
Abstract
The cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway is a central regulator of innate immunity and a promising target for cancer immunotherapy. However, the clinical translation of STING agonists is limited by suboptimal response rates and dose-limiting toxicities, particularly in the liver. These challenges highlight the presence of endogenous inhibitors of STING signaling and underscore the need for strategies that enable tissue-specific modulation of STING activity. Here, we identify proprotein convertase subtilisin/kexin type 9 (PCSK9), a key regulator of cholesterol metabolism, as a negative modulator of STING activation. Mechanistically, PCSK9 competes with STING for binding to a shared cargo receptor, which is critical for STING trafficking. PCSK9 deficiency markedly enhances the immunostimulatory effects of STING agonists. Capitalizing on the elevated expression of PCSK9 in the liver relative to tumors, we develop a formulation that delivers a low-dose STING agonist alongside PCSK9-targeting siRNA, thereby achieving tumor-selective STING activation while minimizing hepatotoxicity. These findings reveal an unanticipated role for PCSK9 in innate immune regulation and establish a therapeutic approach to enhance the safety and efficacy of STING-based immunotherapies, with broader implications for other STING-associated modalities, including radiotherapy and chemotherapy.
Similar content being viewed by others


Introduction
Against an invading pathogen, the body’s first line of defense is the innate immune system, which recognizes foreign pathogens and initiates the appropriate adaptive immune response. As such, the innate immune system plays a crucial role in cancer therapeutics by shaping the tumor microenvironment and determining the therapeutic efficacy of treatments1. One important innate immune pathway is the cGAS-STING pathway2, whose activation produces various cytokines, including type I interferons that promote adaptive T cell-mediated antitumor immunity3,4,5,6,7,8.
While STING agonists have shown great initial success in preclinical studies9,10,11, they suffer from low objective response rates and severe adverse events, including exacerbated liver function in clinical trials12,13. These clinical challenges suggest the existence of key negative regulators of STING signaling, yet clinically translatable modulators remain elusive. Equally pressing is the need to selectively enhance STING activation within tumors while limiting off-target activation in the liver—a goal complicated by the ubiquitous expression of STING across tissues. Although various STING agonist-based cancer immunotherapies have been developed to enhance tumor-targeted drug delivery and improve therapeutic efficacy, they often accumulate in the liver, contributing to hepatic toxicity11,14. Thus, a strategy capable of reprogramming the spatiotemporal activation of STING is urgently needed.
Cholesterol has been shown to impair type I interferon responses in a STING-dependent manner15. One key regulator of cholesterol homeostasis is PCSK9, which is highly expressed in the liver16,17. Interestingly, tumors also express PCSK918, although at a lower level than the liver. Moreover, intratumoral PCSK9 levels negatively correlate with survival and type I interferon responses across multiple cancers19,20. These findings motivate us to hypothesize that PCSK9 may negatively regulate STING signaling. Importantly, because PCSK9 expression in the liver is higher than in tumors21, a formulation containing low-dose PCSK9 siRNA and STING agonists may completely knock down PCSK9 in the tumor to amplify STING activation, while the remaining PCSK9 in the liver can still suppress STING activation. These features will ultimately lead to enhanced therapeutic efficacy and reduced toxicity that can not be achieved by STING agonists alone.
Here, we demonstrate that PCSK9 is an endogenous suppressor in STING signaling. Mechanistically, the STING signaling involves the anterograde trafficking of STING from the endoplasmic reticulum (ER) to the Golgi apparatus, which requires surfeit locus protein 4 (SURF4), a bidirectional cargo receptor that mediates protein transport. PCSK9 competes with STING for binding to SURF4 during their anterograde trafficking and, therefore, limits STING signaling. These findings reveal a critical and multifaceted role for PCSK9 at the interface of maintaining metabolic and innate immune homeostasis, uncovering a rate-limiting mechanism in STING signal transduction. Strikingly, systemic administration of nanoparticles (NP) containing a low-dose STING agonist and PCSK9-siRNA induces potent antitumor immunity without inducing hepatic immune toxicity that is typically seen for STING agonists alone at the effective dose. Because PCSK9-siRNA has already been approved to treat hyperlipidemia16, we envision that our strategy may have immediate and broad clinical applications for cancer immunotherapy in the future.
Results
PCSK9 silencing potentiates STING activation
To initially explore the potential correlation between PCSK9 and innate immune signaling, we analyzed its mRNA expression alongside six immune-related genes in tumors from 42 rectal cancer patients (GEO: GSE15781, Fig. 1a)22. PCSK9 levels negatively correlate with IFN-β or ISG20, but not with STING, mitochondrial antiviral signaling protein (MAVS), IL-6, or IL-1β (Fig. 1b–g). These findings suggest that elevated PCSK9 expression may impair type I interferon responses, but this effect is not associated with the expression of key adapter proteins (STING or MAVS).

a Schematic illustrating the aim and strategy of the clinical data analysis. b–g Correlations between PCSK9 and indicated innate immune signaling-related genes. Shown are mRNA levels of b PCSK9 and IFN-β, c PCSK9 and ISG20, d PCSK9 and STING, e PCSK9 and MAVS, f PCSK9 and IL-6, and g PCSK9 and IL-1β (GEO dataset: GSE15781, n = 42 patients). h, i BMDCs were incubated with siPCSK9 (h) or siCtrl (i) and/or different concentrations of Mn2+. After 24 h, IFN-β secretion was quantified by ELISA (n = 3 independent experiments). j, k Dose-response curves of the IFN-β response in BMDCs after siPCSK9 or siCtrl treatment (n = 3 independent experiments). The EC50 values are shown for the indicated formulations. l Raw264.7 ISG luciferase reporter cells were exposed to Mn2+(50 μM), and/or siPCSK9 (7.5 µg/ml) for 24 h before measuring the luciferase signal. m, THP1 ISG luciferase reporter cells were exposed to Mn2+ (50 μM), and/or siPCSK9/siCtrl (7.5 µg/ml) for 24 h before measuring the luciferase signal (n = 3 independent experiments). n THP1-Luica ISG reporter cells, were exposed to c-di-AMP (1 μM), and/or siPCSK9/siCtrl (7.5 µg/ml) for 24 h before measuring the luciferase signal (n = 3 independent experiments). o THP1-Lucia ISG reporter cells were exposed to cGAMP (1 μM), and/or siPCSK9/siCtrl (7.5 µg/ml) for 24 h before measuring the luciferase signal (n = 3 independent experiments). Data represent mean ± SD. Data were analyzed by one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons post-test (l–o) or two-way ANOVA with Bonferroni’s multiple comparisons post-test (j, k). Source data are provided as a Source Data file. Panel (a) was created in BioRender. Kuai, R. (2025) https://BioRender.com/28bogc8.
To confirm whether PCSK9 affects STING activation, we treated bone marrow-derived dendritic cells (BMDCs) with a weak STING agonist23 Mn2+ and PCSK9-siRNA (siPCSK9) or control siRNA (siCtrl), and then measured IFN-β production to assess STING activation. To reduce double-stranded RNA-induced immunostimulation, both siPCSK9 and siCtrl were chemically modified16 in our study. Surprisingly, treating BMDCs with siPCSK9 and Mn2+ (siPCSK9/Mn2+) induced a 40-fold increase in IFN-β production. This effect was not observed in BMDCs treated with siCtrl/Mn2+ (Fig. 1h, i). Mn2+ alone had an EC50 of 5.35 ± 0.72 mM, while combining with 10 or 20 μg/ml of siPCSK9 reduced the EC50 to 0.87 ± 0.42 mM and 0.37 ± 0.17 mM, respectively. In contrast, Mn2+ combined with siCtrl showed no increase in activity, confirming that the enhanced STING activation was PCSK9-dependent (Fig. 1j, k). BMDCs treated with siPCSK9/Mn²⁺ and siCtrl/Mn²⁺, or THP1PCSK9 KO and THP1WT cells treated with Mn²⁺, showed similar levels of cGAMP (Supplementary Fig. 1a, b), indicating siPCSK9 did not change the ability of Mn2+ to catalyze cGAS to produce cGAMP24. Moreover, mouse and human IFN reporter monocytes, including Raw264.7-Lucia ISG and THP1-Lucia ISG cells, exhibited stronger IFN responses upon exposure to siPCSK9/Mn2+ (Fig. 1l, m). This pattern was further validated by combining siPCSK9 with other STING agonists such as c-di-AMP and cGAMP (Fig. 1n, o). These results indicate that PCSK9 plays an important role in limiting STING activation, and silencing PCSK9 amplifies the activity of STING agonists. We also tested the PCSK9 antibody Evolocumab in BMDCs and found that it failed to enhance IFN-β production with either Mn²⁺ or cGAMP (Supplementary Fig. 2a, b).
PCSK9 suppresses type I interferon production in a STING-dependent manner
We next sought to understand whether PCSK9 regulates other innate immune pathways, such as the Toll-like receptor 3 and 4 (TLR3 and 4) pathways, which have also been shown to induce type I interferon secretion25. Interestingly, compared with siCtrl, siPCSK9 only slightly changed IFN response upon mixing with the TLR3 agonist Poly(I:C) (Fig. 2a–c) or TLR4 agonist Lipopolysaccharide (LPS) (Fig. 2d, e). Based on these results, PCSK9 appears to inhibit the STING signaling more substantially. Indeed, knocking out STING in BMDC abrogated the ability of siPCSK9/Mn2+ to induce type I interferon secretion (Fig. 2f), and a similar pattern was observed in THPSTING KO cells (Fig. 2g). Moreover, interferon-stimulated genes (Ifnb, Cxcl10, Ccl5, ISG15, IFIT3, and TNFα) were also substantially decreased in BMDCSTING KO but not in BMDCWT (Fig. 2h–m). Altogether, these results indicate that PCSK9 restrains Mn2+-induced type I interferon responses in a STING-dependent manner.

a BMDCs were incubated with siPCSK9 or siCtrl (4 µg/well) and/or 10 μg/ml Poly (I: C). After 24 h, the IFN-β secretion was quantified by ELISA (n = 3 independent experiments). b THP1-Lucia ISG reporter cells were exposed to 10 μg/ml Poly (I: C) and/or siPCSK9/siCtrl (7.5 µg/ml) for 24 h (n = 3 independent experiments) before measuring the luciferase signal. c THP1-Lucia ISG reporter cells were incubated with siPCSK9 /siCtrl (7.5 µg/ml) and/or 10 μg/ml Poly (I:C). After 24 h, the IFN-β secretion was quantified by ELISA (n = 3 independent experiments). d THP1-Lucia ISG reporter cells were exposed to 500 ng/ml LPS and/or siPCSK9/siCtrl (7.5 µg/ml) for 24 h before measuring the luciferase signal (n = 3 independent experiments). e THP1-Lucia ISG NF-κB reporter cells were exposed to 500 ng/ml LPS and/or siPCSK9/siCtrl (7.5 µg/ml) for 24 h before measuring the luciferase signal (n = 3 independent experiments). f WT or STING−/− BMDCs were incubated with siPCSK9/siCtrl (4 µg/well) and/or Mn2+ (400 µM). After 24 h, the IFN-β secretion was quantified by ELISA (n = 3 independent experiments). g WT or STING−/− THP1-Lucia ISG reporter cells were exposed to Mn2+(50 μM), and/or siPCSK9 /siCtrl (7.5 µg/ml) for 24 h before measuring the luciferase signal (n = 3 independent experiments). h–m Relative expression of h Ifnb, i Cxcl10, j Ccl5, k Isg15, l Ifit3, and m Tnfa mRNA in WT or STING−/− BMDCs treated with siPCSK9/siCtrl (4 µg/well) and/or Mn2+ (400 µM) for 24 h (n = 3 independent experiments). n Representative confocal images of STING (Green) translocation to Golgi (Red) in WT HeLa-hSTING-GFP or PCSK9-knockout HeLa-hST-GFP (HeLa-hSTING-GFPPCSK9KO) treated with 500 µM Mn2+for 3 h (Scale bars 10 μm). This experiment was repeated independently three times with similar results. o Pearson’s correlation coefficient of STING and Golgi colocalization of HeLa cells in (n) (n = 50 cells). This experiment was repeated independently three times with similar results. p Quantification of the number of PCSK9 dots/cell in (q) (n = 50 cells). q Representative confocal images of STING (Green) translocation and PCSK9 (Red) secretion in HeLa-hSTING-GFPWT or HeLa-hSTING-GFPPCSK9KO treated with 500 µM Mn2+for 3 h (scale bar 10 μm). r Representative confocal images of Sec24B (Violet), STING (Green), and PCSK9 (Red) colocalization in HeLa-hSTING-GFPWTorHeLa-hSTING-GFPPCSK9 KOcells treated with 500 µM Mn2+ for 3 h (scale bar 10 μm). This experiment was repeated independently three times with similar results. s, t SURF4, PCSK9, STING, and phosphorylated STING/TBK1 expression in BMDCs treated with siPCSK9/siCtrl (20 µg/ml) and/or Mn2+ (400 µM) for 24 h (s) or in HeLaWT/HeLa-hSTINGPCSK9 KO cells treated with 500 µM Mn2+ for 12 h (t). This experiment was repeated independently three times with similar results. Data represent mean ± SD. NS not significant. Data were analyzed by one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons post-test. In the immunofluorescence images shown in 2n, 2q, and 2r, PCSK9 was labeled with fluorescence by using a commercially available antibody, and STING was tagged with GFP. Source data are provided as a Source Data file.
PCSK9 limits STING signaling by interfering with STING anterograde trafficking
A key rate-limiting step in cGAS-STING signaling transduction is STING trafficking from the ER to the Golgi (anterograde trafficking)26,27,28,29,30,31. Thus, we next monitored the formation of STING puncta, a hallmark of trafficked STING on the Golgi, in HeLaWT and PCSK9-knockout HeLa (HeLaPCSK9 KO) cells under Mn2+ stimulation. HeLaPCSK9 KO cells exhibited faster STING puncta formation than HeLaWT cells (Fig. 2n, o), suggesting PCSK9 deficiency promoted the anterograde STING trafficking.
We next sought to understand the mechanism by which PCSK9 regulates STING trafficking. Previous studies have shown that the anterograde STING trafficking was mediated by COP II vesicles, and the retrograde STING trafficking from Golgi to ER was mediated by COP I vesicles to maintain the delicate balance and avoid autoimmunity32,33. Moreover, SURF4 on the COP I vesicle was responsible for binding to STING and facilitating the retrograde trafficking32,34,35,36. Interestingly, SURF4 on the COP II vesicles also mediates the anterograde trafficking of various secretory proteins37, including PCSK938. In light of these observations, we hypothesized that SURF4 might also mediate anterograde trafficking of STING, and PCSK9 might compete with STING for binding to SURF4, thereby limiting the anterograde transport of STING in COPII vesicles.
To confirm the role of SURF4 in STING anterograde trafficking, we generated SURF4-overexpressing HeLa (HeLaSURF4 OE) and SURF4-knockout HeLa (HeLaSURF4 KO) cells. After cGAMP stimulation, we observed strong colocalization of STING with SURF4, and anterograde STING trafficking was enhanced in HeLa-hSTINGSURF4 OE cells (Supplementary Fig. 3a, b). Moreover, cGAMP treatment induced stronger STING activation in THP1SURF4 OE and 293T SURF4 OE cells (Supplementary Fig. 3c, d). In contrast, SURF4-knockout HeLa cells exhibited impaired STING and PCSK9 anterograde trafficking and reduced STING signaling activation compared to HeLaWT or HeLaSURF4 OE cells. Meanwhile, a basal activation of STING was also observed in SURF4-knockout cells (Supplementary Fig. 3e–j). These results indicate that SURF4 is involved not only in the retrograde trafficking of STING but also in the anterograde trafficking and activation of STING.
To directly analyze this possible vesicle transport-mediated signaling crosstalk, we investigated the intracellular distribution of PCSK9 and STING in HeLa cells. Interestingly, the enhanced STING anterograde trafficking appears to be associated with decreased PCSK9 transport, as measured by counting fluorescence antibody-labeled PCSK9 protein puncta (dots) (Fig. 2p). PCSK9 was strongly colocalized with GFP-labeled STING in a vesicle-like structure upon STING activation (Fig. 2q). Further immunostaining analysis confirmed that the vesicle-like structure contained SEC24 Homolog B (Sec24B) (Fig. 2r), a hallmark of COPII vesicles37,39, indicating that PCSK9 and STING are co-packaged and transported by COPII vesicles. The observed changes in STING trafficking were positively associated with the activation of cGAS-STING signaling, as evidenced by the stronger activation of phospho-STING (Ser366) and phospho-TBK1 (Ser172) following STING agonist treatment. Moreover, the unchanged expression of STING and SURF4 further suggests that PCSK9 restricts STING signaling by modulating its anterograde trafficking efficiency (Fig. 2s–t).
PCSK9 limits STING anterograde trafficking through competitive binding to SURF4
We next investigated the interactions among STING, PCSK9, and SURF4. THP1PCSK9 KO, L929PCSK9 KO, and HeLaPCSK9 KO cells exhibited faster and stronger STING/TBK1/IRF3 phosphorylation than the corresponding wild-type cells upon exposure to low-dose STING agonists (Mn2+ or cGAMP) (Fig. 3a–d). We also found increased STING phosphorylation at early time points in THP1PCSK9 KO cells following cGAMP treatment (Supplementary Fig. 4). We further assessed the kinetics of STING puncta formation in HeLa cells by confocal microscopy under the treatment of a low concentration of cGAMP. STING puncta formed rapidly within 60 min in HeLaPCSK9 KO cells, while in HeLaWT cells, it took at least 120 min (Fig. 3e), which is consistent with the pattern of STING signaling activation in Fig. 3d. In contrast, the overexpression of PCSK9 (OE) significantly inhibited STING activation in THP1 cells at 0.5, 1, 2, and 4 h after cGAMP treatment, compared to THP1WT cells (Fig. 3f). Similarly, cGAMP or plasmid-induced cGAS-STING activation was also significantly reduced by transfection with different doses (1.5–5 µg) of PCSK9 plasmids (Fig. 3g and Supplementary Fig. 5a). These results indicate that PCSK9 inhibits STING anterograde trafficking from the earliest stages and throughout the entire process, likely by affecting the initiation of SURF4-mediated COPII vesicle assembly.

a–d SURF4, PCSK9, STING, and phosphorylated STING /TBK1/IRF3 expression in THP1WT/ THP1PCSK9 KO-Lucia ISG reporter cells treated with 50 µM Mn2+, or in THP1WT/THP1PCSK9 KO (b), L929WT/ L929PCSK9 KO (c), HeLaWT / HeLa-hSTINGPCSK9 KO cells (d), cells treated with 1 μM cGAMP for 0, 2, 4, and 6 h. This experiment was repeated independently three times with similar results. e Representative confocal images of STING (Green) translocation in HeLa-hSTING-GFPWT or HeLa-hSTING-GFPPCSK9 KO cells treated with 100 nM cGAMP for 0, 1, and 2 h. This experiment was repeated independently three times with similar results. f THP1WT or THP1PCSK9 OE-Lucia reporter cells were exposed to different concentrations of cGAMP for 24 h (n = 3 independent experiments). g HEK293T cells were transfected with STING, different concentrations of PCSK9 plasmid (1.5, 2.5, and 5 ng), and IFN-β promoter-driven luciferase reporter (IFN-β-luc) together with or without 0.5 μM cGAMP treatment for 24 h (n = 3 independent experiments). h HEK293T cells were transfected with cGAS, STING, SURF4, PCSK9 alone or indicated combinations and IFN-β promoter-driven luciferase reporter (IFN-β-luc) together for 24 h (n = 3 independent experiments). i THP1WT, THP1PCSK9 KO, or THP1SURF4 OE -Lucia ISG reporter cells were exposed to 1 µM cGAMP for 24 h (n = 3 independent experiments). j HEK293T cells were transfected with STING, different concentrations of SURF4 plasmid (10, 20, and 40 ng), and IFN-β promoter-driven luciferase reporter (IFN-β-luc) together with or without 0.5 μM cGAMP treatment for 24 h (n = 3 independent experiments). k Co-IP analysis of SURF4 and PCSK9/STING interactions in lysates from BMDC cells treated with siPCSK9/siCtrl (20 µg/ml) and Mn2+ alone or indicated combinations for 6 h. This experiment was repeated independently three times with similar results. l Co-IP analysis of SURF4 and PCSK9/STING interactions in lysates from THP1WT and THP1PCSK9 KO cells treated with cGAMP 1 µM for 4 h. This experiment was repeated independently three times with similar results. Predicted conformation and Gibbs binding energy of SURF4/PCSK9 complex (m) and SURF4/STING complex (n) via molecular dynamics simulation. o Structural clash model of PCSK9 and STING via molecular dynamics simulation. Data represent mean ± SD. Data were analyzed by one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons post-test or two-way ANOVA with Bonferroni’s multiple comparisons post-test (f). Source data are provided as a Source Data file.
To investigate the potential interaction among PCSK9, SURF4, and STING, we conducted a competitive binding and inhibition assay in HEK293T cells. Here, PCSK9 transfection significantly inhibited the cGAMP-induced STING activation. By contrast, SURF4 expression promoted cGAMP-induced STING activation, which is impaired by PCSK9 overexpression in SURF4-transfected HEK293T cells (Fig. 3h). Next, we compared the effects of SURF4 overexpression and PCSK9 knockout on STING activation in THP1 cells with the same cGAMP stimulation using the IFN reporter assay. Overexpressing SURF4 induced stronger STING activation than knocking out PCSK9, suggesting that adequate SURF4 can counteract the inhibitory effect of PCSK9 on STING signaling (Fig. 3i). Similarly, SURF4 and STING-transfected HEK293T cells also showed enhanced cGAS-STING activation upon cGAMP treatment (Fig. 3j). However, this enhanced activation was abolished in HEK293T cells that received the same treatment but without STING transfection, confirming that this process is STING-dependent (Supplementary Fig. 5b). The co-immunoprecipitation (Co-IP) revealed that in the process of STING activation and anterograde trafficking, PCSK9 competitively interacts with STING, ultimately preventing STING from binding to SURF4. We observed the stronger binding of SURF and STING, both in siPCSK9/Mn2+-treated BMDC (Fig. 3k) and cGAMP-treated THP1PCSK9 KO cells (Fig. 3l). Notably, HeLaPCSK9 KO cells and THP1PCSK9 KO cells had similar levels of intracellular cholesterol compared with their WT counterparts (Supplementary Fig. 6a, b), thus excluding the potential influence of cholesterol on STING activation40.
To further explain our findings, molecular dynamics (MD) simulations of the SURF4-STING and SURF4-PCSK9 complexes (Fig. 3m) revealed a lower binding free energy (ΔG_bind = −324 kJ/mol) for the SURF4-PCSK9 complex, suggesting a more favorable interaction compared to the SURF4-STING complex (ΔG_bind = −218 kJ/mol) (Fig. 3n). Further analysis identified a steric clash when both PCSK9 and STING bind to SURF4 simultaneously (Fig. 3o). Additionally, AlphaFold2-predicted structures of human and mouse SURF4, PCSK9, and STING dimers showed high similarity across species (Supplementary Fig. 7). AlphaFold3 predictions for the SURF4-PCSK9 and SURF4-STING complexes revealed similar binding patterns in both species, with high confidence (pTM > 0.5) (Supplementary Fig. 8 and 9). However, pTM scores for the SURF4-PCSK9-STING ternary complexes were below 0.5, suggesting the instability and low likelihood of ternary complex formation (Supplementary Fig. 10). Altogether, the above results indicate that PCSK9 negatively regulates the cGAS-STING pathway by competitively binding to SURF4 and limiting STING anterograde trafficking (Supplementary Fig. 11).
NP containing PCSK9 siRNA and STING agonists enable safe and effective cancer immunotherapy
Given the promising results and mechanistic rationale of amplified cGAS-STING signaling following PCSK9 silencing, we sought to translate this effect into a viable therapeutic approach. To this end, we developed a facile method for encapsulating siPCSK9 and STING agonists (e.g., Mn2+ or cGAMP) into hyaluronic acid (HA)-coated lipid NP11,41 (Fig. 4a and Supplementary Fig. 12a). Dynamic light scattering (DLS) showed that NPsiPCSK9/cGAMP had an average diameter of 66 nm (Fig. 4b and Supplementary Fig. 12b). The encapsulation efficiency was over 95% for siPCSK9 and approximately 10% for cGAMP (Supplementary Fig. 12c, d). The sizes and encapsulation efficiencies of NPcGAMP and NPsiPCSK9 were comparable to those of NPsiPCSK9/cGAMP(Supplementary Fig. 12e, f). Additionally, we used a similar method to prepare NPsiPCSK9/Mn2+, which had a size and structure similar to NPsiPCSK9/cGAMP (Supplementary Fig. 12g), with Mn2+ exhibiting a pH-responsive release profile due to its pH-dependent binding to siRNA (Supplementary Fig. 12h). We next analyzed the in vitro activity of these NP. As expected, NPsiPCSK9/cGAMP and NPsiPCSK9/Mn2+ showed significantly enhanced IFN-β release compared to their respective controls (Supplementary Fig. 12i–j), suggesting that siPCSK9 significantly potentiates the effect of low-dose STING agonists. Cryo-EM analysis revealed that NPcGAMP had one lipid bilayer, whereas NPsiPCSK9 and NPsiPCSK9/cGAMP exhibited two lipid bilayers (Fig. 4c and Supplementary Fig. 13). This structural difference was likely due to the strong negative charge of siRNA, which influences lipid organization. RNA-seq analysis revealed that NPsiPCSK9/cGAMP treatment broadly activated the cGAS-STING signaling-related genes in BMDCs (Fig. 4d and Supplementary Fig. 14a, d). Moreover, the expression of structural proteins related to COPI and COPII vesicles was similar for NPcGAMP and NPsiPCSK9/cGAMP (Supplementary Fig. 14e), thus excluding the effect of vesicle number on STING trafficking.

a Schematic showing PCSK9-mediated suppression of STING activation and the design of bioinspired NP. b Size distribution of NPsiPCSK9/cGAMP measured by DLS. This experiment was repeated independently three times with similar results. c Cryo-electron microscopy (cryo-EM) images of NPsiPCSK9/cGAMP, Scale bar = 50 nm. This experiment was repeated independently three times with similar results. d Heatmap analysis of type I interferon, MHC I complex, and co-stimulatory signals from RNA sequencing data (n = 3 independent experiments). e C57BL/6 mice were subcutaneously injected with 300,000 MC38 cells on day 0. On day 8, tumor-bearing mice were intravenously injected with indicated NP formulations containing 100 µg/dose siPCSK9 and/or 1.5 µg/dose cGAMP (n = 5 mice). Shown are (f) average tumor growth curves, g body weight changes, and h tumor growth curves post rechallenge (n = 5 mice). i, j C57BL/6 or C57BL/6Batf3−/− mice were inoculated subcutaneously with 3 × 105 MC38 tumor cells on day 0 and intravenously injected with NPsiPCSK9/cGAMP on day 8. Average tumor growth curves (i) and survival curves (j) for animals treated with NPsiPCSK9/cGAMP and antibodies that deplete indicated populations of immune cells (n = 5 mice). k Percentage of CD86+ cells among CD11C+ dendritic cells, l percentage of CD8+ T cells among CD3+ T cells, m M1/M2 macrophage ratio, n percentage of CD25+Foxp3+ T cells among CD4+ T cells in the tumor microenvironment on day 11 (n = 3 mice). o Percentage of SIINFEKL-specific T cells among CD8+ T cells in the tumor microenvironment on day 11 (n = 3 mice). Data are presented as mean ± SD. Data were analyzed by one-way ANOVA with Tukey’s multiple comparisons test (k–o). Data were analyzed by two-way ANOVA with Dunnett’s multiple comparisons test (f–i). Source data are provided as a Source Data file. Panel (a) was created in BioRender. Kuai, R. (2025) https://BioRender.com/b04l005.
Intravenously injected hyaluronic acid-coated NP accumulated in the tumor as early as 1 h and remained for over 72 h (Supplementary Fig. 15a), indicating these NP are suitable for in vivo therapeutic studies. A single dose of NPsiPCSK9/cGAMP led to complete tumor regression in 100% of treated mice, while NPsiPCSK9 or NPcGAMP failed to inhibit tumor growth (Fig. 4e–g and Supplementary Fig. 16a–c). Notably, animals cured with NPsiPCSK9/cGAMP were protected from rechallenge with MC38 cells (Fig. 4h). NPsiCtrl/cGAMP failed to inhibit tumor growth (Supplementary Fig. 17a, b), further indicating the enhanced therapeutic effect was PCSK9-dependent. Combination of the PCSK9 antibody Evolocumab and NPsiCtrl/cGAMP also failed to inhibit tumor growth (Supplementary Fig. 18), implying that blocking extracellular PCSK9 does not affect the STING activation. Moreover, NPsiPCSK9/cGAMP lost the therapeutic effect in C57BL/6STING−/− mice bearing MC38WT tumor cells (Supplementary Fig. 19a–c), but it maintained the strong therapeutic effect in C57BL/6WT mice bearing MC38STING−/− tumor cells (Supplementary Fig. 19d–g), suggesting that host STING signaling plays a critical role in NPsiPCSK9/cGAMP treatment. The immune cell depletion in C57BL/6 or C57BL/6Batf3−/− mice revealed that dendritic cells, macrophages, NK, CD4+, and CD8+ T cells all contributed to the therapeutic effect (Fig. 4i, j).
A single dose of NPsiPCSK9/cGAMP induced higher levels of mature dendritic cells (mDCs), M1 macrophages, cytotoxic CD8+ T lymphocytes, and natural killer (NK) cells within the tumor microenvironment compared with other groups. In contrast, the frequencies of immune-suppressive regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) were significantly reduced in the NPsiPCSK9/cGAMP group compared to control NP (Fig. 4k–n and Supplementary Fig. 20a–h). Additionally, NPsiPCSK9/cGAMP induced a marked increase in OVA-specific CD8+ T cells (SIINFEKL) in the MC38-OVA model (Fig.4o and Supplementary Fig. 20i–j), indicating enhanced priming and expansion of cytotoxic T lymphocytes (CTLs). Similarly, NPsiPCSK9/Mn2+ induced stronger antitumor responses than control groups (Supplementary Figs. 21–23).
Notably, a single-dose systemic administration of NPsiPCSK9/cGAMP (1.5 µg/dose cGAMP) achieved not only potent antitumor effects but also exhibited excellent safety profiles, without body weight changes at 24 h and 48 h post-administration (Fig. 5a–c). In contrast, NPcGAMP at the effective dose (5 µg/dose) induced over 15% body weight loss at 24 h post intravenous administration, although this dose also induced robust antitumor immune responses and therapeutic effects. It should be noted that lower doses (1.5 µg and 2.5 µg/dose cGAMP) of NPcGAMP failed to show strong therapeutic effects.

a Dosing regimen for the treatment of mice bearing MC38 tumors. b Average tumor growth curves and c body weight changes of MC38-bearing mice treated with NP formulations containing 100 µg/dose siPCSK9 and 1.5 µg/dose cGAMP or 1.5, 2.5, 5 µg/dose cGAMP (n = 5 mice). d CB-17 SCID mice were inoculated subcutaneously with 1 × 106 MDA-MB-231 tumor cells on day 0 and intravenously treated with the indicated formulations on day 8. e Average tumor growth curves for animals treated with indicated formulations (n = 6 mice). f Body weights of animals treated with the indicated formulations. g The survival curves of animals treated with the indicated formulations (n = 6 mice). h Representative images of tumor-bearing mice treated with the indicated formulations on days 8 and 21. i Individual tumor growth curves for animals treated with indicated formulations (n = 6 mice). j C57BL/6 mice were inoculated subcutaneously with 2 × 105 B16-F10 tumor cells on day 0, intraperitoneally injected with αPD-L1 on days 6, 9, 12, and 15, and intravenously injected with the indicated formulations on day 7. k Average tumor growth curves for animals treated with indicated formulations (n = 6 mice). l Body weights of animals treated with indicated formulations. m The survival curves of animals treated with indicated formulations (n = 6 mice). n Individual tumor growth curves for animals treated with indicated formulations (n = 6 mice). o Representative images of tumor-bearing mice treated with indicated formulations on days 6 and 14. Data are presented as mean ± SEM. Data were analyzed by two-way ANOVA with Dunnett’s multiple comparisons test (b, c, e, f, k, and l). Data were analyzed by log-rank (Mantel–Cox) test (g, m) or two-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons test (c). Source data are provided as a Source Data file. Panel (a) was created in BioRender. Kuai, R. (2025) https://BioRender.com/28bogc8.
We next evaluated the therapeutic efficacy and safety of systemic NPsiPCSK9/cGAMP and NPcGAMP in the MDA-MB231 xenograft models. A single dose of NPsiPCSK9/cGAMP resulted in complete tumor regression in 100% of treated mice without significant weight loss. In contrast, high-dose NPcGAMP failed to induce tumor regression in any of the six animals and caused significant weight loss (>15%) at 24 h post-administration (Fig. 5d–i). Moreover, combining NPsiPCSK9/cGAMP with αPD-L1 resulted in complete tumor remission in five out of six B16F10 tumor-bearing mice, without significant body weight loss (Fig. 5j–o). Altogether, these results indicate that a low dose of NPsiPCSK9/cGAMP is highly effective and safe for cancer immunotherapy, while NPcGAMP was only effective at a relatively high dose that induces severe side effects.
NPsiPCSK9/cGAMP induces stronger STING activation in the tumor than in the liver
NPsiPCSK9/cGAMP and high-dose NPcGAMP had similar biodistribution profiles at 24 h post administration, with both formulations demonstrating ~80% liver accumulation (Supplementary Fig. 24a–c), indicating the liver may experience stronger off-target STING activation than other organs. However, the liver has a higher basal level of PCSK9 than the tumor (Fig. 6a), making it possible to silence PCSK9 for achieving differential activation of STING in the liver and tumor. Interestingly, NPsiPCSK9/cGAMP induced a significantly lower liver IFN-β production and serum AST (the liver injury marker) than high-dose NPcGAMP between 12 and 72 h (Fig. 6b–c). Moreover, NPsiPCSK9/cGAMP induced a lower serum IFN-β than NPcGAMP, although IFN-β in the spleen was similar. Compared with NPcGAMP, NPsiPCSK9/cGAMP induced a slightly higher peak concentration of IFN-β, which remained in the tumor for about 72 h (Supplementary Fig. 25a–c). In parallel with these changes, NPsiPCSK9/cGAMP did not induce inflammation or vascular damage in the liver, but high-dose NPcGAMP induced large areas of inflammatory damage in the liver, including inflammation in the hepatic artery and portal vein (Fig. 6d and Supplementary Fig. 26). Meanwhile, NPsiPCSK9/cGAMP treatment induced more extensive inflammatory damage and erythrocyte infiltration in the tumor than NPcGAMP (Fig. 6e and Supplementary Fig. 27). Additionally, NPsiPCSK9/cGAMP and high-dose NPcGAMP treatments exhibited no difference in the recruitment of immune cells to the periarteriolar lymphoid sheaths surrounding the splenic arterioles (Supplementary Fig. 28).

a STING and PCSK9 expression levels in the liver, spleen, and tumor of untreated mice (n = 3 mice). b, c C57BL/6 mice (n = 3 mice) were treated with NPcGAMP and NPsiPCSK9/cGAMP on day 8. Shown are e liver IFN-β levels and f serum concentrations of aspartate aminotransferase (AST) at the indicated time points (0, 12, 24, 48, and 72 h post-treatment, n = 3 mice). d, e Martius Scarlet Blue (MSB) staining of the liver (d) and tumor (e) after 24 h of treatment with indicated NP formulations containing 100 µg/dose siPCSK9 and 1.5 µg/dose cGAMP, or 5 µg/dose cGAMP (n = 3 mice), and expression of PCSK9 and key proteins related to the STING pathway in the liver (f) and tumor (g). h Schematic showing differential activation patterns of STING in the liver and tumor following systemic administration of NPcGAMP and NPsiPCSK9/cGAMP. Systemic delivery of NPcGAMP (high dose) leads to a lack of differential STING regulation in the tumor and liver, providing effective cancer immunotherapy but also causing uncontrollable hepatotoxicity. However, systemic delivery of NPsiPCSK9/cGAMP induces differential STING regulation of low-dose cGAMP and siPCSK9 in the tumor and liver, offering the potential for safe and effective cancer immunotherapy. Data are presented as mean ± SD. AUC data were calculated from the corresponding curves and subsequently analyzed by one-way ANOVA with Tukey’s multiple comparisons test (b, c). Source data are provided as a Source Data file. Panel (h) was created in BioRender. Kuai, R. (2025) https://BioRender.com/obym8jd.
We next sought to understand the mechanism underlying the differential STING activation in the liver and tumor. Low-dose NPsiPCSK9/cGAMP almost completely knocked down PCSK9 in the tumor to amplify STING activation, while the liver still had remaining PCSK9 to suppress STING activation (Fig. 6f, g). This is because the liver has a higher basal level of PCSK9 than the tumor. Although high-dose NPcGAMP-treated mice also had PCSK9 in the liver, PCSK9 failed to fully suppress the STING activation as ~80% of the high-dose NPcGAMP was in the liver (Fig. 6f). Moreover, NPcGAMP-treated mice had intratumoral PCSK9 that suppressed the STING activation, as shown by the lower level of phosphorylated STING and TBK1 (Fig. 6g). The proposed working model is presented in Fig. 6h. The single-cell sequencing data (sourced from the Human Protein Atlas42) revealed high PCSK9 expression in hepatocytes and liver-resident macrophages (Kupffer cells) (Supplementary Fig. 29a). In line with this, both CD45⁺ and CD45− cells exhibited high PCSK9 in the liver. As a result, NPsiPCSK9/cGAMP only partially reduced PCSK9 and exhibited less STING activation compared with the therapeutically effective dose of NPcGAMP. In the tumor, CD45⁺ cells showed stronger STING phosphorylation after NPsiPCSK9/cGAMP treatment, whereas CD45⁻ cells displayed a similar but less pronounced trend (Supplementary Fig. 29b). Interestingly, the structures of PCSK9, STING, and SURF4 were conserved across different species (Fig. 7), suggesting that the inhibition of STING trafficking by PCSK9 may be an ancient and evolutionarily conserved regulatory process. These results suggest that the patterns identified in mice may be relevant for future clinical trials, supporting the potential clinical translation of our findings.

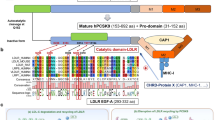
Alphafold2-predicted structures of PCSK9, STING, and SURF4 in Homo sapiens, Pan paniscus, Mus musculus, Cottoperca gobio, and Ornithorhynchus anatinus, spanning a time range from 300,000 years to 110 million years.
Discussion
Our findings reveal that PCSK9 inhibits activation of the cGAS–STING pathway by restricting anterograde trafficking of STING from the ER to the Golgi apparatus. This mechanism not only offers a potential explanation for the limited clinical efficacy of STING agonists but also suggests a strategy to unlock their therapeutic potential for safe and effective cancer immunotherapy. While recent studies have shown that targeting extracellular PCSK9 enhances tumor cell-surface MHC I stability43, the role of intracellular PCSK9 in regulating innate immunity has remained largely unexplored. Our study addresses this gap and underscores the multifaceted functions of PCSK9 across innate and adaptive immune responses.
Moreover, we demonstrate that co-delivery of siPCSK9—an FDA-approved therapeutic—with a low-dose STING agonist via bioengineered NP markedly enhances antitumor efficacy. This approach may help overcome key barriers that have hampered STING agonist-based therapies, providing a broadly applicable and translationally feasible strategy for cancer immunotherapy. Furthermore, this ‘biological switch-like’ strategy for tissue-specific immune modulation may also have broad applications in radiotherapy and chemotherapy, both of which involve STING activation.
Methods
Preparation of immature BMDCs
BMDCs were isolated by flushing the femur and tibia from C57BL/6 or C57BL/6STING−/− mice and cultured in RPMI 1640 media (Gibco) supplemented with 10% FBS (Biological Industry (BI)), 1% penicillin/streptomycin (Gibco), 20 ng/ml of GM-CSF (Genscript), and 50 μM β-mercaptoethanol at 37 °C and 5% CO2. Half of the medium was removed and supplemented with 20 ng/ml GM-CSF-containing medium on days 3 and 6, and the non-adherent cells were harvested on day 7 and used immediately.
Synthesis of siRNA
The sequence and modification of PCSK9 and Ctrl siRNA were designed as described previously16. siRNA was customized from General Biol. (Anhui) Co., Ltd. The synthesis and deprotection of Oligonucleotides were carried out with a standard solid-phase synthesis procedure. Then, single-stranded RNAs were purified from crude oligonucleotides by reversed-phase HPLC. Double-stranded siRNAs were generated by annealing equal amounts of sense and antisense strands.
Preparation of NP containing siRNA and/or STING agonist
The hyaluronic acid-coated lipid nanoparticle (NP) formulation was prepared by using a microfluidic device (INanoTM E, Micro&Nano, Shanghai, China) to mix the ethanol phase containing selected lipids and aqueous phase containing selected siRNA and/or STING agonists. Briefly, to obtain the lipid-containing ethanol phase, 60.84 mM 1,2-Dioleoyl-3-trimethylammonium-propane chloride (DOTAP), 57.12 mM 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and 5.35 mM 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000) (all purchased from AVT Pharmaceutical Tech Co., Ltd, Shanghai) were dissolved in ethanol. To obtain the aqueous phase, 100–500 µg/ml siPCSK9 or siCtrl was dissolved in sterile normal saline (for NPsiPCSK9or NPsiCtrl preparation); or 100–500 µg/ml siPCSK9 or siCtrl, and 20 mM MnCl2 (Sigma, Cat#M3634) were dissolved in sterile normal saline (for NPsiPCSK9/Mn2+ and NPsiCtrl/Mn2+ preparation); or 100–500 µg/ml siPCSK9 and 40 µg/ml cGAMP (MCE, Cat# HY-100564) were dissolved in sterile normal saline (for NPsiPCSK9/cGAMP preparation). The ethanol phase containing indicated lipids and the aqueous phase containing indicated molecules were mixed at a volume ratio of 3:1 (aqueous: ethanol) via the microfluidic device at a flow rate of 8 ml/min for nanoparticle production. Finally, the hyaluronic acid (HA) was added dropwise into the prepared liposome under constant vortexing to achieve a final HA concentration of 1 mg/ml, followed by dialysis for 4–6 h in normal saline at 4 °C to remove ethanol and unloaded molecules. NPBlank, NPMn2+, and NPcGAMP were all prepared using the same methods by adjusting the content of the aqueous phase accordingly. All NP formulations were sterilized through a 220 nm filter before use.
To simplify the in vitro mechanism study, all siRNAs used in Figs. 1–3 were delivered by HA-coated NP: namely, NPsiPCSK9 was used in the siPCSK9 group, and NPsiCtrl was used in the siCtrl group, with STING agonists physically mixed with these NP formulations for in vitro studies. However, to facilitate the codelivery of siRNA and STING agonists in vivo. All the siRNA and STING agonists were encapsulated into NP for in vivo use.
Antibodies and reagents
Rabbit antibodies against p-TBK1 (#5483), STING (#13647), p-STING (#19781), p-IRF3 (#4947), Golgin-97 (#13192S), Sec24B (#12042), and GAPDH (#5174) were purchased from Cell Signaling Technology. Rabbit antibodies against SURF4(#PA5-110450) were purchased from Thermofisher. Rabbit antibodies against PCSK9 (#Ab185194) were purchased from Abcam. Rabbit antibodies against PCSK9 (#A21909, A24478) were purchased from ABclonal Technology. Antibodies against Tubulin (HC101-01) and GADPH (HC301-01) were obtained from TransGen biotech.
LPS (L3024) was purchased from Sigma. Human IFN-β bioluminescent ELISA kit (luex-hifnbv2), 2'3’-cGAMP (tlrl-nacga23), and c-di-AMP (tlrl-nacda) were purchased from InvivoGen. Polyethylenimine (PEI, #23966-2) was obtained from Polysciences. Poly (I: C) LMW (#tlrl-picw-250) was purchased from Invivogen.
Cell culture and treatment
Throughout the studies, all cells were tested negative for mycoplasma contamination and morphologically confirmed. THP1-Lucia ISG cells (InvivoGen, No. thpl-isg) and THP1-Lucia NF-κB Cells (InvivoGen, No. thpl-nfkb) were cultured in RPMI 1640 medium (Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS, Gemini), 1% (v/v) penicillin (100 U/ml, Solarbio), and streptomycin (100 μg/ml, Solarbio). Raw264.7 (ATCC, TIB-71), HEK293T cells (ATCC, CRL-3216), HeLa cells (ATCC, CCL-2), and L929 cells (ATCC, CCL-1) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco) containing 10% FBS and penicillin-streptomycin. All these cells mentioned above were maintained in a humidified incubator containing 5% CO2 at 37 °C. To facilitate cGAMP delivery in vitro, perfringolysin O (PFO, 0.02 μg/mL) was utilized to improve the entry of the indicated concentrations of cGAMP into HeLa, HEK293T, and L929 cells. After treatment, cells were incubated at 37 °C with 5% CO2 for 24 h for the Luciferase reporter assay. For western blotting, cells were treated and cultured for the indicated lengths of time, as shown in the figure legends.
Construction of stable cell lines
To generate stable cell lines overexpressing SURF4 or PCSK9, human Surf4 and Pcsk9 genes were inserted into the pCDH-CMV-MCS-IRES vector, followed by virus packing and lentiviral plasmid transduction of parent cells described below. To generate PCSK9-knockout L929 cells, PCSK9-knockout THP1-Lucia ISG, SURF4-knockout THP1-Lucia ISG, PCSK9-knockout HeLa-hSTING-GFP cells, PCSK9-knockout-HeLa-hSTING cells, SURF4-knockout HeLa-hSTING cells, or SURF4-knockout HeLa-hSTING-110GFP cells, the parent cells were transduced by lentiviral plasmids consisting of gRNA and plentiCRISPR v2 (Addgene Plasmid). For virus packaging, 14 μg of constructed plasmids with the two packaging plasmids, psPAX2 (6 μg) and pMD2.G (4 μg) were co-transfected into HEK293T cells by the PEI transfection reagent. After 72 h, the supernatant was collected and filtered through a 0.22-μm membrane filter. The filtrate was then added to the indicated cells with polybrene (Sigma Aldrich) for another 48 h. The transfected cells were selected by 2 µg/ml puromycin or 5 µg/ml blasticidin (Thermo Fisher Scientific), determined by the selection markers on the plentiCRISPR backbone. The purified cells were used for further western blotting or immunostaining. All gRNA sequences for gene knockout and primer sequences for gene overexpression are provided in Supplementary Tables S1 and S2. The gRNAs demonstrating optimal knockout efficiency, as determined by functional screening, are indicated in bold, and the corresponding knockout cell lines generated with these gRNAs were used for subsequent experiments.
Enzyme-linked immunosorbent assay (ELISA)
For human IFN-β detection after PCSK9 silencing, THP1 cells were seeded in 12-well plates and treated with formulations containing 7.5 μg/ml siCtrl or 7.5 μg/ml siPCSK9 or 10 μg/ml Poly (I: C) (transfected with PEI), or indicated combinations and incubated for 24 h at 37 °C in 5% CO2. Then the supernatant was collected and the concentration of secreted IFN-β was measured by the human IFN-β bioluminescent ELISA kit following the manufacturer’s instructions. The optical density (OD) in each well was read at 450 nm.
For murine IFN-β detection after PCSK9 silencing, BMDCs seeded in 96-well plates were treated with siCtrl or siPCSK9 (10 and 20 µg/ml), or Mn2+ (0–1600 µM), or indicated combinations for 24 h. In some experiments, BMDCs were seeded in 96-well plates and treated with 20 µg/ml siCtrl or siPCSK9, or 10 μg/ml Poly(I: C) (transfected with PEI) alone, or indicated combinations for 24 h. The supernatant was collected, and the concentration of secreted IFN-β was measured by the LumiKine™ Xpress mIFN-β 2.0 (InvivoGen, Cat# luex-mifnbv2) following the manufacturer’s instructions.
To assess the cGAS-STING pathway activation induced by siRNA and STING agonists co-delivered by NP. BMDCs were seeded in 96-well plates and treated with NPBlank(Blank vesicle), NPMn2+ (NP containing 400 µM Mn2+), NPcGAMP (NP containing 100 nM cGAMP), NPsiCtrl (NP containing 20 μg/ml siCtrl), NPsiPCSK9 (NP containing 20 μg/ml siPCSK9), NPsiCtrl/Mn2+ (NP containing 20 μg/ml siCtrl and 400 µM Mn2+), NPsiPCSK9/Mn2+ (NP containing 20 μg/ml siPCSK9 and 400 µM Mn2+), andNPsiPCSK9/cGAMP (NP containing 20 μg/ml siPCSK9 and 100 nM cGAMP) for 24 h. The supernatant was collected, and the concentration of secreted IFN-β was measured by the LumiKine™ Xpress mIFN-β 2.0 (InvivoGen, Cat# luex-mifnbv2).
Luciferase reporter assay
THP1-Lucia ISG cells are derived from THP1 cells by stably transfecting the luciferase reporter gene under the control of interferon-stimulated response elements (ISRE). THP1-Lucia NF-κB cells were derived from THP1 cells by stably integrating the NF-κB-inducible Luc reporter element for monitoring the NF-κB activation. RAW-Lucia ISG cells were derived from Raw264.7 cells. For the luciferase assay, THP1-Lucia ISG, STINGCRISPR−/− THP1-Lucia ISG, and RAW-Lucia ISG cells were seeded in 96-well plates at a density of 3 × 106 cells/well and treated with 50 μM Mn2+ with or without 7.5 μg/ml NPsiPCSK9 at 37 °C for 24 h. Then, 20 μl cell suspension was mixed with 60 μl luciferase buffer (50 mM NaCl, 50 mM HEPES pH 7.0, 10 mM EDTA, 0.05% CHAPS, 1 μM Coelenterazine) and incubated in darkness for 5 min at room temperature. Luminescence was measured by Cytatio3TM Cell Imaging Reader (BioTek).
THP1-Lucia ISG cells were also treated with another two STING agonists (1 μM cGAMP, 10 μM c-di-AMP) or TLR agonists (500 ng/ml LPS, 10 μg Poly(I: C)) with or without 7.5 μg/ml siCtrl or siPCSK9 at 37 °C for 24 h. THP1-Lucia NF-κB cells were treated with 500 ng/ml LPS and/or siCtrl or siPCSK9. The luciferase reporter activity was determined following the standard protocol.
RNA isolation and quantitative RT-PCR
BMDCs were treated with 400 μM Mn2+ or 20 µg/ml siCtrl or siPCSK9 for 24 h. Total RNA was extracted by M5 HiPer Total RNA Extraction Reagent (MF034-01, Mei5 Biotechnology) according to the manufacturer’s protocol. Then, 1 μg RNA was reverse transcribed into cDNA by utilizing the M5 Super plus qPCR RT kit (MF166-plus-01, Mei5 Biotechnology). Real-time quantitative PCR was performed using HiPer SYBR Premix EsTaq (MF787-T, Mei5 Biotechnology). The primer sequences detecting the indicated genes are shown in Table S3. All the indicated siRNAs we used in this study were delivered by HA-coated NP as described above.
Western blotting
Cells were lysed by lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 0.1% SDS, 1 mM EDTA-Na2, 1 mM Na3VO4, 25 mM β-glycerophosphate) supplemented with 0.5% PMSF and 0.05% Leupeptin on ice for 30 min and then centrifuged at 13,000 rpm for 15 min at 4 °C. Supernatant was retained and boiled with 4X SDS loading buffer at 95 °C for 10 min. The denatured protein samples were separated by 10% or 12% SDS-PAGE gels and transferred to PVDF membranes (Millipore). These membranes were blocked with 5% skim milk for 1 h at room temperature and incubated with the specific primary antibodies at 4 °C overnight. After being washed by TBS-T for 30 min, the membranes were incubated with appropriate HRP-conjugated secondary antibodies at room temperature for 1 h. The bands were visualized by the automatic chemiluminescence/fluorescence Imaging System (Tanon 5200).
Interferon β-luciferase reporter assay
HEK293T cells (seeded in 96-well plates) were transfected with IFN-β-luciferase (50 ng/well), human STING (50 ng/well) plasmids, different doses of human PCSK9 plasmid (1.5, 2.5, 5 ng/well) or human SURF4 plasmid (10, 20, 40 ng) for 12 h. The HEK293T cells were stimulated with 0.5 μM cGAMP in the presence of 0.02 mg/mL PFO for another 12 h. Alternatively, HEK293T cells were transfected with IFN-β-luciferase (50 ng/well), human cGAS (30 ng/well), and STING (50 ng/well) plasmids with vector (20 ng), PCSK9 (5 ng or 20 ng), and SURF4 (20 ng) for 24 h. The IFN-β reporter activity was detected according to the standard protocol.
Immunofluorescence microscopy
For immunofluorescence staining, HeLa cells stably expressing STING-110-GFP or STING-GFP were cultured on coverslips in 24-well plates and treated as described in the figure legends. After removing the medium, cells were fixed with 4% paraformaldehyde (Solarbio) for 10 min at room temperature and then washed with PBS three times. Cells were blocked by staining buffer containing 3% BSA and 0.05% Triton X-100 for 1 h and then incubated with specific primary antibodies overnight at 4 °C. After washing three to four times in PBS, cells were incubated with secondary antibodies conjugated to Alexa Fluor 488 or 568 (Invitrogen) and 4’,6-diamidino-2-phenylindole (DAPI) for nuclear staining for 2 h at room temperature. The confocal images were acquired with a Zeiss LSM980 laser scanning microscope. Zen Blue 3.2 software and Image J software were used for image processing.
Co-IP of SURF4
BMDCs were seeded at 3 × 106 cells/well in 6-well plates. After overnight culture, BMDCs were pretreated with 20 μg/ml siRNA siPCSK9 or siCtrl for 24 h and then treated with 400 µM Mn2+ for 3 h. SURF4 Co-IP samples were prepared by PierceTM Direct Magnetic IP/Co-IP Kit (Thermofisher, Cat# 88828) following previously reported methods with slight modifications34. Briefly, cells were lysed by IP buffer in IP/Co-IP Kits supplemented with 1% 3-[(3-Cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS), and beads were washed by wash buffer IP/Co-IP Kits supplemented with 0.7% CHAPS (MCE, Cat# HY-15435) for stabilized protein-protein complexes and retained the biochemical activity of proteins in solution.
Protein-protein molecular docking and MD simulation
The molecular docking assays of human PCSK9 (PDB id:6U2F) and human STING (PDB id:6NT5) with human SURF4 (AlphaFold:AF-B7Z1G8-F1) were performed by HDOCK, and the top three structures with the best docking scores were chosen for subsequent molecular dynamics (MD) simulation. The protein–protein complex was placed in a cubic box filled with solvent (TIP3P water molecules), and the complex was simulated by Gromacs 2022.1 under constant temperature (310 K, V-rescale temperature coupling method), pressure (1 bar, Berendsen method), and Charmm36 force field for a 50 ns MD simulation, and finally visualized by Pymol.
MM-PBSA (Molecular Mechanics Poisson Boltzmann Surface Area) methods were used to calculate the binding free energy of the docked complexes between PCSK9/STING and SURF4. A stable segment of the RMSD (Root mean square deviation) was selected as a frame for PCSK9 (11.5 ns to 37.5 ns) and STING (6.5 ns to 32.5 ns) for the MM-PBSA estimation. As SURF4 is a membrane protein, the membrane protein parameters were used for the free energy calculation in this system. The membrane center position was set as T36, the upper side of the membrane was L25, and the lower side was F46.
Cryogenic electron microscopy (Cryo-EM)
Copper carbon grids (Quantifoil R 1.2/1.3, 300 mesh) were pre-treated with a 30-s glow discharge at a medium level using a Harrick plasma cleaner (PDC-32G-2). Liposome samples were applied to the grids in a Vitrobot Mark IV (Thermo Fisher) under conditions of 100% humidity and 8 °C. After a 10-s equilibration, an excess sample was blotted on both sides of the grid using filter paper with a blot force of −1 to 1 for 3.5 s. The grids were then vitrified by plunging into liquid ethane and subsequently stored in a liquid nitrogen-cooled container until further evaluation. Grid quality was assessed using a Talos Arctica 200 kV electron microscope equipped with a K2 camera (Gatan).
Animal studies
The mice used in this research include C57BL/6 mice (6–8 weeks, purchased from Laboratory Animal Resources Center of Tsinghua University), CB-17 SCID mice (6 weeks, purchased from Vital River, China), and BALB/c mice (6-8 weeks, purchased from Vital River, China). All animals were housed under controlled conditions (constant temperature: 22 ± 2 °C; constant humidity: 50 ± 10%; 12 h dark/light cycle).To evaluate the tissue distribution of indicated formulations, tumor-bearing mice were intravenously injected with the NPsiPCSK9/Mn2+, NPsiPCSK9, or NPMn2+ (labeled by DiR, 20 µg/ml, MCE, Cat# HY-D1048) on day 3 post inoculation of 3 × 105 tumor cells in the right flank of C57BL/6 mice. The mice were monitored using the IVIS Spectrum 3D system (Caliper Life Sciences) at 0, 1, 6, 24, 48, and 72 h after intravenous injection. After whole body imaging at the last time point, animals were euthanized, and the DiR fluorescence signals in the heart, liver, spleen, lung, kidney, and tumor were imaged by the IVIS Spectrum 3D system (Caliper Life Sciences).
To evaluate the therapeutic efficacy of siPCSK9 and STING agonist-containing NP with right flank MC38 tumors, female C57BL/6 mice of age 6–8 weeks or female C57BL/6STING−/− mice (GemPharmatech) of age 6–8 weeks were subcutaneously inoculated with 3 × 105 MC38 cells on the right flank on day 0. Tumor-bearing mice were intravenously injected with NPsiPCSK9/Mn2+, NPsiPCSK9, or NPMn2+ (containing 100 µg/dose siPCSK9 and/or 40 µg/dose Mn2+), or intravenously injected with the control formulation NPsiPCSK9/Mn2+ (containing 100 µg/dose siCtrl and 40 µg/dose Mn2+) on days 8, 11, and 14. In some experiments, animals were treated with NPsiPCSK9/cGAMP, NPsiPCSK9, or NPcGAMP (containing 100 µg/dose siPCSK9 and/or 1.5 µg/dose cGAMP) on day 8. Tumor size was monitored every 3 days, and tumor volume was calculated by the following equation: tumor volume = length × width2 × 0.5.
To evaluate the safety of siPCSK9 and STING agonist-containing, MC38 tumor-bearing mice were intravenously injected with indicated formulations NPBlank, NPsiPCSK9, NPMn2+, or NPsiPCSK9/Mn2+ on day 8, and serum samples were collected at 24, 48, and 96 h post-injection. Serum aspartate transaminase (AST), alanine aminotransferase (ALT), creatinine (CR), and creatine kinase (CK) levels were detected by an automatic clinical chemistry analyzer (Rayto Chemary-800). Plasma cytokines (IL-6, IFN-β, and TNFα) were detected by mouse IL-6 ELISA Kit (Cat# GEM0001), mouse IFN-beta ELISA Kit (Cat# GEM0018), and mouse TNF-alpha ELISA Kit (Cat# GEM0004), respectively. Animals were euthanized, and major organs such as the heart, liver, spleen, lung, and kidney were collected at 72 h for H&E staining.
For studies with right flank B16F10 tumors, female C57BL/6 mice of age 6–8 weeks (From the Laboratory Animal Resources Center of Tsinghua University) were subcutaneously inoculated with 2 × 105 B16F10 tumor cells on the right flank on day 0. When tumor volumes reached approximately 50 mm³ (day 6), mice were intravenously administered with NPcGAMP (containing 5 µg/dose cGAMP) or NPsiPCSK9/cGAMP (containing 100 µg/dose siPCSK9 and 1.5 µg/dose cGAMP). In some experiments, animals receiving NPsiPCSK9/cGAMP on day 7 were intraperitoneally injected with 200 µg/dose anti-mouse PD-L1 antibody (10 F.9G2, Selleck, Cat# A2115) on days 6, 9, 12, and 15. Body weight was recorded on days 9, 10, and 11, and subsequently every 3 days thereafter. Tumor size was measured every 3 days to evaluate tumor progression.
For studies using the xenograft model, CB-17 SCID mice were subcutaneously inoculated with 1 × 106 MDA-MB-231 cells on day 0. On day 8, the mice were intravenously administered a single dose of NPsiPCSK9/cGAMP (containing 100 µg/dose siPCSK9 and 1.5 µg/dose cGAMP) or NPcGAMP (containing 5 µg/dose cGAMP), along with the indicated formulations. Mice were monitored for tumor growth and body weight throughout the study. Body weight was recorded on days 9, 10, and 11, and subsequently every 3 days thereafter. Tumor size was measured every 2-3 days to evaluate tumor progression.
For the evaluation of the safety and efficacy of NPsiPCSK9/cGAMP, 3 × 10⁵ MC38 cells were subcutaneously inoculated into C57BL/6J mice. On day 8, the mice were intravenously administered a single dose of NPsiPCSK9/cGAMP (containing 100 µg/dose siPCSK9 and 1.5 µg/dose cGAMP) and various doses of NPcGAMP (containing 1.5, 2.5, and 5 µg/dose cGAMP), along with the indicated formulations. Mice were monitored for tumor growth and body weight throughout the study. Body weight was recorded on days 9, 10, and 11, and subsequently every 3 days thereafter. Tumor size was measured regularly to assess tumor progression. The liver, spleen, tumor, and serum were collected at 0, 12, 24, 48, and 72 h post-treatment for IFN-β detection. Additionally, the liver, spleen, and tumor tissues were collected at 24 h post-treatment for Western blot analysis, MSB staining, and H&E staining.
To examine the in vivo antitumor immune responses, 5 × 105 MC38 cells were inoculated into C57BL/6J mice. When the tumor size reached ~150 mm³, NPsiPCSK9/cGAMP, NPsiPCSK9, or NPcGAMP (containing 100 µg/dose siPCSK9 and/or 1.5 µg/dose cGAMP) were intravenously injected into the mice. Tumor tissues were harvested on the indicated days and cut into small pieces, followed by dissociation using the digestion buffer (1 mg/ml collagenase and 100 μg/ml deoxyribonuclease I in serum-free RPMI) for 30 min at 37 °C with gentle shaking. The obtained suspension was passed through a 70-μm cell strainer to obtain the single-cell suspension. For surface marker staining, the cells were incubated with anti-mouse CD16/32 (92, Biolegend, Cat# 101302) for 10 min at room temperature, and then incubated with PerCP anti-mouse CD45 (30-F11, Biolegend, Cat# 103130), APC anti-mouse CD11c (N418, Biolegend, Cat# 117310), APC/Cy7 anti-mouse F4/80 (BM8, Biolegend, Cat# 123118), FITC anti-mouse CD80 (16-10A1, Biolegend, Cat# 104706), PE/Cy7 anti-mouse CD86 (GL-1, Biolegend, Cat# 105014), PE anti-mouse CD206 (MMR) (C068C2, Biolegend, Cat# 141706), FITC anti-mouse CD3 (17A2, Biolegend, Cat# 100204), APC/Cy7 anti-mouse CD4 (GK1.5, Biolegend, Cat# 100414), PE anti-mouse CD8α (53-6.7, Biolegend, Cat# 100708), FITC anti-mouse CD45 (30-F11, Biolegend, Cat# 103108), PE/Cy7 anti-mouse NK-1.1 (PK136, Biolegend, Cat# 108714), BV605-anti-mouse CD69 (H1.2F3, Biolegend, Cat# 104530), APC anti-mouse/human CD11b (M1/70, Biolegend, Cat# 101212), PE anti-mouse Ly-6G/Ly-6C (Gr-1) (RB6-8C5, Biolegend, Cat# 108408), PE anti-mouse CD3 (17A2, Biolegend, Cat# 100206) and PerCP anti-mouse CD25 (PC61, Biolegend, Cat# 102028) at 4 °C for 1 h. For intracellular marker staining, the single-cell suspension was fixed and permeabilized using the Foxp3/Transcription Factor Fixation/Permeabilization kit (eBioscience), and then stained with PE anti-mouse FOXP3 antibody (MF-14, Biolegend, Cat# 126404) at 4 °C for 1 h. Cells were washed twice before flow cytometry analysis (BD LSRFortessa SORP). Data were collected using BD FACSDiva Software v8.0 and analyzed using BD FlowJo™ Software (v10.6.2).
To investigate which population of immune cells contributed to the therapeutic efficacy of NPsiPCSK9/cGAMP, 200 µg/dose anti-mouse CD4 (GK1.5, Selleck, Cat# A2101), anti-mouse CD8α (2.43, Selleck, Cat# A2102), anti-mouse NK1.1 (PK136, Selleck, Cat# A2114) and clodronate liposomes (LIPOSOMA, Cat# C-005) were intraperitoneally injected into MC38 tumor-bearing mice on day-1, 1, 3, 5, 7 post the first injection of NPsiPCSK9/cGAMP (2 injections in total at 3-day intervals) to specifically deplete endogenous CD4+ T cells, CD8+ T cells, NK cells, and macrophages, respectively. Batf3−/− mice were used to test the contribution of cDC1s in the therapeutic effect of NPsiPCSK9/cGAMP. Rat IgG2b (LTF-2, Selleck, Cat# A2116) was used as the isotype control for those in vivo depleting antibodies.
To measure antigen presentation on dendritic cells, female C57BL/6 mice of age 6–8 weeks (Vital River) were subcutaneously inoculated with 1.5 × 106 MC38-OVA tumor cells on the right flank on day 0. On day 10, tumor-bearing mice were intravenously injected with the indicated formulations. On day 12, tumor tissues were harvested and cut into small pieces, followed by dissociation using a digestion buffer (1 mg/ml collagenase and 100 μg/ml deoxyribonuclease I in serum-free RPMI) for 30 min at 37 °C with gentle shaking. The obtained suspension was passed through a 70-μm strainer to obtain the single-cell suspension. Then the cells were incubated with anti-CD45 (30-F11), anti-CD11c (N418), and anti-mouse SIINFEKL/H-2Kb antibody (25-D1.16) on ice for 20 min before flow cytometry (BD LSRFortessa SORP). Flow cytometric data were collected using BD FACSDiva Software v8.0.The frequency of antigen-specific CD8α+T cells in the tumor was analyzed following previously reported protocols. Briefly, female C57BL/6 mice of age 6–8 weeks (Vital River) were subcutaneously inoculated with 1.5 × 106 MC38-OVA tumor cells on the right flank on day 0. On day 10, tumor-bearing mice were intravenously injected with the indicated formulations. On day 16, animals were euthanized, and tumors were harvested and prepared into single-cell suspensions as described above. The cells were incubated with CD16/32 (1:20) for 10 min and then incubated with peptide-MHC tetramer (H-2Kb-restricted SIINFEKL) for 30 min at room temperature and stained with anti-CD45 (30-F11), anti-CD3 (17A2), and anti-CD8α (53-6.7) on ice for 20 min. Cells were washed twice with FACS buffer and resuspended in DAPI before flow cytometry.
Statistical analysis
All quantitative data are presented as mean ± standard deviation (SD). Statistical analyses were performed using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons post hoc test, unless otherwise specified. The n number indicates the number of independent experiments, animals, or cell samples, as defined in each figure legend. For all representative images (e.g., immunofluorescence or blots), the number of independent experiments with similar results is indicated in the legend. Assumptions of normality and homogeneity of variance were confirmed prior to statistical analysis. Exact p-values are provided in the figures, legends, or Source Data file. Independent experiments rather than technical replicates were used for all statistical analyses unless otherwise stated.
Ethics statement
All animal experiments were in accordance with and approved by the University Committee on Use and Care of Animals at Tsinghua University (Approved Animal Protocol 22-KR-1). Animals were euthanized when the tumor reached 15 mm in any dimension or when they became moribund with severe weight loss (defined as >20% body weight loss) or unhealing ulceration. This limit was not exceeded at any point.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that data supporting the findings of this study are available within the article, Supplementary, or Source data files. RNA sequencing datasets have been deposited to the NCBI-Sequence Read Archive (SRA) under accession code SRP607162. The link to this project is https://www.ncbi.nlm.nih.gov/sra/PRJNA1301533. Source data are provided with this paper.
Code availability
Protein structure predictions in this study were generated using AlphaFold2, which was run locally with standard structure databases and is freely accessible for non-commercial use (https://alphafold.ebi.ac.uk/). For protein complex modeling, predictions were performed using AlphaFold3 via the on-demand server(https://deepmind.google/science/alphafold/alphafold-server/).
References
Demaria, O. et al. Harnessing innate immunity in cancer therapy. Nature 574, 45–56 (2019).
Samson, N. & Ablasser, A. The cGAS-STING pathway and cancer. Nat. Cancer 3, 1452–1463 (2022).
Cao, L. L. & Kagan, J. C. Targeting innate immune pathways for cancer immunotherapy. Immunity 56, 2206–2217 (2023).
Chen, Q., Sun, L. J. & Chen, Z. J. J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149 (2016).
Chin, E. N., Sulpizio, A. & Lairson, L. L. Targeting STING to promote antitumor immunity. Trends Cell Biol. 33, 189–203 (2023).
Deng, L. F. et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852 (2014).
Woo, S. R. et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors (vol 41, pg 830, 2014). Immunity 42, 199–199 (2015).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Chin, E. N. et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science 369, 993–999 (2020).
Pan, B. S. et al. An orally available non-nucleotide STING agonist with antitumor activity. Science. https://doi.org/10.1126/science.aba6098 (2020).
Yu, J. et al. Single-dose physically cross-linked hyaluronic acid and lipid hybrid nanoparticles containing cyclic guanosine monophosphate-adenosine monophosphate eliminate established tumors. ACS Nano 18, 29942–29955 (2024).
Aval, L. M., Pease, J. E., Sharma, R. & Pinato, D. J. Challenges and opportunities in the clinical development of STING agonists for cancer immunotherapy. J. Clin. Med. https://doi.org/10.3390/jcm9103323 (2020).
Meric-Bernstam, F. et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J. Clin. Oncol. https://doi.org/10.1200/JCO.2019.37.15_suppl.2507 (2019).
Dane, E. L. et al. STING agonist delivery by tumour-penetrating PEG-lipid nanodiscs primes robust anticancer immunity. Nat. Mater. 21, 710–720 (2022).
York, A. G. et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell 163, 1716–1729 (2015).
Frank-Kamenetsky, M. et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. USA 105, 11915–11920 (2008).
Lancellotti, P. & Oury, C. A. Highly durable RNAi therapeutic inhibitor of PCSK9. N. Engl. J. Med. 376, e38 (2017).
Wong, C. C. et al. The cholesterol uptake regulator PCSK9 promotes and is a therapeutic target in APC/KRAS-mutant colorectal cancer. Nat. Commun. 13, 3971 (2022).
Yang, Q. C. et al. Targeting PCSK9 reduces cancer cell stemness and enhances antitumor immunity in head and neck cancer. iScience 26, 106916 (2023).
Sun, C. et al. Identification and validation of PCSK9 as a prognostic and immune-related influencing factor in tumorigenesis: a pan-cancer analysis. Front. Oncol. https://doi.org/10.3389/fonc.2023.1134063 (2023).
Bao, X. et al. Targeting proprotein convertase subtilisin/kexin type 9 (PCSK9): from bench to bedside. Signal Transduct. Target Ther. 9, 13 (2024).
Snipstad, K., Fenton, C. G., Kjaeve, J., Anderssen, E. & Paulssen, R. New specific molecular targets for radiochemotherapy in colorectal cancer. Febs J. 276, 121–121 (2009).
Lv, M. Z. et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. 30, 966–979 (2020).
Wang, C. G. et al. Manganese increases the sensitivity of the cGAS-STING pathway for double-stranded DNA and is required for the host defense against DNA viruses. Immunity 48, 675–687.e7 (2018).
Wang, H. Z. et al. IFN-β production by TLR4-stimulated innate immune cells is negatively regulated by GSK3-β. J. Immunol. 181, 6797–6802 (2008).
Dobbs, N. et al. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18, 157–168 (2015).
Ogawa, E., Mukai, K., Saito, K., Arai, H. & Taguchi, T. The binding of TBK1 to STING requires exocytic membrane traffic from the ER. Biochem. Bioph. Res. Co. 503, 138–145 (2018).
Jeltema, D., Abbott, K. & Yan, N. STING trafficking as a new dimension of immune signaling. J. Exp. Med. https://doi.org/10.1084/jem.20220990 (2023).
Srikanth, S. et al. The Ca(2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol. 20, 152–162 (2019).
Ishikawa, H. & Barber, G. N. Sting is an endoplasmic reticulum adaptor that facilitates innate immune signaling. Cytokine 48, 128–128 (2009).
Hopfner, K. P. & Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 21, 501–521 (2020).
Deng, Z. et al. A defect in COPI-mediated transport of STING causes immune dysregulation in COPA syndrome. J. Exp. Med. https://doi.org/10.1084/jem20201045 (2020).
Lepelley, A. et al. Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling. J. Exp. Med. https://doi.org/10.1084/jem.20200600 (2020).
Mukai, K. et al. Homeostatic regulation of STING by retrograde membrane traffic to the ER. Nat. Commun. 12, 61 (2021).
Shen, Y., Gu, H. M., Qin, S. & Zhang, D. W. Surf4, cargo trafficking, lipid metabolism, and therapeutic implications. J. Mol. Cell. Biol. https://doi.org/10.1093/jmcb/mjac063 (2023).
Yan, R., Chen, K., Wang, B. & Xu, K. SURF4-induced tubular ERGIC selectively expedites ER-to-Golgi transport. Dev. Cell 57, 512–525.e518 (2022).
Zanetti, G., Pahuja, K. B., Studer, S., Shim, S. & Schekman, R. COPII and the regulation of protein sorting in mammals. Nat. Cell Biol. 14, 20–28 (2011).
Emmer, B. T. et al. The cargo receptor SURF4 promotes the efficient cellular secretion of PCSK9. Elife. https://doi.org/10.7554/eLife.38839 (2018).
Brandizzi, F. & Barlowe, C. Organization of the ER-Golgi interface for membrane traffic control. Nat. Rev. Mol. Cell Biol. 14, 382–392 (2013).
Zhang, B. C. et al. Cholesterol-binding motifs in STING that control endoplasmic reticulum retention mediate anti-tumoral activity of cholesterol-lowering compounds. Nat. Commun. 15, 2760 (2024).
Luo, Z. J., Dai, Y. & Gao, H. L. Development and application of hyaluronic acid in tumor targeting drug delivery. Acta Pharm. Sin. B 9, 1099–1112 (2019).
Uhlen, M. et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 28, 1248–1250 (2010).
Liu, X. et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 588, 693–698 (2020).
Acknowledgements
The work was supported in part by grants from the National Natural Science Foundation of China (82173751, 32070875), the National Key Research and Development Program of China (2023YFC3403100), the National High-Level Young Talent Program, Tsinghua University Initiative Scientific Research Program (2024Z11DSZ001, 2025Z11DSZ001, and 2022Z11QYJ036), start-up packages from Tsinghua University, support from Tsinghua-Peking Center for Life Sciences, and support from the Key Laboratory of Innovative Drug Research and Evaluation. We thank Prof. Meng Xu (School of Medicine, Tsinghua University) for kindly providing the murine colon adenocarcinoma cells (MC38). We would like to acknowledge Bingyu Liu from the Imaging Core Facility, Technology Center for Protein Sciences, Tsinghua University, for her assistance with the Zeiss LSM980 laser scanning microscopy. We would like to acknowledge Weihua Wang at the Center of Pharmaceutical Technology, Tsinghua University, for her assistance with liquid chromatography-mass spectrometry (LC-MS) analysis. We would like to acknowledge Xiangjie Ge and Yanjie Li from the Cryo-EM Facility of the China National Center for Protein Sciences (Beijing), Tsinghua University, for their support on the FEI Tecnai Arctica TEM D683.
Author information
Authors and Affiliations
Contributions
P.S., F.H., C.Z., and R.K. designed the experiments. P.S., F.H., X.L., C.W., T.D., and J.H. performed the experiments. P.S., F.H., C.Z., and R.K. analyzed the data. All authors have given approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
A patent application (2023107003368) has been filed based on the findings described in the manuscript, with R.K., P.S., C.Z., and F.H. as inventors. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, P., Han, F., Li, X. et al. Silencing PCSK9 reshapes the spatiotemporal activation of STING for safe and effective cancer immunotherapy. Nat Commun 16, 11622 (2025). https://doi.org/10.1038/s41467-025-66630-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66630-x
