
Abstract
Prostaglandins (PGs) are bioactive lipids that regulate inflammation, reproduction, and vasopermeability. Timely clearance of excessive PGs is critical to prevent potential damages caused by prolonged signalling. The high-affinity prostaglandin transporter (PGT) plays an essential role in this termination process by efficiently re-uptaking PGs into cells. Here, we report human PGT structures in different oligomerization and substrate-binding states. Beyond the canonical MFS-fold, PGT harbours a cystine-rich extracellular segment that incorporates a Kazal-like domain crucial for PGT localization and activity. Two distinct PGE2-bound structures revealed essential elements for substrate recognition, elucidating a PGE2-flipping process during the multi-stop translocation cycle. Notably, our data suggest that PGT may dimerize in detergent micelles and lipid nanodiscs. This dimerization would likely increase the mobility of the extracellular region and induces a profound rotation of the C-domain transmembrane helices, an interesting observation for MFS transporters. These functionally distinct snapshots thus shed light on prostaglandin clearance and reveal intriguing features of MFS transporters.
Similar content being viewed by others

Introduction
Prostaglandin E2 (PGE2), along with PGD2, PGF2, prostacyclin (PGI2), and thromboxane A2 (TXA2), constitutes the five major essential prostanoids1. Clinical applications of Prostaglandins (PGs) and their metabolically stable analogs include inducing labor and abortions, treating ocular and pulmonary hypertension, and preventing gastric ulcers2,3. The biosynthesis and metabolism of PGs are tightly regulated to maintain physiological homeostasis. In response to diverse stimuli such as tissue damage, infection, and chemical irritants, PGs are locally and temporally produced from plasma membrane phospholipids through a metabolic cascade of phospholipases, cyclooxygenases (COX1 and COX2) and dedicated prostaglandin synthases. Upon secretion, PGs potently activate their cognate G-protein-coupled receptors, initiating downstream signaling cascades. While essential for numerous bodily functions, such as inflammation and vascular permeability, prolonged PG signaling leads to cellular stress and toxicity, necessitating timely deactivation. This inactivation process, orchestrated by transporters and metabolic enzymes, confines PGs’ bioactivity within the immediate vicinity of their release in an autocrine or paracrine manner4.
Dysregulated PGs signaling underpins a spectrum of pathological conditions, including periodontal diseases, chronic pain, hypertension, atherothrombosis, and carcinogenesis5. Notably, conventional nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin and ibuprofen, targeting COXs to inhibit PGs synthesis, have long been practiced in managing and treating acute inflammation, chronic pain, and fever6. However, owing to their broad-spectrum impact on PGs production, these drugs are often insufficient for certain patients and are accompanied by side effects over long-term prescription, urging safer and more specific intervening strategies.
Under physiological conditions, PG lipids are negatively charged and incapable of diffusing across the biogenic membranes, thus requiring transporters for translocation. Seminal work by the Schuster group has identified the solute carrier organic anion transporter OATP2A1 as the primary high-affinity prostaglandin transporter (PGT, also known as SLCO2A1/SLC21A2) in most tissues and organs7,8. PGT mediates the active cellular reuptake of excessive PGs, an obligatory initial step preceding the oxidative inactivation by 15-ketoprostaglandin dehydrogenases (15-PGDH). This enzymatic conversion to 15-keto PGs terminates signaling (Fig. 1a). PGT belongs to the major facilitator superfamily (MFS) transporters, and its PG transporter activity is sodium-independent and potentially powered by an outwardly directed lactate gradient9. In addition, PGT is implicated in shuttling PGs into intracellular acidic compartments, thus facilitating the exocytotic secretion of intact PGE210.

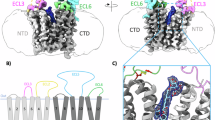
a Schematic of PGs production, function, and clearance. PGs are generated from membrane phospholipids via a series of enzymes, including COXs. Released PGs bind to their cognate GPCRs in an autocrine or paracrine manner. PGT reuptakes released PGs back into cells in exchange with a possible outward lactate flux. Subsequently, PGs are enzymatically inactivated by 15-PGDH. PGT is also suggested as a central component of the Maxi-Cl channel through oligomerization. b Topology diagram of full-length human PGT used in the study. Transmembrane helices of N-domain (green) and C-domain (blue), the Kazal-like domain within ECL5 (purple), and ECLs 1, 2, 3, 6 are labeled appropriately. The eight conserved intra- (yellow) and inter-loop (green) disulfide bridges are indicated by lines. c PGT structure in the absence of substrate viewed from the membrane plane (top) and from the extracellular domain (bottom). The coloring scheme is the same as in (b). N-glycosylation modification is shown as pink sticks. d 3H-PGE2 uptake activity of PGT variants with truncations of ECL2 (Δ135-165) and ECL5 (Δ425–508, Kazal-like domain) in stably transfected HEK293T cells. Activity values (mean ± SEM, n = 3 biologically independent experiments with 3 technical replicates each) are normalized to that of WT based on their surface protein expression level measured by surface biotinylation (Supplementary Fig. 8). e Transport kinetics of WT PGT (black) and the ECL2-truncated mutant (Δ135–165) (orange). Activity values are normalized to those of WT based on the relative expression level, as in (d). The inset shows the expanded plot for WT. f Binding affinity for the WT and the ECL2-truncated mutant with PGE2 measured using microscale thermophoresis assay (mean ± SEM, n = 3 independent experiments). Source data are provided as a Source Data file.
Recently, PGT has emerged as the long-sought core component of the maxi-anion channels (Maxi-Cl)11. These widely distributed channels permit highly efficient fluxes of inorganic and organic anions, including chloride ions and ATP, in response to diverse stimuli, including osmotic, oxidative, mechanical, and temperature stresses. However, the transition between PGT’s dual role as a lipid transporter and a channel component remains obscure.
Mounting evidences suggest PGT’s significance as a pharmacological target for health management and disease treatment. PGT exhibits protective effects in bleomycin-induced lung fibrosis12. Depletion of the mouse Pgt gene resulted in elevated systemic PGE2 levels and persistent opening of the ductus arteriosus, ultimately leading to neonatal mortality13. Altered PGT expression has been reported in tissues with chronic inflammation and cancers, potentially contributing to persistent activation of wound-healing pathways14. Moreover, recessive loss-of-function PGT mutations are related to refractory diseases, including primary hypertrophic osteoarthropathy (PHO)15,16,17,18, and chronic enteropathy associated with the SLCO2A1 gene (CEAS)19,20,21. PHO, characterized by pachydermia, periostosis, digital clubbing, and painful joint enlargement, is a rare disease that affects skin and bones15. CEAS is a new intractable type of enteropathy different from Crohn’s disease, and features chronic nonspecific multiple ulcers of the small intestine and persistent gastrointestinal bleeding20. Notably, these conditions typically characterize an elevated plasma PGE2 level, likely due to the insufficient clearance mediated by PGT mutants, which causes prolonged inflammation. On the other hand, PGT modulators could also be beneficial. For example, inhibition of PGT has been shown to have potential in alleviating hypertension in rats and mice models22, promoting perfusion, vascularization, and wound healing in rats23. Furthermore, considering that PGE2 clearance by PGT during mid-pregnancy triggers parturition onset24, PGT inhibitors could potentially offer a nonhormonal contraception strategy25.
In this study, we determined several cryo-EM structures of human PGT in monomer and dimer states. Together with biochemical analysis and molecular dynamics (MD) simulations, these structures reveal crucial features of substrate recognition and transport.
Results
Structure determination and overall architecture
PGT is predicted to be an N-glycosylated MFS transmembrane protein with large extracellular loops (molecular weight ~70 kDa, Fig. 1b). Proper glycosylation is crucial for its cell surface expression26. To obtain a functional protein sample for biochemical characterization and structural studies, plasmids harboring full-length human wild-type (WT) PGT were transiently transfected into HEK293F suspension cells (Supplementary Fig. 1a). Overexpressed PGT was purified to near homogeneity using Strep affinity chromatography followed by gel filtration (Supplementary Fig. 1b). Using cryo electron microscopy (cryo-EM) single particle analysis, we reconstructed a 3.14 Å map of the monomeric PGT in the absence of substrate (PGTapo) (Supplementary Fig. 2 and Supplementary Table 1). Well-resolved map densities permitted unambiguously model building of most regions (Supplementary Fig. 3a).
The right-trapezoidal zither-like architecture of PGT can be divided into three parts (Fig. 1c) to contain a large extracellular domain (ECD), a transmembrane domain (TMD), and a small intracellular domain. A distinct feature of PGT structure is the striking ECD “umbrella” that shields the substrate translocation funnel from the wide opening facing the extracellular space (Supplementary Fig. 4a). Within this domain, conserved N-glycosylation sites (Asn478 and Asn491) were identified. The TMD adopts a canonical MFS fold (Fig. 1c), consisting of two pseudo-symmetrical helix bundles: N-domain TMs 1–6, and C-domain TMs 7–12. Our PGTapo structure captures an outward-open conformation, characterized by a large funnel facing the extracellular space ending at one-third of the membrane close to the cytosol. This state is overall similar to the outward-facing structures of other lipid-transporting MFS members, including MFSD2A, Spns2, and the organic cation transporter OCT1-3 (with a Cα RMSD of approximately 1.1 Å, Supplementary Fig. 4c)27,28,29,30,31. The N-domain and C-domain of PGTapo are connected via a long cytoplasmic linker that has two intracellular helices (ICH1 and ICH2). Notably, the amphipathic ICH2 parallelly embedded in the membrane latches the cytoplasmic tips of TM2 and TM11, potentially facilitating the adoption of the outward-facing state (Fig. 1c).
The disulfide bond-rich ECD features a Kazal-like domain crucial for localization and function
The distinctive ECD “umbrella” is mainly formed by two extracellular regions, ECL2 and ECL5, emanating respectively from the N- and C-halves of PGT. To compensate for the flexibility of the loops, eight conserved intra- and inter-loop disulfide bonds are employed to presumably stabilize ECD (Fig. 1c and Supplementary Fig. 5a). The elongated ECL5 (~95 residues) connecting TM9 and TM10 makes the bulk of the Kazal-like domain, which sits atop the C-domain. In contrast to the fold featuring a central α-helix and a two-stranded β-sheet (Supplementary Fig. 5b) observed in the well-studied Kazal-type proteinase inhibitor (turkey ovomucoid protein OMTKY3 domain, PDB: 1YU6), the PGT counterpart displays two short α-helices and a four-stranded anti-parallel β-sheet fortified by four disulfide bonds (i.e., Cys444-Cys494, Cys450-Cys470, Cys459-Cys507, and Cys474-Cys492). This feature is conserved in the recent structures of SLCO family members OATP1B1, OATP1B3, and SLCO6C1 (Supplementary Fig. 4). An additional intra-loop disulfide bridge (Cys420-Cys511) forms within the juxtamembrane regions of ECL5, tethering the terminal ends of flanking loops and likely conferring rigidity. Notably, a homozygous C420F mutation has been identified in PHO patients16, underscoring the significance of this cystine linkage. ECL5 is further buttressed by a short, twisted ECL6, which wedges between the membrane plane and ECL5, and pinpoints several long side-chain residues (e.g., Arg582 and Tyr596) into a deep concavity of the Kazal-like domain (Supplementary Fig. 5c).
Kazal domain in soluble proteins typically functions as a serine proteinase inhibitor32. However, such inhibitory effects are absent for the Kazal-like domains found in membrane proteins. The Kazal-like domain is highly conserved among PGT/SLCO2A1 orthologs across diverse species (Supplementary Fig. 6). Additionally, it is a recurring feature within all eleven SLCO family paralogs that transport various organic anion substrates (Supplementary Fig. 7). To dissect the role of the Kazal-like domain, a stable HEK293T cell line with this domain excised (PGTΔ425–508) was generated. This deletion mutant exhibited impaired cell-surface localization, coupled with a markedly reduced expression level (Fig. 1d and Supplementary Fig. 8), a phenomenon also reported for the human estrone-3-sulphate and heme transporter SLCO2B133. Moreover, the PGE2 reuptake activity of PGTΔ425–508 cells was completely abolished (Fig. 1d and Supplementary Fig. 8). This underscores the critical role of the glycosylated Kazal-like domain in both PGT stability and surface trafficking.
The deletion of the ECL2 region (PGTΔ135–165) did not change surface localization or protein expression level (Supplementary Fig. 8a). Surprisingly, despite showing a WT-level uptake activity at 80 nM of PGE2, PGTΔ135–165 exhibited a 3-fold increase of PGE2 uptake activity compared to WT PGT at 500 nM PGE2 (Fig. 1d and Supplementary Fig. 8b). Michaelis–Menten kinetic analysis revealed that PGTΔ135–165 exhibited an 8-fold higher Vmax, as well as an 8-fold higher KM (Fig. 1e). The higher KM value likely reflects a weakened interaction between PGE2 and PGTΔ135–165, since the binding affinity was reduced by approximately 2 folds in the deletion mutant (Fig. 1e). Consistently, MD simulations showed increased mobility of the Kazal-like domain in PGTΔ135-165 than the WT PGT (Supplementary Fig. 9a, b). We hypothesize that ECL2 restrains the overall conformational changes of PGT. While weakening the substrate binding, the increased flexibility in PGTΔ135–165 lowers the energetic barriers to conformational changes that govern the transporting cycle, thus showing an elevated KM and uptake rate.
Structural elements crucial for substrate recognition and transport at the central PGE2 binding site
PGE2, the most abundant PG in the body, has been extensively studied due to its pivotal role in inflammation and labor induction. To elucidate the recognition and transportation mechanism of PGE2, we incubated PGT with 0.14 mM of PGE2 for 20 min prior to cryo-EM vitrification. The analysis yielded a 2.96 Å map. In addition, a second monomeric map of global 4.01 Å-resolution was obtained upon 3D classification without image alignment (Supplementary Fig. 2 and Supplementary Table 1).
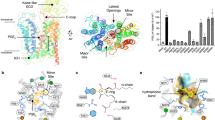
Compared to the PGTapo map obtained in the absence of PGE2, the 2.96 Å map exhibited additional density that fits well for a PGE2 at the center of the TMD (Fig. 2a, dubbed PGTcen). Despite having a PGE2 bound, the TMD remains unchanged compared to PGTapo, suggesting the transporter exists in a state primed for substrate binding. The PGT translocation funnel, similar to most MFS transporters, is composed of TM1, TM4, TM5, TM7, TM8, TM10, and TM11. The funnel is lined with hydrophobic and charged/polar residues, which form neutral and positively charged surfaces, suitably accommodating the modestly hydrophobic and negatively charged PGE2 (Fig. 2b, c). Similar composite hydrophobic/electropositive substrate binding sites have been observed in the structures of the PGE2 exporter MRP4 and the PGE2 G-protein coupled receptor EP3 (Supplementary Fig. 10), albeit with different residue identities34,35,36,37.

a Overview (left) of the PGE2-bound, outward-facing PGTcen structure and with the expanded view (right) of the L-shaped density that fits a PGE2 molecule (green sticks). b Cutaway side-view of the electrostatic surface potential (negative in red, positive in blue) shows PGE2 lipid occupying the composite hydrophobic/positive binding site. c Residues lining the central substrate binding cavity that holds PGE2 (green). Transmembrane helices are numbered. d 3H-PGE2 uptake activity of PGT mutants at central binding site under 80 nM of PGE2. Data for residues involved in direct PGE2 interaction were shown in dark gray, while surrounding residues were in light gray. Activities are normalized to that of WT PGT (mean ± SEM, n = 3 biologically independent experiments with 3 technical replicates each), based on the relative surface expression level assessed by surface biotinylation (Supplementary Fig. 8). Source data are provided as a Source Data file.
PGE2 sits snugly in the PGT central cavity, adopting a compact L-shape similar to that found in MRP4 and EP3 (Supplementary Fig. 10). The 20-carbon PGE2 comprises a cyclopentane ring and two flanking hydrocarbon tails with carbonyl, carboxyl, and hydroxyl groups at various positions (Fig. 2a). The carboxyl group forms a salt bridge with the positively charged Arg561. Residue Phe212 on TM5 extends towards the middle of the translocation funnel and upholds the middle cyclopentane, which is buttressed by surrounding residues Ser215, Pro219, and Met380. The ω alkyl tail locates in a hydrophobic niche formed by Phe557, Met560, and Trp565 (Fig. 2c). In line with these findings, perturbations in these interactions by mutagenesis, such as Q42A, Q188A, M560A, R561E, and W565A, resulted in severe reduction or complete loss of PGE2 uptake activity (Fig. 2d); Such residues are highly conserved among PGT orthologs (Supplementary Fig. 6). This interaction mode was further supported by our MD analysis, which showed the ligand remained relatively stable in most simulation trajectories (Supplementary Fig. 9c). Notably, mutations of several surrounding residues, including Val216, Ser339, Ile342, and His533, also produced substantial effects on substrate transport (Fig. 2d), although these residues are located relatively distant from the bound substrate (Fig. 2c). However, in a recent structure reported during the revision of our manuscript38, these four residues are involved in PEG2 binding. Thus, it is attempting to propose that the ligand samples distinct sublocations with the relatively spacious binding cavity of PGT. Interestingly, the S346A mutant, which is naturally an alanine in species like zebra fish, exhibited increased PGE2 uptake activity by threefold (which was also reported in the abovementioned paper), despite a decreased binding affinity (KM = 1.14 μM) compared to WT protein (KM = 0.11 μM) (Supplementary Fig. 6c).
Structural elements crucial for substrate selection at the entrance site
In the outward-facing state, the N-domain and the C-domain are not tightly joined at the extracellular interface, resulting in a wide cleft that opens to the bulk membrane between TM5 and TM8 on one side, and TM2-TM11 on the other side. Lipid-like densities were observed in these V-shaped openings in both PGTapo and PGE2-bound PGTcen maps (Supplementary Fig. 13a, b). Plausibly, these lateral paths facilitate PGE2 entrance from either or both sides, as similarly proposed in the cases of MFSD2A, Spns2, and OCT327,30,39.
The aforementioned second map, reconstructed from a PGE2-supplemented PGT sample, resembles the outward-facing PGTapo and PGTcen. Despite its relatively lower resolution, a stretched density spanning from the lateral V-opening between TM2 and TM11 to the center of the translocation funnel was observed. Apart from this different positioning, the density has a rod shape distinct from the previously described L-shaped density at the central binding site. As this population was not observed in the PGTapo sample, we modeled a PGE2 molecule into the density. This structure is referred to as PGTlen (Fig. 3a). Conforming to the rod shape, the PGE2 in this context adopts a straight conformation, similar to that observed in the recent EP2/PGE2 complex structure (Supplementary Fig. 12c, d)40. Detailed substrate-transporter interactions were challenging to depict due to the less well-resolved side chains in this map. Nonetheless, several residues in the close vicinity of PGE2 provided important insights into substrate entrance. The cyclopentane moiety packs against the indole ring of Trp565 on TM11. The hydrocarbon toward the carboxyl end of PGE2 is situated in the V-shaped opening between TM2 and TM11, while the carboxyl is electrostatically fixed by Lys53 on TM1. Although a conserved arginine was well-tolerated at this site, introducing an opposite charge by the K53E mutation dramatically reduced the 3H-PGE2 uptake (Fig. 3c), suggesting the importance of the electrostatic interaction in PGE2 transport. Consistently, Lys53 is conserved across species, except for an arginine in bovine (Supplementary Fig. 6). Finally, the opposite ω alkyl chain inserts deeply towards the midway of transport funnel (Fig. 3b). MD simulations confirmed this pose was unstable, consistent with its poorly defined density (Supplementary Fig. 9f). Intriguingly, however, in one simulation trajectory, the ligand transitioned into a pose resembling that of PGTcen, suggesting a plausible transport pathway (Supplementary Fig. 9g). High-resolution visualization of this lateral pose and potential other intermediate states will help comprehend the PGE2 transport process.

a The elongated density (semi-transparent purple) that fits a PGE2 (purple sticks) spanning the translocation funnel in PGTlen. Sidechains (not well-resolved in the map) in the vicinity are shown as stick representation for illustration purpose. b Top view of the PGT molecular surface colored by residue hydrophobicity. The PGE2 (purple) is shown as stick representation. c 3H-PGE2 uptake activity of mutants of Lys53 at 80 nM of PGE2. Activities are normalized to that of WT PGT (mean ± SEM, n = 3 biologically independent experiments with three technical replicates each), based on their relative surface expression level assessed by surface biotinylation. Source data are provided as a Source Data file.
Taken together, these observations support a mechanism through which the positively charged Lys53 performs the first-line scrutiny for negatively charged PGs at the lateral V-opening between TM2 and TM11.
Structural interpretation of disease-associated mutations
PGT regulates prostaglandin signaling magnitude and duration by clearing prostaglandins from circulation. Altered PGT expression levels have been detected in patients with cancers and/or chronic inflammation. Moreover, multiple autosomal loss-of-function PGT genetic variants, including missense, stop codon loss, indel and nonsense, have been identified in PHO15,16,17,18 and CEAS patients19,20,21.
Our PGT structures provide a framework for comprehending the mechanisms underpinning these recessive variants. The missense mutations mainly occur in the TMD, and specifically, on TMs 4, 5, 8, and 11, which together constitute the translocation pathway (Fig. 4a). For example, Trp565 on TM11 directly interacts with the PGE2 in both configurations described above. Mutating Trp565 to alanine almost abolished the uptake activity (Fig. 2d). Pro219 introduces a kink in TM5, shaping the central cavity and facing the ω alkyl chain of PGE2 (Fig. 4b). Mutating Pro219 to a leucine drastically reduced uptake activity, despite normal protein expression and surface localization (Fig. 4c and Supplementary Fig. 8). In addition, the mutation reduced substrate binding affinity by seven folds (Fig. 4d). Apart from shaping the central binding site, the helix bending of TM5, which can only be achieved by the mainchain-distorting proline, may be required for lowing the energy barrier to the outward-to-inward transition during the transport cycle.

a Structural mapping of genetic variants identified in PHO and CEAS patients. b Expanded view of the region covering G222R and P219L. c Relative activity (%WT) of G222R and P219L at 80 nM of PGE2. Activities are normalized to that of WT PGT (mean ± SEM, n = 3 biologically independent experiments, with three technical replicates in each experiment), based on their relative surface expression level assessed using in-gel fluorescence (Supplementary Fig. 8). d PGE2 binding affinity measured for the two mutants (mean ± SEM, n = 3 independent experiments). Source data are provided as a Source Data file.
Among these mutations are several Gly residues that are substituted by charged Arg, Glu and Asp, with the G222R variant being prevalent in PHO and CEAS patients15,16,19. Although Gly222 was not directly involved in substrate binding, introducing bulky and charged side chains at this site could cause severe electrostatic and spatial clashes with the neighboring residues Leu47, Ala50, and Tyr51 (Fig. 4b). In addition, such mutations at this point may perturb the curved helical structure of TM5, thus affecting PGT protein stability and function. Indeed, the expression of the G222R mutant was threefold lower than the WT, and the transportation activity was nearly abolished (Fig. 4c and Supplementary Fig. 8). Moreover, the affinity of G222R for PGE2 was reduced by over 25-fold (Fig. 4d).
Finally, several mutations (P457L, V458F, G460R, P469L, and R445C) occur on the Kazal-like domain. These mutations may affect PGT folding and surface expression in a similar manner to other mutants in the Kazal-like domain reported here (Supplementary Fig. 8) and previously for the human SLCO2B133.
Dimerization of PGT and potential functional consequences
Aside from the monomeric PGT structures, a substantial population of dimeric PGT particles was observed in both the apo and the PGE2-incubated samples (Supplementary Fig. 2a, b). Initially, we reconstructed separate cryo-EM maps for the two samples. Interestingly, no obvious ligand density was discernible in the transport funnels of the PGE2/PGT dimeric map. Given the striking similarity between the two dimer maps, we merged the two datasets to generate an improved dimer map (PGTdd) with a global resolution of 3.16 Å (Supplementary Fig. 2c–e and Supplementary Table 1). To our surprise, the ECD and TM12 were not resolved despite their overall map resolution being comparable to PGTapo (Fig. 5a and Supplementary Fig. 3). To investigate whether this conformational dynamics was an artifact induced by the detergent micelle, we reconstituted the PGT protein into a lipid nanodisc (Supplementary Fig. 1c). A considerable number of dimeric PGT averages were identified in this lipid environment, suggesting the dimer formation of the transporter in the membrane. Despite having a lower resolution (4.24 Å), the map of the nanodisc sample (PGTdn) shares a similar configuration, including the ill-defined ECDs and the missing TM12 helices, with PGTdd (Supplementary Fig. 12d). Furthermore, to probe oligomerization in a more native membrane context, we attempted extraction using SMA (styrene–maleic acid) copolymer. Thus, we will use PGTdd for discussions below.

a Side-view (left) and top-view (right) of the dimeric PGTdd structure. The two PGT protomers are colored rainbow (blue/red for the N-/C-terminus). The ECD region and TM12 are not resolved. b The electrostatic surface representation of one protomer highlighting the surface involved in the dimerization. c The dimerization interface formed by residues from TM8 and TM10. Other TMs are omitted for clarity. d Structural alignment between PGTapo (gray) and a protomer of the PGTdd dimer (rainbow). Conformational changes including the large rotational movement of TM9 are highlighted. e DSS crosslinking analysis. eGFP-tagged WT and cysteine substitution mutants were treated with increasing DSS concentrations (0 ~ 4 mM), deglycosylated by PNGase F, resolved by SDS-PAGE, and visualized by in-gel fluorescence. Oligomeric states (monomer [*], dimer [**]) were assigned based on molecular size markers derived from in-house prepared GFP-tagged proteins. f Disulfide bond analysis. Deglycosylated WT and cysteine substitution mutants were treated with (+) or without (−) 0.1 M DTT prior to SDS-PAGE and visualized as in (e).
The PGTdd dimerization, primarily mediated by hydrophobic interactions, occurs at a total buried surface area of 2250 Å2 mainly at the membrane-facing sides of TM8 and TM10 (Fig. 5b, c). To further investigate PGT dimerization in a cellular membrane environment, we performed chemical crosslinking experiments using the amine-reactive disuccinimidyl suberate (DSS) on membrane samples or disulfide cross-linking experiments with rationally designed cysteine mutants at the structurally observed dimer interface. In both WT and cysteine substitution mutants at interface residues (P374C-F524C, V371C, A375C, and I385C), DSS treatment produced a ~180 kDa band and higher molecular bands at the cost of decreasing intensity of the 96-kDa monomer (Fig. 5e and Supplementary Fig. 12e, f). Consistent with our design, disulfide-linked dimer bands sensitive to reduction by dithiothreitol (DTT) were observed for the symmetric V371C cysteine pair (self-pairing across the dimer interface) and the double mutant P374C-F524C (Fig. 5f). As a negative control, the single F524C mutant, which lacks a complementary cysteine at the interface, did not form disulfide-linked dimers (Supplementary Fig. 12f). These results further support the physiological existence of PGT dimers.
Notably, the two protomers were packed tightly with not enough transmembrane space to allow ligand passage (Supplementary Fig. 13), suggesting that the dimerization does not create a new transport pathway. The N-domain of PGT dimer aligns well with that of PGTapo, while the C-domain exhibits a remarkable conformational change (Fig. 5d). As mentioned above, TM12 and the extracellular regions, including the Kazal-like domain, became flexible. Besides, TM9 rotates a dramatic 30°, departing from TM10 by ~23 Å (distance measured between Cα atoms of Gly419 and Leu515). This separation may have introduced flexibility in the Kazal-like domain, as the two TMs jointly supported this domain in PGTapo (Fig. 1b). Moreover, although the inner leaflet halves of the TMD remain relatively unchanged, the outer leaflet halves of TM8 and TM10 bend towards TM7 to make enough room for the dimerization (Fig. 5d). Notably, TM7 and TM11 swing 10° away from their original positions towards the membrane boundary, drastically increasing the volume of the transport funnel (Supplementary Fig. 13c). Since residues on TM7 and TM11 interact extensively with PGE2, this extensive distortion likely disrupts the substrate binding site. This disruption may account for the lack of ligand density in the dimer map despite the inclusion of PGE2 in the cryo-EM sample. Taken together, these large movements would transform PGT into a conformation incompatible with its transport function.
Discussion
Prostaglandins are oxygenated derivatives of cellular membrane phospholipids that play vital roles in body homeostasis, smooth muscle contraction, sleep-wake regulation, and immune responses. An important aspect of PGs signaling is its timely clearance of these potent messengers, which necessitates their active reuptake by the PGT transporter. Our results have uncovered the structural basis of PGE2 recognized and transported by human PGT. The central substrate binding site of the PGT transport funnel is composed of clustered hydrophobic and polar/positively charged residues. Similar pockets are also found in structures of PGE2 exporter MRP4 and receptor EP3, suggesting a general recognition mechanism for prostaglandins.
Akin to other lipid-like substrate MFS transporters, PGT is presumed to undergo alternating access-type conformational changes during its transport cycle (Fig. 6). Recent structural studies of MFSD2A27,28,29, have revealed a flipping motion required for translocating the charged substrates across the membrane. Given the shared chemical characteristics between the PGs and the lysophospholipids, both of which are phospholipid derivatives, it is plausible that PG molecules also flip within the PGT pathway.

a Schematic representation of the alternating access transport cycle. The conformational states 1–3 are captured in this study, and the states 4–5 in faint shade are deduced from the AlphaFold2 prediction model and recent structural information of MFSD2A and Spns2. b Schematic representation of the proposed transport-to-channel conversion. Dashed lines in the ECD depict presumed flexibility. Upon assembling with other Maxi-Cl channel components, PGT undergoes conformational changes that expand the translocation funnel, deforming the gate near the extracellular interface (Gate 1), which is required for PGs transport (as depicted in (a)). Near the intracellular interface, Gate 2 is spared for ion passage in the channel-like configuration.
Previous reports identified a histidine residue at the extracellular tip of TM3 as critical for pH-dependent transport in several SLCO members41. Although this histidine’s position is not strictly conserved, and PGT-mediated PGs transport pH-insensitive7, the His533 residue near the PGE2 substrate within PGT’s translocation passage may function as a lactate acceptor. This is supported by reports that a lactate gradient drives PGs transport via PGT9. Our docking analysis demonstrated lactate association with His533 (Supplementary Fig. 9e). Consistent with the importance of His533, substitution with phenylalanine (H533F) reduced transport activity to ~50% of WT levels, while glutamate substitution (H533E) severely impaired PGE2 uptake (Fig. 2d). MD simulations showed comparable or slightly reduced stability of PGE2 bound to either H533F and H533E mutants (Supplementary Fig. 9d). These results suggest the potential role of His533 in either lactate reception or conformational equilibrium, or both.
With the structural information, we propose the following model for PGT transport. In the outward-facing PGT (Fig. 6a, state 1), which is primed for substrate entrance, PGs enter the transport funnel laterally via one of the two V-shape openings (Supplementary Fig. 11). The opening between TM2 and TM11 may be more favorable, as the positively charged Lys53 in the proximity provides attraction to the negatively charged PGs (state 2). Subsequently, PGs undergo a flipping motion, adjusting to the central binding site. This movement is facilitated by the protonated and thus positively charged His533 (state 3). Conformational changes relating to the alternating access model then occur, transforming PGT into an inward-facing state (state 4). Finally, the intracellular lactate may act as the counterion to facilitate the exit of PGs from the transport funnel (state 5), resetting PGT back to the outward-facing state for another transport cycle (state 1). We note that this model may be supported by PGs-bound occluded and inward-facing PGT structures in the future.
Apart from being a PGs transporter, PGT/SLCO2A1 has been recently identified as a key component of the Maxi-Cl anion channel by Okada and colleagues11. This ubiquitous channel facilitates the flow of inorganic and organic anions with a large unitary amplitude of over 300 pS, in response to a variety of stimuli, including cell swelling, ischemia or hypoxia, and heat. Our dimeric PGT structures, obtained in detergent micelles and lipid nanodiscs, may provide some architectural inspiration for the Maxi-Cl channel. While transporters generally operate with sequential gates to control directional substrate passage (Fig. 6b), channels often utilize a single gate to regulate the downhill movement of ions42. In the PGT dimer structure, both the ECD and TM12 exhibit considerable dynamics, and the other C-domain TMs also show increased flexibility. Although we acknowledge that PGT’s transport function might persist to some extent in the dimeric form, the altered substrate binding site due to the shifting C-domain TMs likely impairs PGs binding and hence transport. In line with this hypothesis, the Okada group found that PGE2 only mildly suppressed the Maxi-Cl currents, suggesting weaker PGE2 binding for the channel-oriented PGT11. Moreover, the tight interactions between the C-domains in the PGT dimer, combined with unstable ECDs and TM12, could get the transporter stuck at the current conformation, unable to complete the whole alternating access transport cycle, thus regulating the transport activity. Alternatively, PGT dimerization may hinder the formation of the extracellular gate and result in a configuration more akin to a one-gate channel than a two-gate transporter. Furthermore, the intracellular annexin A2/S100A10 complex has been identified as an important regulator of PGT’s channel activity43. Future structural studies of the PGT-annexin A2/S100A10 complex, particularly in the native membrane environment, are therefore warranted to elucidate the mechanism of Maxi-Cl channel formation and the potential transition between transporter and channel functions.
During the revision of our manuscript, Orlando and colleagues reported a 4.30 Å resolution outward-facing Apo structure of PGT, though PGE2 was included during sample preparation44. Consistent with our structural and MD analyses, their results demonstrate that the six intra-loop disulfides in the ECD are essential for trafficking and activity, while two inter-loop disulfides restrict maximal uptake. More recently, Jiang and colleagues reported structures of PGT in both Apo and PGE2-bound outward-facing states38. These structures share similar architecture, with the extracellular side of TM2 consistently exhibiting weak density across the three studies. Notably, the conformation of TM2 in our structure is similar to that of the Jiang group, but differs from the Orlando group (Supplementary Fig. 16a). These structural observations collectively indicate a dynamic nature of TM2 and suggest a regulatory role. Furthermore, the pose of PGE2 in the central binding site differs between our study and the Jiang group, despite comparable map resolutions (Supplementary Fig. 16b). These differences suggest the flexible PGE2 molecule samples distinct sublocations within the relatively spacious binding cavity of PGT. Together with the lateral entry PGE2 pose observed in our study, these structures paint a landscape of energetically favorable conformational poses during the transport cycle.
In conclusion, our PGT structures and biochemical evidence offer insights into the mechanism underpinning the clearance of exocytotic PGE2, as well as other PGs by reasonable extension, considering their shared chemical characteristics.
Methods
Material and methods
Detergents including dodecylmaltoside, Lauryl Maltose Neopentyl Glycol (LMNG), and cholesteryl hemisuccinate (CHS) were purchased from Antrace Inc. PGE2 was ordered from MedChemExpress LLC (New jersey, US). 3H-labelled PGE2 was sourced from Perkin Elmer (Cat. NET428050UC). Phospholipids were purchased from Avanti Polar Lipids (Alabaster, AL, US). All other chemicals and cell culture supplies were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cloning, expression, and purification
The human full-length PGT sequence (Uniprot ID: Q92959) was codon-optimized for expression in HEK293 cell lines. The PGT sequence was cloned into a modified pcDNA3.1 vector with maltose binding protein (MBP) and TEV cleavage site preceding the PGT, followed by a second TEV site and a Twin-Strep purification tag, with a subsequent enhanced green fluorescence protein (eGFP). Site-directed mutagenesis was performed using homologous recombination PCR, and the constructs were verified by sequencing in Beijing Tsingke Biotech Co., Ltd.
Cell lines and cell culture
Stable polyclonal HEK293T cell lines, expressing WT PGT or mutants, were established as follows: HEK293T cells were transfected with the respective plasmids (pCDH with target genes, pMD2.G and psPAX2) according to the manufacturer's procedures (System Biosciences). Cells were selected for 10 days using 3 μg/mL puromycin.
Cells were maintained in 10-cm cell culture dishes (NEST) at 37 °C and 5% CO2 in an incubator. The day before uptake assays, cells were seeded onto PDL (poly-D-lysine) coated 96-well plates at a density of ~0.4 × 105 cells per well. For live confocal microscopy, cells were seeded onto PDL-coated 20 mm glass-bottom dishes (Zhejiang Sorfa Life Science Research Co., Ltd) at a density of 0.2 × 106 cells per dish.
Radiotracer uptake and kinetic assays
For PGE2, uptake assay, stable HEK293T cell lines recombinantly expressing WT PGT or mutants were used, as endogenous PGT expression is negligible in HEK293T cells. On the day of the experiment, cells in 96-well plates were rinsed twice with prewarmed (37 °C) Waymouth medium (ThermoFisher, US). A PGE2 mix was made by mixing 10 μL of unlabelled (cold) PGE2 with various amounts of 3H-labeled (hot) PGE2 to a total volume of 20 μL in PBS buffer. The PGE2 mix and 80 μL of Waymouth medium were added to the cells to initiate PGE2 uptake. A time-course measurement revealed that 3H-PGE2 uptake in stably transfected cells remained linear over the 5-min incubation period (Supplementary Fig. 14). After 5 min, transport was stopped with ice-cold Waymouth medium, followed by two additional washes with PBS supplemented with 0.2% BSA. Cells were then lysed with 100 μL of 1% (wt/vol) sodium dodecyl sulfate (SDS). Lysate was transferred to a counting 96-well plate, containing 900 μL of scintillation cocktail (Liquiscint, Cat. LS-121). Radioactivity was measured using a Tri-carb 5110 TR Counter (Perkin Elmer, Waltham, USA). A stable cell line expressing eGFP was used as a control in all assays.
To calculate the specific activity of WT PGT, the amount of PGE2 transported into the cells was calculated using the known specific radioactivity of 3H-PGE2 (166.9 Ci/mmol, Cat. NET428050UC, Lot. 3125399, Perkin Elmer) and the dilution factors. The amount of protein was assayed by in-gel fluorescence using a known amount of eGFP on the same SDS-PAGE as calibration standards. Gels were scanned using a Typhoon FLA-9000 imager (Starion FLA-9000, FUJIFILM), and the calibration curve was generated using integrated intensity measured using ImageJ (Fiji, USA). Specific activity was expressed as nmol of PGE2 per minute per milligram of eGFP-tagged PGT (nmol/min/mg). In our stable cell line, PGT had a typical specific activity of 1.58 ± 0.18 nmol/min/mg in the presence of 80 nM PGE2 in the medium (Supplementary Fig. 15).
For the activity assay of mutants, their activity relative to WT PGT (%), rather than specific activity, was measured as follows. The uptake assay was performed as above. The relative transport activity was obtained by normalizing the amount of transported PGE2 using the relative surface expression level determined via surface biotinylation assay. The factors for the normalization are provided in the related figures.
To measure the KM of WT PGT and PGTΔ135–165, assays were performed as above but with various substrate concentrations as indicated in Fig. 1E. The maximal PGE2 concentration used for the PGTΔ135–165 mutant was 2.5 μM, which was higher than that for WT (1.0 μM), as the mutant required a higher concentration for saturation. The transport activity was normalized by in-gel fluorescence as stated above, and the normalized, relative activity (% of wild-type Vmax) was plotted in Fig. 1E.
Cell surface biotinylation and immunoblot analysis
For biotinylation assays, stable HEK293T cell lines expressing either WT PGT or mutants were used. Cells were cultured in 6 cm dishes for 24 h, then washed twice with 2 ml of ice-cold PBS. Subsequently, cells were incubated with 2 ml of 1.0 mg/ml Sulfo-NHS-SS-Biotin(Thermo Fisher Scientific) for 1 h at room temperature. The reaction was quenched by washing three times with ice-cold PBS containing 100 mM glycine, followed by a 10-min incubation in the same glycine solution at 4 °C. Cells were then lysed in 1 ml of lysis buffer (PBS,150 mM NaCl, 0.1% SDS, 1% Triton X-100, pH 7.4, supplemented with protease inhibitors) for 4 h at 4 °C with gentle agitation. Lysates were centrifuged at 10,000 × g for 2 min. The supernatants were incubated with 30 μl of streptavidin-agarose beads (Thermo Fisher Scientific) overnight at 4 °C. Beads were washed five times with ice-cold lysis buffer, then incubated with 20 μl of SDS loading buffer containing 100 mM DTT for 30 min at room temperature.
Proteins were resolved by 10 % SDS–PAGE and transferred to nitrocellulose (NC) membranes at 300 mA for 120 min. Membranes were blocked in 5% non-fat milk/TBST and probed with an anti-SLCO2A1 antibody (1:500 dilution, Cat. NBP2-13349, Novus Biologicals). Following detection, the membrane was stripped for 10 min and reprobed with anti-Na+/K+-ATPase monoclonal antibody (1:10,000 dilution, Cat. T40109M, ABmart). Finally, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Cat. L3012, SAB) for 1 h at room temperature. Protein bands were visualized using Tanon High-sig ECL Western Blotting substrate.
Chemical crosslinking and disulfide bond mutagenesis analysis
WT and mutant proteins were separately expressed in HEK293 cells as described. All subsequent steps were performed at 4 °C. Cells were resuspended in lysis buffer (25 mM HEPES, 150 mM NaCl, 5% glycerol, pH 7.4) and homogenized using a Dounce homogenizer (30 strokes). The amine-reactive crosslinker DSS (Cat. 21655, Thermo Fisher Scientific) was added to the homogenates at various concentrations, followed by incubation for 3 h with gentle agitation. Reactions were quenched by adding 0.1 M Tris-HCl (pH 7.5). Samples were then solubilized in 1% (w/v) n-dodecyl-β-D-maltoside (Cat. D310S, Anatrace) for 1 h and centrifuged at 10,000 × g for 10 min. The supernatant was deglycosylated using homemade PNGase F for 2 h. Deglycosylated samples were centrifuged at 10,000 × g for 5 min before separation on a 6% Tris-acetate SDS-PAGE gel. In-gel fluorescence was visualized using an Amersham Typhoon 5 scanner (Cytiva) controlled by Amersham Typhoon Control software (v4.0.0.4).
For disulfide bond analysis, WT and mutant proteins were lysed and deglycosylated as described above. Lysates were then treated with or without 0.1 M DTT prior to separation by SDS-PAGE. Gels were visualized and analyzed as described for the DSS crosslinking experiment.
Confocal microscopy and image analysis
Confocal microscopy images were taken on a Leica SP8 LSCM+ laser scanning confocal microscope system with a 60xNA 1.4 oil immersion objective (Leica, Germany). eGFP fluorescence was excited with a 488 nm laser line. Emitted light was detected by a HyD detector.
Protein expression and purification
The plasmids containing genes of recombinant proteins at 1 mg/mL were transiently transfected into HEK293F cells with 3 mg/mL PEI (Biohub International Trade Co., LTD) when HEK293F-PGT cells were grown in suspension at 37 °C to a density of ~2.0 × 106 mL−1 in Gibco® FreeStyleTM 293 Expression Medium (ThermoFisher Scientific). The cells were harvested after 48 h growth by centrifugation at 1500 × g for 15 min and stored at −80 °C until the day of the experiment.
For purification, the cell pellets collected from 1 L of cell culture were thawed and resuspended in buffer A (50 mM Hepes, 150 mM NaCl, 5% glycerol, pH 7.3) supplemented with protease inhibitors (1 μg/mL leupeptin, 1 μg/mL aprotinin, 1 μg/mL pepstatin, and 1 mM PMSF) and lysed using a Dounce homogenizer. The lysate was then solubilized in buffer A supplemented with 1% LMNG and 0.1% CHS at 4 °C for 2 h in a rotator. Cell debris in the lysate was removed by centrifugation at 20,000 x g for 40 min. The supernatant was then incubated with 2 mL of Strep-Tactin beads at 4 °C for 2 h. The Strep-Tactin beads were then collected in a gravity column and washed with 30 column volumes of buffer A containing LMNG and CHS at a concentration ranging from (0.1% LMNG + 0.01% CHS) to (0.001% LMNG + 0.0001% CHS). The target protein was eluted with 2.5 mM D-desthiobiotin. MBP and eGFP tags were cleaved with TEV protease. The protein sample was then concentrated with a 100 kDa cut-off Amicon Ultra spin (Millipore) and subjected to size-exclusion chromatography (SEC) on a Superose 6 Increase 10/300 GL column (GE Healthcare) equilibrated in buffer B (50 mM Hepes, 150 mM NaCl, 0.001% LMNG and 0.0001% CHS, pH 7.3). The fractions corresponding to purified PGT were collected and concentrated to ~1.0 mg/mL for subsequent assay. The protein purity was assessed using a 12% SDS-PAGE gel and visualized by Coomassie brilliant blue staining.
Nanodisc reconstitution
The membrane scaffolding protein (MSP) 1E3 was expressed and purified as described45. Briefly, to prepare the lipid stock, 7.5 mg of mixtures of POPC: POPS (3:1), 6 mg of sodium cholate, and 3 mg of LMNG were co-dissolved in chloroform/methanol (3:1, vol:vol). The solution was dried under nitrogen and dissolved in 0.3 mL of buffer A without glycerol. For nanodisc reconstitution, PGT, MSP1E3, and lipids were incubated at 1:4:200 molar ratio with a total volume of 2 mL. Biobeads (Cat. 152-3920, Bio-Rad) were included evenly to a final concentration of 150 mg/mL in three steps with 2 h incubation at first two additions, and a 16 h incubation after the second addition. The biobeads were then removed from the reconstitution mixture by centrifugation, and the supernatant was concentrated to 1.8 mg/mL using a 100-kDa cutoff membrane concentrator (Cat. UFC810096, Merck Millipore) before being applied onto a Superose 6 increase 10/300 GL column (Cat. 29-0915-96, Cytiva) with buffer B without detergents as the running buffer. Peak fractions as shown in Supplementary Fig. 1B were collected and concentrated to ~3 mg/mL of total protein for cryo-EM sample preparation. Protein concentration was quantified by absorbance at 280 nm measured using a Nanodrop machine with the theoretical molar extinction coefficient of 141,650 M−1 cm−1 assuming a molar ratio of 1:2 (PGT:MSP1E3).
Styrene maleic acid (SMA) copolymer solubilization
Cells were resuspended in lysis buffer (25 mM HEPES, pH 7.5, 300 mM NaCl), lysed by sonication, and centrifuged at 6000 × g for 20 min to remove debris. The supernatant was subjected to ultracentrifugation at 60,000 g for 1 h. Membrane pellets were resuspended in resuspension buffer (25 mM HEPES, pH 7.5, 300 mM NaCl, 5% glycerol, protease inhibitors) and homogenized with 30 strokes in a Dounce homogenizer. SMALP 300 copolymer (Cube Biotech, #18201) was added to the membrane suspension to a final concentration of 2.5% (w/v). The mixture was stirred gently at room temperature for 1 h. After centrifugation at 14,000 × g for 1 h, the supernatant was incubated with Strep-Tactin resin overnight at 4 °C. Subsequent purification steps were performed as previously described, without introducing additional SMA copolymer or detergents.
Microscale thermophoresis
The eGFP-tagged full-length PGT and PGT mutants were purified as described above without cleavage. Peak fractions were collected and diluted to about 85 nM for further use. Ligand stock (14 mM PGE2) was diluted to the highest concentration used in the assay buffer (50 mM Hepes, 150 mM NaCl, 0.001% LMNG + 0.0001% CHS, pH 7.3). Measurements were carried out with a Monolith NT.115 device at 20–40% LED power and 40% MST power. Kd was initially determined using the MO. Affinity Analysis software (version 2.2.4, NanoTemper Technologies, Germany). Points from dose response curves (fraction bound data) were fitted and presented by Graphpad Prism 8.4.3 (GraphPad Software Inc., San Diego, USA). Data of at least three independently pipetted measurements were analyzed. Kd is expressed as mean ± SEM.
Cryo-EM sample preparation and data collection
Freshly purified PGT samples were used for vitrification. To prepare the PGT/PGE2 complex, final ~140 μM PGE2 was added to SEC-purified PGT proteins, and incubated for 20 min at room temperature. Quantifoil Au R1.2/1.3 grids (300 mesh) were briefly glow-discharged in a PELCO easiGlow (Ted Pella) glow discharge cleaning system. 2.5 μL protein solutions were then applied to glow-discharged grids, blotted for 3 ~ 5 s, and plunged into liquid ethane using Vitrobot Mark IV (Thermo Fisher Scientific) with 100% humidity at 4 °C. The frozen grids were transferred under cryogenic conditions and stored in liquid nitrogen for subsequent screening and cryo-EM data collection.
All datasets were collected on a Titan Krios G4 cryo-electron microscope, equipped with a Falcon G4i direct electron detector with a Selectris X imaging filter (Thermo Fisher Scientific) operated with a 20 eV slit. Movie stacks were acquired using the EPU software (Thermo Fisher Scientific) in super-resolution mode with a defocus range of −1.2 to −2.0 μm and a final calibrated pixel size of 0.932 Å. The total dose per EER (electron event representation) movie was 50 e−/Å2.
Cryo-EM data processing
All datasets were processed similarly in cryoSPARC (v.3.3.2)46 and RELION (v.3.1.4)47. For the PGTapo detergent sample, a total of 8145 EER movies were collected. Each EER movie of 1080 frames was fractionated into 40 subgroups, and beam-induced motion was corrected with a MotionCor2-like algorithm implemented in RELION. Exposure-weighted micrographs were then imported to cryoSPARC for CTF (contrast transfer function) estimation by patch CTF. Particles were blob-picked and extracted with a box size of 240 pixels, and subjected to multiple rounds of 2D classification. The monomeric and dimeric particles were selected from 2D averages and processed separately in later steps. Several rounds of heterogeneous refinement (3D classification) were conducted using ab initio reference maps reconstructed with 2D averages of nice features. The good particles were then converted for Bayesian polishing in RELION, which was subsequently imported back to cryoSPARC for one more round of ab initio reconstruction and several rounds of heterogeneous refinement to remove residual contaminants or poor-quality particles. Final 3.21 Å monomeric PGTapo map from 238,917 particles, and 3.34 Å dimeric apo map from 113,734 particles were obtained by CTF refinement and local refinement. Resolution of these maps was estimated internally in cryoSPARC by gold-standard Fourier shell correlation using the 0.143 criterion.
For the PGE2-supplemented detergent sample, a total of 12,630 EER movie stacks were processed in a similar way as PGTapo data. Briefly, monomeric and dimeric 2D averages from cryoSPARC were picked and subjected to the following process separately. The good monomeric PGE2/PGT particles were converted back to RELION for Bayesian polishing, and 3D classification without alignment was performed to improve ligand density quality and sort out possible ligand poses. Two different positions for PGE2 ligand density were obtained. After ab initio reconstruction and additional heterogeneous refinement, two final maps were reconstructed 2.96 Å (PGTcen: PGE2 at central binding site) and 4.01 Å (PGTlen: possible PGE2 close to lateral entry site), respectively. Similarly, the dimeric particles were processed to a final 3.54 Å map, which were combined with the apo dimer particles for an improved final dimeric PGTdd map (3.16 Å with C2 symmetry applied), since the two dimeric maps were nearly identical.
The above procedure was also applied to 8635 EER movies collected for PGE2/PGT sample reconstructed in lipid nanodisc. Obvious dimeric particles were chosen for extensive classification, which led to a final 4.24 Å map.
Model building and refinement
Initial PGT model was retrieved from AphaFold48 database, which is predicted as inward-open conformation (ID: AF-Q92959). The predicted model was rigid-body docked into PGTapo cryo-EM density map in ChimeraX (v.1.6)49, followed by iterative manual adjustment in COOT (v.0.9.8)50 and real-space refinement in Phenix (v.1.19)51. PGE2 model and geometry restraints were generated by the eLBOW tool from Phenix. The model statistics were validated by Molprobity52. Sidechains that do not have well-defined density were trimmed to alanine. The final refinement statistics are provided in Supplementary Table 1. Structural figures were prepared in ChimeraX or PyMOL (PyMOL Molecular Graphics System, v.2.3.4, Schrödinger) (https://pymol.org/2/).
Molecular dynamics simulations
We performed all-atom MD simulations in explicit solvents. The chain termini were neutralized by capping groups (acetylation and methylation) to avoid termini-charge-dependent effects. PropKa was used to determine the dominant protonation state of all titratable residues at pH 7.353. The CHARMM-GUI Membrane builder module54 was used to place each protein in a 1:1 POPC membrane patch with 20 Å of water above and below and 0.15 mM NaCl in the solution. The final systems had ~112 POPC lipids, ~23,000 water molecules, and initial dimensions of 95 × 95 x 129 Å3. The CHARMM36m force field was adopted for lipids, proteins, sodium and chloride ions, and the TIP3P model for waters55. Ligands were modeled with the CHARMM CGenFF small-molecule force field56.
Simulations were performed using Gromacs 2020.757. For each condition, three to five independent simulations were run. All systems were energy minimized and equilibrated in six steps consisting of 2.5 ns long simulations, while slowly releasing the position restraint forces acting on the Cα atoms. Initial random velocities were assigned independently to each system. Production simulations were performed for 1.0 µs. The Verlet neighbor list was updated every 20 steps with a cutoff of 12 Å and a buffer tolerance of 0.005 kJ/mol/ps. Non-bonded van der Waals interactions were truncated between 10 and 12 Å using a force-based switching method. Long-range electrostatic interactions under periodic boundary conditions were evaluated by using the smooth particle mesh Ewald method with a real-space cutoff of 12 Å58. Bonds to hydrogen atoms were constrained with the P-LINCS algorithm with an expansion order of four and one LINCS iteration59. The constant temperature was maintained at 310 K using the v-rescale (τ = 0.1 ps) thermostat60 by separately coupling solvent plus salt ions, membrane, and protein. Semi-isotropic pressure coupling was applied using the Parrinello–Rahman barostat61, using 1 bar and applying a coupling constant of 1 ps. Finally, a restraint-free production run was carried out for each simulation, with a time step of 2 fs. The parameters were provided in Supplementary Table 2. Initial coordinate and simulation input files, and a coordinate file of the final output are provided as Supplementary Data 1.
Data and statistical analysis
KM and Kd values were calculated and graphs plotted with GraphPad Prism 8.0 (GraphPad Software Inc., San Diego, USA). Michaelis-Menten kinetics were determined via Y = Vmax*X/(KM + X). All data are from at least three biologically independent experiments (n = 3), in triplicate and portrayed as mean ± SD. For representative results such as chemical cross-linking assays, at least three biological repeats were conducted independently.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The coordinates for PGTapo, PGE2-bound at central site (PGTcen), and PGT dimer in detergent (PGTdd) have been deposited in the Protein Data Bank under accession codes 8KGV, 8KGW, and 8KGI, respectively. The cryo-EM density maps for PGTapo, PGTcen, PGE2-bound close to lateral entry (PGTlen), PGTdd, and PGT dimer in nanodisc (PGTdn) have been deposited in the Electron Microscopy Data Bank with accession codes EMD-37233, EMD-37234, and EMD-37222. Source data are provided with this paper.
References
Miller, S. B. Prostaglandins in health and disease: an overview. Semin. Arthritis Rheum. 36, 37–49 (2006).
Alexander, C. L., Miller, S. J. & Abel, S. R. Prostaglandin analog treatment of glaucoma and ocular hypertension. Ann. Pharmacother. 36, 504–511 (2002).
Yount, S. M. & Lassiter, N. The pharmacology of prostaglandins for induction of labor. J. Midwifery Women’s. Health 58, 133–144 (2013).
Biringer, R. G. The enzymology of the human prostanoid pathway. Mol. Biol. Rep. 47, 4569–4586 (2020).
Smyth, E. M., Grosser, T., Wang, M., Yu, Y. & FitzGerald, G. A. Prostanoids in health and disease. J. Lipid Res. 50, S423–S428 (2009).
Qureshi, O. & Dua, A. COX inhibitors. in StatPearls (StatPearls Publishing, 2023).
Kanai, N. et al. Identification and characterization of a prostaglandin transporter. Science 268, 866–869 (1995).
Schuster, V. L., Chi, Y. & Lu, R. The prostaglandin transporter: eicosanoid reuptake, control of signaling, and development of high-affinity inhibitors as drug candidates. Trans. Am. Clin. Climatol. Assoc. 126, 248–257 (2015).
Chan, B. S., Endo, S., Kanai, N. & Schuster, V. L. Identification of lactate as a driving force for prostanoid transport by prostaglandin transporter PGT. Am. J. Physiol. Ren. Physiol. 282, F1097–F1102 (2002).
Shimada, H., Nakamura, Y., Nakanishi, T. & Tamai, I. OATP2A1/SLCO2A1-mediated prostaglandin E2 loading into intracellular acidic compartments of macrophages contributes to exocytotic secretion. Biochem. Pharmacol. 98, 629–638 (2015).
Sabirov, R. Z. et al. The organic anion transporter SLCO2A1 constitutes the core component of the Maxi-Cl channel. EMBO J. 36, 3309–3324 (2017).
Nakanishi, T. et al. Prostaglandin transporter (PGT/SLCO2A1) protects the lung from bleomycin-induced fibrosis. PLoS ONE 10, e0123895 (2015).
Chang, H.-Y., Locker, J., Lu, R. & Schuster, V. L. Failure of post-natal ductus arteriosus closure in prostaglandin transporter-deficient mice. Circulation 121, 529–536 (2010).
Kasai, T. et al. Role of OATP2A1 in PGE2 secretion from human colorectal cancer cells via exocytosis in response to oxidative stress. Exp. Cell Res. 341, 123–131 (2016).
Zhang, Z. et al. Exome sequencing identifies SLCO2A1 mutations as a cause of primary hypertrophic osteoarthropathy. Am. J. Hum. Genet. 90, 125–132 (2012).
Diggle, C. P. et al. Prostaglandin transporter mutations cause pachydermoperiostosis with myelofibrosis. Hum. Mutat. 33, 1175–1181 (2012).
Busch, J. et al. Mutations in the prostaglandin transporter SLCO2A1 cause primary hypertrophic osteoarthropathy with digital clubbing. J. Investig. Dermatol. 132, 2473–2476 (2012).
Sasaki, T. et al. Identification of mutations in the prostaglandin transporter gene SLCO2A1 and its phenotype–genotype correlation in Japanese patients with pachydermoperiostosis. J. Dermatol. Sci. 68, 36–44 (2012).
Umeno, J. et al. A hereditary enteropathy caused by mutations in the SLCO2A1 gene, encoding a prostaglandin transporter. PLoS Genet. 11, e1005581 (2015).
Umeno, J. et al. Clinical features of chronic enteropathy associated with SLCO2A1 gene: a new entity clinically distinct from Crohn’s disease. J. Gastroenterol. 53, 907–915 (2018).
Hamon, A. et al. Crohn-like disease affecting small bowel due to monogenic SLCO2A1 mutations: first cases of chronic enteropathy associated with SLCO2A1 gene [CEAS] in France. J. Crohns Colitis 17, 816–820 (2023).
Chi, Y. et al. Inhibition of the prostaglandin transporter PGT lowers blood pressure in hypertensive rats and mice. PLoS ONE 10, e0131735 (2015).
Liu, Z., Benard, O., Syeda, M. M., Schuster, V. L. & Chi, Y. Inhibition of prostaglandin transporter (PGT) promotes perfusion and vascularization and accelerates wound healing in non-diabetic and diabetic rats. PloS ONE 10, e0133615 (2015).
Inagaki, M. et al. Contribution of prostaglandin transporter OATP2A1/SLCO2A1 to placenta-to-maternal hormone signaling and labor induction. iScience 23, 101098 (2020).
Yerushalmi, G. M. et al. The prostaglandin transporter (PGT) as a potential mediator of ovulation. Sci. Transl. Med. 8, 338ra68–338ra68 (2016).
Nakamura, Y., Aizawa, C., Kawata, H. & Nakanishi, T. N-glycosylation modifies prostaglandin E2 uptake by reducing cell surface expression of SLCO2A1. Prostaglandins Other Lipid Mediat. 165, 106714 (2023).
Wood, C. A. P. et al. Structure and mechanism of blood–brain-barrier lipid transporter MFSD2A. Nature 596, 444–448 (2021).
Cater, R. J. et al. Structural basis of omega-3 fatty acid transport across the blood–brain barrier. Nature 595, 315–319 (2021).
Nguyen, C. et al. Lipid flipping in the omega-3 fatty-acid transporter. Nat. Commun. 14, 2571 (2023).
Khanppnavar, B. et al. Structural basis of organic cation transporter-3 inhibition. Nat. Commun. 13, 6714 (2022).
Suo, Y. et al. Molecular basis of polyspecific drug and xenobiotic recognition by OCT1 and OCT2. Nat. Struct. Mol. Biol. 30, 1001–1011 (2023).
Schlott, B. et al. Interaction of Kazal-type inhibitor domains with serine proteinases: biochemical and structural studies. J. Mol. Biol. 318, 533–546 (2002).
Hänggi, E., Grundschober, A. F., Leuthold, S., Meier, P. J. & St-Pierre, M. V. Functional analysis of the extracellular cysteine residues in the human organic anion transporting polypeptide, OATP2B1. Mol. Pharmacol. 70, 806–817 (2006).
Huang, Y. et al. Structural basis for substrate and inhibitor recognition of human multidrug transporter MRP4. Commun. Biol. 6, 1–11 (2023).
Pourmal, S. et al. Structural basis of prostaglandin efflux by MRP4. Nat. Struct. Mol. Biol. 31, 621–632 (2024).
Bloch, M., Raj, I., Pape, T. & Taylor, N. M. I. Structural and mechanistic basis of substrate transport by the multidrug transporter MRP4. Structure 31, 1407–1418.e6 (2023).
Morimoto, K. et al. Crystal structure of the endogenous agonist-bound prostanoid receptor EP3. Nat. Chem. Biol. 15, 8–10 (2019).
Xia, Z. et al. Structure and transport mechanism of the human prostaglandin transporter SLCO2A1. Nat. Commun. 16, 8124 (2025).
Chen, H. et al. Structural and functional insights into Spns2-mediated transport of sphingosine-1-phosphate. Cell 186, 2644–2655.e16 (2023).
Qu, C. et al. Ligand recognition, unconventional activation, and G protein coupling of the prostaglandin E2 receptor EP2 subtype. Sci. Adv. 7, eabf1268 (2021).
Leuthold, S. et al. Mechanisms of pH-gradient driven transport mediated by organic anion polypeptide transporters. Am. J. Physiol. Cell Physiol. 296, C570–C582 (2009).
Minor, D. L. Channel surfing uncovers a dual-use transporter. EMBO J. 36, 3272–3273 (2017).
Nakamura, Y. et al. Modulation of prostaglandin transport activity of SLCO2A1 by annexin A2 and S100A10. Am. J. Physiol. Cell Physiol. 326, C1042–C1053 (2024).
Yu, P., Orlando, M. A. & Orlando, B. J. Structure and post-translational modification of the prostaglandin transporter. Commun. Biol. 8, 1066 (2025).
Ritchie, T. K. et al. Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 (2009).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. Publ. Protein Soc. 27, 14–25 (2018).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. Sect. Struct. Biol. 74, 531–544 (2018).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 66, 12–21 (2010).
Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M. & Jensen, J. H. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 (2011).
Wu, E. L. et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 35, 1997–2004 (2014).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Yu, W., He, X., Vanommeslaeghe, K. & MacKerell, A. D. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 33, 2451–2468 (2012).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015).
Steinbach, P. J. & Brooks, B. R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 15, 667–683 (1994).
Hess, B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4, 116–122 (2008).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Acknowledgements
We thank the Center of Cryo-Electron Microscopy and the Core Facility of Shanghai Medical College, Fudan University for technical support and the staff member of the Core Facilities of Molecular Biology and Cell Biology at the Shanghai Institute of Biochemistry and Cell Biology for technical support. This work was supported by the National Key R&D Program of China (2023YFA0915000 to Q.Q.), the National Natural Science Foundation of China (32171194 & 32371256 to Q.Q.; W2412096 & 32471246 to D.L.; 32401007 to Z.Zhou), Noncommunicable Chronic Diseases-National Science and Technology Major Project of the Ministry of Science and Technology of China (2023ZD0503203 to Q.Q.), the Strategic Priority Research Program of CAS (XDB37020204 to D.L.), Science and Technology Commission of Shanghai Municipality (22ZR1468300 to D.L.), and the China Postdoctoral Science Foundation (2022M720805 to Z.Zhou).
Author information
Authors and Affiliations
Contributions
Y.Li. and Z.Zhu purified proteins, performed biochemical and cellular assays, and participated in cryo-EM grids preparation and data processing. H.X. conducted 3H-PGE2 uptake assays. C.Y. performed molecular dynamics simulations. Z.Zhou collected cryo-EM data and built models with the assistance of Y.C. Q.Q., D.L., Q.B., and Y.Li. oversaw the project. Q.Q. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Fabio Polticelli and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, Z., Li, Y., Xia, H. et al. Molecular basis of prostaglandin E2 reuptake by organic anion transporter PGT. Nat Commun 17, 315 (2026). https://doi.org/10.1038/s41467-025-67025-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67025-8
