
Abstract
Loss-of-function mutations in AKAP11 (a protein kinase A (PKA)-binding protein) greatly increase the risk of bipolar disorder and schizophrenia. To determine the neurobiological functions of AKAP11, we conduct multi-omic and neurobiological analyses of Akap11 mutant mouse brains. We find that AKAP11 is a key regulator of PKA proteostasis in the brain whose loss leads to dramatically increased levels of PKA subunits and phosphorylated PKA substrates, especially in synapses. Akap11 mutant mice show extensive transcriptomic changes throughout the brain, including prominent decreases in synapse-related genes sets. Gene expression is highly impacted in spiny projection neurons of the striatum, a brain region implicated in motivation, cognition and psychotic disorders. Real-time measurements of PKA activity reveal elevated basal PKA activity in the striatum of Akap11-/- mice, with exaggerated additional response to dopamine receptor antagonists. Behaviorally, Akap11 mutant mice show abnormally prolonged locomotor response to amphetamine, deficits in associative learning and contextual discrimination, as well as depression-like behaviors. Our study connects molecular changes to circuit dysfunction and behavioral disturbance in a genetically valid animal model of psychotic disorder.
Similar content being viewed by others


Introduction
Bipolar disorder (BD) and schizophrenia (SCZ) are debilitating psychiatric conditions within the psychosis spectrum whose biological basis remains unknown. BD is typically characterized by episodes of mania alternating with periods of depression. SCZ is characterized by positive symptoms including hallucinations and delusions that occur alongside negative symptoms that include emotional impairment, and is often associated with cognitive symptoms. Together, BD and SCZ account for a substantial fraction of the global mental health crisis: the lifetime prevalence of BD is 1–2%1 and SCZ is approximately 0.7%2. Both diseases have substantial heritability (60-80%)3,4, suggesting that gene-derived molecular programs contribute toward disease risk.
Recent landmark exome sequencing efforts have led to the discovery of several genes whose loss of function greatly increase the risk for SCZ5 and BD6. These rare variant mutations are associated with large odds ratios (3–50), conferring much more substantial risk than common variants previously identified by genome-wide association studies (GWAS) (odds ratios <1.1)7,8. In further contrast to GWAS-associated loci, these mutations occur in coding sequences of specific genes and can be inferred to be loss of function in nature.
Mutations in AKAP11 (A-kinase anchoring protein 11) are associated with greatly elevated risk of both SCZ and BD5,6,9,10. AKAP11 has not been associated with autism spectrum disorder, epilepsy or intellectual disability6 and therefore is a relatively specific risk gene for the SCZ–BD psychotic disorder spectrum. In contrast, previously studied large-effect SCZ risk variants11,12,13 are also associated with intellectual disability and developmental disorders5, making Akap11 animal models particularly interesting to study. In our previous neurophysiologic study, we reported that Akap11 loss-of-function mutant mice recapitulate several electroencephalogram (EEG) abnormalities found in schizophrenic patients, including increased power of resting gamma oscillations, non-rapid eye movement (NREM) sleep reduction, and gene-dose dependent reduction in sleep spindles14. The molecular, cellular and circuit mechanisms by which Akap11 deficiency results in brain dysfunction have not been studied.
The Akap11 gene encodes a member of the A-kinase anchoring proteins (AKAPs), which bind Protein Kinase A (PKA) and are typically localized to compartments within a cell, targeting PKA to specific subcellular domains15. In non-neuronal cell lines, AKAP11 is reported to localize to punctate structures, including multivesicular bodies16 and peroxisomes17. In addition to PKA, AKAP11 is reported to interact biochemically with the protein kinase GSK3β18, Protein Phosphatase 119, IQGAP120, GABAC receptors21, and vesicle-associated proteins A and B (VAPA, VAPB)22, each of which is implicated in signaling and trafficking pathways that influence synaptic plasticity and neuronal development. Intriguingly, AKAP11 not only binds but also mediates inhibition of GSK3β18, a target of the mood-stabilizing drug lithium that is used to treat BD. AKAP11 also functions as an autophagy receptor for the regulatory subunit 1 of PKA (PKA R1α) which promotes protein-level degradation of PKA R1α in the context of cellular stress23.
Because AKAP11 has not been studied in the brain, there is insufficient knowledge to bridge the cell biology of AKAP11 and psychiatric disease risk. In this study, we use unbiased multi-omic approaches to explore the consequences of Akap11 loss-of-function across widespread brain regions and cell types, spanning several postnatal ages and within synaptic compartments. To follow up on multi-omic analyses, we carry out targeted real-time measurements of net striatal PKA activity in the context of dopaminergic signaling using in vivo fiber photometry. Our data uncover a fundamental role of AKAP11 in synapse function and highlight the striatum as a nexus of circuit dysfunction in a model of psychosis risk with human-genetics validity.
Results
Akap11 mutant mice exhibit activity, cognitive and affective behavioral phenotypes
To investigate the effects of Akap11 deficiency on mouse behavior, we conducted a multi-level set of behavioral experiments encompassing long-term naturalistic observations in the home cage, machine learning based analysis of complex motor behavior, and specific tests that may be relevant to psychiatric symptom domains. We first examined the general activity patterns of Akap11+/-, Akap11-/-24, and wild-type (WT) littermate controls continuously in their home cages from 4 weeks to 16 weeks of age using an automated home cage activity monitoring system25,26,27. Akap11 mutants were less active than WT littermates during their night-time (active) and daytime (resting) periods, in a gene-dose dependent manner (Fig. 1a, b). This hypoactivity was apparent at 4 weeks of age (at the beginning of monitoring) and remained until 12 weeks (Fig. 1c). When a running wheel was introduced for 1 week during the assay, Akap11-/- mutant mice showed less engagement than WT28 (Fig. 1d).

a Heatmap of average activity of each genotype across 24 h in a homecage locomotor activity monitoring system. Each row is 24 h, and 60 rows representing 60 consecutive days are shown. Yellow and black bars indicate lights-on and lights-off periods respectively. ZT (Zeitgeber Time) refers to the number of hours past the lights-on time in light-dark scheduled conditions. b Averaged locomotor activity of each WT and Akap11 mutant mice, across 60 days of the assay. c Locomotor activity averaged across 24 h, aligned according to animals’ postnatal ages. d Running wheel usage during a 1 week test period. e Syllable usage of WT and Akap11 mutant mice (n = 11 mice for WT, n = 11 mice for Akap11+/-, and n = 10 mice for Akap11-/-), sorted by usage difference between WT and Akap11-/-. Color codes on the x-axis represent annotated behaviors. * Represents represent syllables with FDR < 0.1. Data are represented as mean ± 95% confidence interval. f Relative syllable usage of WT and Akap11 mutant mice sorted by velocity. g Syllable transition matrix showing the upregulated (red lines) and downregulated (blue lines) transition probability between Akap11+/- and WT (left) or between Akap11-/- and WT (right). h Average usage of significantly downregulated rearing syllables (top), grooming syllables (middle), and significantly upregulated pausing syllables (bottom) between Akap11 mutants and WT (n = 11 mice for WT, n = 11 mice for Akap11+/-, and n = 10 mice for Akap11-/-). The box plots display the median (white dot) and interquartile range (25–75th percentile); the whiskers represent upper and lower adjacent values; the outer bounds represent the data distribution. i Schematic diagram of the associative learning and sensory discrimination task. j During fear conditioning (Day 1), freezing time (%) to CS+ (left) and CS- (middle) by WT and Akap11 mutant mice. Bar graph (right) represents the freezing time (%) during trials 6–10. k During fear recall (day 2), freezing time (%) and to CS+ and CS- by WT and Akap11 mutant mice (left). Discrimination index (right) is defined as: (CS+ freeze - CS- freeze) / (CS+ freeze + CS- freeze). b,c,d,j,k data are represented as mean +/- SEM, and bar graphs as mean + SEM. b,d,h One-way ANOVA with Tukey’s post hoc test. See Supplementary Data 1 for statistics. j,k Repeated measures two-way ANOVA with Tukey’s post hoc test for CS+ and CS- freezing time (%), and one-way ANOVA with Bonferroni post hoc test for discrimination index. See Supplementary Data 1 for statistics.
To examine volitional (“exploratory”) behavior at finer resolution, we applied motion sequencing (MoSeq), an unsupervised machine learning based tool that decomposes continuous 3D video recordings into discrete, repeated behavioral motifs (“syllables”)29,30. During a 30 min open field trial, Akap11-/- mice displayed significant differences from WT littermates (P < 0.05) in their usage of a wide range of behavioral syllables (26 of 51 syllables) (Fig. 1e, Supplementary Fig. 1a) including those related to locomotion, pausing, grooming and rearing (Fig. 1e, h). Many of these same syllables (10 of 26 syllables significant in Akap11-/-) were differentially utilized in Akap11+/- mice but to an intermediate degree. Gene-dose dependent alterations in syllable usage were also apparent in the proportions of low/high velocity syllables (Fig. 1f) and transition probabilities between syllables (Fig. 1g). Akap11 mice were hypolocomotive in home cage and MoSeq assays, consistent with prior open-field measurements14,24. In the rotarod test, Akap11 mutant mice did not differ from WT in latency to fall, suggesting that their low activity was not due to grossly impaired motor coordination (Supplementary Fig. 1b).
We next measured mouse behaviors that are commonly used as models of positive and negative symptoms31. Akap11 mutant mice performed similarly to WT in prepulse inhibition (Supplementary Fig. 1c), suggesting intact sensory gating. Akap11 mutant mice also had normal social behaviors – their social preference to a novel mouse over an object (Supplementary Fig. 1d), or preference to a novel mouse versus a familiar mouse (Supplementary Fig. 1e), did not differ from WT controls. In the forced swim test (often used to infer “depressive-like” states32), Akap11 mutant mice displayed greater immobility in a gene-dose dependent fashion (Supplementary Fig. 1f). Akap11 mutants performed normally in the sucrose preference test if they were tested as a “naïve” cohort; interestingly, however, if the sucrose preference test followed a forced swim test on the preceding day, then Akap11-/- mutants showed robustly impaired sucrose preference compared with WT controls (Supplementary Fig. 1g), a pattern that could be interpreted as anhedonia33. Together with the greater immobility in the forced swim test, this pattern could be interpreted as depressive-like and anhedonia-like behavior that is induced by prior stress33.
Cognitive deficits are highly prevalent in SCZ, including attention deficits, memory impairments, and reduced executive control34. We designed a sensory discrimination task using auditory fear conditioning to simultaneously test Akap11 mutants for associative learning and sensory/context discrimination (Fig. 1i). Mice were presented with a certain tone (Conditioned Stimulus + [CS + ]) that is associated with footshock (Unconditioned Stimulus, US) and a different tone (Conditioned Stimulus -, CS-) that is not associated with any US. During the conditioning day (Day 1), WT, Akap11+/-, and Akap11-/- mice all displayed increased freezing during CS+ compared with during CS-, indicating successful learning of fear conditioning and CS discrimination (Fig. 1j). During the recall test (Day 2), Akap11+/- and Akap11-/- had impaired fear recall as measured by less freezing time in response to CS+ compared with WT (Fig. 1k, left). While WT and Akap11+/- mice showed increased freezing to CS+ than to CS-, Akap11-/- mice froze similarly to both CS+ and CS- (Fig. 1k, right). Thus Akap11+/- and Akap11-/- mutant mice had gene-dose dependent impairment in fear conditioning, whereas Akap11-/- mice also exhibited impaired ability to discriminate CS+ and CS- one day after conditioning, suggesting a generalization of fear memory in the homozygous mutant. Together, these data show behavioral abnormalities in Akap11 mutant mice in domains (mood, cognition) that are relevant to neuropsychiatric disease.
Identifying the AKAP11 interactome in brain
Little is known about AKAP11 in the brain, so we characterized the protein interactome of AKAP11 by immunoprecipitation of AKAP11 from cortical extracts followed by mass spectrometry (coIP-MS, Fig. 2a, b; Supplementary Fig. 2a). Of the ~2000 proteins identified by mass spectrometry, 68 proteins were enriched (P < 0.05) in AKAP11 coIP from Akap11 WT cerebral cortex versus coIP from negative control Akap11-/- (knockout) mice24; these were considered specific interactors of AKAP11 (Fig. 2c; Supplementary Data 2). This approach yielded putative AKAP11-associated proteins that were largely overlapping with those found by comparison of AKAP11 coIP vs control IgG coIP from WT brains (Supplementary Fig. 2b). Of the 68 proteins that were specifically associated with AKAP11 in the brain, only 7 were previously reported in cell lines or other tissues, including PKA subunits, GSK3β, and VAPA and VAPB (Fig. 2d)18,22. The biochemical association of AKAP11 with PKA catalytic and regulatory subunits and several other interactors (VAPA, VAPB, GSK3β, DRYK1A) was confirmed by coIP-immunoblot experiments (Figs. 2e–j). AKAP11 did not associate with the cytoskeleton protein Vinculin, indicating specificity for the above results (Fig. 2e).

a Schematic of AKAP11 immunoprecipitation (IP) and its most notable, known interactors. b Immunoblot of AKAP11 in IPs of anti-AKAP11 and control IgG in 12-week-old (12wk) WT or Akap11-/- cortical tissue. This experiment was repeated in >5 independent experiments, with similar results. c Volcano plot comparing Log2 Fold-Change (Log2FC) against P value for AKAP11 IP in WT vs Akap11-/-, displaying all proteins detected by coIP-MS. A moderated two-sample t-test was applied to the sample group datasets. Proteins that were enriched (red) by anti-AKAP11 IP from WT versus Akap11-/- cortical extracts. Enriched proteins, defined by having a nominal P value < 0.05 and Log2FC > 0, are colored red. Downregulated proteins are mostly antibody-related artifacts and can be found in Supplementary Data 2. d Venn diagram comparing previously published AKAP11 interactions with the brain interactome in this study. Full results are displayed in Supplementary Data 1. e Immunoblot of AKAP11, PKA subunits and control proteins, in IPs of AKAP11, GluA1 or IgG control in WT or Akap11-/- cortical lysate. This experiment was repeated in >3 independent experiments, with similar results. f Immunoblot of AKAP11 in IPs of PKA subunits, in WT cortical lysates. This experiment was repeated in >2 independent experiments, with similar results. g–j Immunoblot with indicated antibodies, in IPs of anti-GSK3α, GSK3β, VAPA, VAPB, AKAP11, P62 or DYRK1a in WT or Akap11-/- cortical lysate. These experiments were repeated in at least 2 independent experiments, with similar results. k Gene set enrichment analysis of all proteins displayed in 1c, showing selected gene ontologies with a positive normalized enrichment score (NES) and adjusted P-value (FDR) < 0.01. Redundant (similar) pathways were removed for clarity. The top most enriched proteins within each ontology are listed within the histograms. GSEA uses a Kolmogorov-Smirnov test, two tailed, with Benjamini-Hochberg (B-H) False Discovery Rate (FDR) correction applied.
As expected for an AKAP protein15, we observed high enrichment of multiple subunits of PKA in AKAP11 immunoprecipitates (Fig. 2c, e). PKA exists as a tetrameric holoenzyme with regulatory subunits that bind and suppress the kinase activity of catalytic subunits. In AKAP11 coIPs, there was greater degree of enrichment for regulatory subunits PKA R1α and PKA R1β (encoded by Prkar1a and Prkar1b) than the catalytic subunits PKA Cα, PKA Cβ (encoded by Prkaca and Prkacb), which is consistent with AKAP11 directly binding to regulatory subunits of PKA35. Adenylyl cyclase 10 (encoded by Adcy10) was also enriched, and this enzyme produces cyclic AMP, which binds and activates PKA. There was only modest enrichment for type 2 regulatory subunits (PKA R2α, PKA R2β. Supplementary Fig. 2c) even though AKAP11 has high affinity binding sites for R235. We confirmed that AKAP11 also associates with GSK3β in the brain, consistent with prior studies in cell lines18,35,36 and did not detect an association with GSK3α (Fig. 2h).
Our coIP-MS experiments from brain uncovered multiple novel interactors of AKAP11 including DYRK1A, SPHKAP, Synaptic Vesicle Protein 2B (SV2B), and P62 (also known as SQSTM1) (Fig. 2c, d). By coIP-immunoblot, we confirmed AKAP11 binding to DYRK1A (Fig. 2i, j), a tyrosine kinase that is genetically linked to autism spectrum disorders37 and is implicated in neurodevelopment, neurogenesis and neurite outgrowth38,39,40,41. Association with VAMP-associated Proteins A and B (VAPA/VAPB) and SPHKAP is suggestive of localization near endoplasmic reticulum (ER). VAPA/B are ER-resident proteins that facilitate inter-organelle contact and vesicular transport, and SPHKAP interacts with VAP proteins to regulate calcium signaling from ER stores42. We also explored the AKAP11 interactome by coIP from the brains of Akap11+/- (heterozygote) mutants, which mimics the human heterozygous genotype that is associated with greatly increased risk for SCZ/BD6. Proteins coIP’d with AKAP11 were largely similar in WT and Akap11+/- (Supplementary Fig. 2d,e).
To gain further insight into molecular pathways in which AKAP11 might play a role, we conducted gene-set enrichment analysis (GSEA)43 on AKAP11-associated proteins (Fig. 2k, Supplementary Data 3). In addition to expected gene ontology (GO) pathways like “Phosphorylation”, we observed enrichment in AKAP11 coIP of the “Ubiquitin like protein ligase binding” GO term, which includes the selective autophagy receptor P62 and RPL40 (encoded by Uba52) (Supplementary Fig. 2f). In cell lines, AKAP11 interacts with autophagosomes, intracellular vesicular structures involved in protein degradation44, through the selective autophagy receptor LC323,45. In our coIP experiments, we found an association of AKAP11 with P62, but not LC3 (Supplementary Fig. 2c), suggesting that in the brain, AKAP11 also interacts with autophagosomes but possibly by distinct molecular mechanisms.
Accumulation of PKA in Akap11 mutant mice
Given our finding that AKAP11 could interact with autophagosomes, coupled with previous studies showing its role in targeting proteins for degradation, we tested how the abundance of AKAP11-interacting proteins was altered by loss of AKAP11. By immunoblotting of total cortical lysate, we found a striking elevation of PKA protein levels (regulatory (~6x) and catalytic (~9x) subunits in Akap11-/- mice) (Fig. 3a, Supplementary Fig. 3a). The association of AKAP11 with P62 in the brain (Fig. 2c) suggests the possibility that AKAP11 regulates degradation of PKA by receptor-mediated selective autophagy. Levels of other AKAP11 interactors (GSK3β, VAPA/B, P62 and DYRK1A) were not substantially altered in Akap11 mutant brains.

a Immunoblots and quantifications of 12wk cortex total lysate using antibodies against AKAP11-interacting proteins. Measurements are normalized to β-Actin and to WT controls for each antibody. P < 0.05 comparisons are shown. b Immunoblots and quantifications of 4wk, 12wk, and 28wk cortex total lysate using antibodies against AKAP11 and PKA subunits. Measurements are normalized to β-Actin and to WT controls within an age group. c Immunoblots and quantifications from microdissected tissue total lysate from 4wk mice using antibodies against AKAP11 and PKA subunits. Measurements are normalized to β-Actin and to WT controls within a region. a-c Data are represented as mean ± SEM. Two-way ANOVA with Tukey’s post hoc test. See Supplementary Data 1 for exact P-values, statistical tests and sample sizes for each comparison.
The marked elevation of PKA subunits in Akap11-/- mice was apparent at all ages examined (4, 12, 28 weeks of age, Fig. 3b). PKA subunits were increased not only in the cortex, but across all brain regions examined: prefrontal cortex (PFC), somatosensory cortex (SSC), striatum (Str), hippocampus (Hipp), thalamus (Thal) and substantia nigra (SN) (Fig. 3c). Compared with the multiple-fold increases in the homozygous (Akap11-/-) mutant, changes in PKA protein levels in the heterozygous (Akap11+/-) mutant brain were much more modest (Fig. 3a–c). Nonetheless, comparison with WT animals showed that PKA R1α and Cα subunits were consistently elevated by 20%-75% in Akap11+/- (Supplementary Fig. 3b, c). In the course of these immunoblotting experiments, we noted that levels of AKAP11 protein itself were abolished in the homozygous mutant (Akap11-/-) but minimally reduced in heterozygous (Akap11+/-) mutants (Fig. 3a, b, Supplementary Fig. 3a,d), despite gene-dose dependent reduction of Akap11 RNA (Supplementary Fig. 3e). This pattern is suggestive of post-transcriptional homeostatic compensation of Akap11 expression when just one copy of the Akap11 gene is deleted, and might explain the relatively modest effect on PKA in Akap11+/- vs. Akap11-/-. Quantitative immunofluorescence imaging of PKA R1α protein in brain sections showed increased intensity across all brain regions of Akap11-/- mice (Supplementary Fig. 3f). Level of PKA R1α RNA was not significantly altered in Akap11-/- (Supplementary Fig. 3g), suggesting that AKAP11 regulates the stability or turnover of PKA at the protein level.
Protein changes and elevated PKA at synapses of AKAP11 deficient mice
PKA plays a key role in regulation of synaptic plasticity46. To understand the consequences of Akap11 deficiency and PKA elevation in synapses, we investigated protein changes in purified synaptic (“postsynaptic density (PSD)”) fractions of Akap11 mutant mice by quantitative mass spectrometry-based proteomics and phosphoproteomics using Tandem Mass Tag (TMT) labeling (see methods). Since the onset of SCZ occurs typically in adolescence and early adulthood47, we examined 4 week (juvenile) and 12 week (young adult) mice. ~9000 proteins were identified in the PSD fraction purified from cortex, including AKAP11 itself which was modestly (~2x) enriched in the PSD fraction vs total lysate (Supplementary Fig. 4a,b). The number of differentially abundant proteins (DAPs, nominal P < 0.05) was greater in Akap11-/- than Akap11+/- mutants at 12 weeks but similar at 4 weeks, with approximately equal proportion of upregulated and downregulated DAPs overall (Fig. 4a, Supplementary Data 2). The DAPs were well correlated between Akap11+/- and Akap11-/- genotypes (Supplementary Fig. 4c) and weakly correlated between 4 and 12 weeks (Supplementary Fig. 4d).

a Number of DAPs across different ages and genotypes in synapse proteomics. Shading indicates upregulated (red) and downregulated (blue) DAPs, respectively. 4wk synapse proteomics data also appears in (Aryal et al., 2023)51. b Volcano plots of 12wk synapse proteomics in Akap11 mutants relative to WT. A moderated two-sample t-test was applied to the sample group datasets. Upregulated and downregulated DAPs are colored red and blue, respectively. c Immunoblotting of synapse fractions with antibodies against AKAP11, PKA subunits, and SIK2. Right, quantifications for 4wk and 12wk animals. Data are represented as mean ± SEM. Two-way ANOVA with Tukey’s post hoc test. See Supplementary Data 1 for statistics. d A selection of GSEA analysis of synapse proteomics. Pathways displayed were significant at FDR < 0.05 in at least two age/ genotype conditions. e Sunburst plots showing SynGO Biological Process pathways and the number of P < 0.05 proteins within these ontologies. Enrichment analysis is conducted with 1-sided Fisher’s exact test with all detected proteins as a background set52. For selected ontologies, representative DAPs in the ontology are shown.
In Akap11-/- synapses, the catalytic subunits PKA Cα and PKA Cβ were the two most upregulated proteins, at both 12 weeks (Figs. 4b) and 4 weeks, with PKA regulatory subunits upregulated to a lesser extent (Supplementary Fig. 4e, f). Immunoblotting confirmed strong accumulation of PKA Cα (~5x) and R1α subunits (~2-3x) in homozygous Akap11-/- PSD fractions (Fig. 4c) and modest elevation of PKA Cα ( ~ 1.3x −1.6x) in heterozygous Akap11+/- synapses (Supplementary Fig. 4g). Besides PKA, many proteins showed changes in Akap11-/- synapse proteomics. Levels of SIK2, a PKA-regulated kinase involved in mood and circadian rhythms48 and ARHGAP20, a Rho GTPase-activating protein49, were significantly decreased in Akap11-/- synapses, at both 4 weeks and 12 weeks (Fig. 4b, Supplementary Fig. 4e, f). Notably, levels of AKAP11 itself were significantly decreased in Akap11+/- synapse fractions (Fig. 4b, c; Supplementary Fig. 4e), which is in contrast to the minimally altered levels of AKAP11 in Akap11+/- crude lysate (Fig. 3; Supplementary Fig. 3). This suggests that AKAP11 haploinsufficiency results in synapse-specific alterations to cellular signaling that are not apparent in bulk tissue readouts.
GSEA analysis of the synapse proteomic changes (Fig. 4d, Supplementary Data 3) revealed upregulation of “Vesicle tethering complex” and “Ubiquitin ligase complex” processes at 4 weeks (Fig. 4d, Supplementary Fig. 4h), which resembles the pathway changes observed in synapses purified from post mortem BD and SCZ human brain tissues50. These findings may relate to our observation that AKAP11 interacts with proteins involved in ER-Golgi transport and proteostasis (Fig. 2c, k). We further analyzed DAPs using GO terms from SynGO51 to dissect the “sub-synaptic” processes that are altered in Akap11 mutant synapses (Fig. 4e). DAPs in Akap11-/- mutant synapses were represented among pathways, such as “Postsynaptic modulation of chemical transmission”, and “Postsynaptic neurotransmitter receptor endocytosis”. Relatedly, proteins in the “Clathrin coat” GO were downregulated in 4wk and 12wk Akap11+/- synapse proteomes (Supplementary Fig. 4i).
Hyperphosphorylation of synapse proteins in Akap11 mutant mice
Within Akap11-/- synapses, the levels of several protein kinases and phosphatases were altered, most prominently the accumulation of PKA catalytic subunits Cα and Cβ (Fig. 4b, c, Supplementary Fig. 4e). To quantify phosphorylation changes in synaptic proteins in an unbiased fashion, we analyzed PSD fractions purified from Akap11 mutant cortex by quantitative phosphoproteomics. Akap11 mutants showed a large number of differentially abundant phosphoproteins (DAPPs, nominal P < 0.05), which was higher in Akap11-/- than Akap11+/-, and higher at 4 weeks than 12 weeks (Fig. 5a, Supplementary Fig. 5a, Supplementary Data 2). We mapped changes in the synapse phosphoproteome against a reference of known and predicted kinase effectors52 to infer the kinases responsible. The vast majority of PKA phospho-substrates detected in the synapse phosphoproteomics data were increased in abundance in Akap11-/-; and a number of these phosphoproteins were also highly upregulated in Akap11+/- heterozygous mutant synapses (Fig. 5b). The sequences flanking the phosphosites showed strong enrichment of the basophilic RRXS*/T* motif, particularly in the upregulated DAPPs of Akap11-/- synaptic fractions at both 4 and 12 weeks (Fig. 5c), which matches the consensus amino acid sequence that is preferred for phosphorylation by PKA53. Together, the synapse proteomics and phosphoproteomics data show that PKA kinase activity as well as protein levels are abnormally elevated in the synapses of Akap11 mutant mice.

a Number of phosphoproteins reaching a significance threshold of nominal P < 0.05. Fractions of upregulated and downregulated DAPPs are highlighted in red and blue, respectively. b Volcano plots of 12wk synapse phosphoproteomics data for Akap11+/- and Akap11-/- vs. WT controls. Phosphoproteomic measurements are normalized to MS-proteomics measurements from the same samples. Known PKA Cα substrates from PhosphoSitePlus are labeled green. AKAP11 phosphopeptides are omitted from Akap11-/- plots, for clarity. a,b A moderated two-sample t-test was applied to the datasets to compare WT, Akap11+/- and Akap11-/- sample groups. c Motif analysis of peptides flanking the phosphorylation site of P < 0.05 phosphosites in A. n(fg) are the number of foreground peptides, and n(bg) are the number of background peptides. Red lines indicate FDR significance, calculated by log odds enrichment. d PTM-SEA of synapse phosphoproteomics data, using known kinase substrates from PhosphoSitePlus. Black borders indicate FDR significance. e ELISA-based PKA activity measurements comparing total lysate and synapse fractions of cortex (left) and striatum-enriched tissue (right). One Unit (U) is defined by the manufacturer as the quantity of PKA that catalyzes the transfer of 1.0 pmol phosphate from ATP to substrate. Two-way ANOVA with Tukey’s post hoc test. f Immunoblotting and quantification of AKAP11, PKA subunits, p-GluA1S845 and GluA1 in 12wk cortical total lysate. One-way ANOVA with Tukey’s post hoc test. e,f Data are represented as mean ± SEM. See Supplementary Data 1 for detailed statistical information.
Post-Translational Modification Signature Enrichment Analysis (PTM-SEA), which measures enrichment of kinase substrate sets (akin to GSEA using GO terms)54, also indicated elevation of phosphorylated PKA substrates in Akap11-/-, and to a lesser degree, Akap11+/- synapses (Fig. 5d). Phosphorylation of substrates by CaMK2A, a kinase involved in synaptic potentiation55 was significantly downregulated in Akap11-/- synapses at 12 weeks. Since AKAP11 is reported to inhibit GSK3β, it was intriguing to find that phosphorylation of β-Catenin (a well-known GSK3β substrate, encoded by Ctnnb1) was increased at Ser37 in Akap11 mutants56,57 (Supplementary Fig. 5c). Phosphorylation of GSK3β itself on S9, a PKA phosphosite that inhibits GSK3β activity58, was significantly elevated in Akap11 mutants (Supplementary Fig. 5f). Regulation of brain GSK3β by Akap11 is especially relevant because lithium, an inhibitor of GSK3, is a mainstay treatment of BD59 and GSK3β overactivity has been implicated in SCZ animal models60.
To further test whether Akap11 deficiency altered PKA activity in a compartment-specific fashion, we compared net PKA activity in total lysate versus synapse fractions, using a colorimetric enzyme-linked immunosorbent assay (ELISA) that measures phosphorylation of a PKA substrate. PKA activity was increased in both total lysates and synapse-enriched fractions of cortex and striatum (see Methods, Fig. 5e, Supplementary Fig. 5e). Notably, the relative increase of PKA activity is greater (~3.9x) in synapse fractions than in total lysate (~1.6x).
Among the most highly changed phosphoproteins in Akap11-/- synapses was AMPA receptor subunit GluA1 (Gria1), which is phosphorylated by PKA at S845 (annotated as S863 in the UniProt mouse reference; Fig. 5b, f; Supplementary Fig. 5a, b). Phosphorylation of GluA1 at S845 is a well-known correlate of AMPA receptor trafficking to the synapse61,62,63,64,65. We confirmed by immunoblotting that GluA1S845 phosphorylation was sharply increased (~4x) in brain lysates of Akap11-/- mutants, while total GluA1 protein showed little change (Fig. 5f, Supplementary Fig. 5g). Additional DAPPs in Akap11-/- synapses that are known to be regulated by PKA include Snap25T138, a phosphosite required for presynaptic vesicular release66 and RIM1S413, phosphorylation of which is induced during synaptic potentiation67,68,69 (Fig. 5b). In Akap11+/- synapses, PKA substrate DAPPs included RIMS1S413 and ITPR2S937, a phosphosite that regulates intracellular calcium release70. The strength of synaptic transmission is regulated in large part by dynamic phosphorylation of synaptic proteins, including postsynaptic glutamate receptors subunits, such as GluA171. By mapping our phosphoproteomics data onto the phosphosites that are induced with chronic neuronal activation or silencing72, we found that many phosphosites associated with upscaling or downscaling of synaptic strength were also highly changed in Akap11 mutants (Supplementary Fig. 5d), reflecting potential changes in synaptic function.
Aberrant synaptic function in Akap11 mutants
To directly examine synaptic function and plasticity in Akap11 mutants, we performed electrophysiology recordings from acute hippocampal slices (6-8 week old mice). Under basal conditions, the stimulation input/output relationships (stimulation of Schaffer collaterals to CA1) of Akap11-/- mice were indistinguishable from WT (Supplementary Fig. 6a). The amplitude of early long-term potentiation (LTP) induced by theta burst stimulation (TBS) was modestly but significantly reduced in Akap11-/- mice (Supplementary Fig. 6b). We speculate this phenotype could be due to partial occlusion of LTP by hyperphosphorylated GluA1 at S84564,71,73. Akap11-/- slices also showed heightened paired-pulse facilitation (Supplementary Fig. 6c), suggesting reduced presynaptic release probability, which might relate to altered RIM1S413 and Snap25T138 phosphorylation seen in the synapse phosphoproteomics (Fig. 5b). The electrophysiologic phenotype in heterozygous Akap11+/- hippocampus was generally not significantly different from WT, though there was a trend toward reduced magnitude of early LTP (Supplementary Fig. 6b).
Altered synaptic genes and striatal circuits revealed by transcriptomic analysis
Akap11 mRNA is broadly expressed across the brain and across postnatal development in WT mice (Supplementary Fig. 7a). To query the brain regions/circuits and molecular pathways that are impacted by Akap11 loss of function, we conducted bulk RNA-seq on PFC, SSC, Str, Thal, Hipp, SN, as well as single-nucleus RNA sequencing (snRNA-seq) on PFC and Str, at 4 weeks and 12 weeks (Fig. 6a, Supplementary Fig. 7b, c). By bulk RNA-seq, we found more differentially expressed genes (DEGs; defined as genes that changed in either direction with FDR < 0.05, Supplementary Data 4) in Akap11-/- than Akap11+/-, and at 4 weeks than at 12 weeks of age (Fig. 6b). Based on DEG count, the most impacted regions in Akap11-/- were striatum, thalamus and PFC, whereas thalamus was the most affected brain region in Akap11+/-. The striatum had by far the most DEGs, at either age, in Akap11-/-. Only a few DEGs were common across brain regions (Supplementary Fig. 7d), and the correlation of Log2-Fold-Changes of nominally significant genes was low between different brain regions, and modest between 4 and 12 weeks for the same brain region (Supplementary Fig. 7e). Thus the transcriptional consequence of Akap11 mutation varies with the brain region and developmental context.

a Schematic of brain regions collected for RNA sequencing. b Number of differentially expressed genes (DEGs) in bulk RNA sequencing across brain regions and ages. Experiments are n = 4 to 6 per condition (see Methods). Area of circles represents DEG counts, defined by FDR adjusted P-values < 0.05, for Akap11 mutants compared to WT controls. Two-tailed Wald test, with B-H FDR correction. c GSEA of bulk RNA sequencing, showing MsigDB pathways of interest, genes associated with psychiatric and neurological conditions by GWAS12,83, and a curated set of activity-regulated genes80. Black outline indicates FDR < 0.05 significance. GSEA uses a Kolmogorov-Smirnov test, two tailed, with B-H FDR correction applied. d Schematic of snRNA-seq experiments in PFC and striatum, with UMAP plots of major cell types identified at 12w by snRNA-seq. Experiments are n = 4 per condition, with the exception of 12w Str, with n = 5 WT. e Number of DEGs in snRNA-seq experiments, defined by having an FDR adjusted P-value < 0.05. Comparisons were calculated using two-tailed likelihood ratio test, which were FDR corrected with B-H adjustment. f Heatmap showing Log2FC values for a selection of neuromodulation-related genes, plotted across transcriptomics, proteomics, and phosphoproteomics measurements. Genes indicated in the phosphoproteomics columns reflect the most significantly changed phosphosite per gene. P < 0.05 proteins and FDR < 0.05 transcripts are outlined with a black box.
Nonetheless, GSEA of bulk transcriptomic changes revealed consistent dysregulation of biological pathways related to synapses, mitochondria/oxidative phosphorylation, ribosomes, and RNA processing/splicing, which was particularly evident in 4wk Akap11+/- (Fig. 6c, Supplementary Data 5). The “Synapse” GO term was consistently downregulated in PFC and SN at both 4 and 12 weeks in Akap11+/- and Akap11-/- mutants (Fig. 6c). Immune-related pathways were upregulated in the 4wk PFC and SN, which is notable in the context of synaptic pruning by glia during development and disease74 and SCZ associations with immune-related genetic loci75,76. Two pathways required for cellular energetics and upkeep, “Oxidative phosphorylation” (Ox-phos) and “Ribosome”, were changed in correlated fashion across brain regions, similar to observations in another SCZ rare variant mouse model (GRIN2A)12; these pathways are also altered in human SCZ postmortem samples77,78. The “ribosome” and “oxidative phosphorylation” pathway (a mitochondrial process that generates ATP) were down-regulated at 4 weeks in the PFC of both Akap11+/- and Akap11-/- mutants (Fig. 6c). We investigated changes to neuronal activity at the brain region level in Akap11 mutant mice. As a surrogate measure of neuronal activity, we conducted GSEA of the bulk RNA-seq data using a set of activity-regulated genes (ARGs, Fig. 6c)12,79. At 4 weeks, ARGs were upregulated in the striatum of Akap11+/- and downregulated in the SSC and PFC of Akap11-/- (though the latter did not reach significance). The consistent downregulation of the “Synapse”, “Ox-phos” and “ARGs” GO terms in PFC is suggestive of hypofunction of PFC that is reported in fMRI studies of humans with SCZ and BD80,81.
We queried the bulk RNA-seq changes in Akap11 mutants for enrichment of SCZ- and BD-associated risk genes identified from human GWAS (Fig. 6c). The most significant enrichments occurred for BD risk genes, and interestingly, risk genes associated with multiple psychiatric disorders were most enriched in Akap11+/- heterozygous mutants at 4 weeks of age (Fig. 6c). Though reaching lower statistical significance across fewer brain regions, the pattern of SCZ risk gene enrichment had an overall similar pattern to BD. Besides SCZ and BD, we also observed similar GSEA signals for genes associated with ASD and neurodevelopmental disorders (NDD)37,82,83,84. Overall, these results support that Akap11 functionally engages with other SCZ and BD and neurodevelopmental risk genes, and are consistent with the known genetic overlap between SCZ, BD, and NDD5,85.
Major disturbances to striatal neurons revealed by single nucleus RNA-seq
In WT mice, Akap11 mRNA is expressed in excitatory and inhibitory neurons in all brain regions, as well as in glia (Supplementary Fig. 8a). To investigate the cell type-specific effects of Akap11 deficiency, we conducted single-nucleus RNA sequencing (snRNA-Seq) in the PFC and striatum at 4 and 12 weeks (Fig. 6d, Supplementary Fig. 8b, Supplementary Data 6). There were no major changes in cell type proportions in Akap11 mutant PFC or striatum (Supplementary Fig. 8b). Although all cell types in PFC and striatum showed transcriptomic changes in Akap11 mutants, the largest numbers of DEGs (FDR < 0.05) were found in the major neuron-types of the striatum of Akap11-/-, namely the inhibitory direct- and indirect-pathway spiny projection neurons (dSPNs, iSPNs; Fig. 6e). Transcriptomic alterations in Akap11-/- mutants were highly correlated between iSPNs and dSPNs (Supplementary Fig. 8c,d) and between 4 and 12 weeks of age (Supplementary Fig. 8d, e). Consistent with bulk RNA-seq findings (Fig. 6b), Akap11+/- had much less effect than Akap11-/- on SPNs in snRNA-seq (Fig. 6e, Supplementary Fig. 8d, f). Nearly all PFC neuron subtypes showed downregulation of the “Synapse” GO term (Supplementary Fig. 9a, Supplementary Data 7), which aligns with bulk RNA-seq (Fig. 6c). Unlike striatal neurons, which are much more impacted by homozygous than heterozygous Akap11 mutation, glia in the PFC and striatum (astrocytes, oligodendrocytes and microglia) showed comparable numbers of DEGs genes between Akap11+/- and Akap11-/- (Fig. 6e). Akap11 mutant astrocytes showed downregulation of the “Sterol Biosynthesis” pathway, particularly in the 4 week Akap11+/- PFC and Akap11-/- striatum (Supplementary Fig. 9b). Across regions, ages and genotypes, decreases in synapse-related gene expression in neurons often occurred alongside decreased expression of cholesterol biosynthesis genes in astrocytes (Supplementary Fig. 9c), suggesting coordinately disturbed neuron-glia gene expression, as has been observed in postmortem human SCZ brain tissue86.
The largest changes in the Akap11 mutant SPNs highlighted several neuromodulatory systems, such as serotonin (example genes: Htr2a, Htr4), adenosine (Adora1), acetylcholine (Chat, Chrna3), and dopamine (Cartpt) (Fig. 6f; Supplementary Fig. 10a, b). Intriguingly, the serotonin receptor HTR2A, which is a target of hallucinogenic drugs87,88, was downregulated in both Akap11+/- synapse proteomics and snRNA-seq of striatum SPNs, and Htr4 was robustly downregulated in bulk RNA-seq of striatum and in snRNA-seq of striatal SPNs (Fig. 6f; Supplementary Fig. 10a, b). Adenosine receptor ADORA1, a G-protein coupled receptor that inhibits cAMP production, was downregulated in Akap11-/- synapse proteomics as well as in bulk RNA-seq and snRNA-seq data in striatum and SPNs. Choline acetyltransferase (Chat) was also prominently downregulated in the striatum of Akap11-/- (Fig. 6f). Chat participates in the synthesis of acetylcholine, and signaling through muscarinic acetylcholine receptors is implicated in SCZ pathophysiology89,90. Transcellular signaling factors Epha10, Cdh9, and Igsf21 were among the most altered genes in the Akap11-/- striatum (Fig. 6f). Epha10 is a receptor tyrosine kinase that acts as an axon guidance molecule91,92. Cdh9, which decreased consistently across proteomics and transcriptomics measurements of Akap11-/-, is involved in calcium dependent cell-cell adhesion93. Together, these alterations in neuromodulation and trans-cellular signaling indicate potentially aberrant cell-to-cell interactions in the striatum of Akap11-/- mice.
Dopamine is a key neuromodulator in the striatum94. Excessive or aberrant dopaminergic signaling in the striatum is a long-standing hypothesis in pathophysiology of SCZ and other psychiatric disorders, underpinned by the antipsychotic efficacy of D2 dopamine receptor antagonists95.We found several dopamine-related genes, proteins, and posttranslational modifications that were altered in Akap11 mutants (Fig. 6f), including: (i) increased phosphorylation of tyrosine hydroxylase (TH) at S40, which positively regulates its dopamine biosynthesis activity96,97; (ii) reduction of protein and increased phosphorylation of dopamine receptor DRD1; (iii) increased phosphorylation of RASGRP2S116 in 12 week Akap11-/- synapses (Fig. 5b), which is consistent with enhanced DRD1 signaling98; (iv) elevated MAO-B protein in synapse fractions, an enzyme that degrades dopamine, v) increased proportion of Drd2-expressing (Drd2+) neurons in Akap11+/- relative to WT at 4 and 12 weeks (Supplementary Fig. 10c). One dopamine-regulated transcript, Cart (Cocaine- and amphetamine-regulated transcript)99,100 was highly elevated in the Akap11-/- striatum (bulk RNA-seq) and in striatal SPNs and PFC excitatory neurons (snRNA-seq) (Supplementary Fig. 10d) which may reflect elevated dopamine signaling in striatum and in PFC of Akap11 mutants.
In vivo striatal PKA dynamics and dopamine signaling are regulated by AKAP11
Two of the most striking phenotypes of Akap11 mutant mice are highly elevated PKA protein (Figs. 3–4) and strong transcriptomic changes in the striatum and SPNs (Fig. 6). PKA is a central signaling molecule in the striatum, acting in the signal transduction pathway downstream of dopamine receptors94. To assess real-time dynamics of PKA activity in live animals, we used fluorescent lifetime fiber photometry measurements of FLIM-AKAR, a fluorescence lifetime reporter of net PKA activity101,102 that was virally expressed in the nucleus accumbens (NAc; within the ventral striatum) (Fig. 7a, Supplementary Fig. 11a–c). In awake, behaving Akap11-/- mice, PKA activity was elevated across 10 min recording sessions in an open field arena (Fig. 7b), which was consistent across weeks of measurement (Supplementary Fig. 11d). As a negative control, there were no differences across Akap11 genotypes in the lifetime of FLIM-AKART391A, a mutated variant of that is insensitive to PKA activity (Fig. 7c).

a Schematic overview of the viral injection strategy. b Fluorescence lifetime of FLIM-AKAR in WT or Akap11 mutant mice. The y-axis is inverted to reflect that decreased fluorescence lifetime corresponds to increased net PKA activity. c Fluorescence lifetime of FLIM-AKART391A in WT or Akap11 mutant mice. d Fluorescence lifetime changes of FLIM-AKAR in response to D2R antagonist. e Fluorescence lifetime changes of FLIM-AKAR in response to D2R antagonist, normalized to each genotype’s baseline level of activity. f Fluorescence lifetime changes of FLIM-AKAR in response to D1R antagonist. g Fluorescence lifetime changes of FLIM-AKAR in response to D1R antagonist, normalized to each genotype’s baseline level of activity. h Distance traveled after administration of amphetamine (2 mg/kg) or saline to WT or Akap11 mutants. i Quantification of distance traveled during baseline (0–90 min), 90–120 min, and 150–180 min. Repeated measures two-way ANOVA with Tukey’s post hoc test. j Schematic of dopamine to PKA regulation by Akap11. b–i Data are represented as mean ± SEM, with the shaded regions or error bars representing SEM. b,d,e,g Two-way ANOVA with Tukey’s post hoc test. See Supplementary Data 1 for statistics.
Dopamine receptors are enriched in the striatum, and excessive dopamine signaling in striatum may drive positive symptoms (e.g. hallucinations) of SCZ103,104,105, which is supported by the clinical efficacy of dopamine receptor 2 (Drd2) antagonists for positive symptoms95. To evaluate how PKA is dynamically regulated by dopamine in Akap11 mutant mice, we administered dopamine receptor antagonists via intraperitoneal injection. Dopamine acting on D2-type receptors inhibits cAMP production and thereby inhibits PKA94,106. As expected, net PKA activity was increased in striatal neurons following administration of D2 antagonist (eticlopride hydrochloride, 0.5 mg kg−1) (Fig. 7d). Despite the higher baseline activity of PKA activity in Akap11-/- striatum, phosphorylation of the FLIM-AKAR sensor was further enhanced by D2 antagonist in Akap11-/- mutants (Fig. 7e). Dopamine acting on D1-type receptors stimulates cAMP synthesis and activates PKA94,106. As expected, delivery of D1 antagonist (SKF83566, 3 mg kg−1) suppressed net PKA activity in striatum in all 3 genotypes (Fig. 7f). When normalized to the pre-drug baseline level, D1R antagonism caused a more pronounced inhibition of PKA activity in Akap11-/- mice (Fig. 7g). Thus loss of Akap11 results in markedly higher basal PKA activity; however, this increased basal activity does not occlude further activation or block inhibition of PKA by dopamine receptor drugs, but rather expands the dynamic range of PKA activation or inhibition by endogenous dopamine.
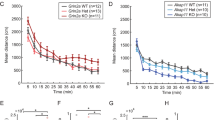
Dopamine in the striatum regulates multiple aspects of behavior, including movement, reward processing, cognition and mood107. Dopamine levels induced by amphetamine are higher in the striatum of schizophrenic patients108. We tested locomotor response to subcutaneous amphetamine administration, which depends on dopamine and is often used as a behavioral measure of dopamine sensitivity in rodents (Fig. 7h, i). WT mice, as expected, showed amphetamine-induced hyperactivity (AIH; measured by distance traveled). During the first 30 minutes after amphetamine administration, Akap11-/- mice displayed less AIH than WT littermates; however, Akap11-/- mice showed a much more sustained hyperactivity response, such that at 60–90 min after amphetamine administration, Akap11-/- mice still showed high levels of locomotion, when WT animals had returned to baseline (Fig. 7h). During this prolonged period of hyperactivity, Akap11-/- mice spent more time in the center of the open field, and exhibited more rearing movements than WT or heterozygous mutants (Supplementary Fig. 12a–f). In conjunction with FLIM-AKAR and behavioral readouts, these in vivo pharmacological experiments suggest that the strength and temporal dynamics of dopamine-to-PKA signaling in striatal SPNs are abnormally high and excessively prolonged in Akap11 mutants (Fig. 7j).
Discussion
Genetic variation contributes strongly toward SCZ and BD risk. Psychiatric symptoms ultimately emerge from dysfunction of specific neural circuits; however the mechanisms by which genetic variants lead to molecular-cellular disturbances and then to circuit dysfunction remain unclear. In this study, we examined mice with mutation in Akap11, a risk gene for SCZ and BD. Our unbiased multi-omic studies revealed that AKAP11 loss of function results in a remarkable elevation of PKA activity in the brain, especially at synapses; and the striatum, the major site of dopaminergic signaling and integration in the brain, is particularly impacted by Akap11 deficiency.
Hyperdopaminergic signaling in striatum is strongly implicated in the positive symptoms of psychotic disorders. Our study focuses attention on factors downstream of dopamine and dopamine receptors—namely, PKA dysregulation in SPNs—as an important mechanism by which striatal dopaminergic signaling can be exaggerated. Since PKA is elevated across the brain in Akap11 mutants, we expect that excessive PKA activity also contributes to pathophysiological disturbance in other brain regions and circuits beyond the striatum, such as the PFC and thalamus, where we also saw major transcriptomic changes.
It is important to note that, in comparison with homozygous Akap11 knockout, the phenotypes of Akap11 heterozygous mutants are relatively subtle. With the exception of transcriptomics changes in thalamus and in glial cell types (astrocytes, oligodendrocytes and microglia), we observed much larger molecular, physiological and behavioral phenotypes in homozygous than in heterozygous mutants. However, the phenotypes that were most prominent in Akap11-/-, such as elevated PKA, were also seen in Akap11+/-, albeit to a lesser degree. The modest phenotype in Akap11 heterozygotes could be explained by the fact that Akap11 haploinsufficiency resulted in only slight changes in AKAP11 protein levels. This latter observation suggests that there are proteostatic mechanisms operating in heterozygous mutants to buffer against sharper losses of AKAP11 protein and function. Since Akap11 regulates autophagy, it would be especially interesting to test whether cellular stressors, which can stimulate autophagy, could exacerbate the reduction of AKAP11 protein and amplify the downstream consequences, such as elevation of PKA activity in Akap11 heterozygous mutants, thereby offering a potential way for environmental triggers to induce a stronger disease phenotype.
Across transcriptomic measurements in varying brain regions and ages, the most consistent alterations were among genes related to oxidative phosphorylation, mitochondrial respiration, ribosomes and RNA processing/splicing. Intriguingly, selective autophagy of PKA regulatory subunit R1α through interactions with AKAP11 increases PKA activity and mitochondrial metabolism in the context of glucose starvation23. We have also uncovered direct evidence for abnormal lipid metabolism in the Akap11 mutant brain109. Future work should explore whether Akap11 mutant brains respond differently to environmental, metabolic and cellular stressors.
There are likely to be hundreds of genetic variants underlying SCZ and BD. Because these genes are diverse in molecular function, their most proximal molecular mechanism would be expected to be varied and perhaps unique to each genetic variant. Thus, it is essential to identify downstream convergent cellular and circuit disturbances that are shared among different SCZ and BD risk genes. Our findings highlight one mechanistic pathway by which a specific genetic variation leads to neural circuit dysfunction. In this case, loss of function of Akap11, which is required for autophagy-degradation of PKA23,45, leads to elevated PKA kinase activity in neurons, resulting in unbalanced dopamine-to-PKA signaling in SPNs of striatum. Another mouse model of a rare variant SCZ gene (GRIN2A)5 also shows evidence of hyperdopaminergic signaling in striatum, but via a mechanism different than exaggerated PKA activation12. In SETD1A mutant cell lines and Trio mutant mice, which are also genetic models for SCZ5, there is elevated PKA activity that results in synaptic dysfunction13,110. Further, the cAMP synthesizing enzyme adenylyl cyclase 2 (Adcy2) is a significant genetic association for BD8. Thus, human genetics and mechanistic studies corroborate a plausible role of PKA dysregulation in psychiatric disorders.
D2 receptor antagonists are efficacious for treatment of SCZ positive symptoms; this seems counterintuitive to the results of our fiber photometry experiments, in which D2 antagonism further exacerbated the PKA hyperactivity phenotype of Akap11-/-. Although used in the clinic for decades, the physiologic mechanism for the efficacy of D2 antagonists in SCZ is still unclear111,112. Clinically effective antipsychotics (including D2 receptor antagonists) better correlate with the suppression of D1-expressing neuron (dSPN) activity, rather than the suppression of D2-expressing neurons (iSPNs)113, suggesting that the activity of D1-expressing neurons is more relevant to SCZ pathophysiology. Since dSPN neuron activity is enhanced by PKA activation98, it is reasonable to predict that dSPN activity is increased in Akap11 mutants. Future work should directly measure neuronal activity in D1 and D2 SPNs, and how their activity levels are affected by antipsychotic medicines and mood-stabilizing drugs like lithium.
A separate study of Akap11 mutant mice also found elevated PKA activity in synapse-enriched fractions, but decreased PKA activity in cytosolic fractions of the brain114. This apparent difference from our results could be accounted for methodological differences, such as full body germline knockout of Akap11 (this study) versus neuron-specific knockout in Lee et al. (2024). Another possibility is that PKA activity is differently regulated by AKAP11 across cellular compartments, which should be the subject of future investigation.
Our study, which started with unbiased -omics approaches, highlights the multi-faceted molecular control of dopaminergic signaling in striatum—already much implicated in psychosis pathophysiology by human and mouse model studies—as a worthy focus of future investigation. Our multi-omic analysis of the Akap11 mouse model, together with similar investigation of other high penetrance SCZ and/or BD genes, should enable additional hypothesis-driven experiments and lead to a more nuanced understanding of the complex and heterogeneous mechanisms underlying psychiatric disease.
Methods
Animals
Experiments were approved by the Broad Institute IACUC (Institutional Animal Care and Use Committee) and conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals (0259-10-19-1, 0288-11-20-1 and 0008-06-14). Heterozygous Akap11 mice (B6.Cg-Akap11[tm1.2Jsco/J]) were obtained from The Jackson Laboratory following cryorecovery (strain #028922). As previously described24, these mice have deletions of exons 6 and 7 of the mouse Akap11 gene to create a knockout allele. Akap11 mutant mice were deposited by the donor lab after six generations of backcrossing to the C57BL/6 J background. We confirmed the genetic background of the animals to be greater than 90% C57BL/6 J through SNP analysis (MiniMuga panel, Transnetyx). To establish the colony, founders were crossed to WT C57/BL6J mice (Jackson Laboratory, #000664) for one generation. Subsequently, heterozygous mice were crossed with each other to produce WT (WT, +/+), heterozygous (Het, +/-) and knockout (KO, -/-) littermates used for experiments. Mice were housed at AAALAC-approved facilities on a 12-hour light/dark cycle, between 30-60% humidity, 67°–73° Fahrenheit, with food and water available ad libitum. For fluorescence lifetime photometry experiments (FLiP), mice were housed in a 12 h/12 h light/dark reverse cycle. FLiP and pharmacological experiments were conducted in compliance with protocols approved by the Harvard Standing Committee on Animal Care (IS-00000571-6), adhering to guidelines outlined in the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Homecage locomotor activity monitoring
Male and female mice aged P23-P34 were transferred to Digital Ventilated Cages (DVC, Tecniplast) and were single housed throughout the duration of the experiment. Briefly, DVC racks use positional sensors beneath each cage that can detect disturbances that correlate with animal movement26,115. The DVC Analytics web platform was used to generate animal locomotor indices, which calculate the percentage of 12 electrodes that measured movement over a 1-minute bin. For the locomotor activity index, a value of 1 means 12 electrodes measured movement, and a value greater than 1 means that there were > 12 detected movements. The experiment included n = 11 WT males, n = 12 WT females, n = 12 Akap11+/- males, n = 14 Akap11+/- females, n = 10 Akap11-/- males, and n = 13 Akap11-/- females. For a subset of animals, a running wheel was introduced for 1-2 weeks during the homecage study. Running wheel usage was assessed during the first 6 days of introduction. For statistical comparisons of locomotor activity index (Fig. 1b, c), locomotor activity index data was excluded on cage change days, and days where the running wheel was present.
Motion sequencing (MoSeq) behavioral recording
Mice were tested in a Motion sequencing (MoSeq) open field assay as previously described12. Briefly, a matte black bucket with a Microsoft Kinect Camera attached overhead was used to perform the recordings. Two days prior to the recording session, mice were habituated for 10 min in the recording arena. On the day of the recording session, all mice being recorded that day were placed in the room 30 min prior to the first recording. Each mouse was individually recorded for 30 min. Prior to the first recording, the bucket was wiped with 70% ethanol and allowed to dry for 5 min. This was repeated after each subsequent mouse. Between mice, the arena was cleaned and the sides of the bucket were sanded down with sandpaper as to limit the glare leading to background noise during analysis. Mice were tested after the lights-off time (Zeitgeber Time 12, ZT12) since mice were nocturnal. All tests occurred between ZT12-ZT18. Mice were 12 weeks (±1 week) of age. Sample sizes were 11 WT, 11 Akap11+/- and 10 Akap11-/- mice. Moseq syllable videos were manually annotated by five independent observers as “Grooming”, “Rearing”, “Pausing”, or “Turning, lunging or Darting”. Moseq data has been uploaded to a public repository (https://zenodo.org/records/17094753).
MoSeq computational analysis
MoSeq is a tool that employs an unsupervised machine learning algorithm to characterize and quantify complex mouse behavior. It breaks down spontaneous mouse behavior into sub-second, regulated, and fine-tuned behavioral motifs called syllables29. Computational analysis of recordings as generated above were analyzed using the Depth MoSeq pipeline available here: http://www.moseq4all.org/. Extraction was performed with default configuration parameters and minimum mouse height set to 12 to reduce visual glare in the recordings. After generating a robust model with selected kappa value (kappa = 599,484, trained with 1000 iterations of Gibbs sampling) as outlined in the pipeline, 51 syllables were produced, which explained 99% of the total frames. These syllables were manually labeled into four larger groups of mouse behavior: pausing, grooming, rearing/climbing, and darting/lunging/turning. After assigning each frame of each video a syllable label, the number of occurrences of each syllable (syllable usage) and the number of transitions from one syllable to another were quantified. Syllables were then ordered and labeled by the fraction of times they were used across all sessions, as seen in Fig. 1e, f, with syllable 0 being the most used syllable and syllable 51 being the least used behavioral motif.
Discriminatory fear conditioning
Mice were habituated to a sound attenuated conditioning chamber, “Context A,” 30.5 × 24.1 × 21.0 cm3 dimensions with an automated freezing analysis software (Med Associates) and were presented with 10X alternating trials of 20 s 3 and 10 KHz auditory tones (75 dB sound pressure level). The next day, mice underwent fear conditioning in the same chamber with the same tones. Mice were exposed to 10X trials of stimulus–unconditioned stimulus pairings (tone-foot shock) with the 3 KHz tone or 10 KHz tone serving as the reinforced conditioned stimulus and the other tone serving as the neutral non-reinforced stimulus (counterbalanced). The aversive unconditioned stimulus was a 0.3 mA foot shock co-terminating with the last 1.5 s of the CS + . Context A was cleaned before and after behavioral sessions with chlorine dioxide sterilant solution (Clidox-S). Memory recall and extinction sessions occurred 24 and 48 hours. later in a novel “Context B” consisting of the same chamber with white contextual inserts (white curved wall and white floors) and vanilla extract to serve as a distinct olfactory cue. Mice were presented with 15X trials of non-reinforced CS+ and CS- tones per session. Context B was cleaned with 70% ethanol before and after each session. All CS+ and CS- tones were presented in an alternating fashion with a 60 s intertrial interval. Memory recall and discrimination to the CS+ and CS- tones was scored as average freezing behavior (cessation of all movement other than respiration for >1 s) during the first 5X tones presentations during the first recall session. To compare sensory discrimination normalized to each animals’ behavioral performance and overall fear level we generated a discrimination index using the percent time freezing, defined as: (CS+ freeze - CS- freeze) / (CS+ freeze + CS- freeze).
Behavioral Battery
Fifty-five AKAP11 mice (n = 19 WT, n = 22 Akap11+/-, n = 14 Akap11-/-) aged between 12 and 16 weeks were used in a battery of behavioral assays including sucrose preference test, forced swim test, prepulse inhibition and social interaction tests. Animals were grouped housed in a maximum of five animals per cage, under normal lighting (lights on 7 am–7 pm) and ad libitum supply of food and water. All procedures were approved by the Institutional Animal Care and Use Committee at Boston Children’s Hospital and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Sucrose preference test
For the sucrose preference test: on day 1, mice were individually housed and were habituated to 2% sucrose for 24 h. All bottles were pre-weighed and each mouse got two bottles filled with 2% sucrose solution. The bottles are retrieved and weighed at the end of the habituation day. On day 2 and 3, mice were individually housed and underwent a sucrose preference test. One of the bottles was filled with 2% sucrose solution while the other was filled with water. The position of the sucrose/water bottle was reversed at the end of day 2, and the bottles were weighed at the end of each day to determine water/sucrose consumption. Sucrose preference test was conducted once at the beginning of the behavioral test battery and was repeated once after other behavioral assays (forced swim test and prepulse inhibition) were completed. The experimenter was blinded to the genotype of the test mice throughout the experiment.
Forced swim test
For the forced swim test, mice were habituated to the behavioral room 30 min prior to the experiment. A 4 L beaker was filled 2/3 of the way with water and was maintained at room temperature (RT) (24 °C ± 1 °C), measured before each experiment with a glass thermometer. Mice were gently placed in water and were closely observed throughout the 6 min test. Mice were immediately removed if they were unable to keep their head above water. After the test, mice were gently dried with a paper towel and were allowed to recover in the home cage for 10 min under monitoring. Time immobile was manually scored with a stopwatch by an experimenter blinding to the animal genotype.
Prepulse inhibition
For prepulse inhibition, mice were habituated to the behavioral room 30 min prior to the start of the experiment. During the experiments, mice were allowed 5 mins to acclimate to the 60 db background noise that was present throughout the experiment. Mice were presented with four variable auditory pre-pulse levels (70, 74, 78, 82 dV; 20 ms), followed by an auditory startle pulse (110 dB, 20 ms) 100 ms after the pre-pulse. A total of 20 trials is delivered for each mouse (5 repeats for each of the four pre-pulse levels) in a random order with a variable inter-trial-interval ranging from 10–30 s. The startle response was measured during the 250 ms period after the onset of the startle pulse by a force meter (Kinder Scientific). The percentage of prepulse inhibition is quantified by the following equation: %PPI = 100 × [(pulse-alone) – (pre-pulse + pulse score)]/pulse-alone score.
Three-chamber social interaction
Mice were habituated in the room 30 min prior to the start of testing. A plastic apparatus (60 × 40 × 20 cm) with an open top divided into 3 chambers (20 × 40 × 20 cm) by 2 transparent walls was used. The testing session starts with a 10 min habituation period, during which chambers are separated by a wall containing a small circular opening. TEST mice are first placed in the middle chamber and are allowed to explore the entire arena. To examine social interaction after the habituation period, an unfamiliar, sex-matched mouse (STRANGER 1) is placed in one of the side chambers. The other side has an empty cage to control for object novelty. STRANGER 1 is enclosed in a small round wire cage, which allows nose contact between the bars, but prevents fighting. The TEST mouse is allowed to explore the entire chamber during the 10 min session. The amount of time spent investigating the mice or the empty cage, time spent in each chamber, and the number of entries into each chamber are automatically scored using Ethovision. To examine social memory, after an interval of 10 min where the TEST mouse is returned to the home cage, the TEST mouse is tested in a second 10 min session to quantify social preference for a new stranger. In this second trial, STRANGER 1 remains in the wire cage and a second, unfamiliar mouse (STRANGER 2) is placed in an identical small wire cage located on the opposite side of the chamber. The TEST mouse now has a choice between the familiar mouse (STRANGER 1) and the novel mouse (STRANGER 2). The amount of time spent investigating each of the mice and the number of entries into each chamber is scored automatically using Ethovision.
Accelerating rotarod
A subset of male and female mice that underwent homecage assay (n = 7 WT, n = 6 Akap11+/-, n = 6 Akap11-/-) were evaluated with the rotarod test (Harvard 76-0770) when they were 6 months old. Biological sex was approximately evenly split between genotypes. Mice were first allowed to acclimate to the testing room in their home cages for at least 30 min prior to beginning testing. Mice underwent three training trials where they acclimated to walking on the rotating rod spinning at 4 rotations per minute (rpm). Two mice (n = 1 Akap11+/-, n = 1 Akap11-/-) were excluded from testing because they fell off immediately during all three training trials. Mice then underwent 4 test trials, with at least 1 min between trials, where the rod accelerated at a constant rate from an initial speed of 4 rpm until the mouse was unable to keep pace with the rotations and fell off of the rotating bar, or until a maximum speed of 40 rpm. This latency to fall was recorded and the data are shown as the average of 4 test trials.
Anti-AKAP11 antibodies
Two custom polyclonal rabbit anti-AKAP11 antibodies were generated for this paper and used for immunoblot experiments. For both polyclonal antibodies, the first 284 amino acids of the mouse AKAP11 protein (Uniprot reference E9Q777) were injected into rabbits, and affinity purified by a company (Genscript). The antigen sequence used was: MAAFQPLRSSHLKSKASVRKSFSEDVFRSVKSLLQSEKELCSVSGGECLNQDEHPQLTEVTFLGFNEETDAAHIQDLAAVSLELPDLLNSLHFCSLSENEIICMKDTSKSSNVSSSPLNQSHHSGMLCVMRVSPTLPGLRIDFIFSLLSKYAAGIRHTLDMHAHPQHHLETTDEDDDDTNQSVSSIEDDFVTAFEQLEEEENAKLYNDEINIATLRSRCDAASQTTSGHHLESHDLKVLVSYGSPKSLAKPSPSVNVLGRKEAASVKTSVTTSVSEPWTQRSLY and were validated by immunoblot comparisons between WT and Akap11-/- brain tissue. For immunoprecipitations, we used a commercial rabbit monoclonal antibody targeting residues surrounding Alanine 1125 of mouse AKAP11 (Cell Signaling Technologies (CST), #77313). All other AKAP11 antibodies we tested, including our own polyclonal antibodies, immunoprecipitated PKA subunits in Akap11-/- brains, indicating insufficient specificity (data not shown but available upon request).
Immunoprecipitation
Twelve week-old (P120) male mice were euthanized using CO2 inhalation, harvested around the lights-off period (Zeitgeber Time 12, ZT12) when mice are active, which was approximately ZT10-ZT14. For coIP-MS, 5 WT, 5, Akap11+/-, and 5 Akap11-/- animals underwent immunoprecipitation with the AKAP11 antibody. Three WT animals underwent immunoprecipitation with IgG control. After sacrifice, cortical hemispheres were rapidly dissected and mechanically homogenized with 12 slow strokes by a Teflon homogenizer and a glass vessel in ice-cold phosphate buffered saline (PBS) with 1% Triton-X with protease (Sigma 4693159001) and phosphatase (Sigma 4906837001) inhibitors. The homogenate was centrifuged at 10,000 g (4 °C) for 10 min and the resultant supernatant was used for immunoprecipitation. Protein concentration was measured with bicinchoninic acid assay BCA (Thermo Fisher Scientific A53225) and samples were normalized to equal protein concentrations and volumes with homogenization buffer in protein LoBind tubes (Eppendorf #022431102). For coIP-mass spectrometry, samples had 4 mg of protein in a volume of 2 mL. For coIP-immunoblot, samples had either 500 μg or 1 mg (which were matched between experimental and control samples), normalized to 1 mg/mL. Lysates were incubated overnight rotating at 4 °C with 1 μg anti-AKAP11 monoclonal antibody (1:20; CST 77313) or 1 μg mAb IgG control (CST 3900) for mass spectrometry experiments. For coIP-immunoblot, lysates were incubated with the following primary antibodies: 1:50 Rabbit anti-AKAP11 (CST 77313), 1:50 Rabbit anti-GluA1 (CST 13185), 1:50 Rabbit anti-PKA Cα (CST 4782), 1:50 Rabbit anti-PKA R1α (CST 5675), 1:50 Rabbit anti-PKA R2β (Sigma ABS14), 1:50 Rabbit anti-VAPA (Proteintech 15275-1-AP), 1:50 Rabbit anti-VAPB (Proteintech 14477-1-AP), 1:50 Rabbit anti-GSK3α (CST 4337), 1:50 Rabbit anti-GSK3β (CST 12456), 1:50 Mouse anti-DYRK1A (Abnova H00001859-M01), 1:50 Mouse anti-P62 (Abcam ab56416). As additional negative controls, Rabbit mAb IgG (CST 3900), Normal Rabbit IgG (CST 2729) or Mouse mAb IgG2a Isotype Control (CST 61656) were diluted according to manufacturer’s instructions and added in lieu of primary antibodies.
The next day, lysate-antibody mixtures were incubated with Dynabead Protein G (Invitrogen 1004D) for 1 h at RT, following the manufacturer’s specifications. Beads were then washed three times for 10 min with PBS containing 0.02% Tween20. For coIP-immunoblotting, proteins were eluted from magnetic beads with 1x Laemmli buffer and beta-mercaptoethanol (BME) mixture (Boston biosciences BP111R, Biorad 1610737, Sigma) and boiled at 98 °C for 8 min and subsequently examined by SDS-PAGE (immunoblot). For coIP-MS, samples were washed twice with ice cold 50 mM Tris-HCl pH 7.5 and transferred to a new tube. Subsequently samples were resuspended in 50 μl ice cold Tris-HCl pH 7.5.
CoIP sample preparation for liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) analysis
Samples were incubated in 80 μl of urea-based digestion buffer (30 mM Tris-HCl pH=8, 0.4 μg trypsin, 1 mM dithiothreitol, 2 M Urea) at 25 °C and 1000 rpm for 1 h. The supernatant was then collected, followed by a second incubation in 80 μl of the same urea-based buffer at 25 °C and 1000 rpm for 30 min. The second supernatant was also collected. The beads were washed with 60 μL of wash buffer (2 M Urea, 50 mM Tris-HCl pH=8 wash buffer) two times, collecting both washes. The on-bead digests (supernatants) and two washes were combined into 280 μl per sample and reduced with 2.3 μl of 500 mM dithiothreitol (DTT) for 30 min at 1000 rpm at 25 °C. This was followed by alkylation with 4.6 μl of 500 mM iodoacetamide (IAA) for 45 min at 1000 rpm at 25 °C. The samples were digested overnight with 0.5 μg of Trypsin at 700 rpm at 25 °C. Reactions were quenched with formic acid (FA) at a final concentration of 1%, then desalted using reverse-phase C-18 StageTips as previously described50 and dried down. The peptides were reconstituted in 80 μl 50 mM HEPES, followed by 8 μl of neat acetonitrile (MeCN), and isobarically labeled using 500 μg of TMT 18-plex (TMT18) reagent (Thermo Fisher Scientific A44520 & A52046) for 1 h at 1000 rpm and 25 °C. After confirming successful labeling (> 95% label incorporation), the reactions were quenched with 4 μl of 5% hydroxylamine. Samples were mixed and dried. The sample was resuspended in 300 μl of 0.1% formic acid to proceed with fractionation by bRP fractionation strategy on StageTips. The tips were packed with three plugs of Empore Extraction Disks SDB-RPS 47 mm material. StageTips were sequentially conditioned with 100 μl of methanol (MeOH) and 100 μl of 50%MeCN/0.1%FA. The sample was loaded onto the StageTips, and washed twice with 100 μl of 0.1% FA. 25 μl of 20 mM ammonium formate was added to the stage tip for buffer exchange. It was then eluted into eight consecutive fractions with increased organic content at 100 μl per elution step (5, 10, 15, 20, 23, 27, 30 and 45% MeCN in 20 mM ammonium formate). The fractions were transferred to HPLC vials and dried.
LC-MS/MS analysis of coIP samples
The samples were analyzed on a Q Exactive HF-X mass spectrometer with a Vanquish Neo UHPLC system (Thermo Fisher Scientific). The solvents used for the HPLC system were solvent A (0.1% FA) and solvent B (0.1% FA/100% MeCN). The samples were injected into a 75 μm ID PicoFrit column packed manually to approximately 28 cm of length with ReproSil-Pur C18-AQ 1.9-μn beads (Dr. Maisch) and heated to 50°C for chromatographic separation.
The bRP fractions were loaded into the column at a flow rate of 200nL/min. The chromatography consists of a 110 min method with a gradient of 1.6–5.4% solvent B for 1 min, 18–27% B for 22 min, 27–54% B for 9 min, 54–72% B for 1 min, followed by a hold at 72% B for 5 min and 72–45% B for 10 min. The mass spectrometer was operated in data-dependent acquisition mode. The MS parameters were set as follows: MS1, r = 60,000; MS2, r = 15,000; MS1 AGC target of 3e6 with maximum injection time of 10 ms; MS2 for the 20 most abundant ions using an AGC target of 5e4 and maximum injection time of 105 ms; and normalized collision energy of 28.
Data analysis of the coIP-LC-MS/MS experiment
Collected data were analyzed using the Spectrum Mill software package (Broad Institute). All extracted spectra were searched against a UniProt database containing mouse reference proteome sequences retrieved on 04/07/2021 and containing 55,734 entrees. Fixed modifications were carbamidomethylation at cysteine. TMT labeling was required at lysine, but peptide N termini could be labeled or unlabeled. Allowed variable modifications were protein N-terminal acetylation and oxidized methionine.
Protein quantification was achieved by taking the ratio of TMT reporter ions for each sample over the median of all channels. TMT18 reporter ion intensities were corrected for isotopic impurities in the Spectrum Mill protein/peptide summary module using the afRICA correction method which implements determinant calculations according to Cramer’s Rule79 and correction factors obtained from the reagent manufacturer’s certificate of analysis for lot numbers XD343709 and XD346675. After performing median-MAD normalization, a moderated two-sample t-test was applied to the datasets to compare WT, Akap11+/- and Akap11-/- sample groups.
Immunoblot
Four week (P30) or 12 week (P120) male mice were euthanized using CO2 inhalation between ZT10-ZT14, after which whole cortices were rapidly dissected and flash-frozen in liquid nitrogen. Single hemispheres were homogenized in a 5 ml ice-cold homogenization buffer (1% SDS in PBS) supplemented with phosphatase (Sigma 4906837001) and protease inhibitors (Sigma 4693159001), using the mechanical homogenization procedure described above. After BCA (Thermo Fisher Scientific, A53225 or 23225) quantification of protein concentration, samples were diluted with 6X Laemmli buffer with BME (Boston Bioproducts, BP-111R) or 2x Laemmli buffer (Biorad, 1610737) with BME (Sigma, M3148) to a final concentration of 1x and boiled in reducing SDS loading buffer at 98 °C for 8 min. After preparation, sample aliquots were frozen at −80 °C. Before running on a gel, samples were thawed, heated at 98 °C for 1 min and centrifuged briefly. Samples were run on NuPAGE 3–8% Tris Acetate gradient gels (Invitrogen) at a constant voltage. Equal amounts of sample were loaded unless otherwise specified (Supplementary Fig. 4a,-b). For Fig. 3a, a subset of smaller molecular weight proteins were tested on 4–20% tris glycine gradient gels (Invitrogen). Proteins were transferred to 0.2 mm nitrocellulose membranes using semi-dry transfer (Bio-Rad Transblot Turbo; 25 V 30 min) and visualized by Coomassie stain (Sigma, P7170). Subsequently, membranes were blocked with 5% milk in Tris-buffered saline with 0.2% Tween 20 (TBST). Membranes were incubated overnight with primary antibodies in 1% milk TBST with gentle rotation at 4 °C. Membranes were incubated with primary antibodies overnight at 4 °C, washed three times with TBST for 10 min, and then incubated with secondary HRP-conjugated antibodies raised against the appropriate species for 1 h at RT. After three additional TBST washes, membranes were imaged on ChemiDoc MP (Bio-Rad) and analyzed using Image Lab Software (Bio-Rad). Enhanced chemiluminescence (Thermo Fisher Scientific, 34580, 34096 and A38556) and exposures were optimized to obtain signals in the linear range. Membranes were then stripped (Thermo, 46430),washed three times, re-blocked with 5% milk in TBST, and re-probed as described above. For cortical lysate immunoblots, 20 μg of protein was loaded. For microdissected brain regions, 15 or 20 μg of protein and 5 or 10 μg synapse fraction protein were used for SDS-PAGE, unless otherwise specified (Supplementary Fig. 4a, b). For all experiments, protein loading amounts were consistent across samples. Uncropped images appear in Source Data Files.
For coIP-immunoblot, the following antibodies were used during SDS-page (after immunoprecipitation): 1:1000 Mouse anti-PKA R1α (BD 610609), 1:1000 Mouse anti-PKA Cα (BD 610980), 1:1000 Mouse anti-PKA R2β (BD 610625), 1:1000 Mouse anti-GluA1 (NeuroMab 75-327), 1:1000 Rabbit anti-Vinculin (CST 4650), 1:1000 Rabbit anti-VAPA (Proteintech 15275-1-AP), 1:1000 Rabbit anti-VAPB (Proteintech 14477-1-AP), 1:1000 Rabbit anti-GSK3α (CST 4337), 1:1000 Rabbit anti-GSK3β (CST 9315), Mouse anti-GSK3α/β (Thermo Fisher Scientific 44-610), 1:1000 Rabbit anti-Dyrk1a (CST 2771), 1:1000 Rabbit anti-P62 (CST 5114). For coIP-immunoblot, we used horseradish peroxidase (HRP)-conjugated secondaries recognizing the host species of the primary antibody (Thermo Fisher Scientific 31460 and 31430 for rabbit and mouse respectively). When the IP and immunoblot antibodies were from the same species (Fig. 2g), we used secondaries that are selective toward non-denatured IgG (1:1000, TrueBlot HRP, Rockland, 18-8816-33 and 18-8817-30).
For immunoblot, the following antibodies were used:
1:1000 Rabbit anti-AKAP11 (Custom R171), 1:1000 Rabbit anti-AKAP11 (Custom R173), 1:1000 Mouse anti-PKA R1α (Thermo Fisher Scientific MA5-24981), 1:1000 Rabbit anti-PKA R1α (CST 5675), 1:1000 Rabbit anti-PKA Cα (CST 4782), 1:1000 Rabbit anti-Iqgap1 (Abcam ab86064), 1:1000 Rabbit anti-Dyrk1a (CST 2771), 1:1000 Rabbit anti-GSK3α (CST 4818), 1:1000 Rabbit anti-GSK3β (CST 9315), 1:1000 Mouse anti-P62 (Abcam ab56416), 1:1000 Rabbit anti-LC3A/B (CST 12741), 1:1000 Rabbit anti-VAPA (Proteintech 15275-1-AP), 1:1000 Rabbit anti-β Catenin (CST 8480), 1:1000 Mouse anti-Protein Phosphatase 1 (Santa Cruz SC-7482), 1:1000 Rabbit anti-Sik2 (CST 6919), 1:1000 Rabbit anti-p-GSK3αS21 / anti-p-GSK3βS9 (CST 8566), 1:1000 Rabbit anti-p-GSK3α / anti-p-GSK3β (CST 5676), 1:1000 Rabbit anti- p-GSK3βS9 (CST 9336), 1:1000 Rabbit anti- p-GluA1S845 (CST 8084), 1:1000 Rabbit anti- GluA1 (CST 13185), 1:1000 Mouse anti- GluA1 (NeuroMab 75-327), 1:1000 Mouse anti-PSD95 (BioLegend 810301), 1:1000 Rabbit anti-Homer1 (Synaptic Systems 160003), 1:1000 Guinea Pig anti-Synaptophysin 1(Synaptic Systems 101004). 1:1000 Mouse anti-PSD95 (clone K28/43, Antibodies Inc 75-028). HRP-conjugated anti-Rabbit or anti-Mouse IgG (Thermo Fisher Scientific 31460 and 31430). β-actin was probed using an HRP conjugated primary antibody after all other proteins were probed (1:10,000. Thermo Fisher Scientific A3854). Otherwise, we used HRP conjugated secondaries recognizing the host species of the primary antibody (Thermo Fisher Scientific 31460, 31430 and A18775 for rabbit, mouse and guinea pig respectively).
Synapse purifications
Synapse fractions were purified as we described previously50,116,117. Flash-frozen whole cortex with subcortical forebrain was mechanically homogenized with 12 slow strokes by a Teflon homogenizer and glass vessel in ice-cold homogenization buffer (5 mM HEPES pH 7.4, 1 mM MgCl2, 0.5 mM CaCl2, supplemented with phosphatase (Sigma 4906837001) and protease inhibitors (Sigma 4693159001). The homogenate was centrifuged for 10 min at 1400 g (4 °C) and the resulting supernatant (S1) was re-centrifuged at 13,800 g for 10 min (4 °C). The resulting pellet (P2) was resuspended in 0.32 M Sucrose, 6 mM Tris-HCl (pH 7.5) and layered gently on a 0.85 M, 1 M, 1.2 M discontinuous sucrose gradient (all layers in 6 mM Tris-HCl pH 7.5) and ultracentrifuged at 82,500 g for 2 h (4 °C). For synapse mass spectrometry experiments, phosphatase inhibitors were added to 1 M and 1.2 M sucrose buffers. The synaptosome fraction, which sediments at the interface between 1 M and 1.2 M sucrose, was collected. An equal volume of ice-cold 1% Triton X-100 (in 6 mM Tris-HCl pH 7.5) was added, mixed thoroughly, and incubated on ice for 15 min. The mixture was ultracentrifuged at 32,800 g for 20 min (4 °C), and the pellet (the synapse fraction) was collected by resuspension in 1% SDS. A small aliquot was taken to measure the protein concentration using the Pierce Micro BCA assay (ThermoFisher Scientific, 23235) or BCA (ThermoFisher Scientific A53225) and the remaining protein was stored at -80 °C until processed for MS or immunoblot. For synapse proteomics, sixteen synaptic fractions extracted from the cortex of wild type, Akap11+/- and Akap11-/- mouse brains were used for the proteomics study using TMT isobaric labeling strategy for quantitation. At 4 weeks, sample sizes were n = 5 WT, 5 Akap11+/- and 6 Akap11-/-. At 12 weeks, the sample sizes were n = 6 WT, 5 Akap11+/- and 5 Akap11-/-.
Processing of samples for proteomics and phosphoproteomics
Synapse fractions from animals at 4 weeks and 12 weeks were processed in two separate TMT16 plexes as described before50. Briefly, all samples were prepared in a 1% SDS buffer and digested using S-TrapTM sample processing technology (Protifi) following manufacturers recommendations. Following digestion, 100 μg of each sample was reconstituted in 20 μl of 50 mM HEPES and labeled with 200 μg TMT16 reagent (Thermo Fisher Scientific A44520). After verifying successful labeling of more than 95% label incorporation reactions were quenched, mixed, desalted and dried down. TMT16 labeled peptides were fractionated by high pH reversed-phase chromatography on a 4.6 × 250 mm Zorbax 300 extend-c18 column (Agilent). One-min fractions were collected during the entire elution and fractions were concatenated into 18 proteome fractions for LC-MS/MS analysis. Five percent of each fraction was removed for proteome analysis. The remaining of each of the 18 fractions were further concatenated to 9 fractions and dried down for phosphopeptide enrichment using immobilized metal affinity chromatography (IMAC).
LC-MS/MS analysis for synapse fractions
One microgram of each proteome fraction and half of each of the IMAC fractions were analyzed on a QE-HFX mass spectrometer (Thermo Fisher Scientific) coupled to an Easy-nLC 1200 LC system (Thermo Fisher Scientific). Samples were separated using 0.1% Formic acid/3% Acetonitrile as buffer A and 0.1% Formic acid /90% Acetonitrile as buffer B on a 27 cm 75um ID picofrit column packed in-house with Reprosil C18-AQ 1.9 mm beads (Dr Maisch GmbH) with a 110 min gradient consisting of 2–6% B in 1 min, 6–20% B in 62 min, 20–30% B for 22 min, 30–60% B in 9 min, 60–90% B for 1 min followed by a hold at 90% B for 5 min. The MS method consisted of a full MS scan at 60,000 resolution and an AGC target of 3e6 from 350–1800 m/z followed by MS2 scans collected at 45,000 resolution with an AGC target of 3e6 from 300–1800 m/z followed by MS2 scans collected at 35,000 resolution with an AGC target of 1e5 and 5e4 for proteome and phosphoprotome fractions, respectively and a maximum injection time of 105 ms. Dynamic exclusion was set to 15 s. The isolation window used for MS2 acquisition was 0.7 m/z and 20 most abundant precursor ions were fragmented with a normalized collision energy (NCE) of 29 optimized for TMT16 data collection.
Database search and MS/MS quantification for PSD fractions
All the datasets were searched on Spectrum Mill MS Proteomics Software (Broad Institute) using mouse database downloaded from Uniprot.org on either 12/28/2017 or 04/07/2021 containing 47,069 or 55,734 entrees for 4 weeks and 12 weeks coIP-MS experiments, respectively118. Search parameters included: ESI Qexactive HCD v4 35 scoring parent and fragment mass tolerance of 20 ppm, 40% minimum matched peak intensity, trypsin allow P enzyme specificity with up to four missed cleavages, and calculate reversed database scores enabled. Fixed modifications were carbamidomethylation at cysteine. TMT labeling was required at lysine, but peptide N-termini could be labeled or unlabeled. Variable modifications were allowed for protein N-terminal acetylation and oxidized methionine as well as phosphorylated serine, threonine, and tyrosine for the phosphoproteome datasets. Protein quantification was achieved by taking the ratio of TMT reporter ions for each sample over the TMT reporter ion for the median of all channels. TMT16 reporter ion intensities were corrected for isotopic impurities in the Spectrum Mill protein/peptide summary module using the afRICA correction method which implements determinant calculations according to Cramer’s Rule and correction factors obtained from the reagent manufacturer’s certificate of analysis (Thermo Fisher Scientific, 90406) for lot numbers VH310017 for both synapse proteomics experiments. Median normalization was applied to both proteome and phosphoproteome datasets prior to statistical analysis. Additionally, phosphosite changes were normalized for changes in protein abundances by fitting a global linear model between the phosphosite and the cognate protein and extracting the residuals. A moderated two-sample t-test was applied to the datasets to compare WT, Akap11+/- and Akap11-/- sample groups. For proteins with multiple isoforms, the isoform with the greatest abundance (number of spectra observed) was displayed in the plot.
Annotations from phosphosite plus
Kinase substrate annotations were downloaded from https://www.phosphosite.org on June 20, 2022. In this database, some kinase substrate sites are inaccurately attributed to a kinase to which it is indirectly related. For example, Grin2bY1472 is annotated as a PKA Cα substrate in phopshopsite.org (see Fig. 5b, but is actually a PKC substrate119. Similarly, Ctnnb1S45 is annotated as a Gsk3β substrate in phosphosite.org, but is actually a CK1 substrate57. These discrepancies were not adjusted for this manuscript in order to maintain unbiased usage of the database.
Phosphoproteomics motif analysis
Motif enrichment analysis was performed to generate motif logos using the pyproteome Python library120. Motif logo enrichment values were calculated according to the method described for pLogo121. Peptide 15-mers around each phosphorylation site were generated by Spectrum Mill “VMsiteFlanks”. Foreground and background N-mers were extracted from protein-aligned sequences, using ‘-’ to pad N-and C-terminal peptides. Quantification values from identical N-mers (mis-cleaved, oxidized, or multiply-phosphorylated peptides) were combined by taking the median value. Log odds enrichment values are equal to \(\left(-\log \frac{{sf}(x-1)}{{cdf}(x)}\right)\), with k = the count of amino acids at that relative position for phosphosites in a foreground collection (P < 0.05; FC > 1.25 or FC < 0.8). Survival function (sf) and cumulative distribution functions (cdf) were calculated using scipy’s hypergeometric distribution model (scipy.stats.hypergeom), with: n = the total number of foreground N-mers; K = the count of amino acids at that position in the background collection; and N=the total number of background N-mers.
PKA activity assay
12 week) male mice were euthanized using CO2 inhalation between ZT10-ZT14. Cortex and subcortical forebrain were separated during dissection, and flash-frozen in liquid nitrogen. The subcortical forebrain is enriched for the striatum but also includes other regions, such as basal forebrain and hypothalamus. For this experiment only, we refer to the subcortical forebrain as “Striatum”. This crude dissection was necessary since the synapse-enrichment protocol requires large amounts of starting material. Single cortical hemispheres and both combined subcortical forebrains from both hemispheres were prepared as described in the “synapse purifications” section. Total lysate and synapse fractions were collected from the same starting material and resuspended in 1% Triton-X-100 and frozen on dry ice and stored at -80. After all samples were collected, samples were thawed and protein concentration was measured using Pierce Micro BCA assay. A colorimetric PKA activity assay (Invitrogen EIAPKA) was conducted according to the manufacturers instructions. 50 ng of cortex and striatum samples were compared in separate plates. Lysate and synapse fractions were compared within the same plate. Curve-fitting functions in Graphpad Prism were used to infer PKA activity from provided standards.
Quantitative immunofluorescence
15–17-week-old male mice were euthanized under isoflurane with transcardial perfusion of ~25 mL with PBS followed by ~25 ml 4% paraformaldehyde (PFA). These mice also underwent an 1 hour open field assays for a separate study14, at 4 weeks and 12 weeks of age. Brains were post-fixed overnight in 4% PFA, and washed the next day four times with PBS for 15 min. Tissues were cryoprotected for 72 hours and stored at 4 °C until sectioning. Brains were sectioned on a vibratome (Leica) at either 40 or 60 μm thickness (consistent within technical replicates) and stored in a freezing buffer (0.1 M PBS, 30% ethylene glycol, 30% glycerol, 0.01% sodium azide).
For immunostaining, free-floating sections were first rinsed 4x with PBS for 10 min. Sections were blocked and permeabilized with 1 h RT incubation in a blocking solution. For the first technical replicate, we used 5% Normal Goat Serum (Sigma, NS02L), with 10% BSA + 0.2% Triton. For the second technical replicate, we used 5% Normal Donkey Serum (Jackson, 017-000-121) + 0.2% Triton in PBS for 60 min. Primary antibodies were diluted in blocking solution and incubated overnight at 4 °C, rotating gently. The following day, sections were rinsed 3x for 15 min in PBS containing 0.2% Triton at RT on a shaker. DAPI (Thermo Fisher Scientific, 62248) was added to PBS at a final concentration of 1:5000 (for the first technical replicate) or 1:10,000 (for the second technical replicate) before mounting. The following primary and secondary antibodies were used: PKA Ca (CST 4782, 1:100), PKA R1a (ThermoFisher MA5-24981, 1:100). Donkey anti Rabbit 488 (Invitrogen A32790), Donkey anti-Mouse 555 (Invitrogen A32773), Goat anti Rabbit 488 (Invitrogen A11034), Goat anti-Mouse 555 (Invitrogen A32727). Whole sections were imaged with a Zeiss Axioscan 7 Slidescanner using exposure and intensity settings that were constant across samples within technical replicates.
PKA R1α was quantified in FIJI using the freehand selection tool and the intensity measurement function. Regions were selected using the Allen Brain Atlas as a reference. Experimenters were blind to conditions during analysis. For representative images, brightness levels were uniformly increased across the whole images with Zen software (Carl Zeiss) and scaled down to 10% to accommodate the large field of view of the stitched image.
Tissue collection for RNA sequencing
For bulk and snRNA-seq, mice were harvested between ZT9 and ZT15, near the animals active period at ZT12. Brain tissues for bulk and snRNA-seq were prepared as described12, and also available at protocols.io (https://www.protocols.io/view/fresh-frozenmouse-brain-preparation-for-single-nu-bcbrism6). At 4 and 12 weeks of age, male WT, Akap11+/- and Akap11-/- mice were anesthetized by administration of isoflurane in a gas chamber. Within each age, four mice per genotype were harvested for snRNA-seq, with the exception of 12 week striatum, which had five WT samples. For bulk RNA-seq, 5-6 mice per genotype within an age were collected, with the exception of 4 week Akap11+/- hipp.
Transcardial perfusions were performed with ice-cold Hank’s Balanced Salt Solution (HBSS, Gibco 14175-095) in mice anesthetized with 3% isoflurane delivered continuously via nose cone. Brains were then frozen gradually over liquid nitrogen vapor and stored at –80 °C with tubes containing O.C.T. (optimal cutting temperature) freezing medium (Tissue-Tek) to prevent desiccation. Brain region microdissections were performed by hand in a cryostat (Leica CM3050S), with a ophthalmic microscalpel (Feather safety razor P-715) or 1.5 mm diameter biopsy punch (Integra 33-31 A). PFC, Hipp and SSC were excised by hand. SN and Thal were collected by biopsy punch. For snRNA-seq studies, both dorsal and ventral striatum were collected together with a microscalpel, while for bulk RNA-seq, dorsal striatum was collected by punch. All brain regions were identified according to the Allen Brain Atlas. Dissected tissues were placed into a precooled 1.5 ml PCR tube and stored at –80 °C.
RNA extraction and bulk RNA-seq library preparation
RNA was prepared from micro-dissected tissues using RNeasy Mini Kit (Qiagen, 74106) following the manufacturer’s instructions. Briefly, tissue samples were lysed and homogenized, placed into columns, and bound to the RNeasy silica membrane. Next, contaminants were washed away. Columns were treated with DNase (Qiagen) to digest residual DNA and concentrated RNA was eluted in water. RNA concentration was measured using a NanoDrop Spectrophotometer and RNA integrity (RIN) with RNA pico chips (Agilent) using a 2100 Bioanalyzer Instrument (Agilent). Purified RNA was stored at -80 °C until library preparation for bulk RNA-seq analysis. Bulk RNA-seq libraries were prepared using a TruSeq Stranded mRNA Kit (Illumina) following the manufacturer’s instructions. 200 ng of isolated total RNA from each sample was used and the concentration of resulting cDNA library was measured with High Sensitivity DNA chips (Agilent) using a 2100 Bioanalyzer Instrument (Agilent). A 10 nM normalized library was pooled, and sequencing was performed on a NovaSeq S2 (Illumina) with 50 bases each for reads 1 and 2 and 8 bases each for index reads 1 and 2.
For snRNA-seq library preparation, brain tissue dissection and nuclei extraction were performed within 16 hours to avoid desiccation and freeze-thaw cycles. A gentle, detergent-based dissociation was used to extract the nuclei, according to a previously published protocol122 also available at protocols.io (https://www.protocols.io/view/frozen-tissue-nuclei-extraction-bbseinbe). Extracted nuclei were then loaded into the 10x Chromium V3.1 system (10x Genomics) and library preparation was performed according to the manufacturer’s protocol. A 10 nM normalized library was pooled, and sequencing was performed on a NovaSeq S2 (Illumina) with 28 and 75 bases for reads 1 and 2, respectively, and 10 bases each for index reads 1 and 2.
Bulk RNA-seq DE analysis
FASTQ files were aligned to a reference, which was derived from the genome FASTA file and transcriptome GTF extracted from the CellRanger mm10 reference. Alignment and quantification were performed using a Nextflow V1 pipeline123 (https://github.com/seanken/BulkIsoform/blob/main/Pipeline). Quantification was performed by Salmon124 (version 1.7.0, with arguments -l A --posBias --seqBias --gcBias --validateMapping), using a Salmon reference built with the salmon index command with genomic decoys included. Two samples - HP_HT_941 and TH_WT_913 - were removed from analysis due to evidence of contamination by aberrant marker gene expression.
Dataset | WT sample size (n) | Akap11+/- sample size (n) | Akap11-/- sample size (n) |
|---|---|---|---|
4wk PFC (Prefrontal Cortex) | 5 | 6 | 5 |
4wk Hipp (Hippocampus) | 6 | 4 | 5 |
4wk SSC (Somatosensory Cortex) | 5 | 6 | 5 |
4wk Striatum (Str) | 5 | 5 | 5 |
4wk Thalamus (Thal) | 5 | 5 | 5 |
4wk Substantia Nigra (SN) | 6 | 6 | 5 |
12wk PFC | 5 | 5 | 5 |
12wk Hipp | 5 | 5 | 5 |
12wk SSC | 5 | 5 | 5 |
12wk Str | 5 | 5 | 5 |
12wk Thal | 5 | 5 | 5 |
12wk SN | 5 | 5 | 5 |
Differential expression (DE)
DE analysis was performed to compare the heterozygous or knockout mutants to the WT mice for each brain region and age using the Wald test in R package DESeq2125 (version 1.34). Salmon output was loaded using R package tximport126 (version 1.22). Genes that had less than 10 counts across all samples in each experiment were removed from our analysis. Due to evidence of dissection variability in striatum by excitatory marker genes, DESeq2’s PC1 was added as a covariate for DE in 1 month striatum. Log2 fold change shrinkage was applied using the “normal” shrinkage estimator in DESeq2. DESeq2’s default results filtering was used and all genes in a given comparison with padj = NA were removed from downstream analysis. For 1 month hippocampus and thalamus, the default filtering removed the majority of genes, so we changed the filtering for the heterozygous vs. WT comparison to match the less aggressive filtering in the knockout vs. WT comparison. The statistical method applied for bulk RNAseq is two-tailed Wald test, with Benjamini-Hochberg (B-H) false discovery rate (FDR) correction.
Single-nucleus RNA-seq analysis
snRNA-seq reads were aligned to an mm10 mouse reference genome containing introns (https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-mm10-2020-A.tar.gz) using the Cell Ranger v3.0.2 pipeline (10x Genomics)127. Cell Ranger count was run with default parameters and the following additional flags: --chemistry=SC3Pv3 and --expect-cells = 15,000. Replicates for the 4-week STR dataset were down-sampled to contain the same number of reads per cell using Cell Ranger aggr with default parameters.
Seurat v4.0.3128 was used to analyze unique molecular identifier (UMI) counts. Nuclei expressing less than 500 genes were removed and the remaining nuclei were normalized by total expression, multiplied by ten thousand, and log transformed. The data were then scaled using Seurat’s ScaleData function. Seurat’s RunPCA function was used to perform linear dimension reduction on the variable genes within the scaled data. Clustering in the PCA space was then run using Seurat’s FindNeighbors and FindClusters functions and the clusters were visualized using Seurat’s RunUMAP function. Doublet identification was performed using the Scrublet v0.2.3129. Python package with default parameters. Clusters and nuclei with a scrublet score above the region-specific thresholds were determined to be doublets and were removed from the data. The table below shows the used scrublet score thresholds for all snRNA-seq datasets:
Dataset | Scrublet Score Threshold |
|---|---|
4wk PFC | 0.36 |
4wk Striatum | 0.2 |
12wk PFC | 0.4 |
12wk Striatum | 0.4 |
Cluster cell type assignment was evaluated using region-specific cell type marker genes. Neuronal subtypes were identified by re-clustering the nuclei of a given major cell type and evaluating expression of subtype-specific marker genes. Marker genes for the more widely classified major cell types and subtypes were identified in the literature130. Rarer cell types and subtypes were identified using Seurat’s FindMarkers function. The clusters were then identified by searching cluster-specific marker genes on mouseBrain.org131 and or dropviz.org132.
Sample size for single-nucleus RNA-seq datasets:
Dataset | WT sample size (n) | Akap11+/- sample size (n) | Akap11-/- sample size (n) |
|---|---|---|---|
4wk PFC | 4 | 4 | 4 |
4wk Striatum | 4 | 4 | 4 |
12wk PFC | 4 | 4 | 4 |
12wk Striatum | 5 | 4 | 4 |
Pseudobulk DE analysis
snRNA-seq DE analysis was performed using the pseudobulk approach. For each dataset, counts were grouped by cell type and replicate, then gene counts were summed across each replicate within a cell type, resulting in one count value per replicate per cell type. Surrogate variable analysis (SVA) was conducted using the sva v3.42.0 R package133 to account for unknown sources of variation within each experiment. DE analysis was then run with the covariates identified by SVA using the EdgeR v3.22.3 R package134. Comparisons were calculated using two-tailed likelihood ratio test, which were FDR corrected with B-H adjustment. DEGs were defined as genes with a B-H adjusted P-value < 0.05.
Gene set enrichment analysis
GSEA was performed with the R Bioconductor package fgsea135 using C5 ontology gene sets obtained from the Molecular signatures database (MSigDB) on September 28, 2022. Genes were pre-ranked using their test-statistics within each comparison, and their Homo sapiens homolog-associated were used for the analysis. For genes or proteins with multiple isoforms, the isoform with the largest effect-size (absolute value of the t-statistic) was used for pre-ranking. Because the enrichment analyses were carried out using the proteins’ gene symbols, we use the terms gene set and protein set interchangeably in this study. For each of these gene sets, mouse gene symbols from transcriptomics and proteomics results were mapped to matching Homo sapiens homolog-associated gene symbols through annotations extracted from Ensembl’s BioMart data service; the human symbols were then used to perform GSEA. GSEA uses a Kolmogorov-Smirnov test, two tailed, with B-H FDR correction applied. A list of all significant GO terms (FDR < 0.05) for all datasets are provided as Supplementary Data. GSEA analysis with custom gene sets was conducted by appending these sets to the C5 MSigDB database.
Curated gene sets
Phosphosites that correlate with homeostatic synaptic scaling were curated from Desch et al.72, including together all phosphosites that were significantly altered (adjusted P-value < 0.05) after 5 min, 15 min or 24 h of either bicuculline or tetrodotoxin treatment. Curated gene sets that we have previously described12 include “ARGss” (ARGs), which include rapid, delayed and slow primary response genes induced by neuronal depolarization in vitro79, GWAS and exome associations for neuropsychiatric traits37,82,83,84, and dopamine-induced genes which are genes with FDR < 0.1 that were upregulated in striatal neuronal culture following 1 hour dopamine treatment136.
Brain slice electrophysiology
6-8-week-old male and female mice were used for brain slice electrophysiology experiments, including n = 9 slices from four mice WT, and n = 8 slices from four mice Akap11-/- for input-output experiments; n = 25 slices from eight mice WT, n = 24 slices from 7 mice Akap11+/-, and n = 23 slices from 8 mice Akap11-/- for early LTP and paired-pulse facilitation experiments. An NMDG-based cutting solution was used and contains (in Mm): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 Glucose, 10 MgSO4, 0.5 CaCl2, 5 Sodium Ascorbate, 2 Thiourea, 3 Sodium Pyruvate. pH = 7.3-7.4, OSM = 300–310. The external artificial cerebrospinal fluid (ACSF) recording solution contains(in Mm): 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 5 HEPES, 12.5 Glucose,1 MgSO4, 2 CaCl2 pH = 7.35, OSM = 300. For brain slice preparation, cardiac perfusion was performed with cold NMDG cutting solution under isoflurane anesthesia. Transverse (horizontal) brain slices containing hippocampus were cut at 400 μm with a Leica VT1200S Microtome when submerged in ice-cold NMDG cutting solution. Brain slices were transferred to NMDG solution at 32 °C for approximately 10 min before being transferred to RT brain slice holding solution. Slices were allowed to recover for at least 1 hbefore starting the recordings.
To obtain hippocampus field electric postsynaptic potential (EPSP), a concentric bipolar stim electrode (FHC Inc) and a borosilicate glass recording electrode (2–3 mOhm resistance) were placed in the classic schaffer collateral to CA1 (SC-CA1) pathway. The signals were low-pass filtered at 2.8 kHz using a MultiClamp 700B amplifier (Molecular Devices) and digitized at 10 kHz using a DAQ board (National Instrument). Baseline field EPSP were evoked by a paired-pulse stimulation (40 ms ISI) every twenty seconds, and a stimulation intensity that evoked 40% of the maximum field EPSP response was used for baseline recording.
For input-output recordings, field EPSP responses were evoked with varying stimulation intensities (20–500 μA). For early LTP recordings, slices were stimulated for approximately 30 mins until a stable baseline field EPSP was obtained. Theta burst stimulation consists of short bursts of four stimulation pulses at 10 Hz delivered at theta frequency (5 Hz, ten epochs) were used to induce LTP. The field EPSP was recorded for another 20 min after LTP induction. Paired-pulse ratio was quantified using the field EPSP collected from the 10 min period before LTP induction and was defined as the ratio between the second and the first field EPSP slope. Field EPSP slope was quantified as the slope during 10–90% of the raise time.
Single-molecule fluorescent in-situ hybridization (smFISH)
Mouse brain characterization of Akap11, Prkaca, and Prkar1a expression was performed using Advanced Cell System’s (ACD) single-molecule in situ hybridization multiplexed fluorescent amplification kits (ACD catalog numbers 323270). 12-week-old male mice, n = 6 per genotype WT, Akap11+/-, and Akap11-/-, were deeply anesthetized under isoflurane, perfused with ice-cold HBSS for 3 min, and decapitated. Brains were extracted and frozen gradually over liquid nitrogen vapors. Frozen brains were stored in tubes with O.C.T. in -80 °C. Brains were sectioned sagittally at 12 um/section using a Leica cryostat. Four sections per animal (two sections per slide) were collected on SuperFrost Charged slides and stored at -80 °C and smFISH was performed the following day. Slides were post-fixed in freshly made, pre-chilled 4% PFA (Electron Microscopy Sciences) in PBS for 1 h on ice. Sections were then dehydrated with 50, 70, and 100% ethanol for 5 min at RT. All slides were outlined with a grease pen and all following smFISH steps were done on a humidified slide rack. Sections were then treated with hydrogen peroxide for 10 min followed by three PBS washes. Sections then underwent permeabilization with “protease IV” for 30 min followed by three PBS washes. Sections were then hybridized using probes to Prkaca (channel 1), Akap11 (channel 2), and Prkar1a (channel 3) at 40 °C for 2 h washed twice with wash buffer, and placed in aluminum foil-wrapped slide holders filled with 5X SSC (Thermo Fisher Scientific, 15557044) overnight at RT. The following day, sections were thoroughly washed in wash buffer and probes were amplified using the AMP reagents from the ACD kit. Slides were washed between each step using the wash buffer. HRP binding was also carried out according to the kit’s protocol with C1, C2, and C3. Fluorophores were diluted at 1:750 in TSA buffer: C1 was in 488; Channel 2 was in Cy3; Channel 3; Cy5. Once the procedure was completed, slides were counterstained with RT Prolong Gold Antifade Mountant with DAPI (Thermo Fisher Scientific, P36935).
Slides were scanned in a Zeiss Axioscan 7 microscope slide scanner with a 40x objective quadruple bandpass filter set with DAPI focus. CZI files were converted to 8-bit TIFF images in FIJI and appropriate brain regions (cortex, thalamus, hippocampus, and striatum) were outlined using the freehand selection tool. To assess levels of Akap11, Prkaca, and Prka1a, the mean intensity was quantified for each outlined brain region with background subtraction using the declive (VI), molecular layer. Four sections per animal were averaged and normalized to the WT to minimize batch effects.
Viruses
FLIM-AKAR and FLIM-AKART391A viruses were packaged in-house into a AAV2/AAV9 vector using plasmids that were available from Addgene (no. 60445 and 60446). The final viral constructs and concentration used in this study were: AAV2/1-FLEX-FLIM-AKAR (8.53 × 1012gc/ml), AAV9-FLEX-FLIM-AKAR (2.14 × 1013 gc/ml), AAV2-FLEX-FLIM-AKART391A (1.23 × 10^13 gc/ml), AAV9-FLEX-FLIM-AKART391A (7.67 × 1012 gc/ml). AAV2rg-Ef1a-Cre (1.39 × 1013 gc/ml) was obtained from Salk institute. While both AAV2 and AAV9 versions of the FLIM-AKAR and FLIM-AKART391A were used in this study, we saw no noticeable difference in their expression level and baseline fluorescence lifetime when expressed in WT mice. During the experiments, FLIM-AKAR or mutant FLIM-AKART391A virus was mixed with the Cre virus in a 4:1 ratio. A final volume of 250 nL was used for intracranial injection.
Stereotaxic Surgery and Virus Injection
Mice were anesthetized with isoflurane and underwent bilateral injection of the virus mix (FLIM-AKAR + Cre or FLIM-AKART391A + Cre). Typically, one side of the brain is injected with FLIM-AKAR + Cre, while the other side is injected with FLIM-AKART391A + Cre while counterbalancing the sides of the brain, with the exception of twelve mice (mixed genotypes) injected with Dlight 3.8 virus instead of FLIM-AKART391A + Cre (data not included). Virus injection was conducted using coordinates for the nucleus accumbens lateral shell (AP + 1.20, ML ± 1.50 from bregma, DV -4.2 from brain surface). Optical fibers (MFC_200/230-0.37_4.5mm_MF1.25_FLT from Doris Lenses) were placed bilaterally and ~ 0.3 mm above the injection site.
Fluorescence lifetime photometry (FLiP)
Thirty-three 12-20 week-old Akap11 mutant mice of mixed genotypes were used for stereotaxic surgeries and subsequent FLiP measurements, including baseline, D1R antagonist and D2R antagonist expriments. Approximately equal number of male and female mice were used (n = 10 WT, n = 11 Akap11+/- and n = 10 Akap11-/-). The fluorescence lifetime and intensity of the FILM-AKAR (or FLIM-AKART391A) was measured in real time at 10 kHz using a Fluorescent lifetime photometry system106. Customized MATLAB software was used to collect the lifetime measurement, and the lifetime data was down sampled to 10 Hz using a rolling mean. This setup has been described in detail with analysis code provided within137.
Animals were habituated into the recording room for at least 30 min before the experiments. Baseline FLIM-AKAR lifetime was measured for 20 mins during the first measurement, and were then collected for 10 mins each during subsequent measurements, while animals were allowed to freely move in an open field (18×18 cm). Drug injections (dopamine D1/D2 antagonists) during pharmacology experiments were conducted after 10 mins baseline lifetime measurement, and the lifetime data were collected for at least another 20 mins following intraperitoneal injections.
The drugs used in our study include: D1R antagonist (SKF 83566 hydrobromide, Cayman Chemical #29480; 3 mg kg−1), D2R antagonist (eticlopride hydrochloride, Cayman Chemical #29112; 0.5 mg kg−1). Drugs were dissolved in sterile saline at appropriate concentrations and were injected at 0.1 ml solution per 10 g of mouse. An average injection including scruffing the mice took ~30 s and animals were habituated to the injection procedure with sterile saline twice before the pharmacology experiments.
The experimenter was blinded to the genotype of the mice during the experiments and subsequent data analysis. Two out of thirty-three FLIM-AKAR measurements were excluded from the study due to the low expression of the FILM-AKAR causing the autofluorescent contribution to the recorded lifetime to be greater than 20% based on our estimation. The exclusion criteria was determined before the experiment while the experimenter was still blinded to the mice genotypes.
Post- FLiP immunofluorescence expression validation
After the FLiP experiments were completed, all mice were perfused transcardially with PFA. Coronal brain sections (80μm) near the optical fiber implants were sliced using a vibratome (Leica VT1200S). The brain slices were mounted with Prolong Gold Antifade Mountant with DAPI (Thermo Fisher Scientific, P36935) and native fluorescence of FLIM-AKAR was imaged with a Zeiss Axioscan 7 slidescanner.
To assess subcellular localization of FLIM-AKAR, additional brain slices were imaged with a Zeiss 900 LSM confocal microscope. Images were acquired at magnifications of 20x with the green-channel laser power set to 0.2 mW. Z-stack images were taken at 63x under oil with a step size of 0.3 um and averaging was set to 4x. Image regions were matched across animals based on coordinates determined on proximity to bregma (y = 1.54 mm) and site of the injection. To facilitate clear visualization of the FLIM-AKAR sensor’s subcellular localization, images were specifically taken in areas of sparse viral expression located toward the lateral edge of the nucleus accumbens core. Schematic diagrams in Supplementary Fig. 11b, c are adapted with permission138.
Amphetamine induced-hyperlocomotion (AIH)
Akap11 mice that were 10-12 weeks old with roughly equal numbers of male and female mice were used for the AIH experiment. n = 18 WT, n = 22 Akap11+/- and n = 14 Akap11-/- were used for this experiment. Mice in each genotype were randomly assigned to the saline or amphetamine groups. Before the experiment, dextroamphetamine sulfate (Millipore Sigma 1180004; salt corrected) was dissolved in water at 0.2667 mg/mL and was pH corrected to 7.3. Mice were weighted and were allowed to habituate in the behavioral room (under white light) for 30 min before the start of the experiment. For the measurement of locomotion, mice were placed in Omnitech Electronics Inc SuperFlex Open Field apparatus (Omnitech Electronics Inc), which was cleaned with 70% ethanol between each experiment. The Fusion software (Omnitech Electronics Inc) was used for data collection and analysis. First, a 90 min baseline locomotion was recorded, followed by subcutaneous injection of amphetamine at a dose of 2 mg/kg or saline. Following drug injection, the locomotion data were collected for another 90 min. Locomotion data were measured by infrared beams and were measured by beam breaks, and were grouped into 5-min bins for analysis.
Statistics
Statistics for immunoblots, fluorescence lifetime photometry (FLiP), and AIH were calculated in GraphPad Prism, with each dot representing an individual animal. Immunoblots and FLiP experiments including the genotype factor and a second factor (Age, Brain region, Protein interactor, or FLIM-AKAR variant) were quantified with two-way ANOVA. For all analyses with a significant main effect of genotype, post hoc Tukey’s multiple comparisons were conducted for all possible comparisons within the same level of the second factor (e.g. WT, Akap11+/- and Akap11-/- were compared within one age, but not between different ages). For Supplementary Figs. 3b, c and 4g, where Akap11-/- was excluded, Sidak’s multiple comparisons test was used. For immunoblot, and FLiP experiments where genotype was the only variable, statistically analysis were conducted by one-way ANOVA followed by Tukey’s post hoc test to compare all genotype pairs. Further statistical information is available in Supplementary Data 1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The original mass spectra and the protein sequence database used for searches have been deposited in the public proteomics repository MassIVE (http://massive.ucsd.edu) and are accessible at ftp://MSV000095852@massive.ucsd.edu. These datasets will be made public upon acceptance of the manuscript. Processed and raw bulk and single nucleus RNA sequencing data have been deposited at Gene Expression Omnibus (GEO) with the following IDs: bulk RNAseq: GSE306677. single nucleus RNAseq: GSE272098. For analyses of Akap11 transcript abundance in WT mice, the following publicly available datasets were analyzed: GSE218573. All software used in the analysis are publicly available. Source data are provided with this paper.
Code availability
No specialized code was developed for this manuscript. Software and packages that were utilized for analysis are publicly available and referenced throughout the methods.
References
Ferrari, A. J. et al. The prevalence and burden of bipolar disorder: findings from the Global Burden of Disease Study 2013. Bipolar Disord. 18, 440–450 (2016).
McGrath, J., Saha, S., Chant, D. & Welham, J. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol. Rev. 30, 67–76 (2008).
Polderman, T. J. C. et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet. 47, 702–709 (2015).
Jauhar, S., Johnstone, M. & McKenna, P. J. Schizophrenia. Lancet 399, 473–486 (2022).
Singh, T. et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 604, 509–516 (2022).
Palmer, D. S. et al. Exome sequencing in bipolar disorder identifies AKAP11 as a risk gene shared with schizophrenia. Nat. Genet. 54, 541–547 (2022).
Trubetskoy, V. et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022).
Mullins, N. et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet. 53, 817–829 (2021).
Liu, D. et al. Schizophrenia risk conferred by rare protein-truncating variants is conserved across diverse human populations. Nat. Genet. 55, 369–376 (2023).
Thorgeirsson, T. E. et al. Rare loss-of-function variants in HECTD2 and AKAP11 confer risk of bipolar disorder. Nat. Genet. 57, 851–855 (2025).
Chen, R. et al. Cell type-specific mechanism of Setd1a heterozygosity in schizophrenia pathogenesis. Sci. Adv. 8, eabm1077 (2022).
Farsi, Z. et al. Brain-region-specific changes in neurons and glia and dysregulation of dopamine signaling in Grin2a mutant mice. Neuron 111, 3378–3396.e9 (2023).
Katrancha, S. M. et al. Trio haploinsufficiency causes neurodevelopmental disease-associated deficits. Cell Rep. 26, 2805–2817 (2019).
Herzog, L. E. et al. Mouse mutants in schizophrenia risk genes GRIN2A and AKAP11 show EEG abnormalities in common with schizophrenia patients. Transl. Psychiatry 13, 92 (2023).
Wong, W. & Scott, J. D. AKAP signalling complexes: focal points in space and time. Nat. Rev. Mol. Cell Biol. 5, 959–970 (2004).
Day, M. E. et al. Isoform-specific targeting of PKA to multivesicular bodies. J. Cell Biol. 193, 347–363 (2011).
Lester, L. B., Coghlan, V. M., Nauert, B. & Scott, J. D. Cloning and characterization of a novel A-kinase anchoring protein: AKAP 220, association with testicular peroxisomes (∗). J. Biol. Chem. 271, 9460–9465 (1996).
Tanji, C. et al. A-kinase anchoring protein AKAP220 binds to glycogen synthase kinase-3beta (GSK-3beta) and mediates protein kinase A-dependent inhibition of GSK-3beta. J. Biol. Chem. 277, 36955–36961 (2002).
Schillace, R. V. & Scott, J. D. Association of the type 1 protein phosphatase PP1 with the A-kinase anchoring protein AKAP220. Curr. Biol. 9, 321–324 (1999).
Logue, J. S. et al. AKAP220 protein organizes signaling elements that impact cell migration. J. Biol. Chem. 286, 39269–39281 (2011).
Yang, L., Nakayama, Y., Hattori, N., Liu, B. & Inagaki, C. GABAC-Receptor Stimulation Activates cAMP-Dependent Protein Kinase via A-Kinase Anchoring Protein 220. J. Pharmacol. Sci. 106, 578–584 (2008).
Mikitova, V. & Levine, T. P. Analysis of the key elements of FFAT-like motifs identifies new proteins that potentially bind VAP on the ER, including two AKAPs and FAPP2. PLoS One 7, e30455 (2012).
Deng, Z. et al. Selective autophagy of AKAP11 activates cAMP/PKA to fuel mitochondrial metabolism and tumor cell growth. Proc. Natl. Acad. Sci. USA. 118, e2020215118 (2021).
Whiting, J. L. et al. AKAP220 manages apical actin networks that coordinate aquaporin-2 location and renal water reabsorption. Proc. Natl. Acad. Sci. USA 113, E4328–E4337 (2016).
Voikar, V. & Gaburro, S. Three pillars of automated home-cage phenotyping of mice: novel findings, refinement, and reproducibility based on literature and experience. Front. Behav. Neurosci. 14, 575434 (2020).
Pernold, K. et al. Towards large scale automated cage monitoring - Diurnal rhythm and impact of interventions on in-cage activity of C57BL/6J mice recorded 24/7 with a non-disrupting capacitive-based technique. PLoS One 14, e0211063 (2019).
Fuochi, S. et al. Data repurposing from digital home cage monitoring enlightens new perspectives on mouse motor behaviour and reduction principle. Sci. Rep. 13, 10851 (2023).
Sherwin, C. M. Voluntary wheel running: a review and novel interpretation. Anim. Behav. 56, 11–27 (1998).
Wiltschko, A. B. et al. Mapping sub-second structure in mouse behavior. Neuron 88, 1121–1135 (2015).
Lin, S. et al. Characterizing the structure of mouse behavior using motion sequencing. Nat. Protoc. 19, 3242–3291 (2024).
Villarin, J. M. & Kellendonk, C. An ace in the hole? Opportunities and limits of using mice to understand schizophrenia neurobiology. Mol. Psychiatry 30, 4384–4398 (2025).
Armario, A. The forced swim test: Historical, conceptual and methodological considerations and its relationship with individual behavioral traits. Neurosci. Biobehav. Rev. 128, 74–86 (2021).
Liu, M.-Y. et al. Sucrose preference test for measurement of stress-induced anhedonia in mice. Nat. Protoc. 13, 1686–1698 (2018).
Bowie, C. R. & Harvey, P. D. Cognitive deficits and functional outcome in schizophrenia. Neuropsychiatr. Dis. Treat. 2, 531–536 (2006).
Whiting, J. L. et al. Protein kinase A opposes the phosphorylation-dependent recruitment of glycogen synthase kinase 3β to A-kinase anchoring protein 220*. J. Biol. Chem. 290, 19445–19457 (2015).
Dema, A. et al. The A-kinase anchoring protein (AKAP) glycogen synthase kinase 3β interaction protein (GSKIP) regulates β-catenin through its interactions with both protein kinase A (PKA) and GSK3β *. J. Biol. Chem. 291, 19618–19630 (2016).
Satterstrom, F. K. et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23 (2020).
Shih, Y.-T., Alipio, J. B. & Sahay, A. An inhibitory circuit-based enhancer of DYRK1A function reverses Dyrk1a-associated impairment in social recognition. Neuron 111, 3084–3101.e5 (2023).
Dang, T. et al. Autism-associated Dyrk1a truncation mutants impair neuronal dendritic and spine growth and interfere with postnatal cortical development. Mol. Psychiatry 23, 747–758 (2018).
Levy, J. A., LaFlamme, C. W., Tsaprailis, G., Crynen, G. & Page, D. T. Dyrk1a mutations cause undergrowth of cortical pyramidal neurons via dysregulated growth factor signaling. Biol. Psychiatry 90, 295–306 (2021).
Arranz, J. et al. Impaired development of neocortical circuits contributes to the neurological alterations in DYRK1A haploinsufficiency syndrome. Neurobiol. Dis. 127, 210–222 (2019).
Vierra, N. C. et al. Neuronal ER-plasma membrane junctions couple excitation to Ca2+-activated PKA signaling. Nat. Commun. 14, 1–16 (2023).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. 102, 15545–15550 (2005).
Parzych, K. R. & Klionsky, D. J. An overview of autophagy: morphology, mechanism, and regulation. Antioxid. Redox Signal. 20, 460–473 (2014).
Segura-Roman, A. et al. Autophagosomes anchor an AKAP11-dependent regulatory checkpoint that shapes neuronal PKA signaling. EMBO J. 44, 3150–3179 (2025).
Kandel, E. R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain 5, 14 (2012).
Solmi, M. et al. Age at onset of mental disorders worldwide: large-scale meta-analysis of 192 epidemiological studies. Mol. Psychiatry 27, 281–295 (2022).
Darling, N. J. & Cohen, P. Nuts and bolts of the salt-inducible kinases (SIKs). Biochem. J. 478, 1377–1397 (2021).
Yamada, T., Sakisaka, T., Hisata, S., Baba, T. & Takai, Y. RA-RhoGAP, Rap-activated Rho GTPase-activating protein implicated in neurite outgrowth through Rho. J. Biol. Chem. 280, 33026–33034 (2005).
Aryal, S. et al. Deep proteomics identifies shared molecular pathway alterations in synapses of patients with schizophrenia and bipolar disorder and mouse model. Cell Rep. 42, 112497 (2023).
Koopmans, F. et al. SynGO: an evidence-based, expert-curated knowledge base for the synapse. Neuron 103, 217–234 (2019).
Hornbeck, P. V. et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015).
Bramson, H. N., Kaiser, E. T. & Mildvan, A. S. Mechanistic studies of cAMP-dependent protein kinase action. CRC Crit. Rev. Biochem. 15, 93–124 (1984).
Krug, K. et al. A curated resource for phosphosite-specific signature analysis. Mol. Cell. Proteom. 18, 576–593 (2019).
Lisman, J., Yasuda, R. & Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 13, 169–182 (2012).
Liu, C. et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847 (2002).
Amit, S. et al. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 16, 1066–1076 (2002).
Fang, X. et al. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 97, 11960–11965 (2000).
Malhi, G. S., Gessler, D. & Outhred, T. The use of lithium for the treatment of bipolar disorder: recommendations from clinical practice guidelines. J. Affect. Disord. 217, 266–280 (2017).
Nakao, K. et al. GSK3β inhibition restores cortical gamma oscillation and cognitive behavior in a mouse model of NMDA receptor hypofunction relevant to schizophrenia. Neuropsychopharmacology 45, 2207–2218 (2020).
Banke, T. G. et al. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci. 20, 89–102 (2000).
Ehlers, M. D. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28, 511–525 (2000).
Esteban, J. A. et al. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 6, 136–143 (2003).
Diering, G. H., Gustina, A. S. & Huganir, R. L. PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron 84, 790–805 (2014).
Huganir, R. L. & Nicoll, R. A. AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717 (2013).
Nagy, G. et al. Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron 41, 417–429 (2004).
Simsek-Duran, F., Linden, D. J. & Lonart, G. Adapter protein 14-3-3 is required for a presynaptic form of LTP in the cerebellum. Nat. Neurosci. 7, 1296–1298 (2004).
Lonart, G. et al. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell 115, 49–60 (2003).
Kaeser, P. S. et al. RIM1alpha phosphorylation at serine-413 by protein kinase A is not required for presynaptic long-term plasticity or learning. Proc. Natl Acad. Sci. Usa. 105, 14680–14685 (2008).
Betzenhauser, M. J., Fike, J. L., Wagner, L. E. 2nd & Yule, D. I. Protein kinase A increases type-2 inositol 1,4,5-trisphosphate receptor activity by phosphorylation of serine 937. J. Biol. Chem. 284, 25116–25125 (2009).
Diering, G. H. & Huganir, R. L. The AMPA receptor code of synaptic plasticity. Neuron 100, 314–329 (2018).
Desch, K., Langer, J. D. & Schuman, E. M. Dynamic bi-directional phosphorylation events associated with the reciprocal regulation of synapses during homeostatic up- and down-scaling. Cell Rep. 36, 109583 (2021).
Makino, Y., Johnson, R. C., Yu, Y., Takamiya, K. & Huganir, R. L. Enhanced synaptic plasticity in mice with phosphomimetic mutation of the GluA1 AMPA receptor. Proc. Natl. Acad. Sci. USA 108, 8450–8455 (2011).
Dejanovic, B., Sheng, M. & Hanson, J. E. Targeting synapse function and loss for treatment of neurodegenerative diseases. Nat. Rev. Drug Discov. 23, 23–42 (2024).
Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Sekar, A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016).
Gandal, M. J. et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127 (2018).
Ruzicka, B. et al. Single-cell dissection of schizophrenia reveals neurodevelopmental-synaptic link and transcriptional resilience associated cellular state. Biol. Psychiatry 89, S106 (2021).
Tyssowski, K. M. et al. Different neuronal activity patterns induce different gene expression programs. Neuron 98, 530–546 (2018).
Minzenberg, M. J., Laird, A. R., Thelen, S., Carter, C. S. & Glahn, D. C. Meta-analysis of 41 functional neuroimaging studies of executive function in schizophrenia. Arch. Gen. Psychiatry 66, 811–822 (2009).
Chen, C.-H., Suckling, J., Lennox, B. R., Ooi, C. & Bullmore, E. T. A quantitative meta-analysis of fMRI studies in bipolar disorder: Meta-analysis of fMRI studies in BD. Bipolar Disord. 13, 1–15 (2011).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012 (2019).
Kaplanis, J. et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 586, 757–762 (2020).
International League Against Epilepsy Consortium on Complex Epilepsies Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat. Commun. 9, 5269 (2018).
Cross-Disorder Group of the Psychiatric Genomics Consortium Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 179, 1469–1482 (2019).
Ling, E. et al. A concerted neuron–astrocyte program declines in ageing and schizophrenia. Nature 627, 604–611 (2024).
Glennon, R. A., Titeler, M. & McKenney, J. D. Evidence for 5-HT2 involvement in the mechanism of action of hallucinogenic agents. Life Sci. 35, 2505–2511 (1984).
López-Giménez, J. F. & González-Maeso, J. Hallucinogens and serotonin 5-HT2A receptor-mediated signaling pathways. Curr. Top. Behav. Neurosci. 36, 45–73 (2018).
Cookson, J. & Jonsson, F. A new cholinergic mechanism for antipsychotics: emraclidine and M4 muscarinic receptors. Lancet 400, 2159–2161 (2022).
Kaul, I. et al. Efficacy and safety of the muscarinic receptor agonist KarXT (xanomeline–trospium) in schizophrenia (EMERGENT-2) in the USA: results from a randomised, double-blind, placebo-controlled, flexible-dose phase 3 trial. Lancet 403, 160–170 (2024).
Shamah, S. M. et al. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell 105, 233–244 (2001).
Frisén, J., Holmberg, J. & Barbacid, M. Ephrins and their Eph receptors: multitalented directors of embryonic development. EMBO J. 18, 5159–5165 (1999).
Williams, M. E. et al. Cadherin-9 regulates synapse-specific differentiation in the developing hippocampus. Neuron 71, 640–655 (2011).
Gerfen, C. R. Segregation of D1 and D2 dopamine receptors in the striatal direct and indirect pathways: an historical perspective. Front. Synaptic Neurosci. 14, 1002960 (2022).
Aringhieri, S. et al. Molecular targets of atypical antipsychotics: From mechanism of action to clinical differences. Pharmacol. Ther. 192, 20–41 (2018).
Lew, J. Y. et al. Increased site-specific phosphorylation of tyrosine hydroxylase accompanies stimulation of enzymatic activity induced by cessation of dopamine neuronal activity. Mol. Pharmacol. 55, 202–209 (1999).
Lindgren, N. et al. Regulation of tyrosine hydroxylase activity and phosphorylation at Ser(19) and Ser(40) via activation of glutamate NMDA receptors in rat striatum. J. Neurochem. 74, 2470–2477 (2000).
Nagai, T. et al. Phosphoproteomics of the dopamine pathway enables discovery of Rap1 activation as a reward signal in vivo. Neuron 89, 550–565 (2016).
Kimmel, H. L., Gong, W., Vechia, S. D., Hunter, R. G. & Kuhar, M. J. Intra-ventral tegmental area injection of rat cocaine and amphetamine-regulated transcript peptide 55-102 induces locomotor activity and promotes conditioned place preference. J. Pharmacol. Exp. Ther. 294, 784–792 (2000).
Hubert, G. W., Jones, D. C., Moffett, M. C., Rogge, G. & Kuhar, M. J. CART peptides as modulators of dopamine and psychostimulants and interactions with the mesolimbic dopaminergic system. Biochem. Pharmacol. 75, 57–62 (2008).
Chen, Y., Saulnier, J. L., Yellen, G. & Sabatini, B. L. A PKA activity sensor for quantitative analysis of endogenous GPCR signaling via 2-photon FRET-FLIM imaging. Front. Pharmacol. 5, 56 (2014).
Lee, S. J., Chen, Y., Lodder, B. & Sabatini, B. L. Monitoring Behaviorally Induced Biochemical Changes Using Fluorescence Lifetime Photometry. Front. Neurosci. 13, 766 (2019).
Jentsch, J. D., Roth, R. H. & Taylor, J. R. Role for dopamine in the behavioral functions of the prefrontal corticostriatal system: implications for mental disorders and psychotropic drug action. Prog. Brain Res. 126, 433–453 (2000).
Grace, A. A. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 17, 524–532 (2016).
Thomas, E. A. Molecular profiling of antipsychotic drug function: convergent mechanisms in the pathology and treatment of psychiatric disorders. Mol. Neurobiol. 34, 109–128 (2006).
Lee, S. J. et al. Cell-type-specific asynchronous modulation of PKA by dopamine in learning. Nature 590, 451–456 (2021).
Graybiel, A. M. & Grafton, S. T. The striatum: where skills and habits meet. Cold Spring Harb. Perspect. Biol. 7, a021691 (2015).
Breier, A. et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc. Natl. Acad. Sci. USA 94, 2569–2574 (1997).
Liu, X.-M. et al. Abnormal lipid metabolism and altered neuronal support by astrocytes lacking Akap11, a risk gene for schizophrenia and bipolar disorder. bioRxiv 2025.04.25.650548 (2025).
Wang, S. et al. Loss-of-function variants in the schizophrenia risk gene SETD1A alter neuronal network activity in human neurons through the cAMP/PKA pathway. Cell Rep. 39, 110790 (2022).
Horacek, J. et al. Mechanism of action of atypical antipsychotic drugs and the neurobiology of schizophrenia. CNS Drugs 20, 389–409 (2006).
Roth, B. L., Sheffler, D. & Potkin, S. G. Atypical antipsychotic drug actions: unitary or multiple mechanisms for ‘atypicality’? Clin. Neurosci. Res. 3, 108–117 (2003).
Yun, S. et al. Antipsychotic drug efficacy correlates with the modulation of D1 rather than D2 receptor-expressing striatal projection neurons. Nat. Neurosci. 26, 1417–1428 (2023).
Lee, Y.-K. et al. Bipolar and schizophrenia risk gene AKAP11 encodes an autophagy receptor coupling the regulation of PKA kinase network homeostasis to synaptic transmission. bioRxiv.org (2024).
Iannello, F. Non-intrusive high throughput automated data collection from the home cage. Heliyon 5, e01454 (2019).
Dejanovic, B. et al. Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 100, 1322–1336.e7 (2018).
Dejanovic, B. et al. Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nat. Aging 2, 837–850 (2022).
UniProt Consortium UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480–D489 (2021).
Zhou, X. L. et al. ROR2 modulates neuropathic pain via phosphorylation of NMDA receptor subunit GluN2B in rats. Br. J. Anaesth. 123, e239–e248 (2019).
Morshed, N. et al. Phosphoproteomics identifies microglial Siglec-F inflammatory response during neurodegeneration. Mol. Syst. Biol. 16, e9819 (2020).
O’Shea, J. P. et al. pLogo: a probabilistic approach to visualizing sequence motifs. Nat. Methods 10, 1211–1212 (2013).
Biancalani, T. et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 18, 1352–1362 (2021).
Di Tommaso, P. et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319 (2017).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521 (2015).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 1–12 (2017).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291 (2019).
Ding, J. et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 38, 737–746 (2020).
Zeisel, A. et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014 (2018).
Saunders, A. et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030 (2018).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Phipson, B. et al. propeller: testing for differences in cell type proportions in single cell data. Bioinformatics 38, 4720–4726 (2022).
Savell, K. E. et al. A dopamine-induced gene expression signature regulates neuronal function and cocaine response. Sci. Adv. 6, eaba4221 (2020).
Lodder, B. et al. Absolute measurement of fast and slow neuronal signals with fluorescence lifetime photometry at high temporal resolution. Neuron 113, 3554–3566 (2025).
Paxinos, G. & Franklin, K. B. J. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates. (Academic Press, 2019).
Acknowledgements
We thank Kris Dickson and members of the Sheng and Granger Labs for valuable input; Ben Neale, Tarjinder Singh and Calwing Liao for consultations on human genetic evidence; Kira Brenner and Jacalyn Fahey for assistance with immunohistochemistry; Abraham Eafa and Miriam Kpoyto for help with colony management; Miriam Kpoyto for help with amphetamine-induced hyperlocomotion experiments; Abe Eafa and Nate Hodgson for help with behavioral assays; Guido Gottardo, Reivi Voci, Mara Rigamonti and Tecniplast for assistance with the homecage recording system; Michael Dolan and Chuck Vanderburg for guidance on transcriptome experiments; Michael Sherman for packaging AAVs; Tyler Caron and Matthew Beck for help with mouse colony management and in vivo experiment logistics; the Broad Institute Genomic Platform for assistance with RNA sequencing. Artwork of a saggital mouse brain was adapted from open access artwork created and shared by Federico Claudi (doi:10.5281/ZENODO.3925911). We thank the IDDRC Animal Behavior and Physiology Core, funded by NIH/NICHD P50 HD105351.
Author information
Authors and Affiliations
Contributions
B.J.S. and M.S. conceived and designed the -omics, biochemical, and behavioral experiments. Y.G., M.S., and A.G. conceived and designed fiber photometry, electrophysiology, pharmacological and behavioral experiments. B.J.S. and M.S. wrote the manuscript, with help from Y.G. and A.G. and input from all authors. B.J.S. and Y.G. performed the majority of experiments and analyses in this paper. Specifically, B.J.S. performed immunoprecipitations, immunoblots, immunohistochemistry, synapse purifications, nuclei isolation, RNAseq, homecage activity measurements and analyzed these data. Y.G. performed FLiP measurements, slice electrophysiology, dopamine receptor pharmacology, amphetamine-induced hyperlocomotion and analyzed these data; Y.G. analyzed behavioral data from the IDDRC animal behavior and physiology core. A.N. and K.P.M. processed the transcriptome data, including quality control, reference genome alignment, cell type clustering, annotation and DE analysis, with guidance from S.K.S. and supervision from J.Z.L. M.J.K. performed nuclei isolation and library preparation for 12 week striatum and PFC snSeq datasets. B.L. designed the FLiP recording set up, calibrated the measurements and developed the analysis pipeline with guidance from B.L.S. K.B. performed LC-MS/MS for synapse proteomics and synapse phosphoproteomics and A.V.T. performed LC-MS/MS for immunoprecipitations with guidance from H.K. and S.A.C and analysis help from D.R.M. J.A. conducted motion sequencing tests and D.M. analyzed these data. N.H. conducted associative learning and sensory discrimination assays, with help from J.T. and supervision from Z. Fu. J.A. and C.G. helped with immunoblots. S.N. performed smFISH. I.P. and N.S. assisted with brain tissue collection and immunohistochemistry and N.S. additionally designed and performed rotarod behavioral tests. H.P. and N.M.N. assisted with nuclei isolation and snSeq library preparation with supervision from E.Z.M. E.Z.M. designed the snSeq protocol and guided snSeq analysis. S.A., Z. Farsi, and X.L. helped with data analysis. A.H. conducted confocal microscopy with supervision from P.S.K. N.M. conducted phosphoproteomics motif analysis with supervision from B.S. B.D. purified synapse fractions for proteomics/phosphoproteomics and provided guidance and supervision for biochemical experiments. M.S. and A.G. supervised the project and acquired funding. This work was supported by the Stanley Center for Psychiatric Research and the Harvard Brain Science Initiative BD Seed Grant.
Corresponding authors
Ethics declarations
Competing interests
M.S. is cofounder and scientific advisory board (SAB) member of Neumora Therapeutics and serves on the SAB of Biogen, Proximity Therapeutics and Illimis Therapeutics. S.A.C. is a member of the SAB of Kymera, PTM BioLabs, Seer, and PrognomIQ. B.S. serves on the Scientific Advisory Board of Annexon Biosciences and is a minor shareholder of Annexon. B.D. is a full-time employee of Vigil Neuroscience. E.Z.M. is an academic founder of Curio Bioscience. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Tsuyoshi Miyakawa and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, B.J., Ge, Y., Nicolella, A. et al. Elevated synaptic PKA activity and abnormal striatal dopamine signaling in Akap11 mutant mice, a genetic model of schizophrenia and bipolar disorder. Nat Commun 16, 10793 (2025). https://doi.org/10.1038/s41467-025-66504-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66504-2
