
Abstract
The electrocatalytic synthesis of hydrogen peroxide (H2O2) using carbon-based materials is currently constrained by limited activity levels that fall short of industrially relevant production rates, particularly in acidic electrolytes, as well as a lack of atomic-level understanding of the active motifs. Herein, we utilize well-defined zero-dimensional carbon quantum dots (CQDs) with delicately engineered edge-site oxygen functional groups to elucidate the nature of sp3-hybridized carbon active sites and the promotional effects of aldehyde (–CHO), hydroxyl (–OH), and carboxyl (–COOH) groups in promoting acidic O2-to-H2O2 conversion. Moreover, Ampere-level current densities are successfully achieved by integrating these CQDs into a solid-state electrolyte electrolyzer, resulting in a H2O2 Faradaic efficiency of up to 99.03% and a production rate of up to 3.0 μmol s-1 cm-2 with optimized ionic conduction over CQDs-CHO. Theoretical modeling and calculations reveal that the reconfiguration of carbon edge sites upon functionalization can alter the adsorption behavior of oxygenated intermediates in the 2e− oxygen reduction pathway. Additionally, the combined experimental and theoretical findings underscore the crucial role of electron-withdrawing functional groups in facilitating charge transfer kinetics, thereby enhancing the efficiency of H2O2 electrosynthesis.
Similar content being viewed by others


Introduction
Hydrogen peroxide (H2O2) is among the top ten most energy-intensive chemicals in energy bandwidth studies of U.S. manufacturing sectors1,2,3. However, conventional anthraquinone oxidation process and direct synthesis from a H2 and O2 mixture have significant drawbacks, because they cannot simultaneously achieve safe synthesis and maintain controllable, high concentrations of H2O21,4,5. As a result, decentralized and on-demand electrosynthesis of H2O2 has emerged as a compelling alternative. In this approach, the two-electron oxygen reduction reaction (2e−-ORR) occurs at the cathode of a gas diffusion electrode (GDE)-based electrolyzer to yield H2O26. Substantial efforts have been directed toward the electrochemical production of H2O2 at industrial current densities7,8,9,10. Compared with the extensively investigated 2e−-ORR in alkaline media11,12, the acidic counterparts are garnering increasing attention because of the higher stability of H2O2 over its conjugate base (HO2−)13,14. In this context, several classes of selective 2e−-ORR catalysts15, including noble metal alloys3,16, carbon nanomaterials7,17,18, single-atom catalysts19,20,21, and metal compounds22,23, have been discovered for efficient O2-to-H2O2 conversion in acidic electrolytes. Among these materials, carbon-based nanomaterials stand out owing to their earth abundance, high conductivity, and robust chemical stability12. Notably, Wang and co-workers demonstrated the continuous generation of H2O2 with over 90% selectivity at current densities exceeding 100 mA cm−2 using commercial carbon black catalysts1,7, highlighting the potential of carbon-based platforms for practical H2O2 electrosynthesis.
Despite these progresses on catalyst discovery, the specified structures of the catalytically active sites in carbon-based materials remains unclear. Functional groups, such as epoxy (HC–O–CH), ether (C–O–C), hydroxyl (–OH), and carboxyl (O–C=O) groups, as well as the overall oxygen-dopant content11,12,24 and edge site configurations (zigzag and armchair)25,26,27, have been proposed to influence 2e−-ORR activity and selectivity. In addition, carbon quantum dots (CQDs), a subclass of carbon nanomaterials with well-defined zero-dimensional morphologies, tunable surface functional groups, and narrow size distributions28,29, offer a unique opportunity to elucidate the structural‒performance relationship in 2e−-ORR catalysis. The molecular precision and abundant surface functional groups of H2O2 make it an ideal model system from a fundamental perspective, as well as for scaling up the onsite electrosynthesis of H2O2 from a practical standpoint.
In this study, using CQDs as model catalysts, we investigated how the oxygen functionalization of aldehyde (–CHO), hydroxyl (–OH) and carboxyl (–COOH) groups at the edges of CQDs regulates the acidic 2e−-ORR activity and selectivity. By integrating these CQDs into the cathode of a proton-conducting solid-state electrolyte (SSE) electrolyzer, we achieved disentangled ion conduction and mass transport, resulting in a stabilized triple-phase interface and optimized ionic conductivity for continuous O2-to-H2O2 conversion at a high rate of ~3.0 μmol s−1 cm−2. Theoretical calculations revealed that sp3-hybridized carbon atoms, activated by the electron-withdrawing groups, serve as the true active sites for selective H2O2 production via 2e− ORR, achieving optimal *OOH formation energies and accelerated charge-transfer kinetics.
Results
CQDs synthesis and characterization
Using a solvothermal molecular-fusion strategy, we synthesized three types of CQDs using catechol, phthaldialdehyde, and pathalic acid as carbon precursors (Fig. 1a). The resulting CQDs, designated CQDs-OH, CQDs-CHO, and CQDs-COOH, were characterized via high-resolution (HR-) transmission electron microscopy (TEM) and atomic force microscopy (AFM). As depicted in Fig. 1b–d and Supplementary Fig. 1, the TEM images reveal that all the CQDs exhibit excellent uniformity and monodispersity, with an average diameter of approximately 2 nm. HRTEM images display well-resolved lattice fringes with a spacing of 0.21 nm, which is consistent with the (100) graphite plane. AFM measurements confirmed the ultrathin nature of these CQDs30,31. All three CQDs exhibit bright blue fluorescence under 365 nm UV irradiation (Supplementary Fig. 2 and Supplementary Table 1), with the associated excitation-independent fluorescence behavior illustrated in the three-dimensional fluorescence spectra in Supplementary Fig. 3. The time-resolved photoluminescence spectra further reveal a mono-exponential decay profile for each CQD, indicative of a uniform emissive environment (Supplementary Fig. 4).

a Synthetic strategy for CQDs. TEM and AFM images with their corresponding lateral size distributions and height profiles of b CQDs-COOH, c, CQDs-OH, d CQDs-CHO. Scale bars of TEM, 20 nm; those of HR-TEM, 1 nm. e ATR-IR spectra of the CQDs. f Elemental composition and carbon hybridization as derived from the XPS survey spectra and the deconvoluted C 1 s spectra, respectively. g NEXAFS at the C K-edge, and h deconvoluted C K-edge spectra of the 1 s → π* transition, and i comparison of the sp3-C/sp2-C ratio derived from the deconvoluted NEXAFS spectra. Source data for this figure are provided in the Source Data file.
To further probe their structural features, we employed X-ray diffraction (XRD) and Raman spectroscopy. The XRD patterns revealed characteristic (002) diffraction peaks at 25.6°, 24.5°, and 22.0° for CQDs-CHO, CQDs-OH, and CQDs-COOH, respectively (Supplementary Fig. 5)32. The Raman spectra (Supplementary Fig. 6) exhibit nearly identical intensity ratios of ID/IG (ca. 2.46) for all three types of CQDs, suggesting similar graphitization degrees and/or defect contents. Only a minor difference on the shoulder peak (D*) centered at ~1171 cm−1 is noted, with decreasing peak intensity in the order of CQDs-CHO > CQDs-OH > CQDs-COOH, which is attributed to the sp3-C rich phase of disordered amorphous carbon33,34. The surface functional groups and chemical compositions were analyzed via Attenuated Total Refraction infrared (ATR-IR) spectroscopy and X-ray photoelectron spectroscopy (XPS). The ATR-IR spectra (Fig. 1e) exhibit characteristic absorption bands corresponding to various functional groups: O–H stretching vibrations at 3666 and 3168 cm−1, a carbonyl/aldehyde group at 1777 cm−1, carboxylic acid C = O stretching at 1713 cm−1, C–O stretching at 1058 cm−1, and C–H bending vibrations at 1395 and 1249 cm−1 35,36,37,38. Detailed IR peak assignments are tabulated in Supplementary Table 2. XPS analysis confirmed the exclusive presence of carbon and oxygen in all three types of CQDs (Supplementary Fig. 7 and Supplementary Table 3). The measured C/O atomic ratios are 3.8, 3.5, and 3.0 for CQDs-CHO, CQDs-OH, and CQDs-COOH, respectively (Fig. 1f). The high-resolution C 1 s spectra (Supplementary Figs. 8–10 and Supplementary Table 4) were deconvoluted into four distinct components: sp2-hybridized carbon of C=C (284.0 eV) and carbonyl groups (C=O, 288.5 eV), as well as sp3-hybridized carbon of C–C (284.5 eV) and C–O bonds (286.1 eV)31,35,39,40,41. Similarly, the O 1 s spectra (Supplementary Tables 5, 6) exhibit two primary peaks at 531.8 eV (C=O) and 533.0 eV (C-O)24, further confirming the successful incorporation of the desired oxygen-containing functional groups.
To further ascertain the chemical nature of C active sites and their coordination structure, near-edge X-ray absorption fine structure (NEXAFS) at the C K-edge has been carried out on these CQDs (Fig. 1g). All CQDs share a similar spectroscopic feature of the 1 s → π* transition at the region of 283.0−289.4 eV and the 1 s → σ* transition at 290.0~303.0 eV42,43. After careful deconvolution (Fig. 1h and Supplementary Table 7), the three CQDs are found to share the same π* excitonic feature of π*A (sp3-C, 284.8 eV), π*B (sp3-C, ~286.9 eV), and π*C (sp2-C, 288.0~289.0 eV) but at different contents44,45, as demonstrated in Fig. 1i. Noteworthy, the observed variations in carbon hybridization align well the deconvoluted XPS (Fig. 1f) and Raman results (Supplementary Fig. 6), indicating that adjacent oxygen functional groups indeed introduce different carbon hybridization of sp3-C and sp2-C sites.
H2O2 production performance evaluation
The intrinsic ORR performance of the CQDs was initially evaluated in an aqueous electrolyte of 0.05 M H2SO4 (pH = 1) via a catalyst-cast rotating ring-disk electrode (RRDE). Nevertheless, owing to their high surface energy, the cast CQDs layer tends to aggregate after drying, resulting in a compromised Faradaic current (Supplementary Fig. 11). To better assess the electrocatalytic performance of CQDs, a porous hydrophobic gas diffusion electrode (GDE) substrate46 with a gaseous O2 feed configuration was employed to compare their intrinsic activity toward H2O2 electrosynthesis, as well as their ability to drive industrial-level current densities in the absence of mass transport limitations. The functionalized CQDs were air-brushed onto a polytetrafluoroethylene (PTFE)-reinforced GDE at a constant loading of 0.6 mg cm-2 (Supplementary Fig. 12). These electrodes were subsequently integrated into a homemade flow cell electrolyzer9, which served as the cathode (Methods), and were exposed to a catholyte of 0.05 M H2SO4 + 0.05 M Na2SO4. The quantification of electro-synthesized H2O2 was performed via the KMnO4 titration method (Supplementary Fig. 13). For example, when the CQDs-CHO/GDE cathode was used, the Faradaic efficiency (FE) of H2O2 reached 91.2% at 50 mA cm−2 and was maintained at 88.9% at 100 mA cm−2 (Supplementary Fig. 14 and Supplementary Table 8). However, significant cell voltages are required to drive bulk electrolysis in flow cell reactors, likely because of the dissolution of CQDs into the liquid electrolyte (vide infra).
To stabilize the catalyst layer and the associated triple-phase interface, a proton-conducting solid-state electrolyte (SSE) electrolyzer was deployed1,47, with an anion exchange membrane (AEM) positioned between the SSE layer and the cathodic catalyst layer (Fig. 2a and Supplementary Fig. 15). The same electrolyte, 0.05 M H2SO4 + 0.05 M Na2SO4, was circulated through the middle SSE layer to produce H2O2. Compared with the flow cell configuration, this SSE electrolyzer setup effectively prevents the direct exposure of the CQDs to the aqueous electrolyte, achieving a total current density of 1 A cm−2 within a 5-V cell voltage for all three types of CQDs cast GDEs (Supplementary Fig. 16). Comparative electrochemical impedance spectroscopy (EIS) measurements at 100 mA cm−2 were conducted for both the flow cell and SSE electrolyzers. The resulting Nyquist plots are presented in Fig. 2b and Supplementary Table 9, while the transformed distribution of relaxation times (DRT, Supplementary Fig. 17) analysis is shown in Fig. 2c to discern specific changes in charge transfer and mass transport processes within the two types of electrolyzers48,49. By utilizing the GDE substrate, a largely mitigated O2 mass transport limitation has been achieved, resulting in a minimal mass transport resistance of P0 (<10 Hz). Five distinct peaks, labeled P1 to P5, were successfully identified via DRT at characteristic frequency ranges. The P1 peak (10–300 Hz) indicates the superposition of charge transfer processes within the anodic and cathodic catalyst layers, where a similar charge transfer resistance was observed for both cells. The P2 peak (300–1400 Hz) can be attributed to the competitive hydrogen evolution reaction (HER) at the cathode, which is associated with a significantly lower H2 FE and increased P2 resistance in the SSE cell than in the flow cell setup with a CQD‒electrolyte interface (Supplementary Fig. 18). Moreover, owing to the solid‒solid interface between the cathode and the AEM, an increased ionic conductivity is expected in the SSE electrolyzer compared with that in the flow cell. This results in decreased ionic transport resistance for the P3 (1500–7000 Hz) and P4 (7500–40,000 Hz) peaks in the former. An additional P5 peak (45,000–160,000 Hz) is observed in the SSE electrolyzer, which is most likely related to the contact resistance between the sandwiched SSE layer and the cation/anion exchange membranes.

a Schematic of H2O2 electrosynthesis over CQDs-cast gas diffusion electrodes via a solid-state electrolyte reactor. b EIS spectra with an inserted equivalent circuit and c the corresponding DRT plots recorded during electrolysis at jtotal = 100 mA cm−2 in the SSE (red line) and flow cell (blue line) electrolyzers, respectively. d Recorded FEH2O2 and e corresponding H2O2 production rates over different CQDs cast GDEs at current densities ranging from 10 to 1000 mA cm−2 using an SSE electrolyzer. f Stable evaluation of CQDs-CHO operated at 100 mA cm−2. g H2O2 electrosynthesis performance map of CQDs-CHO compared with those of previously reported catalysts operated in SSE electrolyzers (see Supplementary Table 14 for details). The cell voltage was recorded with non-iR compensation. The error bars correspond to the s.d. of three independent measurements. Source data for this figure are provided in the Source Data file.
Thereafter, we compared the ORR performance and H2O2 selectivity across a broad current density range from 10 to 1000 mA cm−2. For all three types of CQDs cast GDEs, FEH2O2 increases with jtotal in the range of 10 to 200 mA cm−2 and then gradually decreases for both CQDs-OH and CQDs-COOH (Fig. 2d and Supplementary Tables 10, 11). In contrast, CQDs-CHO maintains an FEH2O2 exceeding 95.0% across a wide current density range from 100 to 600 mA cm−2, reaching a maximum of 99.03% at 400 mA cm−2 (Supplementary Table 12). We further plotted the partial current density of H2O2 (jH2O2) in Supplementary Fig. 19, where a plateau at ~600 mA cm−2 is noted for CQDs-CHO, corresponding to a H2O2 production rate of 3.0 μmol s−1 cm−2, which is ~1.6 and 1.9 times as high as that of CQDs-OH and CQDs-COOH, respectively (Fig. 2e). This performance variation validates that the H2O2 selectivity is significantly influenced by the surface oxidation functional groups, as reported in previous studies12,24. The bare GDE-catalyzed ORR in the absence of a catalyst layer of CQDs was also measured for comparison. This delivered a maximum FEH2O2 of 56.1% at a jtotal of 100 mA cm−2 and a maximum jH2O2 of 173.1 mA cm−2 at a cell voltage of 5.5 V (Supplementary Fig. 20), thereby ruling out its contribution to the aforementioned 2e–-ORR performance of the CQDs.
In addition to its excellent electrocatalytic activity and selectivity, the CQDs-CHO also exhibit excellent stability for O2-to-H2O2 conversion, maintaining ~90% FEH2O2 for over 100 hours of continuous electrolysis at 100 mA cm−2, generating ~0.18 mol of H2O2 in a 1 cm2 model electrolyzer (Fig. 2f and Supplementary Fig. 21). At an elevated current density of 400 mA cm−2, ca. 0.24 mol of H2O2 was electrosynthesized during 30-hour of continuous electrolysis (Supplementary Fig. 22). Offsite ATR-IR spectra were collected on the spent CQDs-CHO/GDE after disassembling the SSE cell at different operation time under 400 mA cm−2 (Supplementary Fig. 23), which confirm the satisfied structural stability of -CHO functional groups under the high-rate 2e−-ORR process. Furthermore, a pure H2O2 solution can be generated via this SSE electrolyzer with deionized H2O flowing through the middle layer (Supplementary Fig. 24 and Supplementary Table 13). Considering the H2O2 selectivity, partial current density, operational current density, and long-term stability, the developed CQDs-CHO ranks as the best carbon-based 2e–-ORR catalyst (Fig. 2g and Supplementary Table 14).
Besides, we also collected the electrolyte from the SSE layer after 100 hours of electrolysis and compared its ATR-IR spectra with that of the catholyte from a flow cell electrolyzer operated for 1 and 15 hours, using dissolved CQDs-CHO in the same electrolyte as a reference. As shown in Supplementary Fig. 25a, the characteristic peak of C=O stretching, located at 1710 cm−1, is clearly observed in the flow cell catholyte after 15 h of electrolyte exposure. This observation suggests the dissolution of CQDs-CHO from the catalyst layer into the aqueous electrolyte. In contrast, a -CHO-free feature is noted in the electrolyte after 100 h of operation in the SSE electrolyzer, underscoring the stability of the catalyst layer structure at the cathode–AEM interface within the SSE reactor (Supplementary Fig. 25b, c). As a result, although the SSE electrolyzer results in greater contact resistance than does the flow cell setup (Fig. 2c), its stable catalyst layer structure facilitates lower cathodic polarization, leading to more efficient H2O2 electrosynthesis.
Mechanistic understanding of the effect of oxygen functionalization on H2O2 selectivity
In the literature, carbon atoms adjacent to oxygen functional groups have been identified as the active sites for the 2e−-ORR in alkaline electrolytes11. The overall oxygen content, or the O/C ratio, is proposed as a selectivity descriptor for O2-to-H2O2 conversion12,24. In this context, we plotted the trend of H2O2 selectivity over three types of CQDs against the determined atomic contents of oxygen (Fig. 3a), sp2-C (Fig. 3b) and sp3-C (Fig. 3c), as well as their relative ratios (Fig. 3d) at industrially relevant current densities of 200, 400 and 600 mA cm−2. A clear negative correlation between H2O2 selectivity and both the overall O content and the sp2-C content was observed. For example, CQDs-COOH has the highest O/C atomic ratio (0.32, Fig. 1i) over the other two counterparts, but delivers the lowest H2O2 selectivity at all three current densities applied. Another control experimental using CQDs-CHO with the same O/C atomic ratio but different content of -CHO group also rules out the primary contribution of 2e−-ORR selectivity from the overall oxygen content (Supplementary Figs. 26-29 and Supplementary Table 15). In contrast, the sp3-C content and/or the relative ratio of sp3-C to sp2-C carbon effectively describes the trend in H2O2 selectivity, highlighting the crucial role of sp3-C sites in the acidic 2e− ORR pathway.

The effects of atomic content of a oxygen, b sp2 carbon, c sp3 carbon and d relative ratio of sp3 to sp2 carbon on the H2O2 selectivity at industrially relevant current densities for three types of CQDs. Source data for this figure are provided in the Source Data file.
To elucidate the underlying factors governing the ORR activities as observed among the three functionalized CQDs, density functional theory (DFT) calculations were subsequently conducted. Atomic models (Fig. 4a, Supplementary Videos 1-2) for three CQDs with diameters of 2.2, 2.0, and 1.8 nm were constructed based on HRTEM experimental measurements (Fig. 1b). Moreover, the distribution of functional groups (–COOH, –OH, and –CHO) and their relative contents derived from XPS results are largely reproduced in our simulated models (Fig. 4b–4d), exhibiting excellent consistency with the experimental results (Supplementary Fig. 30 and Supplementary Table 16). Given the known correlation between decreased *OOH formation energies (ΔG*OOH) and enhanced 2e⁻-ORR performance50, we systematically calculated ΔG*OOH for the three CQDs. Noteworthy, the predicted ΔG*OOH values were obtained by averaging the energetics over a sufficiently sampled set of active sites (Supplementary Data 1, Supplementary Figs. 31-33), which reflect statistically averaged behavior rather than the activity of a single model site. As shown in Fig. 4e, significantly lower than those of CQDs-OH (4.62 eV) and CQDs-CHO (4.70 eV), CQDs-COOH exhibited the most favorable ∆G*OOH value (4.49 eV), which is close to the theoretical optimum (~4.22 eV), strongly supporting the experimentally observed enhanced selectivity of 2e⁻- over 4e⁻-ORR. Further sites-specified adsorption analysis (Fig. 4e and Supplementary Fig. 34) demonstrated that that sp3-hybridized carbon sites contribute most to the 2e⁻-ORR activity, as their stronger adsorption stabilizes intermediates more effectively than sp2-C sites. This enhancement originates from electronic modulation by the –CHO group, as evidenced by a systematic 0.46 eV upshift in the C 1 s core-level from CQDs-COOH to CQDs-CHO (Fig. 4f). The linear correlation between C 1 s binding energies and ΔG*OOH (Fig. 4f) highlights the crucial role of electron-withdrawing functional groups in promoting charge-transfer kinetics. Notably, the slightly weaker ΔG*OOH on CQDs-CHO facilitates a stable jH2O2 of ~500 mA cm-2 across a wide operational voltage range (Fig. 2d, 2e), while XPS, Raman and NEXAFS characterizations confirm that it possesses the highest sp3-C site density (Fig. 1 and Supplementary Fig. 6). Consequently, -CHO groups, through their electron-withdrawing effect, optimize the electronic structure of edge carbon atoms, thereby enhancing the 2e⁻-ORR performance.

a Simulation models of CQDs, CQDs-COOH, CQDs-OH and CQDs-CHO. Simulated C 1 s XPS of b CQDs-COOH, c CQDs-OH and d CQDs-CHO. The absolute binding energy values of the calculated C 1 s core-level full spectra of CQDs are calibrated to the well-established C-C peak position at 284.8 eV. e Distribution of *OOH formation free energies (ΔG*OOH) on three CQDs, highlighting differences between sp2-C and sp3-C adsorption sites. The statistics distributions of ∆G*OOH are obtained from the calculation of sp3-C sites on three CQDs, f Correlation between the relative C 1 s core-level shifts and the average ∆G*OOH adsorption energies on three CQD variants. The dashed line denotes the optimal energy for 2e⁻-ORR, while the solid line is a linear fit. For clarity, the absolute C 1 s core-level position of CQDs-COOH is referenced as 0 eV. Source data for this figure are provided in the Source Data file.
Scale-up H2O2 production and its onsite utilization
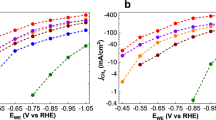
The O2-to-H2O2 performance of CQDs-CHO was further evaluated in an SSE cell with an effective electrode surface area of 100 cm2 (Fig. 5a and 5b) to facilitate large-scale synthesis51. A 100 cm2 IrO2-coated Ti mesh was applied as the anode, and either pure water or an acidic electrolyte of 0.05 M H2SO4 + 0.05 M Na2SO4 was circulated through the middle SSE layer. In line with the 1-cm2 cell electrolysis result, deploying the acidic electrolyte instead of pure water significantly enhanced the ORR rate, as indicated by the overall current (Fig. 5c). Specifically, the cell voltage decreased by 2.72 V at a given current of 20.0 A, attributable to reduced Ohmic resistance. Besides, the maximum H2O2 FE increased from 73.5 ± 1.5% in pure water to 83.2 ± 1.7% in the acidic electrolyte (Fig. 5d), giving rise to a partial current density of 158.3 mA cm−2 and a production rate of 82.9 μmol s−1 (Fig. 5e) for practical H2O2 production.

Digital photos of a SSE elelctrolyzer with an electrode surface area of 1-cm2 or 100-cm2, and b the operational system. c LSV curves recorded in the 100-cm2 SSE electrolyzer at a scan rate of 10 mV s−1 using 0.05 M H2SO4 + 0.05 M Na2SO4 (acidic condition, solid line) or pure water (dashed line) flowing through the middle SSE layer. d Recorded FEH2O2 and e corresponding partial current densities over CQDs-CHO cast GDEs at current ranging from 1.0 to 20.0 A in the 100-cm2 SSE electrolyzer. The error bars correspond to the s.d. of three independent measurements. f Continuous electrolysis at 10.0 A for the degradation of Eosin-Y in pure water. g Time-course UV-Vis spectra of Eosin-Y-containing solution. The concentration of Eosin-Y was 0.1 M, and the volume of electrolyte reservoir was 1 L. Source data for this figure are provided in the Source Data file.
Last but not least, Fig. 5f demonstrates the onsite removal of Eosin-Y using a Fenton process driven by electro-synthesized H2O2 from the CQDs-CHO/GDE-integrated 100 cm2 SSE electrolyzer operated at 10.0 A. During 13.6-h continuous electrolysis, the aqueous Eosin-Y solution gradually changed from pink to colorless, accompanied by a progressive decrease in its characteristic absorbance peak at ~520 nm, as monitored by ultraviolet-visible (UV-Vis) spectroscopy (Fig. 5g)52. These results demonstrate the practical feasibility of this approach for wastewater treatment.
Discussion
In summary, by deploying oxygen-functionalized carbon quantum dots and solid-state electrolyte cells, we investigated the physicochemical nature of the carbon active sites that drive the acidic 2e– oxygen reduction reaction under well-controlled mass transport and ionic conduction conditions. Among the three types of CQDs screened, the sp3-hybridized carbon at edge sites adjacent to the –CHO groups presented the highest H2O2 electrosynthesis efficiency, achieving a Faradaic efficiency of 99.03% at a total current density of 400 mA cm-2 and maintaining continuous electrolysis for over 100 hours with a stabilized triple-phase interface. Furthermore, through a combined experimental and theoretical approach, we reveal that this state-of-the-art H2O2 electrosynthesis performance is closely correlated with the atomic content of sp3 carbon sites and the electron-withdrawing characteristics of oxygen functional groups rather than the simplified overall oxygen content. Given the wide variety of carbon-based materials and their corresponding catalytic reactions, this work provides insights for the rational design of more efficient electrocatalysts and their integration in advanced electrosynthesis systems.
Methods
Materials
Catechol, phthaldialdehyde, pathalic acid and ethanol were purchased from Adamas (Shanghai Titan Scientific Co., Ltd., China) and used without further purification. Sulfuric acid (H2SO4, 96%) and hydrogen peroxide (H2O2, 30%) were purchased from Sinopharm Chemical Reagent. O2 (99.999%) gas was purchased from Shanghai Wetry Standard Gas Analysis Technology Company. Vulcan XC-72 was purchased from Fuel Cell Store. Catechol (RG, 99%), phthaldialdehyde (RG, 99%), pathalic acid (RG, 99%), and ethanol (AR, 95%) were purchased from Adamas (Shanghai Titan Scientific Co., Ltd., China) and used without further purification. Eosin-Y was purchased from Sinopharm Chemical Reagent Co., Ltd. Millipore water at a resistance of 18.2 MΩ·cm was used throughout the experiments. The electrolytes were prepared on a daily basis and stored at ambient conditions.
Synthesis of CQDs
CQDs were synthesized via a refined one-pot solvothermal method on the basis of a previously reported protocol53. Typically, phthalaldehyde (0.5 g) was dissolved in ethanol (50 mL) under magnetic stirring to form a homogeneous solution. This solution was then transferred into a 100 mL Teflon-lined stainless-steel autoclave and heated at 180 °C for 12 h. After naturally cooling to room temperature, the resulting product was filtered through a 0.22 μm microporous membrane to remove insoluble residues. The filtrate was subsequently concentrated under reduced pressure via a rotary evaporator and subjected to dialysis (molecular weight cutoff: 3500 Da) against deionized water for seven days to remove small-molecule impurities. The purified CQDs-CHO were obtained by drying at 80 °C and stored for further characterization. CQDs-OH and CQDs-COOH were synthesized under identical conditions by replacing phthalaldehyde with catechol and phthalic acid, respectively, as the molecular precursors.
For air annealed CQDs-CHO-A synthesis, pristine CQDs-CHO (50 mg) were placed in a tube furnace under air atmosphere. The temperature was raised to 400 °C at a rate of 5 °C min−1, and maintained for 2 h. Alternatively, 50 mg of pristine CQDs-CHO was dispersed in 15 mL dimethylformamide under stirring. The suspension was then transferred into a 50 mL autoclave and heated at 200 °C for 10 h, followed by dialyzed and dried at 80 °C, to prepare the CQDs-CHO-H.
Material characterization
Photographs were captured via a Nikon D7000 digital camera. The Abs, PL, and PLE spectra were recorded via a Horiba Duetta fluorescence spectrometer. HR-TEM measurements were performed on a JEOL JEM-2100F microscope, operated at 200 keV. XRD patterns were collected on a Rigaku D/max-2500 diffractometer. XPS spectra were measured via a Thermo Fischer ESCALAB 250Xi spectrometer. Raman spectra were measured via a Horiba XploRA PLUS spectrometer with an OLYMPUS confocal microscope. Attenuated total reflection infrared spectra were measured via a Thermo Scientific Nicolet iS50 FT-IR spectrometer with a liquid nitrogen-cooled HgCdTe (MCT) detector.
Electrode preparation
GDE was fabricated by air-spraying polytetrafluoroethylene (PTFE) nanoparticles (100 nm) and carbon black (Vulcan XC-72R) onto a Sigracet 28BC to suppress electrowetting54,55,56. PTFE ink was first prepared by adding 30 mg of carbon black and 90 mg of PTFE in 15 mL of ethanol. After sonicating for 30 min, the PTFE ink was air sprayed onto the GDE at 90 °C. After drying, the final C@PTFE loading was 1 mg cm−2. Cathodes were fabricated by air-spraying the cathode ink onto the fabricated GDE at 120 °C. CQDs ink was prepared by mixing 60 mg of CQDs, 300 μL of Nafion solution (5 wt.%), and 30 mL of ethanol. After drying, the final CQDs loading was ~0.6 mg cm−2.
Electrochemical measurements
The RRDE measurements were run in a standard three-electrode system, with signals recorded by a CHI 760e working station. An RRDE component (AFE7R9, Pine Instruments) consisting of a glassy carbon rotating disk electrode (Φ= 5.61 mm) and a Pt ring (Φ= 6.25 mm) was used, with a theoretical collection efficiency of 37%. Experimentally, the apparent collection efficiency (N) was determined to be 39.0% when the ferrocyanide/ferrocyanide half-reaction system was used at a rotation rate between 400 and 2025 rpm. A high-purity graphite rod (99.995%, Sigma‒Aldrich) and a saturated calomel electrode (SCE) was used as the counter and reference electrodes, respectively. All potentials measured against SCE were converted to the reversible hydrogen electrode (RHE) scale in this work using E (V vs. RHE) = E (V vs. SCE) + 0.242 + 0.059×pH. To prepare the CQDs cast RRDE, typically 3.3 mg of CQDs catalyst was first mixed with 985 μL of ethanol (AR, Sinopharm) and 15 μL of Nafion 117 solution (5 wt.%, Sigma‒Aldrich), which was ultrasonicated for 30 min until a homogeneous catalyst ink formed. Then, 7.5 μL of the catalyst ink was pipetted onto a mirror-polished carbon disk (0.247 cm2, ca. 0.1 mg cm−2 mass loading). During measurement, the Pt ring electrode was held at 1.2 V versus RHE to oxidize the H2O2 formed at the disk electrode. The H2O2 selectivity was calculated via the following equation: \({H}_{2}{O}_{2}{selectivity}\left(\%\right)=200\times \frac{{i}_{{ring}}/N}{|{i}_{{disk}}|+\,{i}_{{ring}}/N}\), where iring is the ring current, idisk is the disk current, and N is the collection efficiency. The number of electrons transferred on the disk electrode in the ORR process (n) was calculated via the following Eq. 1:
A customized flow cell-type electrolyzer consisting of a 1-cm2 CQDs-based GDE cathode and an IrO2-coated Ti mesh anode was deployed for O2-to-H2O2 electrolysis. A quaternary ammonia poly(N-methyl-piperidine-co-p-terphenyl) (QAPPT) membrane and a Nafion-115 membrane (Fuel Cell Store) were used for anion and cation exchange, respectively, with a porous styrene-divinylbenzene sulfonated copolymer (Amberchrom 50WX8, hydrogen form) sandwiched between them as the solid ion conductor57. A 0.05 M H2SO4 + 0.05 M Na2SO4 solution or pure water was used as the catholyte, while the anolyte was 0.1 M H2SO4. The catholyte flow rate was 0.5 mL min−1, as controlled by a syringe pump (Longer Pump BT100-2J) without circulation, and the anolyte was circulated through a peristaltic pump (Runze Fluid) at a flow rate of 2.0 mL min-1. A 30 sccm O2 (99.999%) flow was delivered through the cathodic Ti flow field via an Alicat mass flow controller. After operating at each current density for 30 minutes, liquid products were collected to calibrate the H2O2 yield. Potentiostatic electrochemical impedance spectroscopy (PEIS) was performed at frequencies ranging from 200 kHz to 1 Hz with a 4 cm2 working area of the cathode, and distribution of relaxation times (DRT) analysis was performed on the basis of a Tikhonov regularization method implemented in the commercial software Relaxis48,49. All the electrochemical measurements were conducted at room temperature (~295 K) and atmospheric pressure (0.1 MPa) using a Biologic VSP-300 potentiostat. For the flow cell electrolyzer tests without a PSE layer, a bipolar membrane (BPM, FBM-PK, Fumasep) was used to separate the cathodic and anodic chambers. A titanium plate with an interdigital flow panel was applied at the cathodic side to further enhance O2 mass transport9,58.
The H2O2 production performance in a scaled-up SSE electrolyzer was evaluated using the same procedure as the 1-cm2 system, except with a 100-cm2 CQDs-CHO/GDE cathode and a 100-cm2 IrO2-coated Ti mesh anode, supplied with 200 scmm of cathodic O2 flow.
Notably, the SSE did not use a reference electrode; the measured cell voltage is the potential difference between the anode and cathode, and non-iR compensation was performed. For the flow cell and RRDE, 80%-iR compensation was applied.
Products quantification
The amount of H2O2 generated was determined via permanganate titration (0.02 M KMnO4) according to the following equation: 2 MnO4− + 5 H2O2 + 6 H+ → 2 Mn2+ + 5 O2 ↑ + 8 H2O. The calibration curve for H2O2 quantification was measured by adding ∼16.65 ppm H2O2 solution dropwise into different volumes of a 0.02 M permanganate solution, as shown in Supplementary Fig. 13.
The FEH2O2 was calculated via the following Eq. 2:
where cH2O2 is the concentration of H2O2 as determined by permanganate titration, υ1 is the catholyte flow rate set as 0.5 mL min−1, F is the Faradaic constant, and jtotal is the total current density.
The H2 produced from the electrochemical cell was analyzed via a Shimadzu 2014C gas chromatograph (GC) equipped with a thermal conductivity detector (TCD). UHP Ar was used as the carrier gas, and the constituents of the gaseous sample were separated via two Porapak N80/100 columns packed with molecular sieve-13X. The signal responses of the TCD were calibrated by analyzing a series of standard gas mixtures (Wetry, H2 concentrations of 50.6, 505, 7557, and 101 000 ppm). The Faradaic efficiency of H2 was calculated as:
where xH2 is the volume fraction of H2 as determined by online GC, υ2 is the flow rate monitored by an Alicat mass flow controller, F is the Faradaic constant, V is the molar volume of ideal gas under ORR operation conditions (V = 24.5 L mol−1 at 298 K), and jtotal is the total current density.
Computational methods
The ab initio quantum mechanical (QM) calculations are performed using Vienna Ab initio Simulation Package (VASP) at a version of 5.4.4 with the projector augmented wave (PAW) method and a plane wave basis set. The method is density functional theory (DFT) with generalized gradient approximations (GGA) of Perdew-Burke-Ernzerhof (PBE) functional59. A dispersion correction, DFT-D3 method with Becke-Jonson damping60 was included in the calculations. The energy cut-off is set to 400 eV. Reciprocal space was sampled by Γ-centered Monkhorst-Pack scheme with a grid of 3 × 3 × 1. The partial occupancies for each orbital are set with the first-order Methfessel-Paxton scheme within a broadening factor of 0.2 eV. The dipole moment corrections for the total energy are considered in the direction normal to the surface. The self-consistent electronic step is considered converged when the change of total energy and eigenvalues between two steps is both smaller than 1e-6 eV. All structures were fully relaxed until the force components were less than 0.01 eV/Å. The Gibbs free energy G can be derived as follows:
where EZPE is the zero-point energy, kB is the Boltzmann constant and S is the entropic contribution. The above correction is obtained by using the VASPKIT code61 for a post-processing of the VASP output data.
The XPS simulations were conducted to predict the core-level energies of 1 s orbital for all carbon atoms of CQDs implemented in VASP at the PBE-D3 level using the initial state approximation method to predict core-level shift because previous studies have shown that such an approach is reliable in reproducing the relative binding energy change as measured experimentally62.
Data availability
The data generated in this study are provided in the Supplementary Information. Source data are provided in this paper. Source data are provided with this paper.
References
Xia, C., Xia, Y., Zhu, P., Fan, L. & Wang, H. Direct electrosynthesis of pure aqueous H2O2 solutions up to 20% by weight using a solid electrolyte. Science 366, 226–231 (2019).
Tang, J. et al. Selective hydrogen peroxide conversion tailored by surface, interface, and device engineering. Joule 5, 1432–1461 (2021).
Deng, Z., Choi, S. J., Li, G. & Wang, X. Advancing H2O2 electrosynthesis: enhancing electrochemical systems, unveiling emerging applications, and seizing opportunities. Chem. Soc. Rev. 53, 8137–8181 (2024).
Edwards, J. K. et al. Switching off hydrogen peroxide hydrogenation in the direct synthesis process. Science 323, 1037–1041 (2009).
Yang, S. et al. Toward the decentralized electrochemical production of H2O2: a focus on the catalysis. ACS Catal. 8, 4064–4081 (2018).
Wen, Y. et al. Electrochemical reactors for continuous decentralized H2O2 production. Angew. Chem. Int. Ed. 61, e202205972 (2022).
Zhang, X. et al. Electrochemical oxygen reduction to hydrogen peroxide at practical rates in strong acidic media. Nat. Commun. 13, 2880–2890 (2022).
Fan, M. et al. N-B-OH site-activated graphene quantum dots for boosting electrochemical hydrogen peroxide production. Adv. Mater. 35, e2209086 (2023).
Ni, B. et al. Second-shell N dopants regulate acidic O2 reduction pathways on isolated Pt sites. J. Am. Chem. Soc. 146, 11181–11192 (2024).
Gu, Y. et al. Industrial electrosynthesis of hydrogen peroxide over p-block metal single sites. Nat. Synth. 4, 614–621 (2025).
Kim, H. W. et al. Efficient hydrogen peroxide generation using reduced graphene oxide-based oxygen reduction electrocatalysts. Nat. Catal. 1, 282–290 (2018).
Lu, Z. et al. High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials. Nat. Catal. 1, 156–162 (2018).
Zhang, J.-Y., Xia, C., Wang, H.-F. & Tang, C. Recent advances in electrocatalytic oxygen reduction for on-site hydrogen peroxide synthesis in acidic media. J. Energy Chem. 67, 432–450 (2022).
Siahrostami, S. et al. Enabling direct H2O2 production through rational electrocatalyst design. Nat. Mater. 12, 1137–1143 (2013).
Siahrostami, S. et al. A review on challenges and successes in atomic-scale design of catalysts for electrochemical synthesis of hydrogen peroxide. ACS Catal. 10, 7495–7511 (2020).
Zhang, X. et al. Optimizing the Pd sites in pure metallic aerogels for efficient electrocatalytic H2O2 production. Adv. Mater. 35, 2211512 (2023).
Iglesias, D. et al. N-Doped graphitized carbon nanohorns as a forefront electrocatalyst in highly selective O2 reduction to H2O2. Chem 4, 106–123 (2018).
Zhang, C. et al. A pentagonal defect-rich metal-free carbon electrocatalyst for boosting acidic O2 reduction to H2O2 production. J. Am. Chem. Soc. 145, 11589–11598 (2023).
Shen, R. et al. High-concentration single atomic Pt sites on hollow CuSx for selective O2 reduction to H2O2 in acid solution. Chem 5, 2099–2110 (2019).
Jung, E. et al. Atomic-level tuning of Co–N–C catalyst for high-performance electrochemical H2O2 production. Nat. Mater. 19, 436–442 (2020).
Zhao, J. et al. Manipulating the oxygen reduction reaction pathway on Pt-coordinated motifs. Nat. Commun. 13, 685–694 (2022).
Sheng, H. et al. Stable and selective electrosynthesis of hydrogen peroxide and the electro-Fenton process on CoSe2 polymorph catalysts. Energy Environ. Sci. 13, 4189–4203 (2020).
Yu, Z. et al. Selective and durable H2O2 electrosynthesis catalyst in acid by selenization induced straining and phasing. Nat. Commun. 15, 9346–9357 (2024).
Guo, Y. et al. Ultrahigh oxygen-doped carbon quantum dots for highly efficient H2O2 production via two-electron electrochemical oxygen reduction. Energy Environ. Sci. 15, 4167–4174 (2022).
Tang, P., Hu, G., Li, M. & Ma, D. Graphene-based metal-free catalysts for catalytic reactions in the liquid phase. ACS Catal. 6, 6948–6958 (2016).
Ding, Y. & Qiao, Z. A. Carbon surface chemistry: new insight into the old story. Adv. Mater. 34, 2206025 (2022).
Jia, Y. et al. Identification of active sites for acidic oxygen reduction on carbon catalysts with and without nitrogen doping. Nat. Catal. 2, 688–695 (2019).
Wang, D.-W. & Su, D. Heterogeneous nanocarbon materials for oxygen reduction reaction. Energy Environ. Sci. 7, 576–591 (2014).
Hu, B. et al. Graphene quantum dot-mediated atom-layer semiconductor electrocatalyst for hydrogen evolution. Nano-Micro Lett. 15, 217–231 (2023).
Guo, H. et al. Nitrogen-rich carbon dot-mediated n→π* electronic transition in carbon nitride for superior photocatalytic hydrogen peroxide production. Adv. Funct. Mater. 34, 2402650 (2024).
Guo, H. et al. Machine learning-guided realization of full-color high-quantum-yield carbon quantum dots. Nat. Commun. 15, 4843–4851 (2024).
Shen, X. et al. Solvent engineering of oxygen-enriched carbon dots for efficient electrochemical hydrogen peroxide production. Small 19, 2303156 (2023).
Lee, K. et al. Structure-controlled graphene electrocatalysts for high-performance H2O2 production. Energy Environ. Sci. 15, 2858–2866 (2022).
Wang, W., Zheng, Y., Hu, Y., Liu, Y. & Chen, S. Intrinsic carbon defects for the electrosynthesis of H2O2. J. Phys. Chem. Lett. 13, 8914–8920 (2022).
Guo, Z. et al. Multi-stimuli-responsive carbon dots with intrinsic photochromism and in situ radical afterglow. Adv. Mater. 36, 2409361 (2024).
Xie, L. et al. Isomerization engineering of oxygen-enriched carbon quantum dots for efficient electrochemical hydrogen peroxide production. Small 20, 2401253 (2024).
Jiang, L. et al. Photoactivated fluorescence enhancement in F,N-doped carbon dots with piezochromic behavior. Angew. Chem. Int. Ed. 59, 9986–9991 (2019).
Zhang, L. et al. Full-color dynamic afterglow in carbon dot-based materials regulated by dual-phosphorescence resonance energy transfer. Small 20, 2406596 (2024).
Chen, L. et al. Precursor symmetry triggered modulation of fluorescence quantum yield in graphene quantum dots. Adv. Funct. Mater. 34, 2401246 (2024).
Li, Y. et al. Accelerated proton dissociation in an excited state induces superacidic microenvironments around graphene quantum dots. Nat. Commun. 15, 6634–6646 (2024).
Si, W., Li, M., Yan, X., Lv, Q. & Huang, C. Porous nitrogen-doped graphdiyne templated from zinc acetylacetonate for enhanced oxygen reduction reaction. ChemPhysMater 4, 274–279 (2025).
Diaz, J., Monteiro, O. R. & Hussain, Z. Structure of amorphous carbon from near-edge and extended x-ray absorption spectroscopy. Phys. Rev. B 76, 094201 (2007).
Díaz, J. et al. Analysis of the π* and σ* bands of the x-ray absorption spectrum of amorphous carbon. Phys. Rev. B 64, 125204 (2001).
Baio, J. E. et al. NEXAFS imaging to characterize the physio-chemical composition of cuticle from African Flower Scarab Eudicella gralli. Nat. Commun. 10, 4758–4765 (2019).
Schultz, B. J. et al. X-ray absorption spectroscopy studies of electronic structure recovery and nitrogen local structure upon thermal reduction of graphene oxide in an ammonia environment. RSC Adv. 4, 634–644 (2014).
Nguyen, T. N. & Dinh, C.-T. Gas diffusion electrode design for electrochemical carbon dioxide reduction. Chem. Soc. Rev. 49, 7488–7504 (2020).
Zhang, G. et al. Electrifying HCOOH synthesis from CO2 building blocks over Cu–Bi nanorod arrays. Proc. Natl. Acad. Sci. 121, e2400898121 (2024).
Qu, Y. et al. Operando diagnosis of MEA-type CO2 electrolyzer via distribution of relaxation times analysis. Acs Energy Lett. 9, 3042–3048 (2024).
Chen, Q., Kube, A., Kopljar, D. & Friedrich, K. A. Advanced impedance analysis for performance degradation during low-temperature CO2 electroreduction. Acs Energy Lett. 9, 6096–6103 (2024).
Kulkarni, A., Siahrostami, S., Patel, A. & Nørskov, J. K. Understanding catalytic activity trends in the oxygen reduction reaction. Chem. Rev. 118, 2302–2312 (2018).
Zhao, E. et al. Optimization and scaling-up of porous solid electrolyte electrochemical reactors for hydrogen peroxide electrosynthesis. Nat. Commun. 16, 3212–3224 (2025).
Alvarez-Martin, A. et al. Photodegradation mechanisms and kinetics of Eosin-Y in oxic and anoxic conditions. Dyes Pigm 145, 376–384 (2017).
Song, D. et al. Carboxylated carbon quantum dot-induced binary metal–organic framework nanosheet synthesis to boost the electrocatalytic performance. Mater. Today 54, 42–51 (2022).
Crandall, B. S. et al. Kilowatt-scale tandem CO2 electrolysis for enhanced acetate and ethylene production. Nat. Chem. Eng. 1, 421–429 (2024).
DuanMu, J.-W., Gao, F.-Y. & Gao, M.-R. A critical review of operating stability issues in electrochemical CO2 reduction. Sci. China Mater. 67, 1721–1739 (2024).
Xing, Z., Hu, L., Ripatti, D. S., Hu, X. & Feng, X. Enhancing carbon dioxide gas-diffusion electrolysis by creating a hydrophobic catalyst microenvironment. Nat. Commun. 12, 136–146 (2021).
Hua, Y., Ni, B. & Jiang, K. Ambient CO2 capture and valorization enabled by tandem electrolysis using solid-state electrolyte reactor. J. Electrochem. 31, 2504082 (2025).
Wan, S. et al. Improving the efficiencies of water splitting and CO2 electrolysis by anodic O2 bubble management. J. Phys. Chem. Lett. 14, 11217–11223 (2023).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Wang, V., Xu, N., Liu, J.-C., Tang, G. & Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033–108051 (2021).
Ye, Y. et al. Dramatic differences in carbon dioxide adsorption and initial steps of reduction between silver and copper. Nat. Commun. 10, 1875–1883 (2019).
Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFA1505100, 2022YFA1503803), the NSFC (22472038, 92472110, 62474105), the Shanghai Science and Technology Innovation Action Plan (22dz1205500), and the Fundamental Research Funds for the Central Universities (20720250005). H.Y. and T.C. acknowledge support from the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Collaborative Innovation Center of Suzhou Nano Science & Technology, the 111 Project, and Joint International Research Laboratory of Carbon-Based Functional Materials and Devices.
Author information
Authors and Affiliations
Contributions
K.J. and L.W. conceptualized the project. H.G. fabricated and characterized the samples with the help of J.C. B.N. developed and conducted electrochemical tests and related data processing with the help of Y.T. and J.W. H.Y. and T.C. carried out DFT calculations. Z.C. and Y.Y. carried out the NEXAFS measurements. H.W. and W.B.C. provided methodology. All authors discussed the results and commented on the paper. T.C., L.W., and K.J. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Junheng Huang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ni, B., Guo, H., Yang, H. et al. Oxygen functionalization of carbon quantum dots enables efficient acidic hydrogen peroxide electrosynthesis. Nat Commun 17, 221 (2026). https://doi.org/10.1038/s41467-025-66920-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66920-4
