
Abstract
Approximately 50% of breast cancer patients receiving neoadjuvant therapy do not achieve pathological complete response (pCR). Identifying personalized optimal therapeutic strategy is an unmet major challenge. Here, based on 4371 eligible patients from 31 datasets, we develop GDnet, an interpretable deep learning model integrating drug representations and tumor transcriptome profiles to predict the responses to neoadjuvant therapies and aid in selecting optimal treatment strategy. We demonstrate that GDnet significantly outperforms transcriptome-only model in predicting treatment response. Then we conduct two series of in-silico simulated clinical trials based on I-SPY2 trial and test datasets respectively and show that GDnet can significantly increase the pCR rate in all trials. Moreover, the odds ratios for pCR increase from 1.6 to 2.5 linearly as optimization intensifies. Overall, GDnet can function as a digital drug-testing surrogate to optimize treatment decision-making. This approach may have broader applications across various treatment settings and cancer types.
Similar content being viewed by others

Introduction
Breast cancer treatment currently faces many challenges. Approximately 50% of patients receiving neoadjuvant therapy do not achieve pathological complete response (pCR)1,2, and approximately 21.4% patients will experience local recurrence and 38.2% will experience distant recurrence within 15 years3. However, current methods for predicting the efficacy of neoadjuvant therapy in patients with breast cancer are inadequate. Previous models based on genomics4, transcriptomics5, radiomics6, pathology7, or even multiomics8 have been developed based on small sample sizes and cohorts with simplex treatment strategy. However, diverse treatment options, which are critical for cancer response, were not integrated into these models. Although these models can achieve high predictive performance in the treatment scenarios similar to the ones with which the models were built, they cannot be generalized to more complex real-world scenarios well and directly guide treatment regimen selection for patients. Unsurprisingly, physicians continue to select neoadjuvant treatment regimens only using empirical clinical factors such as molecular subtype and tumor stage.
Theoretically, accurately predicting the efficacy of cancer treatment requires addressing two aspects. First, a more comprehensive characterization of the tumor hallmarks is essential to better assess its functional status9. This task requires incorporating as many omics data types as possible, including transcriptomic, genomic, proteomic, and pathological information, to achieve a more comprehensive characterization of tumors and improve the predictive performance of models8,10. However, as the number of features increases, the sample size required for training also grows significantly11. Currently, aside from transcriptomic data, there is limited availability of patients with biologically multi-omics information, and existing large sample-based models are primarily based on transcriptomic data12. Therefore, a comprehensive characterization of transcriptomic status is crucial. Deep learning methods allow for the use of as many genes as possible to optimally characterize the tumor’s state due to the flexibility of neural networks, capturing not only tumor heterogeneity13 but also the tumor microenvironment14,15 information. However, deep learning methods require a large sample size, and their results are more difficult to interpret. Integrating prior biological knowledge can facilitate the construction of sparse, high-performance, and interpretable deep learning models16,17.
The second aspect involves appropriately characterizing drugs to accurately simulate the perturbations in tumors induced by treatments, which is essential for predicting the tumor’s response to treatment. To develop a model that can better guide clinical drug decision-making in precision medicine, incorporating as many diverse treatment types as possible is necessary to learn complex drug‒tumor interaction patterns. To incorporate therapeutic drugs into the model, three important factors need to be considered: (1) The types of anticancer drugs used in clinical practice are relatively limited (fewer than one hundred) and structurally diverse (e.g., small molecules, antibodies). Given the insufficient number of breast cancer therapeutic drug types to learn compound structural characterizations and the difficulty of integrating compound structural data with transcriptomic states, previous strategies that directly characterize compounds on the basis of their structure18 are not suitable for our task. (2) Many chemotherapeutic drugs lack specific targets, and the mechanisms of action for targeted therapies often extend beyond the primary target (e.g., antibody-dependent cell-mediated cytotoxicity related genes in anti-HER2 therapies). Thus, previous characterization of drugs based on specific gene targets cannot fully represent all types of drugs or comprehensively characterize their mechanisms19,20,21. (3) Clinically, drugs are often used in combination, leading to complex synergistic or antagonistic interactions that require flexible model structures (such as deep learning models) to simulate these interactions. Therefore, existing frameworks based solely on late fusion ensembles cannot achieve optimal efficacy predictions22,23. In general, A unified, genome-wide approach that can be applied to characterize different types of drugs and account for the complex interactions in combination therapies is needed.
In this work, we assemble a multi-dataset cohort of breast cancer transcriptomic data paired with diverse treatment records. Through systematic exploration of drug and transcriptome integration strategies to simulate drug-tumor interactions, we develop GDnet, a deep learning model for predicting neoadjuvant treatment response. GDnet demonstrates robust performance across various treatment regimens and breast cancer subtypes, serving as a digital drug-testing surrogate to optimize clinical treatment decision-making. This study will significantly advance precision neoadjuvant treatment selection and improve treatment efficacy in breast cancer.
Results
Schematic of GDnet workflows
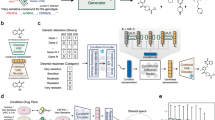
To sufficiently fuse drugs with transcriptomic data, mapping drugs as a series of gene–value pairs is one of the most suitable approaches. The selection of gene–value pairs is the key consideration. The GDSC and CTRP datasets, which contain the transcriptome status of more than 800 cell lines of various cancer types (pan-cancer) and their drug sensitivities to more than 500 molecular drugs, are great reference datasets. The heterogeneous responses of different pan-cancer cell lines in GDSC or CTRP to drugs can be attributed to the heterogeneity of tumor transcriptome states; thus, genes that are significantly correlated with drug sensitivity are more likely to be response- or resistance-related genes and therefore more likely to be genes that are perturbed when tumors are exposed to a specific drug. Therefore, we used the correlation of genome-wide expression with each drug’s half maximal inhibitory concentration (IC50) value (where a lower value indicates greater sensitivity) across different cell lines as the gene–value pairs to represent each drug (Fig. 1a). Notably, antibody or peptide drugs such as anti-HER2 drugs are not included in these drug sensitivity datasets, and IC50 values are not traditionally applied to measure their sensitivity due to complex mechanisms of action24,25,26. Given that the sensitivity of these targeted drugs is strongly positively correlated with targeted gene expression, we used the negative value of the genome-wide expression correlation with drug target expression as the gene–value pairs to represent antibody drug (Fig. 1a). For regimens with multiple drugs, a gene-wise summation of each drug representation in the regimen was performed (Fig. 1b).

a Strategy for representing drugs via gene correlations. Small molecules are represented as significant genome-wide correlations with half maximal inhibitory concentration (IC50) values, while antibody drugs are represented by significant genome-wide correlations with target genes. b Framework of GDnet, a biologically informed deep learning model that fuses transcriptome and regimen representations. The transcriptome data are rank-normalized, and drug representations in a regimen are summed in a gene-wise manner for input into the neural network. Binary masks, containing information on affiliations among genes and (or) pathways, are incorporated into the network to impose constraints on nodes and edges. Created in BioRender. Ge, H. (2025) https://BioRender.com/hybjk0u. Pan-cancer: various cancer types. IC50: half maximal inhibitory concentration. \({\mbox{Gcd}}\): gene correlation with drug sensitivity. \({\mbox{Ge}}\): gene expression.
To better simulate the perturbations in tumors induced by treatments, we input rank-normalized transcriptome and regimen representations in parallel into the gene level layer of the subsequent neural network, which we refer to as GDnet (Gene-Drug net) (Fig. 1b). We further explore three additional model frameworks: (1) GAnet (Gene-Attention net), which integrate drugs and transcriptome utilizing a method imitating key-value attention mechanism (Supplementary Fig. 1a); (2) Gnetens (Gene net ensemble), which employs an ensemble learning method to aggregate predictions across all drugs administered to a patient (Supplementary Fig. 1b); and (3) Gnet (Gene net), which relies solely on transcriptome information (Supplementary Fig. 1c). Next, the gene layer is fed into the biologically informed neural network proposed by Elmarakeby et al.16, which sets nodes, layers, and connections by incorporating the affiliation (child‒parent) relationships between genes, fine pathways, and more complex pathways on the basis of the Reactome biological knowledge dataset (version 2022) (Fig. 1b). In other words, the flow of information from the input to hidden layers are guided by prior biological knowledge. The finally built prediction model is designed to precisely predict the response of breast cancer patients to neoadjuvant therapy, aiding in the selection of regimens among multiple candidates.
Drug representations based on the GDSC and CTRP datasets
Applying the drug representation strategies described in Fig. 1a, we successfully generated the gene-correlation pairs for all drugs used in the included breast cancer neoadjuvant therapy datasets. The representations of all drugs were aggregated (Fig. 2 and Supplementary Data 1). Methotrexate has the largest number of represented gene-value pairs with 5378 pairs in total. Among 22 drug representations, 17 have larger than 1000 gene-value pairs, 5 (ANG, lapatinib, carboplatin, epothilone B, and doxorubicin) have less than 1000 gene-value pairs. Consensus clustering of drugs clustered these 5 drugs with fewer number of pairs together (Fig. 2). For drugs with larger number of pairs, three main clusters were generated, the first cluster contains anti angiogenic therapies, anti HER2 therapies, and anti IGF1R therapies, the second cluster contains chemotherapies, AKT inhibitors, and PARP inhibitors, and the last cluster contains anti PD-1 therapies and HSP90 inhibitors (Fig. 2). Then the gene-correlation pairs of drug representations and gene-expression pairs of the tumor transcriptome were fed into the gene layer of GDnet network framework described in Fig. 1b.

Rows and columns are clustered using consensus clustering, drugs are annotated by type, the numbers of gene correlation pairs are annotated by color and shown in the bracket after drug name. Source data are provided as a Source Data file. PDCD1 represents anti-PD-1/PD-L1 therapy (pembrolizumab and durvalumab). ANG represents an ANG peptide inhibitor (AMG-386). VEGFA represents an anti-VEGFA antibody (bevacizumab). IGF1R represents anti-IGF1R antibody (ganitumab). Capecitabine is represented by 5-fluorouracil. Ganetespib is represented by luminespib. Ixabepilone is represented by epothilone B.
GDnet showed superiority in predicting the outcome of neoadjuvant therapy
We collected 9550 breast cancer samples from 68 independent public datasets. After removing duplicates and filtering out samples with non-pretreatment status or missing key information (outcome and treatment), 31 datasets with 4371 samples and 16,133 genes were filtered out (Supplementary Fig. 2a). All breast cancer subtypes in populations from Northern America, Europe, and East Aisa were included (Supplementary Fig. 3a, b). Seventeen datasets containing the largest number of cases, totaling 3459 samples, were used to train the model, the remaining 14 datasets with the least number of cases, totaling 912 samples, were employed as external test sets, and 2 datasets with 134 patients published after February 15, 2024 were employed as prospective test sets (Supplementary Data 2).
For benchmark comparison, we trained the biologically informed models GDnet, GAnet, Gnetens, and Gnet (informed group), where Gnet was the baseline model with only transcriptome data; the dense neural network (dnet) variants (without biologically informed knowledge designed connections) of GDnet, GAnet, Gnetens, and Gnet, named GDdnet, GAdnet, Gdnetens, and Gdnet (dense group); dense deep neural network variants with hyperparameter optimized nodes and layers (without designed nodes, layers, and connections), named GDdnet-o, GAdnet-o, Gdnetens-o, and Gdnet-o (optimized group); deep learning models using previously applied drug representation strategies with optimized hyperparameters, named GSnet (Gene-SMILES net) and GTnet (Gene Target net) (previous group); and machine learning variants of GDnet with optimized hyperparameters (machine learning group). GDdnet, Gnet, GTnet, GSnet, and the drug-only model Dnet (Drug net) were used for ablation study for GDnet. Tenfold cross-validation was applied for hyperparameter tuning and internal validation using the training set. The results revealed that models with multiple optimized hyperparameters had higher AUROC values in tenfold cross validation than informed and dense group models (Supplementary Fig. 4).
The deep learning models were then retrained with the whole training set 10 times and tested with the external test set, facilitating a robust comparison across different models. In the external test, GDnet significantly performed better (median AUROC = 0.712, median AUPRC = 0.691) than all the other models did (e.g., for the transcriptome-only model Gnet, the median AUROC = 0.676; median AUPRC = 0.632) (Fig. 3a; Supplementary Fig. 5). Notably, GDdnet and GDdnet-o, which have the largest number of parameters (168,977,476 and 200,048,640), exhibited worse performance (AUROC = 0.678, AUPRC = 0.661) than GDnet, which has only 37,127 parameters, demonstrating the importance of prior knowledge in guiding training and increasing performance. The ablation study further showed that deleting either component of GDnet decreased performance (Supplementary Fig. 6).

a The area under the ROC curve (AUROC) of GDnet and other models, evaluated through retraining and testing 10 times on the test set. GDnet significantly outperforms the other models on median. The data are presented in boxplots, where the middle line is the median, the lower and upper hinges correspond to the 1st and 3rd quartiles, respectively and the whiskers correspond to the minimum or maximum values no further than 1.5 × IQR from the hinge (where the IQR is the interquartile range). Any data beyond the whiskers are considered outliers. AUROC comparisons were assessed using two-sided Wilcoxon rank-sum tests based on n = 10 values generated from 10 independent retraining cycles, without multiple testing correction. b Bar plot showing the AUROC of the 10 repetitions ensembled GDnet, ensembled other deep learning models, and machine learning models in the testing set. The bar height denotes the AUROC value in the test set, and the error bars represent the 95% confidence interval derived from 1000 bootstraps. The sample sizes in each bar are thus all equal to n = 1000 bootstraps (biological replicates) from the test set of 912 patients. c ROC curve comparing the ensembled GDnet and Gnet models in the test set (n = 912). Statistical comparison was performed using two-sided DeLong’s test. d Bar plot showing the AUROC of the ensembled GDnet and Gnet in each breast cancer subtype in the test set. The bar height denotes the AUROC value, and the error bars represent the 95% confidence interval derived from n = 1000 bootstrap replicates. Bootstrapping was performed across all molecular subtypes: ER + HER2+ (n = 121), ER- HER2+ (n = 112), ER + HER2- (n = 247), TNBC (n = 197), and unsure status (n = 235) patients. e Bar plot showing the AUROC of the 10 repetitions ensembled GDnet, ensembled other deep learning models, and machine learning models in the prospective test set. The bar height denotes the AUROC value in the prospective test set, and the error bars represent the 95% confidence interval derived from n = 1000 bootstrap replicates of the 134-patient prospective test set. f ROC curve comparing the ensembled GDnet and Gnet models in the prospective test set (n = 134). Statistical comparison was performed using two-sided DeLong’s test. Source data are provided as a Source Data file. ROC: receiver operating characteristic. TNBC: Triple negative breast cancer.
To generate the final robust deep learning model for future application, we performed ensemble learning by averaging the predictions from the 10-times retrained models for the final prediction. Similarly, GDnet (AUROC = 0.725) achieved the highest AUROC (Fig. 3b), and its ROC curve significantly differed from that of transcriptome model Gnet (AUROC = 0.683) (Delong’s Z = 2.734, P = 0.006, ΔAUROC = 0.042, 95% CI [0.012, 0.073]) (Fig. 3c). The AUPRC results were similar to the AUROC results (Supplementary Fig. 7a). GDnet also showed generally high performance in terms of accuracy, F1-score, precision, recall, and specificity when cutoff value was set as 0.5 (Supplementary Fig. 7d–f). Moreover, GDnet demonstrated robust performance across all breast cancer subtypes (Fig. 3d; Supplementary Fig. 8) and outperformed Gnet in most cases. The only exception was TNBC, where GDnet showed slightly worse performance (Fig. 3d; Supplementary Fig. 8). We then tested the performance of GDnet in 13 individual datasets in the test datasets via the 10-times repetitions ensemble models to determine whether GDnet can perform well in limited regimen settings compared with Gnet. The GSE191127 dataset was not included in individual testing because it contains only residual disease (RD) patients. The results showed that GDnet outperformed Gnet, achieving higher AUROCs in 10 out of 13 datasets, while exhibiting only marginally lower AUROCs (difference < 0.02) in the remaining three datasets (Supplementary Fig. 9a). Similar results were observed for the AUPRC values (Supplementary Fig. 9b). The comparison among all models is displayed in Supplementary Fig. 10. These results suggested that GDnet successfully captures the patterns reflecting how treatments perturbate tumor transcriptomes rather than simply overfitting the drug distribution bias among datasets. To further support the reliability of the model, we tested the model performance in the prospective test sets. The comparison results also showed that GDnet had higher performance than most other models (Fig. 3e; Supplementary Fig. 11), with a significantly better ROC curve than Gnet (Delong’s Z = 2.76, P = 0.006, ΔAUROC = 0.059, 95% CI [0.017, 0.1]) (Fig. 3f).
GDnet can optimize the regimen selection for better outcomes
To assess the ability of GDnet to guide regimen selection for clinical use, we then conducted in-silico clinical trials to inspect if GDnet can improve breast cancer neoadjuvant treatment outcome (Fig. 4a). Given the consideration of multiple potential regimens for a patient, we input tumor transcriptome data and drug representation of each regimen into GDnet to computationally predict the pathological complete response (pCR) probability for each regimen. We selected the best regimens with the top N% highest predicted pCR probabilities, which are likely to benefit the patient, and the worst regimens with the top N% lowest predicted pCR probabilities, which are not suitable for patients. If the regimen actually used by the patient was included among the best regimens, we assigned the patient to the optimized group, and if it was included among the worst regimens, we assigned the patient to the control group. To inspect the baseline characteristics of the two groups, we also calculated the predictive score via the transcriptome-only model to evaluate the degree of tumor malignancy between them, and Gnet is priorly considered because its similar structure with GDnet and its more robust performance than GNNet regardless of AUROC (Levene’s W(1, 18) = 5.9, P = 0.026) or AUPRC (Levene’s W(1, 18) = 6.9, P = 0.017) (Fig. 3a, b).

a Schematic of the in-silico clinical trial. Given a patient with multiple optional treatment regimens, the transcriptome data and representations of optional regimens were input into GDnet to predict the pCR probability for each regimen. Regimens with the top N% highest and those with the top N% lowest pCR probabilities were selected as best regimens and worst regimens separately. If the patient’s chosen regimen (indicated with a white star “*” in the plot) was included among the best regimens, then the patient is assigned to the optimized group; if the chosen regimen was included among the worst regimens, then the patient was assigned to the control group. N represents the optimization threshold of the simulated trial. Created in BioRender. Ge, H. (2025) https://BioRender.com/vtl91ag. b The pCR rate in the optimized and control groups at different optimized threshold levels of I-SPY2 trial-based simulation trials after Propensity Score Matching (PSM). The difference in the pCR rate was tested via two-sided Fisher’s exact test without multiple testing correction, with the P value labeled on the green solid line of the optimized group. c The odds ratio for pCR in optimized group compared to control group of I-SPY2 based simulation trials after PSM. The dashed lines are the fitted linear regression function between the y-axis and optimized threshold. The coefficient of determination (R²) was calculated to assess goodness-of-fit, and the statistical significance was determined by an F-test comparing the fitted model to an intercept-only model. d The pCR rate in the optimized and control groups at different optimized threshold levels of the external test dataset-based simulation trials after PSM. The difference in the pCR rate was tested via two-sided Fisher’s exact test without multiple testing correction, with the P value labeled on the green solid line of the optimized group. e The odds ratio for pCR in the optimized group compared to that in the control group of the external test dataset-based simulation trials after PSM. R² was calculated to assess goodness-of-fit, and the statistical significance was determined by a one-sided F-test comparing the fitted model to an intercept-only model. Source data are provided as a Source Data file. Opt: optimized group. Ctl: control group.
We first conducted a series of in-silico trials based on the I-SPY2 trial, one of the datasets used to train our model. A total of 653 patients who received one of 11 regimens in I-SPY2 were enrolled in our simulation study, where these regimens were set as optional choices. We set varying optimization thresholds, starting at 50% and decreasing in 5% increments (e.g., 50%, 45%, 40%, 35%) until either group size dropped below 30 (Supplementary Fig. 12a), to categorize the best and worst regimens, thereby ensuring the robustness of the results. Lower thresholds indicate increased optimization stringency. The results revealed that the pCR rates (Supplementary Fig. 12c) were significantly greater in the optimized group than in the control group in all optimize thresholds. Moreover, the odds ratio (OR) for pCR increased linearly (r(6) = −0.849, P = 0.008, 95% CI [−0.972, −0.358]) as the optimization intensifies (optimize threshold decrease) (Supplementary Fig. 12d). However, a higher Gnet prediction score was observed in the optimized group (Supplementary Fig. 12b), suggesting that tumors in this group were less malignant. This difference may bias the effect of our model. Thus, we performed Propensity Score Matching (PSM) to balance transcriptome profile based on Gnet prediction score (Supplementary Fig. 12e, Supplementary Fig. 12f). Similarly, pCR rates were significantly higher in the optimized group than in the control group (Fig. 4b), with OR increased linearly (r(6) = −0.932, P < 0.001, 95% CI [−0.988, −0.661]) as the optimization intensifies (Fig. 4c), suggesting that GDnet has the ability to optimize treatment regimen choices.
We further performed a series of simulation trials with the external test datasets. Given the limited variety of regimens in each individual test dataset, we combined all available test datasets together, yielding 912 patients and 19 regimens. Similar to what was observed in the above I-SPY2 simulation, the results for the external test set showed that the pCR rates (Supplementary Fig. 13a, Supplementary Fig. 13c) were significantly greater in the optimized group than in the control group in all optimize thresholds, with OR increased linearly (r(6) = −0.824, P = 0.003, 95% CI [−0.957, −0.404]) as the optimization intensifies (Supplementary Fig. 13d), even a lower Gnet prediction score was observed in the optimized group (Supplementary Fig. 13b). PSM analysis based on Gnet prediction score (Supplementary Fig. 13e and Supplementary Fig. 13f) also revealed that pCR rates were significantly higher in the optimized group than in the control group (Fig. 4d), with OR increased linearly (by PSM, r(6) = −0.971, P < 0.001, 95% CI [−0.993, −0.879]) as the optimization intensifies (Fig. 4e), further indicating the excellent ability of GDnet to guide neoadjuvant treatment selection for breast cancer.
Interpretation of GDnet reveals important regimens and genes involved in the treatment response
We then sought to investigate which treatment types contribute to response importantly. With respect to the treatment optimization strategy shown in Fig. 4a, we further inspected the distribution of regimens and drugs used in each group in PSM analysis at different optimization thresholds; the differences in the proportions of regimens and drugs in different groups can reveal those with high contributions to the model. The results revealed that the paclitaxel|doxorubicin|cyclophosphamide|olaparib|PDCD1 and paclitaxel|doxorubicin|cyclophosphamide|ERBB2 | ERBB2 regimens were used much more frequently in the optimized group than in the control group, with an increasing tendency as the optimization intensifies (Fig. 5a). Docetaxel|cyclophosphamide, doxorubicin, and 5-fluorouracil|epirubicin|cyclophosphamide|lapatinib were regimens that were mostly or even exclusively used in the control group, with an increasing tendency as the optimization intensifies (Fig. 5a). Specifically, drugs such as PDCD1 (PD-1 inhibitors), olaparib, and ERBB2 (anti-HER2 antibodies) may play important roles (Supplementary Fig. 14) in improving the pCR rate.

a Proportion of regimens used in the optimized (middle) and control (right) groups and the difference in proportion between the two groups (left) with the external test set-based simulation trials at different optimization thresholds. The optimization thresholds are color-coded. b Visualization of GDnet’s inner layers showing the estimated relative importance of different nodes in each layer based on the dual anti-HER2 regimen in the test set. The nodes in the far left layer represent input types, the nodes in the first hidden layer represent genes, the next layers represent higher-level biological entities, and the final layer represents the model outcome. Nodes with greater lengths are more important. The contribution of a certain source node to the target node, such as each input type (drug or transcriptome) to the importance of each gene, each gene to each pathway, and each pathway to each broader pathway, is depicted via a Sankey diagram, e.g., the importance of the PSMC7 gene is driven mainly by the transcriptome. Source data are provided as a Source Data file. Opt: optimized group. Ctl: control group.
To understand the important genes and pathways that influence the response to the promising regimens, paclitaxel|doxorubicin|cyclophosphamide|olaparib|PDCD1 (indicated as the immunotherapy regimen below) and paclitaxel|doxorubicin|cyclophosphamide|ERBB2 | ERBB2 (indicated as the dual anti-HER2 regimen below), in the optimized group, we visualized the layers of GDnet based on the patients who applying the corresponding regimen in the external test dataset (Fig. 5b and Supplementary Fig. 15). An integrated gradient attribution method27 was applied to obtain the importance score of each node in each layer for each patient in the patients set. The mean importance score across all patients was used as the final score for each node, which was displayed as the node length in the Sankey diagram. For the dual anti-HER2 regimen (Fig. 5b), high-scoring genes included PSMB3 (reported to be co-expressed with ERBB2 in breast cancer28), PSMB7 (reported associated with anthracycline resistance in breast cancer29), PIK3CA (reported associated with anti-HER2 treatment resistance in breast cancer30,31), YWHAH (reported associated with cell migration in breast cancer32), and SRC (reported associated with adhesion, and cell migration in breast cancer33). Pathways at various levels, such as ER-Phagosome pathway in hidden layer 2, which is reportedly linked with innate immune system and phagocytosis34, RHO GTPasc cycle in hidden layer 3, which is reportedly linked with tumorigenesis, invasion, and metastasis35, and immune system in hidden layer 6, were identified, highlighting their critical role in anti-HER2 treatment. For the immunotherapy regimen (Supplementary Fig. 15), high-scoring genes included CCND1 (reported to be linked to enhanced invasiveness, a poorer prognosis, and resistance to drug therapies in ER-positive breast cancer36), SRC, JUN (reported to promote breast cancer progression via the AKT signalling pathway37), TRIO (reported to facilitate breast cancer cell migration38), and DUSP6 (reported to be associated with poor survival in HER2 positive breast cancer patients39). Collectively, we identified multiple key genes implicated in the response or resistance mechanisms to specific treatment regimens.
Discussion
In summary, GDnet successfully learned how treatments perturbate tumors, achieving significantly higher performance to predict breast cancer neoadjuvant treatment outcome than transcriptome-only model. Application of GDnet to two series of simulation clinical trials demonstrated GDnet can function as a digital drug-testing surrogate to optimize the treatment selection of breast cancer patients. Compared with previous models, GDnet has the following 4 advantages. First, most previous machine learning models face issues such as unstable performance due to limited sample sizes and the lack of a sufficient independent external test set4,5,8,40,41. However, we collected a multi-source cohort of 4371 breast cancer samples from 31 datasets, all with paired pretreatment transcriptome and neoadjuvant treatment information, to train and validate GDnet robustly.
Second, we collected data from all types of transcriptomics technologies by applying the rank normalization strategy for each sample. This approach provides robustness against technical artifacts that could otherwise introduce systematic bias in the absolute transcript counts, while maintaining the overall relative ranking of genes within each cell at a more stable level42. Although rank-based encoding has limitations (for example, it does not fully exploit the precise gene expression measurements provided in transcript counts), it makes the model more generalizable to real-world complex conditions. Previous studies have been limited to limited transcriptomics technologies because of the ease of normalization and reducing batch effects; however, these approaches tend to reduce the available sample size and the diversity of datasets, making it difficult to generalize the results to broad scenarios5,43. In addition, unlike previous studies that performed normalization among patients (such as z score transformation) and even among datasets (such as the Combat algorithm) in multiple cohorts43, our method contains only normalization within each patient. This design choice stems from the consideration that the trained normalization parameters across patients or datasets cannot be applied effectively to patients in another dataset with diverse protocol of transcriptomics test, thus resulting in less generalizability. As a result, our model can be directly generalized to new cases tested with any transcriptomics technologies in broader scenarios and complex real-world conditions.
Third, unlike previous models, such as OncotypeDX score, MammaPrint, and Adjutorium5,8,40,44,45,46, which are mainly based on traditional statistical models or machine learning models. GDnet utilizes a deep learning framework that includes a much larger number of genes to maximize the model capacity. Furthermore, by integrating prior biological knowledge, GDnet markedly reduces the number of parameters for learning (parameter number 37,127), leading to better stability and performance than dense models such as GDdnet (parameter number 168,977,476) and GDdnet-o (parameter number 200,052,736), as also demonstrated in previous studies16,17,20,23. Visualization of the importance and attribution of the multilevel genes and biological pathways in GDnet enabled a multilevel view of model interpretation, which may guide researchers in developing hypotheses regarding the underlying biological processes involved in resistance and translate these discoveries into new therapeutic opportunities. Interestingly, the informed GAnet, Gnet, and Gnetens models exhibited generally worse performance than dense GAdnet, Gdnet, and Gdnetens models but generally better performance than optimized GAdnet-o, Gdnet-o, and Gdnetens-o models. By comparing the number of parameters, we found that the informed models include the smallest number of parameters (GAnet-20,373, Gnet-20,373, and Gnetens-101,865), dense models include an intermediate number of parameters (GAdnet-7,301,376, Gdnet-7,301,376, and Gdnetens-36,506,880), and optimized dense models include the highest number of parameters (GAdnet-o-18,270,464, Gdnet-o-18,270,464, and Gdnetens-o-91,352,320). Some possible explanations are as follows: The prior knowledge in this study was derived from biological knowledge database, yet current research on genes, pathways, functions, etc., remains largely incomplete. Thus, existing biological knowledge databases do not fully reflect all actual biological behaviors. Assuming a fixed sample size, in cases with an extremely large number of parameters, prior knowledge represents a guide for the identification of the correct and generalizable learning direction for deep learning models under the limited sample size, thereby achieving better external test performance compared to models not incorporating prior knowledge. However, when the number of parameters becomes smaller (while still sufficient for learning), deep learning can accurately reveal more potential actual biological functions of breast cancer beyond the existing prior knowledge under the same sample size, leading to high external test performance. Thus, there is a trade-off to incorporating prior knowledge. In the scenario of GAnet, Gnet, and Gnetens, the incomplete prior knowledge instead becomes a constraint in the training of deep learning models. Specifically, prior knowledge integration improved GAnet, Gnet, and Gnetens performance over parameter-heavy optimized models, yet these still underperformed relative to the relatively parameter-efficient dense models. In other words, dense models and optimized models represent the opposite sides of the balanced benefit-drawback point of incorporating prior knowledge (informed models) for GAnet, Gnet, and Gnetens. This phenomenon highlights the importance of selecting appropriate scenarios for applying prior knowledge. When the parameter count is large and the sample size is small, prior knowledge can significantly aid deep learning model training. However, this advantage gradually diminishes as the number of parameters decreases or the sample size increases.
Finally, we proposed an innovative method for drug representation using gene-correlation pairs. This approach aids in the fusion with tumor transcriptome, thus simulating the perturbations in tumors induced by treatments. By applying this approach, GDnet not only achieved higher performance than the transcriptome model but also enabled the comparison of all optional treatments regimens before initiating therapy, which cannot be properly achieved by any of the previous models. In other words, we established a model that has the function of drug-testing surrogate and can test the sensitivities of various drugs and regimens, revealing the optimal regimen47. This idea has the potential to change related treatment guidelines. There are many potential scenarios in which GDnet can play an important role: (1) In real-world clinical practice, for a breast cancer patient scheduled for neoadjuvant therapy, we can first list all eligible treatment options based on guidelines and clinical trial evidence. The patient’s tumor transcriptome data and treatment options can then be input into GDnet to predict the probability of achieving a pCR. The regimen with the highest pCR probability can then be recommended as the optimal treatment choice for the patient. The efficacy of this treatment selection strategy is demonstrated by in-silico simulation trials based on I-SPY2 and external test dataset, showing significantly improved pCR rate compared to the control group in our study. (2) In clinical trial design, GDnet can assist in optimizing treatment strategies. Specifically, given a set of candidate monotherapies or combination regimens, GDnet can simulate and predict the pCR probabilities for each regimen option in patient subpopulations that meet the inclusion criteria. The regimen demonstrating the highest overall pCR probability across the patient subpopulations would be the most promising candidate for clinical trial. Even for novel drug combinations not included in GDnet’s training set, the model can still guide trial strategy selection. This is evidenced by GDnet’s strong performance in the external test set, where it successfully predicted the efficacy of drugs that were not included in model training (e.g., durvalumab, bevacizumab, cisplatin, olaparib, and gemcitabine). (3) Beyond trial design, GDnet can further optimize patient selection for clinical trials. By estimating individual patients’ likelihood of responding to a trial regimen, GDnet can identify the subgroup most likely to benefit, enabling selective enrollment and thereby improving trial success rates. Notably, when implementing GDnet in clinical practice, several ethical considerations warrant attention. First and foremost, patient data privacy must be safeguarded; we recommend storing all data and deploying the model exclusively within hospital firewalls to mitigate breach risks. While clinicians should monitor for potential outcome disparities across diverse populations, our model was trained on a multi-ethnic cohort (Americas, Europe, and Asia), ensuring generalizability to diverse populations. Crucially, unlike genomics, where SNP polymorphisms vary by ancestry, transcriptomics capture biological functional states, rendering our model robust to ethnic differences.
There are several limitations to our study. First, regimen-related factors such as drug initiation dose, sequence, and interval are not considered in GDnet, as these factors are more related to the patient’s tolerance level and typically do not have a decisive impact on treatment outcomes. Second, transcriptome cannot fully characterize the complex status of tumor and its microenvironment, high-resolution multi-omics testing methods48,49,50 are required for better tumor representation in the future. Third, evaluating whether our model can guide and optimize trial practices or routine clinical care warrants prospective randomized clinical trials to determine if GDnet-optimized regimens offer greater benefits than those selected by clinicians. Fourth, the predictive performance of GDnet across different patient populations warrants further investigation. While this study included patients from Northern America, Europe, and Aisa, and preliminarily demonstrates GDnet’s robust performance across various breast cancer molecular subtypes, other potential bias factors including age, menopausal status, tumor stage and tumor grade remain to be validated in future studies.
The GDnet architecture and digital drug-testing surrogate proposed in this study can be extended to a broader range of diseases and therapeutic scenarios. While this study primarily focuses on the neoadjuvant treatment setting for breast cancer, the same framework can be adapted to other treatment stages (e.g., adjuvant therapy, advanced-stage therapy) and other cancer types (e.g., lung cancer, pancreatic cancer, prostate cancer) by constructing corresponding digital drug-testing surrogate models based on their respective efficacy evaluation systems. Moreover, the framework can also be tailored to non-oncological diseases, such as diabetes and psychiatric disorders, by developing disease-specific drug representation methods that align with their unique omics profiles and building corresponding models for treatment selection.
In conclusion, GDnet, a biologically informed deep learning approach that fuses drug representations and transcriptomes enables more precise cancer treatment decision-making for breast cancer neoadjuvant therapy. Integrating drug representation and biological omics data represents a promising approach to simulate the perturbations in tumors induced by treatments, providing digital drug-testing surrogate platforms for precision oncology that may be broadly applicable across different treatment settings and diseases.
Methods
Publicly available data collection and processing
Transcriptome data related to breast cancer neoadjuvant therapy were systematically retrieved from PubMed and the Gene Expression Omnibus (GEO) database on 2024.2.15. The search terms used were (“Neoadjuvant Therapy”[MeSH Terms] OR neoadjuvant) AND (“Breast Neoplasms”[MeSH Terms] OR ((breast OR mammary) AND (cancer* OR tumor* OR tumour* OR carcinoma* OR neoplasm* OR oncology OR malignanc*))). “*” is a wildcard, which is used to match any sequence of zero or more characters. For example, tumor* can match tumor and tumors. High-throughput sequencing such as next-generation sequencing (NGS) and microarrays were included, and only datasets containing no less than 20 samples were selected. In total, 9550 breast cancer samples from 68 independent public datasets were accessed. We obtained transcriptomic expression data in both raw and processed forms, encompassing NGS-derived count matrices or normalized (such as FPKM, TPM, or RPKM) value matrices and microarray raw intensities or normalized (such as RMA) values. Probe sets of microarrays are annotated for genes referring to the reference table provided in GEO. For quality control of genes and samples, 16,133 genes occurred in larger than 50% samples and 4580 samples with less than 50% missing genes were retained. Among these datasets, 4371 patients from 31 datasets (GSE194040, GSE25066, GSE16716, GSE41998, GSE180962, GSE20271, GSE34138, GSE50948, GSE149322, GSE22226, GSE32603, GSE22358, GSE32646, GSE16446, GSE130788, GSE231629, GSE123845, GSE4779, GSE173839, GSE22093, GSE21997, GSE42822, GSE66399, GSE181574, GSE23988, GSE41656, GSE8465, GSE122630, GSE21974, GSE207248, GSE191127), covering data on complete pCR, treatment regimens, and sufficient transcriptome information were used for model construction, validation, and test.
Moreover, we collected the breast cancer neoadjuvant therapy transcriptome datasets obtained after February 15, 2024, as prospective test. A total of 134 patients out of 195 patients from two independent datasets (GSE240671, GSE260693) were included.
Rank value encoding of transcriptomes
RNA-seq count or patient-level normalization data (e.g., transcripts per kilobase million (TPM) or fragments per kilobase million (FPKM)), microarray patient-level normalized data (e.g., robust multichip average (RMA) or other method) from GEO were collected. A greater upstream level is maintained if multilevel data are accessed. To facilitate translation among gene expression testing technical platforms, we applied a rank transformation (rank value encoding) to normalize the data. For this purpose, gene expression values were transformed from microarray intensities or RNA-seq counts or their normalization format to their respective ranks. This transformation was conducted in a genewise manner. Therefore, all gene expression values per gene were assigned a rank on the basis of their order from lowest to highest. The rankings were then scaled into quantiles, ranging from 0 (lowest gene expression) to 1 (highest gene expression). Missing values are filled with 0. This rank-based approach may be more robust against technical artifacts that may systematically bias the absolute transcript count value, whereas the overall relative ranking of genes within each cell remains more stable.
Treatment drug and regimen representation
To facilitate the fusion of drug treatments and transcriptomes, we aimed to represent drugs by combining related genes and correlation values. To build a sufficiently large pharmacogenomic dataset for drug representation, raw drug sensitivity data of cell lines to molecular drugs and RMA-normalized or TPM-normalized followed by log-transformed transcriptomes of the cell lines were retrieved from the Genomics of Drug Sensitivity in Cancer database v2 (GDSC) and the Cancer Therapeutics Response Portal v2 (CTRP)51,52,53,54,55,56. We used the IC50 metric, where a high IC50 values corresponds to low cell-killing ability and a low IC50 value corresponds to high cell-killing ability, to represent drug sensitivity. For each drug, we calculated the correlation between the gene expression levels of all the cell lines and the log2(IC50 + 1) of the response of the cell lines to each drug. Due to the fact that the normalized gene expression levels and log2(IC50 + 1) values showed a generally normal distribution, we applied the Pearson correlation method. The significance cutoff of correlation was set to 1*10−6 according to the Bonferroni correction to reduce the false discovery rate, considering nearly 5*104 genes to be tested. Genes identified as significant are more likely to play an important role in predicting the drug response of cancer treatment, and the correlation values are prior knowledge that can inform the importance degree of genes to drug response and thus can reflect the perturbation of drugs to genes. The correlated genes were removed if they were not included in the retained set of 16,133 genes in the patient transcriptome. Therefore, each drug was ultimately represented by its significant genes and correlation values (ranging from −1 to 1). If a drug appeared in both the GDSC and CTRP datasets, the GDSC dataset was considered first.
There are several special cases in drug representation. Some types of drugs are not found in the GSDC or CTRP datasets. Therefore, we chose the most relevant drug with similar mechanism and within the same anti-tumor drug type to represent it. Examples include capecitabine, which is represented by 5-fluorouracil; ganetespib, which is represented by luminespib; and ixabepilone, which is represented by epothilone B. Some drug descriptions in GEO datasets, such as taxane or anthracycline, lack sufficient drug details; therefore, we chose an averaged representation of the drugs within the taxane or anthracycline categories. Some types of drugs are antibody- or peptide-based drugs, which are not included in either dataset and are not commonly represented by the IC50 to measure drug sensitivity due to complex mechanisms of action24,25,26. For these drugs, we used the negative correlation of their target genes with other genes in cell lines because the response of these drugs is strongly associated with target expression, including PDCD1 (pembrolizumab and durvalumab), VEGFA (bevacizumab), and IGF1R (ganitumab). If multiple targets exist for a drug, we performed a genewise summation of the correlations among all the targets as the final representation, including ANG, which contains the ANGPT1 and ANGPT2 genes and represents an ANG peptide inhibitor (AMG-386). In addition, drugs such as T-DM1 (which is an antibody‒drug conjugate) and endocrine therapy have no corresponding drug sensitivity data in the GDSC or CTRP. Considering the small number of patients treated with these drugs in our datasets, patients treated with regimens including T-DM1 or endocrine therapy were excluded. The representations of all drugs are listed in Supplementary Data 1. For patients receiving multiple drugs in a regimen, we performed a genewise summation of the gene correlation representations of each drug to represent the regimen.
Fusion of drug representations and transcriptome
Three approaches to integrate regimens and transcriptome information are tested. The first approach involves the use of both gene expression (represented as \({\mbox{Ge}}\)) and gene-sensitivity correlation (represented as \({\mbox{Gcd}}\)) as inputs, which we refer to as GDnet (Gene Drug net) (Fig. 1b). The second approach uses \({\mbox{Gatt}}={\mbox{Ge}}\times \exp \left({\mbox{Gcd}}\right)\) as the final input, utilizing a key-value attention mechanism like approach (Supplementary Fig. 1a), and named this model GAnet (Gene-Attention net). In this mechanism, for each query (\({\mbox{Ge}}\)), we find the corresponding key (gene) in the drug representation, and the attention (\({\mbox{Gatt}}\)) is calculated as \({\mbox{attention}}={\mbox{query}}\times \exp \left({\mbox{value}}\right)\), where the value represents the \({\mbox{Gcd}}\) of the drug representation. The attention calculation is set to \(\exp \left({\mbox{value}}\right)\) to ensure that \({\mbox{Gatt}}\) can simulate the tumor’s response under drug exposure conditions; in other words, \({\mbox{Ge}}\) is expected to decrease or increase relative to the response (\({\mbox{Gcd}}\) < 0) or resistance (\({\mbox{Gcd}}\) > 0) gene of the drug when no drug is present. For example, ERBB2 is a poor-prognosis gene, but when exposed to an anti-HER2 drug with ERBB2 as the response gene (\({\mbox{Gcd}}\) < 0), the expression of ERBB2 is expected to decrease. In addition, we explored a third approach, which involves the use of an ensemble learning approach. This method focuses on building different transcriptome-only models for each drug and combining the predictions of all transcriptome-only models corresponding to patient-applied drugs by averaging them for the final prediction. This model is referred to as Gnetens (Gene net ensemble) (Supplementary Fig. 1b). In Gnetens, only drugs with available data for more than 10% of the patients were used to construct individual models. Five drug types (anthracycline, microtubule inhibitors, cyclophosphamide, pyrimidine analogs, and anti-HER2 antibodies) were chosen (Supplementary Fig. 3a). Transcriptome-only model named as Gnet (Gene net) is also explored (Supplementary Fig. 1c).
Design of the biologically informed model
We introduced prior knowledge into the training process, drawing inspiration from the P-NET framework built by Elmarakeby HA et al.16 to accelerate model training and enhance both performance and explainability. The complete Reactome dataset57 was downloaded and processed into a layered network. This network includes one layer for features, one layer for genes, and five layers for pathways. The affiliation relationships between nodes in adjacent layers (representing genes or pathways) are represented by six 2D binary matrixes. The constructed layered network originated from 8,377 genes (the first hidden layer) that are available both in our datasets and the Reactome dataset. Pathways that are directly related to these genes form the second layer (846 initial pathways). Subsequently, pathways directly related to those in the second layer constitute the third layer (218 superior pathways), and this layering process continues analogously to the fourth layer (108 pathways), the fifth layer (51 pathways), until the sixth layer with the top 18 pathways representing biological processes is built.
The deep learning models were implemented in PyTorch (Python version 3.12.4; torch version 2.4.0; cuda version 12.2), with the layers and nodes numbers from the prior knowledge network defining the structure of a base feedforward dense neural network model. The six 2D binary matrixes containing affiliation relationships between genes or pathways in adjacent layers were fed into the neural network model as the masks between adjacent layers. This mask is multiplied by weights after each epoch during weight updates, imposing constraints on nodes and edges. This configuration yields a sparse neural network with six layers, comprising 8377-gene nodes (hidden layer1), 846 (hidden layer2), 218 (hidden layer3), 108 (hidden layer4), 51 (hidden layer5), and 18 pathway nodes (hidden layer6) in each hidden layer. Specifically, for GDnet and GDdnet, the input layer before the 8377-gene layer comprised gene expression and drug representation data encompassing 19,300 nodes (half gene expression nodes and half drug representation nodes). L2 regulation was set to 0 since the model is sufficiently sparse. Each node encodes a biological entity (for example, a gene or a pathway), while each edge represents a known relationship between the corresponding entities. The node constraints allow for a better understanding of the state of different biological components. The edge constraints lead to a smaller number of parameters than those of fully connected networks with the same number of nodes and thus potentially fewer computations. Since the dataset is unbalanced, we weighted the classes differently to reduce the network bias toward one class on the basis of the bias in the training set. The model was trained 1000 epochs via the Adam optimizer with default parameters to reduce the binary cross-entropy loss functions for imbalance. To allow each layer to be useful by itself, we added a predictive layer with sigmoid activation after each hidden layer. Since it is more challenging to fit the data using a smaller number of weights in the later layers, we used higher loss weights for later layer outcomes (from the gene & drug layer to the later layers, the weights are 1, 1.2, 1.4, 1.6, 1.8, 2, and 2.5) during the optimization process. The final prediction of the network was calculated by averaging all layer outcomes.
Models applying drug representation strategies in previous studies
Using the drug representation strategy based on SMILES (Simplified Molecular-Input Line-Entry System) molecule structure expressions proposed by Kuenzi et al.18, we introduced GSnet (Gene-SMILES net). In this approach, drugs are hashed into 2048-dimensional vectors to represent molecular drugs. Antibody-based drug representations were not generated due to the lack of SMILES entries. Regimens were represented as the vector summation of involved drugs. The SMILES-based 2048-dimensional vectors and gene expression vectors were subsequently used as input for GSnet.
Using the drug representation strategy based on the targets of drugs proposed by Zhang et al.19, we introduced GTnet (Gene-Target net). Drug target information was obtained from the DrugBank database58. Chemotherapeutic agents with nonspecific targets (e.g., DNA, RNA) that cannot be mapped to specific genes were excluded from drug representation. This approach incorporated the following 10 drugs with clearly mappable gene targets: trastuzumab (ERBB2), pertuzumab (ERBB2), bevacizumab (VEGFA), AMG-386 (ANGPT1, ANGPT2), MK-2206 (AKT1), olaparib (PARP1, PARP2), durvalumab (PDCD1), pembrolizumab (PDCD1), ganitumab (IGF1R), luminespib (HSP90AA1). Regimens were represented as the vector summation of involved drugs. The target-based 10-dimensional vectors and gene expression vectors were subsequently used as input for GTnet.
Hyperparameters optimization
Hyperparameter optimization was performed using 10-fold cross-validation on the training set to maximize the AUROC. The selected hyperparameters and their tuning ranges are described below:
(1) Hyperparameter settings for the informed group models: The GDnet, GAnet, Gnet, and Gnetens models incorporate biological prior knowledge into their architecture. Since their neural network structures (including nodes, layers, and connections) were predefined by this prior knowledge ([8388, 846, 218, 108, 51, 18] for GDnet, and [846, 218, 108, 51, 18] for GAnet and Gnet), no further optimization was required for these parameters. Thus, only the learning rate was tuned with candidates [0.1, 0.01, 0.001, 0.0005, 0.0001, 0.00005, 0.00001]. A learning rate of 0.0005 had the highest mean AUROC across all informed models and thus was set as the optimized learning rate.
(2) Hyperparameter settings for the dense group models: For GDdnet, GAdnet, Gdnet, and Gdnetens, to ensure comparability with prior knowledge models, the same node and layer structures were retained, but connections were converted to fully connected. Thus, only the learning rate was tuned, and the candidate values were the same as described above. A learning rate of 0.0005 was selected as the optimized learning rate.
(3) Hyperparameter settings for the optimized group models and previous group models: The GDdnet-o, GAdnet-o, Gdnet-o, Gdnetens-o, GSnet, and GTnet models employ fully connected architectures, requiring the tuning of layers, nodes, and learning rate. Learning rate used the same candidate values as above. For hidden layers, we used a range of 3 to 6 layers. For nodes, we set nodes following geometric decay of around 2 or 4 fold. Considering GDdnet-o as an example, the candidate architecture combinations include: [[8192, 4096, 2048], [4096, 1024, 256], [8192, 4096, 2048, 1024], [4096, 1024, 256, 64], [8192, 4096, 2048, 1024, 512], [4096, 1024, 256, 64, 16], [8192, 2048, 1024, 256, 128], [4096, 2048, 512, 256, 64], [8192, 4096, 2048, 1024, 512, 256], [4096, 1024, 256, 64, 16, 4], [8192, 2048, 1024, 256, 128, 32], [4096, 2048, 512, 256, 64, 32]]. The optimized learning rate was 0.00001 for GDdnet-o, and 0.0001 for others. The optimized hidden layers and nodes were [8192, 4096, 2048] for GDdnet-o, and [2048, 512, 128] for others.
(4) Hyperparameter settings for machine learning group models: We applied 4 machine learning models including L2-regularized logistic regression, random forest, support vector machine (SVM), and XGBoost in this study. Hyperparameters such as regularization strength (C) in L2 logistic regression, n_estimators, max_depth, and min_samples_split in random forest, regularization strength (C) and kernel in SVM, and n_estimators, max_depth, and learning_rate in XGBoost were tuned with empirical candidate values.
Ablation study for GDnet
The proposed GDnet model comprises three critical modules: the transcriptomic module, drug representation module, and prior knowledge module. To demonstrate the significance of these modules, we designed corresponding ablation experiments: (1) For the transcriptomic module, we developed a drug-representation-only model (Dnet) to validate the importance of transcriptomic data. (2) For the drug representation module, we used the transcriptome-only model (Gnet) and alternative drug representation-based models (GSnet and GTnet) to verify the superiority of our proposed drug characterization approach. (3) For the prior knowledge module, we used GDdnet, a fully connected feedforward neural network model without prior knowledge integration.
Model training and testing
The analysis included datasets that met specific criteria, providing a total of 4371 available patients. The 17 largest datasets, comprising approximately 3459 patients (79% of the total), were designated as the training set. The 14 remaining datasets, comprising approximately 912 (21% of the total), were used as the external test set, assigned without subjective assignment. The model was trained with the training set, and hyperparameters such as the learning rate were optimized via tenfold validation. The trained models based on the whole training set were finally validated with the external test set. The prediction performance of different deep learning models was compared using the test set by calculating the median AUROC and AUPRC across 10 rounds of replicated training using roc_auc_score and average_precision_score implemented in Python sklearn.metrics library separately. Then, we ensembled the 10 repetitions of the deep learning models to derive the final prediction for stability and robustness. Machine learning models were trained only once due to the same outcome across repetitions. The final models were evaluated according to the AUROC and AUPRC, and further evaluated using metrics such as accuracy, F1-score, precision, recall, and specificity with the cutoff value set as 0.5.
Model interpretation
We applied IntegratedGradients27, a gradient-based attribution method implemented in captum.attr library, to calculate the sample-level importance of all nodes in all layers. From 10-repetition GDnet models, we chose a GDnet model with a similar AUROC value (0.727) to that (0.725) of the 10-repetition ensemble GDnet for interpretation. The absolute importance score (always positive) of each layer’s nodes was normalized to facilitate comparability between layers via the following formula:
where \({I}_{j}\) is the normalized absolute importance score for each node in a layer, \({A}_{j}\) is the absolute importance score, and n is the total number of nodes in a layer.
To calculate the total node-level importance, we aggregated the sample-level importance scores across all samples in the test set with a specific regimen (the dataset-level interpretation is represented as the mean value of all patient-level weights in the test set with a specific regimen). The importance score of each node is represented as the node length in the Sankey diagram (Fig. 5a). The importance score of links between the source node (in the former layer) and target node (in the latter layer) was represented by multiplying the normalized (within the absolute weight of all source nodes that point to the same target node) absolute weight of the source node that points to the target node in the model by the importance score of the target node, as shown in the following formula:
\({L}_{{\left(s-t\right)}^{\,j}}\) is the importance score of the link from the j source node (refer to as s) to the target node (refer to as t), which is the normalized absolute importance score of the target node, \({W}_{{sj}}\) is the weight of the j source node pointing to the target node in the deep learning model, and n is the number of source nodes that point to a target node.
Statistical analyses
The change in the area under the ROC curve between GDnet and other models was tested via the DeLong test59. The AUROCs derived from tenfold cross-validation and tenfold repeated training and testing for GDnet and other models were compared via the Wilcoxon rank-sum test. The variance of the AUROCs and AUPRCs between Gnet and GNNet was compared via the Levene’s test to assess the robust of performance. The 95% confidence intervals for AUROC, AUPRC, accuracy, F1-score, precision, recall, and specificity were calculated using 1,000 times of bootstrapping. The odds ratio (OR) was calculated to determine preferences for pathological complete response (pCR) in the simulated trials, and differences were compared via Fisher’s exact test. A linear regression model was fitted to examine the relationships between the degree of optimization and OR. The significance of the regression coefficients was evaluated via t tests, and the R-squared value was used to quantify the proportion of variance explained by the model. To address potential confounding caused by the imbalance of transcriptome profile (inferred by the Gnet prediction score) between optimized and control groups, we employed Propensity Score Matching (PSM). PSM was conducted by estimating propensity scores using the transcriptome-only model Gnet prediction score. Matching was performed using nearest neighbor matching (1:1 ratio) with a caliper of 0.1, without replacement, to minimize imbalance. All tests were two-sided. P values < 0.05 were considered statistically significant unless otherwise specified. All the statistical analyses were performed via R statistical software version 4.3.
Statistics and reproducibility
No statistical method was used to predetermine sample size. We systematically retrieved patient data from the GEO database to maximize cohort size and statistical power, initially accessing 9745 patients. Patients were included regardless of sex or gender. Based on pre-established exclusion criteria, we excluded: (1) 628 duplicate samples to prevent data leakage and over-representation of individual patients; (2) 3805 samples collected after treatment initiation or lacking outcomes, as the model predicts treatment response from pre-treatment data; (3) 209 samples with >50% missing genes to maintain transcriptomic data integrity; and (4) 598 samples without treatment details or treatment representations, which are essential model inputs.
The final training and validation cohort included 3459 patients from 17 datasets, all with complete pCR status, treatment regimens, and sufficient transcriptomic data. The testing cohort comprised 912 patients from 14 datasets, with an additional 134 patients from two subsequent datasets for prospective testing. Patient samples were allocated into experimental cohorts based on their source dataset, not random allocation. This strategy was chosen to evaluate the model’s generalizability across independent studies and different patient populations, which is a key strength of our study design. Blinding of investigators to cohort allocation during data collection and outcome assessment was not applicable to this computational study. The analysis was performed using pre-existing, de-identified data from the GEO database, and all model predictions were generated algorithmically, eliminating the potential for subjective bias.
To ensure computational reproducibility, we implemented: (1) tenfold cross-validation during training for hyperparameter optimization and internal validation; (2) 10 rounds of replicated training across 17 datasets with external testing in 14 datasets for robust comparison; and (3) prospective testing on an independent cohort of 134 patients from two subsequent datasets. All replication attempts successfully confirmed our initial findings, demonstrating model robustness.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The public treatment-associated gene expression data used in this study are publicly available without restriction in the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE194040, GSE25066, GSE16716, GSE41998, GSE180962, GSE20271, GSE34138, GSE50948, GSE149322, GSE22226, GSE32603, GSE22358, GSE32646, GSE16446, GSE130788, GSE231629, GSE123845, GSE4779, GSE173839, GSE22093, GSE21997, GSE42822, GSE66399, GSE181574, GSE23988, GSE41656, GSE8465, GSE122630, GSE21974, GSE207248, GSE191127, GSE260693, and GSE240671. The processed gene expression and clinical data can be accessed via a figshare repository (https://doi.org/10.6084/m9.figshare.27686151)60. Source data are provided with this paper.
Code availability
Custom codes are publicly available without restriction on Code Ocean https://doi.org/10.24433/CO.6069410.v1 under the GNU General Public License version 2 (GPL-2.0)61. Our implementation incorporates and modifies components from the ‘pnet_prostate_paper’ project on github (https://github.com/marakeby/pnet_prostate_paper). As the original project is also licensed under the GNU GPL-2.0, our derivative work retains this same license.
References
Symmans, W. F. et al. Long-term prognostic risk after neoadjuvant chemotherapy associated with residual cancer burden and breast cancer subtype. J. Clin. Oncol. 35, 1049–1060 (2017).
Symmans, W. F. et al. Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J. Clin. Oncol. 25, 4414–4422 (2007).
Asselain, B. et al. Long-term outcomes for neoadjuvant versus adjuvant chemotherapy in early breast cancer: meta-analysis of individual patient data from ten randomised trials. Lancet Oncol. 19, 27–39 (2018).
Tanioka, M. et al. Integrated analysis of RNA and DNA from the phase III trial CALGB 40601 identifies predictors of response to trastuzumab-based neoadjuvant chemotherapy in HER2-positive breast cancer. Clin. Cancer Res. 24, 5292–5304 (2018).
Wang, Y. et al. A generalized non-linear model predicting efficacy of neoadjuvant therapy in HER2+ breast cancer. iScience 26, 106330 (2023).
Shi, Z. et al. MRI-based quantification of intratumoral heterogeneity for predicting treatment response to neoadjuvant chemotherapy in breast cancer. Radiology 308, e222830 (2023).
Ogier du Terrail, J. et al. Federated learning for predicting histological response to neoadjuvant chemotherapy in triple-negative breast cancer. Nat. Med. 29, 135–146 (2023).
Sammut, S. J. et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 601, 623–629 (2022).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Lipkova, J. et al. Artificial intelligence for multimodal data integration in oncology. Cancer Cell 40, 1095–1110 (2022).
Peduzzi, P., Concato, J., Kemper, E., Holford, T. R. & Feinstein, A. R. A simulation study of the number of events per variable in logistic regression analysis. J. Clin. Epidemiol. 49, 1373–1379 (1996).
Wolf, D. M. et al. Redefining breast cancer subtypes to guide treatment prioritization and maximize response: Predictive biomarkers across 10 cancer therapies. Cancer Cell 40, 609–623.e606 (2022).
Liao, J. et al. De novo analysis of bulk RNA-seq data at spatially resolved single-cell resolution. Nat. Commun. 13, 6498 (2022).
Chu, T., Wang, Z., Pe’er, D. & Danko, C. G. Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat. Cancer 3, 505–517 (2022).
Sun, D. et al. Identifying phenotype-associated subpopulations by integrating bulk and single-cell sequencing data. Nat. Biotechnol. 40, 527–538 (2022).
Elmarakeby, H. A. et al. Biologically informed deep neural network for prostate cancer discovery. Nature 598, 348–352 (2021).
Hao, J., Kim, Y., Kim, T.-K. & Kang, M. PASNet: pathway-associated sparse deep neural network for prognosis prediction from high-throughput data. BMC Bioinf. 19, 510 (2018).
Kuenzi, B. M. et al. Predicting drug response and synergy using a deep learning model of human cancer cells. Cancer Cell 38, 672–684.e676 (2020).
Zhang, H., Chen, Y. & Li, F. Predicting anticancer drug response with deep learning constrained by signaling pathways. Front. Bioinf. 1, 639349 (2021).
Deng, L. et al. Pathway-guided deep neural network toward interpretable and predictive modeling of drug sensitivity. J. Chem. Inf. Model. 60, 4497–4505 (2020).
Liu, Q. & Xie, L. TranSynergy: Mechanism-driven interpretable deep neural network for the synergistic prediction and pathway deconvolution of drug combinations. PLoS Comput. Biol. 17, e1008653 (2021).
Park, S. et al. A deep learning model of tumor cell architecture elucidates response and resistance to CDK4/6 inhibitors. Nat. Cancer 5, 996–1009 (2024).
Ren, S., Cooper, G. F., Chen, L. & Lu, X. An interpretable deep learning framework for genome-informed precision oncology. Nat. Mach. Intell. 6, 864–875 (2024).
Zinn, S. et al. Advances in antibody-based therapy in oncology. Nat. Cancer 4, 165–180 (2023).
Paul, S. et al. Cancer therapy with antibodies. Nat. Rev. Cancer 24, 399–426 (2024).
Swain, S. M., Shastry, M. & Hamilton, E. Targeting HER2-positive breast cancer: advances and future directions. Nat. Rev. Drug Discov. 22, 101–126 (2023).
Sundararajan, M., Taly, A. & Yan, Q. Axiomatic attribution for deep networks. in Proc. 34th International Conference on Machine Learning, Vol. 70, 3319–3328 (JMLR.org, 2017).
Dressman, M. A. et al. Gene expression profiling detects gene amplification and differentiates tumor types in breast cancer. Cancer Res. 63, 2194–2199 (2003).
Munkácsy, G. et al. PSMB7 is associated with anthracycline resistance and is a prognostic biomarker in breast cancer. Br. J. Cancer 102, 361–368 (2010).
Ma, F. et al. Phase I study and biomarker analysis of pyrotinib, a novel irreversible Pan-ErbB receptor tyrosine kinase inhibitor, in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J. Clin. Oncol. 35, 3105–3112 (2017).
Cho, Y. B. & Park, K. S. The effect and treatment of PIK3CA mutations in breast cancer: current understanding and future directions. Medicina 61, 518 (2025).
Kim, J. et al. Ketohexokinase-A acts as a nuclear protein kinase that mediates fructose-induced metastasis in breast cancer. Nat. Commun. 11, 5436 (2020).
Mondaca, J. M. et al. Therapeutic potential of GNRHR analogs and SRC/FAK inhibitors to counteract tumor growth and metastasis in breast cancer. Biochim. Biophys. Acta Mol. Basis Dis. 1871, 167826 (2025).
Ghavami, M. & Fairn, G. D. Endoplasmic reticulum-Phagosome contact sites from the cradle to the grave. Front. Cell Dev. Biol. 10, 1074443 (2022).
Sahai, E. & Marshall, C. J. RHO-GTPases and cancer. Nat. Rev. Cancer 2, 133–142 (2002).
Yi, J. et al. A circRNA-mRNA pairing mechanism regulates tumor growth and endocrine therapy resistance in ER-positive breast cancer. Proc. Natl. Acad. Sci. USA 122, e2420383122 (2025).
Huan, J. et al. The RRP9-JUN axis promotes breast cancer progression via the AKT signalling pathway. Biol. Direct. 19, 131 (2024).
Li, Y. et al. HOXC8-dependent cadherin 11 expression facilitates breast cancer cell migration through Trio and Rac. Genes Cancer 2, 880–888 (2011).
Momeny, M. et al. DUSP6 inhibition overcomes neuregulin/HER3-driven therapy tolerance in HER2+ breast cancer. EMBO Mol. Med. 16, 1603–1629 (2024).
Paik, S. et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 351, 2817–2826 (2004).
Li, H. et al. A serum microRNA signature predicts trastuzumab benefit in HER2-positive metastatic breast cancer patients. Nat. Commun. 9, 1614 (2018).
Theodoris, C. V. et al. Transfer learning enables predictions in network biology. Nature 618, 616–624 (2023).
Liu, Z. et al. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat. Commun. 13, 816 (2022).
Cardoso, F. et al. 70-Gene Signature as an aid to treatment decisions in early-stage breast cancer. N. Engl. J. Med. 375, 717–729 (2016).
Alaa, A. M., Gurdasani, D., Harris, A. L., Rashbass, J. & van der Schaar, M. Machine learning to guide the use of adjuvant therapies for breast cancer. Nat. Mach. Intell. 3, 716–726 (2021).
Jiang, Y. Z. et al. Transcriptome analysis of triple-negative breast cancer reveals an integrated mRNA-lncRNA signature with predictive and prognostic value. Cancer Res. 76, 2105–2114 (2016).
Zhao, Z. et al. Organoids. Nat. Rev. Methods Prim. 2, 94 (2022).
Wang, X. Q. et al. Spatial predictors of immunotherapy response in triple-negative breast cancer. Nature 621, 868–876 (2023).
Papalexi, E. & Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 18, 35–45 (2018).
Chen, J., Larsson, L., Swarbrick, A. & Lundeberg, J. Spatial landscapes of cancers: insights and opportunities. Nat. Rev. Clin. Oncol. 21, 660–674 (2024).
Iorio, F. et al. A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754 (2016).
Yang, W. et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955–D961 (2013).
Garnett, M. J. et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575 (2012).
Rees, M. G. et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 12, 109–116 (2016).
Basu, A. et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161 (2013).
Seashore-Ludlow, B. et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 5, 1210–1223 (2015).
Milacic, M. et al. The reactome pathway knowledgebase 2024. Nucleic Acids Res. 52, D672–d678 (2024).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–d1082 (2018).
DeLong, E. R., DeLong, D. M. & Clarke-Pearson, D. L. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837–845 (1988).
Ge, H. Biologically informed deep learning integrating drug representations for breast cancer treatment selection. figshare. Dataset. https://doi.org/10.6084/m9.figshare.27686151 (2025).
Ge, H. Biologically-informed integration of drug representations for breast cancer treatment using deep learning. Biologically-informed integration of drug representations for breast cancer treatment using deep learning. https://doi.org/10.24433/CO.6069410.v1 (2025).
Acknowledgements
This work was supported by The National Key Research and Development Program of China (2021YFF1201300), National Natural Science Foundation of China (82230058), Beijing Natural Science Foundation and Joint Fund of Changping Innovation (L234004), CAMS Innovation Fund for Medical Sciences (CIFMS 2021-I2M-1-014), and The Special Research Fund for Central Universities, Peking Union Medical College (APL23100510010301045).
Author information
Authors and Affiliations
Contributions
Conceptualization: H.G. and F.M. Methodology: H.G. Coding: H.G. Formal analysis: H.G. Investigation: H.G. Writing, original draft: H.G. Writing, review and editing: H.G., H.M., Y.W., J.W., Y.Q., and L.L. Visualization: H.G. Supervision: F.M. Funding acquisition: F.M.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhaoxiang Cai, Saugato Rahman Dhruba, Ming Fan, Yushuai Yu, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ge, H., Mo, H., Wei, Y. et al. Biologically-informed integration of drug representations for breast cancer treatment using deep learning. Nat Commun 17, 10 (2026). https://doi.org/10.1038/s41467-025-66384-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66384-6
