) (adjusted P = 0.013), while those in early-stage polyps associate with CGIs and chromatin state 10 (H3K4me3- and H3K27me3-marked “Bivalent/Poised TSS”) (each adjusted P = 0.0007).
Broader-scale DNA methylation changes were observed across eight differentially methylated regions (DMRs, with multiple constituent DMCs), all of which were hypermethylated in CRC (FDR < 0.05). Consistent with findings from DMC calling with cases grouped by pathology site, DMR-scale profiles of per-group mean methylation levels revealed greatest hypermethylation among CRC cases with rectal pathology, followed by either left-colon or mid-colon cases (Fig. 5a). Per-DMR mean methylation levels across right-colon cases are more comparable to those for controls. We observed a similar gradient of hypermethylation in DMR-scale mean methylation profiles for each clinical category: CRC cases exhibit the most pronounced elevation, followed by participants with late-stage polyps, while levels are more comparable between participants with early-stage polyps and controls (Fig. 5b).

a Mean percent CpG methylation (mCpG) profiles around eight differentially methylated regions (DMRs), calculated across CRC cases grouped by the site of pathology (control n = 88; right-colon CRC n = 14; mid-colon CRC n = 18; left-colon CRC n = 16; rectal CRC n = 29). DMRs are ordered by increasing Benjamini–Hochberg-adjusted P-value (FDR < 0.05). Coordinates for DMRs (dark grey), MANE representative gene promoters (green) and exons (purple)63, and chromatin state segmentations for rectal mucosa reference epigenome E10165 are indicated below the per-group profiles. b As in a, but with participants grouped by clinical category (control n = 88; CRC n = 77; early-stage polyps n = 13; late-stage polyps n = 21). E-Polyps + , Early-stage polyps; L-Polyps + , Late-stage polyps.
To evaluate finer-scale associations, we analysed DMC overlap with (epi)genomic annotations at the level of individual target genes. Ranked by increasing Benjamini–Hochberg-adjusted P-value, the most differentially methylated sites in CRC are hyper-mCpGs in SDC2, LRRC4 and PPP2R5C (Supplementary Data 17). These sites are strongly associated with rectal pathology and CRC Stages II–IV (Fig. 6a). For SDC2, CRC hyper-mCpGs significantly overlap the promoter (adjusted P = 0.019), 5′ UTR, first exon, all exons, CGIs, and chromatin state 10 (“Bivalent/Poised TSS”) (each adjusted P = 0.0003) (Fig. 6b; Supplementary Data 10). These associations are also evident at the SDC2-overlapping hyper-DMR (Fig. 5). SDC2 encodes a transmembrane proteoglycan with a receptor for extracellular matrix proteins, and is involved in cell proliferation, cell migration and cell-to-cell signalling.

a Heatmap showing the most differentially methylated CpG sites in CRC cases relative to controls, with CRC stage, pathology site and clinical category indicated (control n = 88; CRC n = 77; early-stage polyps n = 13; late-stage polyps n = 21). Methylation level is the log2-transformed ratio of methylated to unmethylated read counts at each CpG site, zero-centred and scaled to unit variance. b Volcano plots of target CpG sites in SDC2 evaluated for differential DNA methylation in CRC cases (n = 77) relative to controls (n = 88) (FDR < 0.05), with sites colour-coded by their overlapping genic feature(s) in the MANE representative annotation (top; regions 1–5 kb upstream of the TSS, 1-kb promoters, 5′ UTRs, exons, introns, and 3′ UTRs)63 or CpG island (CGI) feature (bottom; islands, shores (2-kb flanks of islands), shelves (2-kb flanks of shores), and inter-CGI regions (“open sea”)) in the UCSC Genome Browser database61. Permutation tests analysing overlap of CRC differentially methylated CpG sites (DMCs) in SDC2 with MANE representative gene annotations (top) and with CGI features (bottom) are shown below the corresponding volcano plots (Supplementary Data 10). c As in b, but showing target CpG sites in LRRC4. d As in b, but showing target CpG sites in PPP2R5C. e As in b, but showing target CpG sites in RNF217.
CRC hyper-mCpGs in LRRC4 are more concentrated near the TSS, significantly overlapping the promoter, 5′ UTR, first exon, CGIs, and chromatin state 01 (“Active TSS”) (each adjusted P = 0.0003) (Figs. 5 and 6c; Supplementary Data 10). Promoter hypermethylation of CGI-associated genes is a common epigenetic aberration by which tumour suppressor genes are silenced in human cancers67. These trends are therefore consistent with the tumour suppressor activity reported for LRRC4, whose inhibitory effects on glioma cell growth and invasion are abrogated by promoter hypermethylation-mediated inactivation68. Consonant with the tumour-suppressive role of PPP2R5C, including through regulation of TP5369, hyper-mCpGs within or near its transcript variants exhibit significant overlap with chromatin state 01 (“Active TSS”) and CGIs (each adjusted P = 0.0004) (Figs. 5 and 6d; Supplementary Data 10).
CRC hyper-mCpGs in RNF217 associate with the promoter (adjusted P = 0.0003), 5′ UTR (adjusted P = 0.0253), first exon (adjusted P = 0.0068), CGIs (adjusted P = 0.0003), and chromatin state 01 (“Active TSS”; adjusted P = 0.0003) (Figs. 5 and 6e; Supplementary Data 10). The E3 ubiquitin ligase RNF217 regulates intracellular iron homoeostasis through degradation of the iron export protein ferroportin, which depends on TET1-mediated demethylation of the Rnf217 promoter in mouse70. For example, increased ferroportin and dysregulated iron levels have been reported in a conditional knockout line lacking Rnf217 expression in intestinal enterocytes70. Hypermethylation of the RNF217 promoter in CRC may therefore be associated with attenuated ferroportin degradation and impaired iron balance. This is consistent with previously reported aberrant expression and localisation of ferroportin in CRC, leading to intracellular iron accumulation which may induce cell proliferation71.
CRC tumour microbiome signals are detected by representation in the rectal mucosa
Increasing evidence suggests the gut microbiome is associated with risk of CRC development, treatment response and disease recurrence72,73,74,75. This phenomenon is termed a tumour microbiome. We initially performed 16S sequencing on a small cohort of matched rectal swabs, OriCol™-collected rectal mucus samples and stool. The range and trends of α-diversity, for both observed and Shannon diversity metrics, were reflected in both OriCol™ and stool samples (Supplementary Fig. 17). Rectal swabbing had an overall significantly reduced α-diversity and in three incidences resulted in reduced library sizes.
The diversity and abundance of 725 OriCol™ rectal mucus samples were characterised by V3 and V4 16S sequencing to determine associations between microbiome composition and clinical status, where a weak association was observed (all-groups Kruskal–Wallis P = 0.0419) (Supplementary Fig. 18; Supplementary Data 18).
Due to the limitations of 16S sequencing, further work was conducted using whole-genome shotgun (WGS) metagenomic sequencing. This allowed for a more comprehensive assessment of the microbiota community, at species-level resolution and encompassing less abundant taxa. The microbiome diversity and abundance of 420 mucus samples was assessed by WGS to determine microbial species associated with CRC (Fig. 7b, c).

a Bubble plot of the top 60 most abundant species in the cohort, coloured by clinical status (control n = 178; CRC n = 127; early-stage polys n = 18; late-stage polyps n = 47; small bowel cancer n = 1; no CRC n = 8). Each bubble represents a species, with its position on the x-axis corresponding to individual rectal mucus samples and the y-axis indicating different species. The size of each bubble represents the relative abundance of the species in a sample. Bubbles are coloured according to clinical status. The full list of species is available in Supplementary Data 19. b, c The top 20 species composition for controls and CRC cases. Each stacked bar chart represents a sample with the proportion of reads attributed to each microbial species. Different colours distinguish microbial species, with a light grey colour representing “other”, which contains all other species not represented. d Power versus Mann–Whitney–Wilcoxon test Benjamini–Hochberg-adjusted P-value, where each point is a species. The plot is divided into 4 quadrants. Quadrant 1 represents species with the lowest P-values (<0.005) and highest power (>0.4). Quadrant 2 represents species that have higher adjusted P-values (0.05 > P ≥ 0.005) and high power (>0.4). Quadrant 3 represents species that have lower power (<0.4) and low adjusted P-values (<0.005). Quadrant 4 comprises species that have higher adjusted P-values (0.05 > P ≥ 0.005) and low power (<0.4). e, f Hungatella hathewayi and Intestinimonas butyriciproducens relative abundance distributions across CRC cases and controls. E-Polyps + , Early-stage polyps; L-Polyps + , Late-stage polyps; C+ Other, Small bowel cancer; No CRC, Not diagnosed with CRC and not assessed by gold-standard examinations.
Two thresholds have been defined: a minimum abundance level of 1e-04 and a minimum power of 0.4 (Supplementary Fig. 19). In total, 36 species show significant associations with CRC (Fig. 7d). The association landscape was divided into four quadrants, where the first includes the most strongly associated species with the highest power, and the last the least associated species with the lowest power (Supplementary Data 20–23). Quadrant 1, with the lowest Mann–Whitney–Wilcoxon test Benjamini-Hochberg-adjusted P-values (<0.005) and highest power (>0.4), includes two species: Hungatella hathewayi and Intestinimonas butyriciproducens (Fig. 7e, f; Supplementary Data 20). H. hathewayi, also known as Clostridium hathewayi, is present in virtually all samples, and has been reported to be enriched in CRC76,77, together with I. butyriciproducens78,79.
The second quadrant, with higher adjusted P-values (0.05 > P ≥ 0.005) and highest power (>0.4) (Supplementary Data 21), reveals more species that have been extensively reported as associated with CRC. Of interest, we highlight Porphyromonas asaccharolytica (Supplementary Fig. 20) and Clostridium scindens. P. asaccharolytica induces oncogenic stress responses through secretion of the bacterial metabolite butyrate80. Moreover, the invasion of P. asaccharolytica has been observed in CRC tissues and coincides with elevated butyrate levels and senescence-associated inflammatory phenotypes80. The association of a higher relative abundance of C. scindens with CRC has not been previously reported. In fact, according to Arabameri et al.81, C. scindens is an indicator of healthy gut flora, which are reduced in CRC. Quadrant 3 represents species with low adjusted P-values (<0.005) and low power (<0.4) and, like quadrant 2, includes many species previously reported to be associated with CRC, namely Parvimonas micra, Fusobacterium nucleatum and Gemella morbillorum. Interestingly, P. micra and F. nucleatum, alongside Peptostreptococcus stomatis and A. muciniphila (not found to be associated with CRC in this study), have been proposed as part of a four-bacteria biomarker panel for CRC79. Several studies have reported the elevated relative abundance of F. nucleatum in stool and cancer tissue from CRC patients79,82,83, but the role and mechanisms remain unknown. The depth of read coverage across the dataset (Supplementary Data 19) was not high enough to allow for de novo assembly and functional analysis.
Hologenomic mixed integration reveals distinct molecular profiles of colorectal pathology
By combining distinct sources of biological data, multiomics approaches aim to improve model performance beyond what is possible with data from any single omics discipline. The potential value of this is especially clear for CRC given its complex and heterogeneous nature.
To evaluate the potential benefits of combining somatic mutation, DNA methylation and microbiome data, we pursued a mixed integration approach84,85. This involved aggressive omics-specific feature selection to reduce the set of potential biomarkers, with 10 selected from each omics dataset. The intersection of the resulting datasets was then visualised using PCA (Fig. 8b, c) and as a heatmap, with hierarchical clustering across biomarkers and participants (Fig. 8a).

a Heatmap for the tri-omics case (somatic mutation, DNA methylation and microbiome) showing clustering across biomarkers and participants (control n = 30; CRC n = 38; early-stage polyps = 7; late-stage polys = 4). Feature selection was performed before integration, leading to the 30 biomarkers listed in the plot. b Tri-omics PCA analysis showing the data projected onto pairs of principal components (control n = 30; CRC n = 38; early-stage polyps = 7; late-stage polys = 4). Polyps cases are transformed and plotted for comparison. The percent of explained variance for each PC is indicated in parentheses. c As in b, but for the dual-omics case with only mutation and DNA methylation data included (control n = 36; CRC n = 44; early-stage polyps = 12; late-stage polys = 4). E-Polyps + , Early-stage polyps; L-Polyps + , Late-stage polyps.
The biomarkers identified by feature selection (Fig. 8a) are consistent with the results of previous sections. For somatic mutation data, those genes found to be most strongly associated with CRC (Figs. 2 and 3), namely APC, KRAS, BRAF, and TP53, were all selected. Similarly, the selected microbiome species include both H. hathewayi and I. butyriciproducens, which were the only two assigned to quadrant 1 (Fig. 7) owing to their low P-values and high power. Overall, all selected species but one, Selenomonas sputigena, were assigned to quadrants 1–3. For DNA methylation, hyper-mCpGs in SDC2 and LRRC4 together contributed 7 features, along with one hyper-mCpG from each of PPP2R5C, CERS4 and VIM. This agrees with DMC and DMR analyses (Figs. 5 and 6), which identified the most pronounced hypermethylation in SDC2, LRRC4, and PPP2R5C. Furthermore, five of the most differentially methylated hyper-mCpG sites were also chosen by feature selection, showing convergence of results across different methods.
Figure 8 shows the data projected onto pairs of principal components for two combinations of omics, colour-coded by clinical category. Controls tend to cluster together, while CRC cases display more heterogeneous profiles, with many clearly distinct from controls. The locations of early- and late-stage polyps are consistent with their potential progression towards malignancy, with both types tending to cluster at the boundary between classes, while some late-stage polyps have profiles that are indistinguishable from cancers. When labelled by their site of pathology (Supplementary Fig. 21), rectal cancers and, to a lesser extent, left-colon cancers can be most clearly distinguished from controls. Mid- and right-colon cancers are more interspersed with controls. This is consistent with the results of per-omics analyses and indicates that the detectable signal decreases in strength with distance from the pathology. Stage IV CRCs are consistently the most clearly separated from controls, whereas all other stages can be found at varying distances from the control cluster, without any clear patterns of dispersal (Supplementary Fig. 22). Similarly, hierarchical clustering shows a large methylation-driven cluster of CRC cases along with two late-stage polyps, whereas the other polyps cases cluster with a combination of cancers and controls (Fig. 8a). The main clustering by site is found for participants with rectal cancer, with no clear clustering by stage.
We observed a large degree of separation along the first principal component (PC1), which has an explained variance of 44% and 39% in the dual-omics (Fig. 8c) and tri-omics (Fig. 8b) cases, respectively. The PC1 loadings provide an indication of the importance of each selected biomarker to the group stratification (Supplementary Data 24). In the tri-omics case, hyper-mCpGs contribute the largest seven loadings and so are the dominant factor causing separation along PC1 and can therefore be considered the most important omics driving separation. Stratification is also observed along PC2 and PC3, which explain 10% and 8% of the variance, respectively (Fig. 8b, c). The loadings of PC2 are dominated by mutation markers, particularly APC and TP53, while the largest loading of PC3 is the microbiome, particularly Porphyromonas asaccharolytica. Therefore, each principal component is dominated by a specific omics dataset, indicating that all three omics make important contributions to separating cancers from controls.
Discussion
We have demonstrated the application of rectal mucus as a novel biospecimen and important tool for gut biology research, allowing the genetic and epigenetic characterisation of the host bowel and microbiota. Moreover, we illustrate the identification of host-derived pathology-associated signatures and microbial abundance disease indicators, demonstrating the sample utility for in vitro diagnostic applications.
The OriCol™ sampling collection device and its application in the outpatient clinical setting (without bowel preparation in patients who can tolerate a digital rectal examination) adds novelty to this study, with high levels of patient acceptability. Ambient sample management with the tests performed by both clinical and allied healthcare professionals potentially offers a test that can be delivered with minimum infrastructure and process addition, which could be an important addition to cancer diagnostic pathways. Similarly, rectal mucus sampling could be leveraged to facilitate the study and development of translational applications for other conditions of the bowel, such as inflammatory bowel disease.
This work demonstrates that shed pathology-derived material captured locally in the mucus layer migrates to the rectum, where specimens are collected. Analysis of genetic, epigenetic, and microbial datasets allowed the identification of key disease stratification features from each genomic approach, with relative contributions to the separation of clinical categories assessed. Clinical categories were determined by the gold-standard diagnostic procedure, a colonoscopy, or non-gold standard procedure, such as a CT-scan, combined with a one-year follow-up to confirm absence of significant pathology. Analytical findings were benchmarked against clinical diagnosis.
Additionally, we demonstrate that combining genetic and epigenetic information from the host and microbiota, as a composite organism or holobiont, improves detection of disease signals associated with colorectal cancer and adenomatous polyps.
To ensure robust multiomics profiling, the compatibility of rectal-mucus-derived DNA was assessed across multiple technologies for each omics discipline investigated, with the best-performing methodology chosen for in-depth study (Supplementary Figs. 9, 12 and 18).
Somatic mutation profiling detected known pathogenic variants, previously reported in CRC tumours and distal matrices including blood (ctDNA) and stool, that have not been investigated or identified in mucus sampling. Initial analysis of mutation presence/absence and tumour mutation burden showed no significant stratification between clinical categories. Low-frequency pathology-associated mutations were found in all samples, consistent with the notion that no single biomarker reliably indicates disease86. Further examination identified per-gene maximum VAF as a strong correlate of disease state, serving to distinguish pathology-derived signal from non-indicative low-frequency variants87.
Eight genes showed the strongest associations with CRC and adenomatous polyps (APC, TP53, KRAS, BRAF, ERBB2, FBXW7, PIK3CA and SMAD4), with pathogenic variants detected in 15–79% of participants (Figs. 2a and 3). Notably, a site-responsive effect was observed, with maximum VAFs generally decreasing between the rectum and right side of the colon. This is consistent with the hypothesis that more representative and pronounced signals are detected the closer the sampling is to the site of pathology, with detection of right-sided pathology proving more challenging. Moreover, expected mutation frequencies defined across published data for 1605 CRC tumour resections exhibited a strong correlation with observed frequencies in the mucus samples, illustrating a clear representation of pathology (Supplementary Fig. 10).
Interestingly, higher frequencies are observed in APC and KRAS for right-sided CRC compared to TP53, BRAF, ERBB2, FBXW7, PIK3CA and SMAD4 (Fig. 3b, d). These findings support observed differences in right- and left-sided tumour biology identified in resections88, that have not been discernible in alternative sampling methodologies.
Analysis of pathogenic variant allele frequencies highlighted the utility of mucus-based mutation profiling in distinguishing CRC-associated signals across sites and stages of CRC, although stratification is not observed for each case. Additionally, CRC-like signatures were identified in a subset of adenomatous polyps located in the rectum and transverse colon.
DNA methylation profiles have been widely studied as prospective biomarkers for CRC detection. CRC-associated epigenetic marks have previously been identified in tumour-, blood- and stool-based analyses, with little known about these modifications in the gut mucosa.
DNA methylation profiling across 17 known marker genes revealed striking hypermethylation and limited hypomethylation of CpG dinucleotides in CRC and, to a lesser extent, adenomatous polyps, with gradients of hypermethylation observed across pathology sites and clinical categories (Figs. 4 and 5). Consistent with stratification of somatic mutation profiles, CRC cases with rectal pathology exhibited the most pronounced and extensive hypermethylation, followed by left-colon or mid-colon cases, whereas weaker signals were detected for right-colon cases and adenomatous polyps cases.
Hierarchical clustering of participants by DNA methylation levels at the most differentially methylated CpGs (in SDC2, LRRR4, and PPP25RC) showed a general association with clinical category (Fig. 6a). Hypermethylation across these sites is associated with rectal pathology and more advanced disease. Moreover, late-stage polyps are interspersed with CRC cases, which is concordant with DMR-scale profiles of per-group methylation levels (Fig. 5). Additionally, more comparable methylation levels were observed between CRC cases and participants with late-stage polyps, and between those with earlier-stage polyps and controls.
We observed localised hypermethylation in the vicinity of TSSs, primarily associating with CGIs in gene promoters and 5′ ends. CpG-dense promoter hypermethylation is consistent with inhibited transcription factor binding, removal of activating histone modifications like H3K4me3, and epigenetic silencing of tumour suppressor genes67. In contrast, CRC hypomethylation was largely absent from TSS-proximal regions and instead confined to introns in the target genes. This is consistent with the overall decrease in 5-methylcytosine levels frequently observed in cancer genomes, which engenders chromosomal instability by fostering rearrangements and translocations89.
SDC2 is among the target genes with the most pronounced DNA hypermethylation over the promoter and 5′ end (Figs. 5 and 6). This region is characterised by bivalent chromatin in a rectal mucosa reference epigenome. Bivalent chromatin is marked by both activating H3K4me3 and transiently repressive H3K27me3, which together are thought to prime genes for highly coordinated expression during development and cell differentiation. Aberrant DNA methylation and/or expression of SDC2 is tumourigenic in CRC90 and breast cancer91, whereas in vivo tumour suppressor activity has been reported in osteosarcoma92. Poised for transcription, its bivalent chromatin state might reflect tightly orchestrated cell-type-specific expression programmes, with dysregulation of finely balanced transcription promoting cancer in different tissues.
We also observed significant hypermethylation over gene transcribed regions in CRC, which may alter binding affinity and disrupt normal alternative splicing of pre-mRNA. For example, gene-body DNA methylation is known to inhibit CTCF binding and recruit MECP2. Impaired CTCF binding accelerates RNA polymerase (Pol) II processivity, thereby increasing exon skipping93. In contrast, MECP2 binds methylated DNA and recruits histone deacetylases, which reduce the transcription elongation rate of Pol II, promoting exon inclusion94,95. Aberrant alternative splicing events contribute to several malignant processes in CRC96, and their potential diagnostic and therapeutic utility have been highlighted97,98.
Diverging from these trends, intronic overlap of PPP2R5C hyper-mCpGs reflects the structure of the MANE representative transcript. However, several isoforms are encoded by transcript variants with alternative TSSs located ~20–40 kb downstream of the hyper-DMR overlapping intron 2 of the MANE transcript. Intronic hypermethylation has been implicated in gene silencing, in some cases associating with carcinogenesis99,100. DNA methylation has also been proposed to influence intronic splice site selection, changing the balance of expressed isoforms101. Moreover, expression levels of PPP2R5C mRNA variants encoding the three major isoforms correlate with progression-free survival in ovarian cancer patients with residual disease102.
This detection of hypo/hypermethylated states across gene structures has highlighted the ability to detect pathology-associated alterations implicated in a range of dysregulating modalities, including gene-silencing, alternative splicing and genome stability. Hypomethylation signatures were seen in Stage I-IV CRC cases and across all sites of the colon, with a subset of adenomatous polyps exhibiting a CRC-like pathology signature.
Mucosal sampling enables assessment of both host-derived material and the microbiota. Metagenomic analysis identified an average of 782 bacterial species in mucus samples, with the top 20 species in controls and CRC cases highlighted (Fig. 7b, c).
Microbiome diversity and abundance analysis identified 36 bacterial species that are associated with CRC (Fig. 7d). The two most powered and significant species, H. hathewayi and I. butyriciproducens, have previously been implicated in colorectal disease. H. hathewayi has been implicated in treatment response, anti-tumour immune activity, colonic epithelial cell expansion, DNA hypermethylation of tumour suppressor genes and as a potential disease state biomarker76,103,104,105. Correlations between DMR hypermethylation and H. hathewayi were investigated, with no significant associations observed. The butyrate-producing I. butyriciproducens has been implicated in CRC detection and protection, with studies finding that butyrate targets the pyruvate kinase (M2), subsequently reprogramming metabolism and reducing cell growth or causing cell apoptosis directly106,107. Conversely, butyrate-induced senescence has been implicated in tumourigenesis in the context of over-represented Porphyromonas species80.
Additionally, we report several significant bacterial associations with CRC status that are novel or have been reported only in animal models. These include Clostridium scindens, Thomasclavelia ramose and Vescimonas coprocola (Supplementary Data 21). Though previously unidentified as potential clinical indicators, several of these species have plausible mechanisms for dysbiosis in CRC cohorts. For example, Clostridium scindens is known to modify bile acids, with one example being the dehydroxylation of cholic acid into deoxycholic acid108, which in turn has been identified as a promoter of CRC by suppression of CD8 + T cell109.
We hypothesise that the identification of novel microbial associations with CRC may reflect differences between mucus and stool microbiota. Moreover, previous studies have identified bacterial indicators of pathology in mucus samples that were not detected in matched stool analysis110. The underlying functional mechanisms identified by the diversity and abundance analysis are poorly understood and in-depth experimental studies on the mechanisms of action of each organism, or groups of organisms, will be required. This is a grand challenge for the research community and thus is beyond the scope of this work.
These findings highlight the importance of mucosal samples for understanding fundamental biological processes in the gut, exemplified by the identification of novel bacterial associations and dysbiosis with pathology. Furthermore, discoveries localised to mucosal matrices present novel targets for mechanistic research, with potential diagnostic and therapeutic application. Conversely, although microbial CRC associations in the rectal mucus identified in this study demonstrate utility as a proximal indicator of disease state, it remains unproven as to whether these findings are reflective of the microbiome in the tumour itself. Further research is required to determine the concordance of the microbiome of the tumour with that of rectal mucus.
Additionally, a small pilot study was undertaken to assess data quality and concordance of 16S-based microbial analysis between OriCol™-collected rectal mucus and conventional swabbing approaches. Limitations were observed with rectal swabbing, producing a generally poor representation of the host microbiome (Supplementary Fig. 17). The observation of reduced library sizes in 60% of participants assessed suggests insufficiently collected material, a challenge that would be further compounded when assessing host genomics111.
Analysis of each omics category illustrates their potential utility for colorectal disease screening. Genetic, epigenetic, and microbial CRC-associated signals were detected at all stages of cancer. These signals, though variable in strength, are detected across all sites of the colon, consistent with the hypothesis that pathology-derived material captured in the mucosal layer migrate through the colon to the rectum. Although the diagnostic utility of each omics discipline is demonstrated, no single assessment facilities the development of a testing solution to the required standard for adoption in clinical practice, specifically the detection of right-sided lesions.
However, the mixed integration of selected features across all omics into one hologenomic dataset improved the detection of signals of CRC and adenomatous polyps, leveraging the disease-associated features seen across each omic assessment. Bioinformatic and machine learning approaches, such as minimum redundancy maximum relevance, identified key signals driving pathology detection with high concordance. Hierarchical clustering of study participants based on these features resulted in three main clades: CRC and late-stage polyps (right-hand cluster), predominately controls (central cluster), and a population largely comprised of polyps and CRC (left cluster) (Fig. 8a). This two-dimensional representation highlights the additive effect of hologenomic analysis combining genetic, epigenetic and microbial markers.
Contributions to clinical category stratification are detailed in Supplementary Data 24, with the strongest drivers in principal components (one to four) determined by the principal component loading. Interestingly, each principal component is dominated by a specific omics analysis, allowing the identification of clinical categories in the direction of each principal component which would be lost if any one omics technology were removed. For instance, several CRC category samples in the direction of PC2 would be indistinguishable from controls if only features from PC1 were analysed. This observation is recapitulated across the four principal components presented (Fig. 8b).
Further analysis identified the distinguishing power of principal components within the integrated dataset. The weighting of factors for stratification are distinct components, consisting of DNA methylation, mutation, and microbiome respectively, highlighting the contribution of each biomarker type (Fig. 8b, c; Supplementary Data 24).
Challenges in the molecular and biochemical detection of colorectal cancer is well documented, particularly in blood or stool biospecimens. Non or minimally invasive localised sampling is largely confined to stool-based methods, where significantly reduced sensitivity is noted when detecting right-sided pathologies112, in addition to very poor detection of advanced pre-cancerous lesions50. Concordantly, weaker disease-associated signals were identified in rectal mucus biospecimens when genomic analyses were reported individually (Figs. 2 and 4).
However, a combinatory assessment of distinct measures of dysregulation in the rectal mucus, host or biome derived, allowed the broader catchment and identification of disease indicators in a heterogeneous disease, such as colorectal cancer. This integrated hologenomic approach increases signal to noise, driving improved distinguishment across clinical categories, including the site of colorectal cancer and adenomatous polyps.
It is unknown whether this methodology can be applied to stool-based testing approaches, as a combinatory approach requires robust recovery of high-quality DNA from both human and microbial origins, which may be challenging in specimens with low human DNA presence and that are prone to increased DNA degradation113,114. Hologenomics has been utilised to address ecological and evolutionary questions, including invasion, disease transmission, and resistance development, in addition to fundamental systemic host-microbiota interactions115, though these methods have not been exploited for medically translatable research. Here we report the first implementation of hologenomics as a contextualised panoramic analysis for the detection of colorectal pathology-associated features, which can be leveraged for diagnostic applications.
In summary, worldwide large numbers of patients are referred with lower gastrointestinal symptoms and the concern of potential bowel cancer. Current triage tools include qFiT and stool-based genomic analysis, which are limited in diagnostic accuracy and patient engagement. The gold-standard diagnostic test is colonoscopy, which has a low diagnostic yield in symptomatic patients and requires both bowel preparation and can be performed only in patients fit for the test.
In this study, rectal mucus samples were collected from patients without bowel preparation in an outpatient clinical setting, performed by an appropriately trained healthcare professional. Samples were then transported ambiently to the laboratory for storage and subsequent analysis. From a single sample, clinical utility was identified across human genetic, epigenetic, and microbial biomarkers. Sampling of the mucosal layer facilitated the discovery of novel bacterial associations with pathology, which had not previously been reported in stool-based analyses. Lastly, integration of these omics to create a hologenome resulted in unique feature selection and increased resolving power for stratification of clinical categories (Supplementary Data 24).
The focus of this study is to identify innovative means that can be leveraged for colorectal cancer diagnosis, although the technologies and approaches described offer the potential to assess other conditions, such as inflammatory bowel disease, functional and medication-associated symptoms. The health economic benefit from reducing the number of negative colonoscopies, as well as providing early reassurance to patients, has the potential to shorten patient wait times, expedite diagnosis, and drastically reduce the global cost of colorectal cancer diagnostic pathways.
Developing a test in an outpatient clinical setting allows full patient analysis, a controlled environment for testing, and high levels of patient acceptability52. Biobanking of collected specimens allows for wider analysis in future research of clinical applications.
Further developments of this methodology are being conducted in the UK Urgent Suspected CRC pathway (formerly the Two-Week Wait pathway), as part of the TRIOMIC ORI-EGI-04 study, providing an opportunity to enhance diagnostic development, train detection algorithms in a representative cohort, determine performance metrics and further compare with current diagnostic procedures and testing solutions.
Samples collected in the TRIOMIC EGI-ORI-04 study will include a range of additional pathologies identified in the symptomatic pathway. This clinical resource will accommodate future studies to explore the linkage between gut mucosa and pathology, offering opportunities for fundamental research and potential translatable applications.
Methods
Human subjects
Rectal mucus samples and tumour tissues were collected following HRA and EC approvals (IRAS 263745, 19/EM/0266). Written informed consent was obtained from all donors. All samples were collected from patients enroled in the ORI-EGI-02 study; Exploratory Study of Rectal Mucus for Diagnosing Disease (NCT04659590) from four NHS Foundation Trusts. Additionally, for the microbiome study, written consent was obtained from five healthy volunteers who provided matched rectal mucus, stool, and rectal swab samples.
Statistics and reproducibility
All patients had been referred for investigation either through the NHS Colorectal Two-Week Wait Service or were recruited from the Multidisciplinary Team meeting following a diagnosis of colorectal cancer. The study design facilitated the assessment of the heterogeneity of clinical outcomes from participants in the colorectal cancer symptomatic pathway. Additionally, the study permitted the enrichment of samples from participants with confirmed colorectal cancer, augmenting this cohort for a detailed study of colorectal cancer associated genomic features.
Sample size selection was determined by clinical category, study inclusion and exclusion criteria and sample quality control metrics. No statistical method was used to predetermine sample size. No data were excluded from the analyses, unless stated, and the experiments were not randomised. The investigators were not blinded to allocation during experiments and outcome assessment. Analytical processes are detailed in the methods, which utilises open source tools, with the data deposited in a public archive for full reproducibility.
Clinical labels were assigned based on a participant’s NHS diagnostic outcome. CRC+ are histologically confirmed colorectal adenocarcinomas, L-polyps+ are large (>10 mm) or high-grade dysplastic adenomas, E-polyps+ are smaller (≤10 mm) adenomas without high-grade dysplasia, and controls are participants referred through the pathway but without evidence of polyps or colorectal cancer and who have been assessed by the gold-standard CRC diagnostic, colonoscopy, which has reached the caecum or terminal ileum with a fair to excellent quality of bowel preparation. Subjects that were assessed through the Two-Week Wait pathway without the gold-standard diagnostic, but not diagnosed with a cancer by the 12-month follow-up were labelled as No CRC. C+ Other refers to participants diagnosed with cancer that did not involve the large bowel.
Laboratory methods
Mucus sample collection
Rectal mucus was collected from participants prior to bowel preparation and colonoscopy using the proprietary OriCol™ sampling device (Origin Sciences Ltd, Cambridge, UK) according to the manufacturer’s instructions (S1b).
Mucus sample handling
Mucus samples were stabilised at the time of sampling by the addition of 4 mL of stabilisation buffer (Origin Sciences Ltd). Specimens were sealed and transported at room temperature in accordance with UN3373, to the analysis laboratory using standard commercial courier services. Transport times varied between two and five days. Immediately upon receipt specimens were recovered from the device and four 1 mL aliquots of each specimen were frozen at −80 °C until required.
Spin column-based DNA extraction
Two 1 mL aliquots of thawed rectal mucus sample were pooled, and Proteinase K digested (10 min, 65 °C) prior to processing using the QIAamp DNA Blood Midi kit (Qiagen, Hilden, Germany) manual protocol according to the manufacturer’s instructions. Extracted DNA was stored at −80 °C until required.
Magnetic bead-based DNA extraction
DNA was extracted from 500 μL aliquots of rectal mucus in stabilisation buffer. The research use only (RUO) custom rectal mucus nucleic acid extraction kit (NovaCyt, Camberley, UK) was used and efficiency confirmed using qPCR of the provided internal extraction control (IEC) (PrimerDesign), both according to manufacturer’s instructions. Extracted DNA was stored at −80 oC until required.
Ethanol/sodium acetate precipitation
Where required, the calculated equivalent of 0.5 µg ‘human’ DNA (hsDNA) was taken and made to a total volume of 200 µL in ddH20 and 20 µL of 3 M sodium acetate added before vortex mixing for 30 s. 600 µL of ice-cold 100% ethanol was added and the sample mixed by vortexing for 30 s. DNA was precipitated at −80 °C for 16 h and spun for 30 min at 20,000 g in a pre-refrigerated (4 °C) centrifuge. The supernatant was carefully removed without disturbing the DNA precipitate pellet. DNA pellets were washed twice with 1 mL ice-cold 80% ethanol and spun for 10 min at 20,000 g in a pre-refrigerated centrifuge. Supernatants were carefully discarded, and DNA pellets were spun at room temperature at 20,000 g for 1 min to remove residual ethanol. Resultant DNA pellets were air-dried at room temperature for 5 min then resuspended in 35 µL of nuclease-free water and mixed by vortexing for 30 s, followed by pipette mixing. Precipitated DNA was stored at −80 °C until required.
DNA quality control
DNA integrity was assessed by TapeStation 4150 (Agilent Technologies, Santa Clara, USA) using the genomic DNA ScreenTape and reagents. Assays were carried out according to the manufacturer’s instructions. The sample was considered acceptable with a ‘DNA Integrity Number’ (DIN) of ≥3.
Total double-stranded DNA (dsDNA) was quantified using the Qubit flex system (Thermo-Fisher Scientific) and broad range dsDNA (BR dsDNA) reagents according to the manufacturer’s instructions.
Human amplifiable DNA fraction estimation
Estimations of the proportion of Human amplifiable DNA were carried out by comparison of Qubit readings for dsDNA and a qPCR on the LightCycler® 480 II (Roche, Basel, Switzerland) platform using the Taqman® RNAse P assay (Thermo-Fisher Scientific, Waltham, USA) according to manufacturer’s instructions. A six-point human genomic DNA (Promega) standard curve was prepared for each run and deemed acceptable with an r-squared of >0.99, coefficient of variation <1%, and PCR efficiency of 100% ± 10%. Samples, standards, and controls were run in triplicate.
Duplex sequencing
Library preparation
Dual Molecular Index DNA library preparation was carried out using the Duplex Sequencing Universal Kit (TwinStrand Biosciences, Seattle, USA). DNA input quantities for the TwinStrand Library preparation protocol were calculated based on qPCR-estimated human amplifiable DNA fraction, with a target input of ~165,000 genome-equivalents (i.e., equal to that of 500 ng of pure human DNA). Mucus-derived DNA was processed according to the manufacturers protocol. Target enrichment was carried out per manufacturer’s protocol with use of the Cell3™ Target: Cancer 50 hybrid capture panel (Nonacus, Quinton, UK).
Sequencing
DNA sequencing was carried out by CeGaT GmbH (Tubingen, Germany) on the NovaSeq 6000 platform using S4 flow cells (Illumina, San Diego, USA). Eight libraries were pooled and loaded per S4 flow cell with a target duplex depth of ~3000x.
Enzymatic methyl sequencing
DNA shearing
Rectal mucus-derived DNA was sheared using an ML230 focussed ultrasonicator (Covaris, Woburn, USA) and microTUBE-50 AFA Fiber Strips, to an optimal size of 240–290 bp. Successful fragmentation was confirmed by D5000 DNA ScreenTape (Agilent). Assays were carried out according to the manufacturer’s instructions.
The concentration of fragmented sample double-stranded DNA was established using a Qubit Flex Fluorometer (ThermoFisher Scientific, Waltham, USA) and BR dsDNA reagents.
Library preparation
The Targeted Methylation Sequencing (Twist Bioscience, San Francisco, USA), and Enzymatic Methyl-Seq Library Preparation (New England Biolabs) kits were employed according to manufacturer’s instructions. Sample input was normalised to 200 ng in 50uL (4 ng/uL) using 1X TE to dilute. DNA input into library preparation was calculated based on total DNA quantification, with a target input of ~66,000 genome-equivalents (i.e. equal to that of 200 ng of pure human DNA) Samples with <10 ng material were not included. Denaturing was performed using Formamide.
Following library preparation, 190 ng of each indexed, converted libraries were collated into captures, based on indexes used and fragment size. Each capture consisted of no more than eight libraries. All samples were processed with the 17-Gene custom panel designed by Origin (Twist Methyl Custom Panel, SpecReq (103504), Design name: cgitarget-methylseq_v01; Twist Design ID: MTE-91928803). Captures were then taken through hybridisation as outlined in the manufacturers protocol for a minimum of 15 h. Final quality controls of concentration and fragment size were performed before storage at −20 °C. Prepared libraries were then pooled in preparation for sequencing.
Sequencing
DNA sequencing was carried out by CeGaT GmbH (Tubingen, DE) on the NovaSeq 6000 platform using four S4 flow cells (Illumina), and Azenta (Oxford, UK) on the NovaSeq X platform using a 25B flow cell. Libraries were sequenced across five runs to achieve sufficient coverage depth across the target enrichment panel. For rectal mucus-derived library preparations, captures were pooled and loaded across flow cells with a minimum target coverage of 45×.
Microbiome 16S V3-4 and WGS
16S library preparation
Extracted genomic DNA was normalised to 5 ng/µl with 10 mM Tris-HCl. A PCR master mix was made up using 10 µl KAPA 2 G Fast Hot Start Ready Mix (Merck Catalogue No. KK5601), 0.1 µl 100 µM forward tailed specific primer, 0.1 µl 100 µM reverse tailed specific primer and 7.8 µl PCR grade water per sample. 18 µl master mix were added to each well to be used in a 96-well plate followed by 2 µl of DNA and mixed. Specific PCR conditions were 95 °C for 5 min, 30 cycles of 95 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s followed by a final 72 °C for 5 min.
For the second PCR 10 µl KAPA 2 G Fast Hot Start Ready Mix (Merck Catalogue No. KK5601) and 8 µl PCR grade water were mixed per sample and added to a 96 well plate. 1 µl of 10 µM 8 bp Unique Dual Indexes were added to each well. Finally, 1 µl of PCR 1 was transferred into the PCR 2 master mix plate. The second PCR was run using 95⁰C for 5 min, 10 cycles of 95 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s followed by a final 72 °C for 5 min. Final libraries were quantified by Qubit and equimolar pooled together. A single 0.7X SPRI clean-up using sample purification beads (Illumina® DNA Prep, (M) Tagmentation (96 Samples, IPB), 20060059) was performed on the pool. A final Qubit and sizing on a D5000 Screen Tape (Agilent Catalogue No. 5067-5579) using the Agilent Tapestation 4200 was done to calculate the final library pool molarity.
16S sequencing
The pool was run at a final concentration of 12 pM on an Illumina MiSeq instrument using MiSeq® Reagent Kit v3 (600 cycle) (Illumina Catalogue FC-102-3003) following the Illumina recommended denaturation and loading recommendations which included a 20% PhiX spike in (PhiX Control v3 Illumina Catalogue FC-110-3001).
WGS library preparation
Nextera Flex Enzyme kit (Illumina, San Diego, USA) tagmentation master mix was prepared using 0.5 µl Bead-Linked Transposomes (BLT), 0.5 µl Tagmentation Buffer 1 (TB1) and 4 µl H20 for a total volume of 5 µl per reaction. 2 µl of 5 ng/µl metagenomic DNA stock was added to 5 µl of tagmentation master mix for a total reaction volume of 7 µl. Reactions were incubated for 15 min at 55 °C.
12 µl of DNA barcoding mastermix was prepared (10 µl Kapa 2 G Fast HotStart ReadyMix (Roche), 2 µl H2O) per reaction, to which 1ul of barcode primers (10 µM stock of P5 and P7 indexes (Illumina)) were added. Finally, 7 µl of tagmentation reaction from the previous step was added for a total reaction volume of 20 µl. Reactions were carried out using the following amplification profile: 72 °C for 3 min, 95 °C for 1 min, 14 cycles of 95 °C for 10 s, 55 °C for 20 s and 72 °C for 3 min. Finished barcoding reactions were quality controlled using Qbit and Agilent Tapestation as described elsewhere in this manuscript.
Equimolar pooling of libraries was carried out ahead of double-sided size selection using 0.5X and 0.7X ratios of SPRI beads (Beckman Coulter, Brea, USA) sequentially. Pools were characterised using the Qubit HS assay and Tapestation D5000 tape.
WGS sequencing
DNA sequencing was carried out by CeGaT GmbH (Tubingen, DE) on the NovaSeq 6000 platform using S4 flow cells (Illumina). 200 libraries were pooled and loaded per S4 flow cell, with a target output of 63 M 150 bp paired end reads.
Data analysis
Error-corrected NGS bioinformatics
Sequencing run QC was performed by assessing the mean insert size, Q30 (Phred) score and GC content. Demultiplexed FASTQ files were processed through the TwinStrand® DuplexSeq™ FASTQ to VCF Parallel App (Version 3.11.0 to 3.20.1), which comprises bioinformatics processing methods generally as previously described.
Germline matching was performed to confirm identity. This was performed by approximating variants with <0.25 VAF as acquired somatic, between 0.25 and 0.5 VAF as heterozygous and variants >0.75 as homozygous. Acquired somatic variants were disregarded. Iterative pair-wise matching was performed between each sequencing file. Variants that were labelled as homozygous or heterozygous in both sequences were considered a match. Then the fraction match of the total variants was calculated (A ∩ B/A ∪ B). The top 3 matches for each file were evaluated for quality of match and compared with information on sample source. Sequences were considered to contain DNA contamination if they had a generalised downward shift of homozygous VAF and this was confirmed using a combination of index and germline matching. Remaining sequences with a mean duplex depth of ≥1000 were taken forward for analysis.
Variants with less than 3 alternate depth were excluded. The duplex depth was calculated by subtracting no calls from the depth and the VAF was calculated as alternate depth over duplex depth. Statistical outliers were evaluated on the ratios of duplex depth to depth and mean depth minus depth to mean depth. This was used to detect variants with spuriously high no calls or low depth, which would result in artificially inflated VAFs. Where either ratio was more than 3 SD from the mean the VAF was calculated as alternate depth over mean duplex depth.
Variant pathogenicity was annotated using National Library of Medicine ClinVar database (release 20241223). Pathogenic variants were defined as those with ClinVar clinical significance annotations including “Pathogenic” and “Likely pathogenic”, and that are evaluated for a condition linked to neoplastic changes. Putative pathogenic variants were annotated in tumour suppressor genes APC, TP53, FBWX7 and SMAD4. These were either HIGH impact variants (predicted loss of function) or for TP53, FBWX7 and SMAD4, missense variants that had not been labelled as benign. The dataset was filtered for pathogenic and putative pathogenic variants. Of the 50 genes included in our panel, 25 remained after all QCs and filtering across all clinical groups.
Studies available on cBioPortal (Memorial Sloan Kettering, USA) with disease label “Colorectal Adenocarcinoma” were selected to for the multi-study cohort. These studies were collectively queried for the 50 genes used in the ecNGS performed on OriCol™ samples. The reported mutation frequencies were collected for comparison to mutations detected in rectal mucus. For genes reported as mutated in both multi-study and Ori-EGI-02 cohorts, a Spearman’s rank r2 value was calculated on the frequency that the gene was seen as mutated in CRC at any VAF.
Filtered data was grouped by participant and gene, then aggregated by mVAF. This DataFrame was converted into a matrix of participant by gene. Where genes do not have any relevant variants passing filters identified in any participant, they were excluded. Participants without relevant variants detected were included. An oncoprint and both heatmaps were plotted with Complex Heatmap. All boxplots were plotted in python. One-sided Mann–Whitney–Wilcoxon tests analysing per-gene, per-participant mVAF were performed to test the hypothesis that the distribution underlying CRC cases is stochastically greater than the distribution underlying controls. P-values were adjusted for multiple testing using the Benjamini–Hochberg method to control the false discovery rate (FDR), applying a significance threshold of FDR < 0.05.
Enzymatic methyl sequencing bioinformatics
A 161-kb custom target enrichment panel of probes was designed to target loci for hybrid capture followed by EM-seq (Twist Bioscience, San Francisco, USA). Target CpG sites were defined based on GRCh38 coordinates for loci in the Human Methylome Panel (Twist Bioscience, San Francisco, USA) and CGIs in the UCSC Genome Browser database61 that overlap gene-body and promoter (0–1 kb upstream of TSSs) annotations for 17 genes whose DNA methylation levels have been previously implicated in CRC: APC, BMP3, CERS4, CLIP4, FOXA1, LONRF2, LRRC4, MGMT, MLH1, NDRG4, PPP2R5C, RASSF1, RNF217, SDC2, SEPTIN9, SMAD3 and VIM.
Paired-end reads (2 × 100 bp or 2 × 150 bp) from targeted EM-seq libraries were processed with a pipeline written using Nextflow and adapted from nf-core/methylseq116,117. Adapter sequences, low-quality bases (Phred+33-scaled quality <30) and 5 bases from the 5′ and 3′ ends of each read were removed using Trim Galore v0.6.7 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Trimmed reads were aligned to the GRCh38 reference genome assembly (“no_alt_plus_hs38d1_analysis_set”) using bwa-meth v0.2.7118. Alignments were filtered to retain aligned read pairs with MAPQ ≥ 10, discarding secondary and supplementary (chimeric) alignments using SAMtools v1.18119. PCR and optical duplicate reads were identified using Picard MarkDuplicates v3.1.1 (https://broadinstitute.github.io/picard/). Duplicate-marked alignments were filtered to retain those overlapping regions targeted for hybrid capture using SAMtools v1.18119. Methylation bias was evaluated and methylation calls extracted in CpG, CHG and CHH contexts using MethylDackel v0.6.1 (https://github.com/dpryan79/MethylDackel), excluding duplicate alignments, sites covered by <10 reads, and likely variant sites (--maxVariantFrac 0.1 --minOppositeDepth 5).
Non-CpG-derived conversion rate, percent CpG methylation, and depth and breadth of coverage summary metrics were calculated for each library in R v4.3.2. The following criteria were applied for inclusion of libraries in downstream analyses: conversion rate ≥98%, median CpG depth ≥45, and proportion of target CpG sites covered by ≥10 reads ≥80%. Where there were library-preparation replicates among the libraries meeting these criteria, the best replicate library was selected in terms of: (1) depth of coverage, and if there were ties then (2) breadth of coverage, and if there were ties then (3) conversion rate.
To identify potential confounders, clinical labels and DNA methylation levels were evaluated for associations with participant demographic variables (e.g., age, sex, BMI, smoking status, Rockwood frailty) and EM-seq technical variables (e.g., depth of coverage, breadth of coverage, sequencing run). Associations between categorical and continuous variables were evaluated by Kruskal–Wallis tests, those between categorical variables by Fisher’s exact tests, and those between continuous variables by Spearman’s rank-order correlation coefficients (rs). Given that correlation coefficients calculated across large numbers of observations are less likely to be exactly zero, P values for rs were standardised to represent those based on values across 100 samples. P-values were adjusted for multiple hypothesis testing using the Benjamini–Hochberg method, applying a significance threshold of FDR < 0.05.
To test associations with finer-scale methylation levels, mean CpG methylation (M-value) levels for each library were calculated within MANE Select representative promoters (1-kb regions upstream of TSSs) and within gene bodies (TSS–TES) covered by the target panel63. Principal component analysis (PCA) was performed using the resultant mean M-value matrix, and relationships between each principal component and each clinical, demographic and EM-seq variable were evaluated by Kruskal–Wallis tests or Spearman’s rank-order correlation coefficients.
Differentially methylated CpG sites (DMCs) and differentially methylated regions (DMRs) were called in contrasts between OriCol™-derived EM-seq libraries for control participants and CRC cases, and separately for control participants and polyps cases, using the Bioconductor packages DMRcate v2.16.162 and edgeR v4.4.0120, where read counts were normalised to account for differences in library size. The following variables that show significant associations with clinical labels or CpG methylation levels were included as covariates in the model matrix created for each contrast: sequencing run, median CpG depth, CpH methylation and age. DMRcate estimates the mean-variance relationship of log2-transformed methylated and unmethylated read counts using the voom method121, then fits a linear model to the transformed counts via limma122, and applies empirical Bayes shrinkage to generate per-CpG moderated t-statistics and P-values for evaluating CpG-level and regional differential methylation. For edgeR analyses, a negative binomial generalised log-linear model was fitted to the read counts for each CpG site, and quasi-likelihood F-tests were applied to identify significantly differentially methylated sites. The most differentially methylated CpG sites were visualised as a heatmap. For both analyses, per-CpG P-values were adjusted for multiple testing using the Benjamini–Hochberg method, applying a significance threshold of FDR < 0.05 unless indicated otherwise for a given contrast.
Hyper- and hypomethylated CpG sites were analysed for overlap with (epi)genomic annotation categories using permutation tests. These categories included MANE representative gene annotations (regions 1–5 kb upstream of the TSS, 1-kb promoters, 5′ UTRs, exons, introns, and 3′ UTRs) (Morales et al. 2022), CpG island (CGI) features (islands, shores (2-kb flanks of islands), shelves (2-kb flanks of shores), and inter-CGI regions (“open sea”)) in the UCSC Genome Browser database (Perez et al. 2025), and chromatin state segmentations for rectal mucosa reference epigenome E101 (the core 15-state ChromHMM model based on five histone modifications: H3K4me1, H3K4me3, H3K9me3, H3K27me3 and H3K36me3)65. We accounted for annotation biases accompanying CGI-centric target enrichment by comparing observed overlaps with the overlap distribution from permuted sets of eligible CpG sites (within the target regions and evaluated for differential methylation). For each test, 10,000 sets of randomly selected eligible CpG sites of the same number as those within the given DMC set were defined. The overlap distribution from these 10,000 random sets was used to calculate the expected number of overlaps (mean permuted-set overlaps) and the number of overlaps at the significance level (α = 0.05). Permuted overlaps were plotted and compared with the observed number of DMCs overlapping the annotation category of interest to calculate an empirical P-value (minimum P = 0.0001). P-values were adjusted for multiple testing using the Benjamini–Hochberg method, applying a significance threshold of FDR < 0.05.
Microbial 16S bioinformatics
Metataxonomic sequence data targeting the 16S rRNA gene hypervariable regions V3 and V4 on 725 mucus samples from colorectal cancer (CRC) patients and healthy controls was processed (S2a). The quality control analysis was performed on the set of compressed files using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/, version 0.11.9) and MultiQC (version 1.14)123. FastQC is a tool for assessing the quality of sequencing data, while MultiQC is a tool for generating aggregated quality control reports across multiple samples. As criteria for sample inclusion, at least 10 thousand reads must be present. Faecal microbiome data were analysed using QIIME 2 (v2023.5.1)124. Raw paired-end sequences were imported. Sequences were denoised, dereplicated, and chimera-checked using DADA2125 without truncation, producing a feature table and representative sequences. Phylogenetic analysis involved sequence alignment with MAFFT126, masking variable regions, constructing an unrooted tree with FastTree127, and midpoint-rooting. Alpha and beta diversity metrics were computed using the core-metrics-phylogenetic pipeline with a sampling depth of 10 reads per sample and metadata containing health status and covariates. QIIME 2 outputs were further analysed to identify microbiome differences between groups.
Microbial WGS bioinformatics
For the analysis of the microbiome of 420 faecal samples from colorectal cancer (CRC) patients and healthy controls, the following criteria for sample inclusion was as follows: at least 10 million reads must be present; in cases where a duplicated External ID was detected, the sample with the highest similarity to the metataxonomics profile is kept; in cases where disambiguation via taxonomic profiling is ambiguous, the sample with the highest number of reads was kept. Given the source material of the samples, it is expected that a certain amount of host DNA to be present in the samples that underwent sequencing. To remove any human contaminant sequences, the NCBI’s HRRT (https://github.com/ncbi/sra-human-scrubber, version 2.1.0) tool has been employed on the 408 samples that passed initial quality control.
All passing samples were analysed with nf-core/taxprofiler (version 1.0.1)128, which performed quality control with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/, version 0.11.9) and taxonomic classification using Kraken2 (version 2.1.2)129 and Bracken (version 2.7)130. The database provided was k2_standard_16gb_20230605 (https://genome-idx.s3.amazonaws.com/kraken/k2_standard_16gb_20230605.tar.gz). Kraken2 can assign taxonomic labels to DNA or RNA sequences by comparing k-mers (short fixed-length substrings) from the input data to a pre-built database of reference genomes. The database contains k-mer fingerprints for various taxonomic units, such as species, genera, and higher taxonomic ranks. Bracken is a companion tool to Kraken2, designed to improve taxonomic classification and abundance estimation of metagenomic sequencing data. Bracken uses a Bayesian framework to estimate the abundance of taxa in metagenomic samples more accurately. It considers not only the number of k-mers assigned to each taxon by Kraken2 but also the taxonomic composition of reference genomes in the database.
The downstream analysis pipeline employed R version 4.2.3 (2023-03-15) (https://www.R-project.org/) including tidyverse (version 2.0.0), vegan (version 2.6-6.1), coin (version 1.4.3), pheatmap (version 1.0.12) and pwr (version 1.3-0) to produce all statistical analyses and visualisations.
Multiomics analysis
A prevalence threshold was applied to the microbiome data to remove species with greater than 80% sparsity. All datasets were then log-transformed as follows:
where d is an omics data and cst=10−4 is a constant to prevent divergence as d approaches zero.
Polyps cases were excluded, leaving n = 165 methylation, 125 mutation and 305 microbiome samples. Feature selection was then applied to each dataset individually with respect to the binary CRC labels. For the methylation data, reduction of the large feature set was carried out to prevent overfitting downstream; a one-tailed Welch t-test was used to search for hyper-mCpGs, and any sites with P > 0.001 were removed. This filtered feature set was passed through Minimum-Redundancy Maximum-Relevance (mRMR) selection131, which chose the best 10 hyper-mCpGs based on their strong relationship to cancer status, while attempting to avoid selecting features that are too strongly correlated with each other (i.e., redundant). This same combination of methods, a Welch t-test followed by mRMR, was also applied to the microbiome dataset to select 10 species. The mutation data was passed directly to mRMR without a t-test, owing to its small feature set of only 25 genes. This produced three reduced datasets, each with 10 candidate biomarkers. The intersection of these datasets was then taken, leading to 80 samples in the dual-omics case (methylation and mutation), and 68 samples in the tri-omics case (all three omics).
These reduced datasets were visualised using PCA by plotting all combinations of the first four principal components (PCs). This enabled stratification between cancers and controls to be analysed. Polyps cases were transformed and plotted using the learned model, so that their position relative to cancers and controls could be investigated. PC loadings were calculated to learn about the contribution of each biomarker, where the loading of a PC is that PC multiplied by the square root of its explained variance. Finally, the full tri-omics dataset, including polyps, was visualised as a heatmap by hierarchically clustering across both biomarkers and participants.
Data availability
The sequencing data generated in this study have been deposited in the European Genome-phenome Archive database under accession code EGAD50000001803. The sequencing data are available under restricted access for non-commercial research purposes, access can be obtained by contacting the corresponding author or the data access committee (EGAC50000000643) and will be reviewed in accordance with ethical guidelines for the study and participant consent. The raw sequencing data are protected and are not publicly available due to the presence of sensitive personal health information and limitations imposed by participant consent, privacy legislation and institutional ethics approval.
Code availability
All code used is either commercially available (Duplex sequencing, TwinStrand Bioscience) or adapted from open-source publicly available repositories, details of which are provided in the methods section. Additionally, all tools used for this study are described in the methods in detail including reference and versions.
References
International Agency for Research on Cancer. Cancer Today: Global Cancer Observatory. https://gco.iarc.fr/today/en (2022).
Sung, H. et al. Colorectal cancer incidence trends in younger versus older adults: an analysis of population-based cancer registry data. Lancet Oncol. 26, 51–63 (2025).
Ju, W. et al. The occurence of cancer in ageing populations at global and regional levels, 1990 to 2019. Age Ageing 52, afad043 (2023).
Santucci, C. et al. European cancer mortality predictions for the year 2024 with focus on colorectal cancer. Ann. Oncol. 35, 308–316 (2024).
Cancer Survival in England, cancers diagnosed 2016 to 2020, followed up to 2021 - NHS England Digital. https://digital.nhs.uk/data-and-information/publications/statistical/cancer-survival-in-england/cancers-diagnosed-2016-to-2020-followed-up-to-2021.
Murphy, C. C. et al. Decrease in incidence of colorectal cancer among individuals 50 years or older after recommendations for population-based screening. Clin. Gastroenterol. Hepatol. 15, 903–909.e6 (2017).
Zauber, A. G. The impact of screening on colorectal cancer mortality and incidence: has it really made a difference?. Dig. Dis. Sci. 60, 681–691 (2015).
Doubeni, C. A. et al. Effectiveness of screening colonoscopy in reducing the risk of death from right and left colon cancer: a large community-based study. Gut 67, 291–298 (2018).
Preventive Services Task Force, U. S. et al. Screening for colorectal cancer: US preventive services task force recommendation statement. JAMA 325, 1965–1977 (2021).
Anyane-Yeboa, A. et al. The revised colorectal cancer screening guideline and screening burden at community health centers. Sci. Rep. 15, 336 (2025).
Faux, J. W., Cock, K., Bromley, R. & Feldman, M. Colorectal two-week wait service and quantitative FIT: it’s not just about colon cancer. Ann. R. Coll. Surg. Engl. 104, 257–260 (2022).
Obaro, A. E., Burling, D. N. & Plumb, A. A. Colon cancer screening with CT colonography: logistics, cost-effectiveness, efficiency and progress. Br. J. Radiol. 91, 20180307 (2018).
D’Souza, N. et al. Faecal immunochemical test is superior to symptoms in predicting pathology in patients with suspected colorectal cancer symptoms referred on a 2WW pathway: a diagnostic accuracy study. Gut 70, 1130–1138 (2021).
Harnan, S. et al. Faecal immunochemical tests for patients with symptoms suggestive of colorectal cancer: an updated systematic review and multiple-threshold meta-analysis of diagnostic test accuracy studies. Colorectal Dis. 27, e17255 (2024).
Turvill, J. & McAlindon, M. O64 Interim update on the NHS England colon capsule endoscopy evaluation. in Oral presentations A40.1-A40 (BMJ Publishing Group Ltd and British Society of Gastroenterology, 2023). https://doi.org/10.1136/gutjnl-2023-BSG.63.
MacLeod, C. et al. ScotCap - A large observational cohort study. Colorectal Dis. 24, 411–421 (2022).
MacLeod, C. et al. The ScotCap registry: an evaluation of 1000 colon capsule endoscopy procedures carried out in Scotland. Colorectal Dis. 27, e17271 (2025).
Akolkar, D. et al. Circulating ensembles of tumor-associated cells: a redoubtable new systemic hallmark of cancer. Int. J. Cancer 146, 3485–3494 (2020).
Ranade, A. et al. Hallmark circulating tumor-associated cell clusters signify 230 times higher one-year cancer risk. Cancer Prev. Res. 14, 11–16 (2021).
Gaya, A. et al. Evaluation of circulating tumor cell clusters for pan-cancer noninvasive diagnostic triaging. Cancer Cytopathol. 129, 226–238 (2021).
Lawrence, R., Watters, M., Davies, C. R., Pantel, K. & Lu, Y.-J. Circulating tumour cells for early detection of clinically relevant cancer. Nat. Rev. Clin. Oncol. 20, 487–500 (2023).
Liu, H.-N. et al. Diagnostic and economic value of carcinoembryonic antigen, carbohydrate antigen 19-9, and carbohydrate antigen 72-4 in gastrointestinal cancers. World J. Gastroenterol. 29, 706–730 (2023).
Huang, Z. et al. Comparison between Clinical Utility of CXCL-8 and clinical practice tumor markers for colorectal cancer diagnosis. Biomed. Res. Int. 2022, 1213968 (2022).
Cisneros-Villanueva, M. et al. Cell-free DNA analysis in current cancer clinical trials: a review. Br. J. Cancer 126, 391–400 (2022).
Shaw, J. A. et al. Mutation analysis of cell-free DNA and single circulating tumor cells in metastatic breast cancer patients with high circulating tumor cell counts. Clin. Cancer Res. 23, 88–96 (2017).
Sunakawa, Y. et al. Clinical verification of circulating tumor RNA (ctRNA) as novel pretreatment predictor and tool for quantitative monitoring of treatment response in metastatic colorectal cancer (mCRC): A biomarker study of the DEEPER trial. JCO 37, TPS3621–TPS3621 (2019).
Barnell, E. K. et al. Multitarget stool RNA test for colorectal cancer screening. JAMA 330, 1760–1768 (2023).
Cree, I. A. et al. The evidence base for circulating tumour DNA blood-based biomarkers for the early detection of cancer: a systematic mapping review. BMC Cancer 17, 697 (2017).
Zandvakili, I. & Lazaridis, K. N. Cell-free DNA testing: future applications in gastroenterology and hepatology. Ther. Adv. Gastroenterol. 12, 1756284819841896 (2019).
Page, K. et al. Circulating tumor DNA profiling from breast cancer screening through to metastatic disease. JCO Precis. Oncol. 5, 1768–1776 (2021).
Wang, Y. et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc. Natl. Acad. Sci. USA 112, 9704–9709 (2015).
Pascual, J. et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: a report from the ESMO Precision Medicine Working Group. Ann. Oncol. 33, 750–768 (2022).
Kim, H. & Park, K. U. Clinical circulating tumor DNA testing for precision oncology. Cancer Res. Treat. 55, 351–366 (2023).
Chung, D. C. et al. A cell-free DNA blood-based test for colorectal cancer screening. N. Engl. J. Med. 390, 973–983 (2024).
Nicholson, B. D. et al. Multi-cancer early detection test in symptomatic patients referred for cancer investigation in England and Wales (SYMPLIFY): a large-scale, observational cohort study. Lancet Oncol. 24, 733–743 (2023).
Angius, F. PREEMPT CRC: Blood-based screening test for colorectal cancer shows promise. in Medicom Conference Report DDW 2024 (ed. Duijvestein, M.) (Medicom Medical Publishers, 2024). https://doi.org/10.55788/f68304df.
Tébar-Martínez, R. et al. Strategies for improving detection of circulating tumor DNA using next generation sequencing. Cancer Treat. Rev. 119, 102595 (2023).
Vavoulis, D. V. et al. Multimodal cell-free DNA whole-genome TAPS is sensitive and reveals specific cancer signals. Nat. Commun. 16, 430 (2025).
Lee, N. Y. et al. Machine learning unveils an immune-related DNA methylation profile in germline DNA from breast cancer patients. Clin. Epigenetics 16, 66 (2024).
Fortunato, O. et al. Development of a molecular blood-based immune signature classifier as biomarker for risks assessment in lung cancer screening. Cancer Epidemiol. Biomark. Prev. 31, 2020–2029 (2022).
Dyikanov, D. et al. Comprehensive peripheral blood immunoprofiling reveals five immunotypes with immunotherapy response characteristics in patients with cancer. Cancer Cell 42, 759–779.e12 (2024).
Ichiki, Y. et al. Immune profile analysis of peripheral blood and tumors of lung cancer patients treated with immune checkpoint inhibitors. Transl. Lung Cancer Res. 11, 2192–2207 (2022).
Green, E. A. et al. Clinical utility of cell-free and circulating tumor DNA in kidney and bladder cancer: a critical review of current literature. Eur. Urol. Oncol. 4, 893–903 (2021).
Tivey, A., Church, M., Rothwell, D., Dive, C. & Cook, N. Circulating tumour DNA - looking beyond the blood. Nat. Rev. Clin. Oncol. 19, 600–612 (2022).
Smith, C. G. et al. Comprehensive characterization of cell-free tumor DNA in plasma and urine of patients with renal tumors. Genome Med. 12, 23 (2020).
de Vos, W. M., Tilg, H., Van Hul, M. & Cani, P. D. Gut microbiome and health: mechanistic insights. Gut 71, 1020–1032 (2022).
Afzaal, M. et al. Human gut microbiota in health and disease: unveiling the relationship. Front. Microbiol. 13, 999001 (2022).
Wu, Y. et al. Identification of microbial markers across populations in early detection of colorectal cancer. Nat. Commun. 12, 3063 (2021).
Ternes, D. et al. Microbiome in colorectal cancer: how to get from meta-omics to mechanism? Trends Microbiol. 28, 401–423 (2020).
Imperiale, T. F. et al. Next-generation multitarget stool DNA test for colorectal cancer screening. N. Engl. J. Med. 390, 984–993 (2024).
Study Details | Triple Assay of Rectal Mucus in the Diagnosis of Colorectal Cancer | ClinicalTrials.gov. https://clinicaltrials.gov/study/NCT06649123?cond=Colorectal%20Cancer&term=triomic&rank=1.
Humphrey, H. N. et al. An internal pilot study of a novel rectal mucocellular sampling device to allow next-generation sequencing for colorectal disease. Tech. Coloproctol. 27, 227–235 (2023).
Loktionov, A. Cell exfoliation in the human colon: myth, reality and implications for colorectal cancer screening. Int. J. Cancer 120, 2281–2289 (2007).
Loktionov, A. et al. Colorectal cancer detection by measuring DNA from exfoliated colonocytes obtained by direct contact with rectal mucosa. Int. J. Oncol. 34, 301–311 (2009).
Kennedy, S. R. et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc. 9, 2586–2606 (2014).
Lowenfels, A. B. Fecal occult blood testing as a screening procedure for colorectal cancer. Ann. Oncol. 13, 40–43 (2002).
Feng, S., Zhong, Z., Wang, M. & Jacobsen, S. E. Efficient and accurate determination of genome-wide DNA methylation patterns in Arabidopsis thaliana with enzymatic methyl sequencing. Epigenetics Chromatin 13, 42 (2020).
Ponnaluri, V. K. C. et al. Abstract 2099: EM-seq enables accurate and precise methylome analysis of challenging DNA samples. Cancer Res. 81, 2099–2099 (2021).
Vaisvila, R. et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res. 31, 1280–1289 (2021).
Olova, N. N. & Andrews, S. Whole genome methylation sequencing via enzymatic conversion (EM-seq): protocol, data processing, and analysis. Methods Mol. Biol. 2866, 73–98 (2025).
Perez, G. et al. The UCSC genome browser database: 2025 update. Nucleic Acids Res. 53, D1243–D1249 (2025).
Peters, T. J. et al. Calling differentially methylated regions from whole genome bisulphite sequencing with DMRcate. Nucleic Acids Res. 49, e109 (2021).
Morales, J. et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 604, 310–315 (2022).
Tirado-Magallanes, R., Rebbani, K., Lim, R., Pradhan, S. & Benoukraf, T. Whole genome DNA methylation: beyond genes silencing. Oncotarget 8, 5629–5637 (2017).
Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Glancy, E., Choy, N. & Eckersley-Maslin, M. A. Bivalent chromatin: a developmental balancing act tipped in cancer. Biochem. Soc. Trans. 52, 217–229 (2024).
Jones, P. A. & Baylin, S. B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 3, 415–428 (2002).
Li, P. et al. Novel Therapy for glioblastoma multiforme by restoring lrrc4 in tumor cells: LRRC4 inhibits tumor-infitrating regulatory T cells by cytokine and programmed cell death 1-containing exosomes. Front. Immunol. 8, 1748 (2017).
Nobumori, Y., Shouse, G. P., Fan, L. & Liu, X. HEAT repeat 1 motif is required for B56γ-containing protein phosphatase 2A (B56γ-PP2A) holoenzyme assembly and tumor-suppressive function. J. Biol. Chem. 287, 11030–11036 (2012).
Jiang, L. et al. RNF217 regulates iron homeostasis through its E3 ubiquitin ligase activity by modulating ferroportin degradation. Blood 138, 689–705 (2021).
Brookes, M. J. et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut 55, 1449–1460 (2006).
Cao, Y. et al. Intratumoural microbiota: a new frontier in cancer development and therapy. Signal Transduct. Target. Ther. 9, 15 (2024).
Nejman, D. et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980 (2020).
Park, E. M. et al. Targeting the gut and tumor microbiota in cancer. Nat. Med. 28, 690–703 (2022).
Battaglia, T. W. et al. A pan-cancer analysis of the microbiome in metastatic cancer. Cell 187, 2324–2335.e19 (2024).
Zhang, J. et al. Expansion of colorectal cancer biomarkers based on gut bacteria and viruses. Cancers 14, 4662 (2022).
Gutierrez-Angulo, M. et al. Microbiota composition and its impact on DNA methylation in colorectal cancer. Front. Genet. 14, 1037406 (2023).
Kharofa, J., Apewokin, S., Alenghat, T. & Ollberding, N. J. Metagenomic analysis of the fecal microbiome in colorectal cancer patients compared to healthy controls as a function of age. Cancer Med. 12, 2945–2957 (2023).
Osman, M. A. et al. Parvimonas micra, Peptostreptococcus stomatis, Fusobacterium nucleatum and Akkermansia muciniphila as a four-bacteria biomarker panel of colorectal cancer. Sci. Rep. 11, 2925 (2021).
Okumura, S. et al. Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat. Commun. 12, 5674 (2021).
Arabameri, A., Asemani, D. & Teymourpour, P. Detection of colorectal carcinoma based on microbiota analysis using generalized regression neural networks and nonlinear feature selection. IEEE/ACM Trans. Comput Biol. Bioinform. 17, 547–557 (2020).
Ou, S. et al. Fusobacterium nucleatum and colorectal cancer: From phenomenon to mechanism. Front. Cell. Infect. Microbiol. 12, 1020583 (2022).
Pignatelli, P., Nuccio, F., Piattelli, A. & Curia, M. C. The role of fusobacterium nucleatum in oral and colorectal carcinogenesis. Microorganisms 11, 2358 (2023).
Picard, M., Scott-Boyer, M.-P., Bodein, A., Périn, O. & Droit, A. Integration strategies of multi-omics data for machine learning analysis. Comput. Struct. Biotechnol. J. 19, 3735–3746 (2021).
Subramanian, I., Verma, S., Kumar, S., Jere, A. & Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 14, 1177932219899051 (2020).
Venner, E. et al. The frequency of pathogenic variation in the All of Us cohort reveals ancestry-driven disparities. Commun. Biol. 7, 174 (2024).
Tang, Y. et al. Maximum allele frequency observed in plasma: a potential indicator of liquid biopsy sensitivity. Oncol. Lett. 18, 2118–2124 (2019).
Sugai, T. et al. Analysis of molecular alterations in left- and right-sided colorectal carcinomas reveals distinct pathways of carcinogenesis: proposal for new molecular profile of colorectal carcinomas. J. Mol. Diagn. 8, 193–201 (2006).
Watanabe, Y. & Maekawa, M. Methylation of DNA in cancer. Adv. Clin. Chem. 52, 145–167 (2010).
Ryu, H.-Y. et al. Syndecan-2 functions as a docking receptor for pro-matrix metalloproteinase-7 in human colon cancer cells. J. Biol. Chem. 284, 35692–35701 (2009).
Sun, M. et al. RKIP and HMGA2 regulate breast tumor survival and metastasis through lysyl oxidase and syndecan-2. Oncogene 33, 3528–3537 (2014).
Marion, A. et al. Calpain-6 is an endothelin-1 signaling dependent protective factor in chemoresistant osteosarcoma. Int. J. Cancer 130, 2514–2525 (2012).
Shukla, S. et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479, 74–79 (2011).
Maunakea, A. K., Chepelev, I., Cui, K. & Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–1269 (2013).
Kinde, B., Wu, D. Y., Greenberg, M. E. & Gabel, H. W. DNA methylation in the gene body influences MeCP2-mediated gene repression. Proc. Natl. Acad. Sci. USA 113, 15114–15119 (2016).
Xiong, Y. et al. Profiles of alternative splicing in colorectal cancer and their clinical significance: A study based on large-scale sequencing data. EBioMedicine 36, 183–195 (2018).
Manabile, M. A. et al. Alternative splicing events and their clinical significance in colorectal cancer: targeted therapeutic opportunities. Cancers 15, 3999 (2023).
Chen, Y. et al. Alternative splicing of mRNA in colorectal cancer: new strategies for tumor diagnosis and treatment. Cell Death Dis. 12, 752 (2021).
Hoivik, E. A. et al. DNA methylation of intronic enhancers directs tissue-specific expression of steroidogenic factor 1/adrenal 4 binding protein (SF-1/Ad4BP). Endocrinology 152, 2100–2112 (2011).
Zhang, X. et al. Methylation of a single intronic CpG mediates expression silencing of the PMP24 gene in prostate cancer. Prostate 70, 765–776 (2010).
Malousi, A. & Kouidou, S. DNA hypermethylation of alternatively spliced and repeat sequences in humans. Mol. Genet. Genom. 287, 631–642 (2012).
Ramachandran, D. et al. Genome-wide association analyses of ovarian cancer patients undergoing primary debulking surgery identify candidate genes for residual disease. NPJ Genom. Med. 9, 19 (2024).
Huang, Z., Wang, C., Huang, Q., Yan, Z. & Yin, Z. Hungatella hathewayi impairs the sensitivity of colorectal cancer cells to 5-FU through decreasing CDX2 expression. Hum. Cell 36, 2055–2065 (2023).
Zhuang, Y.-P. et al. Gut microbiota interactions with antitumor immunity in colorectal cancer: from understanding to application. Biomed. Pharmacother. 165, 115040 (2023).
Xia, X. et al. Bacteria pathogens drive host colonic epithelial cell promoter hypermethylation of tumor suppressor genes in colorectal cancer. Microbiome 8, 108 (2020).
Luo, Q., Zhou, P., Chang, S., Huang, Z. & Zeng, X. Characterization of butyrate-metabolism in colorectal cancer to guide clinical treatment. Sci. Rep. 13, 5106 (2023).
Hague, A. et al. Sodium butyrate induces apoptosis in human colonic tumour cell lines in a p53-independent pathway: implications for the possible role of dietary fibre in the prevention of large-bowel cancer. Int. J. Cancer 55, 498–505 (1993).
Marion, S. et al. In vitro and in vivo characterization of Clostridium scindens bile acid transformations. Gut Microbes 10, 481–503 (2019).
Cong, J. et al. Bile acids modified by the intestinal microbiota promote colorectal cancer growth by suppressing CD8 + T cell effector functions. Immunity 57, 876–889.e11 (2024).
Glymenaki, M. et al. Compositional changes in the gut mucus microbiota precede the onset of colitis-induced inflammation. Inflamm. Bowel Dis. 23, 912–922 (2017).
Chanderraj, R. et al. The bacterial density of clinical rectal swabs is highly variable, correlates with sequencing contamination, and predicts patient risk of extraintestinal infection. Microbiome 10, 2 (2022).
Hoffman, A., Teubner, D. & Kiesslich, R. Competition in colon cancer screening? what is the role of colonoscopy? Viszeralmedizin 30, 18–25 (2014).
Human Microbiome Project Consortium A framework for human microbiome research. Nature 486, 215–221 (2012).
He, K. et al. A pipeline for faecal host DNA analysis by absolute quantification of LINE-1 and mitochondrial genomic elements using ddPCR. Sci. Rep. 9, 5599 (2019).
Hird, S. M. Evolutionary biology needs wild microbiomes. Front. Microbiol. 8, 725 (2017).
Di Tommaso, P. et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319 (2017).
Ewels, P. et al. nf-core/methylseq: Endless Tofu. Zenodo https://doi.org/10.5281/zenodo.14502249 (2024).
Pedersen, B. S., Eyring, K., De, S., Yang, I. V. & Schwartz, D. A. Fast and accurate alignment of long bisulfite-seq reads. arXiv https://doi.org/10.48550/arxiv.1401.1129 (2014).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Chen, Y., Chen, L., Lun, A. T. L., Baldoni, P. L. & Smyth, G. K. edgeR v4: powerful differential analysis of sequencing data with expanded functionality and improved support for small counts and larger datasets. Nucleic Acids Res. 53, gkaf018 (2025).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 — approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Stamouli, S. et al. nf-core/taxprofiler: highly parallelised and flexible pipeline for metagenomic taxonomic classification and profiling. BioRxiv https://doi.org/10.1101/2023.10.20.563221 (2023).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Lu, J., Breitwieser, F. P., Thielen, P. & Salzberg, S. L. Bracken: estimating species abundance in metagenomics data. PeerJ Comput. Sci. 3, e104 (2017).
Peng, H., Long, F. & Ding, C. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 27, 1226–1238 (2005).
Acknowledgements
We would like to thank Salah Azzi (Illumina) and Nicholas Zoulias (Azenta Life Sciences) for technical contributions and support for Next Generation Sequencing applications. We would like to acknowledge Nuria Melisa Morales Garcia (Science Graphic Design) for the design and creation of main text figures. We would especially like to acknowledge the contributions of Linglan Zhang, who greatly contributed to this study but sadly passed away during the publication of this work.
Author information
Authors and Affiliations
Contributions
A.J.T. and K.S.P. contributed equally. A.J.T. led the bioinformatic analysis and K.S.P led the laboratory processing of clinical samples. E.M.-W. and A.J.P. managed the data analytics team. L.Z., G.M., A.D., and M. Sands managed and coordinated laboratory operations and commercial relationships. C.O., O.E.O., and M. Siew contributed to data analysis. A.D.M., S.N., A.C.-C., and E.L. contributed to experimentation. I.D. and S.S. designed and conducted clinical studies. K.K. and C.H. managed quality and regulatory requirements. H.L led the funding of clinical studies and works completed. D.W. conceived and led the project. I.M. contributed to microbiome analysis. D.J.B completed experiments for microbiome analysis. F.M.D.: clinical study principal investigator. H.N.H., E.T.W.-D., and H.R. enrolled participants. C.C.: clinical study principal investigator. S.B. enrolled participants. A.E.B., K.-V.S., and C.J.P. contributed to biomarker assessment. N.J.B.: clinical study principal investigator. H.S. enrolled participants. J.L.-C.: clinical study chief investigator. G.A.R.J enrolled participants.
Corresponding author
Ethics declarations
Competing interests
The works detailed in this manuscript were funded by Origin Sciences Ltd of which A.J.T., K.S.P., E.M.W., L.Z., C.O., A.D.M., S.N., O.E.O., G.M., M.S., S.S., A.D., K.K., C.H., A.C.C., E.L., M.S., H.L., A.J.P., I.D., and D.W. are employees. No other conflicts of interest are declared by other authors.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tock, A.J., Patel, K.S., Morales-Walker, E. et al. Hologenomic analysis of rectal mucus sampling for detection of adenomatous polyps and colorectal cancer. Nat Commun 16, 10876 (2025). https://doi.org/10.1038/s41467-025-66006-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66006-1
