
Abstract
Rock-salt-structured compounds like lead chalcogenides are promising thermoelectrics, as their high symmetry, strong anharmonicity, and favorable phase behavior collectively lead to high performance by enabling large power factors and ultralow thermal conductivity. Here, we report LiMnSbTe3, a new rock-salt semiconductor stabilized through targeted chemical design by combining hexagonal MnTe with cubic LiSbTe2. Embedded in the high-symmetry matrix, van der Waals-like gaps form due to Sb2Te3 nanoscale segregation, which acts as effective phonon-scattering centers, leading to a low lattice thermal conductivity of 0.37 Wm-1K-1 at 873 K with alloy scattering from disordered cations. The ordered local structure of Sb2Te3-type vdW-like gaps and the cross-gap interaction facilitate the carrier transport. Aided by energy-converged valence bands and a paramagnon drag effect, high Seebeck coefficients and enhanced power factor can be achieved, leading to a high ZT of 1.2 at 873 K. Furthermore, introducing Mn deficiency increases ZT to 1.5, highlighting the potential for higher performance through optimized doping or alloying. A segmented single-leg thermoelectric module achieves an output power density of 0.52 Wcm-2 and an efficiency of 8.7% under ∆T of 478 K, further demonstrating its promising thermoelectric applications.
Similar content being viewed by others

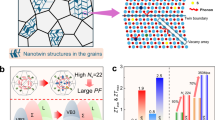
Introduction
Thermoelectric technology enables the conversion of heat energy into electricity via the Seebeck effect and facilitates active cooling through the Peltier effect1,2,3. These dual functionalities have inspired extensive research to develop advanced thermoelectric systems for various applications, including the self-powered sensor systems4. The performance of thermoelectric materials is quantified by the dimensionless figure of merit, ZT, which is related to the electrical resistivity (ρ), Seebeck coefficient (S), thermal conductivity (κ), and absolute temperature (T) via the equation, ZT = (S2/ρ)T/κ5. The S2/ρ is usually referred to as the power factor (PF), which characterizes the electrical transport properties of a material.
With advancements in condensed matter physics, it has become clear that the electrical and thermal transport properties of inorganic compounds are closely linked to the intricate details of their crystal structures6,7. Rock-salt compounds (Fm-3m) have played an important role in thermoelectricity due to their highest-symmetrical structural features. The high symmetry and special bonding character in these structures often yield multiple equivalent pockets at the Fermi surface, enhancing the Seebeck coefficient without significantly compromising electrical conductivity8,9,10. These compounds possess relatively strong anharmonicity, leading to an inherently low thermal conductivity11,12. Consequently, remarkable thermoelectric performance has been reported in rock-salt (distorted rock-salt) compounds, including lead chalcogenide13,14,15, germanium chalcogenides16,17,18, and I-V-VI2 semiconductors19,20. While for non-cubic structures such as SnSe, introducing elements that prefer octahedral coordination or applying high-entropy design can stabilize the cubic phase21,22,23,24,25,26,27. This approach may allow the material to benefit from the advantages of rock-salt compounds and thereby achieve promising thermoelectric performance. However, the strong alloy scattering of carriers caused by the random occupation of crystallographic sites in such compounds needs to be balanced28.
Substructure modifications at the atomic or nanoscale level are effective for enhancing the phonon scatterings, and are particularly valuable in materials with excellent electrical transport properties, such as those with high-symmetry crystal structures. When these substructures are designed to minimally perturb carrier transport, electrical and thermal transport properties can be decoupled, leading to improved thermoelectric performance. The most successful case is to nucleate the nanostructural phase into the matrix, whose valence or conduction bands are energy-aligned to facilitate the transport of charged carriers29,30,31. Therefore, the interface between the nanostructure and matrix can selectively scatter phonons. Another example is the locally discordant atoms, at the atomic level, have been shown to induce low-lying optical phonon modes, which significantly suppress thermal conductivity32,33,34,35,36,37. At the same time, the bonding between the adjacent atoms can maintain the transport of carriers, leading to the selective scattering of phonons38. Moreover, dislocations can scatter medium-frequency phonons through the strain field introduced to the matrix while largely preserving the carrier transport39,40,41,42,43. The rock-salt structure, though simple, exhibits a high tolerance to these substructures, providing substantial opportunities for achieving exceptional thermoelectric performance. Therefore, a promising strategy emerges to develop advanced thermoelectric materials: converting the structure into a high-symmetry form while retaining substructures capable of selectively scattering phonons.
In this study, we combined MnTe, a hexagonal (P63/mmc) compound, with cubic LiSbTe2 to produce a new rock-salt semiconductor, LiMnSbTe3. The high-symmetry structure results in a band structure with multiple energy-aligned valence peaks. Together with the potential paramagnon drag effects, similar to those in MnTe44, high Seebeck coefficients (250 to 300 μV/K) are achieved throughout the measured temperature range. Additionally, spontaneously formed Sb2Te3-type van der Waals (vdW)-like gaps were observed within the high-symmetry matrix. The ordered local structure, in contrast to the disordered cation occupation in cubic matrix, and the strong cross-gap interaction across the vdW-like gaps would facilitate the transport of carriers, realizing the selective scattering of phonons, as illustrated in Fig. 1a. As a result, a low lattice thermal conductivity of 0.37 Wm−1K−1 can be achieved at 873 K, which is lower than most of the typical rock-salt thermoelectric materials, as shown in Fig. 1b. LiMnSbTe3 also exhibits an enhanced weighted mobility, as presented in Fig. 1c, leading to a PF of 7.2 μWcm−1K−2 at 873 K. Therefore, the unqite structural characteristics, vdW-like gaps embedded in the high-symmetry structure, contribute to a high ZT of 1.2 at 873 K, outperforming both MnTe and LiSbTe2, see Fig. 1d. Moreover, Mn deficiency can further enhance the electrical conductivity by elevating carrier concentrations and decrease the lattice thermal conductivity, raising the ZT to 1.5 at 873 K. Based on the developed new thermoelectric materials, we fabricated a segmented single-leg thermoelectric device, which achieves a high output power density of 0.52 Wcm−2 and an efficiency of 8.7% under ∆T of 478 K.

a Schematic of selective scattering of phonons by vdW-like gaps mimicking the structure of Sb2Te3, created partly by VESTA88. b Low lattice thermal conductivity of LiMnSbTe3 and comparison with other typical thermoelectric materials22,24,25,26,27,89,90,91,92. c Enhanced weighted mobility of LiMnSbTe3. d Optimized ZT values of LiMnSbTe3 and the energy conversion efficiency (η) of segmented single-leg under different temperature differences (∆T).
Results
Structure and TE performance of LiMnSbTe3
A series of LiMnmSbTem+2 (m = 1, 2, 3, 5, 10) samples were synthesized via a solid-state reaction using Li pellets, Mn pellets, Sb pieces, and Te chunks as starting materials (details are available in the Method). As shown in Fig. 2a, X-ray diffraction (XRD) patterns indicate that the m = 10 sample exhibits mixed phases of P63/mmc (hexagonal MnTe) and Fm-3m. As m decreases (i.e., the LiSbTe2 content increases), the intensity of the Bragg peaks corresponding to the P63/mmc phase gradually decreases, ultimately resulting in a single Fm-3m phase in the m = 1 sample, LiMnSbTe3. We performed a Rietveld refinement with Fullprof45 to corroborate the structures of LiMnSbTe3 as displayed in Fig. 2b. The fitting parameters, including the lattice parameter and the isotropic displacement parameters, are shown in Table S2. A fine fitting can be achieved with the Fm-3m structure, where the cation (anion) sites are equally occupied by Li, Mn, and Sb (Te). Although the lab-scale X-ray diffraction facilities used in this work are not sufficient for the quantitative determination of Li, the crystal structure of LiMnSbTe3 can be identified as cubic. A relatively large fitting residual at 2θ~70o is also observed, which may arise from local short-range ordering. Such local structures can exist in compounds where multiple elements occupy the same crystallographic site46, as is the case for LiMnSbTe3. The structure evolution of LiMnSbTe3 from its parental binary compounds can be summarized in Fig. 2c. In hexagonal MnTe, the cation, Mn, adopts an octahedral coordination, which is also held by the Mn in the rock-salt LiSbMnTe3 structure. Although Li is a small atom and typically does not adopt octahedral coordination, the strong preference of Sb and Mn for octahedral coordination likely contributes to the cubic structure of LiSbTe2 and LiMnSbTe3. To further verify the stability of LiMnSbTe3, we calculated formation enthalpies of hexagonal MnTe, cubic LiSbTe2, and cubic LiMnSbTe3. As shown in Tables S3 and S4, both LiSbTe2 and LiMnSbTe3 are thermodynamically stable, with LiMnSbTe3 exhibiting a more negative enthalpy. Moreover, the combination of hexagonal MnTe and cubic LiSbTe2 to form LiMnSbTe3 releases an additional 0.325 eV per formula, further supporting the stabilization of cubic LiMnSbTe3.

a X-ray diffraction patterns of LiMnmSbTem+2 (m = 1, 2, 3, 5, 10) after solid state reaction. b Rietveld refinement of LiMnSbTe3 using the Fm-3m structure with the pattern collected from the sintered sample. c The structure evolution of LiMnSbTe3 from its parental materials, MnTe and LiSbTe2, created partly by VESTA88.
The room-temperature electrical resistivities (ρ) of LiMnmSbTem+2 samples are generally lower than those of their parental compounds, MnTe and LiSbTe2, as shown in Fig. 3a. For LiMnSbTe₃, the ρ at 300 K is ~22 mΩ·cm, showing a slight increase with temperature and reaching a peak value of 30 mΩ·cm at 573 K before decreasing at higher temperatures. The initial increase in electrical resistivity with increasing temperature indicates the degenerate semiconducting nature of the material, which corresponds with its high carrier concentration of 2.20 × 1020 cm−3. The m = 10 sample exhibits a substantial amount of MnTe phase. MnTe can be doped with Li to show low ρ of 2 mΩ cm at 300 K44,47, which is the primary reason for the lower ρ of the m = 10 sample compared to LiMnSbTe3. Therefore, the m = 10 sample has a room-temperature carrier concentration of 3.17 × 1020 cm−3, which decreases to 2.20 × 1020 cm−3 of the m = 1 sample, resulting in an increase in electrical resistivity. Consistent with the very high carrier concentration of these mixed-phase samples, the Seebeck coefficients (S) of LiMnmSbTem+2 (m ranges from 1 to 10) samples are lower than those of MnTe and LiSbTe2, as displayed in Fig. 3b. The room-temperature S also increases as m decreases, owing to the decrease in carrier concentrations. In the case of LiMnSbTe3, as the temperature increases, the S, which is 280 μV/K at room temperature, reaches a peak value of approximately 340 μV/K at 573 K before gradually decreasing to 260 μV/K at 873 K. The presence of a maximum in the thermopower can be used to estimate the band gap of LiMnSbTe3 through the Goldsmid-Sharp formula48, which yields a value of 0.39 eV. We also applied the infrared diffuse reflectance spectra to investigate the band gap of LiMnSbTe3, and the results are displayed in Fig. S1. Intense absorption was observed for low-energy photons, which has also been reported in systems with high carrier concentrations, such as GeTe and rhombohedral GeSe17,23. It is likely that the high carrier concentration will impose a certain influence on the infrared diffuse spectroscopy measurement. Nevertheless, it confirms that LiMnSbTe3 is a semiconductor with a relatively narrow band gap. Shown in Fig. 3c is the temperature-dependent power factor (PF). Compared with its parental binary compounds, MnTe and LiSbTe2, LiMnSbTe3 has an enhanced PF of 3.4 μWcm−1K−2 at 300 K and 7.2 μWcm−1K−2 at 873 K due to its high carrier concentration induced lower electrical resistivity.

Temperature-dependence of a electrical resistivity, b Seebeck coefficient, c power factor, d total thermal conductivity, e lattice thermal conductivity, and f ZT.
Figure 3d shows the total thermal conductivities (κ) of LiMnmSbTem+2, which range between those of MnTe and LiSbTe2 across the measured temperature range. As m decreases from 10 to 1, room-temperature κ decreases, reaching a low value of 0.58 Wm−1K−1 for LiMnSbTe3. From 300 K to 873 K, the κ of LiMnSbTe3 remains low, ranging from 0.4 to 0.6 Wm−1K−1. The electronic thermal conductivity (κe) is estimated using the Wiedemann–Franz law, κe = LσT, where L is the Lorenz number determined from the Seebeck coefficients49. Then the lattice thermal conductivities (κL) is calculated by subtracting κe from κ. As shown in Fig. 3e, κL of the LiMnmSbTem+2 series is between those of MnTe and LiSbTe2. With decreasing m, corresponding to reduced MnTe content, κL gradually decreases. This trend for κL with respect to m value is consistent with the fact that MnTe has a higher κL than LiSbTe2. Notably, LiMnSbTe3 exhibits a low κL of 0.56 Wm−1K−1 at 300 K and 0.37 Wm−1K−1 at 873 K, which is even comparable to that of LiSbTe2. While the S, ρ, and κ show the bipolar features at high temperatures, such behavior is absent in κL. This discrepancy may stem from the less accurate Lorenz number used to estimate the κe at high temperatures, as it was determined through a simplified model49. As LiMnSbTe3 exhibits an improved PF over MnTe and LiSbTe2, a superior ZT, shown in Fig. 3f, of 1.2 at 873 K is achieved, higher than either of the two parent compounds.
The thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements have been conducted to gain more insight into the thermodynamic properties of LiMnSbTe3. The TGA signal, displayed in Fig. S2a, shows that the mass ratio is maintained at 100% from room temperature to 873 K, indicating there is no significant evaporation during this temperature range. Meanwhile, the DSC signal in Fig. S2b exhibits no endothermic or exothermic peak, demonstrating that there is no phase transformation or decomposition from room temperature to 873 K. As shown in Fig. S3, the repeat measurement of the same sample also yields similar results to the initial measurement, meaning that there is no significant change in the material’s composition and structure during the first test.
Microstructure of LiMnSbTe3
Because of this higher performance, we further investigated the microstructural characteristics of LiMnSbTe3 to understand the atomic-level and nanoscale structures, as well as the origins of its low lattice thermal conductivity. Its chemical homogeneity was examined at a microscale using scanning electron microscopy (SEM). Figure S4a shows that a uniform contrast was observed on the polished surface of LiMnSbTe3. The elemental mapping, shown in Fig. S4b–S4d, indicated a homogenous distribution of elements that can be reliably detected by EDS (Mn, Sb, and Te) at the scale of tens of micrometers. The measured compositions also match the nominal values as displayed in Fig. S4e, confirming the successful formation of a homogeneous new phase, LiMnSbTe3. A homogeneity analysis along the pressing direction was also performed, considering the concern that alkali ions may become mobile under high electrical current during spark plasma sintering50. As shown in Fig. S5, the morphology across different regions of the sample is generally uniform, and the elemental concentrations are close to the nominal values, confirming that the sample is homogeneous along the pressing direction. We attribute this homogeneity to the relatively low mobility of Li ions in face-centered cubic structures. In such closely packed structures, Li-ion migration paths must pass through both tetrahedral and octahedral sites with significantly different site energies, resulting in a high activation energy for Li-ion motion51.
Then, its nanoscale structural features were examined using scanning/transmission electron microscopy (S/TEM). Figure 4a displays the high-angle annular dark field (HAADF) image over a large spatial area of LiMnSbTe3 along the [110] zone axis, where densely distributed planar defects with bright contrast are present. Additionally, the orientations of these planar defects follow specific crystallographic directions, with the angle between two neighboring planar defects consistently measuring 70.5° or 109.5°. The orientation preference of these defects was further evaluated through electron diffraction. The selected area electron diffraction (SAED) pattern was acquired at the planar defect highlighted in Fig. 4a. As demonstrated in Fig. 4b, diffuse streaks were observed along the \((1\bar{1}1)\) planes, indicating that these bright line features are {111} planar defects. Moreover, the interplanar angles between {111} planes match the measured values of 70.5° and 109.5°, which further corroborates this observation. The contrast difference exhibited by these {111} planar defects suggests the local chemical segregation. Therefore, energy dispersive spectroscopy (EDS) analysis was performed near the planar defects. The EDS maps in Fig. 4c indicate that these planar defects are Sb-rich and Mn-deficient, further suggesting that these defects may form due to Sb2Te3 segregation.

a Low-magnification high-angle annular dark field (HADDF)-STEM image. b Selected area electron diffraction (SAED) pattern on the highlighted area in a. c Elemental mapping of Sb, Mn, and Te in the area highlighted with red rectangular. d High-magnification HADDF-STEM image. e and f Atomic configuration of pseudo vdW-like gaps in different orientations. g Virtual bright-field (BF) image, h differential phase-contrast (DPC) image, and i Electric-field image of areas shown in e. The inset shows the color wheel where the color indicates the direction of the electric field, and the shade corresponds to the intensity.
The atomic arrangement of these {111} planar defects was further investigated using aberration-corrected STEM, and the results are shown in Figs. 4d–4f, which revealed representational planar defects with different configurations. It can be seen that the van der Waals (vdW)-like gaps form in the middle of the planar defects. It is worth noting that light elements, such as Li, produce little HAADF signal, leaving the possibility that Li atoms may be positioned at the ‘gaps’ but cannot be imaged with the HAADF technique. To confirm that the planar defects are indeed vdW-like gaps, we implemented four-dimensional STEM (4D-STEM) to perform differential phase contrast (DPC) phase reconstruction. When the electron probe interacts with the electrostatic potential from the sample, the center of mass (CoM) of the momentum distribution of the beam intensity shifts. DPC uses this momentum change to reconstruct the sample’s electrostatic potential. As the interaction with the nuclei dominates the beam shifts, the DPC images produce contrast roughly linear to atomic number, therefore enabling the visualization of both light and heavy elements52. Figure 4g shows the virtual bright field (BF) image over a planar defect generated from the collected 4D-STEM dataset, where an array of vacancies was observed in the middle. The DPC phase reconstruction performed over the sample region is shown in Fig. 4h. Given the sensitivity of the DPC image to light elements, the absence of atomic columns in the vdW-like gaps confirms that Li is not present in the gap53. Moreover, the local electric field is further calculated from DPC and shown in Fig. 4i, which also reveals a disruption near the vdW-like gaps, further corroborating that the lattice is not continuous.
We further employed atom probe tomography (APT) to investigate the distribution of constituent elements and the vdW-like gaps. As shown in Fig. 5a, the elements Li, Mn, Sb, and Te, exhibit a generally homogenous distribution over tens of nanometers. Here, Li concentrations were excluded from the following quantitative analysis due to potential migration under the high voltage applied during the APT measurement. Despite the generally homogenous distribution, compositional fluctuations are evident in localized regions. As displayed in Fig. 5b, the measured Mn concentration distribution is broader than the theoretical binomial distribution, suggesting compositional inhomogeneity. This is more clearly visualized in the sectional view of Mn distribution in Fig. 5c, where the red dashed line highlights the Mn-poor regions. In Fig. 5d, Mn deficiency is accompanied by Sb and Te enrichment, suggesting the presence of Sb2Te3-rich areas. Due to the resolution limitations and data reconstruction algorithms of APT, the spatial definition of these fluctuations appears less distinct than that observed by STEM. Nevertheless, both STEM and APT consistently reveal the unique vdW-like gaps formed by the phase separation of two-quintuple Sb2Te3.

a Element distributions of the constituent elements. b Comparison between the binomial distribution and the experimentally observed Mn distribution, indicating the inhomogeneity of Mn at the nanoscale c Sectional view of the Mn distribution, where colors from purple to red indicate increasing Mn concentrations, revealing its inhomogenous nature. d Composition profile illustrating fluctuations in Mn, Sb, and Te concentrations. The arrows highlighted Mn-deficiency regions accompanied by enrichment of Sb and Te.
The Sb2Te3-type vdW-like gaps disrupt the periodic atomic arrangement of the rock-salt matrix. Therefore, in the area around the vdW-like gaps, the composition of the system can be represented as LiMnSbTe3-Sb2Te3. However, as Mn can also exhibit different valences like 2+ or 3 + , there is a possibility that the areas near the vdW-like gaps can have a certain amount of Mn, which can replace Sb and also maintain the valence balance. To further check the composition of the areas around the vdW-like gaps, the magnetic response (χ) was measured in an attempt to gauge the valence of Mn. From the magnetic-field-dependence of magnetization (M-H) at 100 K and 300 K, shown in Fig. S6, LiMnSbTe3 exhibits a paramagnetic behavior at both temperatures. Then we measured the zero-field cool (ZFC) magnetic susceptibility (χ) from 10 K to 300 K at 1000 Oe, and the result is displayed in Fig. S7a. The temperature-dependent χ indicates that LiMnSbTe3 exhibits antiferromagnetic behavior with a Néel temperature of 13 K. The effective magnetic moment (μeff) can be determined by the Curie–Weiss law:
where C is the Curie constant and θCW is referred to as the Curie-Weiss temperature54. The Curie constant C can be applied to calculate the μeff by the equation,
where μB is the Bohr magnetons. The μeff of LiMnSbTe3 is determined to be 5.8 μB (Fig. S6b), which is very close to the calculated 5.92 μB of Mn2+ and higher than the 4.9 μB of Mn3+ ion54,55. The X-ray photoemission spectrum (XPS) of the Mn 2p (Mn 2p1/2 and Mn 2p3/2) and 3 s states, shown in Fig. S8, is further measured to corroborate the valence state of Mn. Even with prolonged acquisition time, the signal-to-noise ratio is not satisfactory. As the EDS result, shown in Fig. S4e, indicates that the Mn contents are similar to the nominal one, the reason for the poor XPS signal needs further investigation. Nevertheless, the binding energies for Mn 2p1/2, Mn 2p3/2, and Mn 3 s are closer to that for Mn2+56.
Van der Waals-like gaps are common in GeTe/Se-based materials and GeSbTe-based phase change materials, where ordered vacancies form clusters and evolve into planar vdW-like gaps after heat treatment23,57,58,59,60. In LiMnSbTe3, the formation of Sb2Te3-type vdW-like gaps also indicates a cation deficiency. According to the EDS analysis, shown in Fig. S4, the concentrations of Mn, Sb, and Te are close to the nominal values, which hints that the cation deficiency is most likely due to the loss of Li. Given the high reactivity of Li with the container walls and with the carbon coating, the deficiency of Li is reasonable. For the phase-change material GeSb2Te4, the formation of ordered cation vacancies and vdW-like gaps is calculated to lower the energy of the system59. Therefore, the same mechanism can be displayed in LiMnSbTe3. In principle, the Li deficiency would generate Li vacancies in the rock salt-type lattice. But instead of remaining randomly distributed, these vacancies tend to order and give rise to the thermodynamically-favorable layered vdW-like gaps observed experimentally. Taken together, the lithium deficiency and energetic stabilization via vacancy ordering play central roles in driving the formation of vdW-like gaps in LiMnSbTe3, which accompanies the Sb2Te3 nano-segeration.
These vdW-like gaps form along the {111} crystallographic planes and could act as periodic scattering sources to phonon propagations. The intersection of two {111} planar defects can further induce lattice distortion to accommodate the arrangement of vacancies, as revealed in Fig. 4d. These vdW-like gaps, like vacancy planar defects, disrupt the periodic arrangement of atoms, leading to the scattering of phonons with a broad frequency23,57,58,61. Moreover, Mn, Sb, and Li occupy the cation sites in a disordered fashion, which can lead to alloy scattering of high-frequency phonons62,63,64. To quantitatively estimate the influence of these structural characteristics on phonon transport, we applied the Debye-Callaway model to simulate the lattice thermal conductivity of LiMnSbTe3. The simulation details and parameters can be found in the Supporting Information. From the results shown in Fig. S9a, the inclusion of point defect scattering and vdW-like gap scattering leads to a fine simulation of the lattice thermal conductivity, which indicates that these structural characteristics are important contributors to the low lattice thermal conductivity. A comparison between LiMnSbTe3 and LiSbTe2 can further highlight the role of these structural characteristics. Based on the elastic parameters65, the longitudinal and transverse sound velocities of LiSbTe2 are 3205 m/s and 1682 m/s, corresponding to an average sound velocity of 1881 m/s, which is lower than that of LiMnSbTe3 (2075 m/s). The Grüneisen parameter of LiSbTe2 is 1.84, larger than that of LiMnSbTe3 (1.71). LiSbTe2 shows the strong anharmonicity because both Li and Sb are “rattler-like” or prone to soft bonding environments, a feature that parallels what has been seen in other I–V–VI2 compounds such as AgSbTe2 and AgBiSe266. In LiMnSbTe3, the same types of atoms (Li, Sb) are present, so one would expect some anharmonicity to carry over. However, by introducing Mn into the lattice, the effective concentration of these strongly anharmonic atoms is diluted because Mn–Te bonds are stiffer. Thus, while LiMnSbTe3 still benefits from anharmonic contributions, they are weaker than those in LiSbTe2. This provides a rationale for why LiMnSbTe3 alone could not reach the low thermal conductivity of LiSbTe2 unless additional scattering channels (point-defect disorder, vdW-gap scattering) are invoked.
Furthermore, the cross-gap interaction, induced by multi-center bonding, across the vdW-like gaps in Sb2Te3 has been revealed recently67,68,69,70. The concept of multi-center bonding describes bonding electrons that are not confined between two atoms but instead shared across three or more atomic centers, which was applied to explain the bonding in B2H671,72. It means that the bonding between the Te-Te across the vdW-like gaps is not purely van der Waals forces, but it also contains a substantial electron delocalization, which leads to less disturbance of carrier transport across the vdW-like gap. This effect can be visualized through the anisotropy ratio of electrical conductivity (σin-plane/σout-of-plane) for single-crystal Sb2Te3 (vdW-like gap with substantial cross-gap interaction) and MoS2 (pure vdW gap that can be easily exfoliated by tape). As seen in Fig. S10, the anisotropy ratio for single-crystal Sb2Te3 is 2 to 373, whereas it is 20-30 for single-crystal MoS274. The lower out-of-plane electrical conductivity indicates that the cross-gap interaction in Sb2Te3 is still weaker than the in-plane atomic interaction, which is also reflected by the discontinuous local electrical field across the gap in Fig.4i. However, the comparison between Sb2Te3 and MoS2 demonstrates that the impact of the gap on carrier transport in Sb2Te3 is considerably weakened compared to a pure vdW gap, manifested as multicenter bonding across the vdW-like gap in Sb2Te368. In fact, in layered chalcogenide materials, the degree of two-dimensionality is not fixed but evolves systematically with the chalcogen. Sulfur 3p orbitals are compact and less polarizable, leading to minimal overlap across vdW gaps and strongly two-dimensional electronic behavior. Moving to selenium and tellurium, the 4p and especially 5p orbitals become increasingly diffuse and polarizable, which enhances interlayer orbital overlap. In tellurides, relativistic effects further destabilize and expand the 5p orbitals, allowing for genuine bonding interactions across the layers beyond simple dispersive forces. The combined influence of orbital size, polarizability, and relativistic contributions reduces the strictly two-dimensional character of these systems, and as a result, carrier transport perpendicular to the layers is facilitated more strongly in tellurides than in sulfides or selenides. These interlayer interactions are strong enough that some telluride systems, such as Sb2Te3, hover between a purely 2D vdW material and a fully 3D bonded solid.
Based on our STEM observation, the Te-Te distance across the gap is about 0.37 nm, shown in Fig. S11, which is nearly identical to the value of 0.371 nm in pristine Sb2Te375 and smaller than twice the ionic radius of Te2- (0.442 nm)76. The structural similarity and nearly identical gap distance between LiMnSbTe3 and Sb2Te3 suggest that similar physical transport properties should also be present in the vdW-like gaps of LiMnSbTe3. Moreover, the Sb2Te3-type vdW-like gap represents a locally ordered structure in contrast to the disordered rock-salt matrix, where the cation sites are randomly occupied by Li, Mn, and Sb. Therefore, although the rock-salt matrix in Fig. 4i exhibits a continuous electric field, carrier transport is strongly affected by alloy scattering, in contrast to the suppressed alloy scattering in the ordered vdW-like gaps. Taking into account both the locally ordered structure and cross-gap interaction, the vdW-like gaps in LiMnSbTe3 can facilitate carrier transport while selectively scattering phonons.
Electronic band structure of LiMnSbTe3
We calculated the electronic band structure of LiMnSbTe3 using the density functional theory (DFT). The atomic structure, which best captures the disordered state, was generated using the special quasirandom structure (SQS) method and shown in Fig. S1277. Subsequently, several initial magnetic configurations were imposed on the Mn atoms, including four antiferromagnetic arrangements and one ferromagnetic arrangement, and their relative stabilities were evaluated through total energy calculations. The results, shown in Table S5, indicate that the energy differences among these configurations are rather small (<0.04 eV/formula). As the above magnetic properties measurement indicates an antiferromagnetic nature, the antiferromagnetic configuration with the lowest energy was ultimately adopted as the ground-state structure for subsequent electronic structure calculations. Due to the d5 electronic configuration of Mn2+, the spin-up and spin-down density of states (DOS) were calculated and shown in Fig. 6a. The results show no significant difference between the spin-up and spin-down DOS, despite a slight imbalance at the Fermi level. This slight indifference comes from the relaxation of the magnetic moment of Mn during DFT calculation, indicating a minor degree of ferrimagnetic in the relaxed supercell. The Fermi level lies within the gap between the valence and conduction band DOS, confirming its semiconducting behavior. The partial DOS analysis, displayed in Figs. 6b and S13 shows that the valence band states from the Fermi level to ~2 eV below it mainly arise from the hybridization of Mn 3d, Sb 5s, Sb 5p, Li 2s, and Te 5p orbitals. Among these, the Te 5p orbitals dominate in both spin-up and spin-down channels, which is expected since valence bands in such compounds are typically anion-derived78. Notably, Mn 3 d orbitals also contribute significantly. Because d orbitals are generally less dispersive than s or p orbitals, their contribution tends to flatten the bands. Sb contributes through both 5s and 5p orbitals, though to a lesser extent than Mn. The participation of Sb 5s orbitals is reminiscent of rock-salt PbTe, where Pb 5s states are involved in valence band formation and help reduce the band gap12. By contrast, Li 2s orbitals make only a minor contribution.

a Total density of state and b orbital projected density of state for LiMnSbTe3 in spin up and spin down states. Calculated electronic band structure of LiMnSbTe3 in c spin up and d spin down. The fractional coordinates of high-symmetry k-points are shown in Table S6.
The electronic band structures in momentum space are shown in Fig. 6c for spin up and Fig. 6d for spin down, demonstrating an indirect band gap of 0.21 eV for the spin-up band and 0.28 eV for the spin-down band. These DFT-calculated band gaps are slightly lower than the value estimated via the Goldsmid-Sharp formula (0.39 eV). Both the spin-up and spin-down electronic band structures show multiple-peak valence bands with energy convergence. For the spin-up band structure, the valence band maximum (VBM) is located along the Г-X direction. Notably, there are multiple bands along X-W and W-L directions with energy differences of <0.05 eV compared to the VBM, indicating these bands are energy-aligned and responsible for the transport of carriers. For spin-down band structure, the VBM resides along the W-L direction. Like the spin-up channels, there are second, third, and fourth VBMs along the X-W and W-L directions with an energy difference of <0.04 eV relative to the VBM. These energy-aligned multiple valence bands can be effectively converged to produce a high density-of-state effective mass (m*), which eventually leads to the high Seebeck coefficient in LiMnSbTe3.
The theoretical m* can also be calculated with the obtained band structure. Due to the complex Fermi surface in LiMnSbTe3, it would be tedious to calculate every single valley’s effective mass and multiply it by the valley degeneracy. Instead, the m* can also be calculated with the DOS via the equation79,80,81:
where D(E) is the density of states, D’(E) is the first derivative of DOS with respect to energy, V is the cell volume, and me is the free electron mass. The result is displayed in Fig. S14, which indicates a value of around 3 me at an energy 0.1 eV below the Fermi level. This value is lower than that estimated via the Pisarenko plot in Fig. S15, which is approximately 9 me. Therefore, in addition to the converged energy-aligning multiple valence valleys, there should be other effects functioning. In fact, the m* for MnTe has been estimated to be ~7 me27,82. Some of the MnTe-related compounds also exhibit similarly high m* values, such as approximately 8 me for GeMnTe2 and around 6 me for Ge0.85Mn0.15Te27,36,83,84,85. The inelastic neutron scattering studies on MnTe have demonstrated the presence of paramagnons in the paramagnetic state, which contribute to a high Seebeck coefficient44. Similarly, the paramagnon drag may also contribute to the enhanced m* and Seebeck coefficient in LiMnSbTe3. To further examine the possible presence of paramagnon drag, we measured the low-temperature Seebeck coefficient, as shown in Fig. S16. Near the Néel temperature, the Seebeck coefficient increases sharply, followed by a more gradual rise with increasing temperature. This behavior is reminiscent of that discovered in MnTe44, which suggests that the paramagnon drag also exists in the LiMnSbTe3 system, contributing to the high Seebeck coefficient in the paramagnetic state.
The high m* further results in a high weighted mobility (μW, μW = (m*/me)3/2μ0, where μ0 is nondegenerate mobility)86 of 32 cm2V−1s−1 at room temperature for LiMnSbTe3, as shown in Fig. 1c. Compared to MnTe and LiSbTe₂, LiMnSbTe₃ exhibits consistently higher μW across the measured temperature range, which leads to its superior PF. Therefore, the higher weighted mobility and the unique microstructure-induced low lattice thermal conductivity contribute together to the high ZT in LiMnSbTe3, underscoring its potential as a promising thermoelectric material.
TE performance of LiMn1-xSbTe3
The effects of doping, specifically manganese deficiency, on the thermoelectric performance of LiMnSbTe3 were further investigated. The synthesis and consolidation of LiMn1-xSbTe3 (where x ranges from 0.00 to 0.10) pellets were identical to those used for pristine LiMnSbTe3. As shown in Fig. S17, the XRD patterns reveal that all Mn-deficient samples retain the rock-salt phase structure. However, as x increases, additional peaks corresponding to MnTe2 gradually emerge, indicating the phase’s sensitivity to compositions. Similarly, other compounds such as AgSbTe2 or AgMnSbTe3, where cations exhibit disordered arrangement, also demonstrate composition- and synthesis-dependent phase sensitivity20,24.
As shown in Fig. 7a, with increasing x, the room-temperature electrical resistivity generally decreases, reaching a minimum value of approximately 14 mΩ cm for the x = 0.08 sample. The decrease in electrical resistivity is attributed to a slightly increased carrier concentration resulting from the introduction of Mn vacancies. For instance, the carrier concentration of x = 0.08 sample increased from 2.2 × 1020 cm−3 in pristine LiMnSbTe3 to 3.34 × 1020 cm−3. However, a further increase in x leads to higher electrical resistivity, likely due to the increased amount of less conductive MnTe2 precipitates (approximately 100 mΩ cm at room temperature)87. As displayed in Fig. 7b, the room-temperature Seebeck coefficient follows a trend similar to that of electrical resistivity as x increases. Except for the x = 0.1 sample, other samples generally exhibit lower values of room-temperature Seebeck coefficient than pristine LiMnSbTe3. Overall, these Seebeck coefficients are still considerably high. For instance, the Seebeck coefficient of the x = 0.08 sample ranges from 250 to 320 μV/K over the temperature range of 300 K to 873 K. Owing to the reduced electrical resistivity, the power factor, shown in Fig. 7c, for x = 0.04 and x = 0.06 samples reaches 4.4 μWcm−2K−1 at 300 K and approximately 8 μWcm−2K−1 at 873 K, surpassing that of pristine LiMnSbTe3.

Temperature-dependence of a electrical resistivity, b Seebeck coefficient, c power factor, d total thermal conductivity, e lattice thermal conductivity, and f ZT for LiMn1-xSbTe3 with x ranging from 0.00 to 0.10.
The total thermal conductivity, in Fig. 7d, at room temperature initially decreases as x increases, reaching a minimum value of 0.52 Wm−1K−1 for x = 0.06 samples before gradually increasing again. Similarly, the x = 0.06 sample also exhibits the lowest lattice thermal conductivity, with values of 0.49 Wm−1K−1 at 300 K and 0.3 Wm−1K−1 at 873 K, shown in Fig. 7e. The decrease in lattice thermal conductivity can be attributed to the strengthened phonon scattering caused by Mn vacancies. However, MnTe2 has significantly higher lattice thermal conductivities of 1.8 Wm−1K−1 at 300 K and 0.7 Wm−1K−1 at 850 K87. Therefore, as x increases further, the increased amount of secondary phase MnTe2 starts to dominate the rise in lattice thermal conductivity. Similar vdW-like gaps are also observed in Mn-deficiency samples, as shown in Fig. S18. Therefore, to understand the reduction in lattice thermal conductivity, we applied the Debye–Callaway model, incorporating additional phonon scattering from vacancies based on the simulation for LiMnSbTe3. The result shown in Fig. S9b presents that the model can reproduce the measured lattice thermal conductivity, indicating the role of vacancy in scattering phonons. Finally, a peak ZT value of 1.5 is achieved in the x = 0.06 sample at 873 K as shown in Fig. 7f. The high-performance LiMn0.94SbTe3 sample was annealed at 873 K for 24 hours under vacuum to evaluate its phase and performance stability at high temperature. As shown in Fig. S19, the annealed sample retains the Fm-3m structure and exhibits performance similar to the unannealed sample. These results further highlight the potential of LiMnSbTe3 as a promising mid-temperature thermoelectric material, which could reach even higher ZT values through proper doping or structural modulation.
Single-leg device performance
Based on the optimized thermoelectric performance, a single-leg device was fabricated to demonstrate its thermoelectric potential. As LiMnSbTe3 exhibited high ZTs at the medium temperature, p-type (Bi, Sb)2Te3, which performs better at the low-temperature range, was used to form a segmented LiMnSbTe3/(Bi, Sb)2Te3 single-leg device. The maximization of temperature-dependent ZT of different segments can achieve better performance across the whole temperature range. We first determine the optimal height ratios between (Bi, Sb)2Te3 (HBST) and LiMnSbTe3 (HLMST) by simulation. As shown in Fig. 8a, the maximum efficiency is obtained approximately at a HBST/(HBST + HLMST) value of 0.5, which can also deliver a high output power as demonstrated in Fig. 8b. Therefore, the (Bi, Sb)2Te3 and LiMnSbTe3 sections are designed with equal height.

Simulated a conversion efficiency and b output power with different height ratios of bismuth telluride part (HBST) to the whole leg (HBST + HLMST), and electrical current. Electric-current-dependence of c U-I plot and output power, d conversion efficiency under a cold-side temperature of 295 K and various hot-side temperatures.
The segmented single-leg device is fabricated by one-step sintering of thermoelectric materials, the metalization layer, and the electrode (details are shown in the Methods). The U-I plot and output power of the fabricated device are shown in Fig. 8c. The device achieved a high output power of ~70 mW, corresponding to a power density of 0.52 Wcm−2, under a temperature difference of 478 K (a cold-side temperature of 295 K and a hot-side temperature of 773 K). As displayed in Fig. 8d, a high conversion efficiency of 8.7% has also been achieved under the temperature difference of 478 K. The good performance of the fabricated device demonstrates the promising role of LiMnSbTe3 as a thermoelectric candidate for medium-temperature applications.
Discussion
In summary, we developed a new rock-salt semiconductor LiMnSbTe3 from the combination of hexagonal MnTe and cubic LiSbTe2, resulting in a high-symmetry cubic phase that is especially favorable for high-performance thermoelectrics. This outcome exemplifies a rational chemical design approach for constructing cubic compounds from non-cubic precursors to achieve both enhanced functionality and practical utility. The high-symmetry crystal structure of LiMnSbTe3 leads to the energy convergence of several energy-aligned valence bands, leading to high Seebeck coefficients of 250–300 μV/K from 300 to 873 K with the contribution of paramagon drag effects. It also exhibits an exceptionally low lattice thermal conductivity of 0.37 Wm−1K−1 at 873 K, significantly lower than that of MnTe and comparable to LiSbTe2. This ultralow thermal conductivity originates from two key structural features: cation disorder and locally ordered Sb2Te3-type van der Waals-like gaps, both of which serve as efficient phonon scattering centers. Despite the strong phonon scattering by the vdW-like gaps, the locally ordered structure, in comparison to the disordered matrix, and the cross-gap interaction across the vdW-like gaps help to facilitate good carrier transport. Owing to the synergistic effects of the high-symmetry matrix and the embedded vdW-like gaps, LiMnSbTe3 achieves a high ZT of 1.2 at 873 K. Upon Mn deficiency, the electrical conductivity increases and the lattice thermal conductivity decreases, leading to an enhanced ZT of 1.5 at 873 K. A segmented LiMnSbTe3/(Bi, Sb)2Te3 single-leg device achieved a high output power density of 0.52 Wcm−2 and an efficiency of 8.7%. These results underscore the high thermoelectric potential of LiMnSbTe3 and highlight the effective role of substructure-embedded high-symmetry compounds as efficient thermoelectric materials.
Methods
Sample synthesis
LiMnmSbTem+2 (m = 1,2,3,5,10) and LiMn1-xSbTe3 (x ranges from 0.00 to 0.1) samples were synthesized by the solid-state reaction. Li (pieces, 99.9%, Sigma-Aldrich), Mn (pieces, 99%, Sigma-Aldrich), Sb (pieces, 99.9%, ZhongNuo Advanced Material (Beijing) Technology Co., Ltd) and Te (chunks, 99.99%, ZhongNuo Advanced Material (Beijing) Technology Co., Ltd) were used as starting materials and weighted according to the nominal compositions. Taking LiMnSbTe3 as an example, 0.0306 g Li, 0.2425 g Mn, 0.5371 g Sb, and 1.6897 g Te were weighed and loaded into the quartz tube with a diameter of 13 mm and a wall thickness of 1.5 mm, which was carbon-coated to prevent the reaction between Li and quartz. Then the quartz ampules were vacuum sealed with a residual pressure of ~ 2 × 10−3 torr. The ampules were put into a box furnace for the reaction and heated to 1073 K within 8 h. After soaking at 1073 K for 1 h, the ampules were cooled to 773 K within 2 h and were further annealed at 773 K for 24 h. After annealing, the ingots were ground to powders by a mortar and pestle in the glove box filled with Ar (the concentration of O2 and H2O is less than 0.1 ppm). Finally, the powders were put into a 12.7 mm diameter graphite die in the glove box, which was sintered by spark plasma sintering at 873 K for 5 min with a uniaxial pressure of 40 MPa under dynamic vacuum (SPS 211Lx Fuji Electronic Industrial Co., Ltd, Japan).
Structural characterization
The phase structures of synthesized powders were characterized by X-ray diffraction (D8 Advance, Bruker, Germany, Cu Kα). While for Rietveld refinement, the X-ray diffraction pattern was collected with the spark plasma sintered sample. Finely polished surfaces of the pellet samples were used to identify the morphology and element distribution, respectively, with a scanning electron microscope (SEM, JEOL JSM 7600F, Japan). The scanning/transmission (S/TEM) analysis was performed with an aberration-corrected JEOL ARM200F microscope operated at 200 kV. The atomic probe tomography was performed on needle-shaped specimens, prepared by the site-specific “lift-out” method with a dual-beam scanning electron microscope/focused ion beam (Helios NanoLab 650, FEI). The specimens were measured in a local electrode atom probe (LEAP 5000 XS, Cameca). The APT data were processed using the commercial software package AP Suite 6.3.
Transport properties measurements
The Seebeck coefficient (S) and electrical resistivity (ρ) were measured with a bar-shaped sample using a Seebeck coefficient/electrical resistivity measuring system (ZEM-3, Ulvac-Riko, Japan). The total thermal conductivity (κ) was calculated via the equation κ = DCpd. D is the thermal diffusivity measured by a laser flash method (Netzsch, LFA 457, Germany). Cp denotes specific heat capacity, and a Dulong-Petit value was used in this work. d represents the density obtained using sample dimensions and mass. For samples containing secondary phases (MnTe2), we also applied the Dulong-Petit value of LiMnSbTe3 as the heat capacity, since the heat capacity of MnTe2 is theoretically lower than that of LiMnSbTe3. The electronic thermal conductivity (κe) is estimated using the Wiedemann–Franz law, κe = LσT, where L is the Lorenz number determined from the Seebeck coefficients48. The Hall coefficient (RH) was determined under a reversible magnetic field of 0.52 T (ResiTest 8340DC, Toyo, Japan), and the carrier concentration was calculated using nH = 1/(eRH).
Fabrication and characterization of a thermoelectric device
The device was fabricated through the one-step sintering method. The powders were put into a graphite die in the sequence of Cu-SnTe-LiMnSbTe3-SnTe-Ni-(Bi,Sb)2Te3-Ni-Cu. The Cu is employed as the electrode. SnTe is obtained through the conventional solid reaction and is used as the diffusion barrier for LiMnSbTe3. Commercial (Bi,Sb)2Te3 powders (Macklin Inc.) were purchased and used directly without any further treatment. The Ni was used as the diffusion barrier of (Bi, Sb)2Te3. After loading all the powders, they were sintered by SPS at 773 K for 15 min with a pressure of 40 MPa, after which the die was slowly cooled to 473 K within 30 minutes. Then, the SPS power was turned off, and the die cooled naturally in the furnace. After sintering, the single-leg device was cut out directly from the pellet with a cross-section dimension of 3.8 mm × 3.5 mm. The thicknesses of the LiMnSbTe3 and (Bi, Sb)2Te3 layers were approximately 2.6 mm and 2.7 mm, respectively. Both the SnTe and Ni layers are around 0.3 mm, serving as the diffusion barrier of the thermoelectric materials. The performance of single-leg devices was tested by the commercial Thermoelectric Conversion Efficiency Evaluation System (Mini-PEM, Advance Riko, Japan). Due to the small size of the single-leg device and the high testing temperature, the heat flow will be significantly overestimated by the Mini-PEM. Therefore, the COMSOL Multiphysics software was used to simulate the heat flow, which was used to calculate the efficiency.
Data availability
All data generated in this study are provided in the Supplementary Information or Source Data file. Source data are provided with this paper.
References
Yan, Q. & Kanatzidis, M. G. High-performance thermoelectrics and challenges for practical devices. Nat. Mater. 21, 503–513 (2022).
Mao, J., Chen, G. & Ren, Z. Thermoelectric cooling materials. Nat. Mater. 20, 454–461 (2021).
Yang, D. et al. Multifactor roadmap for designing low-power-consumed micro thermoelectric thermostats in a closed-loop integrated 5G optical module. Inter. Mater. 3, 326–337 (2024).
Chen, Y. et al. Recent progress in self-powered multifunctional e-skin for advanced applications. Exploration 2, 20210112 (2022).
Snyder, G. J. & Toberer, E. S. Complex thermoelectric materials. Nat. Mater. 7, 105–114 (2008).
Chaikin, P. M., Lubensky, T. C. & Witten, T. A. Principles of Condensed Matter Physics. (Cambridge University Press, Cambridge, 1995).
Marder, M. P. Condensed Matter Physics (John Wiley & Sons, New York, 2010).
Pei, Y. et al. Convergence of electronic bands for high performance bulk thermoelectrics. Nature 473, 66–69 (2011).
Zhang, J. et al. High-performance pseudocubic thermoelectric materials from non-cubic chalcopyrite compounds. Adv. Mater. 26, 3848–3853 (2014).
Yu, Y., Cagnoni, M., Cojocaru-Miredin, O. & Wuttig, M. Chalcogenide thermoelectrics empowered by an unconventional bonding mechanism. Adv. Func. Mater. 30, 1904862 (2020).
Delaire, O. et al. Giant anharmonic phonon scattering in PbTe. Nat. Mater. 10, 614–619 (2011).
Zeier, W. G. et al. Thinking like a chemist: intuition in thermoelectric materials. Angew. Chem. Int. Ed. 55, 6826–6841 (2016).
Biswas, K. et al. High-performance bulk thermoelectrics with all-scale hierarchical architectures. Nature 489, 414–418 (2012).
Qin, Y. et al. Grid-plainification enables medium-temperature PbSe thermoelectrics to cool better than Bi2Te3. Science 383, 1204–1209 (2024).
Jiang, B. et al. High-entropy-stabilized chalcogenides with high thermoelectric performance. Science 371, 830–834 (2021).
Jiang, B. et al. High figure-of-merit and power generation in high-entropy GeTe-based thermoelectrics. Science 377, 208–213 (2022).
Li, J. et al. Low-symmetry rhombohedral GeTe thermoelectrics. Joule 2, 976–987 (2018).
Dong, J. et al. Medium-temperature thermoelectric GeTe: vacancy suppression and band structure engineering leading to high performance. Energy Environ. Sci. 12, 1396–1403 (2019).
Roychowdhury, S. et al. Enhanced atomic ordering leads to high thermoelectric performance in AgSbTe2. Science 371, 722 (2021).
Cao, J. et al. Non-equilibrium strategy for enhancing thermoelectric properties and improving stability of AgSbTe2. Nano Energy 107, 108118 (2023).
Sarkar, D. et al. Metavalent bonding in GeSe leads to high thermoelectric performance. Angew. Chem. Ed. 60, 10350–10358 (2021).
Yu, Y. et al. Doping by design: enhanced thermoelectric performance of GeSe alloys through metavalent bonding. Adv. Mater. 35, 2300893 (2023).
Dong, J. et al. High thermoelectric performance in rhombohedral GeSe-LiBiTe2. J. Am. Chem. Soc. 146, 17355–17364 (2024).
Luo, Y. et al. Cubic AgMnSbTe3 semiconductor with a high thermoelectric performance. J. Am. Chem. Soc. 143, 13990–13998 (2021).
Luo, Y. et al. High thermoelectric performance in the new cubic semiconductor AgSnSbSe3 by high-entropy engineering. J. Am. Chem. Soc. 142, 15187–15198 (2020).
Wang, H. et al. Band flattening and phonon-defect scattering in cubic SnSe–AgSbTe2 alloy for thermoelectric enhancement. Mater. Today Phys. 16, 100298 (2021).
Zhou, B. et al. Promising cubic MnGeTe2 thermoelectrics. Sci. China Mater. 62, 379–388 (2019).
Dong, J., Gao, J. & Yan, Q. High entropy strategy on thermoelectric materials. Mater. Lab 2, 230001 (2023).
Tan, G. et al. Non-equilibrium processing leads to record high thermoelectric figure of merit in PbTe–SrTe. Nat. Commun. 7, 12167 (2016).
Tan, G., Zhao, L. D. & Kanatzidis, M. G. Rationally designing high-performance bulk thermoelectric materials. Chem. Rev. 116, 12123 (2016).
Yu, B. et al. Enhancement of thermoelectric properties by modulation-doping in silicon germanium alloy nanocomposites. Nano Lett. 12, 2077–2082 (2012).
Bozin, E. S. et al. Entropically stabilized local dipole formation in lead Chalcogenides. Science 330, 1660–1663 (2010).
Sarkar, D., Bhui, A., Maria, I., Dutta, M. & Biswas, K. Hidden structures: a driving factor to achieve low thermal conductivity and high thermoelectric performance. Chem. Soc. Rev. 53, 6100–6149 (2024).
Xie, H. et al. Hidden local symmetry breaking in silver diamondoid compounds is root cause of ultralow thermal conductivity. Adv. Mater. 34, 2202255 (2022).
Banik, A. et al. Engineering ferroelectric instability to achieve ultralow thermal conductivity and high thermoelectric performance in Sn1-xGexTe. Energy Environ. Sci. 12, 589–595 (2019).
Dong, J. et al. Discordant distortion in cubic GeMnTe2 and high thermoelectric properties of GeMnTe2-x%SbTe. J. Am. Chem. Soc. 145, 1988–1996 (2023).
Dong, J. et al. Off-centering of Ge atoms in GeBi2Te4 and impact on thermoelectric performance. Adv. Funct. Mater. 34, 2314499 (2024).
Xie, H., Zhao, L.-D. & Kanatzidis, M. G. Lattice dynamics and thermoelectric properties of diamondoid materials. Inter. Mater. 3, 5–28 (2024).
Chen, Z. et al. Vacancy-induced dislocations within grains for high-performance PbSe thermoelectrics. Nat. Commun. 8, 13828 (2017).
Kim, S. I. et al. Dense dislocation arrays embedded in grain boundaries for high-performance bulk thermoelectrics. Science 348, 109–114 (2015).
Jiang, Y. et al. Evolution of defect structures leading to high ZT in GeTe-based thermoelectric materials. Nat. Commun. 13, 6087 (2022).
Pan, Y. et al. Melt-Centrifuged (Bi, Sb)2Te3: engineering microstructure toward high thermoelectric efficiency. Adv. Mater. 30, 1802016 (2018).
Wu, R. et al. Strong charge carrier scattering at grain boundaries of PbTe caused by the collapse of metavalent bonding. Nat. Commun. 14, 719 (2023).
Zheng, Y. et al. Paramagnon drag in high thermoelectric figure of merit Li-doped MnTe. Sci. Adv. 5, eaat9461 (2019).
Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B Condens. Matter 192, 55–69 (1993).
Hou, D. et al. Local structures of perovskite dielectrics and ferroelectrics via pair distribution function analyses. J. Eur. Ceram. Soc. 38, 971–987 (2018).
Xin, J. et al. Reinforced bond covalency and multiscale hierarchical architecture to high performance, eco-friendly MnTe-based thermoelectric materials. Nano Energy 57, 703–710 (2019).
Goldsmid, H. J. & Sharp, J. W. Estimation of the thermal band gap of a semiconductor from Seebeck measurements. J. Electron. Mater. 28, 869–872 (1999).
Kim, H.-S. et al. Characterization of the Lorenz number with Seebeck coefficient measurement. APL Mater. 3, 041506 (2015).
Beekman, M. et al. Preparation and crystal growth of Na24Si136. J. Am. Chem. Soc. 131, 9642–9643 (2009).
Wang, Y. et al. Design principles for solid-state lithium superionic conductors. Nat. Mater. 14, 1026–1031 (2015).
Shibata, N. et al. Differential phase-contrast microscopy at atomic resolution. Nat. Phys. 8, 611–615 (2012).
Zachman, M. J., Yang, Z., Du, Y. & Chi, M. Robust atomic-resolution imaging of lithium in battery materials by center-of-mass scanning transmission electron microscopy. ACS Nano 16, 1358–1367 (2022).
Mugiraneza, S. & Hallas, A. M. Tutorial: a beginner’s guide to interpreting magnetic susceptibility data with the Curie-Weiss law. Commun. Phys. 5, 95 (2022).
Figgis, B., Lewis, J. & Wilkins, R. Modern Coordination Chemistry (Interscience, New York, 1960).
Ilton, E. S. et al. XPS determination of Mn oxidation states in Mn (hydr)oxides. Appl. Surf. Sci. 366, 475–485 (2016).
Wu, D., Xie, L., Xu, X. & He, J. High thermoelectric performance achieved in GeTe–Bi2Te3 pseudo-binary via Van der Waals gap-induced hierarchical ferroelectric domain structure. Adv. Funct. Mater. 29, 1806613 (2019).
Yu, Y., Xie, L., Pennycook, S. J., Bosman, M. & He, J. Strain-induced van der Waals gaps in GeTe revealed by in situ nanobeam diffraction. Sci. Adv. 8, eadd7690 (2022).
Jiang, T.-T. et al. In situ characterization of vacancy ordering in Ge-Sb-Te phase-change memory alloys. Fundam. Res. 4, 1235–1242 (2024).
Abdellaoui, L. et al. Density, distribution and nature of planar faults in silver antimony telluride for thermoelectric applications. Acta Mater. 178, 133–145 (2019).
Hong, M. et al. Arrays of planar vacancies in superior thermoelectric Ge1-x-yCdxBiyTe with band convergence. Adv. Energy Mater. 8, 1801837 (2018).
Wang, H., LaLonde, A. D., Pei, Y. & Snyder, G. J. The criteria for beneficial disorder in thermoelectric solid solutions. Adv. Funct. Mater. 23, 1586–1596 (2013).
Toberer, E. S., Zevalkink, A. & Snyder, G. J. Phonon engineering through crystal chemistry. J. Mater. Chem. 21, 15843–15852 (2011).
Dong, J. et al. Reducing lattice thermal conductivity of MnTe by Se alloying toward high thermoelectric performance. ACS Appl. Mater. Interfaces 11, 28221–28227 (2019).
Jain, A. et al. The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Morelli, D. T., Jovovic, V. & Heremans, J. P. Intrinsically minimal thermal conductivity in Cubic I−V−VI2 semiconductors. Phys. Rev. Lett. 101, 035901 (2008).
Hempelmann, J., Müller, P. C., Ertural, C. & Dronskowski, R. The orbital origins of chemical bonding in Ge−Sb−Te phase-change materials. Angew. Chem. Ed. 61, e202115778 (2022).
Wuttig, M., Deringer, V. L., Gonze, X., Bichara, C. & Raty, J. Y. Incipient metals: functional materials with a unique bonding mechanism. Adv. Mater. 30, 1803777 (2018).
Zhang, W. et al. Metavalent bonding in layered phase-change memory materials. Adv. Sci. 10, 2300901 (2023).
Cheng, Y. et al. Understanding the structure and properties of Sesqui-Chalcogenides (i.e., V2VI3 or Pn2Ch3 (Pn = Pnictogen, Ch = Chalcogen) Compounds) from a bonding perspective. Adv. Mater. 31, 1904316 (2019).
Rundle, R. E. Electron deficient compounds. J. Am. Chem. Soc. 69, 1327–1331 (1947).
Jones, R. O. The chemical bond in solids—revisited. J. Phys.: Condens. Matter 34, 343001 (2022).
Langhammer, H. T. et al. Optical and electrical investigation of the anisotropy of Sb2Te3 single crystals. Phys. Stat. Sol. 109, 673 (1982).
Tiong, K. K. et al. Growth and characterization of Rhenium-doped MoS2 single crystals. J. Cryst. Growth 205, 543–547 (1999).
Anderson, T. L. & Krause, H. B. Refinement of the Sb2Te3 and Sb2Te2Se structures and their relationship to Nonstoiehiometric Sb2Te3-ySey compounds. Acta Cryst. B 30, 1307 (1974).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomie distances in halides and chaleogenides. Acta Cryst. A 32, 751 (1976).
Van de Walle, A. Multicomponent multisublattice alloys, nonconfigurational entropy and other additions to the Alloy Theoretic Automated Toolkit. Calphad 33, 266–278 (2009).
Wang, H. et al. Higher mobility in bulk semiconductors by separating the dopants from the charge conducting band– a case study of thermoelectric PbSe. Mater. Horiz. 2, 323 (2015).
Kutorasinski, K., Wiendlocha, B., Tobola, J. & Kaprzyk, S. Importance of relativistic effects in electronic structure and thermopower calculations for Mg2Si, Mg2Ge, and Mg2Sn. Phys. Rev. B 89, 115205 (2014).
Wiendlocha, B. Thermopower of thermoelectric materials with resonant levels: PbTe:Tl versus PbTe:Na and Cu1-xNix. Phys. Rev. B 97, 205203 (2018).
De Boor, J., Berche, A. & Jund, P. Density of states effective mass for p-Type Mg2Si–Mg2Sn solid solutions: comparison between experiments and first-principles calculations. J. Phys. Chem. C. 124, 14987–14996 (2020).
Xu, Y. et al. Performance optimization and single parabolic band behavior of thermoelectric MnTe. J. Mater. Chem. A 5, 19143–19150 (2017).
Dong, J. et al. High-performance electron-doped GeMnTe2: hierarchical structure and low thermal conductivity. J. Mater. Chem. A 7, 27361–27366 (2019).
Zheng, Z. et al. Rhombohedral to cubic conversion of GeTe via MnTe alloying leads to ultralow thermal conductivity, electronic band convergence, and high thermoelectric performance. J. Am. Chem. Soc. 140, 2673–2686 (2018).
Duan, S. et al. Anomalous thermopower and high ZT in GeMnTe2 driven by Spin’s thermodynamic entropy. Research 2021, 1949070 (2021).
Snyder, G. J. et al. Weighted mobility. Adv. Mater. 32, 2001537 (2020).
Xu, Y. et al. MnTe2 as a novel promissing thermoelectric material. J. Materiom. 4, 215–220 (2018).
Momma, K. & Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Dong, J. et al. Lead-free MnTe mid-temperature thermoelectric materials: facile synthesis, p-type doping and transport properties. J. Mater. Chem. C. 6, 4265–4272 (2018).
Chen, Z. et al. GaSb doping facilitates conduction band convergence and improves thermoelectric performance in n-type PbS. Energy Environ. Sci. 16, 1676 (2023).
Xu, L. et al. Enhanced average power factor and ZT value in PbSe thermoelectric material with dual interstitial doping. Energy Environ. Sci. 17, 2018 (2024).
Jia, B. et al. Pseudo-nanostructure and trapped-hole release induce high thermoelectric performance in PbTe. Science 384, 81–86 (2024).
Acknowledgements
The authors thank Prof. G. Jeffery Snyder for helpful discussions. This study was supported at Northwestern University in part by the U.S. Department of Energy, Office of Science Basic Energy Sciences under grant DE-SC0024256, DOE Office of Science (synthesis, transport, and nanoscale characterization). The experiments at Nanyang Technological University were supported by MOE ACRF Tier 2 MOE-T2EP50223-0003, ASTAR MTC programmatic project under grant no. M23L9b0052, MOE Tier 1 RG78/23, awarded to Q.Y. R.V.R., thanks the support from the National Research Foundation, Singapore, under its 29th Competitive Research Program (CRP) Call (Award ID NRF-CRP29-2022-0002). The testing at Tsinghua University was supported by the Basic Science Center Project of NSFC under Grant No. 52388201, and the National Key R&D Program of China No. 2023YFB3809400, awarded to J.-F.L. This study also used the EPIC facility of Northwestern University’s NUANCE Center, which received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS 2025633), MRSEC program (NSF DMR-1720139) at the Materials Research Center, International Institute for Nanotechnology (IIN), Keck Foundation, and State of Illinois (V.P.K.). Y.L. would like to acknowledge the IIN Ryan fellowship. The authors would also like to acknowledge the Facility for Analysis, Characterization, Testing, and Simulation, Nanyang Technological University, Singapore, for the use of their electron microscopy/X-ray facilities.
Author information
Authors and Affiliations
Contributions
J.D., Q.Y. and M.G.K. conceived the research. Q.Y. and M.G.K. supervised the project. J.D. performed the synthesis of materials, performance testing, data analysis, and manuscript writing. Y.L. and V.P.D. conducted the TEM analysis. Y.S. and B.X. performed DFT calculations. Y.Y. and S. performed APT characterization and analysis. Y.J., C.C. and J.-F.L. contributed to the device fabrication and performance measurement. L.P.T. and R.V.R. measured the magnetic properties through PPMS. C.Z., D.V.M.R,. and K.H. measured the low-temperature Seebeck coefficients. P.A. conducted the FTIR measurement. Y.L., Y.S., X.Y.T., Y.Y. and D.Y. contributed to the discussion and data analysis. All authors discussed the results and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hanhwi Jang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dong, J., Liu, Y., Sun, Y. et al. Stabilization of high-performance rock-salt LiMnSbTe3 thermoelectrics with embedded van der Waals-like gaps. Nat Commun 16, 11501 (2025). https://doi.org/10.1038/s41467-025-66518-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66518-w
This article is cited by
-
Tuning Optical and Thermoelectric Response of Strontium Oxide with Ba Substitution for Photovoltaic and Optoelectronic Applications
Journal of Inorganic and Organometallic Polymers and Materials (2026)
