
Abstract
Outer hair cells (OHCs) in the cochlea contain specialized stereociliary structures essential for auditory function. These include horizontal top connectors (HTCs), linking adjacent stereocilia and tectorial membrane (TM)-attachment crowns (ACs), anchoring the tallest stereocilia to the TM. The known molecular components of these structures, such as stereocilin, otogelin, otogelin-like, and tubby, lack transmembrane domains, suggesting the existence of anchoring proteins. This study identifies TMEM145, a transmembrane protein with a Golgi dynamics domain, as a crucial OHC stereocilia component. TMEM145 is expressed in both OHCs and spiral ganglion neurons, with specific localization to TM-ACs and HTCs in OHCs. Tmem145 knockout (KO) mice exhibit profound hearing impairment at 3 weeks of age, with complete loss of distortion product otoacoustic emissions, indicating OHC dysfunction. Immunostaining and scanning electron microscopy reveal the absence of TM-ACs and HTCs in Tmem145 KO mice. In heterologous cell systems, TMEM145 interacts with stereocilin and tubby, facilitating their extracellular secretion. TMEM145 is undetectable in Stereocilin KO and tubby mutant mice, indicating interdependence among these proteins. These findings establish TMEM145 as an essential membrane protein for the structural integrity of OHC stereocilia, providing insights into the molecular architecture of cochlear hair cells and their role in auditory function.
Similar content being viewed by others

Introduction
Hair cells in the cochlea are specialized sensory receptors essential for auditory signal transduction and the conversion of mechanical sound vibrations into electrical signals1,2. Among these, the hair bundles of outer hair cells (OHCs) play a crucial role in amplifying sound vibrations and refining the frequency resolution2. The structural integrity of hair bundles is crucial for detecting and responding to mechanical stimuli with high sensitivity. Therefore, a well-organized hair bundle with precisely arranged actin-rich stereocilia is necessary to maintain the efficiency of these auditory mechanisms3. Disruption of this organization because of genetic mutations can result in hearing impairment, a condition that affects millions of individuals worldwide4,5.
OHC stereocilia are interconnected via molecular complexes associated with extracellular links, such as tip links, horizontal top connectors (HTCs), and tectorial membrane (TM)-attachment crowns (ACs), which maintain the well-organized structure and functional integrity of sensory hair cells6,7. Tip links connect the lateral wall of taller stereocilia to the tips of shorter ones and play a crucial role in opening mechanoelectrical transduction channels by transmitting the tension generated from stereocilia movement8. HTCs are zipper-like structures that interconnect adjacent stereocilia within and across rows, thereby contributing to the structural cohesion and stability of the stereocilia bundle. TM-ACs anchor the tallest row of stereocilia to the TM, ensuring proper mechanical coupling for auditory signal transmission. Various molecular components involved in the formation of these links have been identified. Among them, protocadherin 15 (PCDH15) and cadherin 23 (CDH23) form the tip link9,10,11, whereas stereocilin (STRC), otogelin (OTOG), OTOG-like (OTOGL), and tubby (TUB) constitute the TM-AC and HTC structures12,13,14,15. Mutations in these genes have been shown to impair link integrity, leading to disorganization of the hair bundle structure, which results in hearing loss8,12,13,14,15,16,17,18.
Among the proteins involved in hair cell function, STRC, OTOG, OTOGL, and TUB are key contributors to the formation and maintenance of TM-ACs and HTCs. Mutations in STRC, OTOG, and OTOGL are associated with autosomal recessive nonsyndromic hearing loss DFNB16, DFNB18B, and DFNB84B, respectively12,14,19, underscoring their importance in auditory physiology. TUB encodes the protein TUB, a membrane-anchored scaffolding factor that interacts with multiple extracellular link proteins in OHCs18,20. In mouse models, deletion of Tub (tub/tub) results in severe disruption of TM-AC and HTC structures, accompanied by mislocalization of proteins such as STRC and OTOG18. Notably, in the AKR genetic background, a variant Map1a allele specifically rescued STRC localization at TM-ACs, whereas this rescue effect was not observed in the B6 background21,22,23. Although TUB is not associated with human deafness, its function in these complexes appears to be critical for proper stereociliary organization in mice.
Extracellular link proteins STRC, OTOG, and OTOGL are functionally interdependent; thus, the loss of one often affects the localization or stability of the others within the OHC hair bundle16. Loss of any one of these proteins in mice disrupts the hair bundle localization of the other two proteins, with Strc deletion eliminating OTOG and OTOGL from stereocilia, Otogl deletion reducing OTOG and weakening STRC tip labeling, and Otog deletion reducing OTOGL and abolishing STRC tip labeling and its V-shaped imprints in the TM16. These findings highlight the interdependence of these proteins in maintaining TM-ACs and HTCs16.
However, none of these proteins, TUB, STRC, OTOG, and OTOGL, possess a transmembrane domain, raising important questions about how they are transported and anchored to the stereociliary membrane, and ultimately secreted from the stereocilia. Identifying core proteins that possess transmembrane domains and interact with OTOG(OTOGL)-STRC-TUB is crucial for understanding the formation and maintenance of stereociliary links.
To identify the transmembrane proteins involved in the auditory process, we searched the International Mouse Phenotyping Consortium (IMPC) database24 (https://www.mousephenotype.org/) for candidate genes associated with deafness. This search led to the identification of the TMEM145 protein, which consists of a Golgi dynamics (GOLD) domain and a seven-transmembrane domain. A Tmem145 knockout (KO) mouse model exhibited profound hearing impairment. Although the specific role of TMEM145 in auditory physiology remains unclear, these findings suggest that it plays a crucial role in the structural and functional integrity of cochlear hair cells.
The presence of a conserved GOLD domain in TMEM145 is of particular interest. The GOLD domain, first identified in the Wntless (WLS) protein, is known for its role in facilitating protein trafficking and secretion. In WLS, the GOLD domain mediates the binding and transport of WNT ligands, which are crucial for cellular signaling and development25,26,27. Other GOLD domain–containing proteins, such as TMEDs/p24 and FYCO1, have also been implicated in vesicular transport and intercellular communication28. These observations highlight the versatility of the GOLD domain in mediating interactions between intracellular and extracellular environments.
Given the established role of the GOLD domain in other systems, we hypothesized that TMEM145 is a candidate anchor protein for the lateral links of stereocilia, utilizing its GOLD domain to interact with or regulate the localization of proteins essential for stereociliary links, such as STRC and TUB. In this study, we aimed to investigate the functional relationship between TMEM145, STRC, and TUB, with a focus on elucidating the potential role of TMEM145 in mediating these interactions. By integrating genetic, biochemical, and structural approaches, we provide evidence for the central role of TMEM145 in coordinating the assembly and precise localization of STRC and TUB within OHC stereocilia.
Results
Predicted structure of TMEM145
We searched for deafness-associated candidate genes in the IMPC database24 to identify proteins that had not yet been implicated in auditory functions. The identification of these genes could potentially lead to the discovery of previously unreported deafness-associated genes in humans29,30. We concentrated on genes predicted to encode single or multiple transmembrane domains because we believed that these proteins could potentially act as novel receptors that had not been previously identified in auditory signal transduction. Among these candidates, Tmem145 gained our attention as it was predicted to contain seven transmembrane domains31 (Supplementary Fig. 1a).
First, we aimed to gain functional and structural insights by analyzing the predicted structure of TMEM145. According to the predictions generated by AlphaFold332, TMEM145 comprises an N-terminal domain, followed by seven transmembrane domains. Sequence analysis revealed that the first 29 amino acids at the N-terminus serve as a signal peptide (Fig. 1a, b)33. The DALI server34 was used to assess structural similarities between the TMEM145 and known domains within the human proteome predicted by AlphaFold235,36. Analysis of the N-terminal domain revealed significant similarity to the Golgi dynamic (GOLD) domain found in TMED proteins and the SEC14-like protein family (Fig. 1b, Supplementary Fig. 1b, and Supplementary Table 1). Although sequence alignment of SEC14L4 and TMED7 revealed only limited conservation (Supplementary Fig. 1c), the core β-sheet architecture was preserved. An enlarged structural view in Supplementary Fig. 1d further illustrates the characteristic β-sandwich fold of the GOLD domain and shows clusters of hydrophobic residues that are likely important for maintaining structural stability and mediating potential molecular interactions25,28. The GOLD domain is well known for its role in protein trafficking and secretion25. Further analysis of the seven transmembrane domains revealed significant similarities to class A or F G protein-coupled receptors (Fig. 1b, Supplementary Fig. 1c, and Supplementary Table 2). A comparison of the full-length structure of TMEM145 with other predicted structures revealed that the most significant matches were with seven transmembrane proteins containing GOLD domains—WLS, TMEM87A, TMEM87B, TMEM181, GPR107, GPR108, and GPR180 (Fig. 1b, Supplementary Fig. 1d, and Supplementary Table 3). Among these homologs, WLS is the most thoroughly characterized owing to its crucial function in the trafficking and secretion of WNT proteins26,37,38. TMEM87A is implicated in protein trafficking and ion channel activity39,40,41,42. Other members of this family have also participated in trafficking pathways43,44,45,46. Collectively, these findings indicate that TMEM145 may possess structural and functional similarities to proteins involved in protein trafficking and secretion.

a Schematic of TMEM145, showing the N-terminal GOLD domain (yellow), seven predicted transmembrane segments (blue), and the signal peptide (pink). The epitope recognized by the custom antibody (anti-T145-01) is indicated in red near the C-terminus. b AlphaFold332-predicted three-dimensional structure of full-length TMEM145, color-coded to highlight the GOLD domain (yellow), seven-transmembrane bundle (blue), signal peptide (magenta), and additional linker regions (gray). c UMAP projection of single-cell RNA sequencing data, illustrating Tmem145 expression patterns in various cochlear cell types. Each dot represents an individual cell type and the intensity of the purple shading denotes Tmem145 expression level. TBC Tympanic border cell, SGN Spiral ganglion neuron, SpC Spindle cell, RC Root cell, SC Schwann cell, RMC Reissner’s membrane, OHC Outer hair cell, MC Marginal cell, IC Intermediate cell, ISC Inner sulcus cell, IBC Inner border cell, HC Hensen cell, IHC Inner hair cell, FC Fibroblast, BC Basal cell. d Dot plot comparing the average expression levels of Tmem145 and other related genes (Tmem87a, Tmem87b, Tmem181a, Gpr107, Gpr108, and Gpr180) across multiple cochlear cell populations. The color scale reflects mean expression, and the size of each dot indicates the proportion of cells expressing the gene of interest. e Representative Xenium-based spatial transcriptomics density map of Tmem145 expression in a wild-type mouse cochlea cross-section, visualized with a color scale from low (black) to high (yellow). Regions of interest are marked by dashed boxes. f Enlarged view of the areas outlined in (e), focusing on spiral ganglion neurons (SGNs) and outer hair cells (OHCs). g, h Higher-magnification histological sections corresponding to the same areas shown in (f), highlighting Tmem145-positive cells (orange) against hematoxylin/eosin-stained tissue (pink). In (e–h), experiments were independently repeated twice with similar results. Scale bars: (e–h) 200 μm.
Expression of Tmem145 in the organ of Corti
Subsequently, we examined the expression of Tmem145 in mouse organ of Corti. Single-cell RNA sequencing (scRNA-seq) enables high-resolution transcriptional profiling of individual cells, allowing us to identify the specific cell types in which Tmem145 is expressed. scRNA-seq on postnatal day 28 (P28) revealed that Tmem145 was mostly expressed in OHCs; however, its expression was also detected in spiral ganglion neurons (SGNs)47 (Fig. 1c, d, Supplementary Fig. 2). We also conducted spatial transcriptomics, a technique that maps gene expression within intact tissue sections while preserving spatial information, which indicated Tmem145 expression in OHCs and a subset of SGNs, consistent with scRNA-seq results (Fig. 1e–h, Supplementary Fig. 3). SGNs are categorized as type I, which innervates inner hair cells (IHCs), and type II, which innervates OHCs48. Examination of SGNs categorized from the scRNA-seq data revealed that Tmem145 exhibited higher expression in type I SGNs (Supplementary Fig. 4)48.
Next, we investigated the developmental expression pattern of Tmem145. In situ hybridization performed from P1 to P9 demonstrated that Tmem145 mRNA expression began around P4–P5 in both OHCs and SGNs (Supplementary Fig. 5). Additionally, analysis of publicly available RNA-seq datasets further indicated that Tmem145 expression begins around P4 in cochlear hair cells (Supplementary Fig. 6)49. Collectively, these results indicate that Tmem145 is highly expressed in OHCs and a subset of SGNs, particularly in type I SGNs.
Auditory phenotypes of Tmem145 KO mice
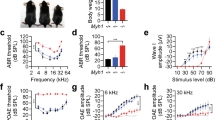
Next, we investigated the role of Tmem145 in auditory function. We generated Tmem145 knockout (KO) mice (Tmem145−/−) by deleting exons 2–12 of Tmem145, located on chromosome 7 of the C57BL6/N strain, using CRISPR-Cas9 (Supplementary Fig. 7). The Tmem145−/− strain was viable and fertile, producing offspring in Mendelian ratios. We then assessed the hearing phenotypes by measuring the auditory brainstem response (ABR). Tmem145−/− mice exhibited severe hearing impairment across all tested frequency ranges by P21, with hearing thresholds significantly increased by 4 weeks of age (Fig. 2a–c; two-sided unpaired t-test in Fig. 2a, **** p < 0.0001). Comparison of wave I amplitude, which represents the synchronized neural response of the auditory nerve to sound stimuli originating from the organ of Corti, revealed that Tmem145−/− mice had a markedly reduced amplitude relative to wild-type (WT) controls, indicating a defect in hair cell- and auditory nerve-mediated sound transmission (Fig. 2d; two-way ANOVA followed by Dunnett’s post hoc test, **** p < 0.0001).

a Auditory brainstem response (ABR) thresholds at P21 and P28 in Tmem145+/+ and Tmem145−/− mice. Red symbols for Tmem145−/− mice indicate significantly elevated thresholds, with some values reaching the upper limit (‘scale out’) of the measuring apparatus. n = 4 for Tmem145+/+ P21; 8 for Tmem145+/+ P28; 6 for Tmem145−/− P21; 9 for Tmem145−/− P28 (p = 3.7e − 6, 3.5e − 5, 4.9e − 7, 2.2e − 6, 9.6e − 9, 7.5e − 6 for P21, and 2.8e − 15, 9.8e − 11, 5.2e − 13, 2.6e − 12, 5.9e − 13, 4.9e − 10 for P28). b, c Representative ABR waveforms at varying stimulus intensities in Tmem145+/+ (b) and Tmem145−/− (c) mice. Each trace represents a 5-dB increase. d ABR input–output (I/O) curves, showing the wave I amplitude as a function of stimulus level in Tmem145+/+ and Tmem145−/− mice at P28. n = 8 for Tmem145+/+; 9 for Tmem145−/− (p = 1.8e − 30). e Distortion product otoacoustic emission (DPOAE) thresholds measured across multiple frequencies in Tmem145+/+ and Tmem145−/− mice at P21 and P28. n = 4 for Tmem145+/+ P21; 6 for Tmem145+/+ P28; 6 for Tmem145−/− P21; 6 for Tmem145−/− P28 (p = 3.7e − 5, 2.0e − 6, 8.4e − 5, 8.8e − 8, 2.2e − 7 for P21, and 5.6e − 5, 1.3e − 6, 2.1e − 5, 2.4e − 6, 4.1e − 5 for P28). f, g Frequency spectra of DPOAEs in Tmem145+/+ (f) and Tmem145−/− (g) mice at a representative stimulus level. Black and red arrows indicate peaks at f1, f2, and their distortion products (2f1-f2 and 3f1-2f2) in Tmem145+/+ and Tmem145−/− mice, respectively. h DPOAE I/O function at 12 kHz, demonstrating reduced responses at P28 in Tmem145−/− compared with Tmem145+/+ mice. n = 6 for Tmem145+/+; 6 for Tmem145−/− (p = 8.2e − 21). i–n Scanning electron microscopy (SEM) images of OHCs in Tmem145+/+ (i–k) and Tmem145−/− (l–n) mice at P21. o, p, r, s Higher-magnification SEM images of OHC stereocilia in Tmem145+/+ (o, p) and Tmem145−/− (r, s) mice at P21. Black arrows indicate preserved tip links. TM-ACs and HTCs are present in Tmem145+/+ mice (o, p, magenta arrows, TM-ACs; white arrows, HTCs) but absent in Tmem145−/− mice (r, yellow arrows). q, t SEM images of imprints on the undersurface of TM in Tmem145+/+ (q) and Tmem145−/− (t) mice at P21. White arrowheads indicate TM imprints in Tmem145+/+ mice. In (i–t), experiments were independently repeated at least three times with similar results. Scale bars: (i–n) 20 µm; (o–t) 5 µm. Statistical analyses in (a) and (e) were conducted using two-sided unpaired t-tests by comparing at the same postnatal day. Statistical analyses in (d) and (h) were conducted using two-way analysis of variance followed by Dunnet’s post hoc test. ****p < 0.0001. Data are presented as mean ± standard error of mean. Source data are provided as a Source Data file.
To further investigate the underlying cause of hearing impairment, we performed distortion-product otoacoustic emission (DPOAE) measurements. DPOAEs are cochlear-generated sound emissions that reflect OHC electromotility, with abnormalities indicating impaired OHC-mediated sound amplification and cochlear dysfunction. Tmem145−/− mice displayed no detectable DPOAE signals across all frequency ranges at P21 (Fig. 2e–g; two-sided unpaired t-test in Fig. 2e, **** p < 0.0001). The analysis of DPOAE amplitude in relation to the input stimulus revealed a significant reduction in Tmem145−/− mice (Fig. 2h; two-way ANOVA followed by Dunnett’s post hoc test, **** p < 0.0001). In contrast, heterozygous mice showed normal hearing phenotypes, indicating that Tmem145 is not haploinsufficient in the auditory system (Supplementary Fig. 8a, b).
Next, we aimed to elucidate the mechanisms underlying hearing loss mediated by OHC defects. Three primary causes were considered: (1) loss of OHCs, (2) impaired mechanical coupling between OHCs and cochlear structures, such as the TMs, which could disrupt sound transmission, and (3) direct or indirect disruption of prestin, the motor protein essential for OHC-driven cochlear amplification50. We performed scanning electron microscopy (SEM) on OHCs from P21 Tmem145+/+ and Tmem145−/− mice to analyze these possibilities. No discernible loss of OHCs was observed in Tmem145−/− mice at low magnification (Fig. 2i, l). A slight loss of OHCs was observed only at P112 in Tmem145−/− mice, suggesting that OHC viability was not the major pathological outcome of TMEM145 loss (Supplementary Fig. 8c). High-magnification SEM images showed disorganized OHC stereocilia bundles in Tmem145−/− mice (Fig. 2j, m), whereas the IHC stereocilia remained structurally intact (Fig. 2k, n). Based on these findings, we further examined the ultrastructure of the OHC stereocilia. Notably, while the tip link was preserved in Tmem145−/− mice (Fig. 2o, r), the TM-ACs and HTCs in OHCs were completely absent in Tmem145−/− mice (Fig. 2p, s). Furthermore, SEM analysis revealed the complete loss of the typical TM imprint in Tmem145−/− mice (Fig. 2q, t), phenocopying Strc KO and tubby mice18. Voltage-clamp recordings of prestin activity in OHCs confirmed comparable nonlinear capacitance between Tmem145+/+ and Tmem145−/− mice (Supplementary Fig. 9a–f; Student’s t-test, p not significant), indicating preserved prestin function in the mutant mice. Consistently, prestin immunostaining revealed similar expression and localization patterns in Tmem145+/+ and Tmem145−/− OHCs (Supplementary Fig. 9g–h).
Single-cell RNA-seq data revealed that Tmem145 is most highly expressed in OHCs, followed by SGNs (Fig. 1d), suggesting a potential role in both cell types. To investigate whether SGNs were affected in Tmem145−/− mice, we examined their histological structure and survival at P21. Hematoxylin and eosin staining of cochlear sections showed no apparent abnormalities in the SGN region of Tmem145−/− mice compared with those in WT mice (Supplementary Fig. 10a–f), and immunostaining with the neuronal marker TUJ1 demonstrated comparable SGN density and organization (Supplementary Fig. 10g–l). These findings indicate that the elevated auditory thresholds observed in Tmem145−/− mice are unlikely to be attributable to SGN degeneration.
The preservation of TM-ACs is essential for the connection between the tallest stereocilia and TM18; thus, among the phenotypes analyzed, the absence of TM-ACs and HTCs in Tmem145−/− mice strongly suggests that their disruption plays a major role in the observed hearing impairment.
TMEM145 localization in the OHC stereocilia
Next, we examined the localization of TMEM145 in OHCs. To validate TMEM145 protein expression, we generated a TMEM145-specific antibody targeting the C-terminal region of the protein (Fig. 1a; anti-T145-01). The evaluation of this antibody in a heterologous system validated its specificity for both human and mouse TMEM145 (Supplementary Fig. 11).
Immunostaining of the organ of Corti samples from P21 Tmem145+/+ and Tmem145−/− mice using anti-T145-01 revealed distinct signals in WT tissue, whereas negligible signals were observed in Tmem145−/− tissue (Fig. 3a, e). Consistent with the SEM findings, we observed substantial disruption in the arrangement of stereocilia in Tmem145−/− mice at P21 (Fig. 3e). Our in situ hybridization results and publicly available RNA-seq datasets suggested that Tmem145 expression in cochlear hair cells begins to increase after P4 (Supplementary Figs. 5 and 6). To further validate and extend this observation with spatial and temporal resolution, we performed whole-mount immunostaining of the cochlea at P5, P9, and P21, analyzing the apex, middle, and basal turns (Supplementary Fig. 12). At P5, the TMEM145 signal was already localized to the stereocilia tips, but its distribution varied across cochlear turns: robustly detected in the basal turn, moderately in the middle turn, and largely absent from the apex (Supplementary Fig. 12a–f). By P9 and P21, TMEM145 localization extended across all cochlear regions, including the apex, consistent with the progression of hair bundle maturation along the base-to-apex gradient (Supplementary Fig. 12g–r). Overall, these results demonstrate that TMEM145 is targeted to OHC stereocilia in a temporally regulated, base-to-apex manner during cochlear development.

a–d Whole-mount immunofluorescence images of stereocilia from Tmem145+/+ mice at P5, P9, and P21. Green signals represent TMEM145 (anti-T145-01), and red signals represent F-actin (phalloidin). a shows whole-mount immunostaining at P21, and b–d are high-magnification views of single outer hair cells at P5, P9, and P21, respectively. In the WT, TMEM145 is evenly distributed along the tips of stereocilia at all stages. e–h Whole-mount immunofluorescence images of Tmem145−/− mice at the same time points. e displays whole-mount immunostaining at P21. f–h High-magnification images at P5, P9, and P21. In the mutant mice, TMEM145 signals are absent, and the stereocilia exhibit disorganized morphology and arrangement compared with controls. i, j Immunogold SEM images of stereocilia from Tmem145+/+ mice at P21. Immunogold labeling was performed using anti-T145-01 (i) or without a primary antibody as a negative control (j). In the anti-T145-01 condition, gold particles were abundantly detected on TM-ACs and HTCs of OHC stereocilia. In (a–j), experiments were independently repeated at least three times with similar results. Scale bars: (a, e) 20 µm; (b–d, f–h) 2 µm; (i, j) 0.5 µm.
A detailed analysis of whole-mount preparations from WT mice at a higher magnification revealed that TMEM145 was localized at the tips of OHC stereocilia, where TM-ACs and HTCs were present (Fig. 3a–d, Supplementary Fig. 13). At P21, its localization to both the single row of TM-ACs and three rows of HTCs was confirmed by phalloidin staining (Supplementary Fig. 13). Sub-immunogold labeling using electron microscopy further confirmed its localization in these structures, providing strong evidence that TMEM145 is specifically enriched in TM-ACs and HTCs of OHC stereocilia at P21(Fig. 3i, j).
To determine when stereocilia disorganization occurs during development, we examined earlier postnatal stages. In mice, OHC stereocilia undergo maturation until P146,51. Analysis of stereocilia morphology and TMEM145 localization at P5 and P9 revealed that TMEM145 was already present at the tips of the tallest stereocilia starting from P5 onward (Fig. 3b, c). This expression pattern of TMEM145 aligns with that of STRC and TUB18. However, the stereocilia arrangement in Tmem145−/− mice remained comparable to that in WT mice at both P5 and P9 (Fig. 3f, g). This suggests that although TMEM145 is present at the tips of the tallest stereocilia from early postnatal stages, it is primarily required for preserving stereocilia organization after they have fully matured.
TMEM145 interacts with STRC and TUB to facilitate their secretion
Next, we investigated the relationship between TMEM145 and other molecular components of TM-ACs and HTCs. Although STRC, OTOG, OTOGL, and TUB have been identified as essential components of TM-ACs and HTCs16,17,18,52, none of these proteins possess a transmembrane domain. Given its membrane-bound nature, we hypothesized that TMEM145 functions as a linker and carrier, facilitating the localization and trafficking of these proteins to the OHC stereociliary membrane.
To test this hypothesis, we investigated whether TMEM145 physically interacts with other TM-AC components. Because of the low expression levels of TMEM145 and TM-AC proteins in Human Embryonic Kidney (HEK) 293 T cells, we heterologously expressed C-terminal FLAG-tagged TMEM145 in HEK293T cells, thereby confirming its robust expression (Supplementary Fig. 14). A cell surface biotinylation assay confirmed the presence of TMEM145 in the plasma membrane (Supplementary Fig. 14a). Furthermore, immunostaining of TMEM145-transfected HeLa cells revealed that TMEM145 was localized to the endoplasmic reticulum (ER), Golgi apparatus, and plasma membrane, showing clear colocalization with the ER and Golgi markers BiP and GM130, respectively (Supplementary Fig. 14b).
We then introduced C-terminal MYC- or V5-tagged STRC and TUB under conditions in which each protein was co-expressed with or without TMEM145 carrying a C-terminal FLAG tag, followed by immunoprecipitation (IP) using FLAG beads. Co-IP (co-IP) of STRC and TUB with TMEM145 revealed physical interactions between TMEM145 and both proteins (Fig. 4a). Subsequent IP experiments using V5 beads to pull down TUB while observing the co-IP of TMEM145 and STRC demonstrated that TMEM145 co-immunoprecipitated with TUB, thus confirming a physical interaction (Fig. 4a). Notably, STRC co-immunoprecipitated with TUB only in the presence of TMEM145 (Fig. 4a). In these experiments, TUB appeared as two distinct bands, both of which co-immunoprecipitated with TMEM145 (Fig. 4a). Western blot analysis of TUB expressed in HEK293T cells after treatment with PNGase F or Endo H showed no change in its expression pattern, indicating that the two bands were not attributable to differential glycosylation (Supplementary Fig. 15). Considering that TUB is a cytosolic protein anchored to the membrane and STRC (with a signal peptide) localizes to the ER lumen or extracellular space, these findings strongly suggest that TUB, TMEM145, and STRC form a physical association in the sequence TUB–TMEM145–STRC.

a HEK293T cells were transfected with TMEM145-FLAG, STRC-MYC, or TUB-V5, followed by immunoprecipitation (IP) using anti-V5 or anti-FLAG affinity matrices. The co-immunoprecipitated proteins (MYC, FLAG, V5) and total cell lysates were analyzed using immunoblotting. Under conditions where TMEM145-FLAG was co-expressed, STRC-MYC and TUB-V5 co-immunoprecipitated, indicating a physical interaction between TMEM145 and STRC/TUB. β-actin (ACTB) was used as a loading control. b Schematic of TMEM145 wild-type (WT) and its deletion mutants (ΔGOLD, Δ7TM). SP indicates the signal peptide (amino acids 1–29). The GOLD domain spans amino acids 31–154 and the seven-transmembrane domain is from amino acids 170–400. ΔGOLD lacks the GOLD domain, whereas Δ7TM lacks the seven-transmembrane region. c HEK293T cells were co-transfected with STRC-MYC and either TMEM145-FLAG, TMEM145-ΔGOLD-FLAG, or TMEM145-Δ7TM-FLAG. Following IP with anti-FLAG, the presence of STRC-MYC in the immunoprecipitates was assessed using immunoblotting with anti-MYC. Deletion of the GOLD domain affects the interaction of TMEM145 with STRC, suggesting that those domains contribute to STRC binding. d Similar co-transfections and immunoprecipitations were performed using TUB-MYC in place of STRC-MYC, with TMEM145-FLAG, TMEM145-ΔGOLD-FLAG, or TMEM145-Δ7TM-FLAG. Deletion of the 7TM domain impaired the interaction of TMEM145 with TUB. e, f Examination of the effect of TMEM145 on the secretion of STRC (e) and TUB (f). Culture supernatants (media) and cell lysates were analyzed using immunoblotting. Secreted STRC or TUB in the media was detected using immunoblotting, and ALDOA served as a loading control for both media and lysates. Both STRC and TUB were secreted only when TMEM145 was co-transfected. In (c–f), input lanes represent 40 μg of total protein lysate loaded to serve as a reference. g Endogenous TMEM145 was detected in Neuro-2a and HEK293T cells using an anti-T145-01 antibody, with ACTB (β-actin) serving as a loading control. In (a–g), experiments were independently repeated three times with similar results. Source data are provided as a Source Data file.
Next, we attempted to identify which region of TMEM145 mediates its interactions with STRC and TUB. Using full-length TMEM145 as a template, we generated two constructs, ΔGOLD and Δ7TM (Fig. 4b). ΔGOLD lacked the GOLD domain but retained the signal peptide and seven-transmembrane domains, whereas Δ7TM lacked the seven-transmembrane and cytoplasmic domains, but contained the GOLD domain. Co-expression of these constructs with C-terminal MYC-tagged STRC followed by FLAG IP showed that STRC interacted with Δ7TM but not with ΔGOLD, indicating that the GOLD domain of TMEM145 is essential for binding to STRC (Fig. 4c). TUB exhibited the opposite behavior, binding only to ΔGOLD and not to Δ7TM, suggesting that the seven-transmembrane domain of TMEM145 mediates TUB binding (Fig. 4d). Collectively, these data indicated that TUB interacts with the transmembrane region of TMEM145 on the cytosolic side, whereas STRC binds to the GOLD domain on the extracellular or luminal side of TMEM145.
Finally, we investigated whether TMEM145 facilitates the secretion of STRC and TUB, similar to the role played by WLS in the trafficking and secretion of WNT37,38. In OHCs, STRC is thought to be secreted to form the extracellular structures of TM-ACs and HTCs17. However, to our knowledge, STRC secretion has never been conclusively proven by showing a substance that regulates its secretion. Co-expression of TMEM145 and STRC in HEK293T cells resulted in the secretion of STRC into the culture medium, whereas STRC remained undetectable extracellularly in the absence of TMEM145 (Fig. 4e). The same phenomenon was observed for TUB, with the lower molecular weight form being secreted at a markedly higher level than the higher molecular weight form (Fig. 4f). Importantly, TMEM145 itself was not secreted into the extracellular space, as shown by western blot analysis of cell lysates and culture media from HEK293T cells overexpressing TMEM145, where TMEM145 was readily detected in cell lysates but was absent from the culture medium (Supplementary Fig. 16).
The overexpression of TUB in Neuro-2a cells has been shown to lead to its secretion53,54, whereas it was not observed in HEK 293 T cells in the present study. This discrepancy prompted us to investigate whether endogenous TMEM145 was responsible for this. Using the anti-T145-01 antibody to detect TMEM145 levels, we discovered that TMEM145 was present in Neuro-2a cells but absent in HEK293T cells, suggesting that cell-type-specific secretion of TUB could be mediated by endogenous TMEM145 (Fig. 4g).
Overall, these in vitro findings confirmed the physical interactions between TMEM145, STRC, and TUB, as well as the role of TMEM145 in promoting the secretion of STRC and TUB, similar to the mechanism by which WLS facilitates WNT secretion.
Structural interactions among proteins that comprise the ultrastructure of OHC stereocilia
Considering the previously demonstrated interactions between TMEM145, STRC, and TUB in a heterologous system, we examined how these elements affect each other in OHC stereocilia. Using validated antibodies from our previous study18, we examined the localization of STRC and TUB in Tmem145+/+ and Tmem145−/− mice. When examining the mid-cochlear region at P5, P9, and P21, both STRC (Fig. 5a–h) and TUB (Fig. 5i–p) were not detected in the TM-ACs and HTCs of Tmem145−/− mice. Furthermore, we conducted additional immunostaining experiments for STRC and TUB in Tmem145+/+ and Tmem145−/− mice at P5, P9, and P21, covering the apex, middle, and basal regions of the cochlea (Supplementary Figs. 17–19). The results showed that STRC expression was completely absent at P9 and P21, whereas at P5, STRC signals were present in the base and mid-base regions (Supplementary Figs. 17 and 18). This finding is consistent with the mRNA data showing that Tmem145 expression begins around P4 (Supplementary Figs. 5 and 6). We therefore suggest that between P1 and P5, other proteins may transiently compensate for STRC localization, but as TMEM145 expression initiates and becomes essential for proper complex formation, its absence leads to defective localization rather than transcriptional downregulation of STRC.

a, b Whole-mount immunofluorescence images of the organ of Corti at P21. Green signals represent STRC, and red signals represent F-actin (phalloidin). c–h Developmental immunofluorescence images of STRC localization in OHC stereocilia in Tmem145+/+ and Tmem145−/− mice at P5, P9, and P21. Compared with Tmem145+/+ mice, STRC signals were absent in the OHC stereocilia of Tmem145−/− mice. i, j Whole-mount immunofluorescence images of the organ of Corti at P21. Green signals represent TUB, and red signals represent F-actin (phalloidin). k–p Developmental immunofluorescence images of TUB localization in OHC stereocilia in Tmem145+/+ and Tmem145−/− mice at P5, P9, and P21. Compared with Tmem145+/+ mice, TUB signals were absent in the OHC stereocilia of Tmem145−/− mice. In (a–p), experiments were independently repeated at least three times with similar results. Scale bars: (a, b, i, j) 20 µm; (c–h, k–p) 2 µm.
We then assessed TMEM145 localization in mouse strains with genetic disruption of other proteins essential for the ultrastructure of OHC stereocilia (Fig. 6a). TMEM145 was absent in the stereocilia of Strc−/− and Otog−/− mice (Fig. 6b, c). To study the role of Tub, we used two different genetic backgrounds. In tub/tub;Map1aB6 mice, in which neither TM-ACs nor HTCs were observed at P2118, TMEM145 was similarly absent (Fig. 6d). In tub/tub;Map1aAKR mice, where TM-ACs were present at P21, but HTCs were not18, TMEM145 was found exclusively at TM-ACs (Fig. 6e). Consistent with previous reports that STRC localization can be rescued in tub/tub mice crossed with the AKR background via a specific Map1a allele, and that this rescue occurs independently of TUB, we found that TMEM145 was localized to TM-ACs even in the absence of TUB. This suggests that, within our complex, TUB may not be as essential for TMEM145 localization as it is for certain other HTC components such as STRC. We also performed whole-mount staining of the P5 cochleae from these mutant mice (Supplementary Fig. 20). TMEM145 failed to localize properly in the absence of STRC, OTOG, or TUB, with the exception of tub/tub;Map1aAKR mice, in which the localization at TM-ACs was largely preserved, consistent with our P21 observations (Fig. 6 and Supplementary Fig. 20).

a Schematic showing the overall structure of OHC stereocilia. The localization of the proteins used in each mouse line is indicated. b–i Whole-mount images of OHC stereocilia from P21 mice immunolabeled for TMEM145 (green) and F-actin (red). Panels depict (b) Strc−/−, (c) Otog−/−, (d) tub/tub;Map1aB6, (e) tub/tub;Map1aAKR, (f) wild-type, (g) Pmca2+/−, (f) Tecta−/−, and (i) Adgrv1−/− genotypes. Each panel shows stereocilia morphology and the localization of TMEM145. All images are shown at similar magnification, with scale bars indicating 2 µm. In (b–i), experiments were independently repeated at least three times with similar results.
To determine whether TMEM145 mislocalization was specifically attributable to the absence of component proteins at TM-ACs or HTCs, we examined its localization in multiple mouse strains deficient in proteins unrelated to these structures (Fig. 6a). These included PMCA2, which is present throughout the stereocilia and functions as a calcium pump55; TECTA, which is specifically localized to the TM and maintains its integrity56,57; and ADGRV1, which establishes ankle links in immature stereocilia58,59. In all three mice lines— Pmca2+/−, Tecta−/−, and Adgrv1−/−—TMEM145 localization was unaffected (Fig. 6f–i). SEM analyses further confirmed the presence of TM-ACs and HTCs in Tecta−/− and Pmca2+/− mice and revealed that although early stereociliary degeneration in Adgrv1−/− mice limited the evaluation of TM-ACs at P5, HTCs were still readily observed (Supplementary Fig. 21). These findings are consistent with the TMEM145 localization data in these mice (Fig. 6 and Supplementary Fig. 20).
Collectively, these results demonstrate that in Tmem145−/− mice, the normal localization of key TM-AC and HTC components was disrupted, and that genetic disruption of these components also caused TMEM145 mislocalization. While the extent of this interdependence may vary among the components, for example, with TUB appearing less critical for TMEM145 localization than STRC, the overall findings underscore the interrelated functions of TMEM145, STRC, TUB, OTOG, and OTOGL in the formation and preservation of OHC stereociliary structures.
Discussion
In this study, we investigated TMEM145, an uncharacterized protein containing a seven-transmembrane protein domain and a GOLD domain. Tmem145−/− mice exhibited severe hearing loss at 3 weeks of age owing to compromised TM-ACs and HTCs in OHCs. Biochemical experiments revealed that TMEM145 physically interacted with STRC and TUB and mediated their secretion (Fig. 7a). In vivo analyses further showed that the absence of TMEM145 impaired the localization of STRC and TUB, and the disruption of these proteins similarly affected TMEM145 localization (Figs. 5 and 6). These findings suggest the potential of TMEM145 as a molecular scaffold for assembling TM-ACs and HTCs and stabilizing their components during or after secretion.

a WNT is trafficked from the endoplasmic reticulum and Golgi to the cell surface with the help of WLS (left panel), and TMEM145 coordinates the proper trafficking of stereociliary proteins, such as STRC and TUB (middle panel). When TMEM145 is absent, these proteins fail to reach the designated location (right panel). b A top view of the organ of Corti with three rows of OHC and one row of IHC (left panel). The 3D illustrations compare Tmem145+/+ (middle panel) and Tmem145−/− (right panel) OHC stereocilia, showing that TMEM145 deficiency causes the loss of TM-AC and HTC with tip link intact and disrupts stereociliary alignment.
Previous studies have suggested that STRC is probably secreted at the tips of the tallest stereocilia, based on the observation that it was detected on the imprints of the lower side of the TM, even when the TM was mechanically detached from the stereocilia17,18. The identification of TMEM145 as a regulator of STRC secretion is an important step in confirming that STRC is a secreted protein. However, the maintenance of STRC at the tip of stereocilia, even after secretion, raises a new question about its mechanism. The first possibility is that the TM anchors STRC to the stereocilia tip and prevents detachment. However, Tecta−/− mice showed the presence of STRC even in the absence of attachment between the stereocilia and TM, dismissing this possibility17 (Supplementary Fig. 22). Throughout this study, the relationship between STRC and TMEM145 was similar to that between WNTs and WLS in the WNT secretion pathway (Fig. 7a). Assuming that the two pairs of molecules share similar regulatory principles, STRC may sit on TMEM145, a transmembrane protein, and dissociate from TMEM145 only when the concentrations of certain molecules are sufficient to displace STRC from TMEM145, just as secreted FZD-related proteins and WNT inhibitory factor 1 release WNTs from WLS in the plasma membrane38. However, the presence of the corresponding extracellular molecules in the endolymph remains unknown. Revealing the identities and changes in various physiological and pathological conditions will further our understanding of the functional stability of secreted STRC in maintaining hearing.
We found that TUB localization to the tip of the stereocilia also requires STRC, suggesting that STRC and TUB are mutually essential for the proper localization of both proteins to the tip of the stereocilia (Supplementary Fig. 23). In addition, OTOG and OTOGL are absent at the tip of stereocilia in Strc−/− mice16. The results of this study further revealed the complexity of these interactions by showing that STRC, TUB, and OTOG all require TMEM145 for their localization to the stereocilia, and vice versa (Figs. 5 and 6). These seemingly reciprocal interactions between the proteins constituting the stereociliary ultrastructure make it difficult to determine which protein plays a more primary and central role. In our previous study18, we observed the transient localization of STRC to the tip of stereocilia at P5 in TUB mice; however, STRC was not present in the stereocilia at P9 or later. This was an unexpected pattern. The disappearance of both proteins in the stereocilia of Tmem145−/− mice at P5 suggests that TMEM145 is the central protein that regulates other secreted proteins, even in immature OHCs. This early organization driven by TMEM145 allows the establishment of more sophisticated regulatory mechanisms in mature OHCs, where each component is strictly interdependent for hair cell functions.
The interactions of these proteins and their arrangement in quaternary structures remain unclear. The information we obtained in this study indicates that TUB and STRC do not directly interact with each other, and that TMEM145 bridges them by interacting with each protein through different domains on TMEM145. The nature of the interactions of OTOG with other proteins remains unknown. However, immunostaining experiments for TUB, STRC, and OTOG provided some clues. To stain these proteins, the TM should be mechanically detached to expose the proteins at the tips of the stereocilia. Although STRC or TUB usually stick to the lower side of the TM in some amounts, in most cases, we can successfully visualize the proteins at the tips of the stereocilia in an even distribution. Conversely, OTOG and OTOGL are not consistently detected at the tips of the OHC hair bundles, with varied immunostaining intensities between the experiments. Tecta−/− mice, where TM is inherently detached from the cochlear epithelium, showed consistent immunostaining of OTOG and OTOGL proteins16, indicating weak adherence of OTOG to the tips of stereocilia during TM detachment. Furthermore, Otog−/− and Otogl−/− mice showed markedly weaker STRC immunostaining signals at the tips than WT mice; however, a significant signal intensity remained16. This is in contrast to the experiment where OTOG and OTOGL were not present in Strc−/− mice16. These data suggest that OTOG and OTOGL may be weakly, but directly associated with TMEM145 or indirectly associated with TMEM145 via TUB or STRC, maintaining TM-ACs.
Our study also presents future opportunities to elucidate the regulation and function of TUB, which remain poorly understood. TUB undergoes unconventional secretion in Neuro-2a, a neuronal cell line53. However, its mechanism of action remains unknown. The well-known molecular features of TUB include a nuclear localization signal at its N-terminus and a phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2)-binding domain at its C-terminal region20. However, the binding of TUB to PI(4,5)P2 has only a partial effect on its secretion from Neuro-2a cells53. Confirmation of TMEM145 expression in Neuro-2a cells (Fig. 4g) suggested that TMEM145 is a major regulator of TUB secretion in Neuro-2a and other cell types. In terms of function, TUB is a MerTK ligand that facilitates the phagocytosis of retinal pigment epithelial cells, macrophages, and microglia54,60. MerTK is a member of the TAM receptor protein tyrosine kinase family (Tyro3, Axl, and MerTK), and its expression in macrophages plays a pivotal role in innate immunity. Although macrophages are not present in the endolymph, they are abundant in the endolymphatic sac, which is connected to the endolymph61. Bone marrow–derived resident macrophages play important roles in immune defense and prevent inflammation of the sensory epithelium61. Secreted TUB in the endolymph may reach the endolymphatic sac and activate MerTK in macrophages. Although we could not find an immunophenotype in TUB or Tmem145−/− mice, the roles of TUB in inner ear immunity may be revealed in TUB or Tmem145−/− mice under pathological conditions, such as infection or acoustic trauma.
The detection of TMEM145 expression in SGNs, particularly in type I SGNs, suggests that its role in auditory physiology extends beyond stereocilia maintenance in OHCs. scRNA-seq analysis revealed that, unlike TMEM145, STRC and TUB were scarcely expressed in SGNs, suggesting that TMEM145 may serve functions distinct from its role in OHCs (Supplementary Fig. 24). Given its structural features—an N-terminal GOLD domain associated with protein trafficking and a seven-transmembrane domain reminiscent of class A G protein-coupled receptors—TMEM145 may be integral to synaptic function in SGNs. It can potentially regulate the trafficking or localization of synaptic proteins, influence synaptic vesicle cycling, and participate in intracellular signaling pathways essential for neural signal transmission. This dual expression profile raises the possibility that TMEM145 contributes to both the structural integrity of hair cell stereocilia and the proper functioning of neuronal synapses, thereby ensuring efficient auditory processing and communication along the auditory pathway.
Overall, this study provides compelling evidence for the central role of TMEM145 in coordinating the assembly and precise localization of STRC and TUB within OHC stereocilia, offering a new perspective on the molecular mechanisms underlying auditory transduction. Future research on the detailed dynamics of TMEM145-mediated protein trafficking and stereociliary organization will significantly deepen our understanding of the cellular processes that support hearing physiology.
Methods
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committee of Yonsei University College of Medicine (Department of Laboratory Animal Medicine, Medical Research Center, Yonsei University College of Medicine, Seoul, Republic of Korea; #2022-0226) and were performed in accordance with institutional and national ethical and animal welfare guidelines. All mouse husbandry was carried out under standard conditions at Yonsei University College of Medicine.
Tmem145 KO mice were generated by Cyagen (Suzhou, China) on a C57BL/6 J genetic background. The Tmem145 gene (NCBI Reference Sequence: NM_183311.3; Ensembl: ENSMUSG00000043843) contains 15 exons, with an ATG start codon in exon 1 and a TGA stop codon in exon 15 (Transcript Tmem145-201: ENSMUST00000108409). Exons 2–12 were selected as target regions. Cas9 protein and two gRNAs (gRNA-A1: GCCAGGCAACCTCCACTTGG-TGG and gRNA-A2: GGCTTTCACGGAGCTAGGCA-AGG) were co-injected into fertilized mouse eggs. The injected embryos were then transferred into pseudopregnant females, and the resulting F0 offspring were screened for mutations. Genotyping was performed using PCR, and the KO allele was confirmed using Sanger sequencing.
Tubby mice in the C57BL/6 J background (B6(AU)-Tubtub/J) were purchased from The Jackson Laboratory (000562; Bar Harbor, ME, USA). Wild-type inbred AKR/N mice were purchased from Japan SLC, Inc. (SLC-M-0248; Shizuoka, Japan). The genotypes of the tubby mice were determined by PCR amplification of the genomic region containing the tubby mutation followed by Sanger sequencing. Tubby mice (tub/tub) in the C57BL/6 background were mated with wild-type (+/+) AKR mice to produce tub/+; Map1aAKR/B6 mice, which contain Map1a alleles from the AKR strain (Map1aAKR) and from the C57BL/6 strain (Map1aB6). These mice were mated among themselves to produce wild-type (+/+; Map1aB6/B6, +/+; Map1aAKR/B6, or +/+; Map1aAKR/AKR), tub/tub; Map1aB6 (tub/tub; Map1aB6/B6), and tub/tub; Map1aAKR (tub/tub; Map1aAKR/B6 or tub/tub; Map1aAKR/AKR) mice. The presence of the Map1aB6 or Map1aAKR alleles was determined by PCR-amplifying the part of the Map1a gene that encodes the Ala-Pro repeat-containing C-terminus18. Strc KO mice were described previously18.
Tecta KO mice (B6.129S-Tectatm1Gpr/H) were obtained from the European Mouse Mutant Archive (Infrafrontier, Munich, Germany). This line carries a targeted mutation of Tecta generated by insertion of a neomycin-resistance cassette into exon 3 using a 129/SvEv-derived targeting construct and had been outbred onto the C57BL/6 J background for 34 generations before archiving.
Adgrv1 KO mice were generated from a conditional knockout line obtained from Cyagen. In this line, loxP sites were inserted on both sides of exon 2 of Adgrv1 using CRISPR/Cas9-mediated genome editing, allowing Cre-dependent deletion of exon 2. For the present study, these conditional Adgrv1flox mice were crossed with a CMV-Cre deleter line to produce germline deletion of exon 2 and global loss of Adgrv1.
Pmca2 KO mice were generated by CRISPR/Cas9 targeting exon 11 of Pmca2 based on the NCBI reference sequence NM_001036684. A single guide RNA (sgRNA) with the sequence 5′-TGAACTCACGTGCATCTGCG-3′ was used to introduce a 1-bp thymidine insertion in exon 11.
Otog KO mice were generated by CRISPR/Cas9 targeting exon 4 of Otog based on the NCBI reference sequence NM_013624.2. Two sgRNAs, 5′-CTTCACGTACCAAGGAGGGG-3′ and 5′-TCTTGCTTCCTACTTCCAGT-3′ (identical to those used in the Jackson Laboratory Otog targeting strategy), were used to produce a 491-bp deletion within exon 4.
TMEM145 antibody production
A synthetic peptide corresponding to the sequence GVPGPGGSQSADK located at the C-terminus of human and mouse TMEM145 was commercially synthesized and subsequently conjugated to a carrier protein, keyhole limpet hemocyanin (KLH), to enhance its immunogenicity. This peptide–carrier conjugate was prepared following established cross-linking protocols.
New Zealand white rabbits were immunized with the peptide conjugates according to a standard immunization regimen. An initial primary injection was administered followed by booster injections at regular intervals. However, additional boosting did not result in a substantial increase in the immune response. Consequently, no further boosting was performed, and the final serum collection was performed based on the immune response obtained from the initial immunization series.
Blood was collected from the rabbits by standard venipuncture and the serum was separated by centrifugation. Serum obtained from individual animals was pooled to generate a uniform final serum sample. The pooled serum was subjected to Protein A affinity chromatography to purify the rabbit polyclonal antibodies. The final antibody preparation was formulated with Proclin 300 and stored at −80 °C for subsequent use. The final antibody product was designated as anti-T145-01. The purified antibody was used at a 1:200 dilution for whole-mount immunostaining and a 1:1000 dilution for immunoblotting.
ABR
ABR thresholds were measured in a soundproof chamber using Tucker-Davis Technologies (TDT) RZ6 digital signal processing hardware and BioSigRZ software (Alachua, FL, USA). Subdermal needles (electrodes) were positioned at the vertex and ventrolateral to the right and left ears of the anesthetized mice. A calibrated click stimulus (10 µs duration) or tone burst stimuli (5 ms duration) were produced at 4, 6, 8, 10, 12, 18, 24, 30, and 42 kHz using SigGenRZ software and an RZ6 digital signal processor and were delivered to the ear canal by a multifield 1 (MF1) magnetic speaker (TDT). The stimulus intensity was increased from 10 to 95 dB SPL in 5-dB increments. The ABR signals were fed into a low-impedance Medusa Biological Amplifier System (RA4LI, TDT), which delivered the signal to the RZ6 digital signal-processing hardware. The recorded signals were filtered using a 0.5–1 kHz bandpass filter and the ABR waveforms in response to 512 tone bursts were averaged. ABR thresholds for each frequency were determined using BioSigRZ software. Peak amplitudes (mV) and peak latencies (ms) were calculated from the waveform signals of the click-evoked ABRs as input/output (I/O) functions with increasing stimulus levels (20–90 dB SPL).
Distortion product otoacoustic emissions (DPOAE)
DPOAE were measured using a TDT microphone–speaker system. Primary stimulus tones were produced using an RZ6 digital signal processor with SigGenRZ software and delivered using a custom probe with an ER 10B+ microphone (Etymotic, Elk Grove Village, IL, USA) and MF1 speakers positioned in the ear canal. The primary tones were set at a frequency ratio (f2/f1) of 1.2 with target frequencies of 6, 8, 10, 12, 16, 18, 24, and 30 kHz. The f2 intensity levels were the same as the f1 intensity levels (L1 = L2). The sounds resulting from the primary tones were received by the ER 10B+ microphone and recorded using an RZ6 digital signal processor. The DPOAE I/O functions were determined at specific frequencies (6 and 30 kHz) with a frequency ratio (f2/f1) of 1.2 and equal intensity levels (L1 = L2). The intensity level of the primary tones was increased from 20 to 80 dB SPL in 5-dB increments. Fast Fourier transform (FFT) was performed at each primary tone for the DP grams and at each intensity for the I/O functions using BioSigRZ software to determine the average spectra of the two primaries, the 2f1–f2 distortion products, and the noise floors.
Immunofluorescence staining and whole-mount assay
The cochleae were isolated from WT, Tmem145−/−, Strc−/−, Otog−/−, tub/tub;Map1aB6, and tub/tub;Map1aAKR mice at P5, P7, and P21. Temporal bones were fixed with 4% paraformaldehyde overnight at 4 °C and decalcified in 250 mM EDTA for at least 2 d. The cochlea was dissected into pieces from the decalcified tissue for whole-mount immunofluorescence. Tissues were permeabilized with 0.3% SDS, blocked with 5% goat serum for 2 h, and then incubated at 4 °C overnight with the following primary antibodies: rabbit anti-T145-01 (1:200 dilution; generated in this study), rabbit anti-STRC (1:200 dilution; used in previous study18), and rabbit anti-TUB (1:200 dilution; used in previous study18). To label IHCs and OHCs, Alexa Fluor 594 phalloidin (1:400 dilution; A12381; Invitrogen) was used to stain F-actin at RT for 1 h. After three rinses with 1× PBS, the specimens were incubated for 1 h with the secondary antibodies goat anti-rabbit Alexa Fluor 488 (1:1000 dilution; A32731; Invitrogen) and goat anti-mouse Alexa Fluor 488 (1:1000 dilution; A32723; Invitrogen). The specimens were mounted in Faramount Aqueous Mounting Medium (S3025; Dako, Glostrup, Denmark) and imaged with a confocal microscope (LSM980; Zeiss, Jena, Germany) at 63x, and images were processed using ZEN software.
Immunohistochemistry
The isolated cochleae were fixed with 4% paraformaldehyde overnight at 4 °C and decalcified in 250 mM EDTA for at least 2 d. The cochleae were transferred to a series of gradient ethanol solutions, immersed in dimethylbenzene, and embedded in paraffin for sectioning. For immunostaining, sections were deparaffinized in xylene and rehydrated in an ethanol gradient. The cells were then incubated in blocking buffer containing 5% goat serum for 1 h at room temperature. The samples were incubated overnight at 4 °C with primary antibodies diluted in blocking buffer. After washing with PBS, the samples were incubated with secondary antibodies and 4′,6-diamidino-2-phenylindole (DAPI) for 30 min at room temperature, washed, and covered with mounting medium and cover slips. Images were captured using a confocal microscope (LSM980; Zeiss).
The following antibodies were used. Primary: mouse anti-Prestin (1:200 dilution; sc-293212; Santa Cruz Biotechnology, Santa Cruz, TX, USA), rabbit anti-MYO7A (1:200 dilution; 25-6790; Proteus Biosciences, Ramona, CA, USA), and mouse anti-TUBB3 (1:200 dilution; MMS-435P; BioLegend, San Diego, CA, USA). Secondary: goat anti-mouse Alexa Fluor 488 (1:1000 dilution; A32723; Invitrogen), goat anti-rabbit Alexa Fluor 594 (1:1000 dilution; A11012; Invitrogen), and DAPI (1 µg/mL; D1306; Invitrogen).
In situ hybridization
In situ hybridization was performed as previously described62. The hemi-sectioned heads of pups from P1, P5, and P9 were fixed in 4% paraformaldehyde overnight and dehydrated in 30% sucrose overnight. The samples were then embedded in Tissue-Tek optimum cutting temperature compound and sectioned at 12 μm thickness using a cryostat (HM 525, Thermo Fisher Scientific, USA). Antisense RNA probes for Nefl, Smpx, and Strc were prepared as previously described63,64,65, Tmem145 was prepared according to the probe sequence of the Genepaint (http://gp3.mpg.de) website. Micrographs of gene expression patterns were acquired using a Leica DM2500 optical microscope. All in situ hybridization figures are representative of at least three different samples from two or more independent experiments.
Scanning electron microscopy
Inner ears were isolated in 1× PBS and initially fixed with 4% paraformaldehyde (PFA) for 40 min, followed by pre-fixation in a solution of 2.5% glutaraldehyde and 2% PFA in 0.1 M sodium cacodylate buffer containing 2 mM calcium chloride for 1 h on ice. The cochlear epithelium was then micro-dissected in 1× PBS and further fixed overnight at 4 °C in a fixative comprising 2.5% glutaraldehyde, 2% PFA in 0.1 M sodium cacodylate buffer supplemented with 3.5% sucrose and 2 mM calcium chloride. Following fixation, the samples were washed three times with 0.1 M sodium cacodylate buffer (containing 2 mM calcium chloride) for 20 min each on a shaker maintained on ice. Post-fixation was carried out using an osmium tetroxide/thiocarbohydrazide (OTO) protocol, which included incubation in 1% osmium tetroxide (OsO₄) for 1 h on ice with shaking, treatment with 1% thiocarbohydrazide (TCH) for 10 min at room temperature, and a subsequent 30 min incubation in 1% OsO₄ with shaking. The specimens were then dehydrated using a graded ethanol series (20%, 40%, 60%, 70%, and 80% for 7 min each at room temperature, followed by 90% ethanol overnight, 95% ethanol for 7 min, and 100% ethanol for 20 min) and dried using a critical point dryer (Leica EM CPD300, Wetzlar, Germany). Finally, the dried samples were coated with a 20-nm thick layer of platinum and imaged using a Schottky field-emission SEM (JSM-7610F; JEOL, Tokyo, Japan).
Sub-immunogold labeling for electron microscopy
Cochlear samples were isolated from the inner ears of mice and fixed in 4% PFA in Hank’s Balanced Salt Solution (HBSS) supplemented with 2 mM CaCl₂ and 0.5 mM MgCl₂ (pH 7.4) at room temperature for 40 min. The dissection buffer was HBSS.
For simultaneous blocking and permeabilization, the samples were incubated in 5% goat serum and 0.1% BSA in HBSS containing 0.3% saponin and 0.3% SDS at room temperature for 1 h, and incubated with primary antibodies diluted 1:200 in the blocking and permeabilization solution overnight at 4 °C. The samples were rinsed once and washed three times for 15 min each with HBSS and then incubated with colloidal gold–conjugated secondary antibodies (12 or 18 nm, Jackson Immuno Research) diluted 1:50 in the blocking and permeabilization solution overnight at 4 °C. After incubation with the secondary antibody, the samples were rinsed once and washed three times for 15 min each with HBSS.
To prepare for fixation, the samples were rinsed twice with 0.1 M sodium cacodylate buffer (pH 7.2) and fixed in a solution of 10% glutaraldehyde and 4% PFA in 0.1 M sodium cacodylate buffer at 4 °C for at least 24 h without agitation. Postfixation of the samples was performed following the OTOTO protocol. The samples were washed three times for 10 min with 0.1 M sodium cacodylate buffer and fixed with 1% osmium tetroxide (OsO₄) in 0.1 M sodium cacodylate buffer for 1 h at room temperature without agitation. The samples were washed four times for 5 min with 0.1 M sodium cacodylate buffer and then three times with distilled water. The samples were incubated with 1% thiocarbohydrazide for 20 min at room temperature without agitation, followed by four washes with water for 5 min each and three washes with 0.1 M sodium cacodylate buffer. This cycle was repeated to complete the OTOTO process.
Following the OTOTO protocol, samples were dehydrated in a graded ethanol series (15%, 30%, 50%, 75%, 90%, 95%, and 100%) for 5 min each on ice. The dehydrated samples were dried using a CPD300 critical point dryer (Leica Microsystems, Wetzlar, Germany) and coated with a 15-nm layer of carbon. Samples were imaged using a field-emission SEM (JSM-IT800; JEOL, Tokyo, Japan).
Spatial transcriptomics
The Xenium workflow, utilizing experimental chemistry alongside a prototype instrument and consumables, started by sectioning 5-μm formalin-fixed paraffin-embedded tissue samples onto a Xenium slide. The sections were deparaffinized and permeabilized to expose the mRNA. The mRNA targets were detected using 313 probes, and two negative controls: (1) probe controls to evaluate non-specific binding and (2) genomic DNA (gDNA) controls to confirm RNA-specific signals. Probe hybridization was carried out at 50 °C overnight at a concentration of 10 nM. Stringent washing was followed to eliminate unbound probes, after which the probes were ligated at 37 °C for 2 h, during which an RCA primer was also annealed. Circularized probes were enzymatically amplified, first at 4 °C for 1 h and then at 37 °C for 2 h, producing multiple copies of gene-specific barcodes for each RNA binding event, enhancing the signal-to-noise ratio. Background fluorescence was chemically quenched to address the autofluorescence caused by lipofuscins, elastin, collagen, red blood cells, and formalin fixation. Finally, the tissue sections were mounted onto an imaging cassette and loaded onto a Xenium Analyzer.
Single cell RNA sequencing analysis
Single-cell RNA-sequencing count matrices for 4-week-old mouse cochleae were obtained from the Gene Expression Omnibus (accession number GSE202920)47. All analyses were performed in R (v4.4.2) using Seurat (v5.1.0)66,67. Feature barcode matrices from each sample were imported using Read10X and converted into Seurat objects. The two Seurat objects were merged into a single object to obtain a unified dataset.
For each cell, the percentage of reads mapping to mitochondrial genes was calculated using the PercentageFeatureSet with the pattern “^MT-“. Low-quality cells, such as those with few detected features or high mitochondrial gene content, were removed according to data-dependent thresholds. These thresholds included having between 200 and 3,000 detected features and fewer than 10% of the reads mapping to mitochondrial genes. Next, we used FindVariableFeatures (method = “vst”) to detect the 2000 most highly variable genes across cells. Identifying highly variable genes helps focus on the genes that capture the greatest biological variability. To account for cell-cycle effects, we applied Seurat’s built-in cell-cycle scoring approach. Gene sets corresponding to the S phase (s_genes) and G2/M phase (g2m_genes) were used for CellCycle Scoring. Each cell was assigned scores for the S and G2/M phases and an overall phase annotation.
We employed the SCTransform workflow for normalization, which regularizes and stabilizes the variance in the single-cell data. We regressed out mitochondrial gene percentage (percent.mt) and cell cycle phase scores (Phase, S.Score, and G2M.Score) to mitigate their confounding effects on downstream analyses. Principal Component Analysis (PCA) was performed on the SCTransformed data using RunPCA, focusing on the variable features identified above. We typically selected the first 20 principal components (PCs) for downstream clustering, guided by inspection of an elbow plot or other metrics. Clustering was performed using a graph-based approach. First, a cell-to-cell similarity graph was constructed using FindNeighbors (dims = 1:20). We then applied Louvain or Leiden clustering using FindClusters (resolution = 1). We performed dimensional reduction via Uniform Manifold Approximation and Projection (UMAP) using RunUMAP (dims = 1:20). Clusters were visualized using DimPlot, and cells were colored according to their cluster identities. We identified cluster-specific marker genes using FindAllMarkers, applying a log-fold change threshold (e.g., 0.25) and focusing on positively differentially expressed genes (only.pos = TRUE). We annotated the clusters by examining known cell-type marker genes and the top differentially expressed genes. Based on these marker genes, we renamed the clusters using RenameIdents to reflect the known cochlear cell types. The final cell-type assignments were stored in the Seurat object metadata for downstream analysis.
To analyze single-cell RNA-sequencing data of SGNs, raw gene-by-cell count matrices for GSE11475948 were downloaded in a tab-delimited (*. txt) format, each containing gene symbols as row names and cell barcodes as column headers, and merged into a single matrix after retaining only genes common to all files. Low-quality cells (fewer than 100 detected genes) and genes detected in 10 or fewer cells were excluded. Cells expressing hemoglobin-related genes (Hba-a1, Hba-a2, Hbb-bh1, Hbb-bs, and Hbb-bt) were also excluded if they comprised over 5% of the total UMI count, thereby mitigating red blood cell contamination. The remaining data were log-normalized using Seurat’s NormalizeData function (scale factor 10,000) and 2000 highly variable genes were identified using FindVariableFeatures. The expression matrix was then scaled (ScaleData), allowing for the optional regression of covariates, such as mitochondrial or cell cycle effects. PCA was conducted on the variable features and an elbow plot guided the selection of 20 PCs. These PCs informed graph-based clustering (FindNeighbors, FindClusters, resolution = 0.5) and two-dimensional visualization with UMAP (RunUMAP, dims = 1:20), which were used to inspect the cluster structure and annotate cell types based on known marker genes.
Cell culture, plasmids, and transfection
HEK293T cells (CRL-3216; American Type Culture Collection, Manassas, VA, USA), HeLa cells (CCL-2; American Type Culture Collection), and Neuro-2a (CCL-131; American Type Culture Collection) were cultured in high-glucose Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Thermo Fisher Scientific). The cells were cultured in a humidified incubator at 37 °C with 20% O2 and 5% CO2, and were cultured every 2–3 d in 90-mm culture dishes.
Human TMEM145 (NM_173633.3) and mouse Tmem145 (NM_183311.3) coding regions were purchased from OriGene (Rockville, MD, USA). Each coding region was amplified using PCR to introduce a C-terminal FLAG tag (DYKDDDK) immediately upstream of the stop codon, and the fragments were subcloned into the pEGFP‑N1 mammalian expression vector (Clontech, USA) using the NheI and NotI restriction sites via In‑Fusion Cloning (Takara Bio, Japan). During this process, the original EGFP coding sequence was removed, leaving only the TMEM145 sequence under control of the CMV promoter.
The mouse Strc (NM_080459.2) and rat Tub (NM_013077.2) coding sequences were provided by Dr. Chul Hoon Kim (Yonsei University College of Medicine). Each coding region was amplified using PCR to introduce a C-terminal MYC tag (EQKLISEEDL) immediately prior to the stop codon. The resulting products were cloned into the pcDNA3.1(+) expression vector (Thermo Fisher Scientific) via In‑Fusion Cloning, using HindIII and NotI for mStrc and KpnI and XhoI for rTub. For some experiments, a C‑terminal V5‑tagged rTub (GKPIPNPLLGLDST) was used. The coding sequences of all plasmids were verified using Sanger sequencing (Macrogen, Seoul, South Korea).
HEK293T cells were seeded in six-well plates 1 d prior to transfection. Transient transfection was performed using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. For single-protein expression, 1000 ng of either human or mouse TMEM145 plasmid was used. For co-expression experiments, the ratio and amount of each plasmid were optimized as follows: 200 ng TMEM145, 300 ng of TUB, and 3000 ng STRC plasmids. Molecular biology experiments were performed at 24–72 h post-transfection.
OHC electrophysiology
OHC electrophysiology was performed as previously described with slight modifications29. The organ of Corti was acutely isolated from mice at P21. The animals were anesthetized using isoflurane (Sigma‑Aldrich) and euthanized by decapitation. The bone surrounding the apical turn of the cochlea was carefully removed and the apical turn of the organ of Corti was dissected using fine forceps and separated from the lateral cochlear wall, stria vascularis, modiolus, and TMs. All dissections were carried out in a standard extracellular solution containing 144 mM NaCl, 5.8 mM KCl, 10 mM HEPES, 5.6 mM glucose, 0.7 mM NaH₂PO₄, 1.3 mM CaCl₂, 0.9 mM MgCl₂, and 10 mM sorbitol, adjusted to pH 7.4 with NaOH. The isolated cochlea was immobilized on a 12-mm-diameter coverslip using a stainless-steel pin and Sylgard and mounted on an upright microscope (ECLIPSE FN1; Nikon, Japan). The extracellular bath solution used during recordings was identical to the dissection solution, while the pipette solution for recording electromotility contained 140 mM CsCl, 10 mM HEPES, 1 mM EGTA, 3 mM Mg‑ATP, and 1 mM MgCl₂, adjusted to pH 7.2 with CsOH.
Whole-cell voltage-clamp recordings were performed at 22–25 °C on OHCs using standard configurations. Microglass pipettes (World Precision Instruments, Sarasota, FL, USA) with a resistance of 2–5 MΩ were fabricated using a PP‑830 single‑stage glass microelectrode puller (Narishige, Japan), and the liquid junction potential was corrected with an offset circuit prior to each recording. Currents were recorded using an Axopatch 700 B amplifier (Molecular Devices, San Jose, CA, USA) coupled with a Digidata 1550 A (Molecular Devices) interface, digitized at 100 kHz, and low-pass filtered at 10 kHz using pClamp software 10.7 (Molecular Devices). The whole‑cell configuration was confirmed by ensuring that the series resistance remained below 10 MΩ, with no series resistance compensation applied.
In the voltage-clamp configuration, recordings were obtained using a sine wave stimulus (10 mV amplitude at 1 kHz) superimposed on a ramp pulse protocol ranging from 150 mV to 100 mV over a duration of 250 ms, with a holding potential of 70 mV. For each cell, at least two recordings were performed to ensure measurement stability, and the nonlinear capacitance (NLC) calculation was performed as described previously29,68,69. The capacitance was fitted to the derivative of the Boltzmann equation
where Vh is the maximal activation voltage, Qmax is the maximal charge transfer between the plasma membrane, α is the slope factor for the voltage-dependent charge transfer, z is the charge valence, e is the electron charge, k is Boltzmann’s constant, and T is absolute temperature. An in-house Python script was used for the NLC calculations and analysis29.
Immunoprecipitation
IP and co-IP experiments were performed as described previously29. Cells were processed and lysed under optimized conditions for the detection of surface and interacting proteins using a lysis buffer containing 150 mM NaCl, 50 mM Tris-Cl, 1 mM EDTA, 1% Triton X-100, and a Protease Inhibitor Cocktail (cOmplete Protease Inhibitor Cocktail tablets; Roche, Basel, Switzerland). The cell lysate was sonicated for 20 s and centrifuged at 20,000 × g for 15 min to remove cellular debris. Protein concentrations across samples were equalized using the Bradford assay (Protein Assay Kit II, Bio-Rad, Hercules, CA, USA). From each sample, a portion was used for IP, whereas the remainder was mixed with 2× Novex Tris-glycine SDS sample buffer (Sigma-Aldrich) and used as a lysate or input control. The lysate was then subjected to IP by incubation for overnight at 4 °C with EZview Red Anti-FLAG M2 Affinity Gel (Sigma-Aldrich), Pierce Anti-c-Myc Magnetic Bead (Thermo Fisher Scientific), or V5-Trap Magnetic Agarose (ChromoTek, Planegg, Germany) on an end-over-end rotator.
Following IP, the affinity matrices were washed five times with lysis buffer at a volume at least 20-fold greater than the matrix volume for each wash to remove nonspecifically bound proteins. For sample elution, 2× sample buffer (Sigma-Aldrich) was mixed with lysis buffer at a 1:1 ratio to achieve a final 1× concentration, and the mixture was incubated with the affinity matrix at 37 °C for 60 min. After elution, samples were collected in a new tube and subjected to immunoblotting.
Surface biotinylation
Surface biotinylation was performed using EZ-Link Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific) and streptavidin (Thermo Fisher Scientific) as previously described70,71. The biotin solution was prepared by dissolving 0.5 mg EZ-Link Sulfo-NHS-SS-Biotin in 1 mL of 1× PBS. Cells were incubated in the dark at 4 °C for 30 min to allow for the biotin reaction. Subsequently, any remaining biotin was quenched by incubating the cells in a blocking solution containing 0.5% bovine serum albumin (prepared by dissolving 0.5 g BSA in 50 mL of 0.5× PBS) in the dark at 4 °C for 10 min. Following biotinylation, the biotinylated proteins were isolated using the IP procedure described above, with a Streptavidin agarose resin used to capture the proteins, followed by sample elution and immunoblotting.
Immunoblotting
Lysate or input samples containing 40 μg of protein were loaded, while IP or biotinylation samples corresponding to the precipitated material from 400 μg of lysate were loaded onto a 4–12% gradient SDS-PAGE gel (KomaBiotech, Seoul, Korea). Electrophoresis was performed at 100 V for 100 min using running buffer composed of 100 mM Tris base, 100 mM Tricine, and 0.1% SDS, adjusted to pH 7.3. Proteins were then transferred onto an Amersham™ Protran® Supported nitrocellulose membrane (Cytiva, Marlborough, MA, USA) at 250 mA for 100 min using a transfer buffer containing 25 mM Tris, 192 mM Glycine, pH 7.3, and 20% methanol, with the transfer performed at 4 °C. The membrane was then blocked at room temperature for 1 h in 5% skim milk and incubated overnight at 4 °C with mild agitation, and the desired primary antibody diluted in a solution containing 5% skim milk and 0.02% sodium azide. The next day, the membrane was washed thrice for 10 min each at room temperature with 1× TBST composed of 20 mM Tris base, 150 mM NaCl, 0.1% Tween-20, adjusted to pH 7.4, incubated with the appropriate secondary antibody diluted in 1× TBST for 1 h at room temperature, and washed thrice for 10 min each with 1× TBST. Finally, immunoreactive bands were detected by developing the blot with ECL solution (Invitrogen) and visualized on a blue film (Agfa, Mortsel, Belgium).
For immunoblotting of secreted proteins from the culture media72, the medium was replaced with serum-free medium 24 h after transfection. The cells were then incubated for an additional 48 h before the culture medium was harvested. The harvested medium was concentrated using an Amicon Ultra-4 filter (Millipore, Burlington, MA, USA). Finally, the concentrated media were mixed with 5× sample buffer and equal volumes of each sample were loaded for subsequent analyses.
The following antibodies were used. Primary: rabbit anti-T145-01 (1:1000 dilution; generated in this study), mouse anti-MYC (1:1000 dilution; 2276; Cell Signaling Technologies, Danvers, MA, USA), rabbit anti-MYC (1:1000 dilution; 2278; Cell Signaling Technologies), mouse anti-FLAG (1:1000 dilution; 8146; Cell Signaling Technologies), rabbit anti-FLAG (1:1000 dilution; 14793; Cell Signaling Technologies), mouse anti-V5 (1:1000 dilution; R960-25; Invitrogen), rabbit anti-ACTB (1:1000 dilution; A284; Abbkine, Atlanta, Georgia, USA), and mouse anti-ALDOA (1:1000 dilution; sc-390733, Santa Cruz Biotechnology). Secondary: goat anti-rabbit IgG polyclonal antibody (HRP conjugate) (1:1000 dilution; ADI-SAB-300-J, Enzo Life Sciences, Farmingdale, NY, USA) and goat anti-mouse IgG F(ab’)2 polyclonal antibody (HRP conjugate) (1:1000 dilution; ADI-SAB-100-J; Enzo Life Sciences).
Immunocytochemistry
Transfected HeLa cells were cultured on 18-mm round coverslips. The cells were washed twice with 1× PBS, fixed with 4% PFA for 10 min, washed twice with 1× PBS, and permeabilized with 0.15% Triton X-100 in 1× PBS for 10 min. The cells were washed twice with PBS and incubated with blocking buffer consisting of 5% donkey serum for 1 h at room temperature. After blocking, the cells were incubated with the appropriate primary antibodies for 1 h at room temperature and washed thrice with 1× PBS. Subsequently, the cells were stained with secondary antibodies conjugated to a fluorescent marker for 1 h at room temperature and washed thrice with 1× PBS73. The samples were mounted onto glass slides using a mounting medium (Dako) and images were captured using a confocal microscope (LSM 700; Zeiss) equipped with a 40× objective lens.
The following antibodies were used. Primary: mouse anti-FLAG (1:200 dilution; 8146; Cell Signaling Technologies), rabbit anti-FLAG (1:200 dilution; 14793; Cell Signaling Technologies), rabbit anti-BiP (1:200 dilution; ab21685; Abcam, Cambridge, UK), and mouse anti-GM130 (1:200 dilution; 610823; BD Biosciences, Franklin Lakes, NJ, USA). Secondary: goat anti-rabbit Alexa Fluor 488 (1:1000 dilution; A32731; Invitrogen), goat anti-mouse Alexa Fluor 488 (1:1000 dilution; A32723; Invitrogen), goat anti-rabbit Alexa Fluor 594 (1:1000 dilution; A11012; Invitrogen), goat anti-mouse Alexa Fluor 594 (1:200 dilution; A11005; Invitrogen), and DAPI (1 µg/mL; D1306; Invitrogen).
Structural modeling
The full-length structure of human TMEM145 was predicted using AlphaFold332. For the other GOLD domain–containing seven-transmembrane domain proteins, predicted structures were obtained by downloading the corresponding models from the AlphaFold2 proteome database35,36. Three-dimensional images of the proteins were generated and visualized using UCSF ChimeraX74.
Statistics and reproducibility
No formal statistical method was used to predetermine sample sizes. Data analyses were performed using GraphPad Prism 9.4.1 (GraphPad Software, San Diego, CA, USA), and Excel (Microsoft, Redmond, WA, USA). Two-sided unpaired t-tests were performed to compare independent groups. For multiple-group and multiple-condition comparisons, two-way analysis of variance (ANOVA) with Dunnett’s post-hoc test was used. Statistical significance was set at p < 0.05. The number of biological replicates was indicated whenever possible. At least three independent animal or biochemical experiments were performed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are provided within the paper, the Supplementary Information, and the Source Data file. Publicly available single-cell RNA-seq datasets used in this study were obtained from GEO under accession numbers GSE202920 and GSE114759. The spatial transcriptomics datasets generated in this study were deposited in the Korean BioData Station (K-BDS, https://kbds.re.kr) with accession number KAP241832. The datasets can also be downloaded directly from the following links: https://kbds.re.kr/KGD10063443 and https://kbds.re.kr/KGD10063444. Source data are provided with this paper.
References
Gillespie, P. G. & Muller, U. Mechanotransduction by hair cells: models, molecules, and mechanisms. Cell 139, 33–44 (2009).
Hudspeth, A. J. Integrating the active process of hair cells with cochlear function. Nat. Rev. Neurosci. 15, 600–614 (2014).
Ashmore, J. et al. The remarkable cochlear amplifier. Hear Res 266, 1–17 (2010).
Rennels, M. & Pickering, L. K. Sensorineural hearing loss in children. Lancet 365, 2085–2086 (2005).
Lieu, J. E., Kenna, M., Anne, S. & Davidson, L. Hearing loss in children: a review. Jama 324, 2195–2205 (2020).
Goodyear, R. J., Marcotti, W., Kros, C. J. & Richardson, G. P. Development and properties of stereociliary link types in hair cells of the mouse cochlea. J. Comp. Neurol. 485, 75–85 (2005).
Avan, P., Buki, B. & Petit, C. Auditory distortions: origins and functions. Physiol. Rev. 93, 1563–1619 (2013).
Qiu, X. & Muller, U. Sensing sound: Cellular specializations and molecular force sensors. Neuron 110, 3667–3687 (2022).
Bolz, H. et al. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat. Genet 27, 108–112 (2001).
Di Palma, F. et al. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat. Genet. 27, 103–107 (2001).
Ben-Yosef, T. et al. A mutation of PCDH15 among Ashkenazi Jews with the type 1 Usher syndrome. N. Engl. J. Med. 348, 1664–1670 (2003).
Verpy, E. et al. Mutations in a new gene encoding a protein of the hair bundle cause non-syndromic deafness at the DFNB16 locus. Nat. Genet. 29, 345–349 (2001).
Cohen-Salmon, M., Mattei, M. G. & Petit, C. Mapping of the otogelin gene (OTGN) to mouse chromosome 7 and human chromosome 11p14.3: a candidate for human autosomal recessive nonsyndromic deafness DFNB18. Mamm. Genome 10, 520–522 (1999).
Yariz, K. O. et al. Mutations in OTOGL, encoding the inner ear protein otogelin-like, cause moderate sensorineural hearing loss. Am. J. Hum. Genet 91, 872–882 (2012).
Heckenlively, J. R. et al. Mouse model for Usher syndrome: linkage mapping suggests homology to Usher type I reported at human chromosome 11p15. Proc. Natl Acad. Sci. USA 92, 11100–11104 (1995).
Avan, P. et al. Otogelin, otogelin-like, and stereocilin form links connecting outer hair cell stereocilia to each other and the tectorial membrane. Proc. Natl. Acad. Sci. USA 116, 25948–25957 (2019).
Verpy, E. et al. Stereocilin connects outer hair cell stereocilia to one another and to the tectorial membrane. J. Comp. Neurol. 519, 194–210 (2011).
Han, W. et al. Distinct roles of stereociliary links in the nonlinear sound processing and noise resistance of cochlear outer hair cells. Proc. Natl Acad. Sci. USA 117, 11109–11117 (2020).
Schraders, M. et al. Mutations of the gene encoding otogelin are a cause of autosomal-recessive nonsyndromic moderate hearing impairment. Am. J. Hum. Genet 91, 883–889 (2012).
Santagata, S. et al. G-protein signaling through tubby proteins. Science 292, 2041–2050 (2001).
Ikeda, A. et al. Genetic modification of hearing in tubby mice: evidence for the existence of a major gene (moth1) which protects tubby mice from hearing loss. Hum. Mol. Genet 8, 1761–1767 (1999).
Ikeda, A. et al. Microtubule-associated protein 1A is a modifier of tubby hearing (moth1). Nat. Genet 30, 401–405 (2002).
Youn, S. Y. et al. Microtubule-associated protein 1 A and tubby act independently in regulating the localization of stereocilin to the tips of inner ear hair cell stereocilia. Mol. Brain 15, 80 (2022).
Munoz-Fuentes, V. et al. The International Mouse Phenotyping Consortium (IMPC): a functional catalogue of the mammalian genome that informs conservation. Conserv Genet 19, 995–1005 (2018).
Anantharaman, V. & Aravind, L. The GOLD domain, a novel protein module involved in Golgi function and secretion. Genome Biol. 3, research0023 (2002).
Port, F. et al. Wingless secretion promotes and requires retromer-dependent cycling of Wntless. Nat. Cell Biol. 10, 178–185 (2008).
Nygaard, R. et al. Structural basis of WLS/Evi-mediated Wnt transport and secretion. Cell 184, 194–206 e114 (2021).
Mendes, L. F. S. & Costa-Filho, A. J. A gold revision of the Golgi Dynamics (GOLD) domain structure and associated cell functionalities. FEBS Lett. 596, 973–990 (2022).
Jung, J. et al. MYH1 deficiency disrupts outer hair cell electromotility, resulting in hearing loss. Exp. Mol. Med 56, 2423–2435 (2024).
Joo, S. Y. et al. Bi-allelic variants of SEMA3F are associated with non-syndromic hearing loss. Molecules and Cells 48, 100190 (2025).
Moller, S., Croning, M. D. & Apweiler, R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 17, 646–653 (2001).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
Holm, L. Dali server: structural unification of protein families. Nucleic Acids Res 50, W210–W215 (2022).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Tunyasuvunakool, K. et al. Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596 (2021).
Banziger, C. et al. Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell 125, 509–522 (2006).
de Almeida Magalhaes, T. et al. Extracellular carriers control lipid-dependent secretion, delivery, and activity of WNT morphogens. Dev. Cell 59, 244–261.e246 (2024).
Shin, J. J. H. et al. Spatial proteomics defines the content of trafficking vesicles captured by golgin tethers. Nat. Commun. 11, 5987 (2020).
Hoel, C. M., Zhang, L. & Brohawn, S. G. Structure of the GOLD-domain seven-transmembrane helix protein family member TMEM87A. Elife 11, e81704 (2022).
Chakrabarti, S. et al. Touch sensation requires the mechanically gated ion channel ELKIN1. Science 383, 992–998 (2024).
Kang, H. et al. GolpHCat (TMEM87A), a unique voltage-dependent cation channel in Golgi apparatus, contributes to Golgi-pH maintenance and hippocampus-dependent memory. Nat. Commun. 15, 5830 (2024).
Carette, J. E. et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science 326, 1231–1235 (2009).
Balazova, L. et al. GPR180 is a component of TGFbeta signalling that promotes thermogenic adipocyte function and mediates the metabolic effects of the adipocyte-secreted factor CTHRC1. Nat. Commun. 12, 7144 (2021).
Mitrovic, S. A. et al. GPR180 is a new member of the Golgi-dynamics domain seven-transmembrane helix protein family. Commun. Biol. 7, 1588 (2024).
Dudek, A. M. et al. GPR108 is a highly conserved AAV entry factor. Mol. Ther. 28, 367–381 (2020).
Xu, Z. et al. Profiling mouse cochlear cell maturation using 10x Genomics single-cell transcriptomics. Front Cell Neurosci. 16, 962106 (2022).
Sun, S. et al. Hair cell mechanotransduction regulates spontaneous activity and spiral ganglion subtype specification in the auditory system. Cell 174, 1247–1263 e1215 (2018).
Scheffer, D. I., Shen, J., Corey, D. P. & Chen, Z. Y. Gene expression by mouse inner ear hair cells during development. J. Neurosci. 35, 6366–6380 (2015).
Zheng, J. et al. Prestin is the motor protein of cochlear outer hair cells. Nature 405, 149–155 (2000).
Wu, X. et al. PKHD1L1 is a coat protein of hair-cell stereocilia and is required for normal hearing. Nat. Commun. 10, 3801 (2019).
Verpy, E. et al. Stereocilin-deficient mice reveal the origin of cochlear waveform distortions. Nature 456, 255–258 (2008).
Caberoy, N. B. & Li, W. Unconventional secretion of tubby and tubby-like protein 1. FEBS Lett. 583, 3057–3062 (2009).
Caberoy, N. B., Zhou, Y. & Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 29, 3898–3910 (2010).
Dumont, R. A. et al. Plasma membrane Ca2+-ATPase isoform 2a is the PMCA of hair bundles. J. Neurosci. 21, 5066–5078 (2001).
Legan, P. K., Rau, A., Keen, J. N. & Richardson, G. P. The mouse tectorins. Modular matrix proteins of the inner ear homologous to components of the sperm-egg adhesion system. J. Biol. Chem. 272, 8791–8801 (1997).
Legan, P. K. et al. A targeted deletion in alpha-tectorin reveals that the tectorial membrane is required for the gain and timing of cochlear feedback. Neuron 28, 273–285 (2000).
McGee, J. et al. The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J. Neurosci. 26, 6543–6553 (2006).
Michalski, N. et al. Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J. Neurosci. 27, 6478–6488 (2007).
Caberoy, N. B., Alvarado, G. & Li, W. Tubby regulates microglial phagocytosis through MerTK. J. Neuroimmunol. 252, 40–48 (2012).
Keithley, E. M. Inner ear immunity. Hear Res 419, 108518 (2022).
Morsli, H., Choo, D., Ryan, A., Johnson, R. & Wu, D. K. Development of the mouse inner ear and origin of its sensory organs. J. Neurosci. 18, 3327–3335 (1998).
Ankamreddy, H. et al. Region-specific endodermal signals direct neural crest cells to form the three middle ear ossicles. Development 146, dev167965 (2019).
Son, E. J. et al. Conserved role of Sonic Hedgehog in tonotopic organization of the avian basilar papilla and mammalian cochlea. Proc. Natl Acad. Sci. USA 112, 3746–3751 (2015).
Son, E. J. et al. Developmental gene expression profiling along the tonotopic axis of the mouse cochlea. PLoS One 7, e40735 (2012).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529 (2021).
Kim, G. D., Lim, C. & Park, J. A practical handbook on single-cell RNA sequencing data quality control and downstream analysis. Molecules Cells 47, 100103 (2024).
Lindau, M. & Neher, E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflug. Arch. 411, 137–146 (1988).
Bavi, N. et al. The conformational cycle of prestin underlies outer-hair cell electromotility. Nature 600, 553–558 (2021).
Roh, J. W., Choi, H. W. & Gee, H. Y. Guidelines for plasma membrane protein detection by surface biotinylation. Mol. Cells 48, 100174 (2024).
Oh, K. S. et al. Overlooked KCNQ4 variants augment the risk of hearing loss. Exp. Mol. Med 55, 844–859 (2023).
Oh, K. S. et al. COCH-related autosomal dominant nonsyndromic hearing loss: a phenotype-genotype study. Hum. Genet 141, 889–901 (2022).
Park, G., Kim, S. S., Shim, J. & Lee, S.-J. V. Brief guide to immunostaining. Molecules Cells 48, 100157 (2025).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Acknowledgements
The authors thank the Yonsei Advanced Imaging Center and Carl Zeiss Microscope. The authors also thank Medical Illustration & Design (MID), a member of the Medical Research Support Services of the Yonsei University College of Medicine, for providing excellent support with graphical illustrations. This work was supported by an MD-PhD/Medical Scientist Training Program grant through the Korea Health Industry Development Institute (KHIDI) (to J.W.R), a grant funded by the Ministry of Health & Welfare (RS-2024-00438709 to H.Y.G.), a National Research Council of Science & Technology (NST) grant by the Korea government (MSIT) (GTL24022-000 to J.B. and H.Y.G), and the National Research Foundation of Korea (RS-2023-00261905 and RS-2025-022214844 to H.Y.G., RS-2022-NR070578 and RS-2025-18362970 to J.B., and RS-2024-00400118 to J.B., H.Y.G., and K.Y.S.), and Samsung Science & Technology Foundation (SSTF-BA2101-11 to C.H.K., J.B., and H.Y.G).
Author information
Authors and Affiliations
Contributions
H.Y.G., C.H.K., and J.B. conceived and supervised the project. H.Y.G., C.H.K., J.B., J.W.R., and K.S.O. designed the experiments. J.W.R. performed biochemical studies, structural analysis, electrophysiological experiments, single-cell RNA sequencing analysis, and mouse phenotype analysis. K.S.O. performed fluorescence microscope imaging, spatial transcriptomics analysis, and mouse phenotype analysis. J.L., Y.C., and H.Y.K. performed fluorescence microscope imaging. S.K. conducted electron microscope imaging. J.W.H. conducted electron microscope imaging and spatial transcriptomics analysis. H.Y.K. performed in situ hybridization experiments. H.Lin and J.J. performed hair cell electrophysiology experiments. Y.K., H.Lew, and S.H.J. performed mouse studies. H.S.S. and K.Y.S. performed mouse studies and contributed to mouse phenotype analysis. J.O. and H.J. contributed to biochemical studies. J.W.R., K.S.O., C.H.K., J.B., and H.Y.G. wrote the initial manuscript. All authors reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Roh, J.W., Oh, K.S., Lee, J. et al. TMEM145 is a key stereociliary component in the link structures of outer hair cells and mediates the secretion of stereocilin and tubby. Nat Commun 17, 307 (2026). https://doi.org/10.1038/s41467-025-67011-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67011-0
