
Abstract
G protein-coupled receptor (GPCR) signaling is one of the most ubiquitous and sensitive forms of cell surface reception. GPCRs stabilize the nucleotide-free state of heterotrimeric guanine-nucleotide binding proteins (G proteins); however, this state is produced at the cost of relieving the G protein of its stable inhibitor, GDP. Upon agonist binding to receptor, the G protein binds GTP and signal transduction ensues. Herein we demonstrate that the agonist can also stimulate the release of GTP. This receptor-mediated mechanism permits dissociation and reassociation of the G protein as the receptor acts as a catalyst for two different reactions. We demonstrate that this mechanism requires a unique, selective active state in addition to the active state that promotes GDP release. The release reaction is competitive with antagonists and we demonstrate operational efficacy. Further, we show that agonists have the potential to preferentially stimulate GTP binding or GTP release. This release selectivity may serve as a form of receptor signaling and reshape our understanding of G protein-coupled receptor signaling. Finally, we demonstrate that these reactions can be recapitulated in human spinal cord dorsal horn, providing an avenue for investigating state selectivity in physiologically significant samples.
Similar content being viewed by others


Introduction
Canonically, G protein-coupled receptors (GPCR) signal through the activation of G proteins1. G proteins are heterotrimeric by nature with monomeric Gα and dimeric Gβ\({{\rm{\gamma }}}\) subunits. The heterotrimer binds guanosine diphosphate (GDP) and, when the receptor is stimulated by an agonist, an interaction between the receptor and the complex prompts the GαGDP subunit to release GDP. The nucleotide-free G protein (Gαapo) has higher affinity for guanosine triphosphate (GTP) and the active GαGTP forms; in this regard the GPCR serves as a guanine-nucleotide exchange factor (GEF). The G protein subsequently returns to the inactive GαGDP state, following GTP hydrolysis, completing the signaling cycle. For the most part, the cycle is believed to be unidirectional where the role of the receptor is limited to stimulating GDP release, however, it is interesting to consider the receptor as a more dynamic partner in the signaling cascade.
Rhodopsin has been extensively studied as a prototypical GPCR and, like other GPCRs, rhodopsin activates a heterotrimeric G protein, transducin (Gt); the release of GDP is considered the rate-limiting step in G protein signaling where the rate of GTP binding is thought to be insaturable2,3,4. In early studies, nonhydrolyzable GTP-analogs such as guanosine-5’-O-[gamma-thio]triphosphate (GTPγS) were useful to study the activated GtαGTP subunit5,6,7. Coincidently, the rate of nonhydrolyzable GTP binding occurs at the same rate that rhodopsin catalyzes the release of GDP8. While the fate of bound GTP was thought to rely on hydrolysis, these early studies showed that the release of a hydrolysis-resistant GTP analog could be catalyzed by photolyzed rhodopsin. It has been hypothesized that active-state GPCRs, including rhodopsin, catalyze the release of both GDP and GTP via a single active state9,10.
More generally GPCRs catalyze the dissociation of GDP from Gα, allowing Gαapo to readily bind GTP; however, some studies have suggested GTP release as a possible explanation for experimental data where the Gαapo species was observed to form, subsequent to the GαGTP species11,12,13. As with rhodopsin, these studies involved measuring both the increase and release of nonhydrolyzable GTP binding over time in the presence of agonist and this has been reproduced in tissue14,15,16,17,18,19,20,21. Notably, these early release studies were performed in both the presence11,12,13 and absence14 of saturating exogenous nucleotide which would suggest that the reaction is permissive under a range of free nucleotide concentrations. Structural biology approaches have captured active state complexes revealing agonist-bound receptor engagement with Gα. In most cases, the G protein is stabilized in the nucleotide-free Gαapo state22,23,24,25,26,27. By contrast, NMR studies of receptor dynamics have revealed a more diversified energy landscape for active-state receptors to wander and receptor-G protein complexes to traverse28,29,30,31,32.
We present here, and in the companion manuscript (Stahl et al.33 in submission), the results of nearly a decade of studies on the role of the mu opioid receptor (MOR) in stimulating 35S-GTPγS release. We have discovered that the mu receptor exhibits active state-selective GTP release and that it is possible for agonist activity to be intrinsically GTP release-selective. Specifically, we present the observation that GEF activity, and therefore active-state affinity, can select for or against a G protein as a function of the nucleotide state (GTP binding or GTP release). This form of selectivity suggests that a requisite second active-state receptor is responsible for, and the selectivity-filter of, the GTP release mechanism. Further, it is possible for an agonist to exhibit a marked preference, or release selectivity, for inducing the dissociation of one or the other nucleotide from the G protein.
Results
Receptor-mediated regulation of GTPγS binding and release
The design of these initial experiments was chosen to mimic a pulse-chase approach34. Cell membranes were prepared from MOR expressing CHO cells and, in the pulse phase, stimulated with 1 μM DAMGO in the presence of 0.1 nM 35S-GTPγS (Fig. 1). At the 1-h time point (t0), this results in an agonist-mediated increase in the population of 35S-GTPγS labeled Giα captured upon rapid filtration. In the subsequent chase phase, the labeling reaction is quenched by a ten-fold dilution into buffer containing 1 µM unlabeled GTPγS. For the next 60 min, the residual 35S-GTPγS binding is captured on filters at the time points shown.

Baseline (t-60) and 1 µM DAMGO (1 h, t0) show the agonist-mediated nucleotide loading in the pulse phase (###p < 0.001, unpaired two-tailed t-test). In the subsequent chase phase, a loss of radiolabeled nucleotide binding relative to t0, is apparent for 100 nM DAMGO(10x dilution) (ns, p > 0.05, *p < 0.05, **p < 0.01 one-way ANOVA with Dunnett’s post-hoc test) and in the presence of 10 µM DAMGO (**p < 0.01, ***p < 0.001; ****p < 0.0001). Notably, 10 µM naloxone completely prevents decay of 35S-GTPγS binding out to 60 min (ns, p > 0.05, all points vs. t0, one-way ANOVA). All data points are presented as the average of three experiments with the bars presenting mean and s.e.m.
In the case where samples were diluted ten-fold a residual 100 nM DAMGO remains from the pulse reaction. At the indicated times, this residual DAMGO produces significant dissociation of radiolabeled GTPγS binding from Giα (Fig. 1). Importantly, the rate of dissociation as well as the magnitude of dissociation is increased by the addition of a saturating 10 µM DAMGO (Fig. 1, 100 nM DAMGO(10x dilution) rate: 0.016 (0.008–0.025) min−1; 10 µM DAMGO rate: 0.048 (0.023–0.086), mean with 95% CI; p < 0.0001 by extra sum-of-squares F test; t-test of 100 nM DAMGO(10x dilution) vs. 10 µM DAMGO at 60 min, p < 0.05); this is in agreement with prior studies35. Inclusion of 10 µM naloxone blocks the release induced by the residual DAMGO in the chase, highlighting the stability of the GTPγS-G protein complex when agonist cannot catalyze the reaction. Further, the naloxone stabilizes the complex as binding does not significantly differ between the t0 and all subsequent time points in the presence of naloxone (Fig. 1, one-way ANOVA of 10 µM Naloxone vs. t0, ns, p > 0.05) arguing against the occurrence of spontaneous release of GTPγS from the complex. The stability of the GTPγS-bound state has previously been reported for MOR stimulated GTPγS binding35. The DAMGO-mediated release prompts the question whether the agonist-mediated dissociation of GTP from Gα could be agonist concentration-responsive.
To assess the concentration sensitivity of the response, the 60-min time point was selected as the logical end to the experiment as this reflected the same amount of time available to load 35S-GTPγS onto Giα during the pulse phase. We note that while DAMGO produces a substantial decrease in 35S-GTPγS binding, the response plateaus by one hour (Fig. 1). The residual 35S-GTPγS binding is not likely due to rebinding of the 35S-GTPγS as the chase reaction includes a final concentration of 0.01 nM 35S-GTPγS and 1 µM unlabeled GTPγS (which is a 100,000-fold excess of unlabeled GTPγS to 35S-GTPγS). The residual binding is therefore interpreted to be a product of the dynamic nature of the receptor system and, in fact, this would be expected under these experimental conditions wherein the unlabeled GαGTPγS becomes a competitor for the receptor to mediate 35S-GTPγS release from Gα35S-GTPγS. This result is a distinct caveat of the single-turnover paradigm used to measure the release following G protein radionucleotide labeling.
As a starting point to assess the concentration-responsive nature of the release function, it was also imperative to be able to load the 35S-GTPγS on Gα without including the agonist to avoid the presence of residual agonist in the chase. Therefore, we pursued an approach that elevates constitutive G protein activity, by removing sodium from the system, and avoids the introduction of agonist until the chase reaction. Sodium ions act as negative allosteric modulators at many GPCRs, including MOR, and the removal of sodium from the binding buffer (– Na+) leads to increased agonist affinity and elevated basal 35S-GTPγS binding36,37,38,39. This effect is demonstrated in Fig. 2A (see Binding Assay schematic) where removal of sodium leads to an increase in 35S-GTPγS binding in untransfected CHO cells (Parental) relative to the baseline in the presence of 100 mM NaCl (Basal); notably, DAMGO has no effect in the presence of sodium in cells lacking MOR. The elevation of 35S-GTPγS binding in the sodium-free conditions likely reflects the activation of all endogenous sodium-regulated GPCRs in the CHO cells38. In the CHO-mMOR cells, both the inclusion of DAMGO and the removal of sodium (without DAMGO) leads to significant 35S-GTPγS binding. Both DAMGO and sodium-free 35S-GTPγS binding are blocked by pertussis toxin pretreatment, implicating the involvement of primarily Giα inhibitory G proteins in both conditions (Fig. 2B). Together, studies in the CHO parental line and using pertussis toxin support the role of both the receptor and Giα in the 35S-GTPγS binding and release reactions. Beyond these controls, we do not rule out the involvement of additional membrane components as these reactions are not prepared or catalyzed in isolation. However, our data suggests that the agonist, receptor, and Giα form at least the minimum ternary complex necessary for these reactions to occur.

A schematic describing the experimental design used to assay 35S-GTPγS binding versus release is shown to the left. A 35S-GTPγS binding in CHO-Parental and CHO-mMOR membranes wherein the Basal and 1 µM DAMGO measures are in the presence of 100 mM NaCl and 0 mM NaCl contains no agonist. (B) Pertussis toxin blocks sodium free 35S-GTPγS binding in both cell lines and the DAMGO effect in CHO-mMOR cells. (C) 35S-GTPγS release following sodium free loading (Basal) is responsive to DAMGO only in the CHO-mMOR membranes. (D) Pertussis toxin treatment prevents loading and subsequent release. **p < 0.01, ***p < 0.001, ****p < 0.0001 (A, B one-way ANOVA within cell line comparing to basal, Dunnett’s post-hoc test: C, D unpaired two-tailed t-test). All data are presented as the mean with s.e.m. of three experiments. BioRender® was used to create the schematic, with license.
We then asked if DAMGO could reverse the increase in 35S-GTPγS binding obtained in the sodium-free condition. For this study membranes (1 mg protein) are preincubated/pulsed for 1 h (see Release schematic in Fig. 2), in the absence of sodium with 0.1 nM 35S-GTPγS and 10 µM GDP to allow for constitutive loading of the G protein with 35S-GTPγS. The reaction is then diluted ten-fold, into buffer containing an excess of unlabeled GTPγS (1 µM) and 100 mM NaCl (to quench the constitutive Giα loading) and allowed to proceed for an additional hour incubation/chase in the absence (Basal) or presence of 1 µM DAMGO. In the MOR expressing CHO cells, DAMGO produces a significant decrease in 35S-GTPγS binding. This effect was observed to be receptor dependent as CHO-Parental membranes did not demonstrate DAMGO sensitivity (Fig. 2C). As was the case with the initial binding/pulse reaction, pretreatment of pertussis toxin blocks any observable release implicating the involvement of Gαi/o proteins (Fig. 2D).
While 35S-GTPγS is generally accepted to be nonhydrolyzable, or highly resistant to hydrolysis, it was a reasonable concern that the decrease in 35S-GTPγS could be due to phosphate hydrolysis. We, therefore, used a thin layer chromatography (TLC) approach to determine if there was cleavage of the radiolabeled thiophosphate group40. Due to the loading limits of the TLC plate (1–3 µL), it was necessary to run these experiments in a much smaller volume at much higher reagent concentrations. For the pulse reaction, the assays were performed as described in the Binding schematic of Fig. 1 but with 300 µg protein and 4.6 nM 35S-GTPγS in the absence of sodium for 1 h in 300 µL assay buffer. Using 30 µL of this reaction, Fig. 3A shows that under these conditions, GTPγS binding is still increased in the absence of sodium. For the chase reaction, the remaining membranes were then rapidly centrifuged to pellet the bound 35S-GTPγS and resuspended two times to wash away residual 35S-GTPγS. This pulsed protein was resuspended into 27 µl of buffer containing 100 mM NaCl and dilute 1:2 into chase reactions containing 1 µM unlabeled GTPγS with and without 1 µM DAMGO for an additional hour. A 2 µL aliquot of the chase reaction was set aside for TLC analysis and the residual bound 35S-GTPγS was measured by rapid filtration. The inclusion of 1 µM DAMGO produces a 50% reduction in bound 35S-GTPγS compared to vehicle (Fig. 3B). This reduction in binding was taken to reflect the same agonist-mediated loss of 35S-GTPγS binding observed in Figs. 1, 2.

A Sodium free conditions still elevate 35S-GTPγS binding when using higher protein and35S-GTPγS concentrations (see “Method” inset), (**p < 0.01, unpaired two-tailed t-test). B DAMGO induces 35S-GTPγS release in the modified reaction (**p < 0.01, unpaired two-tailed t-test). C Aliquots of the release reaction were loaded onto TLC plates and the hydrolyzed thiophosphate was measured (one representative TLC blot is shown with increased brightness saturation). CuSO4 (±Cu2+), in the absence of protein, was used as the positive control to visualize thiophosphate degradation. D 35S-Thiophosphate cleavage was minimal compared to CuSO4 treatment for both the Vehicle and DAMGO treatment (comparing all points: one way ANOVA, Dunnett’s post-hoc, *p < 0.05, **p < 0.01). All data points are presented as the average of three experiments with the bars presenting mean and s.e.m. BioRender® was used to create the schematic, with license.
Aliquots of the chase reactions were loaded onto a TLC plate (Fig. 3C). Since DAMGO produced a substantial reduction in residual 35S-GTPγS binding in these samples, an increase in the radioactivity migrating away from the point of application would occur if the decrease in 35S-GTPγS binding was due to thiophosphate hydrolysis. In a separate volume containing no protein but only buffers, Cu2SO4 was included as a positive control to degrade the thiophosphate, independent of enzymatic hydrolysis41. This is compared to the same sample without Cu2SO4. In all cases, the densitometry indicates only significant hydrolysis occurring in the Cu2SO4 (+Cu2+) lane (one-way ANOVA, Fig. 3D). These observations are in agreement with the findings of others that no significant hydrolysis of the tertiary thiophosphate is occurring during the reaction and, therefore, 35S-GTPγS must be dissociating from the G protein as an intact nucleotide triphosphate.
Together these findings demonstrate that the receptor acts as a control point for binding different states of the G protein. It is well known that the receptor plays a significant role in inducing the GTP binding reaction (receptor-mediated GDP dissociation and subsequent GTP binding). However, the role of the receptor in destabilizing the subsequent G protein-nucleotide interaction (receptor-mediated GTPγS release) is an unappreciated component of the GPCR signaling mechanism. In Fig. 1, we noted that the dilution of DAMGO (100 nM residual) leads to a submaximal decrease in remaining 35S-GTPγS binding relative to the 10 µM DAMGO treatment. Having optimized the system for agonist-independent nucleotide loading it was reasonable to investigate agonist concentration-dependent 35S-GTPγS release.
Ligand-mediated regulation of GTPγS binding and release
To further explore this apparent receptor-mediated mechanism in the GTPγS release paradigm, we compared the concentration-response measures of both the 35S-GTPγS binding and release functions. For these experiments, DAMGO stimulation of 35S-GTPγS binding and release was measured in MOR-expressing CHO cell membranes (Figs. 4A, B). DAMGO induces a concentration dependent increase in 35S-GTPγS binding as well as the release of GTP as revealed by the decrease in 35S-GTPγS bound (Fig. 4A). The saturable nature of the reactions is apparent as sigmoidal concentration-response curves with clear plateaus are presented for both responses. In the release experiments 35S-GTPγS loading was accomplished, as previously described, using the sodium-free approach described in Fig. 2. Nonlinear regression analysis indicates no significant difference in DAMGO potency for binding or release suggesting that, in this cell line, DAMGO does not show selectivity for either of the two responses (Fig. 4A).

A Binding and release are plotted with nonlinear regression analysis; the logEC50 values are compared to the right (ns, p > 0.05 unpaired two-tailed t-test of individual curves). Naloxone shifts the logEC50 of DAMGO in binding (B) and in release (C) in a concentration dependent manner. To the right of each is the Schild Plot with log[Naloxone] v log(Dose Ratio-1) and indicating a linear fit (slope = 1) and a shared X-intercept (logKi), extra sum-of-squares F test F = 1.305 (1, 16), p = 0.2702. All data are presented as the mean with s.e.m. of three experiments.
The competitive antagonist naloxone was applied at increasing concentrations in both experiments to produce a concentration-dependent rightward shift in the DAMGO response curves (Fig. 4B, C). The dextral displacement of DAMGO potency is a hallmark of competitive antagonists42. Using the classical approach, we estimate that naloxone has the same affinity (logKi = −8.7 ± 0.1, the x-intercept in the Schild plot) for MOR in binding and release (Fig. 4B, C) as would be expected for a simple competitive interaction43. Together, the concentration dependency of each of these ligands further supports the receptor-mediated nature of 35S-GTPγS binding and release. It is additionally apparent from the activity of DAMGO and naloxone that their intrinsic efficacy is largely similar between the two responses, with DAMGO demonstrating significant agonist activity and, conversely, naloxone lacking efficacy.
According to a two-state model of receptor activation, this data specifically excludes the possibility that the active state in one (binding or release) response is the inactive state in the other (release or binding) response44. This is because the drugs would need to acquire opposite activities in the two responses, which they clearly do not. That is, DAMGO would become an antagonist in one of the responses, and naloxone may become an agonist in one of the responses. Additionally, the observation that DAMGO potency and naloxone affinity are conserved for both reactions is strong evidence that these drugs act through a shared and accessible receptor population. Otherwise, we may observe a decrease in DAMGO potency or naloxone affinity as a result of the less accessible receptor population, which was also not the case.
Receptor-reserve and the Occupancy-Effector Relationship of GTPγS binding and release
Early on, the possibility that this was a system, or preparation, artifact was a central concern. Essentially, if the reverse reaction was the product of some population of Giα-35S-GTPγS, that remained trapped at the receptor, then the G protein would not be available to perform its physiological responsibility and transduce signal downstream. However, if the release function was the result of the G protein being trapped on the receptor, then it would occur linearly as a product of agonist occupancy on the receptor. (A nonlinear occupancy-response relationship would be impossible.) It is possible to directly determine this relationship using the alkylating agent beta-funaltrexamine (β-FNA)45.
As a MOR irreversible antagonist, β-FNA occludes the orthosteric binding site on the receptor and effectively removes the receptor’s ability to function in the system46,47. Radioligand binding studies show that β-FNA treatment produces a significant loss in 3H-naloxone binding in a concentration-dependent manner (Fig. 5A). To ensure that β-FNA did not lead to a nonselective degradation of receptors, MOR protein density was measured by western blot of the HA-tag on the N-terminal following β-FNA treatment. No difference in protein level was observed indicating that receptor levels remain constant in these preparations (Supplementary Fig. 1). Having demonstrated the selective loss of binding sites, it was reasonable to explore 35S-GTPγS binding and release in these preparations.

A 3H-Naloxone binding in the absence (Control) or presence of increasing β-FNA (one-way ANOVA, *p < 0.05, ***p < 0.001). The fractional degree of 35S-GTPγS (B) Binding and (C) Release relative to control (no β-FNA) run in parallel with each β-FNA concentration used. D The maximum DAMGO response in Binding (B) and Release (C) as a function of both receptor number (A) and the DAMGO potency in each reaction (B, C). For both the Binding and Release reactions, the Effector-Occupancy Relationship is nonlinear (extra sum-of-squares F test p < 0.0001) 3H-Naloxone binding data is the average of five experiments. Data for GTPγS binding and release are the average of three experiments. All data plotted as the mean with s.e.m. in (A–C). Individual data are presented in (D).
As a function of receptor alkylation, the DAMGO potency in both reactions shifts rightward (Figs. 5B, C). Raw data for binding and release following alkylation are included for comparison (Supplementary Fig. 2A, B). Also presented is the Occupancy-Response relationship where the change in 3H-naloxone binding sites is presented on the x-axis and the change in the maximum response for the binding and release function is presented on the y-axis (Supplementary Fig. 2C). In this representation both 35S-GTPγS binding and release have a nonlinear (clearly hyperbolic) occupancy-response relationship indicating that both responses are subject to substantial receptor-reserve. This approach has previously been established as a representation of the receptor-reserve of a system48,49,50.
It would, however, also be possible to demonstrate a nonlinear occupancy-response relationship if receptor alkylation was specific for some receptors that were occluded from the G protein population. (Essentially, if the total receptor population was not the same as the available receptor population.) In this case, 3H-naloxone binding sites would be depleted but 35S-GTPγS binding and release would still be the product of an artifactual MOR-Giα-35S-GTPγS ternary complex. We then hypothesized that the change in potency from the alkylation reaction could be integrated into the occupancy-response analysis to demonstrate that the G protein was also not responding through an occupancy function. Using the analysis in (Fig. 5D), the (10^logEC50|control)/(10^logEC50|alkylation) was integrated into the abscissa (of Supplementary Fig. 2C) to produce the Δbinding*Δpotency relationship. From this representation, a linear relationship would be produced if loss of binding did not affect response potency (as the (10^logEC50|control)/(10^logEC50|alkylation) ratio would equal 1). This would indicate that alkylation was specific for an unavailable fraction of the total population with respect to the G protein.
A nonlinear Δbinding*Δpotency relationship thus implies that either alkylation affects the available fraction of the total population or that the available fraction is the total receptor population. In either case, the G protein must have access to the same available receptor population targeted for alkylation. This, then, demonstrates a mechanism where G proteins are not limited to a linear receptor-occupancy-effector relationship (i.e., pre-coupled) but are able to freely dissociate-reassociate with receptors for both the binding and release reaction. Further, this mechanism is shared for the binding and response function, indicating that receptors are able to interchange between G proteins, and potentially interact with multiple G proteins, not being limited to a static stoichiometry.
Ligand-selectivity for GTPγS binding and release
The clear result of the alkylation experiments is that the G protein is capable of moving in a bidirectional manner onto and off the receptor when bound to either GDP or GTP. Beyond a simple reversible binding reaction, by tracking the binding of the tertiary thiophospate, we see that an active-state of the receptor is capable of coupling with either the GDP- or GTP-bound form of the G protein to induce nucleotide release. This complex relationship between the G protein and the receptor affords the exciting opportunity to investigate the state-selectivity of agonists. The µ opioid receptor has a large and structurally diverse collection of experimental and clinically relevant compounds, therefore, we compared the relative activity for the binding and release function for select agonists. Specifically, we hypothesized that some opioid compounds may demonstrate active state-selectivity for one or the other GTP binding reactions.
In this system, DAMGO did not exhibit selective preference for the binding or release reaction and thereby served as the control for full agonism in each system. The response of all other agonists could then be normalized to this maximum response, allowing for the comparison of changes in potency and efficacy. All agonists tested here exhibit clearly saturable response curves and Hill coefficients are all approximately 1 (Fig. 6). We applied three different models to analyze potency and efficacy in these experiments based on their status as the prevailing means of understanding ligand efficacy in pharmacology. First, the measure of Emax is an empirical estimate of agonist efficacy in a system and is referred to as Stephenson’s Efficacy (ε)51. Secondly, the determination of two-state Efficacy (α) is a mechanistic approach to measuring efficacy that is generally appropriate for partial agonists but incapable of dealing with full agonists52,53. Finally, the Operational Efficacy (τ) is likely the most relevant approach to estimating efficacy in this case as it is also non-mechanistic but incorporates a non-linear occupancy-response relationship which we have good confidence adequately describes both systems (Fig. 5D)54. Although none of these models is capable of correcting for the receptor reserve of a system (absent alkylation), simple estimates of changes in efficacy are still possible and informative of state selectivity. This is especially true for partial agonists or cases where full agonists in one system become partial in another system.

MOR receptor stimulation with different agonists in both response paradigms. In the release reaction, membranes are incubated without NaCl for 35S-GTPγS loading and the release reaction is performed in the presence of 100 mM NaCl. Stephenson’s Efficacy (ε), Two-state Efficacy (α), and Operational Efficacy (τ) are presented for each fit of the data. Where efficacy could not be estimates, maximal bars are presented as without error. Although efficacy cannot be measured directly for full agonists, loss of efficacy can be estimated. All parameter estimates not adequately determined by nonlinear regression are presented as the undefined variable (θ) on the Y-axis. For Stephen’s efficacy, an unpaired two-tailed t-test was used (*p < 0.05, **p < 0.01); for two-state and operation efficacy, an extra sum-of-squares F-test was applied to determine changes in efficacy (*p < 0.05, ****p < 0.0001). DAMGO curves are the average multiple experiments (binding n = 5; release n = 6). All other agonists are the average of three independent experiments run with DAMGO control. Data are plotted as the mean with s.e.m.
When comparing 35S-GTPγS binding and release, several agonists exhibit remarkable differences in both potency and efficacy (Fig. 6). The potency of morphine and fentanyl does not differ between the two responses. Interestingly, both agonists lose efficacy in the release reaction in every model fit. Conversely, nalbuphine exhibits a minimal increase in potency but gains efficacy in the release reaction. The observation that agonists can gain and lose efficacy in a given pathway is inconsistent with a two-state model of activation. Further, all three different models indicate that efficacy decreases for morphine and fentanyl and increases for nalbuphine. Beyond the changes in efficacy, most of the agonists display minimal to no change in potency. Buprenorphine exhibits a dramatic >10-fold increase in potency for the release reaction. This is strong evidence that rank order potency may also be a means of observing selection between the two active states and is consistent with a multi-state model of receptor activation55,56. According to two-state theory, rank order potency and/or efficacy are always conserved between systems if those systems are stimulated by a singular active-state receptor44. Potency and efficacy values are presented in Table 1.
GTPγS binding and release in human spinal cord dorsal horn
Herein we have provided evidence that ligand-induced exchange occurs in transfected CHO cells that overexpress MOR. However, this biochemical approach can also be used to assess agonist activity in endogenous tissue. To demonstrate this, we dissected dorsal horn from the fourth lumbar region of human spinal cord, as this is known to have high MOR expression57 and is a site for epidural delivery of opioid analgesics58. Membranes were prepared and binding was performed in a conventional manner; the release reaction was loaded using 100 nM DAMGO in the “pulse” reaction similar to that shown in Fig. 1 (Fig. 7A). Following the loading of 35S-GTPγS in the pulse reaction, a 15% increase in 35S-GTPγS loading can be achieved (Fig. 7B). Two potent MOR agonists, DAMGO and loperamide, produce a ~ 35% stimulation of 35S-GTPγS binding over baseline; both agonists produce release in the spinal cord preparations, but neither are capable of fully reversing the 15% loading (Fig. 7C, maximum reached is ~6%). (Raw data for DAMGO-mediated binding and release are presented in Supplementary Fig. 3.) Neither compound produced a significant shift in potency between the binding and release, but this may be due to the low sample number (n = 3); regardless, given that these samples were procured as previously frozen tissue, we are encouraged that this approach could be used to compare agonist actions in physiologically relevant samples.

A Dorsal horn was dissected from a previously frozen L4 section of human spinal cord; the “pulse” loading of GTPγS was performed in the presence of sodium chloride as described in Fig. 1. B The loading of 35S-GTPγS in the pulse reaction produces a 15 ± 2% increase in the presence of 100 nM DAMGO (**p < 0.01 paired two-tailed Student’s t-test, n = 3 individual spinal cords). C 35S-GTPγS binding and D. release are demonstrated using DAMGO and loperamide in the same samples presented in B (mean with s.e.m. plotted). The pEC50 (M) and Emax (% stimulation over baseline) are presented in the table inset with 95% CI. BioRender® was used to create the schematic, with license.
Discussion
The observation that agonists can have state selectivity in these two measures is a striking discovery (Fig. 6). Agonist-induced GPCR state selectivity has not previously been considered for two forms of the same primary effector, i.e., the GTP versus the GDP bound state of Gα. Moreover, for the primary signal transducer of receptor signaling (G proteins) to be the direct measure of this selectivity lends support to the broader implications of this finding as the vacillation of the G protein between the two states may dictate its availability to engage with other effectors. Our findings are in agreement with other studies that have reported the ability to release GTP analogs from Gα without hydrolysis and that this event could be affected by the inclusion of hormones13. Herein, we demonstrate that there are different active states of the receptor that will shift the affinity from preferring binding of GTP to preferring release of GTP. Taken together, studies utilizing: the pulse-chase method (Fig. 1), receptor-deficient and pertussis toxin treated cell lines (Fig. 2), and cleaved phosphonucleotide analysis (Fig. 3) indicate that the release mechanism is wholly receptor mediated and quite easily accessible, and it can be measured in relevant human tissue (Fig. 7). Further, the concentration-dependent agonist and antagonist activity highlights the purely pharmacologic mechanism of controlling this reaction (Fig. 4).
In our initial experiments, it was important to validate this mechanism. In particular, using receptor alkylation, we established that each reactant in the substrate-product relationship was given equal access within the system and not unduly constrained by an unknown system limitation. It was initially apparent that both the binding and release reactions were subject to large receptor reserves, which is not unexpected in an overexpression system. In the occupancy-response relationship, more than half of the total receptor population could be depleted by alkylation with a minimal decrease in agonist potency or efficacy (Supplementary Fig. 2C). As discussed, this data became even more substantial when considered for the G protein’s access to the receptor population. When the occupancy-response plot is represented as a fraction of the receptor population available for both function and alkylation, we demonstrate that all G proteins have access to all receptors in the system. This plot specifically highlights the reaction of the G proteins with the receptor population being catalytic, not stochiometric (Fig. 5D), for both the GiαGDP and GiαGTP forms. Similar considerations were applied to estimate the available receptors for the adenylyl cyclase pool in turkey erythrocytes59,60.
It is also possible that the receptor’s role in producing both G protein activation (GDP release) and G protein modulation (GTP release) are energetically favorable. According to the ternary complex model, G protein activation is followed by signaling and hydrolysis where hydrolysis will consume a high energy phosphate bond for each turn of the cycle (Fig. 8). With the introduction of a GTP release function, the G protein signal may be modulated without the direct consumption of the high energy bond. This would permit the receptor to utilize ligand binding-energy and membrane kinetic-energy as a source of a theoretically limitless energy supply. If this is the case, the common view of GPCR signaling as metabotropic, by definition, could in fact be much more complex and energy efficient. Consider that, if signal transduction is a form of membrane transport, receptors may transport the signal not only via primary active membrane transport (through GTPase), but also through passive facilitated diffusion (via GTP release). We speculate that, if this mechanism finds broader application in GPCR signaling, it may be more appropriate to refer to metabotropic GPCR signaling as free-energy accepting signal-transduction Receptors (feastRs). This feastR hypothesis would situate the receptor as a free-energy scavenger that is granted permission, by the agonist, to introduce and modulate energy in the system rather than a metabolic gate-keeper that denies energy expenditure by the system.

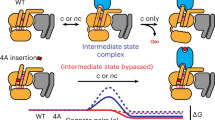
In the two-state model, a single active state receptor is solely responsible for the catalytic activity of the receptor. In the three-state model, the G protein is permitted to cycle between two different active states (R* and R**). Ligands may have different selectivity and/or efficacy for GDP release (α1) compared to GTP release (α2).
We speculate that rhodopsin signaling could offer an even more exciting possibility. Biochemical studies suggest that depletion of transducin may limit the signaling capacity of rhodopsin3. This is because transducin exists in limited pools which rhodopsin must both consume to signal and conserve to be able to signal. In fact, this depletion effect becomes even more substantial when the geometries of the lipid bilayer are considered in the approach of the substrates to the catalyst3. In addition, the concentration of rhodopsin in rod outer segments is ~4-8 mM61; this would surpass the free GTP concentration by approximately five-fold62,63. If the GTP release function were to act as a buffer to the system, it could quantitatively increase the density of transducin (as both GtαGDP and GtαGTP would then be catalytic substrates). This could permit the free nucleotide pool to be maintained, at least fractionally, as not every coupling event would lead to guanosine triphosphate consumption. We note that, in most physiological cases, the free nucleotide concentration would rapidly saturate the displaced nucleotide, but this may still permit the receptor to extend its influence over the shape of the G protein signal. Further, by maintaining the availability of both substrates (GTP and transducin) for rhodopsin the release reaction may provide a control point for rhodopsin to manage energy expenditure of, and impart additional control over, the cellular processing of visual stimulus.
Broadly, it seems reasonable to suggest that the physiological impact of mechanism will be tissue and context dependent. As different tissues have complements of receptors with different densities, as well as potentially different structural geometries, nucleotide densities, and G protein availability, we would expect that not all tissues would benefit or even register changes in release selectivity. We demonstrate in Fig. 6 that the active state of the receptor can differentiate between ligands, whereby some agonists can display a preference comparing binding and release (altered efficacy and/or potency). With optimization, it should be possible to compare such differences between ligands at GPCRs in human tissue, as we demonstrate that binding and release can be measured in human spinal cord dorsal horn membranes (Fig. 7). In this manner, it may be possible to determine “functional selectivity” or “ligand bias” using relevant tissue and the same sample preparation.
Whether or not these differences extend beyond the limits of opioid system will require much more investigation; however, we demonstrate that the release function can be measured for several other Class A GPCRs (Stahl et al.33 in submission). This, at a minimum, suggests that receptors in a number of physiological systems are not absolutely denied access to the release function. In time, as more systems are investigated, a role beyond the Gi/o/t proteins may even become relevant, as other members of the G protein family have not been investigated for this mechanism. It is, however, reasonable to suggest that the release function could contribute to the receptor’s G protein selectivity filter. If receptors have access to the GTP binding and release functions, this would necessarily reduce the apparent affinity of the receptor for other G proteins (for example, GiαGDP and GiαGTP would both complete with GsαGDP) thereby affecting the equilibrium selectivity of the receptor for a given G protein type. However, this type of mechanism would be subject to the free nucleotide concentration as saturation of the apo state would be expected to be rapid at the estimated physiological nucleotide concentration. An additional consideration is the residence time of the Gαapo subunit on the receptor, as the extension of the apo-state lifetime would come at the cost of further delaying both signal propagation and the hydrolytic recovery of the GαGDP ground state.
Beyond issues of receptor effector selectivity, the experiments presented here are fundamentally complicated by the limits of our experimental design. We are able to observe the rate of nucleotide turnover at the G protein. However, these experiments are referred to as single turnover because, as the nucleotide is labeled, we are only able to measure label dissociation. Practically, we assume that, upon 35S-GTPγS release, unlabeled GTPγS occupies the G protein (essentially Giα35S-GTPγS -> GiαGTPγS). We expect that this new, unlabeled species would be equally capable of going through further rounds of nucleotide release and reassociation. This continued reaction would mask our ability to observe the true catalytic rate of the system. We acknowledge that continued release in the chase reaction does complicate our interpretation, but this potentially underestimates the activity of all agonists in the system and, likely, does not change our understanding of the role of the active-states in the system. Moreover, at a minimum, it is entirely reasonable to suggest once 35S-GTPγS is bound in the pulse reaction, it is also subject to the continued release and rebinding reactions. Certainly the release reaction would complicate the interpretation of the pulse experiments as well. Overall, these considerations do not negate the large changes in agonist activity observed.
The activation of the G protein proceeds through changes in the Gα subunit including a requisite movement of the alpha helical domain (AHD) away from the Ras domain and a receptor-interaction with the α5 helix to further destabilize the nucleotide binding site64. However, Ras domain binding of nucleotide has been reported in the absence of the AHD65 and molecular dynamic simulation have estimated that alpha helical domain movement can occur, spontaneously, and independent of nucleotide release4. It is very likely that movement of the AHD is also required for GTP release but, based on the observed changes in rank order agonist efficacy, AHD migration alone is not likely to select for binding or release. Essentially, it is not clear how movement of the AHD would behoove certain agonists, while being disadvantageous to other agonists, in the binding reaction and for this relationship to be inverted in the release reaction. As such, we hypothesize that beyond simply reacting to the difference in nucleotide-bound state of the G protein, the receptor acts as a sensor of the specific nucleotide-bound state of the G protein. This reverse flow of information from the G protein would situate the receptor as a “first and next step” in the G protein signaling cascade, permitting the receptor to exert continued control over the fidelity and texture of the signal.
For clarity, the two-state coupling model (adapted from the ternary complex model) is presented with emphasis on the known and herein defined aspects of the model (Fig. 8)12,66,67,68,69. In particular, the reverse reaction is superimposed into the two-state coupling model to highlight the necessary adaptation required by the present findings. This model has previously been presented, in particular, as a reverse mechanism involving GαGTP reassociation with the receptor70. In fact, a reverse mode was observed to be productive both for nucleotide release and for modifying G protein signaling activity as a coupling mechanism70,71. Importantly this was found to be agonist concentration-dependent as well as nucleotide-specific where the concentration and form of the applied exogenous guanine nucleotide were central to the productivity of the receptor-G protein interaction. Interestingly, in an early study on the β2-receptor, epinephrine/GαppNHp-pulsed stimulation of adenylyl cyclase was reversed by a subsequent epinephrine-chase reaction72. Further, in the reconstitution of the receptor|G protein|adenylate cyclase system, it was observed that epinephrine and GTP, together, could reverse GppNHp-bound G protein-stimulated cyclase activity15,16. As a reconstituted system, this suggests that the receptor and activated G protein are the minimum system requirements necessary to recover the release reaction. These studies coalesce on a mechanism where the receptor continues to convey information to the G protein and, in different ways, may modulate the effectors activity.
Taken together, GαGTP -receptor reassociation does not contradict the two-state coupling model in any way. Rather, it substantially underscores the validity of the model and may add to it in a way that more closely approximates reality. With the inclusion of this reversible and state-selective mechanism, it was possible to integrate the two reactions as the Three-State Coupling Model (Fig. 8). This model highlights the different roles of the two active states (R* induces GαGDP - > Gαapo; R** induces GαGTP - > Gαapo). Formally, we propose the separation of alpha (α) into two efficacy parameters. These efficacy parameters relate the two different active states with their functions: GDP release (α1) and GTP release (α2).
As described in the three-state model, the receptor acts at two different control points in the system. It initially catalyzes GDP dissociation permitting access to GTP and fundamentally transduces signal across the cell membrane by the formation of GαGTP. Beyond signal transduction, in the two-state coupling model, signal propagation is relegated to intracellular partners (G proteins, guanylyl cyclases, adenylyl cyclases, phospholipases, etc). By contrast, if the receptor gains access to catalyze GTP dissociation, it could be considered integral to the signal propagation process. This would allow the receptor to provide texture and potentially shape or contrast the more focal role as the initiation step (GDP dissociation). This capability reintroduces the receptor into the signal transduction pathway in a way where it may act (at every turn of the GTP release cycle) to influence the excitability of the system.
In conclusion, the goal of this work was to investigate the relationship between agonist efficacy and the G protein nucleotide-bound state. In the future, these methods may serve as a starting point for investigating other receptors and other systems for release selectivity. We conclude that agonists and receptors are sensitive to the current state of the G protein and may act accordingly. Specifically, beyond agonist active-state selectivity (via the two-state model), agonists can exhibit microscopic active-state selectivity for different points in G protein signaling (i.e., the three-state coupling model). This signaling mechanism may introduce new avenues for the receptor to act and react to the instantaneous state of the system.
Methods
Materials
Compounds were from the following vendors: Sigma-Aldrich (St. Louis, MO, USA): morphine sulfate pentahydrate (M8777), naloxone hydrochloride (N7758), nalbuphine hydrochloride (N4396), and GDP (G7127). Cayman Chemical (Ann Arbor, MI, USA): fentanyl citrate (22659) and GTPγS (35098). Tocris Bioscience (Bristol, UK): DAMGO (Cat. 11711) and β-FNA (0926). Cell Signaling Technology (Danvers, MA): rabbit anti-HA (3724S, Lot 8), mouse anti- β-actin (3700S, Lot 21). Li-Cor Biotechnology (Lincoln, NE): goat anti-rabbbit800CW (926-32211, Lot D30627-05), goat anti-mouse680LT (926-68020, Lot D30418-15). 35S-GTPγS (NEG030H001MC) was from Revvity (Waltham, MA, USA). 3H-(-)naloxone was generously provided by the National Institute on Drug Abuse (NIDA) Drug Supply Program (NIDA; Bethesda, MD, USA). Human spinal cord sections (L4) were obtained from Anabios (San Diego, CA). Sections from three male donors (age 18, 22, and 35 years), snap frozen in LN2 upon collection, were used for coupling and release measures. Sections were stored in LN2, shipped on dry-ice, and stored at −80 °C prior to use.
Cell lines
Most biochemistry experiments, except human spinal cord, were performed using membranes from Chinese Hamster Ovary cells (CHO-K1, ATCC CCL-61) expressing a N-terminal haemagluttinin-tagged (HA-tagged) form of the mouse µ opioid receptor (CHO-mMOR)53,73. (The CHO-K1 parental cell line was also used in one series of studies and were handled identically to CHO-mMOR cells.) Cells were cultured for harvesting in DMEM/F-12 supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 1 µg/mL puromycin at 37 °C in 5% CO2. Prior to collection, cells were incubated for two h in serum-free media. Following serum starve, cells were rinsed with phosphate-buffered saline (PBS) and removed from the plate by cell scraping in 5 mM EDTA in PBS. Cells were pelleted at 2000 × g for two min and rinsed once in PBS. Cell pellets were stored at −80 °C until use in biochemical assays.
Pertussis toxin (cat# P2980-50ug, MilliporeSigma, St. Louis, MO) treated cells were incubated in standard media 100 ng/mL pertussis toxin overnight. The following day, cells were rinsed with PBS and incubated in serum free buffer for 30 min prior to scarping and pelleting. Cells were intentionally serum starved for a shorter time to ensure that pertussis toxin treatment did not reverse during the serum starvation.
Kinetic studies
In the initial studies, cell membranes were prepared by Teflon-on-glass homogenization in 10 mM Tris HCl pH 7.4, 100 mM NaCl, 1 mM EDTA. Homogenate was centrifuged twice at 20 K × g for 30 min at 4 °C. The resulting pellet was resuspended in assay buffer containing 50 mM Tris HCl pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA. Protein was quantified using the DC protein assay (BioRad, Hercules, CA). For the Pulse-Chase experiments presented in the pilot, 50 µg/mL cell membranes were incubated with 10 µM GDP, 0.1 nM 35S-GTPγS, and 1 µM DAMGO at 25 °C temperature. The Pulse (loading) reaction was measured at the zero (t-60) and one h (t0) time points. In the Chase reaction, aliquots of the loading reaction were dilute, as described, and incubated for the indicated time frame at 25 °C temperature. All samples were handled in this manner with dilution into a 2 mL volume containing 10 µM GDP, 1 µM unlabeled GTPγS, and drug for the time indicated.
All reactions were quenched by rapid filtration through GF/B fiberglass filters on a 12-well manifold vacuum filtration system (MilliporeSigma, St Louis, MO). Filters were rinsed twice with ice cold diH2O and allowed to dry briefly. Filters were loaded into 7 mL scintillation vials and 2 mL of Safety-Solv scintillation cocktail. Vials were permitted to equilibrate, overnight, and scintillation was counted on an LS6500 liquid scintillation counter (BeckmanCoulter, Brea, CA). All data was acquired and analyzed in disintegrations per minute (dpm).
Thin layer chromatography
35S-GTPγS loading and release studies were prepared specifically for the samples to be suitable for chromatographic processing and analysis. Specifically, it was necessary to process these samples in a manner that retained radioactivity density at levels high enough for detection on a phosphoimager. Initially, protein was processed as previously described. The initial loading reactions were Basal in the standard assay buffer (50 mM Tris HCl pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA) or sodium-free stimulation in 0 mM NaCl buffer (50 mM Tris HCl pH 7.4, 5 mM MgCl2, 1 mM EDTA). The loading reaction was run in a 300 µL final volume (1 mg/mL) with 10 µM GDP and 4.3 nM 35S-GTPγS for one h at 25 °C temperature. Three aliquots of each loading reaction (3 × 10 µL) were filtered and processed as described in the Kinetic Studies. The remainder of the sodium-free loading reaction was rapidly centrifuged (20 K × g, 5 min, 4 °C) and rinsed in 100 mM NaCl assay buffer. The final pellet was resuspended in 27 µL 100 mM NaCl assay buffer. Vehicle and 1 µM DAMGO release reactions were measured by 1:2 dilution into 100 mM NaCl assay buffer containing 10 µM GDP and 1 µM unlabeled GTPγS. Release reactions were allowed to proceed for an additional hour and filtered, again, as described. Filters were loaded into vials and counted by liquid scintillation on a scintillation counter.
The thin layer chromatography experiments were adapted from GTPase assays as the product of interest was the cleaved thiophosphate40. For TLC analysis, 2 µL of the release reaction were dilute 1:2 into TLC running buffer (1 mM KH2PO4, 4 mM Na2HPO4 pH 7.4). Half of each dilution was counted by liquid scintillation on an LS6500 to insure equal plate loading. The other half of each dilution was spotted onto a PEI Cellulose TLC sheet (cat# M1055790001, MilliporeSigma, St. Louis, MO) and allowed to dry for one h. As controls for nucleotide phosphate hydrolysis, 35S-GTPγS (absent protein) was incubated +/- 1 mM CuSO4 for one h in assay buffer. These conditions provided minimal and maximal limits for spontaneous (-Cu2+) and maximum catalytic hydrolysis of the phosphate bonds ( + Cu2+)41. Dried plates were then placed in approx. one cm of running buffer and the front was allowed to migrate about 2/3 the distance of the plate. Following each run, the plate was wrapped in plastic wrap, exposed on BAS-MS Phosphor Screen, and frozen overnight in a dark box. The following day, the screen was developed on a GE Typhoon FLA 9500 phosphoimager (Cytiva, Marlborough, MA). Densitometry was measured using Fiji quantification software74.
35S-GTPγS binding and release studies
In general, 35S-GTPγS binding studies were carried out as previously published and as described in the Kinetic Studies previously described herein39. Cells were homogenized in 10 mM Tris HCl pH 7.4, 100 mM NaCl, and 1 mM EDTA. Membranes were pelleted twice by centrifugation (20 K × g, 4 °C, 30 min) and resuspension. After pelleting, membranes were resuspended in assay buffer (50 mM Tris HCl pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA). 35S-GTPγS binding was measured by incubation of membranes (10 µg/well) with 10 µg GDP, 0.1 nM 35S-GTPγS, and drug as indicated. Reactions were performed for one h at 25 °C in 200 µL volumes in 96-well polypropylene plates.
35S-GTPγS release was measured using marginally different conditions. Specifically, cells were homogenized using the same procedure in sodium-free buffer (10 mM Tris HCl pH 7.4, 1 mM EDTA) and resuspended in sodium-free buffer (50 mM Tris HCl pH 7.4, 5 mM MgCl2, 1 mM EDTA). 35S-GTPγS loading was accomplished in a large volume (20 mL per plate) with 1 mg protein, 10 µM GDP and 0.1 nM 35S-GTPγS for one h at 25 °C. Following the sodium-free pulse conditions, preloaded membranes were diluted ten-fold into sodium containing buffer (50 mM Tris HCl pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA). The release reaction included 10 µM GDP, 1 µM unlabeled GTPγS, and drug (as indicated) and was run for one h at 25 °C in a large volume 96-well plate.
Human spinal cord sections were prepared by gross removal of white matter and the dorsal half was homogenized using a tissue tearor (BioSpec Products, Model 985370, Bartlesville, OK, USA). Homogenate was drawn through a glass-on-glass homogenizer several times and pulled through an insulin needle (28-G). All tissue manipulation and homogenization were performed in homogenization buffer (10 mM Tris (pH 7.4), 1 mM EDTA) on ice. Membranes were pelleted by centrifugation at 20,000 × g for 30 min at 4 °C, resuspended and pelleted twice, followed by resuspension in assay buffer (50 mM Tris (pH 7.4), 5 mM MgCl2, 1 mM EDTA). 35S-GTPγS binding was measured by incubation of membranes (10 µg/well) with 10 µg GDP, 0.1 nM 35S-GTPγS, and drug as indicated. Reactions were performed for one h at 25 °C in 200 µL volumes in 96-well polypropylene plates.
35S-GTPγS release was measured in human spinal cord by loading pulse in a 2 mL volume with 100 nM DAMGO, 1 mg protein, 10 µM GDP and 1 nM 35S-GTPγS for one h at 25 °C. Loading controls, without DAMGO, were run for all samples. Following the pulse conditions, preloaded membranes were diluted 100-fold into assay buffer (50 mM Tris HCl pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA). The release reaction included 10 µM GDP, 1 µM unlabeled GTPγS, and drug (as indicated) and was run in a large volume 96-well plate. The release reaction was allowed to proceed for an addition hour at 25 °C before termination.
All reactions were terminated by rapid filtration through GF/B fiberglass filters on a 96-well Brandel MWXRI-96TI cell harvester (Brandel, Inc, Gaithersburg, MA). Filters were soaked in Microscint-20 (Revvity, Waltham, MA) on white OptiPlate-96 plates and counted on either a PerkinElmer TopCount scintillation counter (PerkinElmer, Downers Grove, IL) or a MicroBeta scintillation counter (Revvity, Waltham, MA). All counts were acquired and analyzed in dpm values. Graphical methods for binding and release are presented in the figures and in the companion manuscript.
Alkylation studies
Alkylation studies required special handling of the protein to ensure complete removal of the alkylating agent. For these experiments, pelleted cells CHO-mMOR cells were Teflon-on-glass homogenized in buffer (10 mM Tris HCl pH 7.4, 100 mM NaCl, 1 mM EDTA) followed by rapid centrifugation (20,000 × g) for 30 min at 4 °C. Following centrifugation, harvested membranes were separated into aliquots and incubated in homogenization buffer or buffer including increasing concentrations of the alkylating agent, β-FNA46,47. Incubation, to permit β-FNA alkylation, proceeded for 30 min at 25 °C. After thirty minutes, all aliquots were centrifuged and resuspended (5 min, 20 K × g, 4 °C) two times in homogenization buffer. After the third centrifugation, membranes were resuspended in assay buffer (50 mM Tris HCl pH 7.4, 5 mM MgCl2, 1 mM EDTA). Following final resuspension, membranes were used for 35S-GTPγS binding, 3H-naloxone binding, and western blot protein quantification.
Radioligand binding studies
µ opioid binding was measured as previously reported39. Briefly, 10 µg of membranes were combined with 6 nM 3H-(-)naloxone in Tris pH7.4, NaCl, 1 mM EDTA. Incubations were run at 25 °C for 1 h. Non-specific binding was determined by the addition of 1 µM cold naloxone in DMSO. Final DMSO concentrations were held constant at or below 0.1%. Bound receptor was harvested by rapid filtration through GF/B fiberglass filters on either a 12-well vacuum manifold or 96-well Brandel MWXRI-96TI cell harvester (Brandel, Inc, Gaithersburg, MA). and rinsed twice with ice cold 10 mM Tris HCl pH 7.4. Filters were dried and loaded into scintillation vials with Safety-Solve or OptiPlate-96 plates with Microscint-20 (Revvity, Waltham, MA). Bound radioactivity was measured using a MicroBeta scintillation counter (Revvity, Waltham, MA). All counts were acquired, analyzed, and presented in dpm.
Western blot protein quantification
Receptor levels were determined by western blot using an anti-HA antibody since the mMOR was expressed with an HA-tag on the N-terminus. Aliquots from each alkylation treatment group were dilute into solubilization buffer (20 mM Tris pH 7.4, 150 mM NaCl, 2 mM EDTA, 0.1%SDS). The subsequent soluble protein was quantitated using the DC protein assay and aliquoted for combination with XT buffer (BioRad, Hercules, CA) with 2-mercaptoethanol. The Precision Plus protein standard was run on all gels. Samples were run on 15 well 10% Bis-Tris pre-cast gels (cat# NP0316BOX, ThermoScientific, Waltham, MA) for three h at 100 V. Gels were mounted and transferred onto nitrocellulose membranes for two h at 20 V on ice.
After transfer, nitrocellulose membranes were rinsed in TBST (10 mM Tris pH 8.0, 150 mM NaCl, 0.1% Tween-20) and blocked with 5% non-fat milk in TBST. Primary antibodies, anti-HA (1:1000 rabbit) and anti-β-actin (1:1000 mouse) in 5% non-fat milk in TBST, were added to the blocked membrane and incubated overnight at 4 °C. Membranes were rinsed three times with TBST, blocked with 5% milk, and incubated for one h with secondary antibody, anti-rabbit800 (1:10,000 goat) and anti-mouse680 (1:10,000 goat). Membrane was rinsed three times with TBST and imaged using a Li-cor Odyssey scanner (LI-COR Biotechnology, Lincoln, NE). Densitometry was measured using Fiji quantification software. HA staining was normalized to β-actin as the protein loading control.
Data analysis
Nonlinear regression for stimulatory GTPγS binding, and normalized release data, was performed using the three-parameter equation:
where, Basal is the system baseline, Emax is the maximum response of the system, EC50 is the midpoint of the agonist response curve in molar units, and X is the molar concentration of the agonist. For full agonists, the Emax was considered the same as agonist efficacy. Partial agonist efficacy is represented as the product of intrinsic efficacy (ε) and Emax (Partial agonist efficacy = ε · Emax). This intrinsic efficacy was fit for comparing changes in efficacy between GTPγS binding and release. For partial agonists where efficacy was compared, intrinsic efficacy is referred to as “Stephenson’s efficacy” as it is an observed efficacy that approximates the stimulus function51.
Alternatively, nonlinear regression for the inhibitory GTPγS release was performed using the three-parameter inhibition equation:
where, Basal is the system baseline, Emax is the maximum response of the system, IC50 is the midpoint of the agonist response curve in molar units, and X is the molar concentration of the agonist.
For the linearization analysis used for competitive Schild analysis43, EC50 and IC50 values were used and the standard (Dose Ratio – 1) was calculated as:
where each Dose Ratio is the ratio of the molar EC50 +antagonist or IC50 +antagonist (for the curve of each antagonist concentration) to the EC50 control or IC50 control curves without antagonist. Each Dose Ratio was then converted to the logarithmic value and plotted Log[Antagonist] on the X-axis and Log(Dose Ratio-1) on the Y-axis. The equilibrium dissociation constant (logKi) was estimated from this analysis as the X-intercept.
For the analysis of receptor alkylation, the loss of receptors was calculated as the ratio of 3H-naloxone binding between the control and alkylated membranes. This ratio was then applied as b in the adaptation of the operational model:
with X the molar agonist concentration, K the molar equilibrium affinity constant, n is the transducer slope, and τ the transduction ratio (or simply agonist efficacy). Of note, generally the Hill slope did not largely differ from one and was held constant as a shared value for each dataset fit. As the transduction constant is defined as τ = RT / KE, it is appropriate to modify τ by the alkylation ratio (i.e., q·τ or q·(RT/KE)). Using this modification, the change in system sensitivity due to alkylation, seen as a rightward shift in agonist potency and an absolute loss of maximal response, is incorporated into the equation as a decrease in τ as q is, by definition, less than one. This method permits the estimate of the agonist’s affinity75.
The occupancy-response relationship was initially plotted using the fractional Bmax binding data from the alkylation groups on the X-axis (i.e., Bmax | alkylation / Bmax | control). The fractional Emax from the three-parameter fit of the normalized data for each alkylation group on the Y axis (i.e., Emax | alkylation / Emax | control). In this case, the control X values of one and zero would result when the total receptor population is either 100% available or 100% alkylated for binding. Likewise, the control Y values of zero and one would result from the population being available or alkylated for the measured response. The data from this representation can be fit to the equation:
where X is the size (fraction) of the receptor population available for binding after alkylation, E is the fractional response that remains after alkylation, and r is the EC50/K. In this representation, when response follows a definite occupancy relationship, r reduces to one, and the equation reduces to E = X. Conversely, as EC50 shifts leftward from K, r becomes smaller than one, and the system exhibits a receptor reserve as the difference between EC50 and K50.
This approach was further adapted to demonstrate the effector-occupancy relationship. In this new analysis, X is the product of fractional binding data from the alkylation groups (i.e., Bmax | alkylation / Bmax | control) and the potency ratio from the alkylation groups (i.e., EC50 | control / EC50 | alkylation). These newly defined X representation was plotted against the same Y values described for the occupancy-response relationship. Equation X was used to fit this representation, and linearity was tested using an F-test.
In the comparison of agonist efficacy, the operational model was used to estimate partial agonist efficacy54:
and all parameter definitions are identical between Eqs. 4 and 5. The only difference is that equation X lacks the alkylation parameter (b). This form of the equation is suitable for fitting and comparing partial agonist efficacy between systems but is not suitable for analyzing full agonists due to the lack of information regarding the occupancy-response relationship.
The two-state model of receptor activation was used to estimate partial agonist efficacy (α), or active-state selectivity, as previously described44,52,53. Data was fit, to determine α, with the equation:
with X the molar agonist concentration, Basal the baseline response, Emax the system maximum response, K the molar inactive-state affinity constant, and L the isomerization constant. In all fitting of the two-state model, L was held constant at 0.01. This value was previously used and considered a reasonable assumption. An L value up to 0.1 (effectively a 10% baseline) produced no change in the relative activity of agonists in the two responses. The active-state selectivity, as presented, was greater than one for agonists, as it represented the active-state affinity as a function of the inactive-state affinity. The active-state affinity is the equilibrium affinity constant divided by alpha (K* = K / α). Further, α is the ratio of inactive- to active-state affinity (α = K / K*) or the agonist intrinsic efficacy53. This analysis was only appropriate for partial agonists as both inactive-state affinity and active-state selectivity cannot be estimated in a system that responds differently from occupancy (e.g., a receptor reserve).
For linear regression, data were analyzed for linearity with a slope constrained to unity. The X-intercept for the linearization was compared by an extra sum-of-squares F-test in Prism. For nonlinear regression, all parameters were shared between data sets when they represented system-dependent variables: Three-parameter equation (Basal, Emax), Two-state model (Basal, Emax, L), Operational model (Basal, Emax, n). However, parameters were permitted to float when they represented drug specific properties (e.g., Emax for a partial agonist). Curves were evaluated and best-fit parameter estimates with s.e.m. were compared for significance by an extra sum-of-squares F test in Prism.
All data were plotted and analyzed using the Prism software v.10.5.0 (GraphPad Software Inc, La Jolla, California). All statistical tests are defined in the figure legends and the results of all statical comparisons are presented in Supplementary Table 1. All data presented are the result of multiple experiments (n ≥ 3). The definition of all asterisks is included in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the paper and its Supplementary Information. Source data are provided with this paper.
References
Helmreich, E. J. & Hofmann, K. P. Structure and function of proteins in G-protein-coupled signal transfer. Biochim Biophys. Acta 1286, 285–322 (1996).
Kohl, B. & Hofmann, K. P. Temperature dependence of G-protein activation in photoreceptor membranes. Transient extra metarhodopsin II on bovine disk membranes. Biophys. J. 52, 271–277 (1987).
Heck, M. & Hofmann, K. P. Maximal rate and nucleotide dependence of rhodopsin-catalyzed transducin activation: initial rate analysis based on a double displacement mechanism. J. Biol. Chem. 276, 10000–10009 (2001).
Dror, R. O. et al. SIGNAL TRANSDUCTION. Structural basis for nucleotide exchange in heterotrimeric G proteins. Science 348, 1361–1365 (2015).
Liebman, P. A. & Pugh, E. N. Jr. Gain, speed and sensitivity of GTP binding vs PDE activation in visual excitation. Vis. Res 22, 1475–1480 (1982).
Fung, B. K. & Nash, C. R. Characterization of transducin from bovine retinal rod outer segments. II. Evidence for distinct binding sites and conformational changes revealed by limited proteolysis with trypsin. J. Biol. Chem. 258, 10503–10510 (1983).
Asano, T., Katada, T., Gilman, A. G. & Ross, E. M. Activation of the inhibitory GTP-binding protein of adenylate cyclase, Gi, by beta-adrenergic receptors in reconstituted phospholipid vesicles. J. Biol. Chem. 259, 9351–9354 (1984).
Kwok-Keung Fung, B. & Stryer, L. Photolyzed rhodopsin catalyzes the exchange of GTP for bound GDP in retinal rod outer segments. Proc. Natl. Acad. Sci. USA 77, 2500–2504 (1980).
Bennett, N. & Dupont, Y. The G-protein of retinal rod outer segments (transducin). mechanism of interaction with rhodopsin and nucleotides. J. Biol. Chem. 260, 4156–4168 (1985).
Lee, T. W., Seifert, R., Guan, X. & Kobilka, B. K. Restricting the mobility of Gs alpha: impact on receptor and effector coupling. Biochemistry 38, 13801–13809 (1999).
Yao, X. J. et al. The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc. Natl. Acad. Sci. USA 106, 9501–9506 (2009).
Ross, E. M., Maguire, M. E., Sturgill, T. W., Biltonen, R. L. & Gilman, A. G. Relationship between the beta-adrenergic receptor and adenylate cyclase. J. Biol. Chem. 252, 5761–5775 (1977).
Lad, P. M., Nielsen, T. B., Preston, M. S. & Rodbell, M. The role of the guanine nucleotide exchange reaction in the regulation of the beta-adrenergic receptor and in the actions of catecholamines and cholera toxin on adenylate cyclase in turkey erythrocyte membranes. J. Biol. Chem. 255, 988–995 (1980).
Cassel, D. & Selinger, Z. Catecholamine-induced release of [3H]-Gpp(NH)p from turkey erythrocyte adenylate cyclase. J. Cycl. Nucleotide Res 3, 11–22 (1977).
Citri, Y. & Schramm, M. Probing of the coupling site of the beta-adrenergic receptor. competition between different forms of the guanyl nucleotide binding protein for interaction with the receptor. J. Biol. Chem. 257, 13257–13262 (1982).
Hekman, M. et al. Reconstitution of beta-adrenergic receptor with components of adenylate cyclase. EMBO J. 3, 3339–3345 (1984).
Kupprion, C., Wieland, T. & Jakobs, K. H. Receptor-stimulated dissociation of GTP[S] from Gi-proteins in membranes of HL-60 cells. Cell Signal 5, 425–433 (1993).
Michel, T. & Lefkowitz, R. J. Hormonal inhibition of adenylate cyclase. alpha 2 Adrenergic receptors promote release of [3H]guanylylimidodiphosphate from platelet membranes. J. Biol. Chem. 257, 13557–13563 (1982).
Ellis, J. & Lenox, R. Receptor Coupling to G proteins: Interactions not Affected by Lithium. Lithium 2, 141–147 (1991).
Hilf, G., Kupprion, C., Wieland, T. & Jakobs, K. H. Dissociation of guanosine 5’-[gamma-thio]triphosphate from guanine-nucleotide-binding regulatory proteins in native cardiac membranes. Regulation by nucleotides and muscarinic acetylcholine receptors. Eur. J. Biochem 204, 725–731 (1992).
Breivogel, C. S., Selley, D. E. & Childers, S. R. Cannabinoid receptor agonist efficacy for stimulating [35S]GTPgammaS binding to rat cerebellar membranes correlates with agonist-induced decreases in GDP affinity. J. Biol. Chem. 273, 16865–16873 (1998).
Rasmussen, S. G. et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011).
Furness, S. G. B. et al. Ligand-dependent modulation of G protein conformation alters drug efficacy. Cell 167, 739–749 e711 (2016).
Koehl, A. et al. Structure of the micro-opioid receptor-G(i) protein complex. Nature 558, 547–552 (2018).
Hua, T. et al. Activation and signaling mechanism revealed by cannabinoid receptor-G(i) complex structures. Cell 180, 655–665 e618 (2020).
Zhuang, Y. et al. Molecular recognition of morphine and fentanyl by the human mu-opioid receptor. Cell 185, 4361–4375 e4319 (2022).
Papasergi-Scott, M. M. et al. Time-resolved cryo-EM of G-protein activation by a GPCR. Nature 629, 1182–1191 (2024).
Susac, L., Eddy, M. T., Didenko, T., Stevens, R. C. & Wuthrich, K. A(2A) adenosine receptor functional states characterized by (19)F-NMR. Proc. Natl. Acad. Sci. USA 115, 12733–12738 (2018).
Ye, L. et al. Mechanistic insights into allosteric regulation of the A(2A) adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 9, 1372 (2018).
Huang, S. K. et al. Delineating the conformational landscape of the adenosine A(2A) receptor during G protein coupling. Cell 184, 1884–1894 e1814 (2021).
Wang, X., Neale, C., Kim, S. K., Goddard, W. A. & Ye, L. Intermediate-state-trapped mutants pinpoint G protein-coupled receptor conformational allostery. Nat. Commun. 14, 1325 (2023).
Conflitti, P. et al. Functional dynamics of G protein-coupled receptors reveal new routes for drug discovery. Nat. Rev. Drug Discov. 24, 251–275 (2025).
Stahl, E. L. et al. GTP release-selective agonists prolong opioid analgesic efficacy. Nature https://doi.org/10.1038/s41586-025-09880-5 (2025).
Meselson, M. & Stahl, F. W. The replication of DNA in Escherichia Coli. Proc. Natl. Acad. Sci. USA 44, 671–682 (1958).
Traynor, J. R., Clark, M. J. & Remmers, A. E. Relationship between rate and extent of G protein activation: comparison between full and partial opioid agonists. J. Pharm. Exp. Ther. 300, 157–161 (2002).
Pert, C. B., Pasternak, G. & Snyder, S. H. Opiate agonists and antagonists discriminated by receptor binding in brain. Science 182, 1359–1361 (1973).
Costa, T., Ogino, Y., Munson, P. J., Onaran, H. O. & Rodbard, D. Drug efficacy at guanine nucleotide-binding regulatory protein-linked receptors: thermodynamic interpretation of negative antagonism and of receptor activity in the absence of ligand. Mol. Pharm. 41, 549–560 (1992).
McLoughlin, D. J. & Strange, P. G. Mechanisms of agonism and inverse agonism at serotonin 5-HT1A receptors. J. Neurochem 74, 347–357 (2000).
Stahl, E. L. et al. G protein signaling-biased mu opioid receptor agonists that produce sustained G protein activation are noncompetitive agonists. Proc. Natl. Acad. Sci. USA. 118, e2102178118 (2021).
Karbstein, K., Jonas, S. & Doudna, J. A. An essential GTPase promotes assembly of preribosomal RNA processing complexes. Mol. Cell 20, 633–643 (2005).
Tetas, M. & Lowenstein, J. M. The effect of bivalent metal ions on the hydrolysis of adenosine di- and triphosphate. Biochemistry 2, 350–357 (1963).
Gaddum, J. H. The quantitative effects of antagonistic drugs. J. Physiol. 89, 7P–9P (1937).
Arunlakshana, O. & Schild, H. O. Some quantitative uses of drug antagonists. Br. J. Pharm. Chemother. 14, 48–58 (1959).
Leff, P. The two-state model of receptor activation. Trends Pharm. Sci. 16, 89–97 (1995).
Furchgott, R. F. The pharmacology of vascular smooth muscle. Pharmacol. Rev. 7, 183–265 (1955).
Ward, S. J., Portoghese, P. S. & Takemori, A. E. Pharmacological profiles of beta-funaltrexamine (beta-FNA) and beta-chlornaltrexamine (beta-CNA) on the mouse vas deferens preparation. Eur. J. Pharm. 80, 377–384 (1982).
Rothman, R. B. et al. beta-FNA binds irreversibly to the opiate receptor complex: in vivo and in vitro evidence. J. Pharm. Exp. Ther. 247, 405–416 (1988).
Herepath, M. L. & Broadley, K. J. Affinities of full agonists for cardiac beta-adrenoceptors calculated by use of in vitro desensitization. Naunyn Schmiedebergs Arch. Pharm. 341, 525–533 (1990).
Limbird, L. E. In Cell Surface Receptors: A Short Course on Theory & Methods. Operational Models of Pharmacological Agonism, 14–17 (Springer, 2005).
Stahl, E., Elmslie, G. & Ellis, J. Allosteric modulation of the M(3) muscarinic receptor by amiodarone and N-ethylamiodarone: application of the four-ligand allosteric two-state model. Mol. Pharm. 80, 378–388 (2011).
Stephenson, R. P. A modification of receptor theory. Br. J. Pharm. Chemother. 11, 379–393 (1956).
Del Castillo, J. & Katz, B. Interaction at end-plate receptors between different choline derivatives. Proc. R. Soc. Lond. Ser. B, Biol. Sci. 146, 369–381 (1957).
Stahl, E. L. & Bohn, L. M. Low intrinsic efficacy alone cannot explain the improved side effect profiles of new opioid agonists. Biochemistry 61, 1923–1935 (2022).
Black, J. W. & Leff, P. Operational models of pharmacological agonism. Proc. R. Soc. Lond. B Biol. Sci. 220, 141–162 (1983).
Leff, P., Scaramellini, C., Law, C. & McKechnie, K. A three-state receptor model of agonist action. Trends Pharm. Sci. 18, 355–362 (1997).
Berg, K. A. et al. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol. Pharm. 54, 94–104 (1998).
Faull, R. L. & Villiger, J. W. Opiate receptors in the human spinal cord: a detailed anatomical study comparing the autoradiographic localization of [3H]diprenorphine binding sites with the laminar pattern of substance P, myelin and nissl staining. Neuroscience 20, 395–407 (1987).
Angst, M. S., Ramaswamy, B., Riley, E. T. & Stanski, D. R. Lumbar epidural morphine in humans and supraspinal analgesia to experimental heat pain. Anesthesiology 92, 312–324 (2000).
Sevilla, N., Tolkovsky, A. M. & Levitzki, A. Activation of turkey erythrocyte adenylate cyclase by two receptors: adenosine and catecholamines. FEBS Lett. 81, 339–341 (1977).
Levitzki, A. The mode of coupling of adenylate cyclase to hormone receptors and its modulation by GTP. Biochem Pharm. 27, 2083–2088 (1978).
Nickell, S., Park, P. S., Baumeister, W. & Palczewski, K. Three-dimensional architecture of murine rod outer segments determined by cryoelectron tomography. J. Cell Biol. 177, 917–925 (2007).
Biernbaum, M. S. & Bownds, M. D. Influence of light and calcium on guanosine 5’-triphosphate in isolated frog rod outer segments. J. Gen. Physiol. 74, 649–669 (1979).
Traut, T. W. Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem 140, 1–22 (1994).
Van Eps, N. et al. Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proc. Natl. Acad. Sci. USA 108, 9420–9424 (2011).
Markby, D. W., Onrust, R. & Bourne, H. R. Separate GTP binding and GTPase activating domains of a G alpha subunit. Science 262, 1895–1901 (1993).
Caron, M. G., Limbird, L. E. & Lefkowitz, R. J. Biochemical characterization of the beta-adrenergic receptor of the frog erythrocyte. Mol. Cell Biochem 28, 45–66 (1979).
De Lean, A., Stadel, J. M. & Lefkowitz, R. J. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 255, 7108–7117 (1980).
Samama, P., Cotecchia, S., Costa, T. & Lefkowitz, R. J. A mutation-induced activated state of the beta 2-adrenergic receptor. extending the ternary complex model. J. Biol. Chem. 268, 4625–4636 (1993).
Sunahara, R. K. & Insel, P. A. The molecular pharmacology of G protein signaling then and now: a tribute to Alfred G. Gilman. Mol. Pharm. 89, 585–592 (2016).
Bondar, A. & Lazar, J. Dissociated GalphaGTP and Gbetagamma protein subunits are the major activated form of heterotrimeric Gi/o proteins. J. Biol. Chem. 289, 1271–1281 (2014).
Hommers, L. G., Klenk, C., Dees, C. & Bunemann, M. G proteins in reverse mode: receptor-mediated GTP release inhibits G protein and effector function. J. Biol. Chem. 285, 8227–8233 (2010).
Sevilla, N., Steer, M. L. & Levitzki, A. Synergistic activation of adenylate cyclase by guanylyl imidophosphate and epinephrine. Biochemistry 15, 3493–3499 (1976).
Schmid, C. L. et al. Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 171, 1165–1175 e1113 (2017).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Ehlert, F. J. et al. In Affinity And Efficacy: The Components of Drug-receptor Interactions. Reverse-Engineering and Response-Clamp Analysis, 454–455 (World Scientific, 2015).
Acknowledgements
We acknowledge Professor Klaus-Peter Hofmann for critical and constructive discussion of our preliminary findings. Funding was provided by the National Institute on Drug Abuse/ NIH to LMB (DA038964) and (DA033073).
Author information
Authors and Affiliations
Contributions
L.M.B. and E.L.S. conceived the studies. L.M.B. supervised the experiments, analyzed data, and cowrote the manuscript. E.L.S. developed the experimental design for the studies and performed all experiments, analyzed data, and co-wrote the manuscript. E.L.S. defined the models.
Corresponding authors
Ethics declarations
Competing interests
The authors have no competing interests to declare.
Peer review
Peer review information
Nature Communications thanks Martin Lohse and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bohn, L.M., Stahl, E.L. Characterization of the GTPγS release function of a G protein-coupled receptor. Nat Commun 16, 11193 (2025). https://doi.org/10.1038/s41467-025-66516-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66516-y
