
Abstract
The hedgehog pathway is implicated in resistance to anticancer therapies in glioblastoma (GB). GEINO1602 (NCT03466450) phase Ib/II study evaluated the safety and efficacy of glasdegib and the Stupp scheme in newly diagnosed GB. Patients received glasdegib with radiotherapy plus concomitant and 6 cycles of adjuvant temozolomide followed by glasdegib monotherapy. The recommended phase 2 dose was 75 mg/day of glasdegib. The primary endpoint was the 15-months overall survival (OS), with a futility threshold of 60% to consider the trial positive; accrual required 70 evaluable patients. 79 patients were enrolled. The 15 m OS rate was 52.1% (95% CI: 41.7-65.2). At 2 years, 29.2% of the patients were still alive. The median progression-free survival (PFS) was 7.1 months (95% CI: 6.2-8.6). Glasdegib plus chemoradiotherapy show preliminary efficacy. Despite not surpassing the futility threshold, 30% lived at the data cutoff. Translational research will help define the molecular traits of long-term survivors.
Similar content being viewed by others
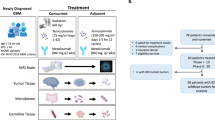


Introduction
Glioblastoma is the most common malignant primary brain tumour1. Maximal safe surgery followed by radiotherapy (RT) with concomitant and adjuvant temozolomide (TMZ) has shown benefits in survival and remains the standard of care for newly diagnosed glioblastoma2,3,4. However, the median overall survival (OS) with the standard chemoradiation scheme is ~15 months (m)2,4. Novel therapeutic approaches over the last decade, most of which are based on the RT-TMZ scheme, have failed to prolong survival. Antiangiogenics and immunotherapy showed limited efficacy in patients with glioblastoma5,6,7,8,9,10.
Given the disappointing results of these widely used treatments, new therapeutic strategies are needed. The Hedgehog (Hh) pathway has been identified as an important factor in the pathogenesis of gliomas11,12. Activation of the Hh pathway leads to glioma stem cell (GSCs) maintenance, one of the key hallmarks of glioblastoma therapy resistance to TMZ and radiation13,14,15,16,17. In addition, the inhibition of the Hh pathway may reverse temozolomide resistance14,18,19,20. Despite its relevant role and molecular rationale, no clinical trials have targeted Hh signalling in patients with glioblastoma.
Glasdegib is a potent small molecule inhibitor of Smoothened (SMO) under development in several cancer types and approved in combination with cytarabine for the treatment of patients with acute myeloid leukaemia21,22,23. Glasdegib, as a monotherapy or in combination with chemotherapy, was well tolerated21,22,23.
This clinical trial aimed to determine the recommended phase 2 dose (RP2D) of glasdegib administered in combination with standard temozolomide during and after RT and to evaluate the pharmacokinetics, pharmacodynamics, and preliminary efficacy of the combination in patients with newly diagnosed glioblastoma.
Results
Patient characteristics
From April 2018 to August 2020, 94 patients were screened, and 79 were enrolled. The first four patients were treated in Phase Ib with glasdegib 100 mg/day, and two (50%) of them experienced DLTs; one patient withdrew consent, and another had an increase in hepatic enzyme levels, which prevented the administration of 80% of the intended glasdegib dose (Fig. 1). Glasdegib was reduced to 75 mg/day for the next six patients in Phase Ib and only one patient (16.7%) experienced a DLT consisting of a grade 4 neutropenia and thrombocytopenia, declaring this as the RP2D for the expansion in Phase II (Primary endpoint phase Ib). In total, 74 patients received 75 mg/day glasdegib. The median age was 55 years (full range: 28-78) and 40 patients (54%) were male. Most patients underwent complete resection (48.6%). Only 8.1% of patients underwent biopsy. MGMT promoter was methylated in 32 patients (43.2%). One patient had an IDH mutation and was excluded from the post hoc sensitivity analysis, given the changes in the WHO 2022 classification (Supplementary Table 1).

Patient distribution showing the number of patients included in each phase of the trial and according to the glasdegib dose. n refers to patients.
Efficacy
With a median follow-up of 14.9 months (95% confidence interval [CI]: 13.7-19), the 15-month OS rate (Primary endpoint Phase II) was 52.1% (95% CI: 41.7-65.2), and the median OS (secondary endpoint) was 15.3 months (95% CI: 14-19.9) (Fig. 2a). The 24-month OS rate (Secondary endpoint) was 29.2% (95% CI: 20.2-42). At the data cutoff (February 2024), 16 patients (21.6%) remained alive, five of whom had an OS longer than 36 months, and 58 (78.4%) had died. The main cause of death was disease progression in 54 patients (72%).

a Kaplan-Meier graph showing overall survival in the treated population. b Kaplan-Meier graph showing overall survival stratified by MGMT promoter methylation status. A Cox hazard model (two sided) was used for statistical comparison between the subgroups (exploratory analysis). The shadowed areas represent the 95% CI. The dotted lines signal the 50% probability of the OS (c) Forest plot showing hazard ratios for death in the analysis of treatment effect in patient subgroups according to baseline characteristics using a Cox hazard model (two sided, exploratory). n indicates the number of patients with events over the total number of patients in each subgroup (N). Whiskers represent 95% CI.
Preclinical data suggest that treatment with Hedgehog inhibitors may be beneficial, irrespective of MGMT promoter methylation status. The median OS in patients with MGMT methylation was 22.9 months (95% CI: 16-not reached [NR]), with a 15-month OS rate of 70% (55.4-88.5) (Secondary endpoint) (Fig. 2b). The median OS in patients with MGMT unmethylated was 14.1 months (95% CI: 12-16), with a 15-month OS rate of 38.5% (95% CI: 25.9-57.3) (Secondary endpoint). Methylation of the MGMT promoter was a prognostic factor for survival (hazard ratio [HR]: 0.43, 95% CI: 0.25-0.76; p = 0.004) (Fig. 2c) (Secondary endpoint). Survival was also correlated with the extent of the surgical resection, as expected based on previous experience showing that achieving greater resection rates correlates with prognosis. Most patients had complete (48.6%) or subtotal (27%) resections. Patients who underwent partial or complete resection had better outcomes than those who underwent biopsy only (HR: 0.22, 95% CI: 0.09-0.56, p = 0.001) (Fig. 2c) (Secondary endpoint).
The median PFS was 7.1 months (95% CI: 6.2-8.6), with 15-month and 24-month PFS rates of 15.1% (95%CI: 8.6–26.4) and 10.6% (95% CI: 5.3–21.1), respectively (Fig. 3a) (Secondary endpoint). The median PFS in patients with MGMT methylation was 7.3 months (95% CI: 6.4–15.5) with a 15-month PFS rate of 29.6% (95% CI: 17–51.7) (Fig. 3b) (Secondary endpoint). The median PFS in patients with unmethylated MGMT was 7.3 months (95% CI: 4.9–9.1), with a 15-month PFS rate of 5.1% (95% CI: 1.3–19.8) (Fig. 3b) (Secondary endpoint). Three patients responded to treatment: one had a complete response, and two had partial responses (Table 1).

a Kaplan-Meier graph showing progression-free survival in the treated population. b Kaplan-Meier graph showing progression-free survival stratified by MGMT promoter methylation status. A Cox hazard model (two sided) was used for the statistical comparison between the subgroups (exploratory analysis). The shadowed areas represent the 95% CI. The dotted lines signal the 50% probability of the PFS. c Forest plot showing hazard ratios for progression or death in the analysis of treatment effect in patient subgroups according to baseline characteristics using a Cox hazard model (two sided, exploratory analysis). n indicates the number of patients with events over the total number of patients in each subgroup (N). Whiskers represent 95% CI.
In the post hoc analysis of patients with IDH wild type, the 15-month OS rate was 52.9% (95% CI: 42.4-66), and the median OS was 15.7 months (95% CI: 14-21.2) (Supplementary fig. 1a). The median PFS was 7.3 months (95% CI: 6.2-8.9) (Supplementary fig. 1b).
Most patients remained on stable or decreasing doses of corticosteroids throughout the study period. Among the 19 patients receiving corticosteroids at baseline, the corticosteroid dose was reduced in 14 (73.6%) throughout the study (Secondary endpoint). The cognition improved during treatment, reaching a statistically significant improvement in the Barthel index at the start of the maintenance phase compared to baseline (mean score 98.12 vs. 94.92; p = 0.03; n = 20) (Fig. 4) (Secondary endpoint).

a Box plots showing the evolution of corticosteroid dependency during the study period. b Barthel index throughout the study period. c Mini-mental index throughout the study period. The box plot shows the median and interquartile ranges 25–75% (box area) and the whiskers represent the 95% CI. The number of patients is depicted below each box. The asterisk indicates time points which showed significant differences from the baseline using the paired Wilcoxon test (two sided); for Barthel maintenance visit 1 (p = 0.03).
Safety
In total, 22 patients (29.7%) experienced grade ≥3 toxicities, the most common of which were thrombocytopenia (9.5%) and neutropenia (8.1%) (Fig. 5a) (Secondary endpoint). Most of these toxicities occurred during the concomitant and adjuvant phases. During maintenance, the only grade ≥3 toxicity was alopecia, which was reported in three patients (4.1%). The most common non-haematological toxicities of any grade were nausea and vomiting (37%), dysgeusia (22%), alopecia (18.9%), and muscle cramps (14.9%) (Secondary endpoint).

a Most frequent treatment-related adverse events (frequency >5%) in the safety population throughout the study period on the left in red (n = 74) and during the maintenance phase with glasdegib monotherapy on the right in yellow (n = 28). b Treatment duration in the overall population and in patients who discontinued treatment due to toxicity. Each dot represents a patient, and the blue areas along the x axis are proportional to the number of patients with that treatment duration. The gray box plot represents the median and 25–75% interquartile range. The horizontal lines represent 95% CIs. Data is shown for the full dataset for any reason (n = 74 patients) and for the subgroup of 13 patients who discontinued due to toxicity (n = 13). c Pharmacokinetic profile of glasdegib after a course of 14 days of oral administration of 75 mg/day glasdegib in concomitance with 5 mg/m2/day TMZ and 1.8-2 Gy/day RT. The blue line represents the median, and the gray bars represent the standard error for each time point (n = 8 patients).
The concurrent phase was completed by 72 patients (97.3%), and the six cycles of the adjuvant phase were completed by 33 patients (51.6%). Maintenance with glasdegib monotherapy was initiated in 28 patients (37.8%) (Secondary endpoint). The main reason for not continuing the scheduled scheme until maintenance was disease progression. The median duration of glasdegib treatment was 6.9 months (95% CI: 5.1–8.6) (Fig. 5b). AEs led to glasdegib dose delays in 22 patients (29.7%) and dose reductions in four patients (5.4%). Five patients (6.8%) completed the 2 years of treatment as scheduled, and 69 patients (93.2%) ended glasdegib prematurely at the data cutoff. The main reasons for premature end of treatment were disease progression (64.9%), toxicity (17.6%), withdrawal (4.1%), AEs not related to study treatment (2.7%), death (2.7%), and investigator decision (1.4%). Most glasdegib discontinuations due to toxicity occurred during the first 6 months of treatment (Fig. 5b).
Pharmacokinetics and pharmacodynamics
Glasdegib showed a fast metabolization, reaching a mean Cmax of 960 ng/mL after a mean tmax of 2 h (Fig. 5c). The mean area under the curve (AUC) of glasdegib over a 24-h time window was 16400 arbitrary units (AU) (Secondary endpoint).
Discussion
To our knowledge, GEINOGLAS was the first clinical study to evaluate the inhibition of the Hh pathway in patients with newly diagnosed glioblastoma. This combination was safe and achieved good maintenance of quality of life and baseline cognitive status. Glasdegib plus RT and TMZ showed a 15-m OS rate of 52.1% (95% CI: 41.7–65.2), over the null hypothesis estimated at 45%, but failed to meet its primary endpoint of 60%. Nevertheless, the median OS with glasdegib was in range with benchmark studies2,4, and approximately one-third of the sample showed OS over 2 years. Therefore, molecular analysis is necessary to answer this question. Moreover, caution is required when interpreting the results, as our study population may have a different prognosis than that of benchmark studies. Our sample included mainly patients with extensive surgical resections, so this factor was not expected to be determinant of the modest efficacy reported.
The toxicity profile was manageable and similar to that in previous reports. Most toxicities were attributable to concurrent chemotherapy. The administration of glasdegib was associated with a higher rate of alopecia, dysgeusia, and muscle cramps. Nevertheless, these events were mild and did not require special interventions for management.
The lack of a parallel control group represents a major limitation of this study, which is a common trait of most phase II trials in patients with glioblastoma. This precluded direct comparisons with current standard-of-care treatments, confining our analysis to indirect comparisons with findings from prior research. The main strengths of our trial are its prospective, multicentre nature, with an easily evaluable primary objective, such as OS.
The Hh pathway is implicated in the maintenance of stem/progenitor cells in many adult tissues24. Dysregulation of the Hh pathway is implicated in the pathogenesis of tumours. Medulloblastoma and basal cell carcinoma are notable examples of tumours that have been strongly linked to the aberrant activation of this pathway25. Three primary mechanisms have been postulated for the aberrant activation of the Hh signalling pathway in diverse cancer types: ligand-independent Hh signalling and ligand-dependent oncogenic Hh signalling operating within autocrine or paracrine mechanisms26. One of the key issues with this pathway is that GSCs have emerged as key drivers of both tumour initiation and progression. These cells, characterised by their slow-cycling nature and capacity for rapid proliferation, reside within tumours and function as stem cells27. There is also a strong rationale for the involvement of the Hh pathways in GBM. Preclinical studies have reported a significant increase in sonic hedgehog (SHH) levels in glioblastoma cells compared to normal tissue and have demonstrated its implications in migration, invasion, and angiogenesis28. Multiple studies have shown that the SHH signalling pathway promotes cancer cell plasticity by regulating cell-extracellular matrix interactions, enhancing cell motility and invasiveness, and ultimately contributing to poor patient prognosis. Furthermore, an analysis of the TCGA-Glioblastoma dataset (June 2016) revealed a significant inverse correlation between SHH expression and overall survival29. Emerging evidence strongly suggests that the aberrant activation of developmental signalling pathways, including NOTCH, WNT, and SHH, contributes significantly to tumour resistance and the maintenance of cancer stem cell phenotype30.
By increasing SHH/GLI1 expression, the acquisition of stem-like properties in GSCs may be induced, facilitating the formation and migration of glioblastoma cell spheres12. In addition, the role of HH/GLI1 signalling has been investigated in GB resistance to temozolomide. Silencing of GLI1 results in increased sensitivity to temozolomide and increases apoptosis in glioma stem cell-like cells, a process likely mediated by alterations in multidrug transporter regulation14,31. Other authors have found that activation of Hh-GLI1 signalling led to increased MGMT expression and therefore temozolomide resistance has been enhanced; in contrast, the inhibition of this pathway with cyclopamine decreased MGMT expression13. However, we did not observe a synergistic effect dependent on the MGMT methylation status.
We hypothesise that several factors may have contributed to the limited survival benefit observed. On the one hand, this pathway is interrelated with multiple other molecular pathways, which may interact with its activation. Among others, PI3K/AKT negatively regulates the degradation of GLI2, RAS/RAF/MEK, potentiates Gli1 activity, the ERK pathway positively modulates Hh-GLI signalling, MEK1 increases GLI1 and GLI2 transcriptional activity, crosstalk between epidermal growth factor receptor (EGFR) signalling and Hh-GLI pathway has also been reported, activation of Hh-GLI signalling due to direct induction of GLI1 expression is observed after activation of WNT/β-catenin signalling and TGF-β also induces Kindlin-2, which increases GLI1 protein32. Ptch1, which is upstream in the Hh pathway, could also be induced through Smo-independent pathways involving the transcription factor Meis133. The intricate network of parallel signalling cascades may have contributed to the limited efficacy of adding glasdegib as a single agent, and dual blockade or multikinase inhibitors might be a potential option to explore, as suggested for other tumours34. Other trials attempting to inhibit a single target of the Hh pathway in patients with gliomas have also failed to improve efficacy outcomes35,36.
In contrast, glasdegib acts on SMO, which is upstream in the pathway; inhibition at this location would leave the alterations that develop further down the pathway out of control. It is important to conduct a complete study of this pathway to understand the mechanisms that are altered in the Hh pathway in glioblastoma. We are currently conducting an exploratory biomarker study, including different pathway components (SHH, SMO, Gli0,1 and CD133), which will help identify potential routes or mechanisms of resistance to glasdegib.
Another concern is the ability of this drug to cross the blood-brain barrier. Preclinical rat models showed that glasdegib was capable of crossing the blood-brain barrier after single-dose exposure, despite its penetration being <3% of blood Cmax37. Serial administration may have achieved higher concentration rates; however, it is possible that the concentrations reached at the tumour site were insufficient to interfere with Hh signalling. Tumour PK/PD for glasdegib was not feasible because of the potential increased risk of bleeding. The PK profile of glasdegib at doses of 75 mg/day in combination with TMZ showed a slightly higher drug exposure than that described for doses of glasdegib at 100 mg/day as a single agent. This may suggest a potential interaction between TMZ and glasdegib. Both drugs may share metabolic pathways. During the phase Ib, the dose of glasdegib had to be reduced from 100 mg (which is considered the recommended dose) to 75 mg/day due to persistent hematologic toxicity in two patients which made it impossible to administer 80% of the planned dose. The use of a lower glasdegib dose and the potential limitations of crossing the blood-brain barrier may have limited the efficacy of this combination therapy. In line with this hypothesis, there is a trend towards better survival outcomes in patients with higher glasdegib treatment compliance.
In conclusion, the addition of glasdegib to standard RT and TMZ was safe with no increase in toxicity rates, maintenance of cognition, and symptom control. However, this combination only achieved a modest OS improvement, which was not statistically significant. Identifying biomarkers that allow the selection of patients prone to Hh signalling inhibition pathway inhibitors may enhance further research in this field.
Methods
Study design and treatment
The GEINOGLAS (GEINO-1602) trial was an academic, single-arm, open-label, multicenter study sponsored by the Grupo Español de Investigación en NeuroOncología (GEINO) and conducted in eight hospitals in Spain. The trial was registered in the European register (2017-002410-31) and Clinicaltrials.gov (NCT03466450). The trial was conducted in accordance with the principles of the Declaration of Helsinki and applicable regulations. The protocol was approved by the competent authorities, the ethics committee of the coordinating site (Comité de la Ética de la Investigación con Medicamentos del Hospital Ramón y Cajal; ref: 2017/334, Jan 2018), and all participating centres. All patients provided signed informed consent.
All patients received standard treatments. The concurrent phase consisted of RT at a total dose of 58-60 Gy in 6 weeks to 1.8-2.0 Gy per fraction/day (30 fractions) in combination with TMZ at 75 mg/m2/day for a maximum of 42 days. After a 4-week break, patients started TMZ for the first five days of every 28-day cycle for up to six cycles (150-200 mg/m2 day). Glasdegib was added to the standard treatment starting at the beginning of the concurrent phase and then continuously until progression or up to 2 years. Treatment may be terminated prematurely in the case of disease progression (PD), unacceptable toxicity, non-compliance, or consent withdrawal.
The trial had a Phase Ib, consisting of dose escalation following a 3 + 3 design to determine the safe dose of glasdegib, followed by a Phase II with an expansion cohort.
Patients
The study included patients ≥18 years of age with a histologically confirmed diagnosis of glioblastoma by biopsy or resection no more than 4-6 weeks before registration who were candidates for the combination of RT-TMZ as the first-line treatment (Stupp scheme). Patients with leptomeningeal or extracranial dissemination were excluded. The patient should have recovered from previous surgery for glioblastoma, had a good performance status according to the Eastern Cooperative Oncology Group (ECOG-PS) of 0-1, and had adequate organ and bone marrow function. Patients with gastrointestinal abnormalities that may impair drug intake, cardiovascular disease or arrhythmias, active infections, and cognitive impairment were not eligible. Women of childbearing potential were requested to have a negative pregnancy test and use adequate contraceptive measures during the study.
Endpoints
The primary endpoint for the Phase Ib part was the RP2D of glasdegib, defined as the dose for which < 33% of patients experienced dose-limiting toxicity (DLT). DLT was defined as any of the following occurring within 12 weeks after initiation of treatment: i) grade 4 neutropenia lasting for ≥7 days; ii) febrile neutropenia; iii) grade ≥3 neutropenic infection; iv) grade ≥3 thrombocytopenia with bleeding; v) grade 4 thrombocytopenia lasting for ≥7 days; vi) non-haematologic grade ≥3 adverse events (AE) that had been maximally treated; and vii) failure to deliver ≥80% of planned doses owing to treatment-related AEs.
For Phase II, the primary endpoint was the OS rate at 15 months after the start of the study treatment, defined as the estimated percentage of patients alive at 15 months by Kaplan–Meier. Secondary endpoints included median OS, antitumor activity including progression-free survival (PFS), and objective response rate (ORR) locally assessed by the investigators according to the Response Assessment in NeuroOncology (RANO) criteria38, safety, changes in neurological status and changes in the use of corticosteroids, and biomarker studies. Subgroup analyses were exploratory.
Assessments
Radiographic tumour assessments were performed using contrast-enhanced magnetic resonance imaging (MRI) at baseline, 4 weeks after completion of RT, and every 12 weeks thereafter until PD. AEs were assessed continuously from informed consent signature up to 30 days after the end of treatment according to National Cancer Institute Common Terminology Criteria for AEs (CTCAE) version 4.03. Corticosteroid use was recorded during each visit. Cognition was assessed using the Mini-Mental Test (MMS), and Barthel Index (BI) at baseline, weeks 1 and 5 of the concurrent phase, week 9 during the resting period, and monthly during the adjuvant and maintenance phases.
The O6-methylguanine-DNA methyl-transferase (MGMT) promoter methylation and isocitrate dehydrogenase (IDH) mutational status were assessed by the investigators according to local standard procedures and equipment. Formalin-fixed paraffin-embedded (FFPE) tumour tissue specimens were collected at baseline and shipped for central assessment of molecular biomarkers. Serial blood samples (pre-dose and post-dose at 0.5, 1, 2, 4, 6, and 24 h) were collected on day 14 of the concurrent phase, processed to plasma, and shipped (-80 °C) to the central laboratory to analyse the pharmacokinetics (PK) of glasdegib by liquid-liquid extraction followed by high-performance liquid chromatography (HPLC) with mass spectrophotometry (MS/MS) detection.
Statistical analysis
For the Phase II part of the trial, we hypothesised that the addition of glasdegib would improve the 15-m OS rate from 45% as described in benchmark studies2,4 to 60%, with an alpha error of 0.05 and a power 1-beta of 80%, 70 patients were required. Assuming a 7% dropout rate, a total of 75 patients at the RP2D were required.
Efficacy and safety endpoints were assessed in all patients who received at least one dose of the study treatment. Baseline characteristics were represented by descriptive statistics, frequencies, and percentages for categorical variables and median, full range, or 95% CIs for continuous variables. PFS and OS were estimated using the method, the log-rank and Cox test was used for comparisons between subgroups. Efficacy was assessed by subgroups based on relevant characteristics based on medical criteria, including sex, ECOG-PS, MGMT, surgical outcome, treatment compliance and corticosteroid use. Patients without events were censored at the last follow-up. All statistical analyses were performed with R (version 3.6.3 [2020-02-29] “Holding the Windsock,” The R Foundation for Statistical Computing, Vienna, Austria). Figures and tables were generated using RStudio (Version 1.2.5033 2009-2019 RStudio, Inc., Boston, MA, US).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Individual patient data are not available due to institutional review board constraints and data protection rights. Reasonable data requests (i.e. qualified researchers for non-commercial academic research purposes) should be directed to the corresponding author: mariaangeles.vaz@salud.madrid.org. Requests for data will be evaluated by the corresponding author in a timely manner (i.e. 1 month), and data may be shared after the institutional review board approves its transfer. The protocol and statistical analysis plan are provided in the Supplementary Information. Source data are provided with this paper.
References
Stupp, R., Brada, M., Bent, M. J., van den, Tonn, J.-C. & Pentheroudakis, G. High-grade glioma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 25, iii93–iii101 (2014).
Stupp, R. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 (2009).
Wen, P. Y. et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-Oncol. 22, 1073–1113 (2020).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
Chinot, O. L. et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 370, 709–722 (2014).
Gilbert, M. R. et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 370, 699–708 (2014).
Lassman, A. B. et al. Depatuxizumab mafodotin in EGFR-amplified newly diagnosed glioblastoma: a phase III randomized clinical trial. Neuro-Oncol. 25, 339–350 (2023).
Omuro, A. et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: an international randomized phase III trial. Neuro-Oncol. 25, 123–134 (2023).
Lim, M. et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro-Oncol. 24, 1935–1949 (2022).
Lassman, A. B. et al. Pl02.3.a nrg oncology study bn007: randomized phase ii/iii trial of ipilimiumab (ipi) plus nivolumab (nivo) vs. temozolomide (tmz) in mgmt-unmethylated (umgmt) newly diagnosed glioblastoma (ngbm). Neuro-Oncol. 25, ii2 (2023).
Kenney, A. M. & Rowitch, D. H. Sonic hedgehog Promotes G1 cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol. Cell. Biol. 20, 9055–9067 (2000).
Wang, H. et al. Hedgehog signaling regulates the development and treatment of glioblastoma. Oncol. Lett. 24, 294 (2022).
Wang, K. et al. Hedgehog/Gli1 signaling pathway regulates MGMT expression and chemoresistance to temozolomide in human glioblastoma. Cancer Cell Int. 17, 117 (2017).
Melamed, J. R. et al. Investigating the role of Hedgehog/GLI1 signaling in glioblastoma cell response to temozolomide. Oncotarget 9, 27000–27015 (2018).
Hung, H.-C., Liu, C.-C., Chuang, J.-Y., Su, C.-L. & Gean, P.-W. Inhibition of sonic hedgehog signaling suppresses glioma stem-like cells likely through inducing autophagic cell death. Front. Oncol. 10, 1233 (2020).
Biswas, N. K. et al. Variant allele frequency enrichment analysis in vitro reveals sonic hedgehog pathway to impede sustained temozolomide response in GBM. Sci. Rep. 5, 7915 (2015).
Zhang, L., Zhang, Y., Li, K. & Xue, S. Hedgehog signaling and the glioma-associated oncogene in cancer radioresistance. Front. Cell Dev. Biol. 11, 1257173 (2023).
Azzi, S. et al. Desert Hedgehog/Patch2 axis contributes to vascular permeability and angiogenesis in glioblastoma. Front. Pharmacol. 6, 281 (2015).
Torrisi, F. et al. Connexin 43 and sonic hedgehog pathway interplay in glioblastoma cell proliferation and migration. Biology 10, 767 (2021).
Wu, X. et al. A novel protein encoded by circular SMO RNA is essential for Hedgehog signaling activation and glioblastoma tumorigenicity. Genome Biol. 22, 33 (2021).
Sekeres, M. A. et al. Glasdegib plus intensive or non-intensive chemotherapy for untreated acute myeloid leukemia: results from the randomized, phase 3 BRIGHT AML 1019 trial. Leukemia 37, 2017–2026 (2023).
Heuser, M. et al. Clinical benefit of glasdegib plus low-dose cytarabine in patients with de novo and secondary acute myeloid leukemia: long-term analysis of a phase II randomized trial. Ann. Hematol. 100, 1181–1194 (2021).
Wagner, A. J. et al. A phase I study of PF-04449913, an oral hedgehog inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 21, 1044–1051 (2015).
Petrova, R. & Joyner, A. L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Dev. Camb. Engl. 141, 3445–3457 (2014).
Teglund, S. & Toftgård, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 1805, 181–208 (2010).
Skoda, A. M. et al. The role of the Hedgehog signaling pathway in cancer: a comprehensive review. Bosn. J. Basic Med. Sci. 18, 8–20 (2018).
Peacock, C. D. et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 104, 4048–4053 (2007).
Jiang, K. et al. Fms related tyrosine kinase 1 (Flt1) functions as an oncogene and regulates glioblastoma cell metastasis by regulating sonic hedgehog signaling. Am. J. Cancer Res. 7, 1164–1176 (2017).
Cherepanov, S. A., Cherepanova, K. I., Grinenko, N. F., Antonova, O. M. & Chekhonin, V. P. Effect of hedgehog signaling pathway activation on proliferation of high-grade gliomas. Bull. Exp. Biol. Med. 161, 674–678 (2016).
Takezaki, T. et al. Essential role of the Hedgehog signaling pathway in human glioma-initiating cells. Cancer Sci. 102, 1306–1312 (2011).
Munoz, J. L., Rodriguez-Cruz, V., Walker, N. D., Greco, S. J. & Rameshwar, P. Temozolomide resistance and tumor recurrence: Halting the Hedgehog. Cancer Cell Microenviron. 2, e747 (2015).
Pandolfi, S. & Stecca, B. Cooperative integration between HEDGEHOG-GLI signalling and other oncogenic pathways: implications for cancer therapy. Expert Rev. Mol. Med. 17, e5 (2015).
Busch, A. M. et al. All-trans-retinoic acid antagonizes the Hedgehog pathway by inducing patched. Cancer Biol. Ther. 15, 463–472 (2014).
Kast, R. E. IC Regimen: Delaying Resistance to Lorlatinib in ALK Driven Cancers by Adding Repurposed Itraconazole and Cilostazol. Cells 13, 1175 (2024).
Defer, G. L. et al. All-trans retinoic acid in relapsing malignant gliomas: clinical and radiological stabilization associated with the appearance of intratumoral calcifications. J. Neurooncol. 34, 169–177 (1997).
Phuphanich, S., Scott, C., Fischbach, A. J., Langer, C. & Yung, W. K. All-trans-retinoic acid: a phase II Radiation Therapy Oncology Group study (RTOG 91-13) in patients with recurrent malignant astrocytoma. J. Neurooncol. 34, 193–200 (1997).
Pfizer, Inc and Affiliates. PF-04449913 (Glasdegib) Investigator’s Brochure. (2022).
Wen, P. Y. et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J. Clin. Oncol. J. Am. Soc. Clin. Oncol. 28, 1963–1972 (2010).
Acknowledgements
The study was sponsored by Grupo Español de Investigación en NeuroOncología (GEINO). Funding for the research and glasdegib was provided by Pfizer under the Investigator-Initiated Research program. The funder had no involvement in the conception, design, conduct, or data analysis of this study. The authors thank all patients and families, investigators, and study staff involved in the GEINO-1602 study; the MFAR Clinical Research team, Emilio Pecharroman, M.Sc., for statistical analysis and Pau Doñate, Ph.D. for manuscript and language editing support.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.A.V-S. Formal analysis: M.A.V-S., S.d.B., J.M.S., M.A., E.P, C.B., M.M-G., R.G. Funding acquisition: M.A.V-S., S.d.B., J.M.S., M.A., E.P, C.B., M.M-G., R.G. Investigation: M.A.V-S., S.d.B., J.M.S., M.A., E.P, C.B., M.M-G., R.G. Methodology: M.A.V-S. Project administration: M.A.V-S. Resources: M.A.V-S., S.d.B., J.M.S., M.A., E.P, C.B., M.M-G., R.G. Supervision: M.A.V-S. Validation: M.A.V-S., S.d.B., J.M.S., M.A., E.P, C.B., M.M-G., R.G. Visualization: M.A.V-S., S.d.B., J.M.S., M.A., E.P, C.B., M.M-G., R.G. Writing – original draft: M.A.V-S., S.d.B., J.M.S., M.A., E.P, C.B., M.M-G., R.G. Writing – review & editing: M.A.V-S., S.d.B., J.M.S., M.A., E.P, C.B., M.M-G., R.G.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. MAV declares to receive a grant and act as a speaker for Pfizer, participate in advisory boards for Novocure, act as a speaker, receive congress fees from Pharmamar, participate in advisory boards, and receive congress fees from Servier. MM-G declares to participate in advisory boards for Novocure, act as speaker for Novocure, and receive congress fees from Pfizer. EP declares to participate in advisory boards for Novocure and Servier, and receive congress fees from Pfizer. All other co-authors declare no conflicts of interest regarding this study.
Peer review
Peer review information
Nature Communications thanks Alessandro Lussana, Raffaele Addeo and Anthony Chalmers for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vaz-Salgado, M.Á., del Barco, S., Sepúlveda, J.M. et al. Glasdegib in combination with temozolomide and radiotherapy in adult patients with newly diagnosed glioblastoma: the phase Ib/II GEINO 1602 trial. Nat Commun 17, 86 (2026). https://doi.org/10.1038/s41467-025-66747-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66747-z
