
Abstract
Several Physaria species (Brassicaceae) produce abundant hydroxy fatty acids in their seeds, with industrial applications. Here, we report three chromosome-level genomes of Physaria species: P. lindheimeri, P. pallida and P. fendleri, with sizes of 344 Mb, 329 Mb and 452 Mb, respectively. Comparative genome analysis reveals that these three Physaria species diverged from Arabidopsis thaliana approximately 14.10-14.46 million years ago and underwent two consecutive Physaria-specific whole-genome duplication events. Their centromeres harbor an 111-bp satellite repeat and two retrotransposon classes (Gypsy/CRM, Copia/Ale). Transcriptomic analysis identifies seed-highly expressed lipid synthesis genes potentially underlying unique fatty acid profiles. Furthermore, we pinpoint the two residues in FAH12 variants that cause the disparity in hydroxylation activity among the three Physaria species. Taken together, this study provides important genomic resources for investigating the evolution of Physaria species and developing industrial oil crops for sustainable production of hydroxy fatty acids.
Similar content being viewed by others


Introduction
Plant oils represent an important natural resource of human society, providing food, essential nutrients or industrial feedstock depending on their fatty acid composition. Most oilseed crops produce oils containing common fatty acids, such as palmitic acid (C16:0), stearic acid (C18:0), oleic acid (C18:1), linoleic acid (C18:2), and α-linolenic acid (C18:3), and can be used in food or non-food applications, many species produce unusual fatty acids that may be valuable for industrial applications due to their unique chemical structures, including variations in carbon chain length, positions of double and triple bonds, and the presence of functional groups such as hydroxyl, epoxy, and cyclopropane groups1. For example, castor (Ricinus communis) accumulates approximately 90% ricinoleic acid (12-hydroxyoctadeccis-9-enoic acid; C18:1-OH) in its seed oil2,3. While it provides a vital source of hydroxy fatty acid (HFA) for various industrial applications4, castor is unsuitable for large-scale agricultural production due to the presence of toxin ricin and the hyper-allergic 2S albumins5. A similar HFA, lesquerolic acid (14-hydroxyeicos-cis-11-enoic acid; C20:1-OH), is found at abundance (50–85%) in Physaria seeds6. The oil is a colorless to pale-yellow liquid that exhibits excellent stability and biodegradability, and it may provide an alternative source of HFA for making lubricants, motor oils, desiccants, nylon, plastics, cosmetics, pharmaceuticals, and polishes6,7,8,9,10,11.
Physaria, which was previously classified as Lesqueralla, is a genus of the Brassicaceae family and is closely related to Paysonia12. Some Physaria species exhibit valuable traits, including non-dehiscent seeds, neutral photoperiod response, upright growth habit, and high seed oil yield6 (Fig. 1a). Exploration of the Physaria species found higher (85%) contents of lesquerolic acid in P. lindheimeri and P. pallida13, making them valuable genetic resources to enhance HFA production in agricultural crops. P. fendleri is being developed to provide a safe source of HFA14,15. However, compared to castor with high oil content (38–55% of seed weight) and nearly 90% HFA, P. fendleri seed oil content is generally lower at about 27–33% and contains up to 60% C20:1-OH16. It is necessary to improve the productivity of HFA by plant breeding. Metabolic engineering offers another attractive approach to produce HFA in established agricultural oil crops17. The HFA synthesis occurs on the phosphatidylcholine (PC), where C18:1 is converted into C18:1-OH by an oleic acid Δ12-hydroxylase (FAH12)18,19,20. Ensuing elongation C18:1-OH produces lesquerolic acid (C20:1-OH). This step is catalyzed by the fatty acid condensing enzyme 3-ketoacyl-CoA synthase 3 (KCS3) in a fatty acid elongase (FAE) complex. Physaria KCS3 is highly homologous to AtKCS18 (or AtFAE1) in Arabidopsis but specifically catalyzes the elongation of C18-HFA21. HFA are predominantly incorporated into triacylglycerol (TAG) through the Kennedy pathway and acyl editing reactions22,23,24. Despite the progress in understanding oil biosynthesis pathways, engineering plants with high levels of HFA remains largely unsuccessful25.

a The morphological characteristics of P. lindheimeri, P. pallida and P. fendleri plants, including young plants (left), flowering plants (middle) and flowers (middle, upper right), and seeds (right). Bar = 1 mm. b The circos plot of the distribution of genomic features of three Physaria species using 500 kb per window. GC content (1), Gene density (2), TE density (3), Gypsy density (4), Copia density (5). The number of colored stars represents the number of telomeric sequences per chromosome. Pl, Pp and Pf are the abbreviations for P. lindheimeri, P. pallida and P. fendleri species. c Genome-wide Hi-C interaction matrix of P. lindheimeri, P. pallida and P. fendleri. The color intensity represents the frequency of contact between 150 kb loci. Source data are provided as a Source Data file.
In this study, we assemble chromosome-level genomes of three representative Physaria species (P. lindheimeri, P. pallida, and P. fendleri) by PacBio sequencing and Hi-C technologies. Based on the high-quality genomes, we investigate the evolutionary history of the Physaria genus. Following the divergence of Physaria from Arabidopsis, the species-specific whole-genome duplication event is observed. Transcriptomic analysis unveils the high expression of key genes such as FAH12 and KCS3 that are involved in HFA synthesis in seeds. We show that the activity of PfFAH12 is lower than that of P. lindheimeri and P. pallida, which may explain the varying contents of lesquerolic acid in these three species. Taken together, the high-quality assembly and analysis of three chromosome-level genomes, as well as the study of representative enzymes, would provide valuable insights into the evolutionary history of Physaria and genetic tools for engineering plants for HFA production and for establishing Physaria species as economically significant oil crops.
Results
High-quality assembly and annotation of three Physaria genomes
Multifaceted sequencing approaches were used to assemble the genomes of Physaria genus. About 30.46 Gb (~92.58×) PacBio high-fidelity (HiFi) reads for P. pallida, 28.81 Gb (~65.28×) for P. fendleri, and 167.78 Gb (~488×) PacBio continuous long reads (CLR) for P. lindheimeri were used to de novo assembly and generate draft genome assemblies. Illumina short reads of 115.43 Gb (~335×) for P. lindheimeri, 110.10 GB ( ~ 335×) for P. pallida and 55.73 Gb (~126×) for P. fendleri were used to correct genome assemblies. In addition, 117.52 Gb (~341×) high-throughput chromosome conformation capture (Hi-C) reads for P. lindheimeri, 50.36 Gb (~153×) for P. pallida and 112.37 Gb (~254×) for P. fendleri were used in genome assembly (Supplementary Data 1). Through these approaches, we assembled 344.42 Mb chromosome-level P. lindheimeri genome with contig N50 size of 6.08 Mb and scaffold N50 size of 53.59 Mb, for which the total length of pseudochromosomes was 324.41 Mb with pseudochromosome lengths ranging from 46.30 Mb to 66.59 Mb. For P. pallida, we generated 329.03 Mb chromosome-level genome with the contig N50 size of 30.42 Mb, scaffold N50 size of 51.55 Mb, and a total length of pseudochromosomes amounting to 314.31 Mb with pseudochromosome lengths ranging from 41.77 Mb to 65.95 Mb. For P. fendleri, 451.62 Mb chromosome-level genome was generated with the contig N50 size of 33.69 Mb, scaffold N50 size of 69.75 Mb, and a total length of pseudochromosomes amounting to 429.99 Mb with pseudochromosome lengths ranging from 66.60 Mb to 81.59 Mb (Fig. 1b, c, Table 1, Supplementary Data 2). The sizes of the three genome assemblies are consistent with the estimated sizes based on k-mer statistics with short reads (Supplementary Fig. 1, Supplementary Data 3). Moreover, we identified 6, 10 and 10 seven-base telomeric (5’-TTTAGGG-3’) repeats in the P. lindheimeri, P. pallida and P. fendleri (2n = 12) genome assemblies (Fig. 1c).
To assess accuracy and completeness of the genome assemblies of P. lindheimeri, P. pallida and P. fendleri, the Illumina pair-end reads were mapped back to three genome assemblies with the alignment rates of 99.28%, 97.50% and 96.26%, respectively (Supplementary Data 4). A total of 98.5%, 99.1% and 98.6% core eukaryotic genes from the Benchmarking Universal Sing-Copy Orthologs (BUSCO) were captured by the three genome assemblies, respectively (Supplementary Data 5). In addition, the long terminal repeat assembly index (LAI) of the genome assemblies was 18.60, 20.80, and 23.79, respectively (Table 1), indicating that all three genome assemblies meet the criteria for high-quality reference genomes. Notably, the genome assemblies of P. pallida and P. fendleri meet the gold standard for genome assembly quality26. In summary, all assessment suggested the high-quality reference genomes of the three Physaria species.
A total of 33,677, 36,997 and 39,517 protein-coding genes were annotated in P. lindheimeri, P. pallida and P. fendleri genomes with an average coding sequences length of 1166, 1208 and 1205 bp, which were similar to other species of Brassicaceae (Supplementary Fig. 2, Supplementary Data 6, Supplementary Data 7). Mapping these genes to multiple protein databases, it was found that 95.95%, 98.72 and 98.80% of the genes were functional in each genome (Supplementary Data 8). Transposable elements (TEs) occupy 57.39%, 53,93% and 62.62% of P. lindheimeri, P. pallida and P. fendleri genomes and most abundant category of TEs was the long-terminal retrotransposon (LTR), with LTR/Copia and LTR/Gypsy accounting 15.17% and 18.22% in P. lindheimeri genome, 12.05% and 12.08% in P. pallida genome, and 13.59% and 23.58% in P. fendleri genome (Supplementary Data 9). Furthermore, 10,394, 10,382 and 13,143 non-coding were predicted in P. lindheimeri, P. pallida and P. fendleri genomes, containing 152, 154 and 155 microRNAs, 1647, 2647 and 1,864 transfer RNAs, 3927, 5457, and 6494 ribosomal RNAs and 4668, 2124 and 4,631 small nuclear RNAs in each genome, respectively (Table 1).
Species-specific whole-genome duplication events in the three Physaria species
A phylogenetic tree was constructed to elucidate the evolution of Physaria genus, based on 276 single-copy gene families from three Physaria species and 15 other representative species (Fig. 2a). This revealed that the genus Physaria is most closely related to A. thaliana, diverging from it about 15.11 million years ago (Mya). Subsequently, P. fendleri diverged from the other two Physaria species approximately 5.36 Mya, followed by P. lindheimeri and P. pallida around 3.39 Mya (Fig. 2a). Analysis of gene family expansion and contraction revealed 769, 884 and 997 expanded gene families, as well as 1181, 628 and 732 contracted gene families in P. lindheimeri, P. pallida and P. fendleri, respectively (Fig. 2a). Further gene ontology (GO) enrichment analysis indicated that these expanded genes were significantly enriched in the terms of ‘regulation of protein ubiquitination’, ‘photosynthetic electron transport chain’, and ‘ATP synthesis coupled with proton transport’ (Supplementary Fig. 3), respectively.

a The phylogenetic tree of Physaria species and 15 other species, alongside the number of gene family expansion (blue numbers) and contraction (orange numbers). The number at the root of the tree (13,676) represents the total number of gene families predicted in the most recent common ancestor (MRCA). b The density distribution of Ks values for homologous gene pairs within Physaria species, within A. thaliana, and between P. lindheimeri and A. thaliana. c Syntenic comparative analysis between A. thaliana and P. lindheimeri. The red dotted frame marks some syntenic blocks at Ks < 0.5, and the syntenic relationship of 1:4 can be clearly seen. Points of different colors represent different Ks values of collinear gene pairs. d Syntenic blocks between P. lindheimeri and P. pallida, as well as P. lindheimeri and P. fendleri (e), share a 1:1 syntenic relationship. Source data are provided as a Source Data file.
To identify the whole-genome duplication event in Physaria genus, Ks values were analyzed using A. thaliana as a representative species of Brassicaceae. Our analysis indicated that, in addition to the previous reported α whole-genome duplication (WGD) event in Brassicaceae (with a peak Ks value around 0.92–0.95), Physaria genus exhibited a distinct peak at a Ks value of 0.42–0.43 (Fig. 2b). These suggested the species-specific WGD in Physaria following its divergence from A. thaliana, estimated to occur approximately 14.10–14.46 Mya. To further investigate WGD specific in Physaria, we identified 1298, 1387, and 1387 syntenic blocks in P. lindheimeri, P. pallida and P. fendleri compared to A. thaliana, respectively. Syntenic blocks at Ks < 0.5 revealed a 1:4 relationship between A. thaliana and Physaria, suggesting that two whole-genome duplicated events occurred following their divergence (Fig. 2c, Supplementary Fig. 4).
The synonymous substitution rates exhibited a unimodal pattern, indicating that two whole-genome duplication events took place within a relatively narrow time frame, making it challenging to distinguish them based on divergence times alone. To differentiate between two whole-genome duplication events within the Physaria genus, A. thaliana was divided into 8 blocks based on syntenic relationships, and homologous gene pairs in each block were used to construct maximum likelihood trees (Supplementary Fig. 5). The aim was to separate 1:4 syntenic blocks from each other based on the topology of the tree. Unfortunately, when we added up the maximum frequency of the tree topology for each block, we found that P. lindheimeri, P. pallida and P. fendleri were only 40.63%, 39.15%, and 39.94%, respectively (Supplementary Data 10). This indicated that it was not possible to separate all pairwise syntenic blocks by this method. This could be due to that the Physaria genus underwent two diploidization events with a short interval, which makes them indistinguishable. Furthermore, analysis of genome-wide synteny revealed that P. lindheimeri and P. pallida, as well as P. lindheimeri and P. fendleri, share a 1:1 syntenic relationship, indicating that three Physaria species underwent the same WGD event and did not experience further WGD event after their divergence (Fig. 2d, e).
By comparing with the ancestral eudicot karyotype (AEK)27, we were able to trace the karyotype evolution route of Physaria genus. After diverging from A. thaliana (which has 5 chromosomes), the chromosomes of Physaria genus underwent multiple fissions and fusions, resulting in the current six chromosomes (Fig. 3a, Supplementary Fig. 6). The long terminal repeat retrotransposon (LTR-RT) is the most abundant type of repetitive sequence in the Physaria genus (Supplementary Data 9). Estimation a burst of LTR-RT insertion times for P. lindheimeri, P. pallida and P. fendleri indicated that they approximately 0.0296 Mya, 0.0219 Mya and 0.0096 Mya, respectively (Supplementary Fig. 7). This implied that the LTR-RTs remain highly active in the Physaria genus, with their activity predominantly localized to chromosome ends (Supplementary Fig. 8). In addition, when comparing the three Physaria species with A. thaliana and Capsella Rubella, we observed that 15,890 gene families were shared among five species, while 375, 357 and 518 gene families were unique to P. lindheimeri, P. pallida and P. fendleri, respectively (Fig. 3b). KEGG enrichment analysis of these unique gene families revealed their significant enrichment in pathway such as “DNA replication”, “DNA repair and recombination proteins”, “Mismatch repair” and “Nucleotide excision repair”. These results suggested that after the recent WGD and LTR-RTs amplification events in Physaria, there gene families may play a key role in maintaining normal cellular function and genome stability (Fig. 3c–e)28.

a Evolutionary scenarios of chromosomal rearrangements and structural evolution from ancestral eudicot karyotype (AEK, n = 7 and n = 21) to three Physaria species and other plants. Chromosomes are marked with different colors to indicate the segmental evolution of the common ancestral eudicot karyotype with seven protochromosomes. b Gene families shared by three Physaria species, A. thaliana and C. rubella. KEGG enrichment analysis results of P. lindheimeri (c), P. pallida (d) and P. fendleri (e) unique gene families. The KEGG pathways related to repair and recombination are marked in red. Statistical analysis was performed using a one-sided hypergeometric test and the Benjamini-Hochberg (BH) method to control the false discovery rate (FDR). Source data are provided as a Source Data file.
Sequence architecture of the centromeric region of Physaria species
Throughout the intricate process of cell division, centrosomes assume a pivotal role in upholding the integrity of chromosomes and ensuring the precise transmission of genetic information29. Functioning as a pivotal constituent essential for firmly anchoring chromosomes to the mitotic spindle during cell division, the assembly of centromere poses inherent challenges, attributed to the abundance of repetitive DNA elements. These repetitive sequences are mainly composed of high copy number tandem repeats (TR) and centromeric retrotransposons (CR). The distinctive satellite repeat units of centromeres have been recognized across a variety of species, including the presence of 155 bp CentO satellite repeat sequence in rice, the abundance of 178 bp CEN180 satellite repeat sequence in Arabidopsis, and the enrichment of 156 bp CentC satellite repeat sequence in maize30,31,32. Based on this characteristic, we identified the centromeric regions of chromosomes in the genomes of P. lindheimeri, P. pallida, and P. fendleri (Fig. 4a and Supplementary Fig. 9). In P. lindheimeri genome, centromeres exhibit a length range of 2.18 Mb to 3.14 Mb, with an average length of 2.62 Mb, and each centromere contains an average of 4308 satellite repeat sequences. In P. pallida genome, centromeres span from 1.52 Mb to 4.96 Mb, with an average length of 3.55 Mb, and each centromere contains an average of 4664 satellite repeat sequences. Similarly, in P. fendleri genome, centromere lengths range from 1.48 Mb to 3.26 Mb, with an average length of 2.36 Mb, and each centromere contains an average of 5439 satellite repeat sequences (Supplementary Data 11). Furthermore, we identified satellite repeat sequence with a length of 111 bp in all three genomes. We designated the centromeric satellite repeat of the three Physaria as CentPl, CentPp, and CentPf (Fig. 4b). It is noteworthy that the satellite repeat units among the three Physaria species are highly similar. The identity between the satellite repeat sequences of P. lindheimeri and P. pallida reaches 94%, while the average sequence identity of centromeric satellite sequences within chromosomes is as high as 96%. This result suggests the significant conservation of satellite repeat units in the centromeric regions of these plant genomes.

a Characterization of the centromere of chromosome 6 in P. lindheimeri. From top to bottom are the sequences of the centromere satellite repeats, Gypsy/CRM type retrotransposons, Copia/Ale type retrotransposons, genes and RNA-Seq data from five different tissues. Centromeric regions extend 1 Mb to the left and right. b Satellite sequence motifs in the centromeric regions of three Physaria species. c Segmental duplications and tandemly duplicated genes in P. lindheimeri. Heatmaps in the circos plot represent gene density. Linkage regions represent SD regions between chromosomes, and colored markers on chromosomes represent centromeric regions. d GO enrichment analysis of tandemly duplicated genes. Statistical analysis was performed using a one-sided hypergeometric test and the Benjamini-Hochberg (BH) method to control the false discovery rate (FDR). Source data are provided as a Source Data file.
The centromeric region is characterized not only by the abundance of high-copy-number tandem repeats but also by a significant presence of specific types of retrotransposons. Notably, the centromeres of rice are abundant in Ty3/gypsy type retrotransposons, while those of Arabidopsis are rich in ATHILA long terminal repeat retrotransposons. Similarly, in maize, the centromeres are abundance of CRM and non-CRM Gypsy type retrotransposons32,33,34. Within species of the Physaria genus, we identified an enrichment of two classes of retrotransposons, Gypsy/CRM and Copia/Ale, in the centromeric regions (Fig. 4a and Supplementary Fig. 9). Notably, Gypsy/CRM-type retrotransposons exhibit the highest abundance, followed by Copia/Ale-type retrotransposons. Interestingly, there is no evident correlation between the copy number of retrotransposons on each chromosome and the length of the centromere (Supplementary Data 12). Among the three Physaria species, the centromeres of the P. lindheimeri genome exhibit the highest copy numbers for both classes of retrotransposons. Specifically, the copy numbers of Gypsy/CRM type retrotransposons range from 387 to 577 on each chromosome, while the copy numbers of Copia/Ale type retrotransposons range from 147 to 297. Despite P. fendleri having the largest genome among three species, we observed the lowest copy numbers of Gypsy/CRM and Copia/Ale retrotransposons in its centromeric regions. Notably, in the centromeric regions of chromosomes 3 and 4, Copia/Ale-type retrotransposons are present only once (Supplementary Data 12).
Despite the enrichment of satellite repeat sequences and specific retrotransposons in the centromeric regions, protein-coding genes have been successfully identified in all three Physaria species. P. lindheimeri contains the highest gene count in its centromeres, totaling 111 genes, followed by P. pallida with a count of 68 genes. Notably, despite P. fendleri having the largest genome, its centromeric region comprises only 19 protein-coding genes (Supplementary Data 13). We established a criterion for gene expression, considering TPM values greater than 1 in at least one tissue. In P. lindheimeri, P. pallida, and P. fendleri, we discovered 48, 9, and 9 expressed genes, respectively (Supplementary Data 13). However, it’s noteworthy that their expression levels are notably low. This phenomenon could be attributed to potential heterochromatin-mediated transcriptional suppression or the presence of pseudogenes that are not actively transcribed35.
Segmental duplications and tandemly duplicated genes of Physaria species
We identified segmental duplications (SD) within the genomes of P. lindheimeri, P. pallida, and P. fendleri, comprising 86.88 Mb, 73.41 Mb, and 166.66 MB, respectively. These sequences constitute 26.78%, 23.38%, and 38.76% of their respective genomes (Fig. 4c, Supplementary Fig. 10, and Supplementary Data 14). Notably, in P. fendleri, the content of SD is significantly higher compared to the other species. Differing from the genomes of rice and asparagus bean, the frequency of SD is relatively uniform across each chromosome in three Physaria specie35,36 (Supplementary Data 14). Within the SD regions of the three Physaria species, a total of 4055, 3813, and 6824 genes were identified, respectively. GO enrichment analysis revealed that the genes within these SD regions are predominantly associated with processes such as “DNA integration” and “DNA metabolic process” (Supplementary Fig. 11). These genes are likely instrumental in preserving genome integrity and stability across the three Physaria species and are essential for normal functioning and survival of cells.
Tandem duplication is a widespread occurrence in plant genomes, exerting a crucial influence on evolutionary processes and adaptation to dynamic environments. Tandemly duplicated genes, linked to specific functions, contribute to the expansion of gene families and lead to an increase in gene dosage in the form of gene clusters37. In P. lindheimeri, P. pallida, and P. fendleri, a total of 1446, 1872, and 1718 tandemly duplicated gene clusters were individually identified, collectively comprising 3700, 5139, and 4617 genes (Fig. 4c and Supplementary Data 15). In order to dissect the functional implications of these tandemly duplicated genes within the genome, GO enrichment analysis was performed. The results unveiled a notable enrichment of these genes in categories related to “defense response,” “response to stress,” and “response to stimulus” (Fig. 4d). This finding suggests their potential role in aiding species to maintain biological homeostasis and adapt to environmental fluctuations. Analyzing the expression patterns of genes within these three GO categories revealed tissue-specific expression, indicating the functional engagement of these genes across diverse tissues (Supplementary Fig. 12).
We conducted an analysis of NLR (nucleotide-binding leucine-rich repeat) resistance genes in the three species. This gene category is distinguished by the abundance of nucleotide-binding leucine-rich repeat sequences (NLR). NLR genes play a pivotal role in host resistance responses against pathogens, as they recognize pathogen proteins and activate mechanisms of resistance38. In P. lindheimeri, P. pallida, and P. fendleri, a total of 177, 221, and 167 NLR resistance genes were identified, respectively. Notably, a significant proportion of these resistance genes were observed to be tandemly duplicated genes. Specifically, 73 (41.24%), 98 (44.34%), and 66 (39.52%) of the NLR resistance genes in the three Physaria species were identified as tandemly duplicated genes.
High expression of key genes drives accumulation of hydroxy fatty acids in seeds of three Physaria species
The seeds of three Physaria species are characterized by their oils with abundant HFA6,13. Greenhouse-grown plants produced seeds containing oils from P. lindheimeri, P. pallida and P. fendleri at 24.23%, 18.32% and 17.96%, respectively (Supplementary Fig. 13 and Supplementary Data 16). Notably, the primary fatty acid is lesquerolic acid (C20:1-OH), which accounts for 80.78%, 84.56% and 51.19% of total fatty acids in P. lindheimeri, P. pallida and P. fendleri, respectively (Fig. 5a and Supplementary Data 17).

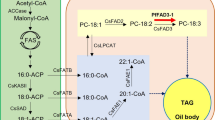
a Contents of different types of fatty acids in seed oils, among which lesquerolic acid (C20:1-OH) accounts for 80.78%, 84.56% and 51.19% in stored triacylglycerol in P. lindheimeri, P. pallida and P. fendleri, respectively. Error bars represent mean values ± SD. For each type of fatty acid, n = 6 biological replicates. b The simplified diagram of the lipid biosynthesis pathway in three Physaria species. The heatmaps represents the expression of each gene and key transcription factors in seeds at different developmental stages (EDS: early developing seed, MDS: middle developing seed, LDS: late developing seed), as well as leaf and stem. The red color of heatmap indicates high gene expression, while blue indicate low expression. ACCase acetyl-CoA carboxylase, FAS fatty acid synthase, KAS Ⅱ β-ketoacyl-ACP synthase Ⅱ, SAD stearoyl-acyl carrier protein desaturase, FATA fatty acid thioesterase A, FATB fatty acid thioesterase B, LACS long-chain acyl-CoA synthetase, LPCAT lysophosphatidyl-choline acyltransferase, FAD2 fatty acid desaturase 2, FAD3 fatty acid desaturase 3, FAH12 oleate 12-hydroxylase, KCS3 3-ketoacyl-CoA synthase 3, PLC phospholipase C, PDCT phosphatidylcholine: diacylglycerol cholinephosphotransferase, CPT CDP choline: diacylglycerol cholinephosphotransferase, PDAT: phospholipid diacylglycerol acyltransferase, GPAT glycerol-3-phosphate acyltransferase, LPAT lysophosphatidic acid acyltransferase, PAP phosphatidic acid phosphatase, DGAT diacylglycerol acyltransferase, ABI3 abscisic acid insensitive 3, WRI1 wrinkled 1, FUS3 fusca 3, LEC1 leafy cotyledon 1, LEC2 leafy cotyledon 2. c Phylogenetic trees and gene expression profiles of FAH12 and FAD2, KCS3 and FAE1 genes in R. communis, A. thaliana, P. lindheimeri, P. pallida and P. fendleri. A. thaliana FAD2 (NP_001319529.1), R. communis FAD2 (QCO64167.1), R. communis FAH12 (ACD39348.1), P. fendleri FAD2 (AFP21683.1), P. fendleri FAH12 (ACQ42234.1), A. thaliana KCS3 (NP_001322003.1), A. thaliana FAE1 (NP_195178.1), P. fendleri KCS3 (AAK62348.1) genes were obtained from NCBI database (https://www.ncbi.nlm.nih.gov/). Source data are provided as a Source Data file.
To elucidate the genetic mechanisms of oil biosynthesis and HFA accumulation in the three Physaria species, we compared transcriptomes between developing seeds at different stages (early developing seed, middle developing seed, and late developing seed) and leaf and stem. A total of 4703 and 5636 up-regulated differentially expressed genes (up-DEGs) were identified in P. lindheimeri, 3194 and 3758 up-DEGs in P. pallida, 3923 and 3370 up-DEGs in P. fendleri between developing seed and leaf (seed vs leaf) or stem (seed vs. stem), respectively (Supplementary Fig. 14). KEGG functional enrichment analysis of these DEGs demonstrated that many were assigned to “lipid metabolism”, “linoleic acid metabolism”, “lipid biosynthesis proteins”, “fatty acid elongation”, “alpha-linolenic acid metabolism” and “biosynthesis of unsaturated fatty acids” (Supplementary Fig. 14). Furthermore, we identified 197, 195 and 182 genes in P. lindheimeri, P. pallida and P. fendleri, respectively, orthologous to a total of 111 fatty acid and lipid biosynthesis genes previously reported in A. thaliana (Supplementary Data 18, Supplementary Data 19).
The unique character of the three Physaria species is the high concentrations of HFA in seed oils, particularly lesquerolic acid (C20:1-OH)2,39,40. The HFA synthesis genes FAH12 and KCS3 in the three Physaria species all showed substantially higher expression levels in middle and late-developing seed as compared to early-developing seed, leaf, and stem, where their expressions were negligible (Fig. 5b, c). Other major genes involved in the lipid synthesis pathway were also predominantly expressed in seeds with low expression in leaf and stem, such as SAD, a key desaturase that catalyzes the conversion of C18:0-ACP to C18:1-ACP in the plastid, and genes encoding TAG assembly enzymes including lysophosphatidic acid acyltransferase (LPAT), diacylglycerol acyltransferase (DGAT) and phospholipid:diacylglycerol acyltransferase (PDAT) (Fig. 5b). LPAT specifically incorporates acyl groups into the sn-2 position of the glycerol backbone41,42, and has been shown to be the main limit for HFA accumulation in the TAG molecules in P. fendleri43. LPAT genes are highly expressed in developing seeds at the early to middle stages (Fig. 5b). DGATs are also highly expressed in middle to late stages of developing seeds (Fig. 5b), which is consistent with the previous reports that DGAT variants with strong preference to HFA contribute to their accumulation in seed oil25,44. Besides these biosynthetic genes, we also identified five transcription factors (TFs), ABSCISIC ACID INSENSITIVE3 (ABI3), WRINKLED1 (WRI1), FUSCA3 (FUS3), LEAFY COTYLEDON1 (LEC1), and LEAFY COTYLEDON2 (LEC2), which play central roles in regulating lipid biosynthesis and the accumulation of seed oil45,46,47,48,49,50. These TFs also showed much higher expression in developing seeds than in leaf and stem (Fig. 5b). The elevated expression of key genes and TFs involved in lipid biosynthesis in seed may contribute to the synthesis of HFA-containing oils in the three Physaria species.
Activities of Physaria FAH12s were substantially affected by two key amino acids
It is notable that the lesquerolic acid content is remarkably higher in P. lindheimeri and P. pallida seeds compared to P. fendleri (Fig. 5a). FAH12 and KCS3 are two key enzymes directly involved in biosynthesis of lesquerolic acid, and they are upstream of acyltransferases, which incorporate lesquerolic acid into TAG. Based on the fatty acid profile of three Physaria species, ricinoleic acid (C18:1-OH) is almost completely converted into lesquerolic acid (Fig. 5a). We speculated that KCS3 is not a limiting metabolic step of lesquerolic acid in three species. Interestingly, in P. fendleri, the expression level of the FAH12 was higher in middle and late developing seeds than in P. lindheimeri and P. pallida (Fig. 6a). To explain this discrepancy, we hypothesized that despite its high transcriptional level, the activity of the PfFAH12 enzyme may be considerably lower than PlFAH12 and PpFAH12. To test this hypothesis, we expressed three FAH12 genes in mammalian cells and compared their abilities to produce HFA (Fig. 6b, and Supplementary Data 20). We purified the FAH12 proteins and observed that the expression of PfFAH12 was higher than PlFAH12 and PpFAH12 (Supplementary Fig. 15a). However, cells hosting PfFAH12 produced much lower amount of ricinoleic acid than PlFAH12 and PpFAH12 (Fig. 6b). No detectable ricinoleic acid was found in negative control purified FAH12 or in the extracts of fatty acid desaturase 2 (FAD2) (Supplementary Fig. 16), which catalyzes the desaturation of oleic acid to produce linoleic acid2. These results suggest that the better performance of PlFAH12 and PpFAH12 than PfFAH12 may play an important role in the much higher contents of lesquerolic acids in P. lindheimeri and P. pallida seeds than P. fendleri.

a The expression of the FAH12 genes across three different tissues of three Physaria species, alongside the proportion of lesquerolic acid to total seed fatty acids in their seeds. The left y-axis represents the expression levels (TPM) of the FAH12 genes in early developing seeds (EDS, yellow folded plot), middle developing seeds (MDS, red folded plot), and late developing seeds (LDS, blue folded plot). Meanwhile, the right y-axis indicates the proportion of lesquerolic acid relative to the total fatty acid content in seeds (gray folded plot). b Activity assay. Assay was performed for three Physaria FAH12s and PfFAH12 mutants in mammalian cells. We analyzed the content of product ricinoleic acid (C18:1-OH) using GC-FID. The data from three independent measurements are averaged, and the error indicates SD. Statistical analysis: unpaired two-tailed t-tests compared with PfFAH12WT. *p < 0.05, **p < 0.01 ***p < 0.001, ****p < 0.0001. n = 3 biological replicates. c Structural visualization of three Physaria FAH12 AlphaFold models in complex with oleic acid, respectively. The docked models are shown in surface and cartoon representation. The histidine motifs in active pocket and substrate are shown in stick representation. d Structural superimposition of these three Physaria FAH12 AlphaFold models. Two pairs of residues predicted to cause differences in the activity of FAH12 among three Physaria are zoomed-in and shown in stick representation. e The ricinoleic acid (C18:1-OH) content in T3 mature seeds. Values are means ± SD (n = 5 biological replicates). Different letters indicate differences at p < 0.05 using one-way ANOVA. Source data are provided as a Source Data file.
Sequence comparison indicated that the PlFAH12 has about 90% sequence identity with PfFAH12, and about 95% sequence identity with PpFAH12 (Supplementary Fig. 17). The residues among the catalytic pockets of PlFAH12 and PpFAH12 are highly conserved. To provide structural context for the substrate binding pocket of FAH12, we ran AlphaFold351 for these three Physaria FAH12, then docked oleic acid into the AlphaFold prediction structures, respectively (Fig. 6c). The docking models52 show that the negatively charged carboxyl head of oleic acid is directed towards the positively charged histidine motifs. Superposing these models reveals highly similar architectures among them (Fig. 6d). Based on sequence comparison and structure prediction, we suspected that the weaker activity of PfFAH12 might be associated with its residues H111 and Q225 (Fig. 6d, Supplementary Fig. 17) instead of Q111 and H225 in PlFAH12 or PpFAH12 (Fig. 6d, Supplementary Fig. 17).
To test whether any of these two residues affect PfFAH12 activity, site-directed mutagenesis was performed by replacing either one residue or both residues of PfFAH12 (PfFAH12H111Q, PfFAH12Q225H, PfFAH12H111Q/Q225H). Visual inspection of the SDS-PAGE analyses of all mutants revealed similar expression as the wild type (Supplementary Fig. 15a). However, the activities of all PfFAH12 mutants significantly increased and the highest was observed in PfFAH12H111Q/Q225H which ricinoleic acid content increased by nearly 2.5-fold compared to that of wild type (Fig. 6b). These results demonstrated that both Q111 and H225 played crucial roles in PfFAH12. The results, together with the genome and evolution analyses, indicated that the divergence of FAH12s among the three Physaria species might have determined the rate of ricinoleic acid synthesis and led to different contents of lesquerolic acid. Additionally, the replacement of two key residues can enhance the activity of PfFAH12.
To further test the function of FAH12 for the biosynthesis of ricinoleic acid, we generated transgenic Arabidopsis lines overexpressing three Physaria FAH12s driven by the seed-specific napin promoter. We obtained three independent homozygous overexpression (OE) lines for each FAH12. The results showed that PlFAH12 had the highest expression level compared with PfFAH12 and PpFAH12. PfFAH12 and PpFAH12 generally had comparable expression levels (Supplementary Fig. 18 and Supplementary Data 21). The ricinoleic acid content in T3 mature seeds was quantified in these FAH12-OE lines. Compared with PfFAH12-OE seeds, those expressing PlFAH12 and PpFAH12 exhibited a significantly higher ricinoleic acid content (Fig. 6e and Supplementary Data 22), consistent with the higher in vitro enzymatic activity of these two enzymes. These results demonstrate that variations in ricinoleic acid accumulation are mainly influenced by the specific FAH12 gene expressed, highlighting differences in enzyme activity among these three FAH12s.
Discussion
Plant oils with unusual structural features such as hydroxy groups, are valuable resources for bio-based products. To provide sustainable supplies of these raw materials, new crops that naturally produce such fatty acids are under domestication and breeding. Also, extensive efforts have been made to produce unusual fatty acids in established oilseed crops by genetic engineering. Physaria fendleri has attracted notable attention due to its potential as a viable substitute for castor to provide a safe source of HFA6. Limited genomic resources in Physaria species greatly hinder the progress of crop improvement. Here, we presented the chromosome-level genomes of P. fendleri and two other HFA-producing Physaria species (P. lindheimeri and P. pallida). These genomes provide powerful genetic tools to study and improve agronomics of P. fendleri.
Comparing multiple Physaria genomes may also help us understand the evolution of Brassicaceae plants. Through an evolutionary analysis of the genomes of three Physaria species, we revealed that the divergence of the Physaria genus from A. thaliana occurred approximately 15.11 Mya. Furthermore, Physaria genus not only experienced the α and β WGDs shared by Brassicaceae species but also underwent two consecutive species-specific WGDs around 14.10–14.46 Mya. These frequent whole-genome duplication events have substantially contributed to the diversity of Brassicaceae species. The presence of a 1:4 synteny block between A. thaliana and Physaria suggests that two whole-genome duplication events occurred in Physaria following divergence from A. thaliana within a relatively condensed timeframe similar to that observed in Amorphophallus konjac, Colocasia esculenta, and Spirodela Polyrhiza53,54,55. Despite attempts to distinguish homologous gene pairs through phylogenetic methods to differentiate the timing of two whole-genome duplication events experienced by the Physaria genus, our efforts were unsuccessful. However, we are optimistic that with the advancement of sequencing technology and algorithms, there will be better methods for unraveling the evolutionary trajectory of Physaria.
Compared to P. lindheimeri and P. pallida, P. fendleri seeds contain much lower lesquerolic acid contents (Fig. 5a). This may be explained by two major causes in P. fendleri. First, it has been reported that the HFA are mostly found at the sn-1 and sn-3 positions of TAG molecules in P. fendleri25, thus limiting the level of lesquerolic acid to ~60%. The exclusion of C20:1-OH at the sn-2 position was presumably due to the substrate specificity of the LPAT in P. fendleri, and expression of the castor LPAT2 indeed enhanced ricinoleic acid at the sn-2 position and therefore increased the content of tri-hydroxy-TAG from 5% to 14%43. We found that the transcriptional expression levels of LPAT were similar, at least between P. fendleri and P. pallida, among the three species (Fig. 5b). Since expression levels alone could not explain the differential accumulation of lesquerolic acid, we then asked whether there were functional differences in LPAT2 proteins. Sequence analysis identified differences in amino acids among PfLPAT2, PpLPAT2, and PlLPAT2. Substrate docking models indicated that the variable amino acids are not located near the active pocket (Supplementary Figs. 19, 20), suggesting that they are unlikely to affect enzyme activity. However, further biochemical characterization such as substrate specificity is required to provide direct evidence that LPATs contribute to different levels of lesquerolic acids in these Physaria species.
Another major cause for the lower production of HFA may be attributed to PfFAH12. This enzyme has been shown to possess bifunctional activities of both hydroxylation and desaturation39, therefore, even a higher expression level of PfFAH12 (Fig. 5c and Fig. 6a) was unable to achieve the amount of HFAs in the other two species. Apart from this possibility, we conducted detailed sequence analysis and discovered two residues, Q111 and H225, at the histidine motifs in PfFAH12 that may play a crucial role in modulating its activity. This was consequently supported by site-directed mutagenesis and in vitro enzyme assays (Fig. 6b, c), and the PfFAH12H111Q/Q225H mutant exhibited increased binding affinity with its substrate (Supplementary Fig. 21). Consistent with this observation, structural modeling of the docked complexes shows that the hydroxylation site Δ12 carbon of PfFAH12H111Q/Q225H is closer to the active sites and the fatty acyl chain penetrates deeper into the internal tunnel56. The addition of varying degrees of polar residues with hydrogen bonding may have also likely induced significant conformational changes. These results therefore indicated that lower activity of PfFAH12 contributed to the lower-level HFA accumulation in P. fendleri seed.
Attempts to engineer crop species for HFA production have yielded limited success thus far. For instance, both RcFAH12 and PfFAH12 were able to synthesize HFAs when their genes were introduced into Arabidopsis or Camelina sativa, but only 17% of HFA accumulated in the seeds20,39,57,58. It was hypothesized that the high levels of HFA accumulation in those native plants may require additional genes and enzymes that have co-evolved with the FAH1258. Several key genes in the oil biosynthesis pathways in castor, such as RcDGAT2, RcPDAT1, RcGPAT9, RcLPAT2, and RcPDAT1, were identified and increased the HFA content to about 28% when co-expressed with FAH1259,60,61,62,63,64,65. Comparing transcriptomes in P. fendleri, engineered HFA-producing camelina, and wild-type camelina resulted in a suite of genes and pathways that may potentially relieve the bottlenecks of the HFA production in transgenic seeds66. Transgenic experiments by co-expressing those genes, including PfKCS367 and a TAG lipase (PfTAGL1)25 that are highly expressed in P. fendleri developing seeds, significantly increased HFA production in transgenic camelina seed. However, to achieve the levels of the native species or those high enough for commercial production, multiple genes need to be incorporated into transgenic seeds. Besides those genes involved in incorporating HFA into TAG and preventing TAG degradation mentioned above, the initial HFA synthesis is also important. Previously, the beneficial effects of WRI1 have been shown by rescuing feedback inhibition of fatty acid synthesis68. Here, we demonstrate that high FAH12 activity can also enhance the overall HFA production. Physaria and the host plants, e.g., Arabidopsis and camelina, all belong to the Brassicaceae family and store oils in embryos other than endosperm in the distantly related castor bean; therefore, it is possible that orthologs in Physaria may be more suitable to integrate into the host oil synthesis pathways for HFA accumulation. The genomes of three HFA-producing Physaria species, along with other available genomes and pangenomes of Brassica plants such as B. napus29 and Camelina sativa69 that accumulate only common fatty acids, will facilitate the discovery of beneficial genes.
Methods
Plant growth condition and sample collection
The seeds of P. lindheimeri, P. pallida and P. fendleri were sown in the pots with nutrient soil: vermiculite = 1:1 (v/v) in a growth chamber (Conviron) at 26/24 °C under 16-h light/ 8-h dark. Before the seed germination of P. fendleri, the seeds in pots were treated with vernalization about half month. The plants in pots were transported to Novogene to extract DNA for genome sequencing when they grew big enough but not bolting. P. lindheimeri and P. fendleri are self-incompatibility plants after the stigma exertion. Hence, artificial compulsory pollination was performed before the flower bud opened. The whole siliques with 20-23 (early developing seed, EDS), 32-35 (middle developing seed, MDS), and 45-47 (late developing seed, LDS) days after pollination (DAP) of P. lindheimeri, 15-17 (EDS), 25-27 (MDS) and 35-37 (LDS) DAP of P. pallida, 10-13 (EDS), 20-23 (MDS) and 30-33 (LDS) DAP of P. fendleri were collected for the RNA-seq experiments. The samples of leaf and stem were collected at the same time as the MDS siliques.
Sequencing
For Illumina paired-end reads sequencing, 0.2 μg DNA from young leaves per species was used for DNA library preparations. The NEB Next® Ultra™ DNA Libraries Prep Kit for Illumina (NEB, USA) was used to generate DNA libraries according to the manufacturer’s recommendations. Illumina PE Cluster Kit (Illumina, USA) was used to cluster the index-coded samples performed on a cBot Cluster Generation System, then the DNA libraries were sequenced on the Illumina platform, and 150 bp paired-end reads were generated.
For PacBio sequencing, ~20 kb SMRTbell libraries were constructed from young leaves per species using the SMRTbell Express Template Preparation Kit 2.0 for the CLR module and SMRTbell Express Template Prep Kit 2.0 for the CCS module. Then, Sequel Ⅱ Sequencing Kit 2.0 was used to sequence and generate PacBio long reads.
For RNA sequencing, RNA was extracted from different tissues stated above. The TruSeq PE Cluster Kit v3-cBot-HS (Illumina) was used to cluster the index-coded samples on a cBot Cluster Generation System. The libraries were then sequenced on an Illumina Novaseq platform, and 150 bp paired-end reads were generated.
Estimation of genome size and heterozygosity
The genome size was estimated using k-mer frequency distribution generated from Illumina short reads. The jellyfish (v2.3.0)70 was used to generate 19-mer frequency distribution. Then, GenomeScope71 was used to estimate genome size and heterozygosity.
Genome assembly and assessing genome assembly quality
For P. lindheimeri, Canu (v2.1)72 was used for de novo assembly based on PacBio continuous long reads (CLR) with the parameters: rawErrorRate = 0.3, minReadLength = 1000, corOutCoverage = 40, correctedErrorRate = 0.045, minOverlapLength = 500, corMinCoverage = 4, maxThreads = 200. The raw contigs were polished two rounds using PacBio continuous long reads with Racon73 and polished two rounds using CLR reads with Pilon74. For P. pallida and P. fendleri, HiFiasm (v0.16.1-r375)75 was used for de novo assembly based on PacBio high-fidelity (HiFi) reads, -l 0 parameter was used for P. pallida de novo assembly, -s 0.5 parameter was used for P. fendleri de novo assembly, and one of the phased contigs as the final assembly result. Then the contigs were subjected to dehybridization using purge_haplogs (v1.1.0)76. Finally, the polished contigs were anchored into chromosomes by using HiC-Pro (v3.1.0)77 and ALLHiC (v 0.9.13)78 based on high-throughput chromosome conformation capture (Hi-C) reads, and then the assembled genome was manually correction by using Juicexbox (v1.11.08)79.
To assess the quality and completeness of the assembled genomes, BWA-MEM (v0.7.17-r1188, https://github.com/lh3/bwa) was used to map the Illumina paired-end reads to the assembled genomes, HISAT2 (2.1.0)80 was used to map the RNA sequencing data from multiple tissues to the assembled genomes, and minimap2 (v2.24-r1122)81 was used to map the PacBio long reads to the assembled genomes. Moreover, BUSCO (v4.0.6)82 was used to assess the completeness of the assembled genomes and predicted protein-coding genes based on the Embryophyta Plant database (odb10), and long terminal repeat retrotransposons assembly index (LAI) was used to evaluate the assembly quality using LTR_retriever (v2.9.0)83.
Genome annotation
Transposable elements annotation: First, a de novo transposable elements database was built using RepeatModeler, Piler84, RepeatScout85, Trf86, and LTR_FINDER87, then combined with the Repbase database (http://www.girinst.org/repbase) to generate a consensus library. Finally, RepeatMasker (https://www.repeatmasker.org/) was used to predict repeat sequences based on the consensus library.
The protein-coding genes were predicted by combining the homology-based prediction, de novo prediction, and RNA-Seq-assisted prediction. Firstly, the non-redundant proteins from five species (A. thaliana, Arabidopsis lyrate, Brassica rapa, Brassica oleracea, and Brassica napus) closely related to the Physaria genus were used as the homology annotation library and the input for TBLASTN to predict homologous sequences. Additionally, Augustus88,89, Genscan90, and GilmmerHmm91 were used for de novo gene prediction. Thirdly, RNA sequencing data were mapped to the genome using HISAT280, and transcriptome assembly was done using the StringTie pipeline92. Lastly, MAKER93 and HiCESAP were used to combine all prediction results to produce a non-redundant gene set. Moreover, the protein databases of SwissProt (https://www.gpmaw.com/html/swiss-prot.html), TrEMBL94, NR (https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/), KEGG95, InterPro96 and GO97 were used to predicted the function of genes.
Non-coding RNA annotation: tRNAscan-SE98 was used to annotate tRNA based on the structural features of tRNA, BLASTN was used to predict the rRNA based on homology prediction method by mapping rRNA sequences from closely related species to the genomes. The covariance model of the Rfam family and the INFERNAL that comes with Rfam were used to predict the miRNA and snRNA.
Phylogenetic analysis and gene family expansion/contraction
To elucidate the evolution of the Physaria genus, 15 other species (Amborella trichopoda, Oryza sativa, Zea mays, Solanum lycopersicum, Vitis vinifera, Glycine max, Populus trichocarpa, Jatropha curcas, Ricinus communis, Theobroma cacao, B. rapa, B. oleracea, A. thaliana, Capsella Rubella, and Camelina neglecta) were selected from the Phytozome database (https://phytozome-next.jgi.doe.gov/)99 for analysis. First, Orthofinder (v2.5.4) was used to identify single-copy orthologous genes. Next, the protein sequences and coding sequences of 276 sing-copy orthologous genes were aligned using MUSCLE (v5.1.linux64)100. Then the best-fit model (Blosum62 + I + G + F) was predicted using ProtTest (v3.4.2)101. RAxML (v8.2.12)102 was used to construct a maximum likelihood tree with 1000 replicates bootstrap. Finally, the iTOL website (https://itol.embl.de/)103 was used to visualize the phylogenetic tree.
MCMCTree program in the PAML package (v4.9.j)104 was used to calculate the divergence time for the above 18 species. Three calibration points were selected from the TimeTree database (http://www.timetree.org/)105 to estimate the divergence time, and the three calibration points are Z. mays and O. sative split time (0.45–0.52 Mya), A. thaliana and V. vinifera split time (1.07–1.35 Mya), as well as A. trichopoda and V. vinifera split time (1.73–1.99 Mya). Finally, CAFÉ (v5.0.0)106 was used to calculate the expansion and contraction of gene families based on the phylogenetic tree and gene family statistics.
Whole-genome duplication events analysis
To identify the whole-genome duplicated event in Physaria genus, WGDI (v0.6.2)107 was used for identifying syntenic blocks (-icl parameter) and calculating the Ks values of gene pairs (-ks parameter). In addition, it was used for visualizing the density of Ks distribution (-kf parameter) and for extracting collinear gene pairs from identified blocks (-a parameter). Finally, WGDI was used for constructing phylogenetic trees of the identified collinear gene pairs (-at parameter).
Identification of centromeric regions
Gepard (v2.1)108 was employed for the identification of repetitive sequences within the genome and to generate a dotplot. The dotplot of chromosomes was subsequently analyzed to identify the distinctive “black block” regions, generated as a result of intercomparison of repetitive sequences. These regions are considered characteristic of the centromeric regions, with the following parameters: Word length = 10, Window size = 0, Use matrix = DNA. To discern the position, number, and consistency of conserved tandem repeats (motifs), the Tandem Repeats Finder software (TRF) was employed86. The specific parameters utilized for this analysis were set as follows: 2 7 7 80 10 50 500 -f -d -m. Subsequently, MEME (v5.5.4) was utilized to mine motifs present in the repeats, specifically focusing on satellite repeats within the centromeric regions. The analysis employed the following parameters: -nostatus -time 14400 -mod zoops -nmotifs 3 -objfun classic -revcomp -markov_order 0 -searchsize 3000000.
Identification of segmental duplication and tandem duplication genes
The genomic sequence underwent soft-masking using RepeatMasker (v4.1.0) with the following parameters: -e rmblast -xsmall -gff -pa 1. Subsequently, the BiSER software was applied to identify segmental duplications within the masked genome. Fragments exhibiting an identity exceeding 90% and lengths surpassing 1 kb were retained as segmental duplications109. Genomic protein sequences were compared using Diamond, and the results were filtered by applying the following criteria: e-value < 10e-20, protein coverage > 80%, with the distance between two gene pairs not exceeding 10 genes110,111. Instances where gene pairs overlapped were construed as tandem duplicated gene clusters.
Differentially expressed gene analysis
Transcriptomic data from different tissues, including seeds at different developmental stages (early developing seed, middle developing seed, and late developing seed), as well as leaf and stem, were used to find differentially expressed genes. HISAT2 (v2.1.0)80 was used to map RNA-Seq data to the genome. StringTie (v2.1.7)92 was then used to calculate the TPM, and featureCounts112 was used to calculate the reads counts of the genes based on the result of mapping. Finally, R package DESeq2 (v1.38.3)113 was used to identify the differentially expressed genes with an adjusted p-value of 0.05.
Expression and purification of FAH12 in mammalian cell
The FAH12 genes were cloned into the pMLink vector114, containing an N-terminal 3×Flag tag. The site-specific mutations were introduced into the genes by overlapping PCR and were verified by DNA sequencing.
Expi293FTM cells (Invitrogen) were cultured in Union-293 media (Union-Biotech, Shanghai) in a 37 °C ZCZY-CS8 shaker (Zhichu Instrument) with 5% CO2. When the cell density reached 2.0 × 106 cells per mL, the FAH12 genes was transfected into the cells with plasmid, respectively. For 40 mL of cell culture, 80 μg of plasmids were pre-incubated with 240 μg of 4 kDa linear polyethylenimine (PEI) (Polysciences) for 20 min followed by adding the mixture into the diluted cells.
After culturing for another 60 h, the cells were harvested by centrifugation and resuspended in lysis buffer containing 100 mM Tris-HCl (pH 7.4), 150 mM NaCl and 1% LMNG (Anatrace) at 4 °C for 2 h. The insoluble component was removed by ultracentrifugation at 14,000 g for 30 min. The supernatant was incubated with anti-Flag G1 affinity resin (Genscript) at 4 °C for 1 h, and the resin was washed with lysis buffer with 0.01% LMNG. Then eluted by lysis buffer with 0.01% LMNG and 250 μg mL−1 3 × FLAG peptide (GenScript), and subsequent analysis by SDS-PAGE and immunoblot. The primer sequences are provided in Supplementary Data 23.
Determination of oil content and fatty acid content of seeds and mammalian cells
For seed oil analysis, 8–9 mg mature seeds were accurately weighted and used in oil extraction. Seed oil content and fatty acid composition were analyzed with GC-FID115. Six replicates were performed for each plant. To analyze fatty acid composition of mammalian cells, the biomass was pelleted by centrifugation and fatty acids were extracted in 2 mL acid methanol solution (5% H2SO4 in methanol and 1% BHT), supplemented with 40 μL internal standard (C17:0, Sigma). The glass tubes were tightly capped and heated at 85 °C for 2 h. After cooling, 2 mL of ultrapure water and 2 mL of hexane were added, and fatty acid methyl esters (FAMES) were recovered by collecting the hexane phase. Reaction products were analyzed by gas chromatography-flame ionization detector (GC-FID) using oleic acid (C18:1Δ9) (Sigma) and ricinoleic acid (MedChemExpress) as standards. The GC conditions were as follows: oven temperature was increased from 170 °C to 230 °C at a rate of 3 °C min–1. Fatty acid composition in individual cell samples was identified according to their retention times and calculated based on internal standard content with peak area.
Model docking of FAH12 and oleic acid
We predicted three Physaria FAH12 models by AlphaFold351, and used the program AutoDock Vina 1.1.2116 to dock the complex models of FAH12 and substrate oleic acid. The original structure of oleic acid was generated or modified using PyMOL and further prepared with AutoDock tools1117 for the docking process. During the docking calculation, the protein structures were regarded as the rigid body, while the ligand was given complete torsion freedom. The docking box was set around the catalytic area. For each substrate docking, a total of ten models were created, and the model with the best binding affinity was chosen for further analysis. All structural figures were generated using the PyMOL program (http://www.pymol.org/).
Agrobacterium-mediated transformation of Arabidopsis
After the plants reached the flowering stage, agrobacterium-mediated transformation was performed. The constructs were introduced into competent cells of Agrobacterium tumefaciens GV3101 via electroporation. Arabidopsis genetic transformation was carried out using the floral-dip method. Transformed Agrobacterium cells were collected by centrifugation and resuspended in a solution containing 5% (w/v) sucrose and 0.05% (v/v) surfactant SilwetL-77, adjusting the bacterial suspension to an OD₆₀₀ of 0.6–0.8. The suspension was placed in a disposable petri dish, and the inflorescences of flowering Arabidopsis plants were immersed in the bacterial suspension for 30 s. After infiltration, the plants were placed horizontally in the dark for 16–24 h. The infiltration step was repeated one week later. Subsequently, plants were returned to standard growth conditions until seeds were harvested at maturity. T₀ seeds collected after transformation were sown in nutrient soil and covered with plastic wrap for 4–5 days. They were then grown under conditions of 22 °C with a 16-hour light/8-hour dark photoperiod. Positive transformants were selected by screening with glufosinate ammonium. The primer sequences are provided in Supplementary Data 23.
Gene expression analysis
Total RNA was extracted from developing siliques at 10 days using TRIZOL reagent (Vazyme, R401-01). For reverse transcription, the EasyScript® One-Step gDNA Removal and cDNA Synthesis SuperMix (TransGen Biotech, AE311-02) was used to synthesize first-strand cDNA, which subsequently served as the template for PCR amplification. For RT-PCR analysis, the PCR products were separated on a 1% agarose gel stained with ethidium bromide and visualized under UV light. The quantitative real-time PCR (qRT-PCR) reactions were performed using the 2× ChamQ Universal SYBR qPCR Master Mix (Vazyme, Q711). Fluorescence signals were detected with a LightCycler® 480 II instrument, and the data were analyzed using the LightCycler® 480 Software (Version 1.5.1). The transcript levels of the target genes were examined using gene-specific primers. Ubiquitin 5 (UBQ5, At3g62250) was used as an internal control. The primer sequences are provided in Supplementary Data 23.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The P. lindheimeri, P. pallida, and P. fendleri genome sequence data reported in this paper have been deposited in the Genome Warehouse in the National Genomics Data Center118, Beijing Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation, under accession GWHESQS00000000 [https://ngdc.cncb.ac.cn/gwh/Assembly/84821/show], GWHESQT00000000 [https://ngdc.cncb.ac.cn/gwh/Assembly/84822/show], and GWHESQU00000000 [https://ngdc.cncb.ac.cn/gwh/Assembly/84852/show], respectively. The raw sequencing data and transcriptome data generated in this study have been deposited in the National Genomics Data Center (NGDC) under accession PRJCA026095. All genome assemblies and annotations are available at Zenodo [https://doi.org/10.5281/zenodo.17453772]. Source data are provided with this paper.
Code availability
Code used for the analysis is available at Zenodo [https://doi.org/10.5281/zenodo.17453772].
References
Cahoon, E. B. & Li-Beisson, Y. Plant unusual fatty acids: learning from the less common. Curr. Opin. Plant Biol. 55, 66–73 (2020).
Dauk, M., Lam, P., Kunst, L. & Smith, M. A. A FAD2 homologue from Lesquerella lindheimeri has predominantly fatty acid hydroxylase activity. Plant Sci. 173, 43–49 (2007).
Severino, L. S. et al. A Review on the Challenges for Increased Production of Castor. Agron. J. 104, 853–880 (2012).
Singh, S., Sharma, S., Sarma, S. J. & Brar, S. K. A comprehensive review of castor oil-derived renewable and sustainable industrial products. Environ. Prog. Sustain. 42, e14008 (2022).
Chan, A. P. et al. Draft genome sequence of the oilseed species Ricinus communis. Nat. Biotechnol. 28, 951–956 (2010).
Dierig, D. A. et al. Lesquerella: New crop development and commercialization in the U. S. Ind. Crops Products. 34, 1381–1385 (2011).
Barclay, A. S., Gentry, H. S. & Jones, Q. The search for new industrial crops II:Lesquerella (Cruciferae) as a source of new oilseeds. Econ. Bot. 16, 95–100 (1962).
Badami, R. C. & Patil, K. B. Structure and occurrence of unusual fatty acids in minor seed oils. Prog. Lipid Res. 19, 119–153 (1980).
Carlson, K. D., Chaudhry, A. & Bagby, M. O. Analysis of oil and meal from Lesquerella fendleri seed. J. Am. Oil Chem. Soc. 67, 438–442 (1990).
Ogunniyi, D. S. Castor oil: a vital industrial raw material. Bioresour. Technol. 97, 1086–1091 (2006).
Wu, Y. V. & Hojilla-Evangelista, M. P. Lesquerella fendleri protein fractionation and characterization. J. Am. Oil Chem. Soc. 82, 53–56 (2005).
Al-Shehbaz, I. A. & O’Kane, S. L. Lesquerella Is United with Physaria (Brassicaceae). Novon 12, 319–329 (2002).
Jenderek, M. M., Dierig, D. A. & Isbell, T. A. Fatty-acid profile of Lesquerella germplasm in the National Plant Germplasm System collection. Ind. Crops Products. 29, 154–164 (2009).
Hayes, D. G., Kleiman, R. & Phillips, B. S. The triglyceride composition, structure, and presence of estolides in the oils of Lesquerella and related species. J. Am. Oil Chemists’ Soc. 72, 559–569 (1995).
Dierig, D. A., Thompson, A. E., Rebman, J. P., Kleiman, R. & Phillips, B. S. Collection and evaluation of new Lesquerella and Physaria germplasm. Ind. Crops Products. 5, 53–63 (1996).
Isbell, T. A., Mund, M. S., Evangelista, R. L. & Dierig, D. A. Method for analysis of fatty acid distribution and oil content on a single Lesquerella fendleri seed. Ind. Crops Products. 28, 231–236 (2008).
Azeez, A., Parchuri, P. & Bates, P. D. Suppression of Physaria fendleri SDP1 Increased Seed Oil and Hydroxy Fatty Acid Content While Maintaining Oil Biosynthesis Through Triacylglycerol Remodeling. Front Plant Sci. 13, 931310 (2022).
Moreau, R. A. & Stumpf, P. K. Recent studies of the enzymic synthesis of ricinoleic Acid by developing castor beans. Plant Physiol. 67, 672–676 (1981).
Bafor, M., Smith, M. A., Jonsson, L., Stobart, K. & Stymne, S. Ricinoleic acid biosynthesis and triacylglycerol assembly in microsomal preparations from developing castor-bean (Ricinus communis) endosperm. Biochem J. 280, 507–514 (1991).
van de Loo, F. J., Broun, P., Turner, S. & Somerville, C. An oleate 12-hydroxylase from Ricinus communis L. is a fatty acyl desaturase homolog. Proc. Natl Acad. Sci. Usa. 92, 6743–6747 (1995).
Moon, H., Smith, M. A. & Kunst, L. A condensing enzyme from the seeds of Lesquerella fendleri that specifically elongates hydroxy fatty acids. Plant Physiol. 127, 1635–1643 (2001).
Bates, P. D. & Browse, J. The significance of different diacylgycerol synthesis pathways on plant oil composition and bioengineering. Front Plant Sci. 3, 147 (2012).
Chapman, K. D. & Ohlrogge, J. B. Compartmentation of triacylglycerol accumulation in plants. J. Biol. Chem. 287, 2288–2294 (2012).
Vanhercke, T., Wood, C. C., Stymne, S., Singh, S. P. & Green, A. G. Metabolic engineering of plant oils and waxes for use as industrial feedstocks. Plant Biotechnol. J. 11, 197–210 (2013).
Parchuri, P. et al. Identification of triacylglycerol remodeling mechanism to synthesize unusual fatty acid containing oils. Nat. Commun. 15, 3547 (2024).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126 (2018).
Murat, F., Armero, A., Pont, C., Klopp, C. & Salse, J. Reconstructing the genome of the most recent common ancestor of flowering plants. Nat. Genet. 49, 490–496 (2017).
Liu, Y. et al. Insights into amphicarpy from the compact genome of the legume Amphicarpaea edgeworthii. Plant Biotechnol. J. 19, 952–965 (2021).
Song, J.-M. et al. Two gap-free reference genomes and a global view of the centromere architecture in rice. Mol. Plant. 14, 1757–1767 (2021).
Cheng, Z. et al. Functional rice centromeres are marked by a satellite repeat and a centromere-specific retrotransposon. Plant Cell. 14, 1691–1704 (2002).
Wolfgruber, T. K. et al. Maize centromere structure and evolution: sequence analysis of centromeres 2 and 5 reveals dynamic Loci shaped primarily by retrotransposons. PLoS Genet. 5, e1000743 (2009).
Naish, M. et al. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science 374, eabi7489 (2021).
Miller, J. T., Dong, F., Jackson, S. A., Song, J. & Jiang, J. Retrotransposon-related DNA sequences in the centromeres of grass chromosomes. Genetics 150, 1615–1623 (1998).
Chen, J. et al. A complete telomere-to-telomere assembly of the maize genome. Nat. Genet. 55, 1221–1231 (2023).
Yang, Y. et al. A near-complete assembly of asparagus bean provides insights into anthocyanin accumulation in pods. Plant Biotechnol. J. 21, 2473–2489 (2023).
Li, K. et al. Gapless indica rice genome reveals synergistic contributions of active transposable elements and segmental duplications to rice genome evolution. Mol. Plant. 14, 1745–1756 (2021).
Yu, J. et al. PTGBase: an integrated database to study tandem duplicated genes in plants. Database (Oxf.). 2015, bav017 (2015).
Kourelis, J. & van der Hoorn, R. A. L. Defended to the Nines: 25 Years of Resistance Gene Cloning Identifies Nine Mechanisms for R Protein Function. Plant Cell. 30, 285–299 (2018).
Broun, P., Boddupalli, S. & Somerville, C. A bifunctional oleate 12-hydroxylase: desaturase from Lesquerella fendleri. Plant J. 13, 201–210 (1998).
Chen, G. Q. et al. Transcriptome Analysis and Identification of Lipid Genes in Physaria lindheimeri, a Genetic Resource for Hydroxy Fatty Acids in Seed Oil. Int J. Mol. Sci. 22, 514 (2021).
Okazaki, K., Sato, N., Tsuji, N., Tsuzuki, M. & Nishida, I. The significance of C16 fatty acids in the sn-2 positions of glycerolipids in the photosynthetic growth of Synechocystis sp. PCC6803. Plant Physiol. 141, 546–556 (2006).
Maisonneuve, S., Bessoule, J.-J., Lessire, R., Delseny, M. & Roscoe, T. J. Expression of rapeseed microsomal lysophosphatidic acid acyltransferase isozymes enhances seed oil content in Arabidopsis. Plant Physiol. 152, 670–684 (2010).
Chen, G. Q. et al. Expression of Castor LPAT2 Enhances Ricinoleic Acid Content at the sn-2 Position of Triacylglycerols in Lesquerella Seed. Int J. Mol. Sci. 17, 507 (2016).
Burgal, J. et al. Metabolic engineering of hydroxy fatty acid production in plants: RcDGAT2 drives dramatic increases in ricinoleate levels in seed oil. Plant Biotechnol. J. 6, 819–831 (2008).
Giraudat, J. et al. Isolation of the Arabidopsis ABI3 gene by positional cloning. Plant Cell. 4, 1251–1261 (1992).
Parcy, F., Valon, C., Kohara, A., Miséra, S. & Giraudat, J. The ABSCISIC ACID-INSENSITIVE3, FUSCA3, and LEAFY COTYLEDON1 loci act in concert to control multiple aspects of Arabidopsis seed development. Plant Cell. 9, 1265–1277 (1997).
Lotan, T. et al. Arabidopsis LEAFY COTYLEDON1 is sufficient to induce embryo development in vegetative cells. Cell 93, 1195–1205 (1998).
Stone, S. L. et al. LEAFY COTYLEDON2 encodes a B3 domain transcription factor that induces embryo development. Proc. Natl Acad. Sci. USA. 98, 11806–11811 (2001).
Yamamoto, A. et al. Diverse roles and mechanisms of gene regulation by the Arabidopsis seed maturation master regulator FUS3 revealed by microarray analysis. Plant Cell Physiol. 51, 2031–2046 (2010).
Tian, R. et al. Direct and indirect targets of the arabidopsis seed transcription factor ABSCISIC ACID INSENSITIVE3. Plant J. 103, 1679–1694 (2020).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Sperling, P., Ternes, P., Zank, T. K. & Heinz, E. The evolution of desaturases. Prostaglandins Leukot. Ess. Fat. Acids 68, 73–95 (2003).
Wang, W. et al. The Spirodela polyrhiza genome reveals insights into its neotenous reduction fast growth and aquatic lifestyle. Nat. Commun. 5, 3311 (2014).
Yin, J. et al. A high-quality genome of taro (Colocasia esculenta (L.) Schott), one of the world’s oldest crops. Mol. Ecol. Resour. 21, 68–77 (2021).
Gao, Y. et al. A chromosome-level genome assembly of Amorphophallus konjac provides insights into konjac glucomannan biosynthesis. Comput Struct. Biotechnol. J. 20, 1002–1011 (2022).
Shen, J., Wu, G., Tsai, A.-L. & Zhou, M. Structure and Mechanism of a Unique Diiron Center in Mammalian Stearoyl-CoA Desaturase. J. Mol. Biol. 432, 5152–5161 (2020).
Broun, P. & Somerville, C. Accumulation of ricinoleic, lesquerolic, and densipolic acids in seeds of transgenic Arabidopsis plants that express a fatty acyl hydroxylase cDNA from castor bean. Plant Physiol. 113, 933–942 (1997).
Lu, C., Fulda, M., Wallis, J. G. & Browse, J. A high-throughput screen for genes from castor that boost hydroxy fatty acid accumulation in seed oils of transgenic Arabidopsis. Plant J. 45, 847–856 (2006).
Kim, H. U. et al. Endoplasmic reticulum-located PDAT1-2 from castor bean enhances hydroxy fatty acid accumulation in transgenic plants. Plant Cell Physiol. 52, 983–993 (2011).
van Erp, H., Bates, P. D., Burgal, J., Shockey, J. & Browse, J. Castor phospholipid:diacylglycerol acyltransferase facilitates efficient metabolism of hydroxy fatty acids in transgenic Arabidopsis. Plant Physiol. 155, 683–693 (2011).
Hu, Z., Ren, Z. & Lu, C. The phosphatidylcholine diacylglycerol cholinephosphotransferase is required for efficient hydroxy fatty acid accumulation in transgenic Arabidopsis. Plant Physiol. 158, 1944–1954 (2012).
Aryal, N. & Lu, C. A Phospholipase C-Like Protein From Ricinus communis Increases Hydroxy Fatty Acids Accumulation in Transgenic Seeds of Camelina sativa. Front Plant Sci. 9, 1576 (2018).
Lunn, D., Wallis, J. G. & Browse, J. Tri-Hydroxy-Triacylglycerol Is Efficiently Produced by Position-Specific Castor Acyltransferases. Plant Physiol. 179, 1050–1063 (2019).
Shockey, J. et al. Specialized lysophosphatidic acid acyltransferases contribute to unusual fatty acid accumulation in exotic Euphorbiaceae seed oils. Planta 249, 1285–1299 (2019).
Kim, H. U., Park, M.-E., Lee, K.-R., Suh, M. C. & Chen, G. Q. Variant castor lysophosphatidic acid acyltransferases acylate ricinoleic acid in seed oil. Ind. Crops Products. 150, 112245 (2020).
Horn, P. J. et al. Identification of multiple lipid genes with modifications in expression and sequence associated with the evolution of hydroxy fatty acid accumulation in Physaria fendleri. Plant J. 86, 322–348 (2016).
Snapp, A. R., Kang, J., Qi, X. & Lu, C. A fatty acid condensing enzyme from Physaria fendleri increases hydroxy fatty acid accumulation in transgenic oilseeds of Camelina sativa. Planta 240, 599–610 (2014).
Adhikari, N. D., Bates, P. D. & Browse, J. WRINKLED1 Rescues Feedback Inhibition of Fatty Acid Synthesis in Hydroxylase-Expressing Seeds. Plant Physiol. 171, 179–191 (2016).
Bird, K. A. et al. Allopolyploidy expanded gene content but not pangenomic variation in the hexaploid oilseed Camelina sativa. Genetics 229, 1–44 (2025).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinforma. 19, 460 (2018).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101 (2016).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158 (2005).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–358 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, ii215–ii225 (2003).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinforma. 7, 62 (2006).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinforma. 12, 491 (2011).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Zdobnov, E. M. & Apweiler, R. InterProScan-an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096 (2021).
Goodstein, D. M. et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186 (2012).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 (2011).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Kumar, S. et al. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol. Biol. Evol. 39, msac174 (2022).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Sun, P. et al. WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol. Plant. 15, 1841–1851 (2022).
Krumsiek, J., Arnold, R. & Rattei, T. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23, 1026–1028 (2007).
Išerić, H., Alkan, C., Hach, F. & Numanagić, I. Fast characterization of segmental duplication structure in multiple genome assemblies. Algorithms Mol. Biol. 17, 4 (2022).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Belser, C. et al. Telomere-to-telomere gapless chromosomes of banana using nanopore sequencing. Commun. Biol. 4, 1047 (2021).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Lu, P. et al. Three-dimensional structure of human γ-secretase. Nature 512, 166–170 (2014).
Lu, S. et al. Phospholipase Dε enhances Braasca napus growth and seed production in response to nitrogen availability. Plant Biotechnol. J. 14, 926–937 (2016).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput Chem. 31, 455–461 (2010).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput Chem. 30, 2785–2791 (2009).
CNCB-NGDC Members and Partners Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2024. Nucleic Acids Res. 52, D18–D32 (2024).
Acknowledgements
This work is supported by the National Science Fund for Distinguished Young Scholars (32225037, L.G.), Fundamental Research Funds for the Central Universities (2662023PY004, L.G.), and 111 Center (B20051, L.G.). The computations in this study were run on the bioinformatics computing platform of the National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University.
Author information
Authors and Affiliations
Contributions
L.G., L.L.C., C.L., S.L., and E.B.C. designed this study. R.F., M.Z., and S.L. performed the experiments. S.W., R.F., Z.W.Z., and J.P.W. analyzed the data and wrote the manuscript. L.G., L.L.C., C.L., W.X., G.C., and S.L. revised the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, S., Fan, R., Zhang, M. et al. Chromosome-level assembly and analysis of three hydroxy fatty acid-producing Physaria species. Nat Commun 16, 11421 (2025). https://doi.org/10.1038/s41467-025-66337-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66337-z
