
Abstract
Cells in highly crowded environments are exposed to fluctuating mechanical forces. While cells can activate the cortical migration machinery to escape from undesirable compressive stress, the consequence to less motile cells and of prolonged extensive confinement is yet to be uncovered. Here, we demonstrate that nuclear deformation generated by axial confinement triggers a specific form of regulated cell death – ferroptosis. We show that axial confinement is sensed by the nucleus and results in Drp1-dependent mitochondrial fragmentation and mitochondrial ROS accumulation. Meanwhile, we detect cPLA2 translocation to mitochondria. These mitochondrial ROS accumulation and arachidonic acid production concertedly lead to lipid peroxidation and evoke ferroptosis. Interestingly, we find in osteoarthritis, a disease intimately associated with mechanical overloading and inflammation, characteristics of confinement-induced ferroptosis including mitochondrial localization of cPLA2 and high ROS. Together, our findings unveil a pivotal role of cell nucleus and mitochondria in linking mechanical confinement with cell death, highlighting the orchestration of Drp1 and cPLA2 in confinement-induced ferroptosis.
Similar content being viewed by others


Introduction
In multicellular organisms, cells are exposed to mechanical forces including shear stress and compression1,2. In the context of confined migration, the squeezing of cells through narrow constrictions results in high level of nuclear deformation triggering mesenchymal-to-amoeboid migration mode switching3,4. However, not all cells are motile and how adherent and less motile cells respond, adapt to, and survive persistent mechanical confinement at the molecular level is less clear.
Unlike chemical ligands that rely on cell surface receptors to transduce signals into the cell, mechanical force can propagate throughout cells via deformation of intracellular structures5,6. Within cells, the cytoskeleton and organelles are mechano-responsive, acting in combination with molecular-scale alterations in force-sensitive proteins to modify cellular responses to mechanical force7,8. Accumulative evidence in recent years has suggested a role of the nucleus as a mechano-responsive organelle - external force leads to nuclear translocation of certain transcription factors9,10, changes chromatin architecture to prevent DNA damage11. Recently, nuclear deformation is shown to activate contractility of the actomyosin cortex, facilitating the modulation of cell migration in response to force12,13. Further, studies have found that cells undergo nuclear rupture and DNA damage when migrating through narrow pores that herniate the nucleus, which is linked to carcinogenic and inflammogenic status14,15,16. Interestingly, ferroptosis has recently been observed in osteoarthritis (OA), a disease intimately associated with mechanical overloading and inflammation17,18.
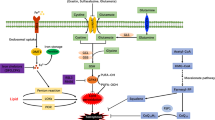
Here we find that nuclear deformation under confinement triggers ferroptosis. Using in-vitro devices to implement axial confinement, we observe enhanced Drp1 liquid-like droplets formation followed by mitochondrial fragmentation and accumulation of mitochondrial ROS (mtROS). Nuclear confinement also triggers cPLA2 translocation to mitochondria, followed by arachidonic acid (ARA) generation. These concertedly lead to lipid peroxidation and ferroptosis. Finally, we have demonstrated that OA patient samples show characteristics of this mechano-stress related cell death response.
Results
Mechanical confinement triggers ferroptosis
First, to understand how axial confinement affects cell morphology and survival, we performed axial confinement of HeLa cells to 3 µm height using microfabricated PolyDiMethylSiloxane (PDMS) micropillars bonded to glass coverslips19. At this height, the nucleus is sufficiently deformed3,12. When cells were subjected to continuous axial confinement for a duration of 9 h, cells began to rupture and die during the subsequent 3 h, with continuous increases in cell death, as indicated by nuclear propidium iodide (PI) influx (Fig. 1a, b and Supplementary Movie 1). In the absence of confinement height control (beneath the PDMS micropillars without spacing protection from the micropillars), cells instantly ruptured, exhibiting acute influx of nuclear PI signal (Supplementary Fig. 1a). In addition, we used a second method20 with 3 μm microsphere spacers and a machined stainless steel weight to confine cells (Supplementary Fig. 1b), and similar observations were made with this new device equipped with spacers (more than 80% cells were PI positive) (Supplementary Fig. 1c, d). These suggest that cells exposed to controlled ~3 µm axial confinement undergo a slower, likely regulated, process of cell death. In addition to HeLa cells, we also found that prolonged confinement induced cell death of human fibrosarcoma cells HT1080 (Supplementary Fig. 1e), suggesting that regulated cell death may be a common cellular response to mechanical confinement.

a Representative time-lapse images of HeLa cells during 9 h confinement mediated by PDMS micropillars in the presence of propidium iodide (PI). Nuclei labeled with Hoechst (5 μg/mL for 20 min). White arrow: large plasma membrane blebs. Scale bar = 20 μm. b Quantification of the percentage of cell death in HeLa cells upon confinement, related to (a) (n = 8 independent fields, N = 3 biologically independent experiments, mean ± SD, ***p = 0.0002, ****p < 0.0001, unpaired one-way ANOVA with Tukey’s test). The cell death is measured by the PI positive nuclei/total nuclei * 100. c Representative time-lapse images of DHE staining in HeLa cells during 6 h confinement mediated by microspheres. Scale bar = 40 μm. d Quantification of total ROS level in HeLa cells upon confinement, related to (c) (n = 50 cells, N = 3 biologically independent experiments, ****p < 0.0001, unpaired one-way ANOVA with Tukey’s test). Violin plot center lines denote the median, and dashed lines represent the 25th and 75th percentiles. e Representative time-lapse images showing BODIPY-C11 581/591 staining in HeLa cells during 6 h confinement mediated by microspheres. Non-oxidative state (C11non-ox), oxidative state (C11ox). Scale bar = 40 μm. f Quantification of lipid ROS level in HeLa cells upon confinement, related to (e) (n = 50 cells, N = 3 biologically independent experiments, ****p < 0.0001, unpaired one-way ANOVA with Tukey’s test). Data was calculated by fluorescence intensity ratio of C11ox/C11non-ox. g qRT-PCR assay showing the COX2 mRNA levels in HeLa cells in the absence (Control) and presence (Confined) of 4 h confinement mediated by microspheres (n = 3 technical repeats, N = 3 biologically independent experiments, mean ± SD, ***p = 0.0004, two-sided unpaired Student’s t test). h Representative time-lapse images of HeLa cells treated with DMSO or ferroptosis inhibitors during 12 h confinement mediated by PDMS micropillars in the presence of PI, including DFOM (100 μM), Ferrostatin-1 (Fer-1, 1 μM), Liproxstatin-1 (Lip-1, 0.5 μM). Scale bar = 20 μm. i The percentage of cell death in HeLa cells treated with DMSO or ferroptosis inhibitors upon confinement, related to (h) (n = 8 independent fields, N = 3 biologically independent experiments, mean ± SD, ns represents not significant, *p = 0.0264, ****p < 0.0001, ####p < 0.0001, unpaired two-way ANOVA with Sidak’s test).
To test whether this cell death may be caused by hypoxia or nutrient starvation during confinement, we firstly performed confinement using 20 μm microspheres as control spacers where cells were not in direct contact with the confinement surface19. Under 20 μm axial confinement height, no observable cell death was detected (Supplementary Fig. 1f). However, we did not observe a significant difference in the fluorescence intensity of the hypoxic probe between 3 μm and 20 μm confinement in either HeLa cells or HT1080 cells, indicating that the degree of hypoxia in these two conditions is comparable (Supplementary Fig. 1g, h). Moreover, we failed to detect HIF1α protein under 3 μm or 20 μm confinement in both HeLa cells and HT1080 cells (Supplementary Fig. 1i). To assess the potential contribution of nutrient starvation to confinement-induced cell death, we exposed HeLa cells to glucose deprivation, serum deprivation, or combined nutrient deprivation. None of these conditions recapitulated the elevated cell death observed under mechanical confinement (Supplementary Fig. 1j). To further validate these findings, we employed a 10 μm height condition as a control and extended the treatment duration to three times compared to that of the 3 μm confinement. Notably, after 27 h under 10 μm confinement, the level of cell death remained significantly lower than that observed after only 9 h under 3 μm confinement (Supplementary Fig. 1k). Collectively, these results indicate that the cell death we observe specifically from prolonged confinement rather than hypoxia or nutrient starvation.
We observed large cell membrane distention during axial confinement, which is in line with the characteristic membrane blebbing during pyroptosis21,22. However, when we screened for activation of the classical effectors of pyroptosis, we failed to detect the cleavage of the pore-forming effectors, GSDMD or GSDME (Supplementary Fig. 2a). Moreover, the pan-caspase inhibitor Z-VAD-FMK that stalls pyroptosis23,24, failed to block this process (Supplementary Fig. 2b). In addition, we detected no changes in MLKL phosphorylation25, eliminating the possibility of confinement induced necroptosis (Supplementary Fig. 2c). Consistently, NSA-dependent inhibition of necroptosis failed to block confinement-induced cell death (Supplementary Fig. 2d). Interestingly, alongside PI influx of confined cells, we noticed time-dependent increase in total and lipid ROS as revealed by the fluorescent probes DHE and BODIPY-C11 581/591, respectively. Total and lipid ROS accumulated at 2 h under 3 μm but not 20 μm confinement (Fig. 1c–f, Supplementary Fig. 2e). As lipid peroxidation is a hallmark of ferroptosis26,27, we examined additional markers of ferroptosis activation. RNA expression of the ferroptosis biomarker, cyclooxygenase-2 (COX-2) increased after confinement (Fig. 1g). Remarkably, confinement induced-cell death was significantly diminished when cells were confined in the presence of ferroptosis inhibitors, including DFOM, Ferrostatin-1 (Fer-1), Liproxstatin-1 (Lip-1), and Trolox (Fig. 1h, i, Supplementary Fig. 2f and Supplementary Movie 2). Together, our data demonstrate that cells subjected to prolonged confinement exhibit ferroptosis.
cPLA2 is activated and translocates to mitochondria upon confinement
Next, to understand how confinement triggers ferroptosis, we asked whether mechano-sensitive ion channels28 and cell-adhesion molecules8 may play a role. When we inhibited Piezo channels with GsMTx4, we found no changes in cell death. Similar observation was made using an siRNA library to knock down a panel of integrins (Supplementary Fig. 3a, b).
Interestingly, we noticed that nuclear deformation occurred with 3 μm confinement, observing a 35% increase in the nuclear projected surface area (Fig. 2a, b). The nucleus has recently been reported as a cellular mechano-ruler12,13,29. Its deformation has been shown to induce translocation of cytosolic phospholipase A2 (cPLA2, also called cPLA2α or PLA2G4A) to the nuclear envelope to catalyze arachidonic acid (ARA) production12,13,30, which contributes to lipid hydroperoxides accumulation for ferroptosis31. We then examined the role of nucleus during this process. However, when we performed live-cell imaging of cPLA2-eGFP dynamics in cells subjected to confinement, we did not observe its nuclear translocation (Supplementary Fig. 3c), but instead, we detected cPLA2 translocation to mitochondria in HeLa and HT1080 cells upon 3 μm but not 20 μm confinement (Fig. 2c, d, Supplementary Fig. 3d–f and Supplementary Movie 3). cPLA2 localizing to and activated at mitochondria have been documented in the literature in astrocytes following sphingolipid lactosylceramide treatment32. Thus, we asked whether cPLA2 may play a catalytic role to produce ARA once translocated to mitochondria. First, intracellular ARA level gradually increased after confinement for 3 h (Fig. 2e), suggestive of cPLA2 activation under confinement. Next, when we knocked down cPLA2 using shRNA (Supplementary Fig. 3g), we observed a decrease in lipid ROS level and abrogation of cell death in confined cells (Fig. 2f–h). Consistent with these findings, cPLA2 knockdown significantly reduced ARA level under confinement (Fig. 2i). Similarly, these results were recapitulated with the cPLA2 inhibitor CAY10650 (Fig. 2j, k), supporting a catalytic role of cPLA2 in confinement-induced ferroptosis.

a xy views of the Hoechst-stained nucleus before and after 20 μm (Up) or 3 μm (Down) confinement mediated by microspheres. Scale bar = 10 μm. b Quantification of projected nucleus surface area upon confinement, related to (a) (n(20 μm) = 14 nuclei, n(3 μm) = 10 nuclei, ns represents not significant, ****p < 0.0001, paired two-way ANOVA with Sidak’s test). c Representative time-lapse images of cPLA2-eGFP expressing HeLa cells co-stained with Mito Red (200 nM, 30 min) during 2 h confinement mediated by microspheres. Scale bar = 10 μm. d The level of colocalization between cPLA2 puncta and mitochondrial networks in HeLa cells under confinement for 0 h or 3 h mediated by microspheres (n = 20 cells, ****p < 0.0001, two-sided unpaired Student’s t test). e ELISA assay of arachidonic acid (ARA) levels in HeLa cells upon confinement mediated by microspheres (n = 6 samples from 3 biologically independent experiments, mean ± SD, ns represents not significant, *p = 0.0345, ***p = 0.0002, unpaired one-way ANOVA with Tukey’s test). f Representative images of BODIPY-C11 581/591 staining in control (shCtrl) and cPLA2α knockdown (shcPLA2α) HeLa cells in the absence (Control) and presence (Confined) of 6 h confinement mediated by microspheres. Scale bar = 40 μm. g Quantification of lipid ROS level in shCtrl and shcPLA2α HeLa cells, related to (f) (n = 50 cells, N = 3 biologically independent experiments, ns represents not significant, ****p < 0.0001, unpaired two-way ANOVA with Sidak’s test). h The percentage of cell death in shCtrl and shcPLA2α HeLa cells upon 9 h confinement mediated by microspheres (n = 8 independent fields, N = 3 biologically independent experiments, mean ± SD, ****p < 0.0001, unpaired one-way ANOVA with Tukey’s test). i ARA levels in shCtrl and shcPLA2α HeLa cells in the absence (Control) and presence (Confined) of 5 h confinement mediated by microspheres (n = 6 samples from 3 biologically independent experiments, mean ± SD, ns represents not significant, ****p < 0.0001, unpaired one-way ANOVA with Tukey’s test). j Lipid ROS levels in HeLa cells treated with DMSO or CAY10650 (150 nM, cPLA2α inhibitor) upon 6 h confinement mediated by microspheres (n(Control) = 60 cells, n(Confined) = 66 cells, N = 3 biologically independent experiments, ns represents not significant, ****p < 0.0001, unpaired two-way ANOVA with Sidak’s test). k The percentage of cell death in HeLa cells treated with DMSO or CAY10650 upon 6 h confinement mediated by microspheres (n = 8 independent fields, N = 3 biologically independent experiments, mean ± SD, ****p < 0.0001, two-sided unpaired Student’s t test). l Representative images of cPLA2-eGFP and mitochondria (Mito Red) spatio-temporal localization in intact HeLa cells or cytoplasts upon confinement mediated by microspheres. Scale bar = 10 μm.
To explore whether nucleus can sense and trigger cPLA2 translocation from the cytoplasm to the mitochondria, we used centrifugation-based enucleation of cells to produce cytoplasts33,34 (Supplementary Fig. 3h). In cytoplasts exposed to axial confinement, cPLA2 translocation to the mitochondria was abolished (Fig. 2l and Supplementary Fig. 3i).
It has been proposed that changes of both Ca2+ and membrane tension contribute to cPLA2 translocation onto the nuclear membrane. We thus wondered whether similar players may work in confinement-induced mitochondrial membrane location of cPLA2. Firstly, imaging of intracellular calcium with the rGECO calcium biosensor revealed a strong increase in cytosolic calcium of HeLa and HT1080 cells under confinement (Supplementary Fig. 4a). Secondly, since the binding of cPLA2 to membrane phospholipids has been reported to rely on the C2 domain (a Ca2+ sensitive domain) and is in a Ca2+-dependent manner, we generated different cPLA2 truncations to explore the mechanism for cPLA2 translocation to mitochondria (Supplementary Fig. 4b). We found that truncations of the C2 domain (ΔC2) or the entire phospholipid binding region (ΔPL) lost the ability to translocate to mitochondria (Supplementary Fig. 4c). Lastly, cPLA2 translocation was inhibited through chelation of intracellular calcium with BAPTA-AM (Supplementary Fig. 4d). Collectively, these results demonstrate that nuclear deformation triggers cPLA2 translocation to mitochondria and this process depends on Ca2+.
Confinement elevates mitochondrial fission and mtROS
While observing cPLA2 mitochondrial translocation, we noted that the majority of cells (87%) exhibited mitochondrial fragmentation when exposed to 3 μm confinement, with some toroidal (ring-shaped) mitochondria in a subset of cells (Supplementary Fig. 5a, b). We thus performed live-cell time lapse imaging of mitochondria labeled cells (Tom20-mCherry) under axial confinement, and indeed observed a progressive conversion of tubular mitochondrial networks into fragmented, spherical structures (Fig. 3a and Supplementary Movie 4), indicative of fission events or breakdown of mitochondrial integrity. Quantitative analysis revealed a significant increase in mitochondrial number per cell, concurrent with expansions in mitochondrial perimeter and circularity (Fig. 3b). These morphological changes indicate both mitochondrial fragmentation and swelling under confinement. Furthermore, we also detected mitochondrial membrane depolarization (Fig. 3c, d), as indicated by tetramethylrhodamine methyl (TMRM). Importantly, we did not observe Parkin translocation to mitochondria (Supplementary Fig. 5c), indicating that mitophagy may not take place.

a Representative time-lapse images of mitochondria in HeLa cells transfected with Tom20-mCherry during 90 min confinement mediated by PDMS micropillars. Scale bar = 5 μm. b Quantification of mitochondrial morphology upon confinement, related to (a) (n = 12 cells, mean ± SD). Magnified images are areas outlined (i), black dashed boxes of (A). Scale bar = 1 μm. c Representative time-lapse images of TMRM staining (100 nM, 20 min) in HeLa cells during 60 min confinement mediated by microspheres, fluorescence intensity presented as a heatmap (low-high). Scare bar = 10 μm. d Quantification of TMRM fluorescence intensity per cell upon confinement, normalized by data at 0 min, related to (c) (n = 6 cells, N = 3 biologically independent experiments, mean ± SD, *p = 0.0488, **p(0 min vs 15 min) = 0.0073, **p(30 min vs 60 min) = 0.0029, paired one-way ANOVA with Tukey’s test). e Representative time-lapse ratiometric images showing mtROS level in HeLa cells stably expressing Mito-roGFP during 90 min confinement mediated by microspheres. Scare bar = 10 μm. f Quantification of the relative mtROS level per cell in HeLa cells upon confinement, normalized by data at 0 min, related to (e) (n = 44 cells, N = 3 biologically independent experiments, ns represents not significant, **p = 0.0015, ****p < 0.0001, unpaired One-way ANOVA with Tukey’s test). g Total ROS level in HeLa cells treated with DMSO or MitoTEMPO (10 μm) in the absence (Control) and presence (Confined) of 4 h confinement mediated by microspheres (n = 50 cells, N = 3 biologically independent experiments, ns represents not significant, ****p < 0.0001, unpaired two-way ANOVA with Sidak’s test). h Lipid ROS level in HeLa cells treated with DMSO or MitoTEMPO in the absence (Control) and presence (Confined) of 5 h confinement mediated by microspheres, normalized by data at Control of DMSO group (n = 50 cells, N = 3 biologically independent experiments, ns represents not significant, ****p < 0.0001, unpaired two-way ANOVA with Sidak’s test). i The percentage of cell death in HeLa cells treated with DMSO or MitoTEMPO upon 9 h confinement mediated by microspheres (n = 8 independent fields, N = 3 biologically independent experiments, mean ± SD, ****p < 0.0001, two-sided unpaired Student’s t test). j Total ROS level in parental and “ρ0” HeLa cells upon confinement mediated by microspheres (n = 50 cells, N = 3 biologically independent experiments, ns represents not significant, ****p < 0.0001, unpaired two-way ANOVA with Sidak’s test). k Representative images of BODIPY-C11 581/591 staining in parental and “ρ0” HeLa cells upon confinement mediated by microspheres. Scale bar = 40 μm. l Quantification of lipid ROS level in parental and “ρ0” HeLa cells, related to (k) (n = 50 cells, N = 3 biologically independent experiments, ns represents not significant, ****p < 0.0001, unpaired two-way ANOVA with Sidak’s test). m The percentage of cell death in parental and “ρ0” HeLa cells upon 9 h confinement mediated by microspheres (n = 8 independent fields, N = 3 biologically independent experiments, mean ± SD, ****p < 0.0001, two-sided unpaired Student’s t test).
As mitochondrial defects are responsible for increased mitochondrial ROS (mtROS)35, which has been implicated in lipid peroxidation and ferroptosis onset36, we next examined whether mtROS level increased during confinement-induced mitochondrial fission. Indeed, Mito-roGFP2 revealed a significant increase of mtROS when cells underwent axial confinement (Fig. 3e, f). When we inhibited mtROS by MitoTEMPO, a mitochondria-targeted superoxide dismutase mimetic (Supplementary Fig. 5d), or by depleting mtDNA through culturing cells in ethidium bromide (EtdBr) (Supplementary Fig. 5e), we found significantly reduced total ROS and lipid ROS levels, as well as decreased cell death of confined cells (Fig. 3g–m). Similar effect was seen with the antioxidant EUK134 or NAC (Supplementary Fig. 5f, g), suggesting that mtROS may play a key role in modulating confinement-induced ferroptosis. In addition, pre-treating cells with the electron transport chain (ETC) complex I inhibitor rotenone plus the complex III inhibitor antimycin A to increase mtROS level (Supplementary Fig. 5h) can enhance lipid ROS elevation and cell death under confinement (Supplementary Fig. 5i–k). Collectively, we demonstrate that mtROS is a potent driver of ferroptosis in cells under confinement.
Our observation of confinement-induced mitochondrial fission recapitulates reports of altered mitochondrial morphology in ferroptotic cells26,37. To better understand the temporal dynamics of mitochondrial fission in response to nuclear compression, we performed atomic force microscopy (AFM) with a flat tipless cantilever to deform single cells above the nucleus, while monitoring mitochondrial morphology by live-cell imaging. Notably, we observed rapid mitochondrial fission when the cantilever deformed the nucleus (Fig. 4a). Fission was absent in mitochondria-containing cytoplasts, in line with our observation of compression-induced fission being a nuclear-dependent process (Fig. 4b and Supplementary Fig. 6a).

a Representative time-lapse images of mitochondria (Mito Green) in a HeLa cell compressed by AFM. The position of the tipless probe is indicated by the yellow dotted box. Insets are magnified images of areas indicated by white boxes. Scale bar = 10 μm. b Representative time-lapse images of mitochondria (Mito Red) in intact HeLa cells or cytoplasts during 90 min confinement mediated by microspheres. Insets are magnified images of areas indicated by white boxes. Scale bar = 10 μm. c Representative live-cell images of mitochondria (Mito Red) in control (shCtrl) and Drp1 knockdown (shDrp1) HeLa cells upon confinement mediated by microspheres. Scale bar = 10 μm. d The percentage of cells with fragmented mitochondria in shCtrl and shDrp1 HeLa cells upon 90 min confinement, related to (c) (n(shCtrl) = 56 cells, n(shDrp1) = 49 cells, N = 3 biologically independent experiments, ****p < 0.0001, two-sided Fisher’s exact test). e Representative time-lapse images of mitochondria (Mito Green) in HeLa cells expressing mCherry-Drp1-K38A during 60 min confinement mediated by microspheres. Non-transfected HeLa cells in the same sample were used as a control. Scale bar = 10 μm. f The percentage of cells with fragmented mitochondria in HeLa cells expressing mcherry-Drp1-K38A or in the control group upon 60 min confinement, related to (e) (n(K38A) = 50 cells, n(Control) = 47 cells, N = 3 biologically independent experiments, ****p < 0.0001, two-sided Fisher’s exact test). g Total ROS level in shCtrl and shDrp1 HeLa cells in the absence (Control) and presence (Confined) of 4 h confinement mediated by microspheres (n = 50 cells, N = 3 biologically independent experiments, ns represents not significant, ****p < 0.0001, unpaired two-way ANOVA with Sidak’s test). h Lipid ROS level in shCtrl and shDrp1 HeLa cells in the absence (Control) and presence (Confined) of 5 h confinement mediated by microspheres (n = 40 cells, N = 3 biologically independent experiments, ns represents not significant, ****p < 0.0001, unpaired two-way ANOVA with Sidak’s test). i The percentage of cell death in shCtrl and shDrp1 HeLa cells upon 9 h confinement mediated by microspheres (n = 8 independent fields, N = 3 biologically independent experiments, mean ± SD, ****p < 0.0001, two-sided unpaired Student’s t test).
Canonically, mitochondrial fission is executed by the dynamin-like GTPase Drp138. We perturbed Drp1 function by (1) shRNA mediated knockdown of Drp1 (Supplementary Fig. 6b, c) and by (2) over-expressing the dominant negative mutant Drp1-K38A (Drp1-K38A OE)39. In both approaches, we observed inhibited mitochondrial fragmentation (Fig. 4c–f) and reduced total and lipid ROS levels as well as decreased cell death (Fig. 4g–i). Taken together, these results indicate that in confined cells, Drp1 triggers mitochondrial fission and elevated mtROS levels, participating in confinement-induced ferroptosis cascade.
Nuclear confinement increases Drp1 liquid-like droplets formation
We next asked how Drp1 achieves spatio-temporal activation in cells under axial confinement. First, we examined whether ER constriction of mitochondria triggered Drp1-dependent mitochondrial fission40. As the rough ER membrane is continuous with the outer nuclear membrane, we speculated that ER-mitochondria contacts may be affected by nuclear deformation during confinement. To address this, we used the contact FP system tagging dimerization-dependent fluorescent proteins (ddFP) to ER and mitochondria to visualize organelle contact sites41. However, we observed that the number of ER-mitochondria contacts remained unchanged during confinement, even after mitochondrial fragmentation (Supplementary Fig. 7a, b).
We next monitored spatio-temporal dynamics of endogenously tagged Drp1 in confined HeLa cells. We used CRISPR-Cas9 knock-in of eGFP at the N-terminus of Drp1 locus (Supplementary Fig. 7c), and found that consistent with the literature39, endogenous Drp1 was mainly diffusive in the cytoplasm and occurred as puncta co-localized with mitochondria (Supplementary Fig. 7d). When cells were subjected to confinement, we observed an increase in both the number of eGFP-Drp1 puncta as well as the association with mitochondria (82.75% at 40 min vs. 74.42% at 0 min) (Fig. 5a, b). The colocalization ratio between Drp1 puncta and mitochondria measured by Mander’s coefficient also increased upon confinement. We demonstrated that this colocalization was not random, since only a small percentage of Drp1 puncta overlapped with the mitochondria after rotating the mitochondria image 90° (Fig. 5c). Specifically, when we focused on mitochondrial fission sites, the majority exhibited eGFP-Drp1 puncta localization (90.79%, Fig. 5d and Supplementary Movie 5). In addition, we found exogenous eGFP-Drp1 in HT1080 cells under confinement also formed droplets, followed by mitochondrial fragmentation (Supplementary Fig. 7e). The Drp1 puncta were reminiscent of phase separated condensates. We then employed an optogenetic approach based on the optoDroplet system42, and observed reversible formation of Opto-Drp1 droplets upon blue light exposure (Fig. 5e). When we purified Drp1 to carry out in-vitro phase separation assays, we found instantaneous droplet formation (Fig. 5f, g and Supplementary Fig. 7f, g). These results demonstrate liquid-like condensation property of Drp1, raising the possibility that the Drp1 puncta observed in confined cells may involve liquid-liquid phase separation. To extensively explore the role of Drp1 liquid-like droplets on confinement-induced mitochondrial fragmentation and ferroptosis, we firstly performed super-resolution Structure Illumination Microscope (SIM) to image mitochondria stained with PK Mito Orange in eGFP-Drp1 knock-in HeLa cells under confinement. We observed events that, when under confinement, two Drp1 puncta underwent fusion followed by mitochondrial fission (Fig. 5h). While the formation of Drp1 puncta within cells was not dependent on its GTPase domain (Supplementary Fig. 7h), a series of disease-related Drp1 point mutants (A395D, R403C, C431Y)43, shown to abrogate Drp1 function, were defected in droplets formation when expressed in Drp1 knock-out cells. The wild-type Drp1 could form condensates co-localizing with mitochondria to promote mitochondrial fragmentation, but these point mutants abrogated droplets formation and prevented mitochondrial fragmentation (Fig. 5i, j and Supplementary Fig. 7i, j), resisting cell death induced by confinement (Fig. 5k).

a Representative time-lapse images of endogenous eGFP-Drp1 knock-in HeLa cells co-stained with Mito Red during 40 min confinement mediated by microspheres. Insets are magnified images of areas indicated by white boxes. Scare bar = 10 μm. b Quantification of the number of Drp1 puncta per region of interest (ROI) upon confinement (n = 30 ROIs from 18 cells, ****p < 0.0001, unpaired One-way ANOVA with Tukey’s test) (Up); Quantification of the proportion of Drp1 puncta localized to mitochondria upon confinement (n = 30 ROIs from 18 cells, **p = 0.0015, ***p = 0.0002, unpaired One-way ANOVA with Tukey’s test) (Down), related to (a). c The level of colocalization between endogenous Drp1 puncta and mitochondrial networks in HeLa cells upon confinement mediated by microspheres (n = 30 ROIs from 24 cells, *p = 0.0171, ****p < 0.0001, unpaired two-way ANOVA with Sidak’s test). d Pie chart showing the proportion of fission sites with Drp1 puncta (n = 152 events from 7 cells). e Representative images of HeLa cells expressing Opto-Drp1 in the absence and presence of blue light (off/on). Insets are magnified images of areas indicated by white boxes. Scale bar = 10 μm. f Representative DIC and fluorescence images of purified Drp1-eGFP (20 μM) diluted in 200 mM NaCl containing 8% dextran. Scale bar = 10 μm. Data are representative of three biologically independent experiments. g Representative time-lapse images demonstrating small Drp1-eGFP droplets fusing to form larger droplets. Scale bar = 2 μm. h Representative live-cell images of endogenous eGFP-Drp1 knock-in HeLa cells co-stained with PK Mito Orange (250 nM, 15 min) upon confinement mediated by microspheres. Insets are magnified images of areas indicated by white boxes (Up); Time-lapse images showing the dynamic interactions between two Drp1 puncta during Drp1-mediated mitochondrial fission events, indicated by the white arrow (Down). The images were captured by Polar-SIM super resolution microscope. Scale bar = 5 μm. i Representative time-lapse images of eGFP-tagged wild-type Drp1 or Drp1 mutants (A395D, R403C, C431Y) expressing Drp1 knock-out HeLa cells co-stained with Mito Red upon confinement mediated by microspheres. Insets are magnified images of areas indicated by white boxes. Scale bar = 10 μm. j The percentage of cells with fragmented mitochondria in eGFP-tagged wild-type Drp1 or Drp1 mutants (A395D, R403C, C431Y) expressing Drp1 knock-out HeLa cells co-stained with Mito Red upon confinement mediated by microspheres. (n(eGFP) = 114 cells, n(WT) = 88 cells, n(A395D) = 116 cells, n(R403C) = 110 cells, n(C431Y) = 84 cells, ns represents not significant, ****p < 0.0001, two-sided Fisher’s exact test). k The percentage of cell death in Control, Drp1 KO, KO+WT (Drp1 KO rescued with wild-type eGFP-Drp1), KO+A395D (Drp1 KO rescued with eGFP-Drp1 A395D mutant) and KO+R403C (Drp1 KO rescued with eGFP-Drp1 R403C mutant) HeLa cells upon 6 h confinement mediated by microspheres (n = 6 independent fields, N = 3 biologically independent experiments, mean ± SD, ns represents not significant, ****p < 0.0001, unpaired One-way ANOVA with Tukey’s test).
In cytoplasts, eGFP-Drp1 failed to form biomolecular condenstates and their mitochondria maintained tubular network (Supplementary Fig. 7k), indicating that nucleus is critical for confinement-induced Drp1 liquid-like droplet formation and mitochondrial fragmentation. Taken together, these data show that axial confinement induces endogenous Drp1 puncta/droplets formation, which is critical for confinement-induced mitochondrial fragmentation.
mtROS and ARA concertedly trigger ferroptosis under nuclear confinement
We observed that mitochondrial fragmentation occurred prior to the mitochondrial translocation of cPLA2 (Fig. 2c). We wondered if, and how, these two events were mechanistically linked. To understand this, we firstly monitored cPLA2 localization in Drp1-depleted cells and found that cPLA2 could still translocate to mitochondria under confinement (Fig. 6a, b). Furthermore, eliminating mtROS through MitoTEMPO cannot hinder the cPLA2 relocation (Fig. 6c). These findings suggest that mitochondrial translocation of cPLA2 is not due to Drp1-mediated mitochondrial fragmentation or ROS accumulation. Secondly, considering that cPLA2 activation can promote ROS production44, we thus explored the role of cPLA2 on mitochondrial fragmentation and ROS production. We found that cPLA2 knockdown had no effect on ROS level at steady state, and did not prevent ROS elevation under confinement (Fig. 6d). Moreover, cPLA2 knockdown did not affect Drp1 liquid-like droplet formation (Fig. 6e) or mitochondrial fragmentation (Fig. 6f, g). Consistent with above results, cPLA2 activity inhibition in confined cells did not prevent mitochondrial fragmentation (Supplementary Fig. 8a, b) nor total ROS increase (Supplementary Fig. 8c, d). Thus, we speculate that Drp1-dependent mitochondrial fragmentation and cPLA2-dependent ARA generation are not inter-dependent in confinement-induced ferroptosis, although they are both required to concertedly drive this process.

a Representative live-cell images of the cPLA2-eGFP expressing shCtrl or shDrp1 HeLa cells co-stained with Mito Red in the absence (Control) and presence (Confined) of confinement mediated by microspheres. Insets are magnified images of areas indicated by white boxes. Scale bar = 10 μm. Data are representative of three biologically independent experiments. b The level of colocalization between cPLA2 puncta and mitochondrial networks in shCtrl and shDrp1 HeLa cells under confinement for 3 h mediated by microspheres (n = 10 cells, ns represents not significant, two-sided unpaired Student’s t test). c Representative time-lapse images of cPLA2-eGFP and mitochondria (Mito Red) in HeLa cells treated with DMSO or MitoTEMPO upon confinement mediated by microspheres. Scale bar = 10 μm. d Total ROS level in shCtrl and shcPLA2α HeLa cells in the absence (Control) and presence (Confined) of 4 h confinement mediated by microspheres (n = 54 cells, N = 3 biologically independent experiments, ns represents not significant, unpaired two-way ANOVA with Sidak’s test). e Representative live-cell images of eGFP-Drp1 in shCtrl and shcPLA2α HeLa cells upon confinement mediated by microspheres. Insets are magnified images of areas indicated by white boxes. Scale bar = 10 μm. f Representative live-cell images of mitochondria (Mito Red) in shCtrl and shcPLA2α HeLa cells upon confinement mediated by microspheres. Insets are magnified images of areas indicated by white boxes. Scale bar = 10 μm. g Quantification of the percentage of cells with fragmented mitochondria in shCtrl and shcPLA2α HeLa cells upon 90 min confinement mediated by microspheres (n(shCtrl) = 117 cells, n(shcPLA2α−1) = 116 cells, n(shcPLA2α−2) = 128 cells, ns represents not significant, two-sided Fisher’s exact test). h Representative bright field and fluorescence images of PI staining in HeLa cells treated with DMSO, H2O2 (400 μM) alone, ARA (400 μM) alone, and H2O2+ARA for 12 h. Scale bar = 40 μm. i Quantification of the percentage of cell death in HeLa cells, related to (h) (n = 6/7 independent fields, N = 3 biologically independent experiments, mean ± SD, ***p = 0.002, ****p < 0.0001, ####p < 0.0001, unpaired one-way ANOVA with Tukey’s test). j The percentage of cell death in HeLa cells treated with DMSO, H2O2 (400 μM)+ARA (400 μM), H2O2+ARA + Lip-1 (10 μM), and H2O2+ARA + Fer-1 (10 μM) for 12 h (n = 6 independent fields, N = 3 biologically independent experiments, mean ± SD, ns represents not significant, ***p = 0.0001, ****p < 0.0001, ####p < 0.0001, unpaired one-way ANOVA with Tukey’s test).
Generation of lipid hydroperoxides requires ARA and cellular ROS31. We thus wondered whether it was possible to recapitulate the confinement-induced ferroptotic cell death by adding H2O2 and ARA into the culture medium to respectively simulate the ROS production and cPLA2 activation under confinement. Indeed, treating cells with ARA or H2O2 individually had negligible impacts on cell lipid ROS level or cell death. Only when cells were exposed to combined treatment, we observed a potent induction of cell death, which were antagonized by treating cells with the ferroptosis inhibitors (Fig. 6h, i and Supplementary Fig. 8e, f). These results suggest that ferroptosis cannot be induced solely by effects of cPLA2 translocation and activation or ROS accumulation.
Confinement-induced ferroptosis may be clinically relevant in osteoarthritis
Osteoarthritis (OA) is a disease associated with excessive mechanical loading of chondrocytes. Inflammation, cell death and ferroptosis are reported to occur in OA17. When we isolated relatively normal chondrocytes from human knee cartilage and examined whether chondrocytes could also undergo ferroptosis under confinement, indeed, we observed similar increase of both total and lipid ROS (Fig. 7a–d), as well as subsequent enhanced cell death in human chondrocytes under confinement (Fig. 7e). In addition, we found that human chondrocytes also underwent Drp1 droplets formation and mitochondrial fragmentation in the early stage of confinement (Fig. 7f). Moreover, the cPLA2 was diffusive in the cytoplasm at the beginning of confinement and partially co-localized with mitochondria after 3 h confinement (Fig. 7g, h), indicative of its translocation to mitochondria in chondrocytes under confinement. Further, using normal and OA human cartilage samples, we compared total ROS and lipid ROS levels, and observed higher levels of total (Fig. 7i, j) and lipid ROS (Fig. 7k, l) in OA regions. Finally, immunofluorescence of cPLA2 and Tom20 in chondrocytes of OA tissue revealed partial colocalization (Fig. 7m, n). Our findings suggest that the same molecular players we find responsible for confinement-induced ferroptosis in-vitro may be also present clinically in OA.

a Representative live-cell images of DHE staining in human chondrocytes upon confinement mediated by microspheres. Scale bar = 40 μm. b Quantification of total ROS level in human chondrocytes upon confinement, related to (a) (n = 100 cells, N = 3 biologically independent experiments, ****p < 0.0001, two-sided unpaired Student’s t test). c Representative live-cell images of BODIPY-C11 581/591 staining in human chondrocytes upon confinement mediated by microspheres. Scale bar = 40 μm. d Quantification of lipid ROS level in human chondrocytes upon confinement, related to (c) (n = 50 cells, N = 3 biologically independent experiments, ****p < 0.0001, two-sided unpaired Student’s t test). e The percentage of cell death in chondrocytes upon 9 h confinement mediated by microspheres (n = 10 independent fields, N = 3 biologically independent experiments, mean ± SD, ****p < 0.0001, two-sided unpaired Student’s t test). f Representative time-lapse images of mitochondria (Mito Red) in chondrocytes expressing eGFP-Drp1 upon confinement mediated by microspheres. Insets are magnified images of areas indicated by white boxes. Scale bar = 10 μm. g Representative live-cell images of the cPLA2-eGFP expressing chondrocytes co-stained with Mito Red upon confinement mediated by microspheres. Scale bar = 10 μm. h Line scan plot showing the distribution of cPLA2 and Mito Red signal along the direction of the white lines related to (g) upon confinement for 0 h (Up) and for 3 h (Down). i Representative scan images of DHE staining in relative normal and OA tissues. Insets are magnified images of areas. The paired normal and OA tissues are from the same patients. Scale bar = 100 μm. j Quantification of total ROS level in normal and OA tissue, related to (I) (n = 150 cells, ****p < 0.0001, two-sided unpaired Student’s t test). k Representative scan images of BODIPY-C11 581/591 staining in normal and OA tissues. Insets are magnified images of areas. Nuclei (DAPI). Scale bar = 100 μm. l Quantification of lipid ROS level in normal and OA tissue, related to (k) (n = 88 cells, ***p = 0.0004, two-sided unpaired Student’s t test). m Representative images of cPLA2 and Tom20 in normal and OA tissue. Scale bar = 5 μm. Data are representative of three biologically independent experiments. n Line scan plot showing the distribution of cPLA2 and Tom20 along the direction of the white arrowheads related to (m) in normal (Left) and OA tissue (Right).
Discussion
Here, we show that prolonged axial confinement of non-motile adherent cells results in cell death via ferroptosis, thereby contributing to the understanding of clinically relevant pathological processes in OA. Mechanistically, we show that when cells and their nucleus are mechanically confined, they undergo ferroptosis driven by Drp1-mediated mitochondrial fragmentation and cPLA2 relocation. Interestingly, these two events concertedly drive confinement-induced ferroptosis, but they are not inter-dependent in this process. We notice that inhibiting mitochondrial fragmentation and ROS production through Drp1 knockdown does not prevent cPLA2 relocation to mitochondria, and cPLA2 knockdown also does not affect Drp1 liquid-like droplets formation or mitochondrial fragmentation. However, these mechanically activated pathways are closely related to mitochondria. Aside from mitochondrial fragmentation, there is also a mitochondrial swelling process under confinement, and it can still happen even when Drp1 is perturbed. It has been reported that cPLA2 can translocate in response to increased membrane tension - a property of swelled organelles12,13,30. Indeed, our data define that Ca2+-dependent C2 domain may be responsible for cPLA2 relocation to mitochondria, as previously reported that hypotonic-induced osmotic cell swelling triggers cPLA2 translocation to the inner nuclear membrane through increasing Ca2+ signals30. Thus, we would like to further test this mitochondrial membrane mechanics-induced lipid metabolic rewiring hypothesis in our future study using more biophysical and in vitro reconstitution assays.
Our work shows that the nucleus acts as the central mechanosensor in confinement-induced mitochondrial fission, resulting in ferroptosis. Whilst we show insight into the molecular transduction of nuclear deformation, leading to mitochondrial fragmentation, it will be important to investigate the possible role that organelle-mitochondrial membrane contact sites may play. While some membrane contacts are better studied (such as ER-mitochondria contacts), others like mitochondria-nuclear membrane contacts have not been well described in detail, which could explain our findings. Although changes in nuclear shapes often directly modulate gene expressions in cells45, and over 95% of mitochondrial proteins are encoded by nuclear DNA46, it is unlikely that the nucleus regulates mitochondrial fragmentation under confinement at the transcriptional or translational levels as fragmentation occurs early during confinement (30 to 60 min). Thus, further research to elucidate the underlying interaction between the nucleus and mitochondria is warranted to define the ruler roles of nucleus in regulating cell biological processes under confinement.
Our study revealed that Drp1 exhibits confinement-induced condensation behavior in cells, forming discrete puncta with liquid-like properties, which we demonstrate via endogenous Drp1 tagging. These structures are morphologically similar to the higher-order Drp1 oligomers that have been previously characterized47. Curiously, when utilizing the optoDroplet system to induce Drp1 foci formation, we find that when we co-express the eGFP tagged cytoplasmic adaptor proteins MID49 or MID51, these adaptor proteins also condense into droplets that co-localized with Drp1 puncta (Supplementary Fig. 8g). These results suggest that the Drp1 may assemble into complexes with adaptor proteins required for mitochondrial fission via phase separation. However, future work is needed to elucidate the specific outer mitochondrial membrane adaptors responsible for Drp1 recruitment under confinement. Biomolecular condensation is governed by multiple physicochemical parameters (concentration, pH, osmotic pressure) and post-translational modifications (PTMs) regulating multivalent interactions48,49. While phosphorylation at Drp1 most characterized regulatory sites including S616/S637 was dispensable for liquid-like droplets formation based on phospho-mutant studies (Supplementary Fig. 8h), point mutations in middle domain (A395D, R403C, C431Y) completely abolished this process. Notably, R403 methylation has been previously shown to be essential for Drp1 self-assembly50, suggesting other PTM-dependent regulation like methylation may occur under confinement. Beyond protein concentration, cytoplasmic viscosity emerge as key determinant of biomolecular condensation. Utilizing genetically encoded multimeric nanoparticles (GEMs) as viscosity probes, we observed a progressive reduction in the effective diffusion coefficient of GEMs under confinement, indicative of increased cytoplasmic viscosity (Supplementary Fig. 8i). These results hint at that confinement may facilitate Drp1 condensation through enhanced macromolecular crowding effects.
Our data establish a direct role for nuclear deformation in mechano-induced ferroptosis-dependent cell death, which may lead to inflammation. These findings may provide a framework for further comprehensive understanding and targeting of ferroptosis in not only treating OA but may also give insight into other diseases related to mechanical stresses and inflammation.
Method
Cell culture
Human cervical carcinoma HeLa cells were a generous gift from Dr. Yong Liu (Xuzhou Medical University). Human embryonic kidney 293T (HEK293T) cells were a generous gift from Dr. Yuxin Yin (Peking University). Human fibrosarcoma HT1080 cells were a generous gift from Dr. Fuping You (Peking University). Cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Corning, 10-013-CRVC) supplemented with 10% fetal bovine serum (FBS; PAN-Biotech, P30-3302), 100 μ/mL penicillin and 100 μg/mL streptomycin (Macgene, CC004) at 37 °C with 5% CO2. For passage, the cells were washed with PBS (Macgene, CC010) and detached by trypsin-EDTA (Macgene, CC012). For long-term live cell imaging, cells were plated on fibronectin (Sigma-Aldrich, F1056-5MG, 10 μg/mL)-coated 35 mm glass-bottom dishes (Cellvis, D35-10-1-N) and maintained in CO2-independent DMEM (Gibco, 18045-088) supplemented with 10% FBS, 100 μ/mL penicillin and 100 mg/mL streptomycin at 37 °C throughout the imaging process.
Isolation and culture of human chondrocytes
Human chondrocytes were isolated from cartilage fragments dissected from knee joint cartilage of OA patients that were discarded at the time of total joint replacement surgeries. During isolation, the cartilage fragments were minced and digested with 0.2% type II collagenase at 37 °C for 4 h. After centrifugation at 300 × g, the cells were re-suspended in Minimum Essential Medium Alpha modification medium (MEM Alpha; HyClone, SH30265.01) containing 10% FBS, 100 μ/mL penicillin and 100 μg/mL streptomycin, and maintained at 37 °C with 5% CO2. Above procedure was approved by the Human Ethics Committee of Peking University Third Hospital, and we have complied with all relevant ethical regulations for work with human patients. Informed consent was obtained from all patients.
Plasmid construction and DNA transfection
The full-length human Drp1 was cloned using plasmid containing Drp1 from Dr. Yuhui Zhang (Huazhong University of Science and Technology) and subcloned into a lentiviral vector (pLVX-AcGFP-N1) with N-terminal tagged eGFP using an ABclonal MultiF Seamless Assembly kit (ABclonal, RK21020). The Drp1 truncations and single-point mutations were amplified and cloned into the same vectors. The FL-Drp1 (1-699 aa), GTPase domain (1-302 aa), ΔGTPase domain (303-699 aa), FL-Drp1-A395D, FL-Drp1-R403C, FL-Drp1-C431Y, FL-Drp1-S616A, FL-Drp1-S616D, FL-Drp1-S637A and FL-Drp1-S637D were constructed. The FL-Drp1-K38A was cloned from the pcDNA-Drp1K38A plasmid, a gift from Alexander van der Bliek & Richard Youle (Addgene plasmid # 45161; http://n2t.net/addgene:45161; RRID: Addgene_45161), and subcloned into pLVX-AcGFP-N1 with N-terminal tagged mCherry. Plasmid containing Cry2 was a gift from Chandra Tucker (Addgene plasmid # 60032; http://n2t.net/addgene:60032; RRID: Addgene_60032) to generate the optoDroplet optogenetic system (Opto-Drp1). The full-length human cPLA2α was cloned from the HeLa cell-extracted cDNA library and subcloned into pLVX-AcGFP-N1 with C-terminal tagged eGFP. The FL-cPLA2α (1-749 aa), ΔC2 domain (140-749 aa), ΔPL region (179-749 aa) were constructed. The Tom20 coding sequence was cloned from a Drosophila melanogaster cDNA library and subcloned into pLenti6-H2B-mCherry (Addgene plasmid # 89766; http://n2t.net/addgene:89766; RRID:Addgene_89766), replacing the H2B coding sequence using Gibson assembly. The Contact-FP constructs (GA and GB) were cloned from GA-NES (a gift from Robert Campbell; Addgene plasmid # 61018; http://n2t.net/addgene:61018; RRID:Addgene_61018) and GB-NES (a gift from Robert Campbell; Addgene plasmid # 61017; http://n2t.net/addgene:61017; RRID:Addgene_61017), and were subcloned into the pLVX-AcGFP-N1 with C-terminal tagged full-length sequence of Sec61β (pLVX-GA-Sec61β) or mitochondrial localization sequence of MAVS (pLVX-GB-MAVS), respectively. The 40 nm GEM-Sapphire was cloned from pCDNA3.1-pCMV-PfV-GS-Sapphire, a gift from Liam Holt (Addgene plasmid # 116933; http://n2t.net/addgene:116933; RRID:Addgene_116933), and subcloned into pLVX-AcGFP-N1 backbone. pLVX-IRES-Hygro_RGECO1.2 was a gift from Joseph Hyser (Addgene plasmid # 164592; http://n2t.net/addgene:164592; RRID: Addgene_164592).
For transient transfection, cells seeded on 6-well plates were transfected with 2 μg midi-prep quality plasmid DNA in Opti-MEM (Invitrogen, 31985-070) containing 2 μL Neofect™ DNA transfection reagent (TF20121201) following the protocol, for 24–48 h. For viral particle generation, HEK293T cells seeded on 10 cm culture dishes (Nest, 704201) were co-transfected with 5 μg expression plasmid, 3.75 μg packaging plasmid psPAX2, and 1.25 μg pCMV-VSV-G, in Opti-MEM containing 10 μL Neofect™ DNA transfection reagent. The primer sequences used to construct above plasmids are listed below.
FL-Drp1 - F:
5′-GGCATGGACGAGCTGTACAAGATGGAGGCGCTAATTCCTGC-3′;
FL-Drp1 - R: 5′-CGGTAGAATTATCTAGAGTCGCGGTCACCAAAGATGAGTCTCCCGGATTTCA-3′;
FL-cPLA2α - F: 5′-CGAGCTCAAGCTTCGAATTCATGTCATTTATAGATCCTTACCAGCACATTATAGT-3′;
FL-cPLA2α - R:
5′-CTCGCCCTTGCTCACCATTGCTTTGGGTTTACTTAGAAACTCCTTGT-3′;
Tom20 - F: 5′-ATGATTGGAGTCTCGGGAACTTTCAAGGTG-3′;
Tom20 - R: 5′-AAGGCTACTGGTAGCCTCATCATCCAACT-3′.
Antibody and reagents
The following antibodies were used in this study: Rabbit anti-Drp1 (EPR19274, dilution 1:1000 for Western blotting), rabbit anti-cleaved N-terminal GSDMD (EPR20829-408, dilution 1:1000 for Western blotting), rabbit anti-GSDME (EPR19859, dilution 1:1000 for Western blotting), rabbit anti-MLKL (EPR9514, dilution 1:1000 for Western blotting), rabbit anti-GAPDH (EPR16891, dilution 1:4000 for Western blotting) and mouse anti-β-actin (ab8226, dilution 1:2000 for Western blotting) were purchased from Abcam; Rabbit anti-Drp1 (A21968, dilution 1:1000 for Western blotting), rabbit anti-full-length GSDMD (A17308, dilution 1:1000 for Western blotting), rabbit anti-cPLA2 (A0394, dilution 1:1000 for Western blotting), rabbit anti-Tom20 (A19403, dilution 1:200 for immunofluorescence staining) and mouse anti-α-tubulin (AC012, dilution 1:2000 for Western blotting) were purchased from Abclonal; Mouse anti-cPLA2 (sc-454, dilution 1:100 for immunofluorescence), anti-mouse (sc-516102, dilution 1:4000) and anti-rabbit (sc-2004, dilution 1:4000) horseradish peroxidase (HRP)-conjugated secondary antibodies were from Santa Cruz Biotechnology. Rabbit anti-HIF1α (NB100-479, dilution 1:1000 for Western blotting) was purchased from Novus Biologicals. Anti-mouse Alexa Fluor 488 (A-21202) and anti-rabbit Alexa Fluor 555-conjugated secondary antibody (A-31572) were from ThermoFisher Scientific.
For commercial reagents, Z-VAD-FMK (S7023), DFOM (S5742), Liproxstatin-1 (S7699), Ferrostatin-1 (S7243), and MitoTEMPO (S9733) were from Selleckchem. Trolox (ab120747) was from Abcam. FCCP (HY-100410), CAY10650 (HY-10801), BAPTA-AM (HY-100545), MitoSOX Red (HY-D1055), Rotenone (HY-B1756), and Arachidonic acid (HY-109590) were from MedChemExpress. NSA was a gift from Dr. Huang Wang (Peking University). Antimycin A was a gift from Dr. Ye Tian (Institute of Genetics and Developmental Biology, Chinese Academy of Sciences). TNFα was a gift from Dr. Fuping You (Peking University). Smac was a gift from Dr. Xiaodong Wang (National Institute of Biological Sciences, Beijing). Image-iTTM Green Hypoxia Reagent (I14834), MitoTrackerTM Red CMXRos (M7512), Mito TrackerTM Green FM (M7514), TMRM (T668), and ProlongTM Diamond Antifade with DAPI (P36962) were from ThermoFisher Scientific. Other drugs used in this study included EUK134 (TargetMol, T6495), NAC (Shanghai Yuanye Bio-Technology, S20137), and H2O2 (Aladdin, H112515).
Cell confinement
Two-cell confinement systems allowed for the monitoring of multiple fields of live cells over the entire duration of prolonged axial confinement were used in our study. The first one was a bespoke, in-house device consisting of stainless-steel weights, optical glass, and microsphere spacers of different sizes (Bangs Laboratories, USA) (Supplementary Fig. 1b). The device mass is around 220 g, and the dimension of optical glass is 24.7 × 24.7 × 3 mm. For biochemical studies, the mixture of cells and microspheres with appropriate volume was plated into the 35 mm culture dish (Corning, 430165) and incubated overnight. Then, weighted optical glass was carefully placed on top of the culture substrate. The diameters of the microspheres (20 μm versus 3 μm) determined the height for spatial confinement of cells between the glass and the substrate. Notably, a version of the cell confiner adapted to 35 mm glass-bottom dishes was used to perform high-resolution live cell imaging.
The second one was composed of a custom designed PDMS (Slygard 184, Dow Corning) piston based on a previous design19. Briefly, to actuate the piston downwards and initiate confinement, a 1 ml syringe was connected to the piston via 18G blunt needles connected via silicon tubing (5065-9978, Agilent). To engage confinement, the syringe piston was drawn back and locked in place by friction at the furthest point in the syringe barrel. To control confinement height, custom-made PDMS micropillars fabricated at 3.7 µm height, which were passivated with non-adhesive pLL(20)-g[3.5]-PEG(2) (SuSoS). The PDMS micropillars are about 500 μm in diameter and their spacing is 1 mm.
The top surfaces of both two confinement systems that directly contact with cell apex are coated with non-adhesive pLL-PEG. To perform high-resolution imaging, the 35 mm glass-bottom dishes are coated with fibronectin or poly-D-lysine to create an adhesive environment.
Microscopy imaging of cell death
Cell death in confinement was evaluated by propidium iodide (PI, Sigma-Aldrich, P4170-10MG) uptake of cells measured by fluorescence imaging. PI was added to the culture medium at a final concentration of 5 μg/mL before confinement. Static images of PI positive cells were captured using a Plan Apo 10× air objective lens on a Spinning Disk Confocal Microscope (Andor Dragonfly). Time-lapse imaging of cell death was imaged via epifluorescence microscopy (LIFE EVOS FL Auto fluorescent microscope, Life Technologies, USA). Raw full bit depth images were processed using ImageJ software (https://imagej.nih.gov/ij/). Image data shown were representative of at least three randomly selected fields.
Measurement of ROS
To detect total ROS, cells were pre-stained with 10 μM DHE (Beyotime, S0063) in culture media at 37 °C for 30 min, before washing twice with PBS. To detect lipid ROS, cells were pre-incubated with 5 μM BODIPY-C11 581/591 probe (Abclonal, RM02821) at 37 °C for 1 h and washed twice with PBS. After labeling, cells were subjected to confinement and fluorescence images were taken by a 10× air objective lens on Andor Dragonfly Microscope at different time points. To detect mtROS, cells were stably expressed with Mito-roGFP, and imaging was acquired using a 63× objective lens on Andor Dragonfly Spinning Disk Confocal Microscope with the excitation wavelength of 405 nm or 488 nm, and the emission wavelength of 500–550 nm. mtROS level was indicated by the roGFP ratio, which was calculated as the fluorescence intensity under the excitation wavelength of 405 nm divided by the intensity under the excitation wavelength of 488 nm (405/488 Ratio).
Western blotting
For Western blotting, cells were washed with PBS and lysed with appropriate volumes of commercial 1× SDS-PAGE sample loading buffer (Beyotime, P0015A) for 5–10 min. The protein samples were boiled for 15 min at 95 °C and were separated on SDS-PAGE gels before being transferred onto nitrocellulose membranes by wet electrophoretic transfer. After blocking in PBS containing 5% milk, blots were incubated with primary antibodies at 4 °C overnight or for 2 h at room temperature, followed by the secondary antibody incubation for 1 h at room temperature. Blots were visualized and recorded using SH-Compact 523 (SHST). The images were processed by ImageJ software.
Quantitative real-time PCR (qRT-PCR)
Briefly, total RNA from HeLa cells were isolated using Trizol (Life Technologies, 15596026). RNA was reverse transcribed using a Transcript One-Step gDNA Removal and cDNA Synthesis SuperMix Kit (Transgene, AT311-02). mRNA levels of the COX2 and α-tubulin genes were analyzed by qRT-PCR amplified using SYBR Green Fast qPCR Mix (ABclonal, RK21203). Data shown were the relative abundance of the COX2 mRNA normalized to α-tubulin. The primer sequences are listed below.
COX2 - F: 5′-CGGTGAAACTCTGGCTAGACAG-3′;
COX2 - R: 5′-GCAAACCGTAGATGCTCAGGGA-3′;
α-tubulin - F: 5′-CGGGCAGTGTTTGTAGACTTGG-3′;
α-tubulin - R: 5′-CTCCTTGCCAATGGTGTAGTGC-3′.
Arachidonic acid measurement
The control and confined cells were gently washed with cold PBS for 3 times, and then directly lysed by appropriate volumes of PBS-diluted 10× lysis buffer (Promega, G182A). Lysed supernatants and cell debris were collected and centrifuged at 1500 × g for 10 min at 4 °C. After centrifugation, the supernatants were collected for detection using the Arachidonic acid (ARA) ELISA kit (Elabscience, E-EL-0051) in accordance with the manufacturer’s instructions.
Colocalization analysis
For cPLA2 and mitochondria, the colocalization level was determined by calculating the Mander’s coefficient of percentage of cPLA2 overlapping with the whole mitochondrial network in a single cell. For Drp1 and mitochondria, the colocalization level was determined by calculating the Mander’s coefficient of the percentage of Drp1 puncta overlapping with mitochondria within regions of interest (ROIs) (M1). To determine whether above overlap was due to chance, the mitochondria ROl was rotated 90° clockwise and the Mander’s coefficient of the percentage of Drp1 puncta overlapping with the rotated mitochondria ROl was calculated (M190°). The segmentation of cPLA2, Drp1 puncta and mitochondria was through manual threshold, and final calculation were accomplished by the JACoP plugin of ImageJ software (https://imagej.net/plugins/jacop).
Cellular enucleation
Enucleation was performed following an established protocol34 with some modifications. A detailed description of the experimental procedures is provided below. Briefly, small plastic slides that fit into 1.5 mL Eppendorf tubes were cut from 10 cm cell culture dishes, followed by sterilization in 95% ethanol for 10 min followed by ultraviolet irradiation for 30 min. Before seeding cells, the plastic slides were coated with PBS containing 10 μg/mL fibronectin for 30 min at 37 °C. Cells on plastic slides were pre-incubated with DMEM containing 2 μM Cytochalasin D (CytD, Sigma-Aldrich, C2618-200UL) in 1.5 mL Eppendorf tubes for 1 h at 37 °C, then the cells were centrifuged at 16,000 × g in a GeneSpeed microcentrifuge (Gene Company Limited, 1524) for 30 min. After centrifugation, the remaining cells and cytoplasts on the plastic slides were placed in 10 cm cell culture dishes containing fresh medium without CytD to spread and recover from the effects of CytD fully. About 3 to 4 h later, the cells were washed with PBS, trypsinized, and reseeded on fibronectin-coated glass-bottom dishes for live cell imaging.
Mitochondrial morphodynamics analysis
Segmentation of the mitochondrial outer membrane (Tom20-mCherry signal) was done using the auto-context workflow from Ilastik51. First, raw images were converted into HDF5 files and imported in Ilastik. For the first round of training, the features selected were color/intensity, edge and texture. The σ value for all features was set to 0.7 px. The algorithm was trained for two labels: mitochondrial outer membrane and background. As minor bleaching occurred during the acquisition, randomly chosen images were selected throughout the entire acquisition to account for visually different parts of the dataset. For the second round of the auto context workflow, the σ values for all features were set to 0.7 px. Then, segmentation was performed identically as during the first round. After simple segmentation predictions were deemed satisfactory, the simple segmentation masks were exported as multipage tiff files. These multipage tiff files were then imported to ImageJ and despeckled using the despeckle function. Images were then inverted and binarized before particles analysis. Datasets were binned into the 10 first frames (first 20 min) and the following 35 frames (last 70 min of acquisition). The size threshold was 9–150 (Pixel units) and circularity 0.3–1.0 to exclude areas between distinct mitochondria for the first 10 frames. Size and circularity thresholds were set to 9–600 (Pixel units) and 0.3–1.0 for the second part of the acquisition. Regions of interests (ROIs) around individual cells were drawn for each timepoint and particles for each cell were analyzed. Average perimeter, average circularity, and total number of particles per cell were calculated and plotted in a XY graph. Data was imported and plotted using Graphpad Prism.
Generation of mtDNA-depleted “ρ0” cells
HeLa cells were cultured in DMEM supplemented with 10% FCS, 450 ng/ml ethidium bromide (EtdBr; Generay, GA328), 100 μg/ml sodium pyruvate (Gibco, 11360070), and 50 μg/ml uridine (HARVEYBIO, NL1121) for 10 days. To measure the efficiency of mtDNA depletion, total DNA was extracted using Animal Tissues/Cells Genomic DNA Extraction Kit (Solarbio, D1700), and the expression of mtDNA content (Dloop) and genomic DNA (Tert) was measured by qRT-PCR using the following primer pairs:
Dloop - F: 5′-CTTGACCACCATCCTCCGTG-3′;
Dloop - R: 5′-GGGGAACGTGTGGGCTATTT-3′;
Tert - F: 5′-TCACACAGGTGGATGTGACG-3′;
Tert - R: 5′-ACACACATCCTGGACACGAC-3′.
Atomic force microscopy (AFM) coupled with live cell imaging
To confine a single cell, we used a Bioscope Resolve AFM (Bruker, USA) with the HA_FM tipless silicon probe (force constant: 3 or 6 N/m, ScanSens), and the AFM was mounted on the stage of an inverted fluorescence microscope (ECLIPSE Ti, Nikon) for imaging. Before confinement and imaging, HeLa cells were plated on fibronectin-coated 35 mm glass-bottomed dishes (Cellvis, D35-20-1-N). Experiments were initiated 4–6 h after cell plating to allow for cells to fully spread. During imaging, cells were maintained in CO2-independent DMEM supplemented with 10% FBS, 100 μ/mL penicillin, and 100 mg/mL streptomycin. The probe was mounted in the AFM head and the 6 N/m microcantilever was lowered on the cell until a 0.8 V setpoint was reached, at which cells were significantly confined. Meanwhile, fluorescence images were taken every 0.2 s using a 100× oil immersion objective to record the whole process of microcantilever lowering. After cells were stably compressed by the cantilever, images were taken every 30 s to observe changes in mitochondrial morphology. All microscopy equipment was placed in an acoustic enclosure on the top of the vibration isolation platform.
Generation of eGFP-Drp1 knock-in cells
HeLa cell line with an endogenous N-terminal eGFP insertion in the Drp1 gene locus was generated via the CRISPR-Cas9 system. The following sgRNA was generated via the online CRISPR design tool (CRISPR tefor.net) and cloned into a lentiCRISPR-V2 vector (a gift from Feng Zhang; Addgene plasmid # 52961; http://n2t.net/addgene:52961; RRID: Addgene_52961). The donor template contained a left homologous arm (840 bp before the start code), eGFP sequence, and right homologous arm (1000 bp after the start code). The homologous arms were amplified from the HeLa cell-extracted genome DNA. Cells were co-transfected with sgRNA and relative donor template plasmid for 24 h and then treated with puromycin (Sigma-Aldrich, P8833) at 2 μg/mL for 24 h. Surviving cells were sorted by Fluorescence Activated Cell Sorting (FACS) to isolate fluorescence-positive cells. The candidate single cell clones were cultured and verified by Western blotting.
sgRNA for Drp1 N-terminal knock-in in HeLa cells:
sgRNA: 5’-CGCCGGCCACGGCAATGAAT-3’.
Protein expression and purification
For recombinant protein expression in E.coli BL21-CondonPlus (DE3)-RIPL (AngYu Biotechnologies, AYBIO-G6028), DNA fragments encoding Drp1 were cloned into the modified pET28a (+) vector with an N-terminal His6-tag and a C-terminal eGFP tag. Bacteria transformed with plasmid were cultured in LB medium with 50 mg/L kanamycin at 37 °C to an OD600 of 0.8–1.0. After that, protein expression was induced with 0.1 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) for 16 h at 18 °C.
For protein purification, cell pellets were collected by centrifugation at 4000 × g and were re-suspended in TBS buffer containing 20 mM Tris (pH 8.0), 140 mM NaCl, 30 mM KCl, 10% glycerol, 20 mM imidazole, 0.5 mM TCEP, and protease inhibitor mixture (Complete EDTA-free, Roche). The suspension was lysed by sonication and cleared by centrifugation twice at 4 °C, 13,000 × g for 30 min. The supernatant was applied to a Ni-IDA column (Smart-Lifesciences) and washed three times using buffers containing 20 mM Tris (pH 8.0), 500 mM NaCl, 0.5 mM TCEP, and different concentrations of imidazole (0, 20, 40 mM). Bound Drp1 proteins were eluted with buffer containing 20 mM Tris (pH8.0), 500 mM NaCl, 0.5 mM TCEP and 300 mM imidazole, and PreScission protease was added to the elution to cleave the N-terminal His6-tag. Finally, proteins were purified by size exclusion chromatography using Superose 6 Increase 10/300 (GE Healthcare) in buffer containing 20 mM Tris (pH 8.0), 500 mM NaCl and 0.5 mM TCEP. Fractions containing Drp1 were collected, concentrated, and flash frozen in liquid nitrogen. Protein concentrations were measured with NanoDrop 2000 spectrophotometry using the theoretical molar extinction coefficients at 280 nm. Protein purity was evaluated with Coomassie brilliant blue staining of SDS-PAGE gels.
Quality control data of the recombinant Drp1-eGFP protein were shown in Supplementary Fig. 6f, g.
In vitro phase transition assay
Preparing coverslips and glass slides
Glass slides were rinsed with ethanol twice and dried with air. A flow chamber for imaging with two openings on opposite sides was then made by attaching a coverslip (Fisherbrand, 12541016) to the glass slide using double-sided tape placed parallel to each other.
Phase separation assays
Full-length Drp1 and its mutants were labeled by eGFP tags. The purified proteins were saved in a high salt solution containing 500 mM NaCl, 20 mM Tris (pH = 8.0), and 0.5 mM TCEP after protein production. According to different protein and NaCl concentrations for phase separation assay, they were diluted into the corresponding lower salt experimental buffers at a final concentration of 8% dextran to induce phase separation. The protein-phase buffer mixtures were mixed gently. Then, 5 μL mixtures were injected into the homemade flow chamber for fluorescent imaging using the Andor Dragonfly confocal imaging system by putting the chamber upside down on the objective. Notably, the last layer (closest to the lens) of droplets was averted when acquiring the images to minimize the interaction between droplets and the glass surface.
Generation of knock-down and knock-out cells
For Drp1 and cPLA2 knockdown in HeLa cells, the related shRNA were cloned into pLKO.1 vector. After lentivirus infection and puromycin selection, the pooled cells were collected and verified by Western blotting.
For Drp1 knockout, sgRNA was cloned into the LentiCRISPR-V2 vector, followed by transfection into HeLa cells. After puromycin selection, the single-cell clones were cultured and verified by RNA sequence and Western blotting.
The primes used to knockdown are listed below:
shScramble: 5′-AACGCTGCTTCTTCTTATTTA-3′;
shDrp1: 5′-GCTACTTTACTCCAACTTATT-3′;
shcPLA2α-1: 5′-CCTTGTATTCTCACCCTGATT-3′;
shcPLA2α-2: 5′-CCTCCGTTCAAGGAACTTCTA-3′.
sgRNA for Drp1 knockout in HeLa cells:
sgRNA: 5′-AAATAGCTACGGTGAACCCG-3′.
Sequencing primer:
Drp1-seq-F: 5′-GATGCATCACATCAGAGATTGTT-3′;
Drp1-seq-R: 5′-TTGCCAAAGTGAAGGTGTCG-3′.
For integrin knockdown, the siRNAs targeting to different subunits (AV/A1/A3/A5/A8/B1/B3/B5/B6/B8) were purchased from Sangon Biotech.
Measurement of intracellular viscosity
To detect intracellular viscosity upon confinement, HeLa cell stably expressing 40 nm-GEMs were confined and imaged by a 63× 1.4 NA objective lens on Andor Dragonfly Spinning Disk Confocal Microscopy. Fluorescence images were taken every 10 ms with 488 nm excitation wavelength and 525 nm emission wavelength at 100% exposure intensity. To analyze the images, tracking of single GEM particle was performed with the Mosaic suite of ImageJ software, using the following typical parameters: radius = 3, cutoff = 0, 15% of fluorescence intensity, a link range of 1, a maximum displacement of 12 px, and assuming Brownian dynamics. Following tracking, the time-averaged mean-square displacement (MSD), as well as the ensemble-average of the time-averaged MSD were calculated for curve fitting and obtaining the effective coefficient of diffusion (Deff). There is a linear relationship between MSD and Deff in a short time52:
Finally, the distributions of Deff were plotted and the statistical differences were assessed at confinement for 0, 15, and 60 min using MATLAB and GraphPad Prism 8.0.
Polar-structure illumination microscopy (polar-SIM)
The eGFP-Drp1 Knock-in HeLa cells were plated on fibronectin-coated 35 mm glass-bottomed dishes (Cellvis, D35-20-1.5-N) and cultured overnight. To label mitochondria, cells were incubated with 250 nM PK Mito Orange (GENVIVO, PKMO-2) in DMEM at 37 °C for 15 min followed by PBS washing for 3 times. Images were collected through the Polar-SIM system (Airy Technology Co., Ltd, China). 488 and 561 nm laser were respectively used to excite eGFP-Drp1 and PK Mito Orange with 2DSIM modality. The SIM reconstruction process was conducted using the Polar-SIM software with pre-processing (Dark) and post-processing (MRA).
Immunofluorescence of human cartilage samples
Human cartilage samples were obtained from Peking University Third Hospital. The relative normal and OA cartilage samples from the same patient with OA were frozen in OCT and sectioned into slices attached onto glass slides. Slides were washed by PBS for 5 min and were incubated with PBS containing 10% FBS and 0.3% Triton X-100 for 1 h at room temperature. Then, the slides were incubated with the primary antibodies mix solution containing mouse anti-cPLA2 (1:100), rabbit anti-Tom20 (1:200) and 0.3% Triton X-100 for 2 h at room temperature. After three times of PBS washing for 5 min each, the slides were incubated with anti-mouse Alexa Fluor 488- and anti-rabbit Alexa Fluor 555-conjugated secondary antibodies diluted in 1:200 for 2 h at room temperature. The secondary antibodies mix solution also contained 0.3% Triton X-100. Following another three times washing with PBS, a coverslip was mounted using ProLong™ Diamond with DAPI. Images were captured on an Andor Dragonfly confocal imaging system after mounting medium was solidified.
To detect total ROS or lipid ROS levels in normal and OA cartilage samples, the slides were washed by PBS for 5 min and stained with DHE probe (50 μM) for 30 min or BODIPY-C11 581/591 probe (20 μM) for 1 h at 37 °C. Following washing three times with PBS for 5 min each, and then further stained with DAPI (5 μg/mL) for 10 min at room temperature. Finally, slides were washed with PBS for three times, and coverslips were mounted using ProLong™ Diamond. Slides were scanned with Multi-mode intelligent living cells imaging analysis system after mounting medium was solidified.
Above procedure was conducted according to guidelines approved by Medical Science Research Ethics Committee of Peking University Third Hospital (ethical approval No.: 2013003). Informed consent was obtained from all patients and the experiments were conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
Statistics and reproducibility
All data were representative of two or more independent experiments and were expressed as mean ± SD (standard deviation). Unless stated otherwise, statistical analysis was determined using two-sided unpaired or paired Student’s t-test for two-group comparison, or one-way ANOVA analysis with Tukey’s test for more than two-group comparison, or two-way ANOVA analysis with Sidak’s test for multiple pairwise comparisons. All calculations of significance were performed with GraphPad Prism 8.0 Software (ns represents not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the findings of this work are available within the paper and its Supplementary Information files. Source data are provided with this paper.
References
Balzer, E. M. et al. Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J. 26, 4045–4056 (2012).
Uhler, C. & Shivashankar, G. V. Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat. Rev. Mol. Cell Biol. 18, 717–727 (2017).
Liu, Y. J. et al. Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 160, 659–672 (2015).
Pagès, D. L. et al. Cell clusters adopt a collective amoeboid mode of migration in confined nonadhesive environments. Sci. Adv. 8, eabp8416 (2022).
Ekpenyong, A. E. et al. Mechanical deformation induces depolarization of neutrophils. Sci. Adv. 3, e1602536 (2017).
Niethammer, P. Components and mechanisms of nuclear mechanotransduction. Annu. Rev. Cell Dev. Biol. 37, 233–256 (2021).
Fletcher, D. A. & Mullins, D. Cell mechanics and the cytoskeleton. Nature 463, 485–492 (2010).
Humphrey, J. D., Dufresne, E. R. & Schwartz, M. A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 15, 802–812 (2014).
Dupont, S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011).
Yang, S. et al. ETV4 is a mechanical transducer linking cell crowding dynamics to lineage specification. Nat. Cell Biol. 26, 903–916 (2024).
Nava, M. M. et al. Heterochromatin-driven nuclear softening protects the genome against mechanical stress-induced damage. Cell 181, 800–817.e822 (2020).
Lomakin, A. J. et al. The nucleus acts as a ruler tailoring cell responses to spatial constraints. Science 370, 310 (2020).
Venturini, V. et al. The nucleus measures shape changes for cellular proprioception to control dynamic cell behavior. Science 370, 311 (2020).
Denais, C. M. et al. Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358 (2016).
Raab, M. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 353, 1507–1507 (2016).
Nader, G. P. F. et al. Compromised nuclear envelope integrity drives TREX1-dependent DNA damage and tumor cell invasion. Cell 184, 5230–5246 e5222 (2021).
Wang, S. et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J. Adv. Res. 41, 63–75 (2022).
Sanchez-Lopez, E., Coras, R., Torres, A., Lane, N. E. & Guma, M. Synovial inflammation in osteoarthritis progression. Nat. Rev. Rheumatol. 18, 258–275 (2022).
Le Berre, M., Aubertin, J. & Piel, M. Fine control of nuclear confinement identifies a threshold deformation leading to lamina rupture and induction of specific genes. Integr. Biol. 4, 1406–1414 (2012).
Logue, J., Logue, J., Chadwick, R. & Waterman, C. A Simple Method for Precisely Controlling the Confinement of Cells in Culture (Protocol Exchange, 2018).
Wang, Y. P. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99 (2017).
Liu, X., Xia, S., Zhang, Z., Wu, H. & Lieberman, J. Channelling inflammation: gasdermins in physiology and disease. Nat. Rev. Drug Discov. 20, 384–405 (2021).
Shi, J. J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015).
Zhang, Z. et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420 (2020).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282 (2021).
Hung, W. C. et al. Confinement sensing and signal optimization via Piezo1/PKA and myosin II pathways. Cell Rep. 15, 1430–1441 (2016).
Kalukula, Y., Stephens, A. D., Lammerding, J. & Gabriele, S. Mechanics and functional consequences of nuclear deformations. Nat. Rev. Mol. Cell Biol. 23, 583–602 (2022).
Enyedi, B., Jelcic, M. & Niethammer, P. The cell nucleus serves as a mechanotransducer of tissue damage-induced inflammation. Cell 165, 1160–1170 (2016).
Gaschler, M. M. & Stockwell, B. R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 482, 419–425 (2017).
Chao, C. C. et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 179, 1483 (2019).
Graham, D. M. et al. Enucleated cells reveal differential roles of the nucleus in cell migration, polarity, and mechanotransduction. J. Cell Biol. 217, 895–914 (2018).
Efremov, Y. M., Kotova, S. L., Akovantseva, A. A. & Timashev, P. S. Nanomechanical properties of enucleated cells: contribution of the nucleus to the passive cell mechanics. J. Nanobiotechnol. 18, 134–144 (2020).
Weindel, C. G. et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 185, 3214 (2022).
Lei, G., Zhuang, L. & Gan, B. Y. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 22, 381–396 (2022).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2016).
Kraus, F., Roy, K., Pucadyil, T. J. & Ryan, M. T. Function and regulation of the divisome for mitochondrial fission. Nature 590, 57–66 (2021).
Smirnova, E., Griparic, L., Shurland, D. L. & van der Bliek, A. M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256 (2001).
Friedman, J. R. et al. ER tubules mark sites of mitochondrial division. Science 334, 358–362 (2011).
Gregory, E. et al. and Sarah Cohen Contact-FP: A dimerization-dependent fluorescent protein toolkit for visualizing membrane contact site dynamics. Contact 7, 1–12 (2024).
Shin, Y. et al. Spatiotemporal control of intracellular phase transitions using light-activated optoDroplets. Cell 168, 159 (2017).
Bauer, B. L., Rochon, K., Liu, J. C., Ramachandran, R. & Mears, J. A. Disease-associated mutations in Drp1 have fundamentally different effects on the mitochondrial fission machinery. Hum. Mol. Genet. 32, 1975–1987 (2023).
Zhou, X. L. et al. Interactions between cytosolic phospholipase A2 activation and mitochondrial reactive oxygen species production in the development of ventilator-induced diaphragm dysfunction. Oxid. Med. Cell Longev. 2019, 2561929 (2019).
Alisafaei, F., Jokhun, D. S., Shivashankar, G. V. & Shenoy, V. B. Regulation of nuclear architecture, mechanics, and nucleocytoplasmic shuttling of epigenetic factors by cell geometric constraints. Proc. Natl Acad. Sci. USA 116, 13200–13209 (2019).
Quirós, P. M., Mottis, A. & Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 17, 213–226 (2016).
Koirala, S. et al. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc. Natl Acad. Sci. USA 110, E1342–E1351 (2013).
Boeynaems, S. et al. Protein phase separation: a new phase in cell biology. Trends Cell Biol. 28, 420–435 (2018).
Banani, S. F., Lee, H. O., Hyman, A. A. & Rosen, M. K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017).
Cho, Y. & Kim, Y. K. ROS-mediated cytoplasmic localization of CARM1 induces mitochondrial fission through DRP1 methylation. Redox Biol. 73, 103212 (2024).
Berg, S. et al. ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232 (2019).
Delarue, M. et al. mTORC1 controls phase separation and the biophysical properties of the cytoplasm by tuning crowding. Cell 174, 338–349.e320 (2018).
Acknowledgements
We thank National Center for Protein Sciences at Peking University in Beijing, China, for assistance with operation of AFM and Dr. Siying Qin for technical help. This work was supported by funding from the National Key R&D Program of China (2022YFC3401100), the Australian Research Council fellowship (FT190100516), the Regional Innovation Joint Fund of the National Natural Science Foundation of China (Integrated Project) (No.U23A6009), Beijing Advanced Center of Cellular Homeostasis and Aging-Related Diseases (C.Y.W.), the National Key R&D Program of China (2022YFA1304500) to Y.A., and the National Nature Science Foundation of China (32321004 and 32422023) to Y.A.
Author information
Authors and Affiliations
Contributions
C.W. and F.Z. conceived the project and designed the experiments. F.Z. conducted the majority of the experiments. F.Z. and R.J. performed the cell death inhibitors experiment and mitochondria imaging. F.Z., C.K., and J.L. constructed plasmid and performed western blotting. F.Z., J.L., A.Y., and L.L. performed the protein purification and phase separation assay. A.L. developed the mitochondrial morphodynamics analysis. F.Z. and Y.S. assisted with the atomic force microscopy imaging. Y.A. provided advice and reagents for the generation of mtDNA-depleted “ρ0” cells. C.W., S.S., and X.H. supervised the project. F.Z., C.W., S.S., and X.H. wrote the manuscript. All authors participated in discussion and editing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yu-Hui Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, F., Ju, R.J., Kang, C. et al. Mechanical confinement induces ferroptosis through mitochondrial dysfunction. Nat Commun 16, 11717 (2025). https://doi.org/10.1038/s41467-025-66353-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66353-z
This article is cited by
-
Organelle-specific regulation of ferroptosis
Biology Direct (2026)
