
Abstract
Fucosylation is a ubiquitous glycosylation event that shapes cellular communication and immunity. Catalyzed by fucosyltransferases (FUTs), this reaction encompasses diverse substrates, mechanisms, and biologic consequences. In this Review, we explore the structural and functional landscape of FUTs primarily from higher eukaryotes, with focus on the mechanistic determinants of regioselectivity, donor/acceptor coordination, and domain modularity. We highlight advances in structural biology, modeling, and enzyme engineering that clarify how FUTs decode glycan topology and specificity. Phylogenetic and structural analyses reveal two major clades of human FUTs that differ in GDP-Fuc recognition and conformational flexibility, providing a molecular rationale for their mechanistic divergence. Drawing from mammalian FUT studies, we propose a conceptual framework in which distinct family members exploit strategies including donor-induced conformational changes, exosite interactions, or local peptide cues to achieve specificity and catalytic efficiency. We also examine their roles in physiology, inflammation, immune regulation, and cancer, and summarize current FUT inhibitors and enzyme-based therapeutic strategies.
Similar content being viewed by others


Introduction
Glycosylation, one of the most structurally diverse post-translational modifications in nature, involves the covalent attachment of glycans to proteins, lipids, or other biomolecules, giving rise to a vast repertoire of glycoconjugates. In eukaryotic cells, it predominantly occurs in the endoplasmic reticulum (ER) and in the Golgi apparatus, although certain forms, such as O-GlcNAcylation, take place in the cytoplasm, and, more rarely, in prokaryotes1,2. Apart from its remarkable structural variety, glycosylation governs a wide array of essential biological processes, including cell–cell adhesion, signal transduction, immune modulation, molecular recognition, and the stabilization of structural proteins. It also fine-tunes critical physicochemical attributes such as solubility, thermal stability, and protease resistance, thereby shaping both protein behavior and functional fate3,4.
Among the various forms of glycosylation that regulate the structure and function of proteins and lipids, fucosylation has emerged as a particularly versatile and biologically impactful modification, implicated in critical processes such as embryonic development, immune regulation, host-pathogen interactions, and cancer progression5,6. This process involves the enzymatic incorporation of an L-fucose (Fuc) residue into proteins, glycolipids, or N- and O-linked glycan chains and is catalyzed by a family of fucosyltransferases (“FUTs”, also called “FTs”). These enzymes utilize guanosine diphosphate-fucose (GDP-Fuc) as a donor substrate and exhibit diverse catalytic specificities, installing Fuc in α1,2-, α1,3/4-, and α1,6-linkages onto glycan acceptors. In addition to these glycan modifications, some FUTs, such as protein O-fucosyltransferases (POFUTs) 1 and 2, and the recently described POFUT3 and POFUT4 (formerly known as FUT10 and FUT11, respectively) mediate direct O-fucosylation of serine and threonine residues within specific protein domains, including epidermal growth factor EGF-like repeats, thrombospondin type I repeats (TSRs), and elastin microfibril interface (EMI) domains, thereby expanding the functional landscape of fucosylation5,7. Dysregulation of fucosylation has been associated with a wide spectrum of pathological conditions, including cancer, infectious diseases, and congenital disorders of glycosylation, underscoring the biomedical relevance and therapeutic potential of FUTs5,8,9.
FUTs have been the subject of numerous studies highlighting their roles in development, immunity, and disease10,11. Others have focused on specific biological contexts or glycomic patterns, without dissecting the molecular principles that govern FUT specificity, catalysis, and inhibition1,5. Herein, we bridge this gap by providing a chemically grounded perspective on higher eukaryotic FUTs, emphasizing their classification, catalytic mechanisms, substrate specificities, structural diversity, regulatory features, and translational applications. We further examine how different FUTs accommodate chemically diverse acceptors within highly specialized catalytic pockets and how this structural understanding informs the rational development of selective inhibitors. While some inhibitors, such as glycomimetics11,12, directly exploit features of the active site, others like FDW028 (which targets the GDP-Fuc binding site in FUT8) have been identified through high-throughput screening approaches, offering alternative ligand-independent strategies to modulate FUT activity13.
Throughout this review, we summarize recent advances in the study of FUTs, encompassing their classification, biological roles, structural features, catalytic mechanisms, and evolutionary diversification in eukaryotes. We examine how subtle differences in substrate conformation, solvation, and active-site architecture shape FUT specificity and catalytic efficiency, and how these nuances are being elucidated through complementary experimental and computational techniques, including X-ray crystallography, molecular dynamics (MD), QM/MM simulations, and metadynamics. We also explore the implications of FUT dysregulation in major human diseases, surveying current strategies for FUT inhibition and highlighting emerging clinical applications. Particular emphasis is given to their translational potential, the persistent technical and biological challenges that hinder therapeutic development, and the opportunities they present as targets for precision medicine. By bringing together knowledge about FUT structure, how these enzymes work, and how they can be targeted in disease, this review provides a valuable resource for glycoscientists, chemists, and biomedical researchers working to understand and exploit FUTs for scientific and medical applications.
Classification and main characteristics of human FUTs
The human genome encodes thirteen FUTs, designated FUT1–FUT9 and POFUTs POFUT1–POFUT414,15, which are classified into four subfamilies according to the acceptor specificity, as well as the stereospecificity and regiospecificity of fucose installation catalyzed by these enzymes: (i) the α1,2-FUTs (FUT1, FUT2); (ii) the α1,3/4-FUTs (FUT3–FUT7, FUT9); (iii) the α1,6-FUT (FUT8); and (iv) the POFUTs (POFUT1–POFUT4). In all cases, eukaryotic FUTs operate with inversion of configuration at the anomeric center, transferring the fucosyl moiety from GDP-Fuc in its β-configuration to acceptors in the α-configuration, whether on glycans, glycolipids, or directly on protein domains.
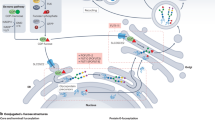
FUT1-FUT9 are localized within the Golgi apparatus, where they act on N- and O-linked glycans, modifying terminal sugar residues to fine-tune glycan structures. These FUTs catalyze the transfer of Fuc residues to form α1,2-, α1,3/4-, or α1,6- adducts on previously assembled glycan structures. Importantly, in the hominids (i.e., humans and the great apes (gorillas, chimpanzees, bonobos, and orangutans)), a cluster of fucosyltransferase genes exist on the short arm of chromosome 19 comprising the α1,3/4 FUTs known as “FUT3” and “FUT5” and the α1,3-FUT called “FUT6”; this genetic cluster arose from old world primate-specific gene duplications that result in considerable functional FUT redundancy, whereas expression of the α1,3-FUTs FUT4, FUT7, and FUT9 is a characteristic of all mammals and has more ancient origins16. In contrast to FUTs 1-9, POFUTs are localized in the ER, where they catalyze the transfer of Fuc moieties to serine or threonine residues within specific protein motifs17 (Fig. 1).

This figure illustrates the ER and Golgi compartments where fucosylation takes place. In the ER, the resident FUTs POFUT1, POFUT2, and POFUT3/4 catalyze the addition of Fuc to specific consensus motifs within certain domain structures: EGF-like domains, TSRs, and EMI domains, respectively. In the Golgi, FUTs mediate Fuc transfer with linkage specificity determined by enzyme subfamilies: FUT1 and FUT2 (α1,2-fucosylation) generate H-type epitopes; FUT3 and FUT5 (α1,3/4-fucosylation) produce various Lewis antigens including LeA, LeB, LeX, LeY, sLeA, and sLeX; FUT4, FUT6, FUT7, and FUT9 (α1,3-fucosylation) synthesize LeX, sLeX, and LeY; and FUT8 (α1,6-fucosylation) adds core Fuc to the chitobiose core of N-glycans. Symbols are used in accordance with the Symbol Nomenclature for Glycans (SNFG)293. The differential subcellular localization of FUTs, coupled with their stringent substrate specificity, constitutes a finely tuned regulatory system that minimizes enzymatic cross-reactivity. This spatial and functional compartmentalization enables precise modulation of the structure and function of fucosylated glycoconjugates across distinct cellular contexts. As a result, disruptions in the intracellular trafficking or localization of FUTs, despite preserved catalytic activity, can lead to aberrant glycosylation patterns with significant pathological consequences. Created in BioRender. Sanz Martínez, N. (2025) https://BioRender.com/pby9649.
The α1,2-FUTs, comprising FUT1 and FUT2, are members of the glycosyltransferase family GT11, as classified by the Carbohydrate-Active enZymes (CAZy) database15. These enzymes catalyze the formation of the glycan epitope called the “H antigen” by transferring Fuc in an α1,2-linkage to galactose located within terminal “Type 1” or “Type 2” lactosamine units (i.e., Gal-β1-3/4-GlcNAc, where “Gal” is galactose and “GlcNAc” is N-acetylglucosamine). The H antigen, when unmodified, defines blood type “O”, whereas further modification of the terminal Gal within the H antigen, either by addition of an N-acetylgalactosamine (GalNAc) or by addition of another Gal (in each case, in α1,3-linkage to Gal, yielding mutually exclusive products), creates the A and B blood group antigens, respectively. As such, FUTs 1 and 2 are critical determinants of cell surface and secretory antigenicity18. Though catalyzing the same chemical reaction, FUT1 and FUT2 differ markedly in their tissue-specific expression and glycan substrate preference: FUT1 is predominantly expressed in erythrocytes and vascular endothelial cells, with a strong affinity for type 2 terminal lactosamines (Gal-β1,4-GlcNAc-R); in contrast, FUT2 is highly expressed in epithelial tissues and exocrine secretions, where it preferentially acts on type 1 terminal lactosamines (Gal-β1,3-GlcNAc-R)19,20 (Fig. 1).
The α1,3/4-FUTs constitute a critical enzyme family involved in the biosynthesis of diverse glycoconjugates, particularly in the generation of Lewis antigens such as Lewis x, y, a, b (LeX (CD15), LeY (CD174), LeA, LeB) and sialyl Lewis X and A (sLeX (CD15s) and sLeA, respectively)21. These enzymes attach Fuc onto GlcNAc within lactosamine units and belong to the CAZy GT10 glycosyltransferase family. While most family members (i.e., FUT4, FUT6, FUT7, and FUT9) exclusively catalyze Fuc transfer onto GlcNAc within Type 2 lactosamine units via α1,3 linkages, FUT3 and FUT5 exhibit both α1,3- and α1,4-FUT activity on Type 2 and Type 1 lactosamines, respectively22 (Fig. 1). Thus, the FUTs FUT3, FUT4, FUT5, FUT6, FUT7, and FUT9 fucosylate Type 2 lactosamine units, but FUT3 and FUT5 can also fucosylate a Type 1 lactosamine unit.
Each FUT displays a unique tissue-specific expression pattern that correlates with distinct functional roles. FUT3 synthesizes LeA and LeB antigens, with its activity regulated epigenetically through DNA methylation22,23. FUT4, which has been implicated in mediating adhesive interactions in early embryonic development24,25, predominantly creates the trisaccharide LeX, but also contributes to sLeX biosynthesis in rodent (but not primate) leukocytes. FUT5 is a close homolog of FUT3 that displays lower enzymatic activity and a more restricted expression, particularly in the gastrointestinal tract and mammary glands26. FUT6 accounts for the bulk of plasma α1,3-FUT activity in humans27, and, as will be discussed below, is prominently expressed on liver and gastrointestinal cells and is a major driver of sLeX expression in malignant cells. On all mammalian leukocytes, FUT7 plays a dominant role in the biosynthesis of sLeX (NeuAc-α(2,3)-Gal-β(1,4)-[Fuc-α(1,3)]-GlcNAc-β1-R, where “NeuAc” is neuraminic acid (also called “sialic acid”)). This tetrasaccharide, along with its Type 1 lactosaminyl glycan isomer sLeA (NeuAc-α(2,3)-Gal-β(1,3)-[Fuc- α(1,4)]-GlcNAc- β1-R), are the prototypical binding determinants for the selectin family of adhesion molecules (a family of Ca2+-dependent lectins which includes E-selectin (CD62E), P-selectin (CD62P), and L selectin (CD62L))28. As such, FUT7 expression drives leukocyte tethering and rolling interactions on vascular endothelial cells that express E-selectin and/or P-selectin; this FUT7-dependent process results in leukocyte extravasation and, thereby, is a principal mediator of host defense/immune surveillance and of all inflammatory responses24. Lastly, FUT9 exclusively modifies “neutral” (i.e., unsialylated) Type 2 lactosamines to create the trisaccharide LeX. FUT9 is expressed in a variety of tissues, with especially high levels in the brain, where it dominantly governs LeX biosynthesis and may influence neural development and behavior29 (Fig. 1).
Beyond the fact that only the hominids express the fucosyltransferases FUT3, FUT5, and FUT6, there is another key difference among mammals that impacts the biology of fucosylated glycans as pertains to selectin receptor/ligand interactions. In all mammals, E-selectin is expressed uniquely on endothelial cells (hence “E”), and P-selectin is expressed both on platelets (hence, “P”) and on endothelial cells (for this reason, E- and P-selectin are called “vascular selectins”). With the exception of dermal and marrow microvessels that constitutively express E-selectin (and to a lesser extent P-selectin), under steady-state conditions, the vascular selectins are not expressed on any other microvessels. In all mammals with the exception of primates, expression of both E- and P-selectin is transcriptionally upregulated in post-capillary endothelial cells by inflammatory cytokines (principally by tumor necrosis factor (TNF) and by interleukin-1 (IL-1)), by microbial products (e.g., lipopolysaccharide (LPS)), by ischemia, and by trauma. However, in primates, the promoter elements within the P-selectin gene are unresponsive to transcriptional induction by inflammatory cytokines and by microbial products30. Indeed, whereas TNF strikingly induces P-selectin expression in murine endothelial cells, TNF conspicuously decreases P-selectin expression in human endothelial cells31. Therefore, in humans, E-selectin expression uniquely controls cell trafficking patterns, a crucial distinction that must be understood when attempting to extrapolate implications for human biology based on findings derived from rodent studies. Notably, because TNF is characteristically expressed at high levels within tumors, microvessels within human tumors and mouse tumors are typically laden with E-selectin, but P-selectin is not displayed on endothelial beds within human tumors.
FUT8 is unique in being the sole FUT in mammals capable of catalyzing the addition of a Fuc residue via an α1,6-linkage to the innermost GlcNAc moiety (i.e., within the “chitobiose core”) of N-glycans14,32. This modification, known as “core fucosylation”, represents a pivotal step in N-glycan maturation and profoundly influences the biological activity, stability, and receptor binding of diverse glycoproteins33 (Fig. 1). FUT8 belongs to the GT23 glycosyltransferase family and is ubiquitously expressed across mammalian tissues, with particularly high levels detected in the liver, kidney, brain, lung, and spleen34.
POFUTs’ activity is highly dependent on the structural context of the target domain and plays essential roles in cell development and cellular homeostasis. POFUT1 and POFUT2 act on distinct protein substrates: EGF-like domains and TSRs, respectively, which are recognized through the consensus sequences C2-X4-(S/T)-C3 for POFUT1 and C1,2-X2-(S/T)-C2,3 for POFUT2. These modifications are integral to key biological pathways such as Notch signaling and extracellular matrix (ECM) remodeling (Fig. 1)10,35,36. More recently, FUT10 and FUT11, now reclassified as POFUT3 and POFUT4, have been identified as novel POFUTs that specifically target EMI domains. Emerging evidence suggests that these enzymes contribute to the maintenance and regulation of embryonic and neural stem cell populations7 (Fig. 1).
Evolutionary divergence and phylogenetic architecture of FUTs
The extensive genomic information currently available enables the analysis of the FUTs phylogeny. In order to understand the FUTs evolutionary dynamics from prokaryotes to eukaryotes. we provide a phylogenetic tree (cladogram) with representative proteins of different kingdoms: archaea, bacteria, fungi, plants and metazoan (which includes mammals) (Supplementary Table 1). We verified the sequence composition of each protein so as to discard incomplete proteins. Two major distinct clades are distinguished in the phylogenetic cladogram (Fig. 2): Clade 1 with POFUT1, POFUT2, FUT1, FUT2 and FUT8; and Clade 2 with POFUT3, POFUT4, FUT3-7 and FUT9 (bootstrap > 95). The cladogram displays fundamental evolutionary divergence among FUTs, characterized by differences in GDP-fucose recognition and conformational flexibility. This divergence likely underpins distinct functional adaptations and substrate specificities across different taxa.

Phylogenetic cladogram including 114 sequences of Archaea (6), bacteria (34), fungi (14), viridiplantae (28) and metazoan (32) (Supplementary Table 1). The sequence profiles were globally aligned with ClustalOmega (https://www.ebi.ac.uk/Tools/msa/clustalo/) and trimmed following the protocol of the trimAL software294. A maximum likelihood phylogenetic tree using the Subtree Pruning and Regrafting (SPR) method was constructed with PhyML (https://ngphylogeny.fr295,296). The tree and cladogram were midpoint-rooted and plotted with FigTree (http://tree.bio.ed.ac.uk/software/figtree/). The approximate Likelihood-Ratio Test (aLRT) and bootstrap analyzes with a value of 100 and SH-like branch supports were performed.
In more detail, POFUTs form separated monophyletic subgroups (bootstrap > 93) in Clades 1 and 2, indicating they have evolved independently and form two independent subclasses: POFUT1-2 and POFUT3-4. Similarly, FUTs have evolved independently and form two separate subclasses: FUT1-2 and FUT8 (in Clade 1), and FUT3-7 and FUT9 (in Clade 2). In addition, two distant paraphyletic subgroups (bootstrap >95) appear in Clade 1 suggesting that subgroups POFUT1-2 and FUT8 diverged later than subgroup FUT1-2. Note that archaea and bacteria homologs do not cluster together with POFUT1-2 and FUT8. Interestingly, plants are only present in Clade 2, indicating they evolved independently of FUT1-2 and FUT8; and homologs of archaea appear with POFUT3 and POFUT4 in the same paraphyletic subgroup (bootstrap > 80), suggesting a common ancestor. They form a subgroup separated from FUT3-7 and FUT9. In addition, human FUT3, FUT5 and FUT6 appear in a separated subgroup from human FUT4, FUT7 and FUT9 in agreement with Dupuy et al.16.
This analysis emphasizes the significance of specific taxonomic distributions within each Clade, and extends our understanding of the evolution and diversification of FUTs in eukaryotes. This analysis also highlights the biological and phylogenetic relationships among FUTs.
Biological functions of fucosylation
Fucosylation exerts widespread influence across biological systems by modifying glycoconjugates involved in essential cellular processes. Its functional impact extends beyond structural decoration, playing active roles in the regulation of cell–cell communication, protein maturation and stability, and intracellular signal transduction. These effects are mediated through the specific activity of FUTs acting on distinct substrates within diverse cellular compartments37. In the following sections, we describe aspects of how fucosylation shapes biological function across three key dimensions. First, we will summarize the main FUT-dependent modulation of cell–cell interactions. Second, we will discuss the implications of FUTs in the regulation of protein folding and stability, and finally we will mention their major role as regulators of signaling pathways relevant to development, immunity, and disease.
Cell-Cell interactions
Fucosylation plays a pivotal role in regulating cell–cell interactions by modifying glycan structures that mediate recognition, adhesion, and communication processes across various physiological and pathological contexts, including immune responses, inflammation, embryonic development, and tumor progression38,39,40,41.
The α1,2-FUTs FUT1 and FUT2 contribute to the biosynthesis of key glycoconjugates such as the H antigen, LeY, and Globo H, structures known to modulate cellular adhesion and signaling42. Their expression is upregulated in response to inflammatory stimuli, enhancing the endothelial presentation of glycan ligands that facilitate leukocyte adhesion and extravasation43. In addition, these enzymes modify receptors such as nucleolin, thereby influencing endothelial cell adhesion and proliferation44. In the intestinal epithelium, FUT1 and FUT2 help shape dynamic fucosylation patterns that regulate epithelial plasticity and local immune responses45. Beyond immunological roles, FUT1 also contributes to the organization of axonal pathways in the developing olfactory system, indicating broader involvement in neural development46.
The α1,3/4-FUTs are critical mediators of selectin-dependent interactions through their role in the biosynthesis of sLeX and sLeA. The engagement of sLeA and/or sLeX on the surface of circulating cells with E-selectin on endothelial cells is the pivotal “Step 1” adhesive interaction (within the “multi-step model" of transendothelial migration) that enables extravasation/tissue colonization of blood-borne cells, a key process in host defense, in engraftment of hematopoietic stem cells within marrow following hematopoietic stem cell transplantation, and in cancer metastasis28. Suppression of FUT3 and FUT5 reduces sLeA expression and significantly impairs E-selectin-mediated adhesion in gastric cancer cells47. In addition to its role in adhesion, FUT5 has been implicated in sperm–oocyte binding, highlighting a role in fertilization26. FUT6 has been associated with enhanced cell proliferation, colony formation, and metastasis in various cancer cell types48,49,50,51. Indeed, serum levels of FUT6, which was originally called the “plasma fucosyltransferase”, was the first diagnostic blood biomarker identified for cancer52,53,54,55. Importantly, FUT6 is the most potent α1,3-FUT in the creation of sLeX21; therefore, this FUT has been utilized extensively to enable cell-based therapeutics by converting cell surface sialylated Type 2 lactosamines into sLeX, thereby engendering E-selectin ligands to program migration of vascularly administered cells to tissues whose vascular beds express E-selectin28,56,57,58.
FUT7 contributes to the biosynthesis of sLeX on glycoconjugates in endometrial cells, thereby facilitating embryo implantation59. Notably, FUT7 is the principal FUT expressed on leukocytes, and it plays a dominant role in leukocyte expression of sLeX on various glycoconjugates, thereby programming leukocyte trafficking to inflammatory sites60, and it also enables homing of hematopoietic stem cells to marrow61. In rodents, FUT4 cooperates with FUT7 in the creation of sLeX to enable leukocyte-endothelial cell adhesive interactions in inflammatory responses24,62. Their combined expression in rodent models has also been shown to promote tumor cell adhesion to brain endothelium, potentially affecting the integrity of the blood–brain barrier and contributing to metastatic progression63. Notably, though FUT9 cannot create sLeX, it can fucosylate “internal” GlcNAc moieties within a2,3-sialylated Type 2 polylactosamines (with putative preference for polylactosamines on glycolipids versus glycoproteins) and may thereby contribute to E-selectin ligand activity64, and it plays important roles in the nervous system by modulating cell adhesion, neuronal differentiation, and neurite outgrowth65.
FUT8 is essential for mediating cell–cell interactions through core fucosylation of glycoproteins. In epithelial cells, FUT8 stabilizes adhesion molecules such as E-cadherin, thereby enhancing intercellular cohesion. Overexpression of FUT8 in colon carcinoma models increases cell–cell adhesion, whereas loss of its catalytic activity disrupts this effect66. In the immune system, core fucosylation catalyzed by FUT8 sustains phosphorylation of the receptor tyrosine kinases EGFR and c-Met67, and is indispensable for the T-cell receptor to form an effective contact with peptide–Major Histocompatibility Complex class II (pMHC-II) at the immunological synapse with antigen-presenting cells68, thereby coordinating the cell-to-cell interactions that launch the adaptive immune response. In addition, FUT8 deficiency has been shown to enhance microglial and astrocytic activation under inflammatory conditions, underscoring its role in cell signaling and communication within the central nervous system69. FUT8 has been reported to drive metastasis and tissue invasion of melanoma70. Recent work has further demonstrated that FUT8 is upregulated in high-grade and metastatic prostate tumors, where it drives cell proliferation, motility, and invasion through transcriptional and signaling rewiring; importantly, inhibition of fucosylation using small-molecule inhibitors effectively suppresses FUT8 activity and tumor growth in preclinical models71. Together, these findings reinforce the centrality of FUT8-mediated core fucosylation in both physiological adhesion and pathological progression.
The POFUTs regulate cell-cell communication by catalyzing the direct fucosylation of specific protein domains. POFUT1 modifies EGF-like domains of the Notch receptor, a modification essential for Notch signaling during embryonic development and cellular differentiation72,73,74. POFUT2 targets TSR repeats on proteins such as thrombospondins and ADAMTS family members, contributing to the regulation of cell adhesion, ECM interactions, and tissue architecture75. POFUT3 and POFUT4 have been identified as FUTs that specifically modify EMI domains. While their precise contributions to cell–cell communication remain under active investigation, emerging evidence suggests roles in the regulation of ECM dynamics and stem cell signaling7.
In summary, fucosylation is a critical regulator of cell-cell interactions, influencing a wide spectrum of physiological and pathological processes through the precise modification of glycan structures. The coordinated activity of various FUTs enables fine-tuning of adhesion, communication, immune response, and tissue development. By modifying key glycoproteins and signaling receptors, these enzymes not only shape cellular behavior but also maintain the structural and functional integrity of tissues. Understanding the complexity and specificity of fucosylation in this context underscores its importance as both a biological modulator and a potential therapeutic target in disease settings.
Protein stability and folding
Beyond its role in mediating cell–cell interactions, fucosylation plays a critical role in the folding, stability, and quality control of glycoproteins, particularly within the ER. Core fucosylation, catalyzed by FUT8, is essential for the proper processing, trafficking, and structural integrity of many glycoproteins. In several cancer types, core fucosylation of membrane-associated proteins has been shown to promote immune evasion and tumor progression by stabilizing glycoprotein structure and simultaneously reshaping downstream signaling cascades76,77.
POFUT1 and POFUT2 function as conformation-sensitive enzymes within the ER, modifying only properly folded substrates. The modification of EGF-like domains by POFUT1 contributes to the maturation and stabilizing the structure of Notch78,79. The catalytic activity of POFUT2 promotes correct folding, structural stability, and efficient secretion of thrombospondins and ADAMTS family proteins80,81,82,83. More recently, POFUT3 and POFUT4 have been implicated in the stabilization of additional glycoproteins. Their deletion leads to reduced expression of multimerin-1 (MMRN1), an adhesive, and multimeric protein primarily involved in blood coagulation, and other members of the EMI Domain ENdowed (EDEN) protein superfamily, suggesting that O-fucosylation extends beyond classical EGF and TSR domains7.
While α1,2- and α1,3/4-FUTs have not been directly linked to protein folding or stability in humans, studies in other organisms indicate that these modifications can modulate glycoprotein half-life and structural integrity84. Furthermore, engineered modulation of α1,3/4-fucosylation has been applied to enhance the stability and reduce the immunogenicity of recombinant proteins in biotechnology applications85.
Together, these insights reveal how fucosylation contributes not only to glycoprotein maturation but also to the fine-tuning of their functional lifespan and secretion, linking this modification to broader mechanisms of cellular quality control and adaptation across physiological and pathological states.
Signal transduction
Fucosylation serves as a crucial regulatory mechanism in cell signaling, directly influencing the maturation, activation, and functionality of membrane-bound receptors and their interactions with ligands. By altering glycan structures, fucosylation affects receptor dimerization, ligand-binding affinity, and the efficiency of signal transduction. These changes, in turn, shape fundamental cellular processes such as proliferation, differentiation, migration, and plasticity.
Among the α1,2-FUTs, FUT1 and FUT2 are key modulators of major signaling cascades, including the epidermal growth factor receptor/mitogen-activated protein kinase (EGFR/MAPK) and the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) pathways, in both normal and pathological contexts. FUT1 is notably overexpressed in multidrug-resistant chronic myeloid leukemia, where its silencing leads to reduced EGFR activation and downregulation of the resistance-associated glycoprotein P-gp86. In breast and squamous cell carcinomas, FUT1 inhibition diminishes EGFR and ERK1/2 phosphorylation, thereby impairing mitogenic signaling. Both FUT1 and FUT2 have also been implicated in promoting cell migration and the acquisition of stem-like phenotypes in tumors through the activation of PI3K/Akt pathways42.
The α1,3/4-FUTs constitute another class of potent signaling regulators, particularly in cancer, immunity, and development. These enzymes modify glycan architectures on surface receptors, altering signal strength and duration across pathways such as Ras/ERK, PI3K/Akt, and Wnt, thereby fostering invasive behavior and stemness in tumor cells50. For instance, FUT3 enhances TGF-β signaling in pancreatic cancer, driving epithelial–mesenchymal transition (EMT) and metastatic progression87, reprograms cellular metabolism via NF-κB in lung cancer88, and contributes to immune escape through TRAIL resistance89. FUT5 plays a significant role in cellular signaling and cancer progression. It promotes PI3K/Akt pathway activation in colorectal cancer90 and is crucial for glycosylating key proteins like versican and β3-integrin in intrahepatic cholangiocarcinoma cells91. Similarly, FUT6 has been implicated in TGF-β–driven EMT in colorectal cancer92.
FUT4 and FUT7 function cooperatively to regulate both glycan-dependent adhesion and intracellular signaling, particularly in inflammatory and metastatic settings. These enzymes contribute to the biosynthesis of selectin ligands and modulate PI3K/Akt and ERK signaling pathways. Notably, FUT4 can activate Ras/ERK signaling independently of classical oncogenic mutations, suggesting a non-genetic mechanism of pathway activation in cancer24,93. FUT7, in addition to its role in leukocyte trafficking, enhances insulin receptor signaling in hepatocellular carcinoma94 and facilitates embryo implantation through modulation of adhesion and signaling at the maternal–fetal interface59.
FUT9 is also involved in both immunity and neurobiology: it is the dominant FUT in the creation of CD15 (LeX) in leukocytes95 and it suppresses Notch signaling in neurons, thereby promoting neuronal differentiation and post-injury regeneration96.
FUT8 plays a critical role in proper signal transduction by modulating the activity of multiple cell surface receptors, including EGFR, c-Met, the TGF-β1 receptor, and integrins. This occurs by influencing their ligand binding and, in some cases, receptor complex formation, which impacts downstream signaling cascades such as MAPK/ERK and SMAD97,98,99,100,101. In the central nervous system, FUT8 deficiency impairs long-term potentiation (LTP), synaptic plasticity, and learning102.
POFUTs also play central roles in intracellular signaling. POFUT1 is indispensable for the activation of the Notch receptor; its loss prevents receptor cleavage and signaling, disrupting neuronal differentiation, angiogenesis, and cardiac development73,78,103,104,105,106. POFUT1 additionally supports urokinase (uPA) pathway activity by enhancing uPA-uPAR interactions, thereby influencing vascular remodeling and embryo implantation107. POFUT2, by modifying TSR domains in proteins such as thrombospondins, ADAMTS family proteases, and CCN proteins, regulates TGF-β and Wnt signaling, ECM organization, and bone development36,108. In addition, POFUT2 modulates neuronal development by adding O-fucose to the TSR3 domain of BAI1 (ADGRB1), which directly interacts with the RTN4 receptor109.
Taken together, the data indicate that FUTs are emerging as key orchestrators of diverse signaling pathways. By modulating the glycosylation of receptors, cofactors, and ECM components, these enzymes influence signaling dynamics across physiological contexts, and FUT dysregulation contributes to the pathogenesis of cancer, inflammation, and developmental disorders, which are discussed in more detail below.
Structural properties of FUTs
A detailed understanding of the structural properties of FUTs is essential for elucidating the mechanisms that underlie their substrate specificity, catalytic activity, and regulation. Despite their functional diversity, these enzymes share core architectural features that place them within well-defined structural families, while also exhibiting unique adaptations that enable their biological specialization.
All biochemically characterized FUTs adopt the canonical GT-B fold, comprising two opposing Rossmann-like domains, an N-terminal acceptor-binding domain and a C-terminal GDP-Fuc-binding domain, separated by a central cleft (Fig. 3a). Crystal structures, peptide-sequence analysis, augmented by predictive models generated with Protenix110, reveals two distinct modes of GDP-Fuc recognition that coincide with phylogenetic clustering: Clade 1 enzymes (FUT1, FUT2, FUT8, POFUT1 and POFUT2) employ one binding strategy, whereas Clade 2 enzymes (FUT3–7, FUT9, POFUT3 and POFUT4) utilize an alternative mechanism. Notably, unlike many other glycosyltransferases (GTs) that adopt a GT-A fold and require divalent metal cofactors for catalysis37, FUTs operate independently of metal ions and instead rely on a network of strategically positioned amino acid side chains within the active site to mediate substrate recognition and drive glycosidic bond formation. Nevertheless, divalent cations can modulate FUT activity in vitro in a isoenzyme-specific manner: Mn²⁺ markedly enhances the catalytic activity of α1,3-FUTs (FUT3, FUT4, FUT5, FUT6, FUT7) but inhibits FUT9111; high MnCl₂ also increases human POFUT2 activity, with modest effects on the C. elegans ortholog112. Although the structural basis of this differential sensitivity remains unresolved, current evidence supports a view that these outcomes might arise from isoenzyme-specific electrostatic forces and conformational dynamics rather than a conserved metal-binding requirement. Thus, “metal independence” denotes absence of an obligatory divalent metal cofactor, not insensitivity to divalent metals; nevertheless, most notably, in many cases where the α1,3-FUTs FUT3-FUT7 have been isolated from cells and/or produced as recombinant proteins, the pertinent protein has been found to display little to no catalytic activity in the absence of divalent metal cofactors. Within Clade 1, the C-terminal Rossmann-like domain maintains a highly conserved secondary-structure topology despite its low primary-sequence identity (Fig. 3b, c). Adjacent to a conserved β-sheet, the invariant H-X-R-X₂-D motif establishes an extensive hydrogen-bonding network with the GDP-Fuc ligand, while a universally retained Asp side chain mediates polar contacts with the guanine base. Complementary Ser/Thr/Gln residues further stabilize the enzyme–substrate complex by engaging the pyrophosphate moiety (Fig. 3b, c).

a Superposition of the catalytic domains from human FUT8 (PDB: 6TKV), FUT9 (PDB: 8D0Q), POFUT1 (PDB: 5UXH), and POFUT2 (PDB: 4AP6) reveals a conserved GT-B fold shared by all structurally characterized FUTs. b Secondary-structure conservation mapped onto the FUT8 catalytic domain for clade 1 FUTs. The heat map gradient (blue → red) reflects the degree of structural conservation, using FUT8 as the reference. To the right, a close-up view of the GDP-Fuc binding site highlights fully conserved and functionally analogous residues. c PROMALS3D-derived alignment297 of the C-terminal catalytic domains from human FUT1, FUT2, FUT8, POFUT1 and POFUT2 (UniProt IDs P19526, Q9BYC5, Q9H488, Q10981 and Q9Y2G5, respectively). Conserved secondary-structure elements are annotated as helices (h) or β-strands (e), and consensus positions are classified by residue properties. Fully conserved residues are indicated with black arrows, and residues with functional equivalence are marked with blue arrows. In the alignment, amino acid residues are color-coded by predicted secondary structure: red, α-helices; blue, β-strands; green, coil/loop regions. Consensus positions are classified by residue character: aliphatic (l), aromatic (@), hydrophobic (h), alcohol (o), polar (p), tiny (t), small (s), bulky (b), positively charged (+), negatively charged (–) and charged (c). Heat maps integrate experimental crystal structures when available and rely on predictive models otherwise. GDP-Fuc is rendered opaque when positioned from crystallographic data and semi-transparent when model-derived. For clarity of visualization, side chains located behind the ligand are displayed with partial transparency to avoid occlusion.
By contrast, Clade 2 FUTs display a distinct yet likewise conserved secondary-structure topology in their C-terminal domains (Fig. 4a, b). In all homology models, an α-helix from the C-terminal region extends into the N-terminal Rossmann fold, recapitulating the interdomain packing observed in the human FUT9 crystal structure111 (Fig. 4a, b). These enzymes share an invariant E-N-X₅-Y-X-T-E-K-X3-(N/R) motif, which forms an extensive hydrogen-bonding network with every region of the GDP-Fuc donor, thereby stabilizing its binding. Additional fully conserved interactions include a Ser side chain and the main-chain of a Val or Leu with the guanine base, besides an Arg contacting the pyrophosphate group. In some family members, non-conserved Arg, Gln, or Asn residues adopt analogous roles in further stabilizing the donor substrate (Fig. 4a, b).

a Secondary-structure conservation heat map for clade 2 FUTs, mapped onto the FUT9 reference structure, with an inset showing conserved contacts at the GDP-Fuc binding pocket. b PROMALS3D alignment of the C-terminal catalytic domains from human FUT3, FUT4, FUT5, FUT6, FUT7, FUT9, POFUT3 and POFUT4 (UniProt IDs P21217, P22083, Q11128, P51993, Q11130, Q9Y231, Q6P4F1 and Q495W5, respectively). Annotation of secondary-structure elements, consensus positions, and residue coloring follows the same PROMALS3D scheme described in Fig. 3. The region of high conservation across all clade 2 FUTs are specifically highlighted in yellow. Heat maps integrate experimental crystal structures when available and rely on predictive models otherwise. GDP-Fuc is rendered opaque when positioned from crystallographic data and semi-transparent when model-derived. Methodological details regarding the integration of experimental structures with predictive models, the rendering of GDP-Fuc, and the use of transparency for side chains are described in Fig. 3.
Despite the overarching conservation shared by the two principal FUT clades, several members have diverged to accommodate structurally distinct acceptor substrates, leading to the emergence of enzyme-specific adaptations that fine-tune catalytic architecture and reactivity. Biochemical characterization of representative subtypes has uncovered unique mechanistic features and substrate preferences that illuminate the functional breadth of the family, yet significant gaps persist in our understanding of the precise catalytic logic and selectivity rules governing many FUT variants.
α1,2-FUTs
Although high-resolution structures of human FUT1 and FUT2 remain unresolved, structural insights have been gleaned from homologous enzymes such as the α1,2-FUT from Crassostrea gigas (CgFUT2). Like other members of the FUT family, CgFUT2 adopts the canonical GT-B fold, and its α1,2-linkage specificity is governed by a well-defined catalytic cavity that accommodates terminal galactose residues on type II glycan substrates113.
α1,3/4-FUTs
Among the human α1,3-FUTs, only FUT9 has been structurally characterized by X-ray crystallography. Structural analysis reveals that its specificity for non-sialylated (i.e., “neutral”) LacNAc arises from a distinctive surface cavity, sculpted by four flexible loops (residues 69–81, 136–153, 157–169, and 326–330), which collectively shield the catalytic site from accommodating sialylated modifications. Key residues involved in acceptor recognition include Phe73, Glu137, Tyr168, and Trp330, which are conserved across all α1,3/4-FUT isoenzymes. His141 is also conserved, but only among α1,3-specific enzymes; in bifunctional α1,3/4-FUTs, this position is instead occupied by an Asn residue, potentially contributing to differences in acceptor specificity. Additional residues (Gln75, Leu136, and Phe329) are uniquely present in FUT9 and may represent determinants of FUT9’s distinct acceptor recognition profile. All of these residues contribute to substrate stabilization through a combination of hydrophobic packing and hydrogen bonding. Among them, Glu137 is likely to act as the catalytic base, while the GDP pyrophosphate moiety adopts a distorted conformation that may lower the activation barrier for Fuc transfer (Fig. 5a). Notably, a rare cis-peptide bond at Phe329 contributes to donor substrate stabilization, highlighting a structural adaptation that may fine-tune catalytic precision111.

a Superposition of the Michaelis complex (MC) of FUT9 (derived from the crystal structure, PDB: 8D0Q, shown in opaque red) with computational models of its α1,3/4-FUT isoenzymes (FUT3, cyan; FUT4, yellow; FUT5, hot pink; FUT6, green; FUT7, slate, all shown with transparency). Conserved disulfide bonds (top left) and the glycosylation site common to all FUT-isoenzymes (bottom left) are highlighted. (right panel) Key residues involved in acceptor recognition are shown, distinguishing between those conserved across FUT-isoenzymes and those specific to FUT9. b Representation of the MC of FUT8 (derived from the crystal structure, PDB: 6TKV), displaying the N-terminal coiled-coil domain (dark salmon), the catalytic domain (lime green), and the C-terminal SH3 domain (orange). Interdomain linkers are shown in navy, light blue, and the C-terminal loop in blue. (left panel) The conformational change of the mobile loops (loop 1 in red and loop 2 in purple) is shown in comparison to the apoenzyme (APO) form (PDB: 6DE0, pink). (right panel) Key interactions involved in substrate recognition are depicted. c Model of POFUT1 (violet), assembled from crystallographic structures (PDBs: 5UXH and 5KXH), with disordered loops in the binary complex highlighted in yellow. (right panel) The three-dimensional structure of the EGF domain (orange) is shown, featuring the fucosylation motif (gray), the three disulfide bonds, and the interaction interface with the enzyme (polar regions in blue and apolar regions in brown). The position of the acceptor Ser/Thr residue and the Asn involved in the proton shuttle are also indicated. d Model of POFUT2 (brick red), derived from crystallographic structures (PDBs: 4AP6 and 5FOE), highlighting its unique structural loop (yellow). On the right, the characteristic disulfide bonds of the TSR domain (lime) are shown, as well as the fucosylation motif (gray), the acceptor Ser/Thr residue, and the catalytic residue Glu54 (represented as sticks). Water molecules observed in the crystal structure (5FOE), displayed as sticks and in DOT surface format, reveal the hydrogen-bonding network that mediates substrate recognition.
Post-translational modifications appear to play a critical role in modulating FUT9 enzymatic activity. FUT9 contains three predicted N-glycosylation sites (Asn62, Asn101, and Asn153), among which Asn153 is essential for function. The N153Q mutation results in a greater than 95% reduction in catalytic activity, while simultaneous mutation of all three glycosylation sites leads to a near-complete loss of function111,114. Remarkably, Asn153 is also the only site conserved across the α1,3/4-FUT family, suggesting this position could represent a shared functional determinant within the broader enzyme family (Fig. 5a).
All α1,3/4-FUTs share four conserved cysteine residues, which, based on the crystal structure of FUT9, form two disulfide bonds anchoring a C-terminal helical segment into the catalytic N-terminal Rossmann fold (Fig. 5a). This interdomain linkage appears to play a crucial role in maintaining the overall structural integrity of the enzyme family. In addition, FUT9 and FUT7 possess an additional disulfide bond that may further stabilize key regions within the catalytic domain111,115.
Although high-resolution structures are not yet available for other α1,3-FUTs, comparative mutagenesis has shed light on key residues governing their enzymatic behavior. For instance, FUT3 D336A drastically lowers affinity and activity on H-type 1 (α1-4) without increasing α1-3 fucosylation. The aromatic residue Trp111, part of the conserved V-X2-H-H-W-(D/E) motif in all α1,4-FUTs, is thought to be critical for dual α1,3/α1,4 activity (Fig. 5a). Substituting this Trp with an Arg, as seen in strictly α1,3-specific enzymes, shifts FUT3 activity toward α1,3-restricted fucosylation116.
α1,6-FUTs
FUT8 exhibits a unique modular architecture among GTs. In addition to its canonical GT-B catalytic domain (residues 204–491), it incorporates an N-terminal α-helical domain (residues 109–173) and a C-terminal Src homology 3 (SH3)-like domain (residues 504–562), both absent in related enzyme families32,117. The stem region (residues ~ 30–108) is essential for enzyme oligomerization within the Golgi, although its deletion does not impair intrinsic catalytic activity118. In contrast, the α-helical and SH3 domains act cooperatively to promote stable multimer formation and enhance enzymatic performance (Fig. 5b)117.
The catalytic mechanism of FUT8 is further refined by a conformational switch that occurs upon GDP-Fuc binding. This ligand-induced transition involves coordinated movement of two flexible elements, loop 1 (residues 428–444, encompassing helix α10) and loop 2 (residues 365–378), which reposition the catalytic residues and reshape the donor-binding pocket. This dynamic rearrangement aligns Glu373 to function as the catalytic base and facilitates precise orientation of both donor and acceptor for efficient glycosidic bond formation (Fig. 5b)14,119,120.
The substrate-recognition interactions are in part mediated by the core-chitobiose GlcNAc moieties of the acceptor N-glycan, which is positioned for fucosylation and engages key residues such as Glu373 (the catalytic base), Lys216 and Asp295. In contrast, the α1,6 arm of the biantennary N-glycan is only modestly recognized, forming a few contacts with Gly501 and Gln502. By comparison, the α1,3-arm is extensively engaged via an auxiliary exosite, composed of the β10–β11 loop and part of the SH3 domain, which directly contacts the α1,3-branch of biantennary N-glycans through Asp494, Asp495, Val531 and His535. This exosite interaction provides the molecular basis for FUT8’s strict preference for complex and hybrid N-glycan structures over high-mannose counterparts (Fig. 5b)14,119. Consistently, recent biochemical studies showed that FUT8 preferentially fucosylates complex N-glycans, and that catalytic efficiency is governed not only by GDP-Fuc–induced conformational rearrangements, but also by the accessibility of the innermost GlcNAc and the local peptide/protein environment, rather than the glycan type alone121.
POFUTs
POFUT1–4 constitute a structurally conserved family of enzymes that share the canonical GT-B fold, which features two Rossmann-like domains forming a cleft that accommodates both the GDP-Fuc donor and the folded peptide acceptor7,122,123. In contrast to most GTs, POFUTs do not act on linear peptide sequences but instead require fully folded protein domains as substrates, enabling them to function as conformation-sensitive regulators during protein maturation in the ER10.
POFUT1 selectively modifies EGF-like domains that harbor the conserved C²-X₄-(S/T)-C³ fucosylation motif, where C² and C³ correspond to the second and third cysteine residues within the canonical six-cysteine framework that forms three disulfide bonds (Fig. 5c). In Mus musculus, substrate binding to POFUT1 induces ordering of previously disordered regions (residues 79–87, 255–267, and 275–281), enhancing shape complementarity with the EGF domain. In the human enzyme, the homologous loop (residues 74–82) is unresolved in the GDP-bound crystal structure, suggesting intrinsic flexibility. Given the high sequence and structural conservation, similar substrate-induced rearrangements are likely to occur in the human ortholog (Fig. 5c)124,125.
The fucosylation site within EGF domains presents invariant backbone and side-chain atoms that interact with key catalytic residues of POFUT1. Based on sequence homology and the crystallographic work of Li et al. on the Mus musculus POFUT1, the recognition of EGF domains by this enzyme is shown to rely on a set of conserved, direct interactions concentrated within the canonical fucosylation motif. In addition to this core recognition module, the interaction surface extends to structurally defined elements of the EGF fold, such as the C1–C2 loop and the C5–C6 subdomain. These regions do not contribute uniformly across substrates, but instead are accommodated with variable affinity, suggesting that POFUT1 recognition balances strict specificity at the catalytic center with broader structural adaptability (Fig. 5c)124. This dual mode of recognition allows POFUT1 to act selectively on properly folded EGF-type domains while tolerating a range of sequence and conformational diversity outside the catalytic core.
Although POFUT1 lacks a conventional basic residue in its active site, metadynamics simulations have demonstrated that the β‑phosphate of GDP‑Fuc serves as the ultimate catalytic base, with Asn51 functioning as a proton shuttle that transfers the proton from the acceptor Thr to the β‑phosphate during the Fuc transfer (see below)122,124,126.
POFUT2 acts on TSRs, which display distinct consensus sequences depending on their disulfide bonding pattern: C¹-X₂-(S/T)-C² in group 1 TSRs, and C²-X₂-(S/T)-C³ in group 2 TSRs10. Although it retains the canonical GT-B fold, POFUT2 features a unique structured loop (residues 140–156), absent in POFUT1, that helps define the inner wall of the N-terminal domain. This element likely contributes to stabilizing the substrate binding (Fig. 5d)127.
Subsequent studies revealed that substrate recognition relies not primarily on extensive direct contacts but rather on an intricate hydrogen-bonding network mediated by water molecules. This hydration layer stabilizes the binding of structurally conserved TSR domains, allowing the enzyme to maintain specificity despite limited sequence conservation. Furthermore, high-resolution crystal structures identified Glu54 as the catalytic base (Fig. 5d)112,127.
POFUT3 and POFUT4, the most recently described members of this family, further expand the ER’s glycosylation repertoire. These enzymes recognize the EMI domains, structurally similar to TSRs, and fucosylate specific Ser/Thr residues in a conformation-dependent manner. Although their crystal structures are yet to be resolved, computational models indicate that both enzymes adopt the same GT-B fold and catalytic organization as their paralogs, suggesting a conserved catalytic mechanism across the FUT family7.
Collectively, these structural studies reveal a unifying GT-B scaffold across the fucosyltransferase family, with catalytic activity dependent on conserved motifs and specific amino acid residues rather than on metal cofactors. Enzyme-specific features, such as flexible loops, glycosylation sites, exosites, and accessory domains, fine-tune substrate selectivity and function. This structural diversity underlies the ability of FUTs and POFUTs to recognize and modify a broad array of glycan and protein targets with remarkable precision.
Catalytic mechanisms of FUTs
Despite the structural diversity of their acceptor substrates, all mammalian FUTs characterized to date operate through a shared catalytic principle: a bimolecular nucleophilic substitution (SN2) reaction that results in inversion of configuration at the anomeric carbon of the donor Fuc. In this mechanism, the hydroxyl group of the acceptor acts as the nucleophile, attacking the anomeric carbon and displacing the GDP moiety as the leaving group. In FUT9, a similar SN2 mechanism has been proposed but not yet validated. Glu137 is well-positioned to serve as the catalytic base, and the GDP-Fuc donor is observed in a distorted conformation that may facilitate bond cleavage and transition state formation. In addition, Arg202 and Lys256 stabilize the negatively charged diphosphate moiety, while Asn195 and Asn246 contribute to transition state stabilization through hydrogen-bonding interactions with the donor substrate (Fig. 6)111. The absence of (obligatory) metal cofactors is a hallmark of the GT-B family, in which the dual Rossmann-like fold and a dense network of polar interactions compensate for the lack of catalytic metals37.

a Catalytic mechanism of POFUT1126. b Catalytic mechanism of POFUT2, the nucleobases is shown as G since the diphosphate moiety is presented as an explicit structure129. c Proposed catalytic mechanism of FUT8119. d Proposed catalytic mechanism of FUT9111. MC represents the Michaelis complex, TS the transition state, and P the reaction products. Hydrogen bonds are shown as dotted blue lines, and key bond formations as dashed gray lines. In panels (c) (FUT8) and (d) (FUT9), the TS are shown in gray to indicate that the mechanistic validation of the proposed TS has not been reported.
In FUT8, crystallographic and mutational studies have confirmed a concerted SN2 mechanism. Upon GDP-Fuc binding, loop 2 (residues 365–378) undergoes a conformational rearrangement that positions Glu373 to function as the general base, deprotonating the acceptor hydroxyl. Simultaneously, Arg365 and Lys368 (loop2 residues) stabilize the negative charge developing on the leaving diphosphate group. This rearrangement also excludes the solvent from the catalytic site, preventing hydrolysis and increasing reaction fidelity (Fig. 6)117,119,128.
Among POFUTs, mechanistic variations reflect adaptations to different acceptor chemistries and environments, particularly regarding nucleophile activation and diphosphate stabilization. In POFUT1, the apparent absence of classical basic residues in the catalytic site initially raised doubts about its catalytic mechanism. However, computational studies have revealed that the β-phosphate of GDP-Fuc acts as the ultimate catalytic base, while Asn51 plays a supportive role by facilitating proton transfer from the acceptor hydroxyl to the phosphate group during the SN2-like reaction. Arg245 and Ser362 help neutralize the developing negative charge on the departing diphosphate, thereby facilitating its release (Fig. 6)126. In POFUT2, Glu52 acts as the catalytic base, directly deprotonating the acceptor hydroxyl to enable Fuc transfer, and Arg294 coordinates the pyrophosphate unit. Notably, unlike other GTs that shield the sugar nucleotide from solvent to avoid hydrolytic side reactions, POFUT2 features a highly solvent-exposed active site. In this context, water molecules do not participate in nucleophile activation but instead play a dual catalytic role by stabilizing the departing GDP and contributing to acceptor substrate recognition. This dual involvement of solvent molecules highlights a distinctive mechanistic adaptation within the FUT family (Fig. 6)112,129.
The catalytic strategies employed by FUTs reflect a conserved chemical logic centered on SN2 displacement with inversion of stereochemistry, yet each enzyme achieves this through distinct structural solutions. Differences in base activation, donor distortion, loop rearrangements, and solvent engagement illustrate how FUTs have adapted to the specific constraints of their substrates and cellular environments, expanding the mechanistic versatility of this enzyme family.
FUTs in Health and disease
FUT enzymatic activities shape the intricate glycan landscape of cells and tissues, modulating a wide array of biological functions. FUTs play critical roles in structural protein stability, cellular communication, immune recognition, and other key biological processes, underscoring their profound impact on human health. In this section, we will explore the multifaceted roles of FUTs in various disease states, focusing on the consequences of aberrant FUT expression in critical pathologies. Specifically, we will first examine their prominent involvement in cancer progression, followed by their significant roles in inflammatory processes and infectious diseases.
FUTs implications in cancers
FUTs are increasingly recognized as critical regulators of cancer progression, influencing key malignant processes such as cell proliferation, invasion, metastasis, and therapy resistance. By modulating glycan structures on cell surface receptors and ECM components, FUTs affect both cell-intrinsic signaling pathways and interactions within the tumor microenvironment.
The α1,2-FUTs are key mediators of aggressive tumor phenotypes across a variety of cancer types130. These enzymes catalyze the biosynthesis of glycoconjugates, such as Lewis antigens and Globo H, which are implicated in enhanced cell migration, invasion, and metastatic spread. In breast cancer and other malignancies, overexpression of FUT1 and FUT2 has been shown to promote tumor aggressiveness. In studies of human breast cancer cell lines, Lai et al.42 observed that RNA interference-mediated inhibition of FUT1 and FUT2 reduced cell proliferation and migration in vitro and significantly decreased tumorigenicity and metastasis in vivo. Aberrant expression of FUT1 has also been implicated in the development of multidrug resistance in human chronic myeloid leukemia cells86. In addition, Loong et al.131 reported that FUT1 inhibition sensitized hepatocellular carcinoma cells to sorafenib, a frontline treatment for hepatocellular and renal carcinomas. Furthermore, FUT1 overexpression was associated with activation of resistance-related signaling pathways86. Kawai et al.132, found that FUT1 suppression attenuated HER2-mediated signaling and reduced cell proliferation. This finding was corroborated by Zhang et al.133, who observed that suppression of both FUT1 and FUT4 inhibited the HER2 pathway and reduced cancer cell proliferation. In addition, FUT1 and FUT2 contribute to autophagy regulation in breast cancer cells through the fucosylation of lysosome-associated membrane protein 1 (LAMP1), which was correlated with increased autophagic activity134. Targeted inhibition of these FUTs partially reversed this phenotype, further supporting their potential as therapeutic targets. In gastric cancer, FUT1 has also been shown to promote angiogenesis by upregulating LeY antigen expression in endothelial cells, thereby facilitating neovascularization and enhancing cell motility43.
Overexpression of FUT2 has been identified as a key contributor to lung cancer development, often in conjunction with FUT8135. In colorectal cancer, FUT2 plays a multifaceted role in tumor progression and metastasis by modulating several molecular pathways. Recent studies have demonstrated that FUT2 contributes to human colorectal cancer by reprogramming fatty acid metabolism136, activating Wnt signaling pathways137, and enhancing EMT through the fucosylation of low-density lipoprotein receptor-related protein 1 (LRP1), thereby promoting tumor cell extravasation138. Conversely, studies of murine colorectal cancer models and of human colorectal cancer cell lines by Wang et al.139 provided evidence that FUT2 deficiency reduces the fucosylation of the melanoma cell adhesion molecule (MCAM), leading to increased cell proliferation and invasion in colorectal cancer. This finding highlights the complexity of FUT2’s role in colorectal cancer, suggesting that both overexpression and loss of FUT2 activity can contribute to tumor progression via distinct mechanisms.
As mentioned above (in the sections “Classification and main characteristics of human FUTs” and in “Cell-Cell interactions”), the α1,3/4-FUTs play essential roles in metastatic dissemination by enabling “Step-1” (selectin-dependent) adhesive interactions between circulating malignant cells and endothelial cells. These enzymes catalyze the synthesis of sLeX and its isomer sLeA (known as the CA19-9 antigen). Expression of these glycan motifs fuels hematogenous metastasis by enhancing tumor cell interactions with E-selectin expressed on the vascular endothelium that both enables extravasation of the tumor cells and the subsequent intraparenchymal lodgment within specialized E-selectin-expressing endothelial beds comprising tumor “growth niches” that support malignant cell proliferation and resistance to chemotherapy51,140,141. Heightened expression of these FUTs has been shown to significantly impact both hematologic malignancies and adenocarcinoma progression. sLeA (the “CA19-9 antigen”) is characteristically expressed on pancreatic, biliary, and gastrointestinal (GI) adenocarcinomas. Monitoring serum CA19-9 levels is the most widely used biomarker for diagnostic and prognostic assessment of pancreatic cancer and of biliary cancer. This fact, alone, implicates sLeA has a driver of these malignancies, and draws attention to the development of therapeutic strategies to interrupt the biosynthesis of this glycan determinant.
In solid malignancies, FUT6 and FUT3 upregulation, in particular, promotes sLeX expression and enhances binding to E-selectin, facilitating metastatic spread in prostate, GI and lung adenocarcinomas142,143,144. Moreover, in renal cancer, FUT3 mediates EMT145, a mechanism also observed in pancreatic87, colorectal146. bone50, and gastric147 cancers. In breast cancer, high FUT3 expression has been correlated with reduced survival rates148. Overall, FUT3 has emerged as a valuable biomarker for the detection of aggressive tumors and metastatic potential. A retrospective study by Tanaka et al.149, demonstrated that standard biomarkers such as CA19-9 and DUPAN-2 (which recognizes Type 2 α2,3-sialylated LacNAc) that are commonly used to monitor pancreatic cancer treatment responses could be normalized against FUT2 and FUT3 expression levels. This adjustment significantly improved patient stratification, optimized the timing of surgical intervention, and enhanced both clinical outcomes and healthcare resource utilization.
FUT4 has shown significant implications in colorectal cancer progression150 and has also been explored as a potential biomarker for breast cancer diagnosis151. Analyzes of serum and tissue samples have demonstrated that FUT4 offers superior diagnostic performance with receiver operating characteristic (ROC) values surpassing commonly used biomarkers such as CA15-3 and CEA. In colorectal cancer, Lv et al.152, identified FUT4 as a core regulatory gene with prognostic value, particularly due to its role in modulating the immune response within the tumor microenvironment. This immune-modulatory function was further supported by Liu et al.153, who reported a strong correlation between FUT4 expression and PD-1 levels in lung adenocarcinoma models. Additional implications in lung cancer have been documented by the Pan group154, who demonstrated that FUT4 inhibition reduced chemoresistance, enhancing the efficacy of therapeutic treatments. Similarly, Lu et al.93, showed that aberrant expression of FUT4 and FUT6 activates oncogenic signaling pathways such as PI3K/Akt through fucosylation of EGFR and related receptors. Elevated FUT4 expression was associated with increased invasion, migration, and EMT in lung adenocarcinoma. The FUT4-mediated promotion of EMT has also been observed in other cancers, including breast155,156 and gastric cancer157,158, reinforcing FUT4’s potential as a therapeutic target across multiple tumor types. Furthermore, FUT4 and FUT7 have been shown to synergistically support metastatic progression in non-small cell lung cancer, one of the most common sources of brain metastasis. In this context, overexpression of fucosylated LeX/sLeX epitopes enhanced tumor cell adhesion to cerebral endothelium via interactions with E-selectin. This interaction facilitates blood–brain barrier disruption, promoting metastatic infiltration into the brain63.
Although relatively few studies have explored the role of FUT5 in cancer, emerging evidence suggests it may contribute to tumor development and progression. Liang et al.90, reported that FUT5 and FUT6 are regulated by microRNA-126-3p (miR-126-3p), a microRNA enriched in endothelial cells known for its role in angiogenesis and vascular homeostasis. Their findings indicated that FUT5 and FUT6 promote colorectal cancer development through miR-126-3p–mediated modulation of the PI3K/Akt signaling pathway. The study also highlighted miR-126-3p as a potential predictive biomarker and therapeutic target in colorectal cancer. Additional studies have shown that FUT5 upregulation is associated with enhanced tumor cell proliferation and migration through modification of extracellular matrix (ECM) proteins. This suggests that FUT5 may contribute to tumor microenvironment remodeling, further supporting its potential relevance in cancer pathogenesis91.
FUT6 appears to play context-dependent roles in cancer development, with both tumor-suppressive and tumor-promoting functions reported in different cancer types. In head and neck squamous cell carcinoma, downregulation of FUT6 has been associated with enhanced tumor aggressiveness. Wang et al.159, reported that reduced FUT6 expression correlates with increased cell proliferation, migration, and invasion in vivo. Conversely, FUT6 overexpression inhibited tumor growth and metastasis, likely through modulation of the EGFR/ERK/STAT signaling pathway. These findings were further supported in a parallel study by Yao et al.160, reinforcing the tumor-suppressive role of FUT6 in this context. In contrast, FUT6 overexpression has been shown to promote tumor progression in colorectal cancer. Specifically, FUT6 contributes to increased proliferation and migration of cancer cells via activation of the epithelial EMT process92. These findings were corroborated in studies of human breast cancer and prostate cancer lines xenografted in mice via intravascular administration, where transduction of the administered cells with human FUT3 or FUT6, but not of any other α1,3/4-FUTs, promoted bone metastasis by inducing mesenchymal-epithelial transition (MET) and triggering WNT signaling50. Recent work161 further supports FUT6’s oncogenic role in colorectal cancer, underscoring the complexity of FUT6’s function, which may vary depending on tissue type and tumor microenvironment. Importantly, as mentioned in the section “Cell-Cell interactions”, the fact that circulating FUT6 levels served as the first serum biomarker of cancers underscores a critical role for sLeX, its principal biosynthetic product, in fueling the proliferation of cancer.
FUT7 has emerged as a potential therapeutic target across several cancer types, with strong evidence linking its overexpression to proliferation and aggressivity of acute leukemias162, lung cancer63,135,163,164,165, breast cancer166, bladder urothelial carcinoma167, and follicular thyroid carcinoma168. In the case of follicular thyroid carcinoma, FUT7 overexpression promotes EGFR fucosylation, which in turn activates EMT, a critical process for tumor progression and metastasis.
FUT8 exerts a broad influence across several tumor types169, contributing to neoplastic transformation, tumor metastasis, and immune evasion. Recent reviews76,77,170,171 summarize how elevated FUT8 expression and activity are linked to various human cancers, including lung, breast, liver, and others. Here, we highlight some of the most recent studies from 2023–2025 that expand on FUT8’s role in cancer. The Munkley group reported that FUT8 regulates gene expression and signaling pathways associated with prostate cancer progression, showing that FUT8 inhibition suppresses tumor progression71. Similarly, Ito and colleagues demonstrated that elevated FUT8 expression correlates with metastasis in papillary thyroid carcinoma172. FUT8 overexpression has also been correlated with poor survival and increased aggressiveness in a wide range of cancers, including: diffuse large B-cell lymphoma173, leukemia174, meningioma175, intrahepatic cholangiocarcinoma176, lung177,178,179, cervical180, esophageal181, colorectal13,182,183,184,185, kidney186, ovarian187, breast188, liver189,190,191, bone192, brain193, and oral squamous cell cancer194. Notably, FUT8 downregulation has been associated with HPV-associated cervical cancer, indicating possible context-dependent roles195.
FUT8 is also key in regulating antibody-mediated immune responses. By “core” fucosylating the asparagine-linked glycan (at “N297”) of the Fc region of IgGs, FUT8 overexpression dampens engagement of Ig Fc to natural killer (NK) cell FcyRIIIa receptors, leading to decreased NK cell-mediated cytotoxicity (“antibody-dependent cell-mediated cytotoxicity” (ADCC)). This reduced efficacy of antibody-mediated immune responses against cancer cells enables immune evasion196,197,198. Globally, the level of IgG fucosylation profoundly impacts antibody efficacy, prompting the development of novel glycoengineering strategies aimed at modulating IgG Fc core fucosylation to enhance mAb-mediated disease treatment170, which is further discussed below.
From a broad perspective, aberrant FUT8 expression enhances cell proliferation and metastasis by modifying key cell-surface receptors such as EGFR, TGF-β receptor, and integrins. This abnormal fucosylation triggers dysregulated signaling pathways that promote EMT, a crucial process for cancer cell invasion and metastasis. Furthermore, FUT8 facilitates immune evasion by regulating the expression and function of immune checkpoint proteins such as PD-1/PD-L1 and B7-H3 on both tumor and immune cells. This results in suppression of T-cell-mediated anti-tumor immunity and fosters an immunosuppressive tumor microenvironment, characterized by increased infiltration of M2 macrophages and disruption of antibody-dependent cell-mediated cytotoxicity. In conclusion, FUT8 upregulation and the resultant aberrant core fucosylation contribute to cancer development by promoting cell proliferation, invasion, and immune escape. These multifaceted roles position FUT8 as a promising therapeutic target in cancer treatment.
FUT9 overexpression promotes the progression of several cancers, including ovarian199, gastric200,201, and esophageal202 cancers. In addition, FUT9 plays a critical role in colorectal cancer stemness by inducing the expression of pluripotency markers and chemoresistance-associated genes, positioning it as a potential therapeutic target for eradicating tumor-initiating cells150,203. For example, Auslander et al.204, demonstrated that FUT9 inhibition attenuates tumor-initiating cells and suppresses tumor development in a mouse xenograft model.
Many studies have unveiled the emerging role of POFUT1 as a central modulator of oncogenic signaling205. The primary mechanism by which POFUT1 exerts its oncogenic effects is through hyperactivation of the Notch signaling pathway. Elevated POFUT1 levels increase the O-fucosylation of Notch receptors, leading to excessive Notch signaling. This aberrant activation drives uncontrolled cell proliferation, inhibits apoptosis, promotes angiogenesis, and facilitates EMT, thus enabling tumor cell invasion and metastasis. Nonetheless, context-dependent roles for POFUT1 in cancer progression have also been reported. For example, POFUT1 can act as a tumor suppressor in certain cancers, including muscle-invasive bladder cancer (MIBC)206. Conversely, in hepatocellular carcinoma207 and glioblastoma208, POFUT1-mediated Notch activation promotes cell proliferation and invasion. Similarly, in colorectal cancer, high POFUT1 expression correlates with advanced stages and metastasis driven by an overactive Notch pathway209,210. Dysregulated POFUT1 expression is also associated with increased proliferation and invasiveness in multiple cancers, including breast211, head and neck212, liver213, and colorectal214 cancers. In the latter, silencing POFUT1 inhibited proliferation and migration and induced apoptosis in colorectal cancer cells. Moreover, Li et al.215, reported that POFUT1 acts synergistically with PLAGL2 to promote colorectal tumorigenesis by maintaining the stemness of colorectal cancer stem cells through both Wnt and Notch pathways. This finding corroborates the initial evidence from Chen et al.216, showing that downregulation of POFUT1 in PLAGL2−/− mice supports a shared promoter region between the two genes217. Within this context, POFUT1 and PLAGL2 have also been linked to MUC2 expression, a glycoprotein biomarker for colorectal mucinous adenocarcinoma. Beyond Notch, POFUT1 influences other crucial tumorigenic pathways such as Wnt/β-catenin signaling. For instance, in gastric cancer, POFUT1 promotes cancer progression by activating both Notch and Wnt pathways218. Due to its widespread implication across multiple cancer types, POFUT1 has been highlighted as an independent and promising biomarker for prognosis205,206,210,219,220, underscoring its significant clinical relevance.
On the other hand, the implications of POFUT2 in cancer remain poorly studied10, partly due to its crucial role in embryonic development. Notably, Pofut2 knockout results in embryonic lethality in mice by blocking the secretion of key target proteins such as ADAMTS9108. Despite this, Jia et al.221, reported that aberrant POFUT2 expression serves as a prognostic marker across various cancer types, emphasizing its potential as a valuable biomarker, particularly for the detection of adrenocortical carcinoma.
POFUT3 and POFUT4, previously classified as FUT10 and FUT11, respectively, are essential for protein secretion7. Zhang et al.222, reported that POFUT3 expression in clear cell renal cell carcinoma correlates with the clinical stage of the disease, indicating its potential as a prognostic biomarker. Similarly, POFUT4 upregulation has been identified as a biomarker for renal carcinoma progression223 and has shown significant implications in the progression of hepatocellular224 and ovarian cancers225. In pancreatic226 and brain227 cancers, hypoxic conditions were found to stimulate POFUT4 expression, enhancing cancer cell proliferation and invasiveness. POFUT4 is also significantly overexpressed in gastric cancer, where it affects the infiltration of immune cells into the tumor microenvironment. A study by Huang et al.228, further supports the idea that POFUT4 expression is induced under hypoxia. In addition, Zhang et al.229, found that POFUT4 inhibits ferroptosis in gastric cancer cells, thereby promoting cell proliferation. On the other hand, POFUT4 knockdown significantly inhibited cancer cell proliferation, a result corroborated by Cao et al.230, who also demonstrated that POFUT4 acts through the PI3K/AKT signaling pathway, underscoring its potential as a prognostic biomarker for gastric cancer detection.
Collectively, these findings position FUTs as pivotal players in tumor biology. Their enzymatic activities not only define specific glycan signatures but also rewire cellular signaling networks, enabling tumors to adapt, evade immune surveillance, and develop resistance to therapies. Consequently, the detection of aberrant FUT expression offers a promising avenue for discovering novel cancer biomarkers, which can be utilized as early diagnostic tools as well as prognostic indicators to facilitate timely treatment and dramatically improve patient outcomes. Moreover, as well described further below, the inhibition of several FUTs has emerged as a primary therapeutic strategy for mitigating various cancers. Selective inhibitors targeting specific FUT enzymes hold significant potential as potent and precise anticancer agents, marking an exciting frontier in cancer treatment development.
Inflammatory diseases
FUTs play a pivotal role in the regulation of immune responses and tissue homeostasis in inflammatory disorders. By modifying glycoconjugates on immune effectors as well as on epithelial cells, FUTs shape the dynamics of leukocyte trafficking, mucosal barrier integrity, and local cytokine environments.
The α1,2-FUTs catalyze the synthesis of α1,2-fucosylated glycans on the apical surface of intestinal epithelial cells, which in turn influence microbiota composition, epithelial barrier function, and innate immune signaling. The Kiyono group demonstrated that FUT2 deficiency disrupts the gut microbial ecosystem and enhances susceptibility to gastrointestinal infections and colitis, as demonstrated in murine models231. In parallel, the same group showed how both FUT1 and FUT2 contribute to inflammatory signaling cascades initiated at the mucosal interface, further amplifying epithelial immune responses45.
As mentioned above in the sections “Cell-cell interactions”, by regulating sLeX expression, the α1,3-FUTs are critical in mediating leukocyte–endothelial interactions, and, therefore, these enzymes are essential for host defense. Importantly, mammalian hematopoietic cells do not express Type 1 lactosamines, and, therefore, all mammalian leukocytes display only sLeX (i.e., not sLeA). Notably, in all mammalian leukocytes, FUT7 expression dominates in the biosynthesis of creating sLeX, and therefore, FUT7 serves as a principal mediator of immune surveillance and host defense232. However, importantly, high sLeX expression is also characteristic of regulatory T cells (“Tregs”) and, therefore, upregulated E-selectin in endothelial beds of inflamed tissues recruit both innate host defense cells (granulocytes and monocytes), as well as cells comprising both limbs of adaptive immunity (i.e., effector lymphocytes and immunoregulatory cells)39; this tapestry of sLeX expression on subsets of leukocytes ensures that host defense is achieved without evolution of a life-threatening inflammatory reaction. As one example of how this balance of sLeX-mediated cell recruitment affects the inflammatory response, up-regulation of FUT7 expression in Tregs alleviates colitis in mouse models233. Moreover, in murine models, combined genetic knock-out of both FUT4 and FUT7 significantly reduces leukocyte infiltration and local proinflammatory cytokine production24.
FUT9 contributes to immune regulation in a more context-specific manner. In viral encephalitis models, FUT9 deficiency intensifies neuroinflammation and impairs cytokine production, indicating a critical role in controlling innate immune responses in the central nervous system, indicating that LeX structures are involved in host responses against viral or bacterial infections234.
FUT8 have also emerged as a critical modulator in central nervous system inflammation and pulmonary diseases. In murine models, FUT8 deficiency markedly increases glial activation in response to inflammatory stimuli, exacerbating neuroinflammatory processes by promoting microglial and astrocytic activation and upregulating proinflammatory markers69. In the pulmonary system, reduced FUT8 activity promotes the development of emphysema. Murine models showed that FUT8-deficient mice spontaneously develop an emphysema-like phenotype, including increased matrix metalloproteinase activity and inflammatory infiltration following elastase exposure235.
Collectively, these findings highlight FUTs both as active pro-inflammatory enzymes and as immunomodulatory enzymes that govern both local and systemic inflammation. Their roles span barrier protection, leukocyte recruitment, and cytokine regulation, with direct implications for gastrointestinal, pulmonary, neurological, and cardiovascular inflammatory diseases.
POFUTs have not been directly linked to severe inflammatory conditions however, their role in the Notch signaling pathway was shown to play a pivotal role in sepsis236. POFUT1 down-regulation and consequent Notch inactivation contributed to the development of gastrointestinal inflammatory processes such as enterocolitis in mice models237,238.
Infectious diseases
The human condition known as “Leukocyte Adhesion Deficiency Type II” (“LADII”; also known as “Congenital Disorder of Glycosylation-IIc” (CDG-IIc”)) has provided direct and unequivocal evidence of the fundamental role of leukocyte sLeX expression in human host defense. LADII is a congenital disease in which there is a mutation in the gene SLC35C1, which encodes the protein that transports GDP-fucose (that is created primarily de novo from GDP-mannose) from its site of biosynthesis in the cytosol into the Golgi. Accordingly, the decreased Golgi levels of GDP-fucose leads to a deficit in fucosylation of all pertinent glycolipids and glycoproteins. Importantly, though LADII results in deficiencies in α1,6-fucosylation and in α1,2-fucosylation (which leads to absence of the α1,2-fucosylated blood groups (including the ABO antigens) resulting in the “Bombay” blood type), the greatest pathobiologic impact of LADII is recurrent bacterial infections due to absence of α1,3 fucosylation, and, as such, diminished sLeX display239,240. Circulating leukocytes in LADII are conspicuously deficient in sLeX expression, resulting in reduced adhesive interactions with endothelial E-selectin, thereby hindering extravasation of leukocytes. This deficit leads to chronic, severe bacterial infections (due to inability to generate pus) despite compensatory marked elevations in blood leukocyte levels (“leukocytosis”). The deficient leukocyte sLeX levels can be partially corrected by administering high doses of oral L-fucose, which drives biosynthesis of GDP-fucose via the “salvage pathway” which utilizes free L-fucose to create GDP-fucose; the increased levels of GDP-fucose in the cytoplasm then enable GDP-fucose diffusion into the Golgi and therefore enable sLeX biosynthesis241,242.
FUTs also play a role as central modulators of host–pathogen interactions. Through their modification of cell surface glycans, FUTs influence pathogen binding, immune recognition, and the clinical features of various infectious diseases243. Numerous studies have shown that the interaction between H-type glycans expressed on mucosal surfaces and the host microbiota contributes to the development of intestinal diseases. For instance, FUT1 controls protein glycosylation in the mucosal layer of the intestine, particularly O-glycan structures that are potential bacterial receptor sites as well as energy sources for both pathogens and beneficial bacteria244. Within this context, Wang et al.245 studied the implications of FUT1 mutation in porcine intestinal infections caused by bacterial pathogens, providing evidence that FUT1 mutations alter the activity of the E. coli adhesion factor ECF18, influencing the resistance to bacterial infections.
In the case of FUT2, the absence of H-type glycans in individuals with the FUT2 non-secretor phenotype is considered a risk factor for inflammatory bowel disease. However, since H-type glycans serve as attachment sites for norovirus and rotavirus246, individuals with inactivating FUT2 mutations lack functional H antigens on epithelial cells and exhibit natural resistance to common norovirus strains, such as Norwalk virus247,248,249,250. Beyond direct pathogen binding, FUT2 also shapes gut microbiota composition251,252,253, which in turn modulates host immunity and susceptibility to infection19,231. In addition to FUT2, FUT3 influences susceptibility to microbial infections through modulation of surface glycan structures, and animal models indicate that FUT3 polymorphism correlates with bacterial infection caused by H. pylori254.
FUT9 has been implicated in neurotropic viral infections. In a murine model of murine hepatitis virus (MHV) infection, FUT9 deficiency led to increased neuroinflammation and impaired cytokine signaling, suggesting a regulatory role in central nervous system immune responses234. Notably, FUT8 stands out for its broad and multifaceted involvement in viral infections. Several viruses, including hepatitis C255, hepatitis B256, HIV257, and human papillomavirus258, exploit FUT8 to enhance replication and persistence. These viruses apparently increase FUT8-mediated fucosylation to modulate the glycosylation of host receptors such as EGFR, suppress interferon signaling, and promote immune evasion255,259. In addition, FUT8 plays critical roles in regulating adaptive immunity, influencing B and T cell activation and affecting both antibody production and effector function260,261.
Finally, POFUTs such as POFUT2 are crucial for the virulence of parasitic protozoa. In Plasmodium berghei and Toxoplasma gondii, genetic disruption of POFUT2 impairs O-fucosylation of key adhesive proteins like TRAP and MIC2, resulting in defective protein folding, trafficking, and dramatically reduced invasive capacity81,82. However, this view has been contested by subsequent studies in both P. berghei and P. falciparum, in which POFUT2-null parasites completed the mosquito stages and established blood-stage infections in mice without overt defects in motility or host invasion, thus calling into question the essentiality of O-fucosylation in these life cycle stages262. These conflicting results underscore the complexity of POFUT2 function in apicomplexan parasites and suggest that its contribution to virulence may be context-dependent or compensated by alternative mechanisms.
Collectively, these findings reveal a dual role for FUTs in infectious disease biology. On one hand, they function as innate immune gatekeepers, shaping glycan landscapes that mediate resistance or tolerance. On the other, their transcriptional upregulation is exploited by pathogens to enhance infectivity and immune evasion. Notably, several viruses, including herpes simplex virus, varicella-zoster virus, and cytomegalovirus, upregulate FUT3, FUT5, and FUT6 expression to facilitate infection263,264,265. Cytomegalovirus also induces FUT1, FUT7, and FUT9 transcription265, further illustrating the breadth of this strategy. The enzymatic versatility and immunomodulatory potential of FUTs position them as attractive targets for therapeutic intervention and biomarker development in the context of infectious diseases.
FUT inhibition and modulation
The inhibition or modulation of FUTs has recently emerged as a promising therapeutic strategy to target key pathological processes in which aberrant fucosylation plays a central role, including chronic inflammation, tumor progression, and autoimmune disease. While the therapeutic relevance of this enzyme family is increasingly recognized, the development of selective and effective FUT inhibitors faces substantial hurdles. These include the limited availability of three-dimensional structural data for most FUTs, inherent difficulties in achieving large-scale enzyme production necessary for structural and biophysical studies, significant limitations within current high-throughput screening methodologies, and the complex challenge of ensuring effective subcellular drug delivery to Golgi-resident enzymes. Another obstacle that complicates the development of FUT inhibitors consists in the high degree of structural conservation in FUT active sites, which complicates the design of inhibitors targeting a specific FUT-isoenzyme.
FUT inhibitors are broadly categorized into two main groups: those that disrupt GDP-Fuc biosynthesis, leading to widespread fucosylation inhibition, and those designed to directly inhibit specific FUT activity, with the ambition of achieving targeted inhibition. The results reported to date encompass the design and development of a diverse array of compounds, including donor and acceptor substrate analogs, bisubstrate inhibitors, novel glycomimetics, and various non-sugar derivatives.
Previous reviews provide a comprehensive discussion of FUT inhibitors reported in literature11,12,140. Therefore, in this section, we will mention the current strategies employed for the development of FUT inhibitors and discuss the major advantages and drawbacks, focusing on selective FUT inhibitors and the relevant examples reported in the literature.
Donor/Acceptor substrate-based inhibitors
Historically, the majority of FUT inhibitors developed so far focused on the development of mimetics based on the structure of the GDP-Fuc donor substrate, or on mimetics of the acceptor substrates. Researchers reported the synthesis of GDP-Fuc mimetics where small key modifications hampered the enzymatic reaction and inhibit the FUT activity. On the other hand, the design of Fuc-based prodrugs allows for chemically modified Fuc derivatives to be processed by the cellular GDP-Fuc biosynthetic pathway into their respective GDP-Fuc mimetics. The latter represents a more effective strategy for cellular fucosylation inhibition since small Fuc prodrugs are more likely to permeate through the cellular membrane in comparison with GDP-Fuc mimetics. Upon cellular entry, prodrugs are processed through either the de novo biosynthetic pathway or the salvage pathway. The de novo pathway “builds” the GDP-Fuc through a metabolic enzymatic cascade, converting glucose and mannose precursors into GDP-Fuc. In parallel, the salvage pathway recovers existing L-Fuc produced from glycan degradation, which is then phosphorylated by fucose kinase and converted into GDP-Fuc by the GDP-Fuc pyrophosphorylase266. Another strategy consists of the blockade of the enzymatic cascade responsible for the de novo or salvage GDP-Fuc synthetic pathways. Moreover, once GDP-Fuc is prepared, the cell utilizes the SLC35C1 transporter to transfer GDP-Fuc from the cytoplasm to the Golgi apparatus. Therefore, inhibition of this transporter will also result in reduced FUT activity. Nonetheless, recent studies demonstrating that abrogation of SLC35C1 did not reduce the protein fucosylation level in cells and in in vivo models, deducing that at least one more unknown mechanism of GDP-Fuc transport into the secretory pathway should be present in mammals267.
The hydrogen (H) to fluorine (F) or hydroxyl group (OH) to F substitution in strategic positions of the fucose moiety enabled the synthesis of potent FUT inhibitors. For example, the Wong group268 pioneered the synthesis of the GDP-Fuc mimetic (1), which showed a broad inhibitory activity against a pool of FUTs (FUT3, 5, 6 and 7). Unfortunately, the application of compound 1 and other inhibitors based on as similar scaffold bearing polar phosphorylated derivatives are limited to in vitro studies due to the limited stability and high polarity of the pyrophosphate moiety that hampers the permeation through the cell membrane and its use in cells. A compelling solution was proposed by the Paulson group in 20128, who exploited the intrinsic promiscuity of the cellular salvage pathways by administering membrane-permeable fluorinated sugar analog 2. Specifically, the peracetylation of compound 3 to provide membrane-permeable derivative 2 served to reduce the polarity of 3 and thereby enhance cellular uptake. After cellular internalization esterases catalyze its conversion into 3 (also known as 2-fluorofucose, or SGN-2FF), which is subsequently processed into its active GDP-Fuc analog capable of entering the endogenous glycosylation machinery. Glycan analysis demonstrated that 2 acted as general inhibitor against FUT4, 7 and 8. Unfortunately, this inhibitor was also found to prevent de novo GDP-fucose synthesis. The de novo and the salvage pathways have shown to not be completely independent from each other due to the presence of metabolic feedback loops between the two synthetic pathways. The Boons group developed bis-fluorinated Fuc prodrug which was converted in cells into GDP-2,2-di-F-Fuc. This donor was shown to provide slow transfer of fucose for all FUTs and acted as a global FUT inhibitor targeting both the de novo and salvage pathways269. Remarkably, SGN-2FF (3) showed to possess cell permeability capability without the need of acetylation of the hydroxyl functions. Okeley et al.270, demonstrated that 3 is as an orally bioavailable drug with potent anticancer effect both in vitro and in vivo. In a recent example, Munkley and co-workers showed that it was effective in reducing the activity of FUT8 in vivo in mice prostate cancer xenograft models providing effective suppression of tumor growth71. Moreover, SGN-2FF (3) represent the only FUT inhibitor that entered clinical trials in combination with immune checkpoint inhibitor PD-1 for the treatment of solid tumors (trial ID: NCT02952989, further discussed below). The Boltje group in 2021271, and the Vocadlo group in 2023272, reported novel FUT inhibitors that are targeting the de novo and salvage biosynthetic pathways, respectively. Boltje and co-workers reported the synthesis and application of mannose derivative Fucotrim-I (4), among others, as a functional Fuc precursor that is metabolized into its GDP derivative. In cells, the GDP-mannose is used as precursor in the de novo pathway, in which the mannose moiety is converted into Fuc by the action of GDP-mannose-4,6-dehydratase (GMDS). Blockage of this pathway provided global FUTs inhibition in cells. Vocadlo and co-workers developed a carbafucose derivative 5 in which the endocyclic oxygen is replaced by a CH2 moiety. This compound is uptake inside the cells and is then processed into its GDP derivative through the salvage pathway, leading to a non-glycosidic donor that is recognized by FUTs but cannot transfer the carbasugar mimetic, resulting in global cellular FUT inhibition.
On the other hand, several studies have attempted FUT inhibition through the synthesis of acceptor substrate mimetics mostly based on the structure of the LacNAc acceptor. For example, Galan et al.273, reported one of the few examples in which a FUT inhibitor shows selectivity. In this case, LacNAc derivative 8 was prepared by converting the galactose moiety into a talose derivative through epimerization of the C2 position. Inhibitor 8 showed selectivity for FUT6 over FUT3,4 and 5 isoenzymes with a Ki of 0.475 mM. More recently, The Richichi and Sackstein groups274 reported another case of successful selectivity: the conformationally constrained glycomimetic 6 is an active cellular inhibitor capable of blunting human FUT6 and human FUT7 activity but not human FUT9 activity, and this FUT isoenzyme-selectivity paves the way for opportunities to fine-tune the cell surface expression of specific fucosylated determinants (“glycan-motif editing” of the glycocalyx)140.
Exploiting the strategy of reducing the global inhibitor polarity through the acetylation of the hydroxyl residues to allow better cellular internalization, the Wong group275 reported the synthesis of a U937 cellular active inhibitor. Compound 10 successfully inhibited the expression of sLeX glycan in cells, and in its O-deacetylated form showed to act as a competing substrate showing a Km of 0.076 mM against FUT6, which is a 62-fold higher selectivity over the LacNAc substrate. The authors utilized this high-affinity substrate as a competitive substrate to decoy the biosynthesis of sLeX. However, its selectivity against other FUTs was not studied (Fig. 7).

The panel illustrates donor-based GDP-Fuc mimetics, acceptor-based analogs, bisubstrate inhibitors, and non-sugar scaffolds that target FUTs.
It is worth mentioning that all these strategies that employ inhibitors based on the structure of donor and acceptor substrates that are shared among different FUTs are likely to lead to broad FUT inhibition. While this represents a viable strategy to assess the global role of fucosylation in state of health and disease and in the study of general fucose-mediated recognition processes, these approaches are lacking selectivity toward a specific FUT isoenzyme. In addition, in certain cases, the presence of off-target effects was observed in the feedback loops between different glycosylation pathways, hampering the study of specific FUT implications in diseases8.
Bisubstrate inhibitors
A compelling strategy to provide FUT inhibition selectivity consists in generating molecular entities that merge in one compound the structures of both the donor and acceptor substrates. This family of bisubstrate inhibitors binds to two distinct binding sites and are likely to provide higher affinities and selectivity toward the target enzyme. This approach proved valid for other GTs such as human O-GlcNAc transferases276,277 and GalNAc-Ts278. For example, based on the structure of the LeX fucosylation product, the Wong group synthesized trisaccharide 9 by linking the N-group of β-l-homofuconojirimycin (red moiety of 9) to the 3-OH of LacNAc through an ethylene unit. This compound was designed to mimic the transition state of the enzymatic reaction. In the presence of GDP, the authors found that 9 inhibits FUT5 with an IC50 of 31 µM. Other bisubstrate inhibitors have been designed to incorporate the entire GDP moiety, linked to acceptor mimetics via stable pyrophosphate surrogates to enhance cellular uptake. One example is compound 11, a sulfonyl-linked mimetic developed by Manabe et al.279, which demonstrated inhibitory activity against FUT8. However, its specificity and efficacy against other FUT-subtypes were not evaluated (Fig. 7).
High throughput screening approaches
A successful strategy for identifying novel and selective FUT inhibitors involves high-throughput screening (HTS) of large compound libraries. This approach may lead to non-glycosidic lead compounds capable to induce selectivity toward a specific FUT. In 2010, Nishimura and co-workers280 developed a MALDI-TOF MS-based HTS of synthetic libraries of compounds identifying GDP-derivative 7 as a selective inhibitor (Ki of 293 nM) for FUT5 with respect to FUT8. One year later in 2011 the Paulson group developed a fluorescence-based assay in which libraries of natural compounds were screened identifying the thiadiazole 12 as a selective FUT6 inhibitor (IC50 of 1.8 µM) with respect to FUT7 (IC50 of >500 µM). Other HTS identified selective FUT inhibitors suitable for cellular studies. For example, Manabe et al.281, isolated a selective FUT8 pharmacophore unit that after SAR studies led to inhibitor 13. This compound successfully permeates the cell membrane where esterases release the active inhibitor 14. This compound interacts with FUT8 and undergoes enzymatic activation to generate a reactive naphthoquinone methide intermediate, which subsequently forms a covalent bond with the enzyme. Despite this covalent inhibition, the enzymatic activity was partially restored upon compound removal, indicating that 14 behaves as a quasi-irreversible or slowly reversible inhibitor. Cellular studies confirmed that 13 effectively inhibits core fucosylation by FUT8, underscoring its potential as a chemical probe for functional studies or therapeutic development (Fig. 7).
Recently, Costa et al. reported a study in which a cell-based HTS approach was used to identify inhibitors of sLeX biosynthesis282. Monensin (15), a polyether antibiotic, emerged as a potent bioavailable inhibitor. Treatment of sLeX-positive cancer cells with monensin showed reduced viability, and decreased motility and invasive capacities. In vivo studies in mice xenograft models revealed that monensin treatment suppressed the growth of sLeX-positive tumors, showcasing the importance of targeting cancer-specific glycans as a new source of cancer drug target candidates against colorectal and gastric cancers.
Finally, epigenetic modulation of FUT expression offers a non-enzymatic approach to reducing fucosylation. In lung cancer, for example, overexpression of miR-200b suppresses FUT4 expression, reducing Lewis antigen synthesis and impairing tumor cell invasiveness and migration in breast cancer cell lines as well as in mice xenograft models283. This highlights the therapeutic potential of harnessing endogenous regulatory networks to modulate FUT activity.
Altogether, these strategies illustrate a growing toolbox for targeting fucosylation in disease. Continued structural, computational, and biochemical innovation will be critical for translating FUT inhibition into clinically viable therapies. To fully realize the promise of FUT inhibition in clinical medicine, several critical points must be systematically addressed. First, the requirement for cell-penetrating FUT inhibitors is key to provide lead compounds for the development of in vivo applications. In addition, while many inhibitors developed so far showed to successfully permeate through the cell membrane, selectivity toward cancer cells and tissues has been poorly explored. Therefore, conjugation to antibodies or different carrier capable to direct the payload to the target regions will strongly amplify the efficacy of FUT based inhibition therapies and reduce the off-target effects. Finally, the development of selective FUT inhibitors will be a gamechanger in FUT-based treatments. To date, a very limited number of compounds have shown FUT selectivity, and in most cases, this feature was explored only toward a limited number of FUTs. The development of selective FUT inhibitors would open the path toward a better understanding of FUT activity and implication in diseases. Taken all together, these points will set the base for the future development of FUT inhibitors with high translational potential toward therapeutic applications.
Therapeutic applications
FUTs have emerged as clinically relevant biomarkers and therapeutic targets across oncologic, infectious, and inflammatory diseases, owing to their central role in cell adhesion, immune signaling, and tissue microenvironment remodeling. Alterations in their expression are consistently associated with tumor aggressiveness, therapeutic resistance, metastatic capacity, and immune evasion, conferring substantial diagnostic and prognostic value in diverse clinical settings. Therefore, molecular inhibitors or genetic approaches capable of selectively inhibiting the activity of a targeted FUT hold great potential as drug candidates for therapeutic applications. In addition, the aberrant FUT expression with consequent up- or down-regulated fucosylation anomalies could be exploited as biomarkers for the detection and prevention of diseases.
Despite the tremendous implications of aberrant FUT activity in various diseases, a limited number of clinical trials have explored the level of FUT expression as a biomarker or of FUT inhibition as a therapeutic strategy. The current list of clinical trials are presented in Table 1. From the perspective of biochemical impact, most trials have measured the level of fucosylation of IgG as a reporter of FUT inhibition, in some cases in conjunction with other glycan biomarkers.
For example, the Wildenbeest group is leading a clinical trial (trial ID: NCT05145348) in which the fucosylation level in IgG, together with other glycan biomarkers, is used as a parameter to monitor the efficacy of SARS-CoV-2 vaccination284. Patients with Down Syndrome showed a 3-10-fold higher death risk than healthy people affected by this virus, and the study aims to assess the immunogenicity of SARS-CoV-2 vaccination in people affected by Down syndrome. Another study reported by Winkelhorst et al.285, used the global fucosylation level of IgG as a parameter to identify pregnancies at risk of Fetal and Neonatal Alloimmune Thrombocytopenia, a cause of severe thrombocytopenia in healthy born neonates (trial ID: NCT04067375). The global fucosylation level of IgGs was also used along with other biomarkers to screen the efficacy of the vaccination responses (trial ID: NCT01967238)286,287,288, and in another study was analyzed as a parameter in lupus patients to find a correlation to the effector and pathogenic functions of IgGs in autoimmune disease (trial ID: NCT05394922).
Delayed engraftment is a major limitation of umbilical cord blood-based hematopoietic stem cell transplantation, due in part to deficits in levels of sLeX in umbilical cord blood hematopoietic progenitor cells (UCB-HPCs; i.e., “CD34 + ” mononuclear cells within cord blood). This low sLeX expression then leads to diminished homing to marrow, thereby impeding engraftment of the administered cells. The defect in sLeX expression is due to insufficient α1,3-fucosylation of UCB-HPC surface α2,3-sialylated Type 2 lactosamines. Accordingly, to boost the UCB-HPC binding to E-selectin, which is constitutively expressed by the bone marrow microvasculature and mediates the marrow recruitment of administered cells, α1,3-exofucosylation of the UCB-HPC cell surface via use of FUT6 (or FUT7) together with GDP-Fuc has been employed to enforce sLeX expression. The first registered clinical trial employing this methodology used FUT6 (trial ID: NCT01471067), whereas FUT7 was employed only in preclinical models289,290. This treatment appeared to improve neutrophil and platelet engraftment in high-risk patients with hematologic malignancies who underwent double umbilical cord blood transplantation260.
Based on results of a pivotal preclinical study employing α1,3-exofucosylation to enforce sLeX expression on human mesenchymal stem cells (MSCs) and thereby program their migration to marrow and subsequent generation of marrow osteoblasts56, a Phase 1 clinical study has been undertaken to assess the safety of α1,3-exofucosylated autologous bone marrow-derived MSCs in the treatment of women with advanced osteoporosis (trial ID: NCT02566655). In addition, a Phase 1 clinical trial (trial ID: NCT02423915) has been completed in which α1,3-exofucosylated regulatory T cells (Tregs) were administered intravascularly to prevent acute Graft-versus-Host disease after allogeneic hematopoietic stem cell transplantation. The outcome of this trial indicated that α1,3-exofucosylated Treg administration has no negative impact on engraftment, but high fevers were observed in recipients of the cells, and further studies are still ongoing. Current trials are also querying the effect of intestinal epithelial fucosylation as a modulator of the efficacy of Ustekinumab, an antibody used as for Crohn’s Disease treatment (trial ID: NCT06203158).
A different therapeutic approach reported by Do et al.291, studied how the inhibition of global fucosylation in combination with immune checkpoint blockade may be exploited for the treatment of a broad range of solid tumors. In this phase I trial (trial ID: NCT02952989) the patients were treated with SGN-2FF (2-Fluoro-2-desoxyfucose) a broad FUT inhibitor in combination pembrolizumab, a PD-1 immune checkpoint inhibitor demonstrating a dose-proportional pharmacokinetics, evidence of pharmacodynamic target inhibition of glycoprotein fucosylation, and preliminary antitumor activity. Unfortunately, a higher-than-expected grade of thromboembolic events led to study termination. The authors hypothesized that this relationship was due to the role of fucosylation in leukocyte adhesion and binding to cellular adhesion molecules on activated endothelial cells. The authors remarked how the development of second-generation fucosylation inhibitors that provide a more tumor-specific targeted approach to FUT inhibition may offer an improved therapeutic window for cancer patients.
Collectively, the broad and multifaceted involvement of FUTs in human disease underscores their value as biomarkers and as strategic targets for therapeutic and biotechnological innovation. However, the development of specific FUT inhibitors still represents a bottleneck to the development of targeted FUT-based therapies. Continued investigation into their biological functions promises to yield new avenues for personalized medicine.
Concluding remarks and future directions
Over the past several decades, FUTs have emerged as central orchestrators of glycan-mediated events in health and disease. From their critical involvement in immune cell trafficking, cell–cell adhesion, and tissue homeostasis, to their roles in cancer progression, pathogen interaction, and stem cell biology, FUTs have revealed themselves as enzymatic nodes of remarkable functional versatility. The growing body of evidence connecting FUT activity to pathological processes has catalyzed intense interest in their exploitation as diagnostic biomarkers and therapeutic targets across a wide range of clinical settings.
Despite this progress, significant gaps remain. While the repertoire of acceptor substrates is now well characterized for most FUTs, the regulatory mechanisms governing FUT expression, localization, and activity across diverse physiological and pathological contexts remain poorly defined. Technical hurdles such as the difficulty of expressing these enzymes, the scarcity of high-resolution structural data for many isoenzymes, and the persistent challenge of achieving isoenzyme-selective inhibition continue to constrain the pace of therapeutic translation. Addressing these limitations will be essential to unlock the full potential of FUTs as precision targets in human disease.
Future efforts must therefore focus on the development of more refined tools to probe FUT function in situ, including chemical probes, isoenzyme-specific inhibitors, and genetic models tailored to interrogate tissue- and cell-type–specific roles. Advances in AI-driven structure prediction are expected to illuminate conserved and divergent features of FUT active sites, accelerating rational drug design. Similarly, the integration of single-cell glycomics with spatial transcriptomics will enable high-resolution mapping of human FUT expression and fucosylation activity at cellular and tissue levels, potentially offering unprecedented insight into their roles across physiological and pathological states.
Finally, the therapeutic exploitation of FUTs will likely require context-specific approaches. Whereas broad inhibition of fucosylation may prove useful in oncology or immunotherapy, selective glycoengineering of hematopoietic progenitor cells, mesenchymal stem cells, or immune cells via selective FUT overexpression or FUT-based exofucosylation holds promise for regenerative medicine and immunotherapies. With the continued advancement of glycoscience technologies, FUTs are well-positioned to evolve from fundamental subjects of investigation into precise and actionable targets for therapeutic development.
References
Liang, D.-M. et al. Glycosyltransferases: mechanisms and applications in natural product development. Chem. Soc. Rev. 44, 8350–8374 (2015).
Lairson, L. L., Henrissat, B., Davies, G. J. & Withers, S. G. Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–555 (2008).
Stanley, P. Genetics of glycosylation in mammalian development and disease. Nat. Rev. Genet. https://doi.org/10.1038/s41576-024-00725-x (2024).
Hurtado-Guerrero, R. & Davies, G. J. Recent structural and mechanistic insights into post-translational enzymatic glycosylation. Curr. Opin. Chem. Biol. 16, 479–487 (2012).
Schneider, M., Al-Shareffi, E. & Haltiwanger, R. S. Biological functions of fucose in mammals. Glycobiology 27, 601–618 (2017).
Pekdemir, B. & Karav, S. Exploring the diverse biological significance and roles of fucosylated oligosaccharides. Front. Mol. Biosci. 11, 1403727 (2024).
Hao, H. et al. FUT10 and FUT11 are protein O-fucosyltransferases that modify protein EMI domains. Nat. Chem. Biol. 21, 598–610 (2025).
Rillahan, C. D. et al. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Biol. 8, 661–668 (2012).
Nie, H. et al. Targeting branched N-glycans and fucosylation sensitizes ovarian tumors to immune checkpoint blockade. Nat. Commun. 15, 2853 (2024).
Holdener, B. C. & Haltiwanger, R. S. Protein O-fucosylation: structure and function. Curr. Opin. Struct. Biol. 56, 78–86 (2019).
Lv, Y., Zhang, Z., Tian, S., Wang, W. & Li, H. Therapeutic potential of fucosyltransferases in cancer and recent development of targeted inhibitors. Drug Discov. Today 28, 103394 (2023).
Tu, Z., Lin, Y.-N. & Lin, C.-H. Development of fucosyltransferase and fucosidase inhibitors. Chem. Soc. Rev. 42, 4459–4475 (2013).
Wang, M. et al. FDW028, a novel FUT8 inhibitor, impels lysosomal proteolysis of B7-H3 via chaperone-mediated autophagy pathway and exhibits potent efficacy against metastatic colorectal cancer. Cell Death Dis. 14, 495 (2023).
Boruah, B. M. et al. Characterizing human α-1,6-fucosyltransferase (FUT8) substrate specificity and structural similarities with related fucosyltransferases. J. Biol. Chem. 295, 17027–17045 (2020).
Drula, E. et al. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577 (2022).
Dupuy, F. et al. Alpha1,4-fucosyltransferase activity: a significant function in the primate lineage has appeared twice independently. Mol. Biol. Evol. 19, 815–824 (2002).
Hao, H., Eberand, B. M., Larance, M. & Haltiwanger, R. S. Protein O-fucosyltransferases: biological functions and molecular mechanisms in mammals. Molecules 30, 1470 (2025).
Larsen, R. D., Ernst, L. K., Nair, R. P. & Lowe, J. B. Molecular cloning, sequence, and expression of a human GDP-L-fucose:beta-D-galactoside 2-alpha-L-fucosyltransferase cDNA that can form the H blood group antigen. Proc. Natl. Acad. Sci. USA 87, 6674–6678 (1990).
Hu, M. et al. Fucosyltransferase 2: A genetic risk factor for intestinal diseases. Front. Microbiol. 13, 940196 (2022).
Moreno, A. et al. Analysis of the FUT2 gene and Secretor status in patients with oral lesions. Inmunología 28, 131–134 (2009).
Mondal, N. et al. Distinct human α(1,3)-fucosyltransferases drive Lewis-X/sialyl Lewis-X assembly in human cells. J. Biol. Chem. 293, 7300–7314 (2018).
Elmgren, A. et al. Significance of individual point mutations, T202C and C314T, in the human Lewis (FUT3) gene for expression of Lewis antigens by the human α(1,3/1,4)-fucosyltransferase, Fuc-TIII. J. Biol. Chem. 272, 21994–21998 (1997).
Yang, L. et al. Association analysis of DNA methylation and the tissue/developmental expression of the FUT3 gene in Meishan pigs. Gene 851, 147016 (2023).
Homeister, J. W. et al. The α(1,3)fucosyltransferases FucT-IV and FucT-VII exert collaborative control over selectin-dependent Leukocyte recruitment and Lymphocyte Homing. Immunity 15, 115–126 (2001).
Ponnampalam, A. P. & Rogers, P. A. W. Expression and regulation of fucosyltransferase 4 in human endometrium. Reproduction 136, 117–123 (2008).
Chiu, P. C. N. et al. Glycodelin-A interacts with fucosyltransferase on human sperm plasma membrane to inhibit spermatozoa-zona pellucida binding. J. Cell Sci. 120, 33–44 (2007).
Mollicone, R. et al. Molecular basis for plasma alpha(1,3)-fucosyltransferase gene deficiency (FUT6). J. Biol. Chem. 269, 12662–12671 (1994).
Sackstein, R. Glycosyltransferase-programmed stereosubstitution (GPS) to create HCELL: engineering a roadmap for cell migration. Immunol. Rev. 230, 51–74 (2009).
Kudo, T. et al. Mice lacking α1,3-fucosyltransferase IX demonstrate disappearance of Lewis x structure in brain and increased anxiety-like behaviors. Glycobiology 17, 1–9 (2007).
Yao, L. et al. Divergent inducible expression of P-selectin and E-selectin in mice and primates. Blood 94, 3820–3828 (1999).
Liu, Z. et al. Differential regulation of human and murine P-selectin expression and function in vivo. J. Exp. Med. 207, 2975–2987 (2010).
Ihara, H. et al. Crystal structure of mammalian α1,6-fucosyltransferase, FUT8. Glycobiology 17, 455–466 (2007).
Nagae, M., Yamaguchi, Y., Taniguchi, N. & Kizuka, Y. 3D Structure and function of glycosyltransferases involved in N-glycan maturation. IJMS 21, 437 (2020).
Ihara, H. et al. Fucosyltransferase 8. GDP-Fucose N-Glycan Core α6-Fucosyltransferase (FUT8). in Handbook of Glycosyltransferases and Related Genes (eds Taniguchi, N. et al.) 581–596 (Springer Japan, Tokyo, 2014).
Luo, Y. & Haltiwanger, R. S. O-Fucosylation of notch occurs in the endoplasmic reticulum. J. Biol. Chem. 280, 11289–11294 (2005).
Neupane, S. et al. O-fucosylation of thrombospondin type 1 repeats is essential for ECM remodeling and signaling during bone development. Matrix Biol. 107, 77–96 (2022).
Moremen, K. W. & Haltiwanger, R. S. Emerging structural insights into glycosyltransferase-mediated synthesis of glycans. Nat. Chem. Biol. 15, 853–864 (2019).
Li, J., Hsu, H.-C., Mountz, J. D. & Allen, J. G. Unmasking fucosylation: from cell adhesion to immune system regulation and diseases. Cell Chem. Biol. 25, 499–512 (2018).
Silva, M., Videira, P. A. & Sackstein, R. E-Selectin ligands in the human mononuclear phagocyte system: implications for infection, inflammation, and immunotherapy. Front. Immunol. 8, 1878 (2017).
Sackstein, R. et al. Glycans in Systemic Physiology (Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 2022).
Sackstein, R. et al. Glycans in Acquired Human Diseases. in Essentials of Glycobiology (Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 2022).
Lai, T.-Y. et al. Fucosyltransferase 1 and 2 play pivotal roles in breast cancer cells. Cell Death Discov. 5, 74 (2019).
Moehler, T. M. et al. Involvement of α 1-2-fucosyltransferase I (FUT1) and surface-expressed lewisy (CD174) in first endothelial cell–cell contacts during angiogenesis. J. Cell. Physiol. 215, 27–36 (2008).
Palumberi, D. et al. RNA-mediated gene silencing of FUT1 and FUT2 influences expression and activities of bovine and human fucosylated nucleolin and inhibits cell adhesion and proliferation. J. Cell. Biochem. 111, 229–238 (2010).
Terahara, K. et al. Distinct fucosylation of M cells and epithelial cells by Fut1 and Fut2, respectively, in response to intestinal environmental stress. Biochem. Biophys. Res. Commun. 404, 822–828 (2011).
St John, J. A. et al. Genetic manipulation of blood group carbohydrates alters development and pathfinding of primary sensory axons of the olfactory systems. Dev. Biol. 298, 470–484 (2006).
Padró, M., Cobler, L., Garrido, M. & De Bolós, C. Down-regulation of FUT3 and FUT5 by shRNA alters Lewis antigens expression and reduces the adhesion capacities of gastric cancer cells. Biochim. Biophys. Acta 1810, 1141–1149 (2011).
Guo, Q. et al. Functional analysis of α1,3/4-fucosyltransferase VI in human hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 417, 311–317 (2012).
Li, J. et al. Human fucosyltransferase 6 enables prostate cancer metastasis to bone. Br. J. Cancer 109, 3014–3022 (2013).
Esposito, M. et al. Bone vascular niche E-selectin induces mesenchymal–epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 21, 627–639 (2019).
Srimongkol, A. et al. Sunitinib efficacy with minimal toxicity in patient-derived retinoblastoma organoids. J. Exp. Clin. Cancer Res. 42, 39 (2023).
Sen, U., Guha, S. & Chowdhury, J. R. Serum fucosyl transferase activity and serum fucose levels as diagnostic tools in malignancy. Acta Med Okayama 37, 457–462 (1983).
Ronquist, G. & Nou, E. Serum sialyltransferase and fucosyltransferase activities in patients with bronchial carcinoma. Cancer 52, 1679–1683 (1983).
Yazawa, S. et al. Cancer-associated elevation of alpha(1––3)-L-fucosyltransferase activity in human serum. Cancer 62, 516–520 (1988).
Asao, T. et al. Tumor cells as the origin of elevated serum alpha1,3fucosyltransferase in association with malignancy. Clin. Exp. Metastasis 18, 605–610 (2000).
Sackstein, R. et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med. 14, 181–187 (2008).
Mondal, N., Silva, M., Castano, A. P., Maus, M. V. & Sackstein, R. Glycoengineering of chimeric antigen receptor (CAR) T-cells to enforce E-selectin binding. J. Biol. Chem. 294, 18465–18474 (2019).
Sackstein, R. Enforced HCELL expression: empowering ‘Step 1’ to optimize the efficacy of mesenchymal stem/stromal cell therapy for stroke and other clinical conditions. Stem Cells 42, 1027–1030 (2024).
Zhang, Y. et al. Overexpression of fucosyltransferase VII (FUT7) promotes embryo adhesion and implantation. Fertil. Steril. 91, 908–914 (2009).
Sackstein, R. The lymphocyte homing receptors: gatekeepers of the multistep paradigm. Curr. Opin. Hematol. 12, 444–450 (2005).
Wan, X. et al. Fucosyltransferase VII improves the function of selectin ligands on cord blood hematopoietic stem cells. Glycobiology 23, 1184–1191 (2013).
Wang, H. et al. α(1,3)-Fucosyltransferases FUT4 and FUT7 Control Murine Susceptibility to Thrombosis. Am. J. Pathol. 182, 2082–2093 (2013).
Jassam, S. A., Maherally, Z., Ashkan, K., Pilkington, G. J. & Fillmore, H. L. Fucosyltransferase 4 and 7 mediates adhesion of non-small cell lung cancer cells to brain-derived endothelial cells and results in modification of the blood–brain-barrier: in vitro investigation of CD15 and CD15s in lung-to-brain metastasis. J. Neurooncol. 143, 405–415 (2019).
Buffone, A. et al. Silencing α1,3-Fucosyltransferases in Human Leukocytes Reveals a Role for FUT9 Enzyme during E-selectin-mediated Cell Adhesion. J. Biol. Chem. 288, 1620–1633 (2013).
Brito, C. et al. Increased levels of fucosyltransferase IX and carbohydrate Lewisx adhesion determinant in human NT2N neurons. J. Neurosci. Res. 85, 1260–1270 (2007).
Osumi, D. et al. Core fucosylation of E-cadherin enhances cell–cell adhesion in human colon carcinoma WiDr cells. Cancer Sci. 100, 888–895 (2009).
Wang, Y. et al. Loss of α1,6-fucosyltransferase suppressed liver regeneration: implication of core fucose in the regulation of growth factor receptor-mediated cellular signaling. Sci. Rep. 5, 8264 (2015).
Liang, W. et al. Core Fucosylation of the T Cell Receptor Is Required for T Cell Activation. Front. Immunol. 9, 78 (2018).
Lu, X. et al. Deficiency of α1,6-fucosyltransferase promotes neuroinflammation by increasing the sensitivity of glial cells to inflammatory mediators. Biochim. Biophys. Acta1863, 598–608 (2019).
Agrawal, P. et al. A systems biology approach identifies FUT8 as a driver of melanoma metastasis. Cancer Cell 31, 804–819 (2017).
Bastian, K. et al. FUT8 Is a Critical Driver of Prostate Tumour Growth and Can Be Targeted Using Fucosylation Inhibitors. Cancer Med. 14, e70959 (2025).
Matsumoto, K., Luther, K. B. & Haltiwanger, R. S. Analysis of endogenous NOTCH1 from POFUT1 S162L patient fibroblasts reveals the importance of the O -fucose modification on EGF12 in human development. Glycobiology 34, cwae047 (2024).
Shi, S. & Stanley, P. Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc. Natl. Acad. Sci. USA 100, 5234–5239 (2003).
Okajima, T. & Irvine, K. D. Regulation of notch signaling by o-linked fucose. Cell 111, 893–904 (2002).
Du, J. et al. O-fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation. Dev. Biol. 346, 25–38 (2010).
Bastian, K., Scott, E., Elliott, D. J. & Munkley, J. FUT8 Alpha-(1,6)-fucosyltransferase in cancer. IJMS 22, 455 (2021).
Liao, C. et al. FUT8 and Protein core fucosylation in tumours: from diagnosis to treatment. J. Cancer 12, 4109–4120 (2021).
Stahl, M. et al. Roles of pofut1 and O-fucose in mammalian notch signaling. J. Biol. Chem. 283, 13638–13651 (2008).
Takeuchi, H. et al. O-Glycosylation modulates the stability of epidermal growth factor-like repeats and thereby regulates Notch trafficking. J. Biol. Chem. 292, 15964–15973 (2017).
Berardinelli, S. J. et al. O-fucosylation stabilizes the TSR3 motif in thrombospondin-1 by interacting with nearby amino acids and protecting a disulfide bond. J. Biol. Chem. 298, 102047 (2022).
Srivastava, P. N., Paul, P. & Mishra, S. Protein O-fucosyltransferase is required for the efficient invasion of hepatocytes by Plasmodium berghei sporozoites. ACS Infect. Dis. 10, 1116–1125 (2024).
Lopaticki, S. et al. Protein O-fucosylation in Plasmodium falciparum ensures efficient infection of mosquito and vertebrate hosts. Nat. Commun. 8, 561 (2017).
Zhang, A. et al. Peters plus syndrome mutations affect the function and stability of human β1,3-glucosyltransferase. J. Biol. Chem. 297, 100843 (2021).
Xie, X. et al. Golgi fucosyltransferase 1 reveals its important role in α-1,4-fucose modification of N-glycan in CRISPR/Cas9 diatom Phaeodactylum tricornutum. Micro. Cell Fact. 22, 6 (2023).
Mercx, S. et al. Inactivation of the β(1,2)-xylosyltransferase and the α(1,3)-fucosyltransferase genes in Nicotiana tabacum BY-2 Cells by a Multiplex CRISPR/Cas9 Strategy Results in Glycoproteins without Plant-Specific Glycans. Front. Plant Sci. 8, https://doi.org/10.3389/fpls.2017.00403 (2017).
Che, Y. et al. Critical involvement of the α(1,2)-fucosyltransferase in multidrug resistance of human chronic myeloid leukemia. Oncol. Rep. 35, 3025–3033 (2016).
Zhan, L., Chen, L. & Chen, Z. Knockdown of FUT3 disrupts the proliferation, migration, tumorigenesis and TGF‑β induced EMT in pancreatic cancer cells. Oncol. Lett. https://doi.org/10.3892/ol.2018.8738 (2018).
Lin, L. et al. FUT3 facilitates glucose metabolism of lung adenocarcinoma via activation of NF-κB pathway. BMC Pulm. Med. 23, 436 (2023).
Do Nascimento, J. C. F., De Oliveira Vasconcelos, A., Seabra, M. A. B. L., Beltrão, E. I. C. & Rocha, C. R. C. The challenge of determining the impact of FUT3 tumor-associated polymorphism rs2306969 (-6951 C> T) in invasive breast cancer cells. Mol. Biol. Rep. 46, 3531–3536 (2019).
Liang, L. et al. miR-125a-3p/FUT5-FUT6 axis mediates colorectal cancer cell proliferation, migration, invasion and pathological angiogenesis via PI3K-Akt pathway. Cell Death Dis. 8, e2968–e2968 (2017).
Guo, J. et al. Fucosyltransferase 5 promotes the proliferative and migratory properties of intrahepatic cholangiocarcinoma cells via regulating protein glycosylation profiles. Clin. Med. Insights Oncol. 17, 11795549231181189 (2023).
Hirakawa, M. et al. Fucosylated TGF-β receptors transduces a signal for epithelial–mesenchymal transition in colorectal cancer cells. Br. J. Cancer 110, 156–163 (2014).
Lu, H.-H. et al. Fucosyltransferase 4 shapes oncogenic glycoproteome to drive metastasis of lung adenocarcinoma. EBioMedicine 57, 102846 (2020).
Wang, Q., Zhang, Y., Chen, H., Shen, Z. & Chen, H. Alpha 1,3-fucosyltransferase-VII regulates the signaling molecules of the insulin receptor pathway. FEBS J. 274, 526–538 (2007).
Nakayama, F. et al. CD15 Expression in mature granulocytes is determined by α1,3-fucosyltransferase IX, but in promyelocytes and monocytes by α1,3-fucosyltransferase IV. J. Biol. Chem. 276, 16100–16106 (2001).
Chen, J. et al. Fucosyltransferase 9 promotes neuronal differentiation and functional recovery after spinal cord injury by suppressing the activation of Notch signaling. ABBS 55, 1571–1581 (2023).
Wang, X. et al. Dysregulation of TGF-β1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc. Natl. Acad. Sci. USA 102, 15791–15796 (2005).
Wang, X. et al. Core fucosylation regulates epidermal growth factor receptor-mediated intracellular signaling. J. Biol. Chem. 281, 2572–2577 (2006).
Zhao, Y. et al. Deletion of core fucosylation on α3β1 integrin down-regulates its functions. J. Biol. Chem. 281, 38343–38350 (2006).
Wang, Y. et al. Loss of αl,6-fucosyltransferase inhibits chemical-induced hepatocellular carcinoma and tumorigenesis by down-regulating several cell signaling pathways. FASEB J. 29, 3217–3227 (2015).
Wei, K.-C. et al. Fucosyltransferase 8 modulates receptor tyrosine kinase activation and temozolomide resistance in glioblastoma cells. Am. J. Cancer Res. 11, 5472–5484 (2021).
Gu, W. et al. Loss of α1,6-fucosyltransferase decreases hippocampal long term potentiation. J. Biol. Chem. 290, 17566–17575 (2015).
Müller, J. et al. O-fucosylation of the notch ligand mDLL1 by POFUT1 is dispensable for ligand function. PLoS ONE 9, e88571 (2014).
Takeuchi, H. et al. Variant in human POFUT1 reduces enzymatic activity and likely causes a recessive microcephaly, global developmental delay with cardiac and vascular features. Glycobiology 28, 276–283 (2018).
Liang, C. et al. Role of poFUT1 and O-fucosylation in placental angiogenesis. Biol. Reprod. 108, 553–563 (2023).
Kakuda, S. & Haltiwanger, R. S. Deciphering the fringe-mediated notch code: identification of activating and inhibiting sites allowing discrimination between ligands. Dev. Cell 40, 193–201 (2017).
Zhang, D. et al. poFUT1 promotes uterine angiogenesis and vascular remodeling via enhancing the O-fucosylation on uPA. Cell Death Dis. 10, 775 (2019).
Benz, B. A. et al. Genetic and biochemical evidence that gastrulation defects in Pofut2 mutants result from defects in ADAMTS9 secretion. Dev. Biol. 416, 111–122 (2016).
Wang, J. et al. RTN4/NoGo-receptor binding to BAI adhesion-GPCRs regulates neuronal development. Cell 184, 5869–5885.e25 (2021).
Team, B. A. A. et al. Protenix - advancing structure prediction through a comprehensive alphaFold3 reproduction. Preprint at https://doi.org/10.1101/2025.01.08.631967 (2025).
Kadirvelraj, R. et al. Structural basis for Lewis antigen synthesis by the α1,3-fucosyltransferase FUT9. Nat. Chem. Biol. 19, 1022–1030 (2023).
Valero-González, J. et al. A proactive role of water molecules in acceptor recognition by protein O-fucosyltransferase 2. Nat. Chem. Biol. 12, 240–246 (2016).
Zemkollari, M., Ruprecht, C., Blaukopf, M., Grabherr, R. & Staudacher, E. Cloning, expression and characterisation of a novel mollusc α-1,2-Fucosyltransferase from Crassostrea gigas (CgFUT2). Glycoconj. J. 41, 255–265 (2024).
Seelhorst, K., Stacke, C., Ziegelmüller, P. & Hahn, U. N-Glycosylations of human α1,3-fucosyltransferase IX are required for full enzyme activity. Glycobiology 23, 559–567 (2013).
De Vries, T. et al. Neighboring cysteine residues in human fucosyltransferase VII are engaged in disulfide bridges, forming small loop structures. Glycobiology 11, 423–432 (2001).
Dupuy, F. Structure/function study of Lewis 3- and 3/4-fucosyltransferases: the 1,4 fucosylation requires an aromatic residue in the acceptor-binding domain. Glycobiology 14, 347–356 (2004).
Ihara, H., Okada, T., Taniguchi, N. & Ikeda, Y. Involvement of the α-helical and Src homology 3 domains in the molecular assembly and enzymatic activity of human α1,6-fucosyltransferase, FUT8. Biochim. Biophys. Acta 1864, 129596 (2020).
Tomida, S., Nagae, M. & Kizuka, Y. The stem region of α1,6-fucosyltransferase FUT8 is required for multimer formation but not catalytic activity. J. Biol. Chem. 298, 102676 (2022).
García-García, A. et al. Structural basis for substrate specificity and catalysis of α1,6-fucosyltransferase. Nat. Commun. 11, 973 (2020).
Kötzler, M. P., Blank, S., Bantleon, F. I., Spillner, E. & Meyer, B. Donor substrate binding and enzymatic mechanism of human core α1,6-fucosyltransferase (FUT8). Biochim. Biophys. Acta 1820, 1915–1925 (2012).
García-García, A. et al. FUT8-Directed core fucosylation of N-glycans is regulated by the glycan structure and protein environment. ACS Catal. 11, 9052–9065 (2021).
Lira-Navarrete, E. et al. Structural insights into the mechanism of protein O-fucosylation. PLOS ONE 6, e25365 (2011).
Lira-Navarrete, E. & Hurtado-Guerrero, R. A perspective on structural and mechanistic aspects of protein O-fucosylation. Acta Cryst. F. 74, 443–450 (2018).
Li, Z. et al. Recognition of EGF-like domains by the Notch-modifying O-fucosyltransferase POFUT1. Nat. Chem. Biol. 13, 757–763 (2017).
McMillan, B. J. et al. Structure of human POFUT1, its requirement in ligand-independent oncogenic Notch signaling, and functional effects of Dowling-Degos mutations. Glycobiology 27, 777–786 (2017).
Piniello, B. et al. Asparagine tautomerization in glycosyltransferase catalysis. the molecular mechanism of protein O -fucosyltransferase 1. ACS Catal. 11, 9926–9932 (2021).
Chen, C.-I. et al. Structure of human POFUT2: insights into thrombospondin type 1 repeat fold and O -fucosylation: Structure of human protein O -fucosyltransferase 2. EMBO J. 31, 3183–3197 (2012).
Järvå, M. A. et al. Structural basis of substrate recognition and catalysis by fucosyltransferase 8. J. Biol. Chem. 295, 6677–6688 (2020).
Sanz-Martínez, I., García-García, A., Tejero, T., Hurtado-Guerrero, R. & Merino, P. The essential role of water molecules in the reaction mechanism of protein O- fucosyltransferase 2. Angew. Chem. Int. Ed. 61, e202213610 (2022).
Liu, Y. et al. Comprehensive multi-omics analysis identifies FUT1 as a prognostic and therapeutic biomarker across pan-cancer. Int. J. Med. Sci. 22, 1313–1328 (2025).
Loong, J. H. C. et al. Glucose deprivation–induced aberrant FUT1-mediated fucosylation drives cancer stemness in hepatocellular carcinoma. J. Clin. Investig. 131, e143377 (2021).
Kawai, S., Kato, S., Imai, H., Okada, Y. & Ishioka, C. Suppression of FUT1 attenuates cell proliferation in the HER2-overexpressing cancer cell line NCI-N87. Oncol. Rep. 29, 13–20 (2013).
Zhang, Z. et al. Suppression of FUT1/FUT4 expression by siRNA inhibits tumor growth. Biochim. Biophys. Acta 1783, 287–296 (2008).
Tan, K.-P. et al. Fucosylation of LAMP-1 and LAMP-2 by FUT1 correlates with lysosomal positioning and autophagic flux of breast cancer cells. Cell Death Dis. 7, e2347–e2347 (2016).
Zhou, W. et al. Clinical significance and biological function of fucosyltransferase 2 in lung adenocarcinoma. Oncotarget 8, 97246–97259 (2017).
Dong, C. et al. FUT2 promotes colorectal cancer metastasis by reprogramming fatty acid metabolism via YAP/TAZ signaling and SREBP-1. Commun. Biol. 7, 1297 (2024).
Liu, P. et al. FUT2 promotes the tumorigenicity and metastasis of colorectal cancer cells via the Wnt/β‑catenin pathway. Int. J. Oncol. 62, 35 (2023).
He, L., Guo, Z., Wang, W., Tian, S. & Lin, R. FUT2 inhibits the EMT and metastasis of colorectal cancer by increasing LRP1 fucosylation. Cell Commun. Signal 21, 63 (2023).
Wang, W. et al. Intestinal epithelium-specific Fut2 deficiency promotes colorectal cancer through down-regulating fucosylation of MCAM. J. Transl. Med. 21, 82 (2023).
Richichi, B. & Sackstein, R. Realizing the promise of ‘La Dolce Vita’ via chemical biology: glycan-motif editing of sLeX for precision cancer therapeutics. FEBS J. 292, 3616–3628 (2025).
Blanas, A., Sahasrabudhe, N. M., Rodríguez, E., van Kooyk, Y. & van Vliet, S. J. Fucosylated Antigens in Cancer: An Alliance toward Tumor Progression, Metastasis, and Resistance to Chemotherapy. Front. Oncol. 8, 39 (2018).
St. Hill, C. A., Krieser, K. & Farooqui, M. Neutrophil interactions with sialyl Lewis X on human nonsmall cell lung carcinoma cells regulate invasive behavior. Cancer 117, 4493–4505 (2011).
Jacobs, P. P. & Sackstein, R. CD44 and HCELL: preventing hematogenous metastasis at step 1. FEBS Lett. 585, 3148–3158 (2011).
Barthel, S. R. et al. Alpha 1,3 fucosyltransferases are master regulators of prostate cancer cell trafficking. Proc. Natl. Acad. Sci. USA 106, 19491–19496 (2009).
Meng, L. et al. High expression of FUT3 is linked to poor prognosis in clear cell renal cell carcinoma. Oncotarget 8, 61036–61047 (2017).
He, C. et al. The DDX39B/FUT3/TGFβR-I axis promotes tumor metastasis and EMT in colorectal cancer. Cell Death Dis. 12, 74 (2021).
Wu, F. et al. FUT3 promotes gastric cancer cell migration by synthesizing Lea on ITGA6 and GLG1, affecting adhesion and vesicle distribution. Life Sci. 359, 123193 (2024).
Do Nascimento, J. C. F., Beltrão, E. I. C. & Rocha, C. R. C. High FUT3 expression is a marker of lower overall survival of breast cancer patients. Glycoconj. J. 37, 263–275 (2020).
Tanaka, H. et al. FUT2 and FUT3 -specific normalization of DUPAN-2 and carbohydrate antigen 19-9 in preoperative therapy for pancreatic cancer: multicentre retrospective study (GEMINI-PC-01). Br. J. Surg. 112, znaf049 (2025).
Blanas, A. et al. Transcriptional activation of fucosyltransferase (FUT) genes using the CRISPR-dCas9-VPR technology reveals potent N-glycome alterations in colorectal cancer cells. Glycobiology 29, 137–150 (2019).
Yan, X., Lin, Y., Liu, S., Aziz, F. & Yan, Q. Fucosyltransferase IV (FUT4) as an effective biomarker for the diagnosis of breast cancer. Biomed. Pharmacother. 70, 299–304 (2015).
Lv, C., Luo, K. & Liu, S. Fucosyltransferase 4 Predicts Patient Outcome in Rectal Cancer through an Immune Microenvironment-Mediated Multi-Mechanism. J. Oncol. 2022, 1–38 (2022).
Liu, C. et al. FUT4 is involved in PD-1-related immunosuppression and leads to worse survival in patients with operable lung adenocarcinoma. J. Cancer Res. Clin. Oncol. 145, 65–76 (2019).
Gao, W. et al. FUT4siRNA augments the chemosensitivity of non-small cell lung cancer to cisplatin through activation of FOXO1-induced apoptosis. BMC Cancer 20, 895 (2020).
Yang, X., Liu, S. & Yan, Q. Role of fucosyltransferase IV in epithelial–mesenchymal transition in breast cancer cells. Cell Death Dis. 4, e735–e735 (2013).
Zhao, L. et al. miR-493-5p attenuates the invasiveness and tumorigenicity in human breast cancer by targeting FUT4. Oncol. Rep. 36, 1007–1015 (2016).
Wu, F. et al. MyoD1 suppresses cell migration and invasion by inhibiting FUT4 transcription in human gastric cancer cells. Cancer Gene Ther. 27, 773–784 (2020).
Aziz, F. et al. Partners in crime: The Lewis Y antigen and fucosyltransferase IV in Helicobacter pylori-induced gastric cancer. Pharmacol. Ther. 232, 107994 (2022).
Wang, Q. et al. FUT6 inhibits the proliferation, migration, invasion, and EGF-induced EMT of head and neck squamous cell carcinoma (HNSCC) by regulating EGFR/ERK/STAT signaling pathway. Cancer Gene Ther. 30, 182–191 (2023).
Yao, Y. et al. LncRNA PART1 promotes malignant biological behaviours associated with head and neck cancer cells via synergistic action with FUT6. Cancer Cell Int. 24, 185 (2024).
Zhang, J., Song, Q. & Hu, W. A functional variant rs10409772 in FUT6 promoter regulates colorectal cancer progression through PKA/CREB signaling. Transl. Oncol. 46, 102011 (2024).
Ales, E. & Sackstein, R. The biology of E-selectin ligands in leukemogenesis. Adv. Cancer Res. 157, 229–250 (2023).
Qiao, R. et al. Identification of FUT7 hypomethylation as the blood biomarker in the prediction of early-stage lung cancer. J. Genet. Genom. 50, 573–581 (2023).
Guo, N. L. et al. A predictive 7-gene assay and prognostic protein biomarkers for non-small cell lung cancer. EBioMedicine 32, 102–110 (2018).
Fang, Y. et al. Novel blood-based FUT7 DNA methylation is associated with lung cancer: especially for lung squamous cell carcinoma. Clin. Epigenet. 14, 167 (2022).
Xu, T. et al. Integrated analysis reveals the participation of IL4I1, ITGB7, and FUT7 in reshaping the TNBC immune microenvironment by targeting glycolysis. Ann. Med. 53, 916–928 (2021).
Liu, M., Zheng, Q., Chen, S., Liu, J. & Li, S. FUT7 Promotes the epithelial–mesenchymal transition and immune infiltration in bladder urothelial carcinoma. JIR 14, 1069–1084 (2021).
Qin, H. et al. FUT7 promotes the malignant transformation of follicular thyroid carcinoma through α1,3-fucosylation of EGF receptor. Exp. Cell Res. 393, 112095 (2020).
Aldonza, M. B. D. et al. Multi-targeted therapy resistance via drug-induced secretome fucosylation. ELife 12, e75191 (2023).
Mao, C., Li, J., Feng, L. & Gao, W. Beyond antibody fucosylation: α-(1,6)-fucosyltransferase (Fut8) as a potential new therapeutic target for cancer immunotherapy. Antib. Ther.6, 87–96 (2023).
Zhang, N.-Z. et al. Core fucosylation and its roles in gastrointestinal glycoimmunology. World J. Gastrointest. Oncol. 15, 1119–1134 (2023).
Ito, Y. et al. Expression of α1,6-fucosyltransferase (FUT8) in papillary carcinoma of the thyroid: its linkage to biological aggressiveness and anaplastic transformation. Cancer Lett. 200, 167–172 (2003).
Xu, H. et al. Targeting fucosyltransferase FUT8 as a prospective therapeutic approach for DLBCL. Oncogenesis 14, 1 (2025).
Zhang, R., Jin, W. & Wang, K. Glycolysis-driven prognostic model for acute myeloid leukemia: insights into the immune landscape and drug sensitivity. Biomedicines 13, 834 (2025).
Talabnin, C. et al. Altered O-linked glycosylation in benign and malignant meningiomas. PeerJ. 12, e16785 (2024).
Du, X. et al. Synthetic retinoid sulfarotene selectively inhibits tumor-repopulating cells of intrahepatic cholangiocarcinoma via disrupting cytoskeleton by P-selectin/PSGL1 N-glycosylation blockage. Adv. Sci. 12, 2407519 (2025).
Li, Z. et al. Diagnostic, prognostic, and immunological roles of FUT8 in lung adenocarcinoma and lung squamous cell carcinoma. PLoS ONE 20, e0321756 (2025).
Dong, G. et al. N6 -methyladenosine -modified circFUT8 competitively interacts with YTHDF2 and miR -186-5p to stabilize FUT8 mRNA to promote malignant progression in lung adenocarcinoma. Thorac. Cancer 14, 2962–2975 (2023).
Ohkawa, Y. et al. Core fucosylation is required for the secretion of and the enzymatic activity of SOD3 in nonsmall-cell lung cancer cells. Antioxid. Redox Signal. 38, 1201–1211 (2023).
Yan, A., Wu, H. & Jiang, W. RACK1 inhibits ferroptosis of cervical cancer by enhancing SLC7A11 core-fucosylation. Glycoconj. J. 41, 229–240 (2024).
Rostami Abookheili, A., Asadi, J., Khosravi, A. & Gorji, A. Fucosyltransferase 3 and 8 promote the metastatic capacity of cancer stem-like cells via CD15s and E-cadherin in esophageal cancer. Iran. J. Basic Med. Sci. 27, 985–995 (2024).
López-Cortés, R., Muinelo-Romay, L., Fernández-Briera, A. & Gil Martín, E. High-throughput mass spectrometry analysis of N -glycans and protein markers after FUT8 knockdown in the syngeneic SW480/SW620 colorectal cancer cell model. J. Proteome Res. 23, 1379–1398 (2024).
Zhang, M. et al. Fusobacterium nucleatum promotes colorectal cancer metastasis by excretion of miR-122-5p from cells via exosomes. iScience 26, 107686 (2023).
López-Cortés, R., Correa Pardo, I., Muinelo-Romay, L., Fernández-Briera, A. & Gil-Martín, E. Core fucosylation mediated by the FucT-8 enzyme affects TRAIL-induced apoptosis and sensitivity to chemotherapy in human SW480 and SW620 colorectal cancer cells. IJMS 24, 11879 (2023).
Wang, P. et al. Fucosyltransferases regulated by fusobacterium nucleatum and act as novel biomarkers in colon adenocarcinoma. JIR 16, 747–768 (2023).
Xin, Z., Wen, X., Zhou, M., Lin, H. & Liu, J. Identification of molecular characteristics of FUT8 and alteration of core fucosylation in kidney renal clear cell cancer. Aging https://doi.org/10.18632/aging.205482 (2024).
Xu, X. et al. A signature based on glycosyltransferase genes provides a promising tool for the prediction of prognosis and immunotherapy responsiveness in ovarian cancer. J. Ovarian Res. 16, 5 (2023).
Qu, J., Wang, M.-H., Gao, Y.-H. & Zhang, H.-W. Identification of molecular subtypes and prognostic features of breast cancer based on TGF-β signaling-related genes. Cancer Inf. 24, 11769351251316398 (2025).
Muranaka, M. et al. Vesicular integral-membrane protein 36 Is involved in the selective secretion of fucosylated proteins into bile duct-like structures in HepG2 cells. IJMS 24, 7037 (2023).
Chen, F. et al. FUT8 is regulated by miR-122-5p and promotes malignancies in intrahepatic cholangiocarcinoma via PI3K/AKT signaling. Cell Oncol. 46, 79–91 (2023).
Guo, Y., Liu, B., Huang, T., Qi, X. & Li, S. HOTAIR modulates hepatocellular carcinoma progression by activating FUT8/core-fucosylated Hsp90/MUC1/STAT3 feedback loop via JAK1/STAT3 cascade. Dig. Liver Dis. 55, 113–122 (2023).
Geng, Z., Huang, Y., Wu, S., Zhu, D. & Li, W. FUT8-AS1/miR-944/fused in sarcoma/transcription factor 4 feedback loop participates in the development of oral squamous cell carcinoma through activation of Wnt/β-catenin signaling pathway. Am. J. Pathol. 193, 233–245 (2023).
Pieri, V. et al. Aberrant L-fucose accumulation and increased core fucosylation are metabolic liabilities in mesenchymal glioblastoma. Cancer Res. 83, 195–218 (2023).
Zhao, Y., Shi, J., Zhao, Y. & Lu, Z. SNHG1/miR-186/FUT8 regulates cell migration and invasion in oral squamous cell carcinoma. Oral. Dis. 29, 105–115 (2023).
Thobias, A. R. et al. Transcriptional scenario of altered glycosylation in HPV-associated cervical cancer. Ind. J. Clin. Biochem. https://doi.org/10.1007/s12291-024-01280-2 (2024).
Xu, T. et al. Correction: Site-specific immunoglobulin G N-glycosylation is associated with gastric cancer progression. BMC Cancer 25, 292 (2025).
Kanto, N. et al. A highly specific antibody against the core fucose of the N-glycan in IgG identifies the pulmonary diseases and its regulation by CCL2. J. Biol. Chem. 299, 105365 (2023).
Wang, Q. et al. Application of the CRISPR/Cas9 gene editing method for modulating antibody fucosylation in CHO cells. in Recombinant Protein Expression in Mammalian Cells (ed. Hacker, D. L.) 2810,249–271 (Springer US, New York, NY, 2024).
Escrevente, C. et al. Different expression levels of α3/4 fucosyltransferases and Lewis determinants in ovarian carcinoma tissues and cell lines. Int. J. Oncol. https://doi.org/10.3892/ijo.29.3.557 (2006).
Liu, S. et al. Differentially expressed mRNAs and their long noncoding RNA regulatory network with helicobacter pylori -associated diseases including atrophic gastritis and gastric cancer. BioMed. Res. Int. 2020, 3012193 (2020).
Guo, J. et al. High-throughput sequencing reveals the differential MicroRNA expression profiles of human gastric cancer SGC7901 cell xenograft nude mouse models treated with traditional Chinese medicine Si Jun Zi tang decoction. Evid. Based Complement. Altern. Med. 2021, 1–12 (2021).
Xu, A. et al. ELF4 contributes to esophageal squamous cell carcinoma growth and metastasis by augmenting cancer stemness via FUT9. ABBS 56, 129–139 (2024).
Blanas, A. et al. FUT9-Driven programming of colon cancer cells towards a stem cell-like state. Cancers 12, 2580 (2020).
Auslander, N. et al. An integrated computational and experimental study uncovers FUT 9 as a metabolic driver of colorectal cancer. Mol. Syst. Biol. 13, 956 (2017).
Yu, F., Lou, S., He, H. & Zhou, Y. Potential role of POFUT1 as a prognostic predictor in low-grade gliomas: Immune microenvironment insights from a pan-cancer analysis. Heliyon 10, e27004 (2024).
Wahby, S. et al. POFUT1 mRNA expression as an independent prognostic parameter in muscle-invasive bladder cancer. Transl. Oncol. 14, 100900 (2021).
Ma, L. et al. Overexpression of protein O-fucosyltransferase 1 accelerates hepatocellular carcinoma progression via the Notch signaling pathway. Biochem. Biophys. Res. Commun. 473, 503–510 (2016).
Li, Q., Wang, J., Ma, X., Wang, M. & Zhou, L. POFUT1 acts as a tumor promoter in glioblastoma by enhancing the activation of Notch signaling. J. Bioenerg. Biomembr. 53, 621–632 (2021).
Du, Y. et al. POFUT1 promotes colorectal cancer development through the activation of Notch1 signaling. Cell Death Dis. 9, 995 (2018).
Chabanais, J., Labrousse, F., Chaunavel, A., Germot, A. & Maftah, A. POFUT1 as a promising novel biomarker of colorectal cancer. Cancers 10, 411 (2018).
Wan, G. et al. Overexpression of Pofut1 and activated Notch1 may be associated with poor prognosis in breast cancer. Biochem. Biophys. Res. Commun. 491, 104–111 (2017).
Barlak, N. et al. Overexpression of POFUT1 promotes malignant phenotype and mediates perineural invasion in head and neck squamous cell carcinoma. Cell Biol. Int. 47, 1950–1963 (2023).
Li, Q. et al. Protein O-fucosyltransferase 1 promotes PD-L1 stability to drive immune evasion and directs liver cancer to immunotherapy. J. Immunother. Cancer 12, e008917 (2024).
Zhang, N. et al. Preliminary study on the mechanism of POFUT1 in colorectal cancer. Med. Oncol. 40, 235 (2023).
Li, D. et al. PLAGL2 and POFUT1 are regulated by an evolutionarily conserved bidirectional promoter and are collaboratively involved in colorectal cancer by maintaining stemness. EBioMedicine 45, 124–138 (2019).
Chen, A. C., Donovan, A., Ned-Sykes, R. & Andrews, N. C. Noncanonical role of transferrin receptor 1 is essential for intestinal homeostasis. Proc. Natl. Acad. Sci. USA 112, 11714–11719 (2015).
Germot, A. & Maftah, A. POFUT1 and PLAGL2 gene pair linked by a bidirectional promoter: the two in one of tumour progression in colorectal cancer? eBioMedicine 46, 25–26 (2019).
Dong, S., Wang, Z. & Xiong, W. POFUT1 promotes gastric cancer progression through Notch/Wnt dual signaling pathways dependent on the parafibromin-NICD1-β-catenin complex. J. Chin. Med. Assoc. 86, 806–817 (2023).
Yokota, S. et al. Protein O-fucosyltransferase 1: A potential diagnostic marker and therapeutic target for human oral cancer. Int. J. Oncol. 43, 1864–1870 (2013).
Chen, Z. et al. Identification of novel serum autoantibody biomarkers for early esophageal squamous cell carcinoma and high-grade intraepithelial neoplasia detection. Front. Oncol. 13, 1161489 (2023).
Jia, Z., Liao, P., Yan, B. & Lei, P. Comprehensive pan-cancer analysis of FUTs family as prognostic and immunity markers based on multi-omics data. Discov. Onc 15, 567 (2024).
Zhang, Y., Cui, K., Qiang, R. & Wang, L. FUT10 is related to the poor prognosis and immune infiltration in clear cell renal cell carcinoma. Transl. Cancer Res. 14, 827-842 (2025).
Zodro, E. et al. FUT11 as a potential biomarker of clear cell renal cell carcinoma progression based on meta-analysis of gene expression data. Tumor Biol. 35, 2607–2617 (2014).
Ruan, W. et al. FUT11 is a target gene of HIF1α that promotes the progression of hepatocellular carcinoma. Cell Biol. Int. 45, 2275–2286 (2021).
Chen, Q., Gao, Y. & Yue, W. Delineating the mechanisms of alpha 1-3 fucosyltransferase FUT11 in ovarian cancer. Ann. Oncol. 30, v10 (2019).
Cao, W. et al. Hypoxia-related gene FUT11 promotes pancreatic cancer progression by maintaining the stability of PDK1. Front. Oncol. 11, https://doi.org/10.3389/fonc.2021.675991 (2021).
Adhikari, E. et al. Brain metastasis-associated fibroblasts secrete fucosylated PVR/CD155 that induces breast cancer invasion. Cell Rep. 42, 113463 (2023).
Huang, Y. et al. FUT11 expression in gastric cancer: its prognostic significance and role in immune regulation. Discov. Oncol. 15, 250 (2024).
Zhang, B., Chen, Y., Gu, X., Zheng, Y. & Jiang, Z. H. Fucosyltransferase 11 restrains ferroptosis via upregulation GPX4 expression in gastric cancer. BMC Cancer 25, 923 (2025).
Cao, W. et al. Knockdown of FUT11 inhibits the progression of gastric cancer via the PI3K/AKT pathway. Heliyon 9, e17600 (2023).
Goto, Y., Uematsu, S. & Kiyono, H. Epithelial glycosylation in gut homeostasis and inflammation. Nat. Immunol. 17, 1244–1251 (2016).
Silva, M., Martin, K. C., Mondal, N. & Sackstein, R. sLeX Expression Delineates Distinct Functional Subsets of Human Blood Central and Effector Memory T Cells. J. Immunol. 205, 1920–1932 (2020).
Liu, K. et al. α1,3 Fucosyltransferase VII improves intestinal immune homeostasis in inflammatory bowel disease by enhancing regulatory T-Cell intestinal homing and immunosuppression. Gastroenterology https://doi.org/10.1053/j.gastro.2025.02.041 (2025).
Kashiwazaki, H. et al. Mice lacking α1,3-fucosyltransferase 9 exhibit modulation of in vivo immune responses against pathogens. Pathol. Int. 64, 199–208 (2014).
Kamio, K. et al. α1,6-Fucosyltransferase (Fut8) is implicated in vulnerability to elastase-induced emphysema in mice and a possible non-invasive predictive marker for disease progression and exacerbations in chronic obstructive pulmonary disease (COPD). Biochem. Biophys. Res. Commun. 424, 112–117 (2012).
Gallenstein, N., Tichy, L., Weigand, M. A. & Schenz, J. Notch signaling in acute inflammation and sepsis. IJMS 24, 3458 (2023).
Duan, C., Wu, J., Wang, Z., Hou, X. & Han, C. Fucosylation in digestive inflammatory diseases and cancers: From mechanical studies to clinical translation. Genes & Diseases 101570 https://doi.org/10.1016/j.gendis.2025.101570 (2025).
Guilmeau, S. et al. Intestinal deletion of Pofut1 in the mouse inactivates notch signaling and causes enterocolitis. Gastroenterology 135, 849–860 (2008).
Etzioni, A. et al. Brief report: recurrent severe infections caused by a novel leukocyte adhesion deficiency. N. Engl. J. Med. 327, 1789–1792 (1992).
Marquardt, T. et al. Leukocyte adhesion deficiency II syndrome, a generalized defect in fucose metabolism. J. Pediatr. 134, 681–688 (1999).
Marquardt, T. et al. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood 94, 3976–3985 (1999).
Dauber, A. et al. Congenital disorder of fucosylation type 2c (LADII) presenting with short stature and developmental delay with minimal adhesion defect. Hum. Mol. Genet. 23, 2880–2887 (2014).
Tinoco, R., Carrette, F., Henriquez, M. L., Fujita, Y. & Bradley, L. M. Fucosyltransferase induction during influenza virus infection is required for the generation of functional memory CD4+ T cells. J. Immunol. 200, 2690–2702 (2018).
Hesselager, M. O., Everest-Dass, A. V., Thaysen-Andersen, M., Bendixen, E. & Packer, N. H. FUT1 genetic variants impact protein glycosylation of porcine intestinal mucosa. Glycobiology 26, 607–622 (2016).
Wang, S. J. et al. Effects of FUT1 gene mutation on resistance to infectious disease. Mol. Biol. Rep. 39, 2805–2810 (2012).
Imbert-Marcille, B.-M. et al. A FUT2 Gene common polymorphism determines resistance to rotavirus A of the P[8] genotype. J. Infect. Dis. 209, 1227–1230 (2014).
Hong, X. et al. Association of fucosyltransferase 2 gene with norovirus infection: A systematic review and meta-analysis. Infect. Genet. Evol. 96, 105091 (2021).
Kaur, P., Gupta, M. & Sagar, V. FUT2 gene as a genetic susceptible marker of infectious diseases: A Review. Int. J. Mol. Epidemiol. Genet. 13, 1–14 (2022).
Iqbal, M. W. et al. Exploring deleterious non-synonymous SNPs in FUT2 gene, and implications for norovirus susceptibility and gut microbiota composition. Sci. Rep. 15, 10395 (2025).
Lin, H.-Y. et al. Clinical significance of the fucosyltransferase 2 (FUT2) secretor status in children hospitalized with acute gastroenteritis in Taiwan. J. Formos. Med. Assoc. 120, 212–216 (2021).
Wacklin, P. et al. Faecal microbiota composition in adults is associated with the FUT2 gene determining the secretor status. PLOS ONE 9, e94863 (2014).
Tong, M. et al. Reprograming of gut microbiome energy metabolism by the FUT2 Crohn’s disease risk polymorphism. ISME J. 8, 2193–2206 (2014).
Wacklin, P. et al. Secretor genotype (FUT2 gene) is strongly associated with the composition of bifidobacteria in the human intestine. PLOS ONE 6, e20113 (2011).
Zhou, S. et al. Correlation of FUT3 and FUT6 gene polymorphisms With Helicobacter pylori Infection. Helicobacter 29, e13122 (2024).
Pan, Q. et al. EGFR core fucosylation, induced by hepatitis C virus, promotes TRIM40-mediated-RIG-I ubiquitination and suppresses interferon-I antiviral defenses. Nat. Commun. 15, 652 (2024).
Takamatsu, S. et al. Core-fucosylation plays a pivotal role in hepatitis B pseudo virus infection: a possible implication for HBV glycotherapy. Glycobiology https://doi.org/10.1093/glycob/cww067 (2016).
Van Pul, L. et al. A genetic variation in fucosyltransferase 8 accelerates HIV-1 disease progression indicating a role for N-glycan fucosylation. AIDS 37, 1959–1969 (2023).
Xu, S. et al. Mapping the landscape of HPV integration and characterising virus and host genome interactions in HPV-positive oropharyngeal squamous cell carcinoma. Clin. Transl. Med. 14, e1556 (2024).
Pan, Q. & Zhang, X.-L. Roles of core fucosylation modification in immune system and diseases. Cell Insight 4, 100211 (2025).
Sun, Y., Li, X., Wang, T. & Li, W. Core fucosylation regulates the function of Pre-BCR, BCR and IgG in humoral immunity. Front. Immunol. 13, 844427 (2022).
Liang, W. et al. Ablation of core fucosylation attenuates the signal transduction via T cell receptor to suppress the T cell development. Mol. Immunol. 112, 312–321 (2019).
Sanz, S. et al. Protein O-fucosyltransferase 2 is not essential for Plasmodium berghei development. Front. Cell. Infect. Microbiol. 9, 238 (2019).
Nordén, R., Nyström, K. & Olofsson, S. Activation of host antiviral RNA-sensing factors necessary for herpes simplex virus type 1-activated transcription of host cell fucosyltransferase genes FUT3, FUT5, and FUT6 and subsequent expression of sLex in virus-infected cells. Glycobiology 19, 776–788 (2009).
Nordén, R., Samuelsson, E. & Nyström, K. NFκB-mediated activation of the cellular FUT3, 5 and 6 gene cluster by herpes simplex virus type 1. Glycobiology 27, 999–1005 (2017).
Nyström, K. et al. Virus-induced transcriptional activation of host FUT genes associated with neo-expression of Ley in cytomegalovirus-infected and sialyl-Lex in varicella-zoster virus-infected diploid human cells. Glycobiology 17, 355–366 (2007).
Becker, D. J. & Lowe, J. B. Fucose: biosynthesis and biological function in mammals. Glycobiology 13, 41R–53R (2003).
Lu, L. et al. In vivo evidence for GDP-fucose transport in the absence of transporter SLC35C1 and putative transporter SLC35C2. J. Biol. Chem. 299, 105406 (2023).
Burkart, M. D. et al. Chemo-enzymatic synthesis of fluorinated sugar nucleotide: useful mechanistic Probes for glycosyltransferases. Bioorg. Med. Chem. 8, 1937–1946 (2000).
Liu, Y., Sweet, I. R. & Boons, G.-J. 2,2-Difluoro derivatives of fucose can inhibit cell surface fucosylation without causing slow transfer to acceptors. JACS Au 4, 3953–3963 (2024).
Okeley, N. M. et al. Development of orally active inhibitors of protein and cellular fucosylation. Proc. Natl. Acad. Sci. USA 110, 5404–5409 (2013).
Pijnenborg, J. F. A. et al. Fluorinated rhamnosides inhibit cellular fucosylation. Nat. Commun. 12, 7024 (2021).
Gilormini, P.-A. et al. A metabolic inhibitor blocks cellular fucosylation and enables production of afucosylated antibodies. Proc. Natl. Acad. Sci. USA 121, e2314026121 (2024).
Carmen Galan, M., Venot, A. P., Phillips, R. S. & Boons, G.-J. The design and synthesis of a selective inhibitor of fucosyltransferase VI. Org. Biomol. Chem. 2, 1376 (2004).
Martin, K. C. et al. Fucosyltransferase-specific inhibition via next generation of fucose mimetics. Chem. Commun. 57, 1145–1148 (2021).
Mong, T. K.-K., Lee, L. V., Brown, J. R., Esko, J. D. & Wong, C.-H. Synthesis of N-acetyllactosamine derivatives with variation in the aglycon moiety for the study of inhibition of Sialyl Lewis x expression. ChemBioChem 4, 835–840 (2003).
Borodkin, V. S. et al. Bisubstrate UDP–peptide conjugates as human O-GlcNAc transferase inhibitors. Biochem. J. 457, 497–502 (2014).
Makwana, V., Sood, R., Malde, A. K., Anoopkumar-Dukie, S. & Rudrawar, S. Bisubstrate uridine-mimetic-peptide conjugates as O -GlcNAc transferase (OGT) inhibitors. ChemistrySelect 9, e202400010 (2024).
Compañón, I. et al. Rational design of dual-domain binding inhibitors for N-acetylgalactosamine transferase 2 with improved selectivity over the T1 and T3 isoforms. JACS Au 4, 3649–3656 (2024).
Manabe, Y. et al. Development of α1,6-fucosyltransferase inhibitors through the diversity-oriented syntheses of GDP-fucose mimics using the coupling between alkyne and sulfonyl azide. Bioorg. Med. Chem. 25, 2844–2850 (2017).
Hosoguchi, K. et al. An efficient approach to the discovery of potent inhibitors against glycosyltransferases. J. Med. Chem. 53, 5607–5619 (2010).
Manabe, Y. et al. Development of a FUT8 Inhibitor with Cellular Inhibitory Properties. Angew. Chem. Int. Ed. 63, e202414682 (2024).
Costa, A. F. et al. Monensin as potential drug for treatment of SLeX-positive tumors. Preprint at https://doi.org/10.1101/2024.03.11.24304048 (2024).
Zheng, Q. et al. miR-200b inhibits proliferation and metastasis of breast cancer by targeting fucosyltransferase IV and α1,3-fucosylated glycans. Oncogenesis 6, e358–e358 (2017).
Streng, B. M. M. et al. Decreased antibody response after severe acute respiratory syndrome coronavirus 2 vaccination in patients with down syndrome. J. Infect. Dis. 226, 673–677 (2022).
Winkelhorst, D. et al. HIP (HPA-screening in pregnancy) study: protocol of a nationwide, prospective and observational study to assess incidence and natural history of fetal/neonatal alloimmune thrombocytopenia and identifying pregnancies at risk. BMJ Open 10, e034071 (2020).
Wang, T. T. et al. Anti-HA glycoforms drive B cell affinity selection and determine influenza vaccine efficacy. Cell 162, 160–169 (2015).
Maamary, J., Wang, T. T., Tan, G. S., Palese, P. & Ravetch, J. V. Increasing the breadth and potency of response to the seasonal influenza virus vaccine by immune complex immunization. Proc. Natl. Acad. Sci. USA 114, 10172–10177 (2017).
Chakraborty, S. et al. Proinflammatory IgG Fc structures in patients with severe COVID-19. Nat. Immunol. 22, 67–73 (2021).
Robinson, S. N. et al. Fucosylation with fucosyltransferase VI or fucosyltransferase VII improves cord blood engraftment. Cytotherapy 16, 84–89 (2014).
Popat, U. et al. Enforced fucosylation of cord blood hematopoietic cells accelerates neutrophil and platelet engraftment after transplantation. Blood 125, 2885–2892 (2015).
Do, K. T. et al. First-In-Human, First-In-Class, Phase I Trial of the Fucosylation Inhibitor SGN-2FF in Patients with Advanced Solid Tumors. Oncologist 26, 925–e1918 (2021).
Kellner, J. et al. Cord blood regulatory T cells for prevention of graft versus host disease. Blood 130, 3246 (2017).
Neelamegham, S. et al. Updates to the symbol nomenclature for glycans guidelines. Glycobiology 29, 620–624 (2019).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Lemoine, F. et al. NGPhylogeny.fr: new generation phylogenetic services for non-specialists. Nucleic Acids Res. 47, W260–W265 (2019).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Pei, J., Kim, B.-H. & Grishin, N. V. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300 (2008).
Acknowledgements
We thank the Spanish MICINN (grants PID2022-137973NB-I00 to P.M. and PID2022-136362NB-I00 to R.H.-G., and predoctoral FPI fellowship to I.S.-M.) and the Gobierno de Aragón (grant E34_20R) with FEDER (2014–2020) funds for “Building Europe from Aragón” for financial support. M.G. acknowledges the Scientific Foundation of the Spanish Association Against Cancer (INVES246008GHIR) for supporting this work. I.Y. acknowledges funding from the Government of Aragón-FEDER (grant E35_23R) for “Building Europe from Aragón”. R.H.-G. acknowledges support of the European Union’s Horizon Europe research and innovation program under the Marie Skłodowska-Curie Actions Grant (Agreement n. 101119601, “GLYCANDRUG”). R.S. acknowledges support from the United States National Institutes of Health/National Cancer Institute grant NCI U01 CA225730 (“Analysis of E-selectin Ligands of Human Acute Leukemia Cells and their Biology in Leukemogenesis”).
Author information
Authors and Affiliations
Contributions
R.S., I.S.-M., and R.H.-G. primarily wrote the review manuscript, with valuable contributions from I.Y., P.M., and M.G. I.Y. conducted the evolutionary analyses presented, while M.G. authored the section focusing on inhibitors. Figures were prepared by I.S.-M., M.G., and I.Y. All authors participated in discussions and critically revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Robert Sackstein has ownership of intellectual property related to “Glycosyltransferase-Programmed Stereosubstitution” (GPS), a technology to effectuate cell surface glycan engineering/glycocalyx editing. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ghirardello, M., Yruela, I., Merino, P. et al. Structure, function, and implications of fucosyltransferases in health and disease. Nat Commun 16, 11279 (2025). https://doi.org/10.1038/s41467-025-66871-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66871-w
