
Abstract
Atherosclerosis is mediated by circulating monocytes and lesional macrophages through chronic inflammation accompanied by metabolic reprogramming. Although methionine metabolism enhances the proinflammatory capacity of monocytes/macrophages, its role in atherosclerosis remains unclear. Here, we use untargeted metabolomics and mass spectrometry to demonstrate that monocyte methionine metabolism is associated with vulnerable plaque (thin-cap fibroatheroma [TCFA]) identified by pancoronary optical coherence tomography. Methionine adenosyltransferase Ⅱ alpha (MAT2A), the key enzyme of methionine metabolism, is highly expressed in atherosclerosis. Further epigenetic profiling of inflammatory and migratory gene promoters reveals MAT2A-mediated enrichment of the transcriptional permissive chromatin mark H3K4me3. Myeloid-specific MAT2A ablation and pharmacological inhibition, or a low-methionine diet, reduce monocyte/macrophage inflammation and migration, thereby attenuating plaque vulnerability. Mechanistically, norepinephrine activates the mTOR-c-MYC axis to upregulate MAT2A expression. The combination of norepinephrine and methionine metabolism is associated with TCFA presence and 5-year clinical prognosis. Consequently, MAT2A-mediated methionine metabolism represents a potential therapeutic target for atherosclerosis.
Similar content being viewed by others

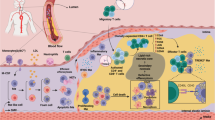

Introduction
Atherosclerosis is a chronic inflammatory disease in which monocytes/macrophages play a central role1. The landmark CANTOS (Canakinumab Antiinflammatory Thrombosis Outcome Study), COLCOT (Colchicine Cardiovascular Outcomes Trial), and LoDoCo2 (Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease) trials demonstrated the clinical relevance of inflammatory risk and the potential of anti-inflammatory therapies for preventing adverse cardiovascular events2,3,4. Clinical studies have consistently shown that the presence of the prototypical vulnerable, rupture-prone plaque phenotype, namely thin-cap fibroatheroma (TCFA), often coincides with elevated circulating monocyte levels and intraplaque macrophage infiltration5,6,7. These findings highlight the pivotal role of monocytes and macrophages as mediators of atherosclerosis progression8. Notably, monocytes and macrophages exhibit remarkable diversity and plasticity, allowing them to acquire different functional profiles9. When exposed to pro-atherogenic stimuli, circulating monocytes give rise to lesional macrophages that maintain a proinflammatory status through profound metabolic reprogramming10,11. Consequently, therapeutic strategies that focus on modulating monocytes/macrophages metabolic reprogramming represent a promising approach for combating atherosclerosis.
Methionine metabolism, which drives epigenetic reprogramming, provides a dynamic biochemical underpinning for inflammatory macrophages12. Within the methionine cycle, homocysteine (Hcy), a metabolite downstream of S-adenosyl-methionine (SAM), has recently been suggested to act as a bystander rather than a causative factor in atherosclerosis13,14,15. In particular, the enzyme methionine adenosyltransferase Ⅱ alpha (MAT2A) primarily mediates SAM synthesis and directly alters the SAM/S-adenosyl-homocysteine (SAH) ratio, known as the “methylation index”, which is a hallmark of cellular methylation capacity16,17. Dysregulation of methionine metabolism, often mediated by MAT2A, can lead to the accumulation of methylated substrates. This disruption alters the delicate balance of cellular methylation, leading to aberrant inflammatory gene expression12,18,19. Accumulating evidence indicates that MAT2A regulates macrophage inflammation, reactive oxygen species accumulation during ferroptosis, and SAM-dependent, hypomethylation-driven DNA damage, all of which are core drivers of atherogenesis12,20,21. However, the potential contribution of methionine metabolism in orchestrating pro-atherogenic monocytes/macrophages by influencing epigenetic regulation in driving atherosclerotic plaque progression and vulnerability remains largely unknown.
In this study, we demonstrate that the activation of methionine metabolism, manifested by high SAM/SAH levels, correlates with atherosclerotic plaque vulnerability in patients. Furthermore, we found that MAT2A drives methionine metabolic reprogramming through H3K4me3 modifications, which are crucial for endowing monocytes/macrophages with inflammatory and migratory capabilities during atherosclerosis development in murine models. Inhibition of MAT2A or dietary methionine restriction attenuated atherosclerosis. Our findings demonstrate a pivotal causal role for methionine metabolism in proinflammatory responses and migration in monocytes/macrophages, and identify MAT2A as a promising therapeutic target for atherosclerotic disease.
Results
MAT2A-mediated methionine metabolism is enhanced in monocytes/macrophages from patients and mice with advanced atherosclerotic plaques
Between January 2017 and December 2017, we enrolled a series of 387 stable patients aged 18 years or older with de novo coronary artery disease and an indication for percutaneous coronary intervention who underwent pancoronary optical coherence tomography (OCT) imaging of all three major epicardial coronary arteries (i.e., left anterior descending coronary artery, left circumflex coronary artery, and right coronary artery). OCT is the most reliable intravascular imaging modality for the detection of TCFA, which is an in vivo equivalent of a vulnerable plaque that places patients at high risk for adverse cardiovascular events22. Among these patients, 102 (26.4%) had at least one TCFA in the entire coronary vasculature, whereas the remaining 285 (73.6%) had no evidence of TCFA (Fig. 1A). Baseline characteristics of patients with and without TCFA are presented in the Supplementary Table 1. Patients with TCFA had more traditional coronary risk factors and higher serum lipid profiles compared with patients without TCFA. To avoid the influence of confounding variables, we selected 38 patient pairs matched for age, gender, traditional coronary risk factors (i.e., diabetes mellitus, dyslipidaemia, current smoking, hypertension, and chronic kidney disease [CKD]), and concomitant statin therapy. The clinical characteristics of these matched pairs were balanced (Supplementary Table 2). The matched cohort was considered to be representative of the main study cohort (Supplementary Table 3). A higher prevalence of vulnerable plaque characteristics was observed in TCFA-positive patients compared with TCFA-negative patients (Supplementary Table 4).

A The enrollment of TCFA-positive patients defined by OCT and schematic representation of the patients screened in the discovery cohort. This figure was created using images from PowerPoint and BioRender. Created in BioRender. wan, p. (2025) https://BioRender.com/dmkgnf0. B Partial least-squares discrimination analysis (PLS-DA) 3D score plots. C Volcano plot of significantly differential metabolites in monocytes. D Detection of monocytes methionine metabolism concentrations (n = 38 subjects). E The final FCT was determined by averaging three measurements performed at the thinnest part of the fibrous cap. F Vulnerable plaque characteristics between patients with high and low SAM/SAH levels (n = 38 subjects). G Multivariate logistic regression analysis revealing the relationship between high SAM/SAH levels and TCFA (n = 76 for total subjects). H Representative OCT images of a normal coronary artery, a vulnerable plaque, and a stable plaque. Scale bar = 5 mm. I Schematic of the methionine metabolism pathway. J The mRNA levels in monocytes isolated from TCFA-positive and TCFA-negative individuals (n = 38 subjects). UMAP visualization (K) and MAT2A expression (L) of human atherosclerotic carotid artery cells (n = 15 patients, GSE253903). UMAP visualization (M) and MAT2A expression (N) of human atherosclerotic carotid artery myeloid cells. Representative immunofluorescence images (O) and quantification (P) of MAT2A and CD11b in carotid plaques from symptomatic and asymptomatic patients (n = 6 subjects. Scale bar = 50 µm). C Two-tailed P-values were indicated. D The bar bands defined as 25th and 75th percentiles; the plot top line as median; two-tailed Mann–Whitney P-values are indicated. F The box shows the 25th, 50th and 75th percentiles of the data. The whiskers represent minimum and the maximum; Chi-squared test and two-tailed Mann–Whitney P-values was used. G The error bars represent the 95% CI for the OR, with their centers (red squares) indicating the point estimates; two-tailed P-values are shown. (J, P) Data are presented as the mean ± SD; two-tailed P-values are shown. L, N Log normalized using the ‘NormalizeData’ function in Seurat with default parameters. CKD chronic kidney disease, FCT fibrous cap thickness, MAT2A methionine adenosyltransferase Ⅱ alpha, OCT optical coherence tomography, SAH, S-adenosyl-homocysteine, SAM S-adenosyl-methionine, TCFA thin-cap fibroatheroma, UMAP uniform manifold approximation and projection. Source data are provided as a Source Data file.
In the matched cohort, we performed untargeted metabolomic profiling on human monocytes. TCFA-positive patients exhibited a distinct metabolic profile compared to TCFA-negative patients (Fig. 1B). Specifically, one-carbon metabolites SAH, methionine, and serine were significantly elevated in TCFA-positive patients (Fig. 1C, and Supplementary Fig. 1A). Pathway analysis further unveiled that methionine metabolism pathway was significantly enriched in the presence of TCFA (Supplementary Fig. 1B). Subsequent targeted mass spectrometry revealed that TCFA-positive patients had altered methionine metabolic profiles (i.e., methionine, SAM, and SAH) compared with their counterparts (Fig. 1D, and Supplementary Fig. 1C). The SAM/SAH ratio, which serves as an index of methylation potential that controls methyltransferase-mediated methylation reactions12, caught our attention (Fig. 1D). We then divided the aforementioned 76 patients into two groups according to the monocyte SAM/SAH ratio. Qualitative and quantitative OCT analysis showed that patients with high SAM/SAH levels had a higher prevalence of TCFA, thinner fibrous cap thickness (FCT), larger mean lipid arc, and smaller minimal lumen area (Fig. 1E, F). In the multivariate analysis, high SAM/SAH levels emerged as an independent predictor of the presence of TCFA (odds ratio [OR]: 4.422; 95% CI: 1.530-12.781; P = 0.0061) (Fig. 1G). Representative images illustrating vulnerable plaque characteristics in patients with high SAM/SAH levels are shown in Fig. 1H. Similar differences in monocyte methionine metabolism were observed in ApoE-/- mice fed a high-fat diet (HFD) (Supplementary Fig. 1D).
Methionine has been reported to be transported into cells by members of the solute carrier (SLC) family23. We next compared the expression of SLC transcripts in monocytes from TCFA-positive and TCFA-negative individuals but found no significant differences between the groups (Supplementary Fig. 2A). Similar results were observed in monocytes from HFD-fed ApoE-/- and control mice (Supplementary Fig. 2B). To further clarify the cause of increased monocyte methionine metabolism, we measured the levels of methionine metabolism-related enzymes, including MAT2A, SHMT2, SAHH, GLDC, MTHFR, and MTR in circulating monocytes (Fig. 1I). Of note, MAT2A mRNA levels demonstrated the highest fold-change in TCFA-positive patients compared with the TCFA-negative patients (Fig. 1J). Meanwhile, monocytes from ApoE-/- mice fed an HFD showed increased MAT2A expression, with similar promotion observed along the atherosclerosis progression (Supplementary Fig. 2C, D). By analyzing single cell RNA sequencing (scRNA-seq) data from human atherosclerotic carotid artery cells24, we visualized the expression of MAT2A in atherosclerotic plaques and confirmed its predominant localization to myeloid cells (Fig. 1K, L). Furthermore, MAT2A expression was highest in proinflammatory monocytes/macrophages, particularly those from symptomatic patients (Fig. 1M, N, and Supplementary Fig. 2E–G). In addition, an increased accumulation of MAT2A was observed in CD11b+ myeloid cells and CD68+ macrophages in carotid plaques from symptomatic patients (Fig. 1O, P, Supplementary Fig. 2H). Similarly, elevated MAT2A expression in CD68+ macrophages was observed in aortic sinus lesions of ApoE-/- mice with advanced atherosclerotic plaques. (Supplementary Fig. 2I). Therefore, these findings indicate that the MAT2A-mediated methionine metabolism is enhanced in monocytes/macrophages during atherosclerosis development.
MAT2A in monocytes/macrophages is indispensable for mediating the progression of atherosclerosis
To confirm the functional role of macrophage MAT2A in atherosclerotic plaque progression, we generated homozygous myeloid-specific MAT2A knockout on an ApoE-/- mice (MAT2Afl/flLyz2CreApoE-/-) by crossing MAT2Afl/flApoE-/- mice with Lyz2-Cre mice, hereafter referred to as MAT2ACKOApoE-/- mice and fed them an HFD as indicated (Fig. 2A and Supplementary Fig. 3A, B). Indeed, the monocyte methionine catabolic products were significantly reduced following MAT2A deletion (Fig. 2B and Supplementary Fig. 3C). Additionally, methionine can be recycled through the methionine salvage pathway following the SAM-dependent polyamine biosynthesis. Using 13C-labeled methionine, we examined the effects of MAT2A knockout on the methionine cycle and polyamine levels. We observed that upon MAT2A depletion, methionine was preferentially channeled into methylation processes rather than the polyamine synthesis pathways (Supplementary Fig. 4A–C). After 16 weeks of HFD feeding, the atherosclerotic lesion area in the whole aortas and aortic roots of MAT2ACKOApoE-/- mice was significantly smaller than that of MAT2Afl/flApoE-/- mice (Fig. 2C–E). Moreover, atherosclerotic plaques from MAT2ACKOApoE-/- mice exhibited ameliorated signs of plaque vulnerability compared with MAT2Afl/flApoE-/- mice, characterized by smaller plaque area, lower necrotic core, but higher collagen content (Fig. 2F, G). A more detailed assessment of aortic composition revealed that MAT2ACKOApoE-/- mice had reduced infiltration of CD68+ macrophages and a significantly lower vulnerability index (an established score of several morphological markers indicating the risk of plaque rupture25) (Fig. 2H, I, Supplementary Fig. 5A). No significant differences were observed in body weight or serum lipid profiles (total cholesterol, triglycerides, LDL-C, and HDL-C) between the two genotypes (Supplementary Fig. 5B). Furthermore, we administered an adeno-associated virus (AAV8) expressing MAT2A to mice and found that it exacerbated atherosclerosis progression. However, this effect not reversed by folic acid (FA) supplementation, despite effective reduction of Hcy levels (Supplementary Fig. 6A–D). Meanwhile, the weight and lipid profiles were comparable across all groups (Supplementary Fig. 6E), consistent with the conclusion that the pro-atherogenic effect of MAT2A is independent of Hcy.

Both groups of 8-week-old female and male MAT2Afl/flApoE-/- and MAT2ACKOApoE-/- mice were fed an HFD for 16 weeks. A Schematic figure showing the experimental strategy for the HFD feeding and the subsequent analysis. This figure was created using images from Photoshop and BioRender. Created in BioRender. wan, p. (2025) https://BioRender.com/3iunh0m. B SAM and SAH concentrations as well as SAM/SAH ratio for mice monocyte cell lysate mass spectrometry samples (n = 6). C Representative photographs of atherosclerotic plaques in the aortic arches and their branches in the 2 groups (n = 10 mice per genotype). Representative images and quantification of Oil Red O-stained aortas (D, n = 10 samples. Scale bar = 2 mm) and aortic roots (E, n = 10 independent experiments. Scale bar = 200 µm). F, H, E and Masson’s trichrome staining of representative aortic root sections. Scale bar = 200 µm. G Quantification of plaque area, necrotic core, and collagen (n = 10 samples). H and I, Immunohistochemistry of CD68+ macrophages and vulnerability index in the aortic roots (n = 10 samples). Scale bar = 100 µm. Data are presented as the mean ± SD and comparisons were made using unpaired Student’s t-test; the two-tailed P-values are shown. α-SMA α-smooth muscle actin, HFD high-fat diet. Source data are provided as a Source Data file.
Collectively, these data provide compelling evidence that MAT2A-mediated methionine metabolism in monocytes/macrophages plays a key role in enhancing plaque vulnerability and promoting atherosclerosis development.
MAT2A is required to orchestrate the inflammatory response and migratory capacity of monocytes/macrophages
To further elucidate the underlying mechanism alleviating atherosclerosis mediated by MAT2A deficiency, we used RNA sequencing (RNA-seq) to assess the gene expression profiles in bone marrow-derived macrophages (BMDMs) from MAT2ACKO mice and control MAT2Afl/fl mice. A total of 2561 significantly downregulated differentially expressed genes (DEGs) and 2757 significantly upregulated DEGs were detected in MAT2A-deficient conditions (Supplementary Fig. 7A). Enrichment analysis revealed that the downregulated DEGs were significantly enriched in pathways related to inflammation and chemotaxis (Fig. 3A), suggesting potential involvement upon MAT2A knockdown. Moreover, several proinflammatory and migratory genes were significantly downregulated in the absence of MAT2A (Supplementary Fig. 7B). In macrophages isolated from atherosclerotic plaques, we observed a similar downregulation of inflammatory and migratory genes, as well as alterations in relevant pathways in MAT2ACKOApoE-/- group compared with controls, recapitulating the changes identified in BMDMs (Supplementary Fig. 7C–G). Based on these findings, we focused subsequent investigations on the role of MAT2A in regulating the inflammatory and migratory capacities of macrophages during atherosclerosis progression.

A Bubble chart of GO term enrichment analysis of downregulated DEGs based on RNA-seq in BMDMs of MAT2ACKO compared with MAT2Afl/fl mice. Flow cytometry was performed to quantify CD45+CD11b+CD115+Ly6Chi monocytes from bone marrow or peripheral blood (B, n = 6 independent experiments), MHC Ⅱ and CCR2 (C, n = 6 samples) on CD45+CD68+ macrophages from aortic plaques of MAT2ACKOApoE-/- and MAT2Afl/flApoE-/- mice fed a 16-week HFD. D–J Analysis of BMDMs from MAT2ACKO and MAT2Afl/fl mice treated with or without SAM (200 µM) following LPS and IFN-γ stimulation. Representative flow cytometry plots and quantification of CD45+CD11b+CD86+ macrophages (D, n = 6 samples), transwell (E, n = 6 samples, Scale bar = 100 µm), and wound assay (F, n = 6 samples. Scale bar = 500 µm). The mRNA levels of Il1b, Il6, Nos2 (G, n = 6 samples) and Ccl2, Ccl7 and Cxcl1 (H, n = 6 samples) measured by qRT-PCR. I, J The THP-1 cells were transfected with siNC or siMAT2A followed by LPS and IFN-γ treatment with or without SAM (200 µM). The mRNA levels of Il1b, Il6, Nos2, Ccl2, Ccl7 and Cxcl1 were analyzed by qRT-PCR (n = 6 samples). A P-values were derived from hypergeometric distribution. Data are presented as the mean ± SD. B, C Unpaired Student’s t-test was used; two-tailed P values are shown. D–J One-way ANOVA was used; the adjusted P-values are shown. 8-week-old female and male mice were both used. BL blood, BM bone marrow, BMDMs bone marrow-derived macrophages, DEGs differentially expressed genes, GO gene ontology, IFN-γ interferon-γ, LPS lipopolysaccharide. Source data are provided as a Source Data file.
Analysis of bone marrow and peripheral blood monocytes revealed a significant reduction in Ly6Chi monocytes in MAT2ACKOApoE-/- mice compared with MAT2Afl/flApoE-/- controls (Fig. 3B and Supplementary Fig. 8A). To determine the effect of MAT2A knockout on myelopoiesis, we quantified the hematopoietic stem cells (HSCs) and progenitor cells bone marrow compartment as well as circulating monocytes. Compared with the controls, the MAT2ACKOApoE-/- mice exhibited a reduction in lineage−Sca-1+c-Kit+ cells (LSKs) hematopoietic progenitor cells. Furthermore, the numbers of myeloid progenitors, including granulocyte-monocyte progenitors (GMP) and common myeloid progenitors (CMP), were significantly reduced in the MAT2ACKOApoE-/- mice. Consistently, peripheral blood monocyte counts were reduced in MAT2ACKOApoE-/- mice, indicating that MAT2A regulates bone marrow stem cell proliferation and myeloid bias, thereby promoting the accumulation of circulating monocytes (Supplementary Fig. 8B–F). Additionally, the proportions of MHC Ⅱ+ and CCR2+ macrophages in aortic plaques were lower in the MAT2ACKOApoE-/- mice (Fig. 3C and Supplementary Fig. 9A). We further observed MAT2A deletion reduced the expression of proinflammatory and chemotactic genes in plaque macrophages (Supplementary Fig. 9B). Next, we found that chemotaxis towards CCL2 was attenuated in peripheral blood monocytes from MAT2ACKOApoE-/- mice (Supplementary Fig. 10A, B). Moreover, when we labeled Ly6Chi monocytes with Fluoresbrite YG microspheres. we found significantly fewer recruited into the aortic plaques of MAT2ACKOApoE−/− mice compared with controls (Supplementary Fig. 10C). Subsequently, we injected PKH67-labeled BMDMs into HFD-fed ApoE-/- mice recipients and found significantly fewer that PKH67 positive cells of MAT2ACKO origin in aortic plaques compared with those receiving MAT2Afl/fl BMDMs (Supplementary Fig. 10D, E). Overall, MAT2A deletion diminishes the recruitment of monocytes/macrophages to atherosclerotic plaques.
We then stimulated MAT2ACKO and MAT2Afl/fl BMDMs in vitro with lipopolysaccharide (LPS) and interferon-γ (IFN-γ). MAT2A deficiency significantly reduced the proportion of proinflammatory (CD11b+CD86+) macrophages and intracellular methionine metabolism, which was reversed by SAM supplementation, indicating the regulatory role of MAT2A on proinflammatory macrophages depends on SAM production (Fig. 3D and Supplementary Fig. 10F). Similarly, SAM restored the decreased migration of MAT2ACKO BMDMs into the well or wound areas (Fig. 3E, F), which was consistent with the effects observed upon MAT2A depletion using RNA interference in RAW264.7 cells (Supplementary Fig. 10G, H). Specifically, MAT2A deficiency resulted in lower expression of proinflammatory and chemotactic genes compared with the control, and SAM supplementation partially rescued this suppression (Fig. 3G–J, and Supplementary Fig. 10I, J).
All these results suggest that MAT2A-mediated methionine metabolism is critical to sustaining the proinflammatory and migratory properties of monocytes/macrophages.
MAT2A drives activation and migration of proinflammatory macrophages by modulating H3K4me3
Methionine metabolism has been reported to regulate histone methylation and confer a defined methylation state in proinflammatory macrophages12. According to enrichment analysis of RNA-seq data, the downregulated DEGs were also significantly associated with one-carbon metabolism and histone H3-K4 methylation (Fig. 3A). Consistently, the epigenetic modifiers that cause H3K4me3 deposition (including Setd1a, Setd1b, Kmt2a, Kmt2b, and Wdr5) were significantly downregulated as core components following MAT2A deletion (Supplementary Table 6). In ApoE-/- mice treated with AAV-MAT2A, administration of OICR-9429 (small-molecule inhibitor of MLL-WDR5 complex26,27) attenuated MAT2A-induced H3K4me3 enrichment in inflammatory and migratory genes, thereby alleviating atherosclerosis progression (Supplementary Fig. 11A–C). Thus, targeting H3K4me3 deposition mediated by MAT2A-governed epigenetic modifiers could counteract its pro-atherosclerotic effects. Immunoblot analysis of histone modifications in BMDMs confirmed that MAT2A deficiency, which depleted the intracellular SAM pool, led to a marked decrease in H3K4me3, a more modest reduction in H3K36me3, but no change in H3K27me3 (Fig. 4A). In aortic root plaques, the proportion of macrophage-associated nuclei stained positive for H3K4me3 was similarly reduced in MAT2ACKOApoE-/- mice (Fig. 4B). To identify MAT2A target genes involved in histone methylation during atherosclerosis progression, we isolated BMDMs from MAT2ACKO and MAT2Afl/fl mice and subjected them to CUT&Tag assay using an anti-H3K4me3 antibody. Principal component analysis revealed clear separation based on binding peaks (Supplementary Fig. 12A). We observed differential H3K4me3 binding in MAT2A-deficient conditions, and the data indicated that H3K4me3 were predominantly enriched at the promoter centric (Supplementary Fig. 12B, C). The data also showed that H3K4me3 was enriched within 2 kb upstream or downstream of gene transcription start sites (Fig. 4C). To identify H3K4me3-target genes in MAT2ACKO BMDMs, we integrated the CUT & Tag and RNA-seq data. By overlapping annotated H3K4me3-modified and downregulated DEGs, we found 206 downregulated genes with decreased H3K4me3 modification (Fig. 4D). Further gene enrichment analysis revealed that the downregulated genes with differential H3K4me3 were involved in inflammatory response including IL-1β production and migratory function (Fig. 4E, F). Simultaneously, we observed that H3K4me3 modification at the Il1b promoter was reduced upon MAT2A depletion (Supplementary Fig. 12D), which is consistent with previous reports indicating that SAM regulates IL-1β expression12. Additionally, among these downregulated genes, Aim2, Ccl2, and Mmp9 exhibited substantial H3K4me3 modification based in genomic snapshots (Fig. 4G). Next, we observed that the MAT2A deficiency-mediated reduction in H3K4me3 enrichment at the promoter regions of Aim2, Ccl2, and Mmp9 was partially rescued by SAM supplementation (Fig. 4H). The reduced secretion of the AIM2, CCL2, and MMP9 into the cellular supernatant was also restored by SAM supplementation (Fig. 4I). Furthermore, serum concentrations of these proteins were lower in MAT2ACKOApoE-/- mice and TCFA-negative patients (Supplementary Fig. 12E, F).

A Immunoblots of H3K4me3, H3K36me3, and H3K27me3 levels in BMDMs from MAT2Afl/fl and MAT2ACKO mice (n = 3). B Representative immunofluorescence images and quantification for H3K4me3 and CD68 in aortic root plaque from MAT2Afl/flApoE-/- and MAT2ACKOApoE-/- mice fed a 16-week HFD (n = 7). Scale bar = 100 µm. C–G Analysis of BMDMs between MAT2ACKO and MAT2Afl/fl mice. C Heatmaps for H3K4me3 binding peaks in BMDMs from MAT2Afl/fl and MAT2ACKO mice. D Venn diagram reflecting overlapping downregulated genes with H3K4me3 modification combining the CUT&Tag and RNA-seq data. Bubble charts of GO (E) and KEGG (F) analysis according to downregulated genes with decreased H3K4me3 modification. G IGV tracks revealing the results of CUT&Tag reads (H3K4me3 binding) distributions in indicated genes. H, I Analysis of BMDMs between MAT2ACKO and MAT2Afl/fl mice treated with or without SAM (200 µM) in the presence of LPS and IFN-γ. H ChIP-qPCR validation of H3K4me3 enrichment on the promoter regions of Aim2, Ccl2, and Mmp9 (n = 6 samples). I ELISA analysis of AIM2, CCL2, and MMP9 in cellular supernatant (n = 6 independent experiments). Data are presented as the mean ± SD. A, B Unpaired Student’s t-test was used; two-tailed P values are shown. E P-values were derived from hypergeometric distribution. H and I One-way ANOVA was used; the adjusted P-values are shown. Both 8-week-old female and male mice were used. ChIP-qPCR chromatin immunoprecipitation-qPCR, H3K4me3 histone h3 lysine 4 trimethylation, H3K27me3 histone H3 lysine 27 trimethylation, H3K36me3 histone h3 lysine 36 trimethylation, KEGG kyoto encyclopedia of genes and genomes, TSS translation start site. Source data are provided as a Source Data file.
In summary, we identified H3K4me3 target genes (including Aim2, Ccl2, and Mmp9) modulated by methionine metabolism and demonstrated that MAT2A deletion disrupts H3K4me3 signaling, thereby limiting the proinflammatory and migratory capacities of macrophages and impeding atherogenesis.
Inhibition of MAT2A and dietary intervention in methionine metabolism ameliorates atherosclerosis
To further evaluate the therapeutic potential of targeting MAT2A-mediated methionine metabolism in atherosclerosis, we employed the MAT2A inhibitor FIDAS-5 in ApoE-/- mice starting at 10 weeks of HFD for 6 weeks (Fig. 5A). We found that monocyte methionine metabolism was significantly suppressed by FIDAS-5 treatment (Supplementary Fig. 13A). Notably, FIDAS-5 reduced atherosclerotic lesion area in the whole aortas and aortic roots (Fig. 5B–E). The plaque area and the proportion of necrotic core were effectively decreased in the aortic roots underlying MAT2A inhibition, whereas the collagen content in their plaques was increased (Fig. 5F, G). Furthermore, the CD68+ macrophages content and the plaque vulnerability index were reduced for the FIDAS-5 condition (Fig. 5H, I, Supplementary Fig. 13B). We observed the frequency of Ly6Chi monocyte subsets was lower in the bone marrow and blood following FIDAS-5 treatment (Supplementary Fig. 13C). FIDAS-5 administration also downregulated H3K4me3 levels and reduced transcription of Aim2, Ccl2, and Mmp9 in peripheral blood monocytes (Supplementary Fig. 13D, E), while the differences in body weight and serum lipid levels between the two groups were not statistically significant (Supplementary Fig. 13F). As expected, additional supplementation of SAM under conditions of MAT2A inhibition by FIDAS-5 would further exacerbate atherosclerosis (Supplementary Fig. 14, A–C), confirming that MAT2A promotes atherosclerosis by regulating SAM availability. Further analysis following HCY supplementation in mice with FIDAS-5 management revealed no significant alterations in monocytes/macrophages inflammatory and migratory function, suggesting that MAT2A inhibition regulates macrophage function independently of Hcy (Supplementary Fig. 15A, B).

A–I The HFD-fed ApoE-/- mice were received either FIDAS-5 or vehicle every other day for 6 weeks. A, Flowchart illustrating the experimental procedure. This figure was created using images from Photoshop and BioRender. Created in BioRender. wan, p. (2025) https://BioRender.com/h76jjnw. B Representative photographs of atherosclerotic plaques in the aortic arches and their branches. Representative images and quantification of Oil Red O-stained aortas (C, n = 8. Scale bar = 2 mm) and aortic roots (D and E, n = 8. Scale bar = 200 µm). F Representative images of H&E and Masson’s trichrome-stained aortic roots. Scale bar = 200 µm. G Quantification of plaque area, percentage of necrotic core, and collagen of aortic roots (n = 8 per group). H, I Immunohistochemistry of CD68+ macrophages and vulnerability index in the aortic root plaque of FIDAS-5 treated mice, n = 8 per group. Scale bar = 100 µm. J–R Representative images of female and male mice fed MRD or CD for 6 weeks after a 10-week HFD. J Schematic figure showing the experimental strategy and subsequent analysis. This figure was created using images from Photoshop and BioRender. Created in BioRender. wan, p. (2025) https://BioRender.com/h76jjnw. K Representative images of aortic arches and their branches in the indicated group. Representative images and quantification of Oil Red O-stained aortas (L, n = 8. Scale bar = 2 mm) and aortic roots (M and N, n = 8. Scale bar = 200 µm). O Representative images of H&E and Masson’s trichrome-stained aortic root. P Quantification of plaque area, percentage of necrotic core, and collagen, n = 8. Scale bar = 200 µm. Q and R Immunohistochemical staining CD68+ macrophages and vulnerability index, n = 8. Scale bar = 100 µm. Data are presented as mean ± SD and comparisons were made using unpaired Student’s t-test; two-tailed P values are shown. 8-week-old female and male ApoE-/- mice were used. FIDAS-5, a MAT2A inhibitor; MRD, methionine-restricted diet. Source data are provided as a Source Data file.
As an essential amino acid, methionine is primarily sourced from dietary intake and could be regulated via dietary pattern17,28. To determine the efficacy of dietary methionine intervention against atherosclerosis, ApoE-/- mice were fed either a methionine-restricted diet (MRD)17,28 or a control diet following HFD initiation (Fig. 5J). As anticipated, MRD reduced methionine metabolism in peripheral blood monocytes (Supplementary Fig. 16A). The aortic arches showed less plaque burden in mice with MRD (Fig. 5K). Meanwhile, atherosclerotic lesions across entire aortas and aortic sinuses were diminished by MRD (Fig. 5L–N). Subsequent analysis of lesions in the aortic roots revealed a decrease in the atherosclerotic plaque and necrotic core areas, while MRD feeding increased the collagen content (Fig. 5O, P). Immunohistochemical staining showed that the MRD attenuated CD68+ macrophage accumulation and decreased plaque vulnerability (Fig. 5Q, R, Supplementary Fig. 16B). Bone marrow and peripheral blood monocytes of the MRD group showed a tendency toward reduction of differentiation into Ly6Chi monocytes (Supplementary Fig. 16C). Moreover, we demonstrated that the H3K4me3 levels and the Aim2, Ccl2, and Mmp9 expression were also reduced in peripheral blood monocytes from mice with MRD (Supplementary Fig. 16D, E). In contrast, MRD did not affect body weight, serum cholesterol, and lipoprotein levels (Supplementary Fig. 16F). Interestingly, MRD did not alter the progression of atherosclerosis in MAT2ACKOApoE-/- mice, demonstrating that the atheroprotective effect of dietary methionine restriction is dependent on MAT2A (Supplementary Fig. 17A–C).
Taken together, both pharmacological inhibition of MAT2A and dietary methionine restriction alleviate atherosclerosis by attenuating the migration of proinflammatory monocytes/macrophages.
MAT2A is induced by the norepinephrine-mTOR-c-MYC communication axis in monocytes/macrophages during atherosclerosis progression
Sympathetic activation promotes norepinephrine release in atherosclerosis patients, which is involved in the concomitant state of chronic stress29,30,31. Norepinephrine-cAMP shaft increases MAT2A activity, which may be accompanied by an increase in SAM production32. Of note, we measured serum norepinephrine concentrations and concluded that TCFA-positive patients had significantly elevated levels of norepinephrine (Fig. 6A). To evaluate whether norepinephrine is associated with the upregulation of MAT2A and MAT2A-mediated methionine metabolism in atherosclerosis, we performed an integrative analysis and confirmed that serum norepinephrine levels correlated with the monocytes SAM/SAH ratio (Fig. 6B). Moreover, patients with high levels of both the SAM/SAH ratio and norepinephrine exhibited the highest incidence of TCFA, whereas those with low levels of both had the lowest incidence (Fig. 6C).

A Norepinephrine levels in serum from TCFA-positive and TCFA-negative individuals (n = 38 independent experiments). B Correlation between serum the ratio of SAM/SAH and norepinephrine levels. C The percentage of patients presenting with TCFA in 3 coronary arteries varied according to their levels of the SAM/SAH ratio and norepinephrine. D MAT2A expression in peripheral blood monocytes from 6-OHDA-treated ApoE-/- mice (n = 3, one representative experiment out of three was shown). E Representative images and quantification of H&E and Oil Red O staining in aortic root treated with 6-OHDA (250 mg/kg) and AAV-MAT2A, n = 6. Scale bar = 100 µm. F Immunoblots of aortic plaques from ApoE-/- mice treated with 6-OHDA (n = 3). G, Mat2a mRNA expression in BMDMs pre-treated with 20 nM RAPA followed by 10 mM norepinephrine (n = 6, one representative experiment out of three was shown). H Transcript levels of c-Myc and Mat2a in BMDMs transfected with the indicated siRNAs (n = 6). I Occupancy analysis of c-MYC by ChIP-qPCR in BMDMs treated with DMSO vehicle or RAPA (20 nM) (n = 6). A Two-tailed Mann–Whitney P-values are indicated; Data are shown as median with IQR. B Pearson correlation coefficient test was used; the regression coefficients and two-tailed P-values are shown. C Chi-squared test was used; Data are shown as %. D–F Unpaired Student’s t-test was used; the two-tailed P-values are shown; Data are presented as mean ± SD. G, I Two-way ANOVA was used; The adjusted P-values are shown; Data are presented as mean ± SD. H One-way ANOVA was used; The adjusted P-values are shown. 8-week-old female and male ApoE-/- mice were used. Data are presented as mean ± SD. AAV adeno-associated virus, RAPA rapamycin, 6-OHDA 6-Hydroxydopamine. Source data are provided as a Source Data file.
Subsequently, we evaluated the role of norepinephrine in regulating MAT2A-mediated methionine metabolism during atherosclerosis. We confirmed that serum norepinephrine levels were predominantly elevated in ApoE-/- mice subjected to prolonged atherosclerosis development (Supplementary Fig. 18A). We further administered 6-hydroxydopamine (6-OHDA) to deplete norepinephrine33,34 in ApoE-/- mice and found significantly reduced monocyte MAT2A expression and methionine metabolism (Fig. 6D and Supplementary Fig. 18B). In addition, an intervention model in HFD-fed ApoE-/- mice using both AAV-MAT2A and 6-OHDA (Supplementary Fig. 18C). 6-OHDA treatment markedly attenuated plaque burden, whereas MAT2A overexpression reversed this effect (Fig. 6E), with no differences in body weight and lipid profiles across all groups (Supplementary Fig. 18D). Furthermore, as anticipated, 6-OHDA administration failed to reduce atherosclerosis progression in MAT2A knockout mice, indicating that the pro-atherosclerotic effects of MAT2A in macrophages is dependent on sympathetic activation (Supplementary Fig. 19A–C). Above all, MAT2A is induced by norepinephrine to promote plaque vulnerability.
The mechanistic target of rapamycin (mTOR), a key regulator of metabolic balance and macrophage fate, is activated by neurotransmitter uptake35,36,37. By comparing levels of the mTOR in aortas isolated from ApoE-/-mice fed an HFD with or without 6-OHDA treatment, we found that 6-OHDA inhibited phosphorylation of mTOR and S6. To identify mTOR-dependent transcription factors regulating of MAT2A, we screened candidates and found that c-MYC depletion specifically reduced Mat2a expression (Supplementary Fig. 20A). Consistently, c-MYC was downregulated by 6-OHDA conditions (Fig. 6F). In c-MYC-deficient BMDMs, the insulin-induced increase in MAT2A expression driven by physiological mTOR pathway activation38 was abolished, demonstrating the transcriptional dependence of MAT2A on the mTOR-c-MYC axis (Supplementary Fig. 20B). Furthermore, norepinephrine promoted Mat2a expression, and this effect was blocked by mTOR inhibition with rapamycin (RAPA)39 (Fig. 6G). Moreover, c-MYC depletion reduced MAT2A levels, whereas MAT2A silencing did not induce c-MYC downregulation, demonstrating the transcriptional regulation potential by which c-MYC controls MAT2A expression (Fig. 6H). CHIP-qPCR further suggested that c-MYC binds to MAT2A, and this interaction was prevented by RAPA treatment (Fig. 6I). Additionally, intervention targeting norepinephrine receptors via propranolol administration or ADRB2 genetic deficiency consistently reduced MAT2A expression and downstream molecules during atherosclerosis progression (Supplementary Fig. 21A–I). Therefore, MAT2A is transcriptionally activated by norepinephrine-evoked mTOR-c-MYC pathway.
Taken together, our results establish that MAT2A-mediated methionine metabolism is modulated by the norepinephrine-mTOR-c-MYC axis in atherosclerosis.
The combination of SAM/SAH and norepinephrine is associated with vulnerable plaque and long-term clinical outcomes in the validation cohort
Between January 2018 and December 2019, 845 stable patients were eligible for pancoronary OCT imaging and 100 matched patient pairs were selected as the validation cohort based on age, gender, traditional coronary risk factors, and concomitant statin therapy after pairwise comparison. Circulating monocyte methionine metabolism levels were determined by targeted mass spectrometry, and serum norepinephrine levels were measured by ELISA (Fig. 7A). Patients with TCFA (n = 237, 28.0%) had a higher prevalence of coronary risk factors than those without TCFA (n = 608, 72.0%) (Supplementary Table 7). 100 matched patients had similar clinical characteristics and well balanced as shown by the standardized mean difference (SMDs) of the baseline characteristics (Supplementary Table 8). Clinical characteristics in the matched cohort were representative of the entire cohort (Supplementary Table 9). Patient characteristics of both the discovery and validation cohorts were comparable (Supplementary Table 10). TCFA-positive patients exhibited a higher prevalence of vulnerable plaque characteristics (Supplementary Table 11).

A Study design in the validation cohort. This figure was created using images from PowerPoint and BioRender. Created in BioRender. wan, p. (2025) https://BioRender.com/qbkjhvs. Detection of monocytes SAM and SAH concentrations (B), and SAM/SAH ratio (C) of TCFA-positive and TCFA-negative individuals (n = 100 subjects). D Serum norepinephrine levels from TCFA-positive and TCFA-negative individuals (n = 100 independent experiments). Qualitative and quantitative OCT analysis of vulnerable plaque characteristics between patients with high and low SAM/SAH levels (E) or with high and low norepinephrine levels (F) (n = 100 independent experiments). Pearson correlation analysis of monocytes SAM/SAH ratio (G) or serum norepinephrine levels (H) with the thinnest FCT and mean lipid arc. I Two-tailed Pearson’s linear regression analysis between the SAM/SAH ratio and norepinephrine levels. J Multivariate logistic regression analysis depicting the relationship between the SAM/SAH ratio, norepinephrine levels, and TCFA (n = 200 for total subjects). K Receiver operating characteristic curve of SAM/SAH, norepinephrine, and combined both for TCFA. L HR for incident 5-year MACE based on multivariable Cox proportional hazards regression analysis. Adjusted for age, gender, traditional coronary risk factors, and statin at discharge (n = 200 for total subjects). M A Kaplan-Meier survival curve plots the 5-year MACE-free survival among 4 subgroups. N MAT2A-mediated monocyte methionine metabolism is closely associated with the presence of TCFA in patients. MAT2A, which is induced by the norepinephrine–mTOR–c-MYC axis, is critical for endowing monocytes/macrophages with proinflammatory state and migratory capacity during the development of atherosclerosis through H3K4me3 modification. Myeloid-specific MAT2A ablation, pharmacological blockade with FIDAS-5, or a low-methionine diet attenuate monocyte/macrophage inflammation and migration, thereby reducing atherosclerotic progression and plaque vulnerability. This figure was created using images from PowerPoint and BioRender. Created in BioRender. wan, p. (2025) https://BioRender.com/gylosq5. B–D Two-tailed Mann–Whitney P-values are indicated. E, F The box shows the 25th, 50th and 75th percentiles of the data. The whiskers represent minimum and the maximum. Data are presented as % or median with IQR, Chi-squared test or Mann-Whitney test was used, and two-tailed P-values were calculated. G–I Pearson correlation coefficient test was used; the regression coefficients and two-tailed P-values are shown. J Data points indicate OR, and 95% confidence intervals are represented by line length; two-tailed P-values are shown. K The 95% confidence interval is shown between brackets. L Data points indicate HR and 95% CI were represented by line length. M P-values were calculated with log rank test. HR hazard ratio. Source data are provided as a Source Data file.
Consistent with the discovery cohort, SAM and SAH concentrations, as well as SAM/SAH levels, were elevated in TCFA-positive patients compared with TCFA-negative controls (Fig. 7B, C). Meanwhile, serum norepinephrine levels were significantly higher in TCFA-positive than in TCFA-negative patients (Fig. 7D). We then divided the 200 patients into two groups based on the monocyte SAM/SAH levels and, separately, on serum norepinephrine levels. OCT analysis showed that patients with high SAM/SAH levels had a higher prevalence of TCFA and other quantitative vulnerable plaque features (Fig. 7E). A similar pattern was observed in patients with high versus low serum norepinephrine levels (Fig. 7F). Furthermore, the SAM/SAH ratio showed a negative linear correlation with the thinnest FCT and a positive correlation with the mean lipid arc in plaques (Fig. 7G), whereas norepinephrine levels showed weaker but significant correlations with these vulnerable characteristics (Fig. 7H). Linear correlation analysis revealed a positive correlation between the monocyte SAM/SAH ratio and serum norepinephrine levels per patient (Fig. 7I). After adjustment for other confounding risk factors, both high SAM/SAH levels (OR: 3.779; 95% CI: 1.975–7.228; P < 0.0001) and high norepinephrine levels (OR: 2.572; 95% CI: 1.397–4.732; P = 0.0024) were independently associated with TCFA presence (Fig. 7J). The combination thereof performed significantly better than the SAM/SAH (delta AUC 0.052, 95% CI 0.006–0.098, P = 0.027) or the norepinephrine alone (delta AUC 0.069, 95% CI 0.026–0.113, P = 0.002) for predicting TCFA (Fig. 7K). Patients were followed for up to 5 years (median of 5.0 years [IQR: 4.2–5.0 years]), and major adverse cardiovascular events (MACE) were the composite of cardiac death, nonfatal myocardial infarction, and unplanned coronary revascularization. Multivariate Cox proportional hazards regression analysis showed that high SAM/SAH levels was independently associated with MACE (HR: 2.395; 95% CI: 1.157–4.956; P = 0.0186), whereas high norepinephrine levels increased the odds of MACE, although this was not statistically significant (Fig. 7L). The Kaplan-Meier curves showed that individuals with high SAM/SAH and norepinephrine levels had the highest incidence of MACE (Fig. 7M). Collectively, these findings highlight the potential clinical value of combining the SAM/SAH and norepinephrine for predicting the presence of vulnerable plaque and long-term adverse cardiovascular events.
Discussion
Atherosclerosis is a multifaceted chronic inflammatory disease that is a major contributor to global cardiovascular mortality40. The CANTOS, COLCOT, and LoDoCo2 trials are attempting to combat inflammatory risk and confirm a reduction in adverse cardiovascular events in patients treated with established anti-inflammatory paradigms2,3,4. Previous investigations have highlighted the inextricable link between the immunometabolic epigenetic modification and inflammation in atherosclerosis41. Methionine metabolism plays a pivotal role in modulating the proinflammatory state of macrophages by regulating the epigenetic switch and conferring the metabolic demands12. This study adds a piece to the jigsaw of metabolic-epigenetic regulation in atherosclerosis by providing compelling evidence that interference with MAT2A, a key enzyme in methionine metabolism, can suppress the migration and infiltration of proinflammatory monocytes/macrophages through H3K4me3 modification, thereby ameliorating atherosclerosis (Fig. 7N). We identify MAT2A as a target within the realm of immunometabolism to impede the progression of atherosclerosis and suggest potential for tailored interventions beyond standard anti-inflammatory strategies.
Monocytes/macrophages play a critical role in the initiation, progression, and eventual rupture of vulnerable plaques42. Notably, they exhibit considerable plasticity, coordinating intrinsic cellular pathways to manifest functional phenotypes in response to nutrient metabolic signals41. Methionine, as an essential amino acid, is crucial for maintaining cellular metabolic homeostasis, which sustains immune cell activation and differentiation12,41. However, the involvement of methionine metabolism in monocytes/macrophages in atherosclerosis remains unclear. In the present study, we observed that MAT2A-mediated methionine metabolism was significantly elevated in monocytes from TCFA-positive patients. This is consistent with the scRNA-seq analysis of monocytes in human atherosclerotic plaques, which revealed a high MAT2A expression profile. Following myeloid MAT2A knockout, myelopoiesis and the abundance of proinflammatory monocytes/macrophages was reduced, coupled with diminished migratory capacity. This led to a substantial reduction in both plaque vulnerability and progression. Thus, MAT2A serves to maintain the nuanced regulation of macrophage function, thereby imposing a critical constraint on the progression of atherosclerosis.
Within the intricate methionine metabolic circle, hyperhomocysteinemia has traditionally been considered a potential risk factor for atherosclerosis43. However, both inconsistent results across numerous studies and inconclusive findings from several large-scale intervention trials insinuate that high Hcy may be a concomitant phenomenon of atherosclerotic disease, rather than a causal factor15,44. Moreover, researchers have recently proposed that dietary methionine restriction is relevant to lifespan extension and confers pleiotropic benefits against age-related diseases in several model organisms17. In this study, administration of an MRD during atherogenesis significantly reduced plaque vulnerability and inflammation. In addition, our study demonstrated that the atherogenic effect of MAT2A-mediated methionine metabolism is independent of Hcy. We therefore propose that hyperhomocysteinemia-associated atherosclerotic pathology may result from MAT2A-mediated metabolite accumulation. It is noteworthy that plasma SAH, the precursor of Hcy, has been proposed to correlate with accelerated atherosclerosis45. This association is attributed to the primary loss of SAHH within smooth muscle and endothelial cells comprising the plaque, coupled with a noticeable decrease in the plasma SAM/SAH ratio46,47. Indeed, from the perspective of intracellular metabolism, we confirmed the increased SAM levels in monocytes from patients with vulnerable plaque, which were accompanied by an elevated SAM/SAH ratio. We inferred that the uniquely high expression of MAT2A in monocytes/macrophages, rather than changes in SAHH, accounts for this phenomenon. In addition, the heterogeneous handling of metabolites by distinct cell types explains the pronounced discrepancies in methionine metabolism, which manifest as notable variations between intracellular and plasma environments. Further in-depth clinical evidence is warranted to clarify the role of methionine metabolism in distinct cell types in atherosclerotic patients and to provide a basis for metabolic therapeutic strategies.
MAT2A catalyzes the conversion of methionine to SAM, a major methyl donor18. The alteration of histone methylation patterns occurs as SAM fuels histone methyltransferases17. In this study, we established that MAT2A-regulated methionine metabolism serves as a critical link between cellular immune responses and epigenetic reprogramming. SAM-mediated histone methylation drives metabolic shifts that induce an inflammatory phenotype of macrophages12. Specifically, we observed a significant decrease in the expression of H3K4me3 in BMDMs and plaques after blockade of MAT2A-mediated methionine. Furthermore, increased MAT2A-dependent H3K4me3 modification enhanced the transcription of the Aim2, Ccl2, and Mmp9. These genes have been implicated as proinflammatory or migratory facilitators during atherogenesis48,49,50, thereby demonstrating how macrophage rewiring of methionine metabolism contributes to atherosclerosis. In our study, although H3K36me3 expression decreased following MAT2A depletion, its reduction was less pronounced compared to H3K4me3. It has been suggested that the SETD2 is responsible for maintaining the activity of H3K36me3 through alternative metabolic pathways, thereby conferring greater resistance to SAM fluctuations51,52. Moreover, in contrast to other histone modifications such as H3K36me3 and H3K27me3, H3K4me3 exhibits a stronger dependence on SAM availability and heightened sensitivity to perturbations in methionine metabolism53. This observation is consistent with the elevated SAM levels we observed in both TCFA-positive patients and ApoE-/- mice, underscoring the dependence of macrophages on sufficient methionine metabolism to generate SAM for H3K4me3 methylation. Additionally, this phenomenon may be attributed to H3K4me3 enrichment at promoter regions, enabling more direct regulation of transcriptional initiation, whereas H3K36me3 is predominantly localized to gene body regions and facilitates transcriptional elongation54,55,56. It should be emphasized that our findings deepen the understanding of the intricate interplay between immune function and amino acid metabolism in the context of atherosclerosis. Specifically, our study elucidates the role of the monocyte/macrophage MAT2A-metabolic-epigenetic axis in the pathogenesis of atherosclerosis by revealing its regulation of H3K4me3-driven inflammatory and chemotactic gene expression. Since the genetic information stored in the context of chromatin is subject to various modifications57, further investigation is warranted to elucidate how MAT2A modulates atherosclerosis through its effect on chromatin structure and the precise deposition of specific chemical methylation modifications on chromatin.
Previous studies have established a crucial connection between neuroimmune responses and atherosclerosis, demonstrating that norepinephrine released upon sympathetic activation is critically involved in the disease process29,30,31,58. By combining high-resolution intravascular OCT for identifying vulnerable plaques, targeted mass spectrometry for detecting methionine metabolism in monocytes, and ELISA for measuring serum norepinephrine, we have demonstrated a clinical link between the neuroimmune-metabolic axis and atherosclerosis. In our study, both SAM/SAH and norepinephrine were positively associated with plaque vulnerability. Intriguingly, their combination outperformed either biomarker alone in correlating with the presence of TCFA and the likelihood of MACE during long-term follow-up. Furthermore, we show that norepinephrine upregulates macrophage MAT2A expression via the mTOR-c-MYC pathway during atherosclerosis progression. In addition to its known role in regulating autophagy59, we identify a previously unrecognized mechanism through which mTOR promotes atherosclerosis, namely via a crosstalk of MAT2A-mediated methionine metabolism through non-autophagic pathways. Future studies are needed to decipher the precise role of the mTOR-MAT2A axis in orchestrating macrophage autophagy and its contribution to atherosclerosis progression.
This study has several limitations. First, while we focused on macrophage-specific methionine metabolism, we did not explore the potential contributions of other cell types within the atherosclerotic plaque, such as endothelial cells and vascular smooth muscle cells. These cells likely compete for methionine metabolites, and their complex interactions could significantly influence the metabolic landscape. Moreover, our investigation was confined to the local plaque microenvironment and did not address the potential impact of systemic methionine levels. Circulating methionine, influenced by dietary intake and hepatic metabolism, serves as the primary source of intraplaque methionine. These fluctuations could modulate the metabolic reprogramming of all plaque cells, including macrophages, thereby influencing atherosclerosis progression. Future studies that incorporate cell-type-specific metabolic analysis and investigate the links between circulating methionine levels and intraplaque metabolism will be crucial to fully elucidate the role of this pathway in atherosclerosis. Second, atherosclerosis features a complex plaque immune microenvironment, and plaque immune cells, especially macrophages, are highly plastic and heterogeneous. We confirmed that MAT2A is primarily expressed in proinflammatory monocytes/macrophages by integrating scRNA-seq with in vivo experiments. However, the role of MAT2A in TREM2hi macrophages, a major plaque subtype distinct from proinflammatory macrophages, remains to be investigated. Third, we examined how MAT2A modulates macrophages via histone methylation and identified several proinflammatory and chemotactic genes activated by H3K4me3. However, MAT2A-mediated methionine metabolism regulates multiple biological processes, including protein synthesis, DNA methylation, and RNA methylation, and the broader implications of this regulation throughout atherosclerosis progression remain to be investigated.
In conclusion, our study elucidates that methionine metabolism shapes proinflammatory monocytes/macrophages to promote atherosclerosis progression. We demonstrated that the key enzyme MAT2A of methionine metabolism activates proinflammatory and chemotactic gene programs through H3K4me3 modification.
Methods
Study population
Between January 2017 and December 2019, a series of 1232 stable patients aged 18 years or older with de novo coronary artery disease and an indication for percutaneous coronary intervention underwent pancoronary optical coherence tomography (OCT) of all three main epicardial coronary arteries (i.e., left anterior descending coronary artery, left circumflex coronary artery, and right coronary artery) at the 2nd Affiliated Hospital of Harbin Medical University (Harbin, China). The main exclusion criteria were cardiogenic shock, end-stage renal disease, severe liver dysfunction, and contrast media allergy. Patients with left main disease, chronic total occlusion, or extremely tortuous or heavily calcified vessels were excluded due to the anticipated difficulty in performing OCT.
From this population, 387 patients (31.4%) were enrolled between January 2017 and December 2017 as the discovery cohort. A total of 845 participants (68.6%) were included between January 2018 and December 2019 as the validation cohort.
Among the 387 patients included in the discovery cohort, 102 patients presented with at least one thin-cap fibroatheroma (TCFA) in all 3 vessels, while the remaining 285 patients showed no evidence of TCFA. 38 matched pairs of TCFA-positive and TCFA-negative patients, balanced for age, gender, traditional coronary risk factors (i.e., diabetes mellitus, dyslipidaemia, current smoking, hypertension, and chronic kidney disease [CKD]), and concomitant statin therapy were selected. Similarly, from the 845 patients in the validation cohort, 100 matched pairs of patients with or without TCFA were included in the subsequent analysis. In addition, the effect sizes Cohen’ s d (standardized mean difference [SMD]) for pairwise comparisons was reported to evaluate the magnitude of the effect size d60. The standardized mean differences for sex were 0.07 and 0.02 in the discovery and validation cohorts, suggesting well-balanced matching and minimization of confounding effects due to sex in the two cohorts. Based on this solid statistical evidence, we deliberately utilized both male and female samples in our subsequent mechanistic studies to ensure the generalizability and translational relevance of our findings. This approach ensures that the identified mechanisms are not exclusive to a single sex, thereby strengthening the overall validity and impact of our conclusions for a broader population. Blood samples were collected before the interventional procedures, and isolated monocytes were stored at −80 °C.
This study was approved by the Ethics Committee of the 2nd Affiliated Hospital of Harbin Medical University (approval No. KY2015-182-04). All subjects provided written informed consent before enrollment. The experiments adhered to the principles of the Declaration of Helsinki.
OCT analysis, and inter- and intra- observer agreement
OCT was performed using a commercially available frequency-domain OCT system (ILUMIEN OPTIS System, Abbott Vascular, Santa Clara, CA). OCT imaging was analyzed according to previously established criteria and consensus using an off-line review workstation (Abbott Vascular)61. Coronary lesions were classified as lipidic plaques (low signal region with diffuse border) or fibrous plaques (homogeneous and signal-rich region). For each lipidic plaque, the lipid arc was measured at every 1 mm interval throughout the entire lesion; fibrous cap thickness (FCT) was measured 3 times at its thinnest part, and the average value was calculated. Lipid length was recorded on the longitudinal OCT view. The cross-section with minimum lumen area (MLA) was automatically selected by the ILUMIEN console algorithm along the entire length of the assessed coronary segment. Specifically, TCFA was defined as a lipid plaque with the thinnest fibrous cap thickness (FCT) < 65 μm and maximum lipid arc >90°.
OCT imaging was analyzed by two blinded expert readers (C.F. and S.L. [Shuang Li]), and a consensus reading was obtained from a third independent investigator (J.D.) when there was discordance between the two readers. Eighty patients were randomly selected to evaluate inter- and intra-observer agreement, as assessed by two independent investigators and by the same investigator at two separate time points with at least a two-week interval, respectively. The inter- and intra-observer agreement for TCFA were 0.86 and 0.92, respectively.
Clinical follow-up
Clinical follow-up was conducted at 1, 3, 6, and 12 months after discharge and then annually by telephone interview and/or clinical visit. Follow-up was censored at 5 years or at last known contact, whichever came first. Major adverse cardiovascular events (MACE) comprised cardiac death, nonfatal myocardial infarction, and unplanned coronary revascularization, which were defined according to the Academic Research Consortium guidelines62. Cardiac death was defined as death from myocardial infarction, cardiac perforation or pericardial tamponade, arrhythmia or conduction abnormalities, procedural complications, or any death in which a cardiac cause could not be excluded. Nonfatal myocardial infarction was diagnosed by the detection of raise and fall of cardiac biomarkers (preferably troponin) above the 99th centile of the upper reference limit, together with evidence of myocardial ischemia with at least one of the following criteria: ischemic symptoms; ECG changes indicative of new ischemia (new ST-T changes or new left bundle branch block); development of pathological Q waves in the ECG; and imaging evidence of new loss of viable myocardium or new regional wall motion abnormalities. Unplanned coronary revascularization was defined as any clinically driven (i.e., unstable angina or progressive angina) repeat percutaneous coronary intervention or surgical coronary artery bypass grafting, which initially was not planned after the index angiography and enrollment in the study.
Human atherosclerotic lesion analysis
Carotid atherosclerotic plaques were obtained from patients undergoing carotid endarterectomy at the 2nd Affiliated Hospital of Harbin Medical University. The baseline characteristics of patients with asymptomatic and symptomatic carotid atherosclerotic disease were shown (Supplementary Table 5). No samples were excluded from the analysis. Investigators were blinded to the patients’ clinical or experimental data. This study was performed in accordance with the Declaration of Helsinki and participants provided written informed consent.
Animal and atherosclerosis induction
All animal procedures were performed in accordance with the Principles of Animal Care established by the National Society for Medical Research and the Guidelines for the Care and Use of Laboratory Animals (NIH publication). All animal experiments were conducted in strict accordance with the guidelines of the Animal Care and Use Committee and were approved by the Institutional Animal Ethics Committee (approval No. YJSDW2023-039). All mice, both female and male, were 8 weeks of age at the initiation of experiments. We used sex-matched mice for each individual experiment. ApoE-/- mice (Beijing Vital River Laboratory Animal Technology) were fed a high-fat diet (HFD) containing 42.09% kcal fat, 19.19% kcal protein, and 38.72% kcal carbohydrate from Junke Biotechnology (JKHFD101, Nanjing, China) for 16 weeks, while control mice were fed a chow diet containing 15.8% kcal fat, 20.3% protein, and 63.9% kcal carbohydrate (JKHFD202, Nanjing, China). The mice had unrestricted access to rodent feed and water. All mice were housed under specific pathogen-free (SPF) conditions with a maintained temperature of 23 ± 3 °C, relative humidity of 55% to 60%, and a 12-h light/dark cycle.
Generation of myeloid-specific MAT2A knockout mice
MAT2Afl/fl homozygous and Lyz2-Cre mice were established by Beijing Vital River Laboratory Animal Technology (Beijing, China). Lyz2-Cre mice were crossed with MAT2Afl/fl mice to generate MAT2Afl/flLyz2Cre mice. MAT2Afl/flLyz2CreApoE-/- were generated by mating MAT2Afl/flLyz2Cre with ApoE-/-mice (Beijing Vital River Laboratory Animal Technology). 8-week-old MAT2Afl/flLyz2CreApoE-/-(MAT2ACKOApoE-/-) mice and control MAT2Afl/flApoE-/-mice were fed an HFD for 16 weeks.
Preparations of solutions for in vivo experiments
To establish a pharmacological MAT2A inhibition model63,64, ApoE-/- mice were administered the MAT2A inhibitor FIDAS-5 (10 mg/kg, HY-136144, MedChemExpress, New Jersey, USA) by intragastric gavage. Treatment was initiated after 10 weeks of HFD feeding and continued every other day for 6 weeks.
ApoE-/-mice were fed a methionine-restricted diet (MRD) (0.16% methionine, 0.00% cysteine) purchased from Junke Biotechnology (JK11051301Bi, Nanjing, China) for 6 weeks17,28.
For supplementation with folic acid (FA), animals were randomly assigned to an HFD or an HFD supplemented with FA (20 μg/mL, Sigma-Aldrich, St. Louis, MO, USA) in the drinking water for 16 weeks.
ApoE-/-mice were received standard or DL-Hcy (1.8 g/L, 6038-19-3, Sigma-Aldrich, St. Louis, MO, USA) -containing drinking water for 4 weeks to establish the Hcy supplementation model65.
To inhibit H3K4me3 deposition, ApoE-/- mice were administrated intraperitoneally with OICR-9429 (2.5 mg/kg, HY-16993, MedChemExpres, New Jersey, USA) once per 3 days for 8 weeks.
For SAM supplementation, mice were injected intraperitoneally with 2.5 mg/kg SAM (S5109, Selleck, USA), which was dissolved in sterile PBS daily for 5 weeks.
The beta-blocker propranolol was administered in drinking water at a concentration of 0.5 g/L. Based on average daily water consumption of 2.5 mL, this regimen resulted in an estimated daily propranolol dose of 35-40 mg/kg (HY-B0573B, MedChemExpres, New Jersey, USA).
For AAV-PCSK9DY administration, the virus was delivered via a single tail-vein injection at a dose of 1 × 1011 vector genomes/mouse. From day 1 after injection, the mice were fed either a chow diet or an HFD for 12 weeks.
ADRB2+/− mice were purchased from Cyagen Biosciences, Inc., and intercrossed to generate homozygous ADRB2⁻/⁻ mice.
Chemical ablation of sympathetic nervous fibers
For sympathetic nervous fibers ablation, ApoE-/-mice received intraperitoneal injections of 6-hydroxydopamine (6-OHDA, 250 mg/kg, H4381, Sigma-Aldrich, St. Louis, MO, USA) once weekly for 8 weeks33,34. Peripheral blood monocytes and aortas were collected at the end of the 16-week experimental period. Throughout the experiment, all mice were maintained on an HFD.
Adeno-associated virus (AAV) construction and injection
AAV-MAT2A was produced by JTSBIO (Wuhan, China). The plasmids were separately co-transfected with the pAAV2/8 trans-plasmid carrying the AAV rep and cap genes and the pAAV helper plasmid into HEK293T cells to generate the AAV8 adenoviruses. AAV-GFP was used as a negative control. Viral titers were measured via PCR using vector-specific primers. For AAV-MAT2A administration, the virus was delivered via a single tail-vein injection at a dose of 1 × 1011 vector genomes (vg)/mouse during the last 8 weeks of the 16-week HFD feeding.
Cell lines and primary cell culture
The RAW264.7 (TIB-71) and THP-1 (TIB-202) cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, Commonwealth of Virginia, USA). RAW264.7 cells were cultured in complete DMEM medium (C11995500BT, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% FBS (FND500, ExCell Bio, Suzhou, China). THP-1 cells were cultured in RPMI 1640 medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% FBS (FND500, ExCell Bio, Suzhou, China) and incubated with 10 ng/mL PMA (P1585, Sigma-Aldrich, St. Louis, MO, USA) for 48 h66. All cell lines were authenticated by examination of morphological and growth characteristics and confirmed to be mycoplasma-free.
Cell transfection
The RAW264.7 and THP-1 cells were seeded in 6-well plates to adhere and transfected 12 h later with Lipofectamine 2000 (11668-019, Thermo Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturer’s directions. Opti-MEM serum-free medium (Gibco, Grand Island, NY, USA) was used to dissolve the transfection mixtures. Using a synthesizer and the AUGC sequence, sense and antisense RNAs were synthesized, annealed into dsRNA, and then purified with HPLC and RPC. The following siRNA duplexes were used: Mouse MAT2A siRNA: GGATACAATTTACCACCTA (Ribo, Guangzhou, China); mouse c-MYC siRNA: CCGUACAGCCCUAUUUCAUTT AUGAAAUAGGGCUGUACGGTT (GenePharma, Shanghai, China); mouse HIF1a siRNA: GAUGUGACAUCCACAGACGAA UUCGUCUUGGGAUGUCACUAG (GenePharma, Shanghai, China); mouse PPARγ siRNA: GGCCUCCCUGAUGAAUAAATT UUUAUUCAUCAGGGAGGCCTT (GenePharma, Shanghai, China); mouse SREBP1 siRNA: GAGCAGAGAUGGCUCUAAUTT AUUAGAGCCAUCUCUGCUCT (GenePharma, Shanghai, China); human MAT2A siRNA: GGAUCGAGGUGCU GUGCUUTT AAGCACAGCACCUCGAUCCTT (GenePharma, Shanghai, China). The medium was replaced with conditioned medium 6 h after transfection. For RNA interference, siRNA was transfected into the cell for 72 h, and the transfection efficiency was detected. Cells were maintained until the following experiments.
Preparations of solutions for in vitro experiments
For the indicated experiments, cells were stimulated with 50 ng/mL Interferon-γ (IFN-γ) (315-05, PeproTech, Cranbury, NJ, USA) and 100 ng/mL lipopolysaccharide (LPS) (L2630, Sigma-Aldrich, St. Louis, MO, USA) for 6 h prior to analysis.
To interfere with intracellular SAM levels, cells were cultured in complete medium containing freshly reconstituted 200 μM SAM (A7007, Sigma-Aldrich, St. Louis, MO, USA) for 48 h.
To investigate the effect of norepinephrine, cells were treated with 10 μM norepinephrine (HY-137158, MedChemExpress, New Jersey, USA) for 24 h. The compound was dissolved in DMSO (D8370, Solarbio, Beijing, China) and then diluted in culture medium to the final concentration.
To activate the mTOR signaling pathway, BMDMs were stimulated with insulin (100 nM, PB180432, Procell, Wuhan, China) for 15 h38. To inhibit mTOR activity, rapamycin (RAPA) (553210, Calbiochem, San Diego, CA) was dissolved in pure DMSO to a concentration of 10 mM. This was further diluted in cell medium to a final concentration of 20 nM as described before39. Cells were treated with it for 15 h.
Isolation and culture of BMDMs
Bone marrow cells were isolated using the reported procedures67. In brief, bone marrow cells were flushed out from the femurs and tibias of 8-week-old mice, and incubated for 7 days with complete DMEM medium (C11995500BT, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% FBS (FND500, ExCell Bio, Suzhou, China), 1% penicillin-streptomycin (C0222, Beyotime, Shanghai, China), and murine macrophage colony-stimulating factor (M-CSF, 10 ng/mL, 315-02-1MG, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) to induce BMDMs differentiation. BMDMs were stimulated with 50 ng/mL IFN-γ (315-05, PeproTech, Cranbury, NJ, USA) and 100 ng/mL LPS (L2630, Sigma-Aldrich, St. Louis, MO, USA) for 6 h.
Monocyte Isolation
Peripheral venous blood was collected from enrolled patients into K2-EDTA-coated tubes (Thermo Fisher Scientific, Waltham, MA, USA) and processed within 1 h. The serum and the monocyte-rich component were obtained by centrifugation (CF7D2, HITACHI, Japan). Monocytes were then isolated from the monocyte-rich component according to the protocol of the human peripheral blood monocytes separation kit (TBD2011H05, Tianjin, China).
Mouse monocytes were isolated from peripheral blood using the TBD Mouse Monocyte Isolation Kit (TBD2011M, Tianjin, China).
Untargeted metabolomics analyses
This study analyzed 76 independent biological replicates (38 from TCFA-positive patients and 38 from TCFA-negative patients). The cell samples were spiked with 1 mL of methanol, and added zirconia beads. The samples were centrifuged at 16000 g and 4°C for 15 min. The supernatant was evaporated to dryness under mild nitrogen and reconstituted in 50 μL of 50% acetonitril,e including internal standards, before performing UHPLC-HRMS/MS analysis. Chromatographic separation was performed on a ThermoFisher Ultimate 3000 UHPLC system with a Waters ACQUITY UPLC BEH Amide column (2.1 mm × 100 mm, 1.7 μm) and an Atlantis Premier BEH Z-HILIC column (2.1 mm × 100 mm, 1.7 μm). Untargeted metabolomics analysis was performed using a ThermoFisher Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometry in Heated Electrospray Ionization Positive and Negative mode, respectively. Spray voltage was set to 3.5 kV. Both Capillary and Aux Gas Temperature were 350 °C. Sheath gas flow rate was 40 (Arb). Auxgas flow rate was 10 (Arb). S-Lens RF Level was 50 (Arb). The full scan was operated at a high resolution of 35000 FWHM (m/z = 200) at a range of 60-900 m/z (positive) and 70-1000 m/z (negative) with AGC Target setting at 3 × 106. Simultaneously, the fragment ions information of the top 8 precursors each scan was acquired by Data-dependent acquisition (DDA) with HCD energy at 15, 3,0 and 45 eV, mass resolution of 17500 FWHM, and AGC Target of 1×105.
The raw data of UHPLC-HRMS/MS were preprocessed by Compound Discoverer (version 3.3, Thermo Fisher) with a metabolomics workflow template, where retention times alignment, compound detection, compound grouping, and structural identification were performed. The following parameters were applied in compound identification: (1) accurate mass to charge ratio (m/z) of parent ion, denoted as MS1; (2) mass spectra of fragment ions, denoted as MS2; and (3) chromatographic retention times (RT). Then, we ascertained the classification of these metabolites by cross-referencing each metabolite with the Human Metabolome Database (HMDB, https://hmdb.ca/). To identify metabolites that exhibited significant alterations between groups, a univariate statistical analysis was conducted on the internal standard-normalized peak intensity data. The metabolomics data analysis was performed with all identified metabolites utilizing MetaboAnalystR 6.0 (https://www.metaboanalyst.ca/). Quantitative Enrichment Analysis (QEA) was performed to integrate quantitative changes in their abundance. The metabolites were mapped into the KEGG database, and the statistical significance of the changes in a pathway’s activity between two groups was evaluated by global testing. The abundances of the annotated metabolites are provided in Supplementary Data 1.
13C-labeled isotope metabolic flux assay
Cellular metabolic flux experiments were performed based on a published study68. Methionine is specifically labeled with 13C (200 μm, HY-NO326S5, MedChemExpress, New Jersey, USA), allowing for the tracking of methyl transfer in biochemical processes. The isolated BMDMs were cultured in a methionine-free medium for 24 h. Then, both groups of BMDMs were supplemented with 13C-labeled methionine and incubated for 12 h. After incubation, the cells were lysed for quantitative analysis of 13C-labeled methionine and its metabolites. UHPLC-MS/MS analysis was used for accurate detection of these metabolites.
Mass spectrometry and metabolite quantification
Human monocytes were isolated from the independent discovery (38 TCFA-positive and 38 TCFA-negative patients, n = 76) and validation (100 TCFA-positive and 100 TCFA-negative patients, n = 200) cohorts. Cell lysates were prepared by extraction with an ice-cold mixture of methanol and water (3/1, v/v) for 20 min on dry ice and snap-frozen in liquid nitrogen. On the day of analysis, samples were vortexed and then centrifuged at 13,300 × g for 10 min at 4 °C. A quality control sample was prepared by mixing aliquots of all samples to form a pooled sample. An aliquot of the 800 μL supernatant was transferred to a glass sampling vial for vacuum-dry at room temperature. A 150 μL mixture of acetonitrile and water (3/1, v/v) was added, vortexed for 30 s, ultrasonicated for 3 min, and stored at −20 °C for 2 h. The extract was centrifuged at 15,620 × g, 4 °C for 10 min. Samples were transferred to UPLC-MS/MS analysis using an ACQUITY UPLC system interfaced with a triple-quadrupole mass spectrometer. A Waters ACQUITY UPLC BEH Amide (2.1 × 100 mm, 1.7 μm) was used for analysis. Mass spectrometry was performed on the AB SCIEX API 6500+ Qtrap System, with an electrospray ionization (ESI) source, operating in both positive and negative ion modes. Targeted metabolites were analyzed in Schedule multiple reaction monitoring (SRM) mode. Samples were normalized based on protein concentrations using a BCA protein detection kit (P0012, Beyotime, Shanghai, China). Data acquisitions and further analysis were conducted using Analyst software. SCIEX OS-MQ software was used to quantify the metabolites.
Measurement of aortic root lesion development
To achieve a pain-free state before the experimental endpoint, all mice were placed under deep anesthesia with 1% pentobarbital sodium, with their condition continuously monitored and sacrificed by cervical dislocation. Saline was injected into the left ventricle for in situ perfusion, after which the hearts were embedded in optimal cutting temperature compound (Tissue-Tek, Sakura, Torrance, CA) and stored at −80 °C. Heart tissues were then serially sectioned into 6 μm frozen sections starting from the point where the aortic valves appeared and continuing until the valve structure was no longer intact. Isolated aortas were briefly rinsed in 75% ethanol for 1 min and subsequently stained using an Oil red O staining kit (G1261, Solarbio, Beijing, China). Images were captured under consistent microscope, camera, and lighting conditions. The Oil red O-stained areas were quantified using Image J (Media Cybernetics, USA) to calculate the lesion area per mouse aortic root. The percentage of lesion area was calculated by dividing the lesion area by the aortic root total area. The aortic roots of mice were fixed in 4% paraformaldehyde (PFA) (BL539A, Biosharp, Hefei, China) at 4 °C for 24 h, dehydrated, and embedded in paraffin for precise sectioning into 4 μm slices.
These paraffin-embedded sections were selectively stained with hematoxylin and eosin (H&E) staining kit (G1120, Solarbio, Beijing, China) to evaluate the plaque area. The necrotic core was defined as the acellular non-fibrotic region exceeding 3000 μm2 at the aortic root69. Advanced quantitative analysis was conducted using Image J (Media Cybernetics, USA), and the percentage of necrotic core was determined by dividing it by plaque area. Collagen staining of aortic sinus sections was performed using a Masson’s trichrome staining kit (G1340, Solarbio, Beijing, China). The percentage of collagen content within the plaques was computed.
Immunohistochemistry and vulnerability index analysis
Immunohistochemistry was performed following standardized ABC protocols69. In brief, immunohistochemistry staining was performed on 4 µm paraffin sections of the aortic sinus using a cocktail of primary antibodies against aSMA (1:50000, ab240654, Abcam, UK) and CD68 (1:100, ab125212, Abcam, UK). Consecutive sections were incubated with appropriate secondary antibodies (ZSGB-BIO, Beijing, China). The sections were incubated with the DAB substrate (ZLI-9018, ZSGB-BIO, Beijing, China) to visualize the bound peroxidase enzyme and then counterstained with hematoxylin. Images were captured, coded, and analyzed blind under uniform microscope, camera, and lighting conditions.
The vulnerability index was adapted from Shiomi et al.70 and Hartwig et al.71 and was calculated as the ratio of the sum of CD68+ and necrotic core areas divided by the sum of αSMA+ and collagen areas. Areas were represented as a percentage of the total plaque area, and only mice with plaques advanced enough to display all parameters were considered in the analysis.
Immunofluorescence
Paraffin-embedded human sections were first dewaxed and then fixed in 4% PFA for 5 min. After rinsing with PBS, the sections were permeabilized by incubation with 0.3% Triton X-100 for 30 min at 37 °C, followed by antigen repair. Frozen aortic sinus sections from mice were equilibrated to room temperature (RT) for 10 min and then fixed in pre-cooled acetone (−20 °C) for 10 min. After rinsing thrice in distilled water, the sections were permeabilized with 0.3% Triton X-100 (T8200, Solarbio, Beijing, China) for 30 min, followed by blocking with 5% BSA (AR0004, Boster Bio, Wuhan, China) for 30 min. Sections were then incubated overnight at 4 °C with primary antibodies against MAT2A (1:200, sc-398917, Santa Cruz Biotechnology, USA), CD11b (1:200, ab128797, Abcam, UK), CD68 (1:100, ab125212, Abcam, UK), and H3K4me3 (1:500, 39060, Active Motif, Shanghai, China). On the subsequent day, sections were incubated with appropriate secondary antibodies (ZSGB-BIO, Beijing, China) and then counterstained with DAPI (C1005, Beyotime, Shanghai, China). Finally, images were acquired using a confocal laser scanning microscope (ZElSS LSM 700).
Adoptive transfer of macrophages
100 μL of solution containing 106 PKH67-labeled MAT2ACKO and MAT2Afl/fl BMDMs (green, PKH67GL, Sigma-Aldrich, St. Louis, MO, USA) was injected into ApoE-/- mice with HFD for 16 weeks. After 72 h, aortas were collected for further analysis.
Monocyte labeling in vivo
Circulating Ly6Chi monocytes were labeled in vivo by retro-orbital injection of 1 μm Fluoresbrite YG microspheres (Polysciences Inc., PA, USA) diluted 1:4 in sterile PBS 18 h after tail vein injection of 250 μL clodronate loaded liposomes. The efficiency of fluorescent beads labeling was verified 24 h after injection by flow cytometry of monocytes. After 72 h, the number of labeled beads in aortic root cross-sections was quantified using fluorescence microscopy.
Flow cytometry
Mice were euthanized, and fresh tissues were collected. Primary mouse bone marrow cells were isolated by flushing the tibiae and femurs with ice-cold PBS. Peripheral blood was collected, and erythrocytes were lysed with red blood cell lysis buffer (NH4CL2009, TBD, Tianjin, China). The aortas (including the aortic arch, thoracic and abdominal regions) were digested in an enzyme solution containing 250 U/mL Collagenase I (BS163-100MG Biosharp, Hefei, China) at 37 °C with constant agitation, and were intermittently disaggregated with a pipette until digestion had completed for 40 min. Then, the digested aortas were gently passed through a 70-μm cell strainer to obtain a single-cell suspension.
Single-cell suspensions from the aorta, bone marrow, and peripheral blood were stained with Fixable Live/Dead stain (423105, BioLegend, San Diego, CA, USA) for 20 min at RT. The harvested cells were washed with cold PBS and subsequently resuspended at a concentration of 1 × 106 cells per milliliter. Cells were resuspended in 100 μL of PBS containing antibodies and incubated for 30 min at 4 °C in the dark, after which they were washed twice. Flow cytometry data were acquired using a FACS Canto II flow cytometer (BD Biosciences, CA, US) and analyzed with FlowJo software (TreeStar Ashland, OR, USA). The antibodies are listed in Supplementary Table 12.
Single-cell suspensions were prepared from aortas. Macrophages were then sorted using a FACS Aria III flow cytometer (BD Biosciences, CA, USA). All populations were routinely back-gated to verify gating and purity. F4/80+ macrophages were sorted for subsequent RNA-sequencing (RNA-seq).
RNA extraction, quantitative RT-PCR, and RNA-seq
Total RNA was extracted using TRIzol reagent (15596-026, Thermo Fisher Scientific, Waltham, MA, USA) and then reverse-transcribed using a Transcriptor First Strand cDNA Synthesis Kit (FSQ-301, TOYOBO LIFE SCIENCE, Shanghai, China). qRT-PCR was performed with a Fast Start Universal SYBR Green Master Mix (Q511-02, Vazyme Biotech, Nanjing, China). The data were normalized to the mRNA expression of the control gene encoding β-actin or GAPDH in each sample. The sequences of the primers are listed in Supplementary Table 13.
For RNA-seq, BMDMs and plaque macrophages were isolated for total RNA extraction. Samples from three mice of each genotype were sequenced. RNA was isolated using the RNeasy Micro Kit (74004, Qiagen, Hilden, Germany). The cDNA library was constructed and subjected to sequencing. The RNA-seq reads were mapped by the mouse reference genome (GRCm39) from NCBI using htseq (version 2.2.1.0). Raw counts were generated and quantified using htseq-count (version 0.9.1). Differentially expressed analysis was performed using DESeq2 (version 1.20.0) to identify DEGs.
CUT& Tag
BMDMs were collected from MAT2ACKO and control MAT2Afl/fl mice. CUT&Tag assay was performed using the Hyperactive In-Situ ChIP Library Prep Kit for Illumina (TD901-TD902, Vazyme Biotech, Nanjing, China)72. In brief, prepared concanavalin A-coated magnetic beads (ConA beads) were added to resuspended cells and incubated at RT to bind cells. Cells were then permeabilized with digitonin. Then, the primary H3K4me3 antibody (39060, Active Motif, Shanghai, China), secondary antibody, and the Hyperactive pA-Tn5 Transposase were incubated with the cells that were bound by ConA beads in order. The purified PCR products were evaluated using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Finally, these libraries were sequenced on the Illumina NovaSeq6000 platform, and 150 bp paired-end reads were generated. Library sequencing and data analysis were performed. In brief, raw reads were filtered and mapped to the mouse genome GRCm39 using Bowtie273 and subsequently analyzed by the SEACR to detect genomic regions enriched for multiple overlapping DNA peaks that we considered to be putative binding sites. Peaks were then annotated using ChIPseeker to obtain the genes and gene annotations about peaks.
Chromatin immunoprecipitation (ChIP)-qPCR
The ChIP-qPCR was carried out according to the kit protocol (9003S, Cell Signaling Technology, MA, USA). In brief, cells were fixed and lysed, followed by ultrasonication to shear them into appropriate fragments. The obtained chromatin was then incubated overnight with antibodies against H3K4me3 (39060, Active Motif, Shanghai, China), c-MYC (9402S, Cell Signaling Technology, MA, USA), and an isotype-matched control IgG (2729, Cell Signaling Technology, MA, USA). After washing with the wash buffer, the complexes were eluted and purified. Finally, the purified DNA was quantified by qRT-PCR using specific primers. Data were normalized to the corresponding input DNA controls. The sequences of the primers are listed in Supplementary Table 13.
Western blot
Proteins were extracted from atherosclerotic aortas and BMDMs using RIPA lysis buffer (P0013B, Beyotime, Shanghai, China) supplemented with 1 mM PMSF (ST506, Beyotime, Shanghai, China). Protein quantification was performed using a BCA protein detection kit (P0012, Beyotime, Shanghai, China). Samples were then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS‒PAGE) (Beyotime, Shanghai, China) and transferred onto polyvinylidene fluoride (PVDF) membranes (FFP22, Beyotime, Shanghai, China). Subsequently, the membranes were blocked with 5% skimmed milk in 0.1% Tween 20 in TBS, and then incubated overnight at 4 °C with primary antibodies in 5% BSA. The following day, membranes were washed with TBST and incubated with the secondary antibodies (ZSGB-BIO, Beijing, China). Protein bands were visualized using enhanced chemiluminescence substrate and imaged with a Tanon 5100 system (Tanon, Shanghai, China). The antibodies used are listed in Supplementary Table 14.
Genotyping
Excised mouse tails were digested in 100 μL of tissue digestion solution (B40013, Selleck Chemicals, Shanghai, China) for 15 min at 55 °C, followed by enzyme inactivation at 95 °C for 5 min. The digestive lysate was centrifuged at 8580 × g for 5 min, and the supernatant was used as the PCR template (1000 ng). Next, PCR amplification was performed using specific primers, and the products were analyzed by agarose gel electrophoresis. Gels were visualized using a gel imaging system (B40013, Vazyme Biotech, Nanjing, China). The sequences of the genotyping primers are listed in Supplementary Table 13.
Transwell assay
Mice peripheral blood monocytes (mPBMs) were labeled with Vybrant DiO cell-labeling solution (V22886, Invitrogen, Waltham, MA, USA) and resuspended in warmed HBSS at a density of 5 × 106 cells per mL. Millicell cell culture inserts were inserted into each well, and 100 µL of the mPBMs were added to the upper chamber of each insert. Following transmigration, the inserts were removed and the co-cultures containing BMDMs and mPBMs in the lower chambers were fixed with 4% PFA for 20 min. Representative images of the DiO-labeled mPBMs were captured with a brightfield overlay to confirm equivalent BMDMs numbers across treatment groups.
BMDMs and RAW264.7 cells were seeded into the upper chambers of the transwells containing a polycarbonate membrane with 8-μM pores (Corning, NY, USA), and then the chamber was placed in a 24-well plate at 37 °C with 5% CO2. Cells were allowed to migrate towards 200 ng/mL of CCL2 (Peprotech, Waltham, MA, USA) in the lower chamber. The non-migrated cells on the upper surface of the membrane were removed using a cotton swab. The migrated cells on the lower surface of the membrane were fixed with 4% PFA, stained with crystal violet staining kit (C0121, Beyotime, Shanghai, China) for 20 min, washed with PBS, and then imaged by light microscopy.
Wound healing
BMDMs and RAW264.7 cells were seeded in 6-well plates and allowed to form a confluent monolayer following stimulation and treatment. Three straight lines were scratched vertically in each well using a pipette tip. The wells were then gently washed with PBS to remove detached cells, and the medium was replaced with fresh DMEM (C11995500BT, Gibco, Thermo Fisher Scientific, Waltham, MA, USA). Images of the scratches were captured immediately (0 h) and 24 h later.
Enzyme-linked immunosorbent assay
Peripheral blood samples from patients and mice were collected and centrifuged at 3810 × g for 10 min to remove sediment. Meanwhile, the cell supernatants were also collected. The levels of inflammatory markers (AIM2, CCL2, and MMP9), norepinephrine, and lipid profiles (total cholesterol, triglyceride, LDL-C, and HDL-C). All measurements were performed using ELISA kits (MeiMianBio, Shanghai, China).
Human scRNA-seq data
Data from total cells of human atherosclerotic carotid arteries were analyzed in Seurat (version 5.1.0), starting from the author-provided cell count matrix (Gene Expression Omnibus, GSE253903)24. Quality control involved three steps: retention of genes expressed in at least five cells, exclusion of cells expressing fewer than 200 genes, and elimination of cells expressing over 15% of mitochondrial genes. A total of 10,934 cells were analyzed. Data were normalized using the ‘NormalizeData’ function in Seurat with default parameters. In total, 2,000 highly variable genes were identified using ‘FindVariableFeatures’. Data were scaled using ‘ScaleData’ with default parameters, and principal component analysis was performed using ‘RunPCA’ with default parameters, and clustering was performed at a 0.4 resolution using ‘FindClusters’ (resolution=0.4). Differentially expressed genes in each cluster were identified using the FindAllMarkers function with threshold criteria of FDR < 0.05 and | log2 (fold change) | >0.25. Cell type annotation was performed based on the expression of known cell lineage markers and on cluster annotations. To analyze myeloid cell subsets, cells corresponding to myeloid cells were extracted and re-clustered using a clustering resolution of 0.6.
Statistics
Categorical variables were expressed as counts and percentages, and differences between groups were compared using the χ2 or Fisher exact test. After assessing data distribution using the Kolmogorov-Smirnov test, continuous variables were expressed as mean ± standard (SD) deviation or median (interquartile range [IQR]) and were compared using the Student’s t-test or the Mann-Whitney U test. Generalized estimating equations methodology was used to take into account the within-subject correlation attributable to multiple plaques analyzed in a single patient. Multivariate logistic regression analysis adjusted for age, gender, traditional coronary risk factors (i.e., diabetes mellitus, dyslipidaemia, current smoking, hypertension, chronic kidney disease [CKD]), and concomitant statin therapy was used to identify whether high SAM/SAH levels or high norepinephrine levels were associated with TCFA. Multivariate Cox proportional hazards regression models adjusted for age, gender, traditional coronary risk factors, and statin at discharge were used to assess the association between high SAM/SAH levels or high norepinephrine levels and the risk of MACE. The correlation between the ratio of SAM/SAH and norepinephrine levels and their correlation with the thinnest fibrous cap thickness (FCT) and the mean lipid arc was tested using Pearson’s correlation analysis. Receiver-operating characteristic (ROC) curve analysis was performed to determine the areas under the curve (AUCs) of SAM/SAH alone, norepinephrine alone, and their combination for discriminating TCFA. Time to-first event Kaplan-Meier curves were estimated to assess the cumulative incidence of MACE during follow-up and were compared using the Log-rank test. Multiple group comparisons were evaluated using analysis of variance (ANOVA). Adjusted P-values were calculated by multiplying raw P-values by g, where g indicates the number of comparisons. Statistical analysis was performed using the GraphPad Prism version 9.0.0 or SPSS 26.0 software (IBM).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA-seq data have been deposited to the Genome Sequence Archive with dataset identifier CRA028975. The mass spectrometry dataset generated in this study have been deposited in the OMIX, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences under accession code OMIX011441. The single-cell data cited in this study are sourced from the dataset identifier GSE25390324. The data supporting the findings of this study are available within the article and its Supplementary Information files. Source data are provided with this paper.
References
Tsao, C. W. et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 145, e153–e639 (2022).
Ridker, P. M. et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 377, 1119–1131 (2017).
Tardif, J. C. et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 381, 2497–2505 (2019).
Nidorf, S. M. et al. Colchicine in patients with chronic coronary disease. N. Engl. J. Med. 383, 1838–1847 (2020).
Tearney, G. J. et al. Quantification of macrophage content in atherosclerotic plaques by optical coherence tomography. Circulation 107, 113–119 (2003).
Araki, M. et al. Predictors of rapid plaque progression. Opt. Coherence Tomogr. Study JACC Cardiovasc. Imag. 14, 1628–1638 (2021).
Yamamoto, H. et al. Impact of CD14(++)CD16(+) monocytes on coronary plaque vulnerability assessed by optical coherence tomography in coronary artery disease patients. Atherosclerosis 269, 245–251 (2018).
Tabas, I. & Lichtman, A. H. Monocyte-macrophages and T cells in Atherosclerosis. Immunity 47, 621–634 (2017).
Tabas, I. & Bornfeldt, K. E. Macrophage phenotype and function in different stages of Atherosclerosis. Circ. Res. 118, 653–667 (2016).
Groh, L., Keating, S. T., Joosten, L. A. B., Netea, M. G. & Riksen, N. P. Monocyte and macrophage immunometabolism in atherosclerosis. Semin. Immunopathol. 40, 203–214 (2018).
Kuznetsova, T., Prange, K. H. M., Glass, C. K. & de Winther, M. P. J. Transcriptional and epigenetic regulation of macrophages in atherosclerosis. Nat. Rev. Cardiol. 17, 216–228 (2020).
Yu, W. et al. One-Carbon metabolism supports S-Adenosylmethionine and Histone methylation to drive inflammatory macrophages. Mol. Cell 75, 1147–1160.e1145 (2019).
Miao, L. et al. No causal effects of plasma homocysteine levels on the risk of coronary heart disease or acute myocardial infarction: A Mendelian randomization study. Eur. J. Prev. Cardiol. 28, 227–234 (2021).
Lonn, E. et al. Homocysteine lowering with folic acid and B vitamins in vascular disease. N. Engl. J. Med. 354, 1567–1577 (2006).
Troen, A. M., Lutgens, E., Smith, D. E., Rosenberg, I. H. & Selhub, J. The atherogenic effect of excess methionine intake. Proc. Natl Acad. Sci. USA 100, 15089–15094 (2003).
Kovatcheva, M. et al. Vitamin B(12) is a limiting factor for induced cellular plasticity and tissue repair. Nat. Metab. 5, 1911–1930 (2023).
Sanderson, S. M., Gao, X., Dai, Z. & Locasale, J. W. Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat. Rev. Cancer 19, 625–637 (2019).
Mentch, S. J. et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 22, 861–873 (2015).
Shiraki, N. et al. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 19, 780–794 (2014).
Xia, C. et al. Methionine-SAM metabolism-dependent ubiquinone synthesis is crucial for ROS accumulation in ferroptosis induction. Nat. Commun. 15, 8971 (2024).
Li, F. et al. Blocking methionine catabolism induces senescence and confers vulnerability to GSK3 inhibition in liver cancer. Nat. Cancer 5, 131–146 (2024).
Gallone, G. et al. Coronary Plaque characteristics associated with major adverse cardiovascular events in atherosclerotic patients and lesions. A Syst. Rev. Meta-Anal. JACC Cardiovasc. Imag. 16, 1584–1604 (2023).
Bian, Y. et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585, 277–282 (2020).
Bashore, A. C. et al. High-dimensional single-cell multimodal landscape of human Carotid Atherosclerosis. Arterioscler Thromb. Vasc. Biol. 44, 930–945 (2024).
Cao, J. et al. DNA-sensing inflammasomes cause recurrent atherosclerotic stroke. Nature 633, 433–441 (2024).
Zhang, J. et al. Targeting WD repeat domain 5 enhances chemosensitivity and inhibits proliferation and programmed death-ligand 1 expression in bladder cancer. J. Exp. Clin. Cancer Res. 40, 203 (2021).
Grebien, F. et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPα N-terminal leukemia. Nat. Chem. Biol. 11, 571–578 (2015).
Wu, G. et al. Methionine-restricted diet: a feasible strategy against chronic or aging-related diseases. J. Agric. Food Chem. 71, 5–19 (2023).
Nabi, H. et al. Increased risk of coronary heart disease among individuals reporting adverse impact of stress on their health: the Whitehall II prospective cohort study. Eur. Heart J. 34, 2697–2705 (2013).
Gharios, C. et al. Cortico-limbic interactions and carotid atherosclerotic burden during chronic stress exposure. Eur. Heart J. 45, 1753–1764 (2024).
Tawakol, A. et al. Relation between resting amygdalar activity and cardiovascular events: a longitudinal and cohort study. Lancet 389, 834–845 (2017).
Kim, J. S. et al. Methionine adenosyltransferase:adrenergic-cAMP mechanism regulates a daily rhythm in pineal expression. J. Biol. Chem. 280, 677–684 (2005).
Varešlija, D., Tipton, K. F., Davey, G. P. & McDonald, A. G. 6-Hydroxydopamine: a far from simple neurotoxin. J. Neural Transm. 127, 213–230 (2020).
Lotharius, J., Dugan, L. L. & O’Malley, K. L. Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J. Neurosci. 19, 1284–1293 (1999).
Maity, S., Chandanathil, M., Millis, R. M. & Connor, S. A. Norepinephrine stabilizes translation-dependent, homosynaptic long-term potentiation through mechanisms requiring the cAMP sensor Epac, mTOR and MAPK. Eur. J. Neurosci. 52, 3679–3688 (2020).
Lipton, J. O. & Sahin, M. The neurology of mTOR. Neuron 84, 275–291 (2014).
Byles, V. et al. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 4, 2834 (2013).
Villa, E. et al. mTORC1 stimulates cell growth through SAM synthesis and m(6)A mRNA-dependent control of protein synthesis. Mol. Cell 81, 2076–2093.e2079 (2021).
Jefferies, H. B. et al. Rapamycin suppresses 5’TOP mRNA translation through inhibition of p70s6k. EMBO J. 16, 3693–3704 (1997).
Weber, C. & Noels, H. Atherosclerosis: current pathogenesis and therapeutic options. Nat. Med. 17, 1410–1422 (2011).
Koelwyn, G. J., Corr, E. M., Erbay, E. & Moore, K. J. Regulation of macrophage immunometabolism in atherosclerosis. Nat. Immunol. 19, 526–537 (2018).
Shi, X. et al. Heavy macrophage infiltration identified by optical coherence tomography relates to plaque rupture. Ann. Clin. Transl. Neurol. 10, 2334–2346 (2023).
Wang, H. et al. Hyperhomocysteinemia accelerates atherosclerosis in cystathionine beta-synthase and apolipoprotein E double knock-out mice with and without dietary perturbation. Blood 101, 3901–3907 (2003).
Ebbing, M. et al. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: a randomized controlled trial. JAMA 300, 795–804 (2008).
Xiao, Y. et al. Increased plasma S-adenosylhomocysteine-accelerated atherosclerosis is associated with epigenetic regulation of endoplasmic reticulum stress in apoE-/- mice. Arterioscler Thromb. Vasc. Biol. 35, 60–70 (2015).
You, Y. et al. Epigenetic modulation of Drp1-mediated mitochondrial fission by inhibition of S-adenosylhomocysteine hydrolase promotes vascular senescence and atherosclerosis. Redox Biol. 65, 102828 (2023).
Dai, X. et al. Epigenetic upregulation of H19 and AMPK Inhibition concurrently contribute to S-Adenosylhomocysteine Hydrolase deficiency-promoted Atherosclerotic calcification. Circ. Res. 130, 1565–1582 (2022).
Fidler, T. P. et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 592, 296–301 (2021).
Georgakis, M. K., Bernhagen, J., Heitman, L. H., Weber, C. & Dichgans, M. Targeting the CCL2-CCR2 axis for atheroprotection. Eur. Heart J. 43, 1799–1808 (2022).
Zhang, Y. et al. S-Nitrosylation of Septin2 Exacerbates Aortic Aneurysm and Dissection by Coupling the TIAM1-RAC1 Axis in Macrophages. Circulation, (2024).
Niu, N. et al. Tumor cell-intrinsic epigenetic dysregulation shapes cancer-associated fibroblasts heterogeneity to metabolically support pancreatic cancer. Cancer Cell 42, 869–884.e869 (2024).
Chen, Y. et al. Methyltransferase Setd2 prevents T cell-mediated autoimmune diseases via phospholipid remodeling. Proc. Natl Acad. Sci. USA 121, e2314561121 (2024).
Roy, D. G. et al. Methionine Metabolism Shapes T Helper Cell Responses through Regulation of Epigenetic Reprogramming. Cell Metab. 31, 250–266.e259 (2020).
Wang, H. & Helin, K. Roles of H3K4 methylation in biology and disease. Trends Cell Biol. 35, 115–128 (2025).
Benayoun, B. A. et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 158, 673–688 (2014).
Wagner, E. J. & Carpenter, P. B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 13, 115–126 (2012).
Xiao, Z. & Locasale, J. W. Epigenomic links from metabolism-methionine and chromatin architecture. Curr. Opin. Chem. Biol. 63, 11–18 (2021).
Barrett, T. J. et al. Chronic stress primes innate immune responses in mice and humans. Cell Rep. 36, 109595 (2021).
Martinet, W., Verheye, S. & De Meyer, G. R. Everolimus-induced mTOR inhibition selectively depletes macrophages in atherosclerotic plaques by autophagy. Autophagy 3, 241–244 (2007).
Meteva, D. et al. Toll-like receptor 2, hyaluronan, and neutrophils play a key role in plaque erosion: the OPTICO-ACS study. Eur. Heart J. 44, 3892–3907 (2023).
Araki, M. et al. Optical coherence tomography in coronary atherosclerosis assessment and intervention. Nat. Rev. Cardiol. 19, 684–703 (2022).
Cutlip, D. E. et al. Clinical end points in coronary stent trials: a case for standardized definitions. Circulation 115, 2344–2351 (2007).
Wang, Y. et al. S-adenosylmethionine biosynthesis is a targetable metabolic vulnerability in multiple myeloma. Haematologica 109, 256–271 (2024).
Zhang, W. et al. Fluorinated N,N-dialkylaminostilbenes repress colon cancer by targeting methionine S-adenosyltransferase 2A. ACS Chem. Biol. 8, 796–803 (2013).
Du, X. et al. B cell-derived anti-beta 2 glycoprotein I antibody mediates hyperhomocysteinemia-aggravated hypertensive glomerular lesions by triggering ferroptosis. Signal Transduct. Target Ther. 8, 103 (2023).
Daigneault, M., Preston, J. A., Marriott, H. M., Whyte, M. K. & Dockrell, D. H. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 5, e8668 (2010).
Ying, W., Cheruku, P. S., Bazer, F. W., Safe, S. H., Zhou, B. Investigation of macrophage polarization using bone marrow derived macrophages. J. Vis. Exp. (2013).
Long, C. P. & Antoniewicz, M. R. High-resolution (13)C metabolic flux analysis. Nat. Protoc. 14, 2856–2877 (2019).
Seneviratne, A. N. et al. Interferon Regulatory Factor 5 Controls Necrotic Core Formation in Atherosclerotic Lesions by Impairing Efferocytosis. Circulation 136, 1140–1154 (2017).
Shiomi, M., Ito, T., Hirouchi, Y. & Enomoto, M. Fibromuscular cap composition is important for the stability of established atherosclerotic plaques in mature WHHL rabbits treated with statins. Atherosclerosis 157, 75–84 (2001).
Hartwig, H. et al. Atherosclerotic plaque destabilization in mice: a comparative study. PLoS One 10, e0141019 (2015).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Acknowledgements
This work was supported by National Natural Science Foundation of China grant No. 82322036 (to J.D.), 82072091 (to J.D.), 82471998 (to P.S.), 62135002 (to B.Y.), 82202286 (to C.F.), and Natural Science Foundation of Heilongjiang Province grant No. YQ2024H017 (to P.S.), PL2024H128 (to C.W.), YQ2023H014 (to C.F.).
Author information
Authors and Affiliations
Contributions
P.S., J.D. and Z.D. conceived the project and designed the experiments. Z.D. and P.W. drafted and revised the manuscript. P.S. and J.D. reviewed and provided critical revision of the manuscript for important content. B.Y. provided critical comments on the project. Z.D., M.D. and P.W. performed the data curation and analysis of the present study. Z.D., P.W., M.D., S.L. (Song Li), Q.Y., S.S., C.T., Z.J., S.L. (Shuang Li), G.S., B.M., X.D., S.L. (Shilong Li), and C.W. conducted experiments. J.D. and C.F. contributed to the clinical information collection and outcome assessment. B.Y. and S.F. performed administrative assistance.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xueqing Ba, Partha Dutta, Huiyong Yin and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Du, Z., Wan, P., Du, M. et al. MAT2A promotes atherosclerotic plaque vulnerability by mediating epigenetic reprogramming of macrophages. Nat Commun 16, 11168 (2025). https://doi.org/10.1038/s41467-025-66121-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66121-z
