
Abstract
Gastric cancer (GC) ranks as the third leading cause of cancer-related mortality. Chemoresistance poses a major obstacle to successful treatment for GC patients. Here, we demonstrate that ferroptosis suppressor protein 1 (FSP1) enables chemoresistance in GC by inhibiting ferroptosis, resulting in diminished chemotherapy response and reduced patient survival rate. USP29 and SMURF1 are subsequently discovered as the deubiquitinase and E3 ligase of FSP1, respectively, orchestrating the ubiquitination of FSP1 to modulate ferroptosis and chemoresistance in GC. In the ubiquitination process, SMURF1 is observed to interact with and polyubiquitinate FSP1 within K63/K193 sites. Notably, pharmacological inhibition of FSP1 by iFSP1 is shown to synergize with chemotherapeutic agents across chemoresistant cellular and mouse models. Our findings thus underscore the pivotal role of the USP29 and SMURF1 in regulating GC chemoresistance via FSP1-mediated ferroptosis suppression. FSP1 inhibition may represent a promising therapeutic avenue to overcome chemoresistance in GC.
Similar content being viewed by others


Introduction
Gastric cancer is the fifth most diagnosed malignancy globally and stands out as a leading cause of cancer-related mortality1,2. Diagnosis typically occurs at advanced stages, precluding surgical intervention and contributing to a bleak prognosis3. Chemotherapy remains a cornerstone of gastric cancer treatment. Nevertheless, the emergence of chemoresistance has a marked impact on therapeutic efficacy, potentially resulting in treatment failure4,5. Consequently, there is an imperative need to investigate the precise mechanisms underpinning drug resistance and develop clinical strategies to address this challenge.
Cell survival and cell death are intricately controlled by a myriad of signals and molecules6,7. Regulated cell death (RCD) represents an orchestrated process that holds major implications across various facets of mammalian biology, encompassing development, homeostasis, and pathogenesis8,9,10. In recent years, multiple forms of RCD have emerged as critical players in human health and disease. Among these, ferroptosis stands out as a distinctive mode of regulated cell demise instigated by iron-driven lipid peroxidation11,12. Ferroptosis ensues when perturbations in intracellular iron levels precipitate an excessive buildup of deleterious lipid reactive oxygen species (ROS), overwhelming the cell’s antioxidant defenses and culminating in membranous structural damage13,14. Noteworthy efforts have elucidated numerous proteins and pathways governing the intricate regulation of ferroptosis.
Ferroptosis suppressor protein 1 (FSP1) has emerged as a regulator of ferroptosis inhibition15,16. Operating in concert with the glutathione-GPX4 axis, FSP1 functions as a nicotinamide-adenine dinucleotide phosphate (NADP+)-dependent coenzyme Q (CoQ) oxidoreductase. Within the FSP1-NADPH pathway, CoQ10 catalyzes the direct synthesis of lipid-soluble antioxidants and sequesters lipid peroxyl radicals to mitigate lipid peroxidation within various biological membranes. Recent investigations have elucidated the roles of posttranslational modifications (PTMs) in the regulation of ferroptosis, encompassing phosphorylation, acetylation, ubiquitination, methylation, and SUMOylation17,18,19,20. Ubiquitination entails the addition of ubiquitin moieties to lysine residues of target proteins, dictating their degradation and turnover dynamics21. Notably, recent studies have underscored the crucial regulatory functions of ubiquitination in the context of ferroptosis. Specifically, the OTU deubiquitinase ubiquitin aldehyde binding 1 (OTUB1) has been shown to interact directly with SLC7A11, stabilizing it through deubiquitination22. Additionally, research by Yang et al. unveiled the induction of Nedd4 by erastin, leading to increased ubiquitination and degradation of VDAC2/323. Nonetheless, the precise mechanisms by which ubiquitination and deubiquitination modulate FSP1 stabilization remain largely unexplored.
In this study, our findings show that FSP1 enhances chemoresistance and correlates with inferior survival outcomes in GC patients. Notably, depletion of FSP1 promotes chemosensitivity through the induction of ferroptosis. USP29 and SMURF1 orchestrate the protein homeostasis of FSP1 and thereby modulating ferroptosis and chemoresistance in GC. Importantly, the inhibitor of FSP1, iFSP1, exhibits synergistic effects with chemotherapy, effectively reversing chemoresistance. Therefore, we propose that FSP1 could serve as a predictive biomarker for chemoresistance and adverse survival prognosis in GC, and targeting FSP1 may represent a promising strategy for overcoming chemoresistance.
Results
FSP1 expression serves as an indicator for unfavorable chemotherapy response and survival in GC
To investigate the key genes contributing to chemoresistance in cancer, cisplatin treatment data in Genomics of Drug Sensitivity in Cancer (GDSC) (https://www.cancerrxgene.org/) were analyzed using weighted correlation network analysis (WGCNA)24. Initially, sample clustering was performed to identify outliers, revealing that all cancer cell lines were clustered together (Supplementary Fig. 1a). A soft threshold of beta = 18 was selected to construct a scale-free network (Supplementary Fig. 1b). Subsequently, 7 modules were delineated by grouping genes with similar expression patterns (Supplementary Fig. 1c). Notably, the turquoise module exhibited the strongest positive correlation with cisplatin resistance (Supplementary Fig. 1d), establishing it as a pivotal module for further investigation. Module membership and gene significance related to cisplatin resistance within the turquoise module are illustrated in Supplementary Fig. 1e. Interestingly, we found that the ferroptosis pathway was enriched in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (Supplementary Fig. 1f). Furthermore, the gene expression profiles of cells sensitive or resistant to cisplatin were analyzed. Setting the cut-off criteria as fold change > 2 and P < 0.01, we identified 156 genes with expression changes (94 genes were upregulated and 62 genes were downregulated) in the cisplatin-resistant group compared with cisplatin sensitive group (Supplementary Fig. 1g). Next, we took the intersection of genes in the turquoise module, 94 upregulated genes and genes in the ferroptosis pathway and obtained only one gene, FSP1 (Supplementary Fig. 1h), highlighting FSP1 as a potential key gene driving cisplatin resistance in cancer cells through the ferroptosis pathway. Indeed, we identified a positive correlation between FSP1 expression levels and chemotherapeutics IC50 values in the GDSC dataset (Supplementary Fig. 1i). Notably, this relationship was particularly pronounced in GC cells (Supplementary Fig. 1j), prompting our focus on GC for further investigation.
Although surgery is the main curative approach for resectable GC, the prognosis of patients remains unfavorable post-curative resection. Preoperative chemotherapy offers potential advantages like reducing the primary tumor size to enhance the chances and effectiveness of curative resection, as well as eradicating potential micro-metastases to prevent or lower tumor recurrence and spread, thus enhancing the management of tumor-related symptoms2. Nevertheless, ~15% of GC patients who receive preoperative chemotherapy face the risk of tumor progression3. Thus, accurately predicting patients’ response to preoperative chemotherapy remains particularly crucial.
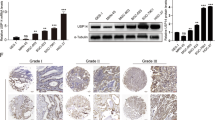
To validate the findings from WGCNA and explore the role of FSP1 in chemotherapy response and outcomes in GC, we conducted a prospective clinical cohort study. A total of 164 GC patients scheduled with preoperative neoadjuvant therapy were enrolled in this study. The expression levels of FSP1 were evaluated in preoperative biopsy samples from the GC cohort, and no substantial differences in clinical characteristics were observed between the FSP1 low and high expression groups (Supplementary Table 1). Remarkably, we found that patients with low FSP1 exhibited favorable response, whereas those with elevated FSP1 expression demonstrated poor responses to preoperative neoadjuvant therapy (Fig. 1a–d and Supplementary Table 1). Subsequently, we performed immunohistochemistry (IHC) staining of 4-HNE to assess cell ferroptosis in GC specimens after preoperative neoadjuvant therapy (Fig. 1e). The results showed that FSP1 expression negatively correlates with 4-HNE levels (Fig. 1f). Furthermore, patients with high FSP1 expression show poorer survival in long-term follow-up (Fig. 1g, h). Collectively, these findings suggest that FSP1 correlates with chemoresistance and indicates poor patient outcomes in GC.

Representative images of patients with low FSP1 expression showing favorable response (a), and high FSP1 expression showing poor response (b) to preoperative neoadjuvant chemotherapy. c Representative images of immunohistochemical (IHC) staining of FSP1 in the GC cohort (n = 164). d Tumor regression grade after preoperative neoadjuvant chemotherapy in the GC cohort with low or high FSP1 expression. American Joint Committee on Cancer tumor regression grade classification criteria was utilized. Representative images of 4-HNE IHC staining (e) and correlation analysis (f) of 4-HNE and FSP1 staining in the GC cohort (n = 164). Progression-free survival (g) and overall survival (h) analysis of the GC cohort with low or high FSP1 expression. Scale bars are indicated in (a–c, e). P values are determined by 2-sided χ2 test in (d, f), or 2-sided log-rank (Mantel–Cox) test in (g, h). Source data are provided as Source Data file.
FSP1 renders chemoresistance in GC via repressing ferroptosis
To explore the causal relationship and underlying mechanism, we utilized two gastric cancer cell lines that acquired resistance to cisplatin for subsequent study (Supplementary Fig. 2a, b). The initial results revealed elevated expression of FSP1, but unchanged expression of other ferroptosis factors, and decreased level of lipid peroxidation induced by cisplatin in chemoresistance cell lines (Fig. 2a, b and Supplementary Fig. 2c–e). We then used two independent single guide RNAs (sgRNAs) to knockout FSP1 in AGS-R and MGC-803-R cells (Fig. 2c and Supplementary Fig. 2f), the results showed that FSP1 depletion markedly enhances the level of cisplatin-induced lipid peroxidation and vulnerability to cisplatin, without affecting DNA damage repair (Fig. 2d, e and Supplementary Fig. 2g–j). Reintroduction of wild-type (WT) FSP1, but not impaired myristoylation and plasma-membrane localization mutant (G2A), or unfunctional CoQ oxidoreductase mutant (E156A), could rescue cisplatin-induced lipid peroxidation and sensitivity to cisplatin contributed by FSP1 deficiency (Fig. 2f–h and Supplementary Fig. 2k–m). Furthermore, only Liproxstatin-1 (Lip-1, a ferroptosis inhibitor), but not Z-VAD-FMK (a pan-caspase inhibitor), 3-Methyladenine (3-MA, an autophagy inhibitor), or necrostatin 2 racemate (Nec-1s, a necroptosis inhibitor) could restore cisplatin susceptibility in FSP1-KO cells (Fig. 2i and Supplementary Fig. 2n).

a Immunoblot of FSP1 in AGS cisplatin sensitive (S) and resistant (R) cells. b Percentage of C11-BODIPY-positive cells in the indicated AGS cells with/without 24-h cisplatin (2 μM) treatment. c Immunoblot of FSP1 in AGS-R cells stably expressing control or two independent FSP1 sgRNAs. d Percentage of C11-BODIPY-positive cells in cells from (c) with/without 24-h cisplatin (2 μM) treatment. e Survival fraction of cells from (c) exposed to cisplatin at the indicated doses for 2 weeks. f Immunoblot of FSP1 in AGS-R cells stably expressing the indicated vectors. g Percentage of C11-BODIPY-positive cells in cells from (f) with/without 24-h cisplatin (2 μM) treatment. h Survival fraction of cells from (f) exposed to cisplatin at the indicated doses for 2 weeks. i Survival fraction of control or FSP1-KO AGS-R cells exposed to cisplatin at the indicated dose combining with/without the indicated reagents for 2 weeks. j Growth curves of the AGS-R xenograft tumor models with the indicated treatment. k Quantification of the weight of the tumors in different groups of the xenograft tumor models. Experiment was repeated 3 times, and representative blots are presented in (a, c, f). Data shown represent mean ± SD from three independent experiments (b, d, e, g–i), or from one representative experiment of 5 mice per group in (j, k). P values are determined by unpaired 2-sided Student’s t test in (b), 1-way ANOVA in (d, g, k), or 2-way ANOVA in (e, h–j). The survival fraction was calculated as the ratio of colony-forming cells with the indicated treatment to those in the untreated control group. NS, not significant. Source data are provided as Source Data file.
The encouraging in vitro results prompted us to explore the impact of FSP1 on the chemoresistance of GC in mouse xenograft models. In vivo growth curves showed that the FSP1 KO tumors exhibited a modest reduction in size compared to tumors derived from parental cells prior to cisplatin treatment, but FSP1 KO was strikingly sensitive to cisplatin treatment (Fig. 2j and Supplementary Fig. 2o). Consistent results were observed upon the sacrifice of the mice and subsequent calculation of tumor weight (Fig. 2k and Supplementary Fig. 2p). Moreover, ferroptosis inhibitor of liproxstatin-1 (Lip-1) effectively reversed the heightened cisplatin sensitivity induced by FSP1 depletion in the in vivo setting (Fig. 2j, k and Supplementary Fig. 2o, p). Taken together, these data demonstrated that FSP1 facilitates gastric cancer chemoresistance in vitro and in vivo by suppressing ferroptosis.
USP29 binds and stabilizes FSP1
Due to the notable elevated protein levels, but not mRNA expression, of FSP1 in cisplatin-resistant cells compared to sensitive cells (Fig. 2a and Supplementary Figs. 2c and 3a), we investigated its stability in these cells. The results revealed a notable increase in FSP1 stability in resistant cells, accompanied by reduced ubiquitination levels (Fig. 3a, b and Supplementary Fig. 3b, c). Next, we set out to investigate the potential deubiquitinases (DUBs) associated with FSP1 by overexpressing a series of Flag-tagged DUBs in HEK293T cells and conducting co-immunoprecipitation (Co-IP) assays. As shown in Supplementary Fig. 3d, DUB3, USP11, USP29, USP33, CYLD, and USP43 exhibited observable interactions with FSP1 among the tested DUBs. To further elucidate the roles of these six DUBs in modulating ferroptosis through their interactions with FSP1, we individually silenced them in HT1080 cells (Supplementary Fig. 3e) and evaluated cell viability following treatment with RSL3 or erastin. The results demonstrated that knockdown of USP29 most substantially sensitized cells to both RSL3 and erastin (Supplementary Fig. 3f, g). We further validated that silencing USP29 with short hairpin RNAs (shRNAs) enhanced ferroptosis in HT1080 induced by RSL3 or erastin (Supplementary Fig. 3h–j). Consequently, our study focuses on USP29 for an initial detailed investigation based on these compelling findings.

a FSP1 protein stability in cisplatin sensitive or resistant AGS cells. b Ubiquitination of FSP1 in cisplatin sensitive or resistant AGS cells. c, d Endogenous co-immunoprecipitation of USP29 and FSP1 in HEK293T cells. e Schematic representation of the structures of USP29 truncation mutants. UCH, ubiquitin carboxyl-terminal hydrolase. f Co-immunoprecipitation of the indicated USP29 truncation mutants and FSP1. The green triangles indicate the band of the specific USP29 truncates. g FSP1 protein level after transduced with lentivirus carrying control or two independent USP29 shRNAs. h Immunoblots of FSP1 and USP29 in HEK293T cells stably expressing control or USP29 shRNAs treated with vehicle or MG132. i Immunoblots of FSP1 and USP29 in HEK293T cells stably expressing the indicated vectors. j, k FSP1 protein stability in cells stably expressing the indicated vectors. l Ubiquitination of FSP1 in cells stably expressing control or USP29 shRNAs. m Ubiquitination of FSP1 in USP29 silencing cells transfected with wild type (WT) or catalytically inactive (CI) USP29 vectors. n Deubiquitination of FSP1 in vitro by USP29. Ubiquitinated FSP1 was incubated with purified GST fusion USP29 WT or USP29 CI in vitro and then blotted with indicated antibodies. CBB staining, Coomassie Brilliant Blue staining. o, p Ubiquitination of FSP1 in cells transfected with the indicated vectors. Experiment was repeated 3 times, and representative blots are presented in (a–d, f–j, l–p). Data shown represent mean ± SD from three independent experiments in (a, k). P values are determined by 2-way ANOVA in (a, k). Source data are provided as Source Data file.
To further confirm the interaction between USP29 and FSP1, endogenous co-immunoprecipitation (Co-IP) assays were conducted, demonstrating that reciprocal immunoprecipitation of endogenous USP29 (or FSP1) effectively precipitated FSP1 (or USP29) (Fig. 3c, d). The binding regions between USP29 and FSP1 were delineated through a series of truncation experiments involving USP29 (Fig. 3e). Notably, the enzymatic domain (UCH domain) of USP29 was identified as crucial for mediating the USP29-FSP1 interaction (Fig. 3f). Consistently, knockdown of USP29 using two distinct USP29-specific shRNAs resulted in a marked reduction in FSP1 protein abundance, without affecting its mRNA levels or the expression of other ferroptosis factors (Fig. 3g and Supplementary Fig. 4a, b), implying a post-transcriptional regulatory role of USP29 on FSP1 expression. Moreover, the diminished FSP1 levels in USP29-depleted cells could be restored by treatment with the proteasome inhibitor MG132 (Fig. 3h), indicating that USP29 regulates FSP1 levels via proteasomal degradation. Furthermore, reintroduction of shRNA-resistant WT USP29 into USP29 knockdown cells successfully restored FSP1 protein levels (Fig. 3i). To further address whether USP29 regulates FSP1 protein stability, cells were treated with cycloheximide (CHX) to determine the half-life of FSP1. The results revealed a notable decrease in FSP1 stability upon USP29 depletion, which could be rescued by ectopic expression of USP29 in the depleted cells (Fig. 3j, k). Collectively, these findings substantiate the role of USP29 in regulating FSP1 protein abundance and stability in a proteasome-dependent manner.
To further elucidate the mechanism by which USP29 mediates FSP1 stabilization, we investigated whether USP29 deubiquitinates FSP1 in cellular contexts. Our results showed that knockdown of USP29 led to a notable increase in polyubiquitination of FSP1 within cells (Fig. 3l). Conversely, ectopic expression of WT USP29, but not the catalytically inactive (CI) mutant USP29 (substitution of cysteine 294 to serine, C294S), resulted in a marked decrease in the polyubiquitination levels of FSP1 (Fig. 3m). To further probe the direct deubiquitination activity of USP29 towards FSP1, in vitro deubiquitination assay were conducted. The results showed that recombinant WT USP29, but not the USP29 CI mutant, demonstrated the ability to deubiquitinate FSP1 in vitro (Fig. 3n).
It is widely recognized that ubiquitin can form diverse types of ubiquitin chains through various lysine residues (Ub-K6, Ub-K11, Ub-K27, Ub-K29, Ub-K33, Ub-K48, and Ub-K63)25. Typically, ubiquitination via K48 of the ubiquitin chain marks proteins for proteasomal degradation, while K63-linked ubiquitination often modulates signaling pathways. To further address whether USP29 specifically deubiquitinates FSP1 on the K48-linked polyubiquitin chains, ubiquitination assays were performed using either K48/K63-intact or K48R/K63R ubiquitin variants. Expectedly, our results revealed extensive ubiquitination of FSP1 primarily through K48 linkages (Fig. 3o). Subsequently, we examined the impact of USP29 depletion on these linkages (Ub-K48) and observed that the loss of USP29 led to increased K48-linked polyubiquitination of FSP1 (Fig. 3p). Overall, these data strongly demonstrate that USP29 deubiquitinates FSP1 K48-linked polyubiquitin chains both in vitro and in vivo.
USP29 enables GC chemoresistance through FSP1-mediated ferroptosis inhibition
To investigate the involvement of USP29-FSP1 signaling in GC chemoresistance, we initially assessed the protein expression levels of USP29 and FSP1 in various cancer cell lines. The results revealed a strong positive correlation between USP29 and FSP1 expression across the majority of these cell lines (Supplementary Fig. 4c, d) as well as within the GC specimens (Supplementary Fig. 4e, f). Moreover, the elevated expression of USP29 also suggests inferior survival in the GC cohort (Supplementary Fig. 4g, h). Next, we knocked out USP29 in AGS-R and MGC-803-R cells (Fig. 4a and Supplementary Fig. 5a), the results showed that USP29 depletion markedly increases the level of cisplatin induced lipid peroxidation and susceptibility to cisplatin (Fig. 4b, c and Supplementary Fig. 5b, c). The heightened cisplatin induced lipid peroxidation and sensitivity to cisplatin observed with USP29 depletion was reversed upon re-introduction of WT USP29, but not the USP29 CI mutant (Fig. 4d–f and Supplementary Fig. 5d–f). Given prior findings indicating that USP29 plays a role in stabilizing AURKB to promote tumor growth in GC26, potentially contributing to drug resistance, we investigated the impact of AURKB on chemosensitivity in the context of USP29 depletion. The results showed that overexpression of AURKB in USP29 knockout cells did not rescue cell viability following cisplatin exposure (Supplementary Fig. 5g–j), suggesting that chemoresistance lead by USP29 is independent of AURKB. On the other hand, re-expressing WT FSP1, but the mutant of G2A or E156A, was able to restore ferroptosis and cisplatin sensitivity contributed by USP29 loss (Fig. 4g–i and Supplementary Fig. 5k–m). Furthermore, only ferroptosis inhibitor could reverse cisplatin susceptibility in USP29-null cells (Fig. 4j and Supplementary Fig. 5n). Consistent results were also observed in mouse models (Fig. 4k, l and Supplementary Fig. 5o, p). Collectively, these data suggest that USP29 promotes chemoresistance in GC through FSP1-medicated ferroptosis suppression.

a Immunoblots of FSP1 and USP29 in AGS-R cells stably expressing control or two independent USP29 sgRNAs. b Percentage of C11-BODIPY-positive cells in cells from (a) with/without 24-h cisplatin (2 μM) treatment. c Survival fraction of cells from (a) exposed to cisplatin at the indicated dose for 2 weeks. d Immunoblots of FSP1 and USP29 in AGS-R cells stably expressing control or USP29 sgRNA transfected with USP29 WT or CI vectors. e Percentage of C11-BODIPY-positive cells in cells from (d) with/without 24-h cisplatin (2 μM) treatment. f Survival fraction of cells from (d) exposed to cisplatin at the indicated dose for 2 weeks. g Immunoblots of FSP1 and USP29 in AGS-R cells stably expressing the indicated vectors. h Percentage of C11-BODIPY-positive cells in cells from (g) with/without 24-h cisplatin (2 μM) treatment. i Survival fraction of cells from (g) exposed to cisplatin at the indicated dose for 2 weeks. j Survival fraction of control or USP29-knockout AGS-R cells exposed to cisplatin at the indicated dose combining with/without the indicated reagents for 2 weeks. k Growth curves of the AGS-R xenograft tumor models with the indicated treatment. l Quantification of the weight of the tumors in different groups of the xenograft tumor models. Experiment was repeated 3 times, and representative blots are presented in (a, d, g). Data shown represent mean ± SD from three independent experiments in (b, c, e, f, h–j), or from one representative experiment of 5 mice per group in (k, l). P values are determined by 1-way ANOVA in (b, e, h, l), or 2-way ANOVA in (c, f, i–k). The survival fraction was calculated as the ratio of colony-forming cells with the indicated treatment to those in the untreated control group. NS, not significant. Source data are provided as Source Data file.
SMURF1 enables FSP1 degradation and chemosensitivity of GC
After elucidating the function of the USP29-FSP1 axis in GC chemoresistance, we sought to identify the specific E3 ubiquitin ligase responsible for this process. Utilizing the UbiBrowser website tool (http://ubibrowser.bio-it.cn/ubibrowser/), we conducted predictive analysis to determine the E3 ubiquitin ligase involved in the degradation of FSP1. Subsequently, the top seven E3 ligases identified through the prediction were selected as potential candidates for further investigation (Fig. 5a). Through Co-IP assays, we confirmed a robust interaction between FSP1 and SMURF1, while no substantial interactions were observed with the other candidate ligases or the paralog SMURF2 (Fig. 5b and Supplementary Fig. 6a, b). Reciprocal immunoprecipitation experiments involving SMURF1 and FSP1 antibodies indicated mutual binding between the two proteins (Fig. 5c, d), suggesting an interaction between FSP1 and SMURF1 in cells. Notably, the WW domain of SMURF1 was identified to be crucial for mediating the SMURF1-FSP1 interaction (Fig. 5e). Silencing SMURF1 increases FSP1 while SMURF1 overexpression decreases FSP1 protein expression level (Fig. 5f, g), without affecting its mRNA level (Supplementary Fig. 6c, d). Additionally, a negative correlation between SMURF1 expression and FSP1 levels was observed in the GC cells and specimens (Supplementary Fig. 6e–g) and high expression of SMURF1 indicates better survival in the GC cohort (Supplementary Fig. 6h, i). Notably, following SMURF1 knockdown, the half-life of FSP1 substantially increased (Supplementary Fig. 6j, k), an effect that could be reversed upon re-expression of WT SMURF1 but not the catalytically inactive mutant (C725A) lacking ubiquitin ligase activity (Fig. 5h, i). Through ubiquitination assays, we found that depletion of SMURF1 led to a marked reduction in the ubiquitination of FSP1, which could be rescued by reintroducing WT SMURF1 but not by the C725A mutant of SMURF1 (Fig. 5j, k). In vitro ubiquitination assay also showed that SMURF1 WT, but not C725A mutant, can ubiquitinate FSP1 (Fig. 5l). Collectively, these findings provide compelling evidence that SMURF1 functions as the E3 ligase responsible for directly ubiquitylating and destabilizing FSP1 protein.

a List of FSP1-associated E3 ligase predicted in UbiBrowser (http://ubibrowser.bio-it.cn/ubibrowser/). b Co-immunoprecipitation of FSP1 and the indicated proteins in HEK293T cells. c, d Endogenous co-immunoprecipitation of SMURF1 and FSP1 in HEK293T cells. e Co-immunoprecipitation of the indicated SMURF1 truncation mutants and FSP1. f Immunoblots of FSP1 and SMURF1 in cells stably expressing control or SMURF1 shRNAs. g Immunoblots of FSP1 and Flag in cells stably expressing empty vector or increased dose of Flag-SMURF1. h, i FSP1 protein stability in cells transfected with the indicated vectors. j, k Ubiquitination of FSP1 in HEK293T cells transfected with the indicated vectors. l In vitro ubiquitination of FSP1 by SMURF1. Purified GFP-FSP1, His-SMURF1 WT and C725A proteins were subjected to in vitro ubiquitination assay in the presence or absence of E1 and E2. CBB staining, Coomassie Brilliant Blue staining. m Immunoblots of Flag and FSP1 in AGS-R cells stably expressing the indicated vectors. n Percentage of C11-BODIPY-positive cells in cells from (m) with/without 24-h cisplatin (2 μM) treatment. o Survival fraction of cells from (m) exposed to cisplatin at the indicated dose for 2 weeks. p Survival fraction of control or SMURF1-OE AGS-R cells exposed to cisplatin at the indicated dose combining with/without the indicated reagents for 2 weeks. q Growth curves of the AGS-R xenograft tumor models with the indicated treatment. r Quantification of the weight of the tumors in different groups of the xenograft tumor models. Experiment was repeated 3 times, and representative blots are presented in (b–h, j–m). Data shown represent mean ± SD from three independent experiments in (i, n–p), or from one representative experiment of 5 mice per group in (q, r). P values are determined by 2-way ANOVA in (i, o–q), or 1-way ANOVA in (n, r). The survival fraction was calculated as the ratio of colony-forming cells with the indicated treatment to those in the untreated control group. NS, not significant. Source data are provided as Source Data file.
Subsequently, we investigated the role of SMURF1 in GC chemoresistance. Ectopic expression of SMURF1 resulted in a substantial enhancement of ferroptosis and sensitivity to cisplatin in drug-resistant GC cells, with susceptibility being rescued only by WT FSP1 or ferroptosis inhibitor treatment (Fig. 5m–p and Supplementary Fig. 6l–o). Consistent conclusions were drawn from in vivo experiments (Fig. 5q, r and Supplementary Fig. 6p, q). Taken together, our findings demonstrate that SMURF1 enhances chemosensitivity in GC by promoting the degradation and inhibition of FSP1-mediated ferroptosis.
SMURF1 regulates GC chemoresistance by destabilizing FSP1 through specific ubiquitination sites
To further understand the molecular basis of how FSP1 is ubiquitinated, we utilized UbiBrowser website tool again to predict the specific lysine sites within FSP1 targeted by the SMURF1 E3 ligase. Predictions identified three potential ubiquitination sites on FSP1 (K63, K193, and K225). Subsequently, we generated mutants for these lysine residues individually (KR) and in combination. Ubiquitination assays revealed that the single-site mutant (K225R) had minimal impact on overall ubiquitination, while single-site mutants (K63R and K193R) or two-site mutants, K63R/K225R (2KR3) and K193R/K225R (2KR2) sharply reduced FSP1 ubiquitination levels (Fig. 6a). Furthermore, the two-site mutant K63R/K193R (2KR1) or three-site mutant K63R/K193R/K225R (3KR) nearly abolished FSP1 ubiquitination (Fig. 6a). Consistently, overexpression of SMURF1 led to the degradation of WT FSP1 protein but not the K63R/K193R mutant FSP1 protein in a dose-dependent manner (Fig. 6b). Moreover, the half-life of K63R/K193R mutant FSP1 protein was notably prolonged compared to WT FSP1 (Fig. 6c, d). These findings collectively suggest that SMURF1 mediates the ubiquitination of FSP1 at the K63 and K193 sites.

a Ubiquitination of the mutated FSP1 in HEK293T cells transfected with indicated vectors. 2KR1, K63R/K193R; 2KR2, K193R/K225R; 2KR3, K63R/K225R; 3KR, K63R/K193R/K225R. b Cells transfected with the indicated vectors, cell lysates were then blotted with the indicated antibodies. c, d Protein stability of WT or K63R/K193R mutant FSP1. e Immunoblots of FSP1 and Flag in AGS-R cells transfected with the indicated vectors. f Percentage of C11-BODIPY-positive cells in cells from (e) with/without 24-h cisplatin (2 μM) treatment. g Survival fraction of cells from (e) exposed to cisplatin at the indicated dose for 2 weeks. Experiment was repeated 3 times, and representative blots are presented in (a–c, e). Data shown represent mean ± SD from three independent experiments in (d, f, g). P values are determined by 2-way ANOVA in (d, g), or 1-way ANOVA in (f). The survival fraction was calculated as the ratio of colony-forming cells with the indicated treatment to those in the untreated control group. Source data are provided as Source Data file.
We proceeded to explore the functional implications of ubiquitination on FSP1 at these specific sites. By overexpressing SMURF1 in AGS-R and MGC-803-R cells and reintroducing either WT or the K63R/K193R mutant FSP1 (Fig. 6e and Supplementary Fig. 7a), we observed that the increased cisplatin-induced lipid peroxidation and sensitivity to cisplatin induced by SMURF1 overexpression were reversed upon re-expression of WT FSP1 (Fig. 6f, g and Supplementary Fig. 7b, c). In addition, reconstitution with the mutant lacking ubiquitination (K63R/K193R) led to a further reduction in cisplatin-induced lipid peroxidation and cell death (Fig. 6f, g and Supplementary Fig. 7b, c). These findings unequivocally demonstrate that SMURF1 regulates the chemosensitivity of GC by promoting FSP1 degradation through ubiquitination at the K63 and K193 sites.
Targeting FSP1 reverses GC chemoresistance
Our data have convincingly illustrated that FSP1 plays a central role in mediating chemoresistance in GC. We next asked whether iFSP1, the first reported inhibitor for FSP1 that is specific for the human enzyme, in combination with chemotherapeutic agents could effectively eradicate tumor cells and reverse chemoresistance. Initially, we observed that iFSP1 synergistically enhanced the effectiveness of cisplatin or 5-FU in both GC cell lines and mouse models (Supplementary Fig. 8a–g). Moreover, in chemoresistant cells, iFSP1 markedly augmented the anti-tumor efficacy of cisplatin in both in vitro and in vivo settings, consequently improving overall survival rates (Fig. 7a–d and Supplementary Fig. 8h–k). Furthermore, we established six cases of GC patient-derived organoids (PDO) to examine the combination efficacy of FSP1 inhibition and chemotherapeutic agents (Fig. 7e, f and Supplementary Table 2). Expectedly, iFSP1 markedly enhanced the sensitivity of PDO to cisplatin or 5-FU (Fig. 7g, h and Supplementary Fig. 8l, m). We also obtained a fresh GC tumor specimen exhibiting high FSP1 expression (Fig. 7i) from a patient who had undergone a second surgical resection due to rapid disease progression following chemotherapy, indicating resistant to chemotherapy (Supplementary Table 2). We successfully established patient-derived xenograft (PDX) models, and the results showed that a combination of iFSP1 and cisplatin worked effectively in killing tumor cells and prolonging survival (Fig. 7j–l). We next examined the body weight and TUNEL staining of the liver and kidneys in mouse experiments to assess any side effects or potential toxicity for a combination of iFSP1 and chemotherapeutic agents. The results indicated that combo drug treatment was well-tolerated, as evidenced by the absence of any notable effects on mouse body weight, as well as a lack of substantial systemic toxicity observed in TUNEL-stained sections of the liver and kidneys (Supplementary Fig. 9a–h). Collectively, these findings from both in vitro and in vivo studies highlight the promising therapeutic potential of iFSP1 in conjunction with chemotherapy for overcoming chemoresistance in GC.

a Cell survival of AGS-R cells treated with vehicle, or cisplatin (1 μM), iFSP1 (10 μM) or their combination for 2 weeks. b Growth curves of the AGS-R xenograft tumor models with the indicated treatment. c Quantification of the weight of the tumors in different groups of the xenograft tumor models. d Survival curves of the AGS-R xenograft tumor models. e Representative bright-field images of the 6 cases of cultured gastric cancer patient-derived organoids (PDO). f Immunoblot showing FSP1 expression levels in the 6 GC PDO. g Organoid viability of PDO #4 with the indicated treatment. h Representative micrographs (left) and quantitation (right) of PDO #4 with the indicated treatment. Organoids were stained with Calcein-AM (live cells, green) and EthD-1 (dead cells, red) for 30 min and then fluorescent signals were captured. i IHC staining of FSP1 in the GC patient. j Growth curves of the patient-derived tumor xenograft (PDX) models with the indicated treatment. k Quantification of the weight of the tumors in different groups of the PDX models. l Survival curves of the PDX models. Scale bars are indicated in (e, h, i). Data shown represent mean ± SD from three independent experiments in (a, g, h), or from one representative experiment of 5 mice per group in (b, c, j, k). Data shown come from one representative experiment of 8 mice per group in (d, l). Experiment was repeated 3 times, and representative blots are presented in (f). P values are determined by 1-way ANOVA in (a, c, g, h, k), 2-way ANOVA in (b, j), or 2-sided log-rank (Mantel–Cox) test in (d, l). NS, not significant. Source data are provided as Source Data file.
Discussion
Chemoresistance in gastric cancer (GC) remains a distinctive obstacle to the effectiveness of chemotherapy and is a key driver of disease progression, contributing to a dismal less than 10% 5-year overall survival rate27,28. Chemoresistance in GC can arise from a dynamic cell death response, where cancer cells switch between different cell death pathways, including apoptosis, autophagy, necrosis, and ferroptosis. This shift enables GC cells evade the effects of chemotherapy, thereby diminishing the efficacy of treatment. While the possibility that mixed cell death contributing to chemoresistance in GC does exist, our findings suggest that FSP1-mediated ferroptosis may play a key role in chemoresistance of GC via WGCNA. Further investigations support the potential of FSP1 as a prognostic indicator and molecular biomarker for predicting chemotherapeutic responses in GC.
Ferroptosis represents a distinct iron-dependent form of cell death, setting it apart from conventional apoptosis, necroptosis, or autophagy12,13. Increasing evidence highlights a notable correlation between ferroptosis and chemoresistance. In colorectal cancer, adipose-derived exosomes diminish susceptibility to ferroptosis, consequently bolstering resistance to oxaliplatin29. Moreover, in pancreatic cancer, ARID3A inhibition of PTEN-induced ferroptosis fosters chemoresistance to gemcitabine30. These findings suggest strong confirmation that ferroptosis is involved in cancer chemoresistance, and ferroptosis-induction treatments might provide a therapeutic strategy to alleviate resistance to chemotherapy. The FSP1/NAD(P)H/ubiquinol system emerges as the principal intracellular antioxidant system driving ferroptosis, notwithstanding the well-established role of the SLC7A11/GSH/GPX4 system. Notably, GPX4 is indispensable for mouse growth and development, while FSP1’s absence does not impede these processes31. Consequently, targeting FSP1 is deemed a safer approach with potentially fewer adverse effects on cardiac physiology and development. In this investigation, we observed a substantial increase in FSP1 expression in cisplatin-resistant GC cells, implicating a probable involvement of FSP1 in GC chemoresistance. Subsequent data confirmed that FSP1 depletion could sensitize resistant GC cells to cisplatin across cellular and mouse models. Furthermore, the small molecular inhibitor of FSP1, iFSP1, exhibited strong synergy with cisplatin, effectively reversing chemoresistance in GC both in vitro and in vivo.
Post-translational modifications (PTMs) play a crucial role in regulating cellular processes by influencing various aspects of protein behavior such as activity, localization, stability, and interactions with other proteins32,33. Among the various types of PTMs, ubiquitination stands out as a prevalent modification involving the covalent attachment of ubiquitin to a specific substrate. E3 ligases are responsible for facilitating the connection between ubiquitin and target proteins, often triggering protein degradation34. In contrast, deubiquitination catalyzed by deubiquitinating enzymes (DUBs) removes ubiquitin from target proteins, thereby maintaining their stability35. Therefore, a delicate balance between ubiquitination and deubiquitination is essential for regulating proper protein levels and functions. In this study, we have identified that the E3 ligase of SMURF1 and the DUB of USP29 exhibit opposing functions in modulating FSP1 protein homeostasis, consequently promoting chemoresistance mediated by ferroptosis in GC.
Previously, USP29 has been reported to stabilize cGAS, promoting cellular antiviral responses and autoimmunity36. In addition, Tu et al. demonstrated that USP29 integrates metabolic processes critical for progression of neuroblastoma and B cell lymphoma by governing the deubiquitination and stabilization of MYC and HIF1α37. In triple-negative breast cancer, increased levels of USP29 have been linked to the stabilization of TWIST1, a pivotal transcription factor in epithelial-mesenchymal transition (EMT) that contributes to the self-renewal of cancer stem-like cells (CSCs), thus driving EMT and conferring resistance to chemotherapy and metastasis38. Our study provides evidence for a potential function of USP29 in stabilizing FSP1 and promoting chemoresistance in GC. Initially, through a series of DUBs screenings, we identify USP29 as a bona fide deubiquitinase of FSP1. USP29 directly interacts with FSP1, reducing its ubiquitination levels through its DUB activity, ultimately leading to the stabilization of FSP1. Subsequently, genetic deletion of USP29 substantially enhances ferroptosis and sensitivity to chemotherapy in cellular and mouse models, an effect not observed in FSP1-silenced cells. Conversely, re-introduction of WT FSP1, but not the non-functional mutants of G2A or E156A FSP1, rescues the ferroptosis induced by USP29 deletion and restores susceptibility to chemotherapeutic agents. Taken together, these findings suggest that the chemoresistance conferred by USP29 in gastric cancer primarily relies on its ability to stabilize FSP1.
Through catalyzing the ubiquitination of substrates for subsequent proteasomal degradation, the E3 ubiquitin ligase of SMURF1 has been proven to be crucial in a variety of processes including bone homeostasis, embryogenesis and viral autophagy39,40,41. We demonstrate here that SMURF1 plays a pivotal role in managing the abundance of FSP1 by ubiquitinating it within K63/K193 sites, promoting its proteasomal degradation and consequently reducing its chemoresistance function. Thus, our results uncovered SMURF1, in conjunction with FSP1 ubiquitination, as a regulatory mechanism governing ferroptosis signaling and chemoresistance in GC.
In summary, our study contributes to understanding the role of USP29 and SMURF1 in controlling the stability of FSP1, highlighting the impact of FSP1-mediated ferroptosis inhibition on chemoresistance in GC (Fig. 8). Importantly, our preclinical data supports the notion that targeting FSP1 can induce ferroptosis and sensitize resistant cancer cells to chemotherapy, proposing a therapeutic approach for overcoming chemotherapy resistance in GC.

SMURF1 interacts with FSP1 and promotes its ubiquitination within Lys63 and Lys193 to induce its degradation, whereas USP29 deubiquitinates and stabilizes FSP1. The elevated expression of FSP1 inhibits ferroptosis and governs chemoresistance in gastric cancer. Pharmacological targeting of FSP1 synergizes with chemotherapy reagents and reverses gastric cancer chemoresistance.
Methods
Study approval
All mouse experiments were approved by the Institutional Animal Care and Use Committee of Mayo Clinic. Collection of the biopsy samples was performed according to the relevant ethical standards and was approved by the Ethics Committee of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology. All registered patients provided informed consent.
Cell culture and plasmids
Cell lines used in this study: HEK-293T (ATCC #CRL-3216), AGS (ATCC #CRL-1739), HT-1080 (ATCC #CCL-121), MGC-803 (Ubigene #YC-C081), SGC-7901 (Ubigene #YC-C055), BGC-823 (Beyotime #C6123). All cell lines were maintained at 37 °C in a humidified atmosphere containing 5% CO2. The culture medium composition for each cell line was consistent with previously described42. Cell line identities were authenticated by the Medical Genome Facility (MGF) at Mayo Clinic (Rochester, MN) using short tandem repeat (STR) profiling. Mycoplasma contamination was routinely monitored every 6 months using the EZ-PCR Mycoplasma Test Kit (Biological Industries). No cell line was used for more than 20 passages after thawing from frozen stocks.
WT USP29 was purchased from Addgene (Plasmid #22599) and subcloned into PLVX3 lentiviral. WT FSP1 was purchased from Horizon (Clone Id: 3506309) and subcloned into PLVX6 lentiviral. WT SMURF1 was purchased from Addgene (Plasmid #11752). WT His-Ub (Plasmid #107392) and WT HA-Ub (Plasmid #17608) were purchased from addgene. All the mutant of FSP1, USP29, HA-Ub, and SMURF1 were generated by site-directed mutagenesis according to standard protocol.
Antibodies and reagents
Antibodies used in this study are described in the Supplementary Table 3. Cycloheximide (CHX) and MG132 were purchased from Selleck. RSL3, erastin, liproxstatin-1, Z-VAD-FMK, 3-Methyladenine, necrostatin 2 racemate, IgG agarose, anti-Flag agarose and anti-HA agarose were purchased from Sigma Aldrich.
Lentivirus production and infection
Short hairpin RNAs (shRNAs) used in this study are listed in the Supplementary Table 4. TransIT-X2 (MIRUS Bio) was used to co-transfect the shRNA vector, the envelope plasmid pMD2.G, and the packaging plasmid psPAX2 into HEK293T cells. The medium was changed at 16 h post transfection and harvested at 48 h after transfection. The medium was filtered through a 0.45 μm syringe filter and supplemented with 8 μg/mL polybrene for infection. The infected cells were selected by 4 μg/mL puromycin.
Generation of CRISPR–Cas9 KO cell lines
To delete the USP29 or FSP1 gene in AGS-R and MGC-803-R cell lines, we designed two independent guide RNAs (gRNAs) using the CRISPR design tool (https://www.genscript.com) for each gene. The gRNA sequences were as follows: USP29 #1: TGTCGAGGTTCATCTTCAAG; and USP29 #2: ATTGAACTCTACGAATGTAT; FSP1 #1: CCTCTTTGCCCGAGTACTTG; and FSP1 #2: GCTCAACAAGTCAGAACTAA. The gRNA sequences were cloned into pSpCas9(BB)-2A-GFP (PX458) all-in-one plasmid (no. 48138, Addgene) according to the standard protocol. The gRNA/Cas9 expression constructs were co-transfected into AGS-R or MGC-803-R cells. At 24 h after transfection, the cells were enriched by fluorescent-based sorting using a FACS Aria SORP (BD Biosciences) and transferred into 96-well plates at ~1 cell per well. Single cells were grown for ~6 weeks and colonies were screened for KOs by genomic sequence and western blotting.
Western blot and immunoprecipitation
We prepared cell lysates, performed immunoprecipitation, and immunoblotting as previously described43. Briefly, cells were lysed with NETN buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40). Whole-cell lysates were centrifuged at 12,000 × g for 15 min. Supernatant containing proteins incubated with 2 μg of antibody and protein A or protein G sepharose beads overnight at 4 °C. The immunocomplexes were then washed with NETN buffer and separated by SDS-PAGE. Immunoblotting was performed following standard procedures.
Protein stability assay
To examine the protein turnover, cycloheximide (CHX, 0.1 mg/ml) was added to cell culture medium and cells were harvested at the indicated time points. Cells were lysed and subjected to western blot. Then, we quantified of the protein levels relative to β-Actin by ImageJ.
Denaturing Ni-NTA pull-down
Cells were harvested and lysed in urea buffer (8 M Urea, 0.1 M NaH2PO4, 300 mM NaCl, 0.01 M Tris (pH 8.0)). Lysates were then sonicated and incubated with Ni-NTA agarose beads for 1–2 h at room temperature. Beads were washed 5 times with urea buffer. The immunocomplexes were boiled in 1× Laemmli buffer and subjected to Western blot.
Deubiquitination in vivo and in vitro
The in vivo and in vitro deubiquitination assays were performed as described previously44,45. Briefly, after transfection indicated vectors and treated with MG132, the cells were lysed in the SDS buffer (62.5 mM Tris-HCl (PH 6.8), 2% SDS, 10% glycerol) containing 20 mM NEM and 1 mM iodoacetamide and boiled for 15 min. Then diluted 10 times with NETN buffer containing 20 mM NEM and 1 mM iodoacetamide. The lysates were immunoprecipitated with the HA beads overnight at 4 °C. After NETN buffer washing, the immunocomplexes were subjected to Western blot. For the in vitro deubiquitination assay, ubiquitinated FSP1 protein was incubated with purified Flag-USP29 WT and USP29 CI mutant in deubiquitination buffer (50 mM Tris-HCl pH 8.0, 50 mM NaCl, 1 mM EDTA, 10 mM DTT, 5% glycerol) for 4 h at room temperature.
In vitro ubiquitination assay
For in vitro ubiquitination, ubiquitination kit (Enzo life science) was used and obtained following manufacturer’s instructions. Briefly, purified GST-FSP1, His-SMURF1 WT and C725A were incubated with 100 nM E1 (UBE1), 1 μM E2 (UbcH7), 5 mM Mg-ATP, 2.5 μM Ub or recombinant Ub variant and 1× ligase reaction buffer at 37 °C for 45 min in 25 μl of reaction mixture, boiled in sample buffer and subjected to Western blot analysis.
Immunofluorescence staining
Cells cultured on coverslips were treated with cisplatin for 24 h. And then washed with PBS, pre-extracted with a solution containing 20 mM HEPES pH 7.4, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose, and 0.5% NP-40 for 2 min on ice, incubated in 3% paraformaldehyde for 15 min, and permeabilized in 0.25% Triton X-100 solution for 5 min at room temperature. Subsequently, samples were incubated overnight at 4 °C with the primary antibody, followed by three times washes and a 1-h incubation with the secondary antibody at room temperature. DAPI staining was performed to visualize nuclear DNA. Finally, coverslips were mounted onto glass slides with an anti-fade solution and observed using a Nikon Eclipse 80i fluorescence microscope.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the cells using Trizol reagent and reversely transcribed into cDNA by utilizing the Takara RR037A kit. The SYBR (Takara, RR820A) and CFX96 Real-Time System (Bio-Rad, USA) were used to conduct the real-time PCR according to manufacturer’s instruction. Expression of β-Actin gene was used as an inner control. Primer sequences used in qRT-PCR are listed in Supplementary Table 5.
Colony formation assays
Cells (500 per well) were seeded in triplicate in 6-well plates. 24 h later, cells were treated with indicated reagents, and cultured for 10–14 days at 37 °C. After the colony formed, washed the colonies with PBS and stained with the Coomassie buffer and counted. Lip-1 50 nM, Z-VAD 10 μM, 3-MA 10 mM and Nec-1s 1 μM were used in the colony formation assays.
MTS cell proliferation assay
Cell proliferation was evaluated using the MTS assay (Promega, USA) according to manufacturer’s instruction. Briefly, cells were seeded in 96-well plates (2000 cells per well) and treated as the indicated. The culture medium was replaced with 100 μL fresh medium containing 10 μL of the MTS solution for each well of the plate. After incubating at 37 °C incubator for 1 h, cell growth was measured in a microplate reader with absorbance at 490 nm.
C11-BODIPY staining
The indicated cells are seed in six-well plates with or without cisplatin treatment. 24 h later, the cells were stained with 5 μM C11-BODPY581/591 (SIGMA ALDRICH, D-3861) at 37 °C for 30 min prior to trypsinization. After trypsinization and resuspension in phosphate-buffered saline (PBS), flow cytometry was employed to analyze the fluorescence emission peak from 590 to 510 nm. The gating strategy for all flow cytometry analyzes is shown in Supplementary Fig. 10.
Culture of human gastric cancer organoids
Gastric cancer tissues were obtained after surgery and minced in the laboratory. After being washed 6 times by DPBS supplemented with 1% of penicillin-streptomycin, the tumor samples were minced into pieces and then digested by tumor tissue digestion solution (bioGenous, K601003) for 15 min at 37 °C. DPBS plus 1% penicillin-streptomycin was added to stop digestion. Samples were then filtered through 100-μm cell strainers, followed by 1800 rpm, centrifugation for 5 min. The centrifugated cells were mixed with Matrigel (BD Pharmingen) and seeded in 24-well-plates. After the Matrigel containing tumor cells was fixed at 37 °C for 10 min, the culturing medium was added into wells. Gastric cancer organoids derived from patients (PDOs) were cultured in gastric cancer organoid kit (bioGenous, K2179-GC), and incubated at 37 °C with 5% CO2.
Organoid viability assay
Approximately 5000 tumor organoids were cultured in 10 µl of Matrigel per well in a 96-well plate. Following 2–4 days of culture, the specified reagents were diluted to various concentrations and introduced into the tumor organoid medium for an additional 3–6 days. At the end of the experiments, CCK8 (Beyotime, C0040) solution at a ratio of 1:100 was applied to each well and allowed to incubate for 60 min at 37 °C and 5% CO2. The absorbance at 450 nm wavelength was measured using a Multiskan Sky with Touch Screen (Thermo Scientific).
GC organoid live/dead staining
PDOs were cultured in a single confocal microscope Petri dish with the specified treatment for 72 h. Following this, the culture medium was removed, and the samples in the dish were rinsed twice with PBS. Subsequently, a staining solution was prepared by adding 1 μl of calcein-AM (1 μM) and 1 μl of EthD-1 (5 μM) to 0.3 ml of PBS. The GC organoids were then stained with calcein-AM and EthD-1 for 20 min at room temperature in dark. Next, the samples in the dish underwent counter-staining with 4,6-diamidino-2-phenylindole (Sigma) and were subjected to confocal microscopic analysis. Images were captured using a Zeiss LSM900 equipped with Z-stack technology. Image analysis was performed using the ZEN-blue (v.2.0) software package. Quantification of live and dead cells was carried out by measuring the total cell area of each dye using ImageJ (v.1.8.0).
Xenograft mouse model
BALB/c female/male mice in the age of 4–6 weeks old were utilized in this study. Indicated cells (2 × 106 cells per mouse) were injected subcutaneously into the left posterior flanks of the mice. Tumor volume were monitored by measurements of tumor length (L), and width (W) (tumor volume = L × W × W × 1/2). When tumors reached a volume of ~100 mm3, mice were randomly allocated into indicated groups. Mouse were treated with cisplatin (8 mg/kg) or 5-FU (10 mg/kg) by intraperitoneal injections 5 times per week, or iFSP1 (10 mg/kg) by daily intraperitoneal injections. For liproxstatin-1 rescue experiment, liproxstatin-1 (10 mg/kg) was intraperitoneally injected once every 3 days for 6 times. At the end of the study, mice were sacrificed and then removed the tumors, weighed and processed. The maximal tumor size has not exceeded the range permitted by the ethics committee (tumor size ≤ 2000 mm3).
Patient-derived xenograft
The PDX experiment was conducted in strict accordance with the ethical guidelines and was approved by the Ethics Committee of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology. Fresh tumor specimens were obtained from a patient with recurrent gastric cancer who was undergoing surgical resection following chemotherapy. Immediately after collection, the tissues were placed in cold sterile physiological saline supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin solution to maintain cell viability. Specimens were minced into 1–2 mm³ fragments using sterile scalpels and washed three times with cold saline to remove excess blood and debris. Minced tumor pieces were then mixed 1:1 with reduced growth factor Matrigel (BD Biosciences, Inc) and injected subcutaneously into 6–8-week-old Balb/c nu/nu mice using an 18-gauge needle. Successful engraftment was defined as sustained tumor growth beyond 100 mm3 for at least 4 weeks. Subsequent passages were performed in a similar manner in Balb/c nu/nu mice. Only F3 or higher-generation models were utilized for experimental studies to ensure genetic stability. The administration of group allocation and treatment followed the same protocol as the xenograft mouse model.
Immunohistochemistry (IHC)
IHC assays were performed as previously described with the indicated antibodies46. The expression scores were assessed based on the German semi-quantitative scoring system according to the staining intensity and the staining region as previously described. The final staining score ≥4 was identified as high expression; otherwise, it was identified as low expression. The Pearson correlation coefficient and χ2 test were used for statistical analysis of the correlation of IHC expression.
Statistical analysis
All data in bar and line graphs are presented as mean ± SD of at least three independent experiments. All the Western blotting data were repeated independently three times with similar results. Differences between two groups were analyzed using unpaired Student’s t test, 1-way analysis of variance (ANOVA), or 2-way ANOVA. All findings were considered significant at a P value threshold of 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are included within the article, Supplementary Information and the Source Data file. Source data are provided with this paper.
References
Siegel, R. L. et al. Cancer statistics. CA Cancer J. Clin. 73, 17–48 (2023).
Smyth, E. C. et al. Gastric cancer. Lancet 396, 635–648 (2020).
Shen, L. et al. Management of gastric cancer in Asia: resource-stratified guidelines. Lancet Oncol. 14, e535–e547 (2013).
Zhou, Y. et al. FAM117B promotes gastric cancer growth and drug resistance by targeting the KEAP1/NRF2 signaling pathway. J. Clin. Invest. 133, e158705 (2023).
Ouyang, S. et al. Inhibition of STAT3-ferroptosis negative regulatory axis suppresses tumor growth and alleviates chemoresistance in gastric cancer. Redox Biol. 52, 102317 (2022).
Newton, K. et al. Cell death. Cell 187, 235–256 (2024).
Yuan, J. et al. A guide to cell death pathways. Nat. Rev. Mol. Cell Biol. 25, 379–395 (2024).
Hadian, K. et al. The therapeutic potential of targeting regulated non-apoptotic cell death. Nat. Rev. Drug Discov. 22, 723–742 (2023).
Peng, F. et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct. Target Ther. 7, 286 (2022).
Cheng, X. et al. Apoptosis propagates through the cytoplasm as trigger waves. Science 361, 607–612 (2018).
Chen, X. et al. Broadening horizons: the role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 18, 280–296 (2021).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Jiang, X. et al. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282 (2021).
Liang, D. et al. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 82, 2215–2227 (2022).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019).
Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019).
Lee, H. et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 22, 225–234 (2020).
Han, Y. et al. IL-1β-associated NNT acetylation orchestrates iron-sulfur cluster maintenance and cancer immunotherapy resistance. Mol. Cell 83, 1887–1902 (2023).
Anandhan, A. et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci. Adv. 9, eade9585 (2023).
Wang, Y. et al. Targeting epigenetic and posttranslational modifications regulating ferroptosis for the treatment of diseases. Signal Transduct. Target Ther. 8, 449 (2023).
Popovic, D. et al. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 20, 1242–1253 (2014).
Liu, T. et al. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res. 79, 1913–1924 (2019).
Yang, Y. et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 11, 433 (2020).
Langfelder, P. et al. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Swatek, K. N. et al. Ubiquitin modifications. Cell Res. 26, 399–422 (2016).
Tu, R. et al. USP29 activation mediated by FUBP1 promotes AURKB stability and oncogenic functions in gastric cancer. Cancer Cell Int. 24, 33 (2024).
Sexton, R. E. et al. Gastric cancer: a comprehensive review of current and future treatment strategies. Cancer Metastasis Rev. 39, 1179–1203 (2020).
Zeng, Y. et al. Molecular pathogenesis, targeted therapies, and future perspectives for gastric cancer. Semin. Cancer Biol. 86, 566–582 (2022).
Zhang, Q. et al. Adipocyte-derived exosomal MTTP suppresses ferroptosis and promotes chemoresistance in colorectal cancer. Adv. Sci. 9, e2203357 (2022).
Mao, X. et al. ARID3A enhances chemoresistance of pancreatic cancer via inhibiting PTEN-induced ferroptosis. Redox Biol. 73, 103200 (2024).
Liang, D. et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell 186, 2748–2764 (2023).
Vu, L. D. et al. Protein language: post-translational modifications talking to each other. Trends Plant Sci. 23, 1068–1080 (2018).
Lee, J. M. et al. Control of protein stability by post-translational modifications. Nat. Commun. 14, 201 (2023).
Fu, J. et al. Legionella maintains host cell ubiquitin homeostasis by effectors with unique catalytic mechanisms. Nat. Commun. 15, 5953 (2024).
Gao, B. et al. USP36 inhibits apoptosis by deubiquitinating cIAP1 and survivin in colorectal cancer cells. J. Biol. Chem. 300, 107463 (2024).
Zhang, Q. et al. USP29 maintains the stability of cGAS and promotes cellular antiviral responses and autoimmunity. Cell Res. 30, 914–927 (2020).
Tu, R. et al. USP29 coordinates MYC and HIF1α stabilization to promote tumor metabolism and progression. Oncogene 40, 6417–6429 (2021).
Guan, T. et al. Phosphorylation of USP29 by CDK1 governs TWIST1 stability and oncogenic functions. Adv. Sci. 10, e2205873 (2023).
Yamashita, M. et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 121, 101–113 (2005).
Wan, L. et al. Cdh1 regulates osteoblast function through an APC/C-independent modulation of Smurf1. Mol. Cell 44, 721–733 (2011).
Huang, C. et al. Talin phosphorylation by Cdk5 regulates Smurf1-mediated talin head ubiquitylation and cell migration. Nat. Cell Biol. 11, 624–630 (2009).
Zhao, J. et al. G3BP1 interacts with YWHAZ to regulate chemoresistance and predict adjuvant chemotherapy benefit in gastric cancer. Br. J. Cancer 124, 425–436 (2021).
Zhou, Q. et al. The bromodomain containing protein BRD-9 orchestrates RAD51-RAD54 complex formation and regulates homologous recombination-mediated repair. Nat. Commun. 11, 2639 (2020).
Deng, M. et al. Deubiquitination and activation of AMPK by USP10. Mol. Cell 61, 614–624 (2016).
Zhu, Q. et al. RNF19A-mediated ubiquitination of BARD1 prevents BRCA1/BARD1-dependent homologous recombination. Nat. Commun. 12, 6653 (2021).
Zeng, X. et al. METTL16 antagonizes MRE11-mediated DNA end resection and confers synthetic lethality to PARP inhibition in pancreatic ductal adenocarcinoma. Nat. Cancer 3, 1088–1104 (2022).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (82303827, 82272757, 82373123 and 82072736), Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2021-RC310-013), Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2021-I2M-1-067, 2023-I2M−2-004, 2024-I2M-ZD-004), Key Projects of Hubei Province (2021BCA116) and Mayo Foundation.
Author information
Authors and Affiliations
Contributions
Conceptualisation: Z.L., M.D. and X.Z.; Methodology: Z.W., X.T., S.Z., Q.J., L.W. and K.T.; Data acquisition and analysis: Z.W., X.T., S.Z. and Q.J.; Writing original draft and revisions: Z.W., X.T., Z.L., M.D. and X.Z.; Supervision: Z.L., M.D. and X.Z.; Funding acquisition: K.T., Z.L., M.D., and X.Z.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhaocai Zhou and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, Z., Tu, X., Zhu, S. et al. USP29 and SMURF1 orchestrate FSP1-mediated ferroptosis suppression to facilitate chemoresistance in gastric cancer. Nat Commun 16, 11439 (2025). https://doi.org/10.1038/s41467-025-66319-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66319-1
