
Abstract
Restoring retinal pigment epithelium (RPE) cells is crucial for treating retinal degenerative (RD) diseases, with chemical reprogramming offering a transformative, scalable solution. However, identifying key chemical compounds for generating functional RPE cells from somatic cells remains challenging. Here, we present a two-step chemical reprogramming strategy to convert fibroblasts into functional chemical induced RPE (ciRPE) cells. Leveraging the Single-Cell Reprogramming Compound Finder (scRCF), which integrates transcriptomics-guided predictions with advanced screening, we identified chemical cocktails that precisely reprogram fibroblasts through an intermediate state into ciRPE cells. These ciRPE cells closely mimic the structure and function of native RPE cells, and upon transplantation into RD rats, they seamlessly integrate into host tissue, protect photoreceptors, and restore visual function. Omics and mechanistic analyses revealed that the identified compounds synergistically activate core transcription factors, including Ascl1 and Olig2, orchestrating the reprogramming process. This study provides a scalable, non-integrative approach for generating functional RPE cells, offering a promising strategy for cell replacement therapies targeting RD diseases.
Similar content being viewed by others

Introduction
The retinal pigment epithelium (RPE) is indispensable for retinal health, playing a critical role in maintaining photoreceptor function and the blood-retinal barrier. RPE dysfunction or loss is a key driver in the pathogenesis of devastating retinal degenerative (RD) diseases, such as age-related macular degeneration (AMD), retinitis pigmentosa (RP), and Stargardt disease, which result in progressive vision loss and, in advanced stages, irreversible blindness1,2. Given the essential role of RPE cells in retinal function, the restoration or replacement of damaged RPE cells represents a transformative strategy for treating these debilitating conditions. However, generating fully functional and therapeutically safe RPE cells remains a substantial challenge in regenerative medicine.
Over the years, two main strategies have been explored to obtain RPE cells: isolation from native tissue and differentiation from pluripotent stem cells (PSCs)1,3,4,5,6. Both approaches have advanced considerably, yet some limitations remain. Tissue-derived RPE cells are relatively scarce and may gradually lose function in culture, which can constrain their use in large-scale applications. PSC-derived RPE cells, while considered an attractive source and already proven safe and feasible in clinical studies, often require lengthy differentiation and rigorous purification to ensure maturity and safety, adding to the complexity and cost of production7,8,9. An alternative method involves direct reprogramming using ectopic transcription factors (TFs), which can convert fibroblasts directly into RPE-like cells, bypassing the pluripotent stage10,11. While promising, this method still faces challenges such as genomic integration risks, low reprogramming efficiency, and high costs. These limitations underscore the need for alternative strategies that are transgene-free, rapid, and compatible with scalable, clinically relevant manufacturing processes.
Several studies have demonstrated the feasibility of using small molecules to reprogram somatic cells into various lineages, such as neurons12,13, astrocytes14, hepatocytes15, and corneal endothelia16. Unlike TF-based methods, chemical reprogramming can offer a non-integrative, non-viral, and cost-effective approach to generating RPE cells. However, despite its potential, achieving precise reprogramming of somatic cells into RPE cells remains a complex and ongoing challenge. Current efforts predominantly rely on high-throughput screening of large chemical libraries, a process that is time-consuming and inefficient17. Furthermore, the incomplete understanding of the complex signaling networks and transcriptional regulation required to induce RPE fate has hindered progress. Thus, there is a critical need for more sophisticated, data-driven platforms that can accurately predict chemical compounds capable of inducing RPE-specific reprogramming.
Recent advances in single-cell RNA sequencing (scRNA-seq) and chemical perturbation databases have revolutionized our ability to study gene expression dynamics and cellular signaling pathways involved in reprogramming18. In particular, databases such as ChEMBL, STITCH, DrugBank, and Reactome provide essential resources for mapping interactions between transcription factors, chemical compounds, and biomolecules. These technological breakthroughs offer unprecedented opportunities to systematically identify key TFs and their interactions with chemical compounds, which could guide cell fate decisions. However, despite these advances, existing predictive platforms still have certain limitations. They rely on static models and pre-existing datasets, which do not fully capture the dynamic nature of cellular reprogramming, particularly in the context of complex cell types like RPE cells19. This highlights the urgent need for more advanced, dynamic, and scalable platforms capable of accurately predicting the chemical compounds required for RPE reprogramming.
Furthermore, identifying suitable intermediate cell states is crucial for enhancing the efficiency and stability of RPE reprogramming. Eye field (EF) or optic vesicle (OV)-derived progenitor cells, which naturally differentiate into RPE cells during retinal development, represent ideal intermediate states. These cells possess remarkable plasticity and differentiation potential, making them a biologically relevant step in guiding somatic cells toward an RPE fate20,21,22,23. Using EF (or OV) as an intermediate state not only increases reprogramming efficiency but also minimizes the risk of generating unwanted cell types, ensuring a more stable and reproducible conversion process.
In this study, we tackle the complex challenge of generating functional RPE cells by developing an innovative two-stage chemical induction protocol, enabled by the Single-Cell Reprogramming Compound Finder (scRCF), an advanced platform that integrates computational predictions guided by single-cell transcriptomics with a sophisticated DRUG-seq screening system. This platform systematically identifies optimal small-molecule combinations that guide fibroblasts to an EF-like state, ultimately generating functional chemical induced RPE (ciRPE) cells. These ciRPE cells closely mirror native RPE cells in morphology, gene expression, and essential functional properties. In an RD rat model, transplantation of ciRPE cells led to seamless integration into the host RPE layer, significant preservation of photoreceptors, and marked restoration of visual function. Multi-omics analyses provided insights into the dynamic transcriptional reprogramming that occurs during this process, while mechanistic studies revealed that chemical compounds synergistically activated endogenous TFs, such as Ascl1 and Olig2, directing fibroblast reprogramming toward the RPE cell phenotype. This study introduces a scalable, non-integrative, and cost-effective chemical approach for generating functional RPE cells, offering a promising innovative strategy for cell replacement therapies targeting RD diseases. Here, we present a two-step chemical reprogramming strategy that efficiently converts fibroblasts into functional RPE cells, bypassing the need for pluripotent intermediates and offering a promising solution for cell replacement therapies.
Results
Systematic screening of small molecules for EF fate reprogramming using scRCF
We established a two-step chemical induction strategy to reprogram fibroblasts into RPE cells. First, the fibroblasts were converted into an intermediate EF-like state and then were followed by differentiation into RPE-like cells. To address the challenge of identifying small molecules for cellular reprogramming, we developed the scRCF (Fig. 1a), which integrates single-cell transcriptomics with gene co-expression networks to identify key TFs and pathways, facilitating the rational selection of compounds. To refine their identification, we incorporated cellular-level sequencing (DRUG-seq2)24,25, enhancing the precision and efficiency of small molecule-driven reprogramming.

a The workflow of scRCF. The input of scRCF comprises scRNA-seq data from both initial and target cell types, in addition to three pre-established databases: (1) A small molecule perturbation database, integrating data from GEO and LINCS L1000, retaining only TFs identified in AnimalTFDB3.0; (2) The small-molecule target database and classification information, incorporating information from STITCH, Drug Repurposing Hub, and MedChemExpress; (3) A signaling network database, derived from Reactome and OmniPath. scRCF contains three major steps: (1) Identify candidate signaling proteins and perform pre-screening using the signaling network; (2) Calculate efficiency scores for small molecules based on the community partitioning results and output the highest-scoring compounds from each category as candidate small molecules; (3) Further screen the candidate small molecules using DRUG-seq2. b UMAP visualization of integrated scRNA-seq data from four MEF datasets and one primary EF (pEF) dataset, used to predict small molecules that facilitate reprogramming between the two cell types. c Volcano plot illustrating the differentially expressed TFs (DETFs) identified from scRNA-seq data of MEFs and EFs, analyzed using Seurat. DETFs were defined by p value < 0.05 and log2 fold change >1. d Candidate small molecules identified by preliminary screening with scRCF for reprogramming MEFs into EF cells. e Bar plot showing the Z-scores for cells treated with LAC + 1 small molecule combinations, based on a gene set of neuroectoderm- and EF-related genes. The Z-scores indicate relative gene expression changes for each drug, with higher values reflecting a stronger effect on the gene set. Bars are ordered from the highest to lowest average Z-score, and the dashed line represents the baseline score of zero. f Heatmap showing the gene expression profiles of neuroectoderm- and EF-related genes in cells after treatment with LAC + 1 small molecule combinations. The color scale represents log2-transformed expression values, with red indicating high expression, blue indicating low expression, and white indicating intermediate levels.
Using this platform, we screened and analyzed scRNA-seq data from mouse embryonic fibroblasts (MEFs) and EF cells (Fig. 1b and Supplementary Fig. 1a)20,26,27,28. Differential gene expression analysis identified 258 TFs associated with cell type transitions (Fig. 1c and Supplementary Data 1). A modified SiPer19 framework calculated the similarity between these TFs and perturbation profiles from a small molecule database, pre-screening 489 signaling proteins. Further refinement via gene co-expression networks reduced the selection to 279 functionally relevant signaling proteins (Supplementary Fig. 1b and Supplementary Data 2). This approach revealed key signaling relationships and transcriptional regulators essential for cell fate transitions. To optimize screening, a random walk algorithm for modular partitioning of the protein network (Supplementary Fig. 1c) enabled effect scoring and prioritized small molecules based on pathway relevance. Ultimately, 41 high-efficacy candidates were identified, including Wnt pathway modulators CKI-7 and CHIR-99021, RTK pathway inhibitors orantinib and rebastinib, epigenetic regulators Trichostatin-A and RG108, TGF-β/Smad inhibitor A 83-01, and NF-κB pathway inhibitor BMS-345541 (Fig. 1d and Supplementary Table 1).
To further identify small molecules targeting EF fate reprogramming, we systematically evaluated 41 candidate compounds using DRUG-seq2. Given the neuroectodermal origin of the EF, we prioritized molecules such as LDN193189 (a BMP type I receptor inhibitor) and A 83-01 (a TGF-β type I receptor inhibitor) for their ability to suppress mesodermal and endodermal differentiation29,30,31. CKI-7 (an ATP-competitive casein kinase 1 inhibitor) was included to promote neuroectoderm formation32,33. Accordingly, LDN193189, A 83-01, and CKI-7 (LAC) were selected as the foundational induction cocktail for generating EF cells, with DRUG-seq2 employed to identify additional synergistic small molecules (Supplementary Fig. 1d). After 14 days of treatment with various small molecule combinations, DRUG-seq2 analysis revealed distinct molecular expression patterns and variations in target gene expression across different treatments, reflecting the specific responses to each condition (Fig. 1e, f). Effect scoring of gene sets associated with neuroectoderm and early EF development identified critical regulatory small molecules facilitating cellular reprogramming. Among these, Hh-Ag1.5, a Hedgehog signaling antagonist known for its role in eye development, achieved the highest score. Additional high-scoring compounds included GSK-3 inhibitors (CHIR-99021, 1-Azakenpaullone, Kenpaullone) and NF-κB pathway inhibitors (BMS-345541, WHI-P154) (Fig. 1e). Based on gene expression activation profiles and the removal of functionally redundant compounds (Fig. 1f), seven small molecules—Hh-Ag1.5, CHIR-99021, Golvatinib, Pirfenidone, BMS-345541, Masitinib, and RG108—were selected from the top-ranked candidates to form an optimized induction cocktail. Using this approach, we identified 10 small molecules from 4319 candidates capable of reprogramming MEFs into EF cells. In summary, we established a systematic, single-cell transcriptomic-driven screening framework integrating computational and experimental analyses. This approach identified a candidate induction cocktail of 10 small molecules with the potential to drive efficient cell fate reprogramming (Supplementary Fig. 1e).
Establishment of a two-stage chemical reprogramming strategy to generate ciRPE cells
First, we optimized serum-free, chemically defined medium containing these 10 small molecules and treated MEFs. For stage Ⅰ, we observed the emergence of epithelial-like cells colonies after 6 days treatment (Fig. 2a). Then, the colonies displayed more defined boundaries and distinct epithelial characteristics by day 12, although cell proliferation started to slow (Fig. 2a). To enhance the expansion of these colonies, we mechanically removed some surrounding cells. As expected, the colonies gradually expanded. By day 18, the cells exhibited tight packing, with some displaying a high nuclear-to-cytoplasmic ratio and typical early RPE characteristics, such as cobblestone or hexagonal shapes (Fig. 2a). Further qRT-PCR analysis revealed a upregulation of genes related to EF fate (Pax6, Sox2, Six3, and Vsx2) and early RPE development (Mitf and Best1) (Fig. 2b). These findings indicated that our identified 10 small molecules induction protocol has the potential to convert MEFs into EF-like cells with early RPE characteristics.

a Representative morphological changes of MEFs exposed to a reprogramming medium (RM) containing 10 small molecules (Stage I) at different time points. Scale bar, 400 μm. b qRT-PCR analysis showing the expression of EF-related genes and early RPE development-associated genes at the indicated time points (n = 3 independent biological samples per group). c Representative morphological images of cells after exposure to differentiation and maturation medium (DM) containing three compounds (Stage II). DMSO, dimethyl sulfoxide, was used as a negative control. M3: NIC, RA, Activin A. Scale bar, 200 μm. d qRT-PCR analysis showing the expression of RPE-associated marker genes at the indicated time points (n = 3 independent biological samples per group). e Schematic diagram illustrating the genetic lineage-tracing strategy and chemical reprogramming of ciRPE cells from MEFs. f Morphological and tdTomato fluorescence expression changes on distinct days during the induction process of ciRPE cells. Scale bar, 400 μm. g Percentages of tdTomato+ cells induced by candidate cocktails on distinct days (n = 5 independent biological samples per group). p values indicate comparisons between adjacent time points. h Percentages of tdTomato+ cells upon treatment with candidate cocktails (all 13 compounds) and after subtraction of the indicated compound from the mixture. - represents withdrawing the indicated component. Each group was compared individually to the “ALL” group (n = 5 independent biological samples per group). i, Percentages of tdTomato+ cells before and after optimization of the reprogramming cocktail at different time points (n = 6 independent biological samples per group). j Schematic diagram illustrating the protocol for reprogramming MEFs into ciRPE cells, accompanied by representative morphological changes at key time points. MM represents the MEF medium, while RM represents the reprogramming medium and DM refers to the differentiation/maturation medium. Scale bar, 200 μm. Data are presented as mean ± SD. Unpaired, two-tailed Student’s t-test was used to assess statistical significance. Source data are provided as a Source Data file.
To promote further differentiation and maturation of EF-like cells into RPE cells, we introduced Nicotinamide (NIC), Retinoic acid (RA), and Activin A (hereafter named M3) during stage Ⅱ induction. NIC facilitates RPE lineage commitment by modulating epigenetic and metabolic states, while suppressing neurogenic differentiation and promoting RPE-specific gene expression34,35,36. RA facilitates RPE differentiation and regulates the expression of RPE-specific genes to enhance pigment production10,37. Activin A promotes the differentiation of EF cells into RPE by activating the TGF-β/SMAD signaling pathway and regulating pigment production to maintain epithelial characteristics10,38,39. After two weeks of treatment, cells exhibited typical hexagonal RPE morphology and pigmentation (Fig. 2c). Subsequent qRT-PCR analysis confirmed significant upregulation of Best1, and mature RPE genes such as Rpe65, Tyr, Lhx2, Pmel, and Otx2 (Fig. 2d), indicating successful generation of ciRPE cells. To further validate the reprogramming effectiveness, we employed a lineage tracing strategy to monitor the reprogramming process (Fig. 2e). The Best1 gene is specifically expressed in RPE cells, and its promoter has been shown to effectively drive the expression of reporter genes in transgenic RPE cells10,40. We used Best1-Cre/ROSA26tdTomato fluorescent reporter mice, which demonstrated stable and specific tdTomato expression in RPE cells (Supplementary Fig. 2a, b). We performed fluorescence-activated cell sorting (FACS) to collect Best1-tdTomato negative (tdTomato-) MEFs (Supplementary Fig. 2c). These tdTomato- MEFs were negative for RPE marker genes such as Mitf, Cralbp, Best1, and Rpe65 (Supplementary Fig. 2d, e), confirming the absence of residual RPE or progenitor cells. We then subjected tdTomato- MEFs to a two-step chemical induction (stage Ⅰ with 10 small molecules, followed by stage Ⅱ with M3). After stage Ⅱ induction (day 32), the proportion of tdTomato-positive (tdTomato+) cells increased, reaching 16.58%, compared to 2.51% after stage Ⅰ induction (Fig. 2f, g). These results confirm that the constructed chemical induction system effectively reprogrammed MEFs into RPE cells.
To minimize potential toxic effects of small molecules on cells, we conducted a minus-one experiment to further optimize the chemical reprogramming system. The results indicated that removing Golvatinib, Pirfenidone, and Masitinib during the stage Ⅰ induction had minimal impact on the positive rate of tdTomato expression (Fig. 2h), achieving a positive rate of 19.66% (Fig. 2i). Consequently, we finalized the reprogramming induction system, consisting of seven small molecules from the stage Ⅰ (M7: LDN193189, A 83-01, CKI-7, Hh-Ag1.5, CHIR-99021, BMS-345541, and RG108) and three compounds from stage ⅠⅠ (M3: NIC, RA, and Activin A) (Fig. 2j). Finally, we validated the robustness of our reprogramming system across MEFs from different batches and different genetic backgrounds, including C57BL/6J and 129S4/SvJae (Supplementary Fig. 2f, g). Furthermore, this M7 + M3 system successfully facilitated the reprogramming of tail-tip fibroblasts (TTFs) from neonatal mice into ciRPE cells (Supplementary Fig. 2f, g). Collectively, these results demonstrate that our optimized M7 + M3 system enables efficient reprogramming of MEFs into functional ciRPE cells.
Characteristics of ciRPE cells
To further confirm the phenotypic and functional properties of ciRPE cells, we first performed immunofluorescence staining to assess the expression of RPE markers. The results demonstrated that ciRPE cells exhibit tight junction structures (ZO-1) and express high levels of RPE markers, including Pax6, Rpe65, Mitf, Best1, and Cralbp (Fig. 3a). Additionally, these cells exhibited polarized morphology, with ZO-1 localized to the apical membrane and Best1 to the basal membrane (Fig. 3b). This structural polarity is crucial for RPE functions, particularly the apical microvilli, which mediate the phagocytosis and clearance of shed photoreceptor outer segments (POSs), ensuring normal photoreceptor turnover41,42. Subsequently, we analyzed the structural characteristics of ciRPE cells using transmission electron microscopy. These cells exhibited distinct apical microvilli, pigment granules, and tight junctions (Fig. 3c), indicating that their morphology closely resembles that of native RPE cells in vivo.

a Representative immunostaining showing that MEF-derived ciRPE cells express ZO-1, Pax6, Rpe65, Mitf, Best1 and Cralbp. Scale bars, 50 μm. b Representative Z-stack confocal micrographs showing ciRPE cells with typical polarized expression of RPE markers. ZO-1 (green) demonstrates apical localization (top), while Best1 (red) shows basolateral localization (bottom). Scale bars, 10 μm. c Representative transmission electron microscopy image of ciRPE cells showing apical microvilli (yellow arrows), melanin granules (red arrows) and tight junctions (black arrows). Scale bars, 1 μm. d Representative confocal micrograph showing phagocytosis of POSs (green) by ciRPEs. The apical sides of ciRPE cells are stained with ZO-1(violet), whereas nuclei are counterstained with DAPI (blue). Scale bars, 50 μm. e Apical and basal secretion of PEDF and VEGF by MEFs, ciRPE, and pRPE cells cultured on Transwells. Each group was compared to the ciRPE group within apical and basal compartments (n = 6 independent biological samples per group). f Representative morphological images showing dome structures formed by ciRPE cells during in vitro culture. The red arrows indicate the dome morphology observed under different phase-contrast microscopy conditions. Scale bars, 50 μm. g TEER measurements of MEFs, ciRPE, and pRPE cells over time in culture (n = 6 independent biological samples per group). Data are mean ± SD. One-way ANOVA was used to assess statistical significance. Three independent experiments were performed with similar results and representative results are shown. Source data are provided as a Source Data file.
To evaluate the phagocytic capacity of ciRPE cells, we introduced fluorescently labeled porcine POSs and latex beads into the ciRPE cell culture medium. After an incubation period, laser scanning confocal microscopy revealed the presence of engulfed POS and latex beads within the ciRPE cells (Fig. 3d and Supplementary Fig. 3a), confirming their phagocytic functionality. Additionally, RPE cells are known to exhibit polarized secretion of growth factors, a process crucial for maintaining homeostasis between the retina and the choroid34,43. We further analyzed the polarized secretion profile of VEGF and PEDF across MEF, ciRPE, and primary RPE (pRPE) cells. As shown in Fig. 3e, ciRPE cells exhibited a characteristic RPE-like pattern, predominantly secreting VEGF from the basal side and PEDF from the apical side. This polarized secretion was consistent with that observed in pRPE cells and distinct from the non-polarized profile of MEFs, supporting the functional maturation of ciRPE cells. Moreover, ciRPE cells formed dome-like structures during in vitro culture (Fig. 3f), suggesting that their epithelial layer is capable of effective fluid transport while maintaining tight junction integrity. Measurements of transepithelial electrical resistance (TEER) further supported the establishment of tight junctions and barrier function in ciRPE cells. As shown in Fig. 3g, ciRPE cells exhibited a steady increase in TEER over the first three weeks, eventually stabilizing around 80 Ω·cm². This value was markedly higher than that of MEFs, which showed no measurable barrier formation, and approached the TEER levels observed in pRPE cells, although somewhat lower in magnitude. Collectively, these data indicate that ciRPE cells exhibit key functional characteristics typical of native RPE cells.
To further validate the proliferative potential of ciRPE cells, we purified and cultured them in an expansion medium containing basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF) for continuous passaging. The results showed that ciRPE cells could be passaged for at least 20 generations while maintaining a stable RPE morphology and normal karyotype (Supplementary Fig. 3b, c). In contrast, pRPE cells gradually lost pigmentation and hexagonal morphology by the sixth passage, with a substantial reduction in growth rate (Supplementary Fig. 3c, d), consistent with previous studies44. Across different passages, ciRPE cells showed higher proliferative capacity compared to pRPE cells, with EdU incorporation rates consistently exceeding those of pRPE cells (Supplementary Fig. 3d). Flow cytometric analysis indicated that the cell cycle distribution of ciRPE cells (P3) was similar to that of pRPE cells, with 70%, 17.4%, and 6.87% of cells in the G0/G1, S, and G2/M phases, respectively (Supplementary Fig. 3e). In summary, our results indicate that ciRPE cells closely resemble native RPE cells in terms of morphology and functionality, exhibiting typical polarized characteristics, phagocytic capabilities, and a polarized secretion profile of growth factors. Additionally, they show strong proliferative capacity and stability.
Finally, we confirmed the reprogramming of MEFs into ciRPE cells using a lineage tracing strategy with Fsp1-Cre and ROSA26-tdTomato mice. MEFs from E13.5 transgenic mice (Fsp1-Cre/ROSA26tdTomato) were sorted by FACS to obtain tdTomato+/Best1- cells (Supplementary Fig. 4a, b). These cells initially lacked RPE marker expression (Supplementary Fig. 4c–e). After chemical reprogramming, ciRPE cells expressed RPE-specific genes and co-expressed tdTomato (Supplementary Fig. 4f, g), confirming their origin from the initial MEFs.
Molecular roadmap of ciRPE chemical reprogramming
To enhance our understanding of the intricate process of reprogramming MEFs into ciRPE cells, we conducted a comprehensive multi-omics analysis (Fig. 4a). RNA sequencing (RNA-seq) was performed on cells at day 0, day 7, day 18, and day 32 of the reprogramming process. Principal component analysis (PCA) of the RNA-seq result revealed substantial transcriptional changes throughout this dynamic process, demonstrating a gradual transition from fibroblasts to ciRPE cells. Notably, the final ciRPE cells exhibited greater transcriptional similarity to pRPE cells compared to earlier stages (Fig. 4b). Time-series fuzzy clustering analysis of differentially expressed genes revealed four distinct gene clusters corresponding to the stages of reprogramming: fibroblasts (day 0), intermediate state (day 7), EF-like cells (day 18), and mature ciRPE cells (day 32) (Fig. 4c).

a Schematic illustrating the multi-omics sequencing strategy employed to analyze the reprogramming process from MEFs to ciRPE cells. b PCA of RNA seq and CUT&Tag data (H3K4me3, H3K27ac, and H3K27me3) were performed on samples collected at days 0, 7, 18, and 32 (ciRPE) of reprogramming, along with pRPE cells serving as the control. c Heatmap showing differentially expressed genes in MEF to ciRPE cell reprogramming samples at indicated time points. The number above heatmap indicates independent biological replicates. Representative genes (left side of the heatmap) and associated gene ontology (GO; right side of the heatmap) for each block are shown. Red and blue indicate upregulated and downregulated genes, respectively. Differential expression was analyzed using the R package limma (v3.58.1) following normalization with edgeR (v4.0.16). Significantly changed genes were defined by |log₂ fold change| > 1.5 and adjusted p < 0.01 (Benjamini-Hochberg correction). d Dynamics of CUT&Tag peaks (H3K4me3, H3K27ac, H3K27me3) associated with cluster-specific genes (C). The red line indicates the median CUT&Tag peaks over time, the blue line represents the median RNA expression levels, and the gray background lines depict individual peak values across time points. e Uniform manifold approximation and projection (UMAP) analysis of integrated scRNA-seq data from cells collected at the indicated time points during the reprogramming process of MEFs to ciRPE cells. f UMAP plot showing identified cell types in samples collected at the indicated time points during the reprogramming process. g Dot plot illustrating the expression of representative marker genes across different cell types during the reprogramming process. h RNA velocity streamline plot illustrating the transitions of cell populations during the reprogramming process using the scVelo method. The arrows represent the flow determined by the ratio of unspliced to spliced transcripts, predicting dynamic changes in cell identity. Black arrows indicate RNA velocity flow based on the unspliced-to-spliced transcript ratio, while the gray to teal arrows are used for visual enhancement to highlight specific trajectories. i Heatmap showing the gene expression similarity between cell types during the reprogramming process and those reported in previous studies. pEF cells represent the data used for small molecule prediction as described above, while pRPE data were obtained from the GSE183572 dataset.
During the initial transition from fibroblasts to the intermediate state, downregulated genes such as Fn1, Itga11, and Runx2 were linked to reduced fibroblast functionality and proliferation activity. In contrast, the upregulated genes including Sox2, Ascl1, Gli2, Gli1, Gbx2 were linked to neuroectodermal processes (Fig. 4c). These results suggest that early chemical induction reprograms gene expression by activating neuroectodermal genes and signaling pathways while suppressing fibroblast characteristics, facilitating the progression towards a neuronal fate. Additionally, transient activation of genes such as Tfap2a, Rorb, Etv5 during the intermediate phase was associated with epithelial proliferation and inflammation, reflecting enhanced cell-cell interactions and adaptation to environmental signals (Fig. 4c). By day 18, the upregulated genes were predominantly related to eye development, such as Pax6, ID4 and Best1. As induction progressed towards mature RPE, early EF-related genes showed a trend of downregulation. By day 32, genes associated with RPE function (e.g., Rpe65, Lhx2, Tyr, Cralbp, Otx2) and pigment cell differentiation (e.g., Pmel, Mlana, Slc24a5) were upregulated (Fig. 4c), confirming the transition to mature, pigmented ciRPE cells.
To confirm successful conversion from fibroblasts to ciRPE cells, we analyzed 16 fibroblast-specific and 18 RPE-specific genes. During reprogramming, fibroblast markers were considerable downregulated while RPE markers were upregulated, indicating that ciRPE cells acquired RPE characteristics (Supplementary Fig. 5a and Supplementary Table 2). Ultimately, the gene expression profile of ciRPE cells closely matched that of pRPE cells, validating the successful attainment of RPE identity (Fig. 4b, c and Supplementary Fig. 5a, b). Notably, the transcriptional trajectory during reprogramming was directed toward ectodermal and eye field lineages without evidence of PSC-like activation (Supplementary Fig. 5c and Supplementary Table 2). Key pluripotency genes such as Pou5f1, Nanog, and Lin28a remained silenced throughout, and Klf4 showed only a mild, transient increase consistent with its physiological role in fibroblasts (Supplementary Fig. 5d). These findings indicate that reprogramming proceeded via direct lineage conversion rather than a pluripotent intermediate.
Epigenetic remodeling plays a crucial role in cell fate reprogramming45,46,47,48. We used CUT&Tag sequencing to track dynamic modifications in H3K4me3, H3K27ac, and H3K27me3 histone marks near gene transcription start sites. PCA analysis highlighted notable chromatin state alterations throughout reprogramming, showing that ciRPE cells closely resembled pRPE cells in histone modification patterns (Fig. 4b). H3K4me3 increased progressively from MEFs to ciRPE cells, peaking at days 18 and 32, linked to RPE gene activation. The loss of H3K4me3 decreased over time, suggesting its role in establishing new gene expression. H3K27ac changes varied across stages, with a decrease from day 7 to 18 indicating gene repression for EF specification. H3K27me3 dynamics reflected gene silencing: substantial loss from MEFs to day 7 facilitated differentiation, while increased acquisition from day 7 to 18 established new silencing. By the ciRPE stage, H3K27me3 levels stabilized, supporting the mature RPE phenotype (Supplementary Fig. 5e). Further analysis showed that the CUT&Tag signals for H3K4me3 and H3K27ac correlated with RNA expression levels, underscoring their role in gene activation (Fig. 4d). Although H3K27me3 generally showed a negative correlation with RNA-seq data, some deviations were observed during the fibroblasts stage, likely due to predominant activating marks. From day 7 to 18, increased H3K27me3 correlated with decreased RNA expression, while a trend toward stabilization at the ciRPE stage reflected balanced gene silencing and activation (Fig. 4d and Supplementary Fig. 5f). These findings underscore the intricate role of dynamic histone modifications in cell type transitions at various stages, highlighting epigenetic regulation’s pivotal role in MEF-to-RPE reprogramming. By modulating gene activation and silencing, cells successfully reprogram towards the RPE lineage.
To accurately delineate the reprogramming trajectory, we performed scRNA-seq at three key time points during the reprogramming of MEFs into ciRPE cells. Single-cell transcriptomes were obtained from samples collected on days 7, 18, and 32, yielding 9679, 13,227, and 11,282 cells, respectively (Fig. 4e). Cluster-specific marker gene analysis revealed that on day 7 of reprogramming, the cell population consisted of MEF cells, neuroprogenitor-like intermediate cells, and a small fraction of EF-like cells. By day 18, the proportion of intermediate cells cells decreased, while that of EF-like cells increased. By day 32, EF-like cells diminished, and a prominent population of ciRPE cells emerged (Fig. 4f, g and Supplementary Fig. 6a, b). To further validate the reprogramming trajectory of ciRPE cells, we performed single-cell pseudotime trajectory analysis. The analysis revealed a sequential transition from MEFs to neuroprogenitor-like intermediate cells, followed by differentiation into EF-like cells and ultimately ciRPE cells (Fig. 4h and Supplementary Fig. 6c). Unsupervised clustering highlighted dynamic molecular events during this process, including fibroblast-to-neuroprogenitor transitions, progression through eye development stages, and the establishment of RPE-specific functions such as pigmentation and retinoid metabolism (Supplementary Fig. 6d). Interestingly, among two identified neuroprogenitor-like subpopulations, the differentiation trajectory of MEFs primarily aligned with one subgroup expressing neurogenic and proliferative markers (e.g., Mki67, Cdk1, Top2a), while the other subgroup lacked these markers (Supplementary Fig. 6e). This indicates that proliferative neuroprogenitor cells are essential for subsequent differentiation into EF-like cells. RNA velocity analysis confirmed the progression of reprogramming toward ciRPE cells, consistent with the trajectory observed in pseudotime analysis (Supplementary Fig. 6f, g). We further performed similarity analysis between the reprogrammed cell population and publicly available single-cell datasets. The induced cells showed a similarity score of 0.80 with RPE cells and 0.64 with EF-like cells (Fig. 4i), indicating that the reprogrammed cells acquired both EF-like and RPE characteristics. In conclusion, our two-step chemical reprogramming system effectively directs MEFs through neuroprogenitor-like intermediate and EF-like stages to generate functional ciRPE cells. This sequential process highlights the precise control of lineage-specific gene expression and epigenetic remodeling.
Transcriptional activation of neural and eye development master factors governs ciRPE reprogramming
To identify the key TFs driving the reprogramming of MEFs into ciRPE cells, we integrated single-cell data with SCENIC analysis to systematically investigate the regulatory factors expressed in each cell type and their dynamic changes throughout reprogramming. Using a logFC>1.0 threshold, we identified 125 TFs across four cell types, highlighting their distinct roles in different reprogramming stages (Fig. 5a and Supplementary Data 3). Using the STRING database, we constructed an interaction network of these TFs, revealing their cooperative roles during reprogramming (Supplementary Fig. 7a and Supplementary Data 4). Network analysis using maximal clique centrality (MCC) identified the 15 key TFs, including MEF-specific factors (Dlx2 and Dlx1), transiently activated factors at day 7 (Sox11, Foxa2 and Pax2), and factors with sustained high expression from days 7 to 18 (Sox2, Sox9, Olig2, Zic1, Ascl1, Atoh1, Pou3f2 and Gbx2), as well as ciRPE-related factors (Lhx2 and Otx2) (Fig. 5b, c and Supplementary Data 5). Notably, the majority of these key TFs are associated with neuroectodermal development, suggesting that neuroectodermal fate is a critical transitional phase in the reprogramming from MEF to EF-like and ciRPE cells.

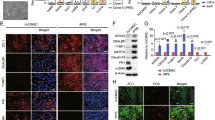
a Volcano plots illustrating TFs with differences in regulon activity across various cell types during reprogramming from SCENIC. b The bar-dash plot shows the MCC scores for the top key 15 TFs. c Heatmap showing the expression of the top key 15 TFs at indicated time points during cellular reprogramming. d Normalized RNA-seq and CUT&Tag sequencing of histone modifications (H3K4me3, H3K27ac, and H3K27me3) at the genome loci of Ascl1, Olig2, Zic1, Pou3f2, and Lhx2 at the indicated time points during cellular reprogramming. Relative reprogramming efficiency, assessed by the proportion of tdTomato+ cells at day 32, following knockdown of Ascl1 (e) or Olig2 (f) at the indicated time points under M7 + M3 induction (n = 5 independent biological samples per group). The reprogramming efficiency at day 32 for M7 + M3-induced cells was set to “1” with DMSO serving as the negative control. WT, wild-type; Control KD, scramble shRNA-mediated gene knockdown. g scRCF network visualization of the putative signaling cascades induced by small molecules targeting Ascl1 and Olig2. Orange rectangles represent perturbagens, blue diamonds represent signaling protein targets, white ellipses represent intermediate signaling proteins, and green hexagons represent query TFs. Data are mean ± SD. One-way ANOVA was used to assess statistical significance. Source data are provided as a Source Data file.
To further explore the regulatory mechanisms underpinning these TFs, we integrated RNA-seq and CUT&Tag-seq data to assess the activity of the top five TFs (Fig. 5d). On day 0, neuroectodermal TFs, including Ascl1, Olig2, Zic1, Pou3f2, displayed weak activation marks (H3K4me3 and H3K27ac) and predominantly repression by H3K27me3, consistent with their suppression in the stable fibroblast state. By day 7, as cells transitioned into a neuroprogenitor-like intermediate state, these TFs showed increased activation marks and RNA expression, indicating the activation of neuroectodermal pathways. This coincided with the suppression of fibroblast characteristics and the initiation of neurogenesis. By day 18, activation of Ascl1 and Olig2 had decreased, whereas Zic1 and Pou3f2 remained highly active. At day 32, during the ciRPE phase, Lhx2 exhibited robust activation and notable RNA upregulation, while earlier neuroectodermal TFs were silenced (Fig. 5d). Notably, while H3K27me3 played a key repressive role during the early phase (days 0-7), its effect diminished in later stages, implying that alternative repressive mechanisms, such as DNA methylation or other histone marks, may regulate TF activity at these stages. These findings underscore the critical role of dynamic epigenetic modifications and TF expression in guiding cell fate transitions during MEF-to-ciRPE reprogramming. Overall, neurodevelopmental TFs act as early “switches” for EF specification, while RPE-associated TFs govern later stages, driving RPE maturation.
Ascl1 and Olig2 have been reported as essential neurodevelopmental TFs that regulate neural progenitor cell fate and reprogramming49,50,51,52. To determine the impact of Ascl1 and Olig2 on fibroblast-to-EF reprogramming, we investigated how the knockdown of these genes individually with small hairpin RNA (shRNA) affected the efficiency of ciRPE induction. As expected, the knockdown of either Ascl1 or Olig2 substantially reduced the reprogramming efficiency, confirming that they positively regulate reprogramming (Fig. 5e, f and Supplementary Fig. 7b-d). Importantly, the knockdown of these genes at the early stage had the most considerable effect on reprogramming efficiency, underscoring their pivotal role in initiating neuroectodermal transitions necessary for subsequent ciRPE differentiation (9.1-, 16.7-, and 3.6- fold reduction when shAscl1 was transduced at days 0, 7 and 18, respectively, Fig. 5e; and 12.5-, 8.3-, and 2.8- fold reduction when shOlig2 was transduced at days 0, 7 and 18, respectively, Fig. 5f). On the other hand, the overexpression of Ascl1 or Olig2 enhanced the efficiency of ciRPE reprogramming mediated by M7 + M3 (Supplementary Fig. 7e), validating that Ascl1 and Olig2 are directly involved in ciRPE reprogramming.
These findings emphasize the importance of transitioning through neuroectodermal and EF-like states for successful MEF-to-ciRPE reprogramming, validating the effectiveness of our first-stage small molecule cocktail in orchestrating these transitions. Using scRCF, we constructed a regulatory network to elucidate how small molecules modulate key TFs during reprogramming (Fig. 5g and Supplementary Fig. 7f). The analysis indicated that the activation of key TFs is a collective contribution of these small molecules, which regulate TFs through their respective target proteins, orchestrating the activation of multiple signaling pathways, including Wnt, TGF-β, and Hedgehog, which collectively guide precise cell fate transitions toward ciRPE reprogramming.
Generation of human ciRPE cells from HEFs by chemical reprogramming
In our efforts to generate human ciRPE (hciRPE) cells, we initially attempted to apply the mouse induction system to reprogramming human embryonic fibroblasts (HEFs). However, the outcomes were suboptimal, likely due to the higher complexity and stricter signaling requirements of human cellular reprogramming compared to murine systems. Consequently, it was necessary to adjust the small molecule combination to enhance reprogramming efficacy and success rates. During the selection of compounds, we focused on the “OV” stage of early human retinal development as a critical intermediate state for guiding human RPE reprogramming. Using single-cell data from human embryos (carnegie stages 12 and 16)21 and 30-day-old retinal organoids22 (Supplementary Fig. 8a, b), we strategically incorporated the top 15 key TFs (Fig. 5b) identified in the mouse ciRPE reprogramming process into scRCF. This approach enabled more accurate identification of small molecules to guide the reprogramming of hciRPE cells. Ultimately, we identified 42 candidate small molecules (Fig. 6a and Supplementary Data 6–8). Notably, LDN193189, CHIR-99021, Hh-Ag1.5, RG108, and BMS-345541 (LCHRB), identified in the murine RPE reprogramming system, were included in the foundational cocktail. We then screened additional compounds using DRUG-Seq2 (LCHRB + 1), ranking the top 10 with a comprehensive scoring system (Supplementary Fig. 8c, d), leading to a final selection of 15 small molecule combinations. We hypothesized that these combinations would promote HEFs convert into OV-like cells.

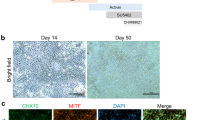
a Candidate small molecules identified by preliminary screening with scRCF for reprogramming HEFs into OV. b Schematic diagram of the protocol for the reprogramming of HEFs into hciRPE cells, along with representative morphological changes at indicated time points. HM represents the HEF medium, while RM represents the reprogramming medium and DM refers to the differentiation/maturation medium. Scale bar, 300 μm. c FACS purification of reprogrammed BEST1-EGFP + hciRPE cells. d Representative optical microscopy and TEM images of hciRPE cells showing melanin granules (red arrows). Scale bars, 1 μm. e qRT-PCR analysis showing the expression of RPE-associated genes at the indicated time points during reprogramming (n = 3 independent biological samples per group). f Representative immunostaining analysis showing positive expression of ZO-1, RPE65, MITF and BEST1 in the BEST1-EGFP- HEFs-derived hciRPE cells. Scale bar, 20 μm. g PCA of samples from day 0, day 12, day 24 and day 38 (hciRPE) of celluar reprogramming, and the control primary hRPE cells. h Heatmap showing differentially expressed genes in HEF to hciRPE cell reprogramming samples at indicated time points. The number above heatmap indicates independent biological replicates. Representative genes (left side of the heatmap) and associated GO (right side of the heatmap) for each block are shown. Red and blue indicate upregulated and downregulated genes, respectively. Differential expression was analyzed using the R package limma (v3.58.1) following normalization with edgeR (v4.0.16). Significantly changed genes were defined by |log₂ fold change| > 1.5 and adjusted p < 0.01 (Benjamini-Hochberg correction). i Polarized secretion of VEGF and PEDF from the apical and basal sides of hciRPE cells grown on Transwells (n = 6 independent biological samples per group). j TEER in hciRPE cells for 30 days. p values indicate comparisons between adjacent time points (n = 5 independent biological samples per group). Data are mean ± SD. Statistical analyses were performed using one-way ANOVA (e) and unpaired, two-tailed Student’s t-test (i, j). Three independent experiments were performed with similar results and representative results are shown. Source data are provided as a Source Data file.
To validate our hypothesis, we developed the BEST1 Pr-EGFP-HEFs tracing cell line to monitor the reprogramming process (Supplementary Fig. 8e). Initially, these cells did not express RPE markers such as MITF, CRALBP, BEST1, and RPE65, nor did it exhibit EGFP expression (Supplementary Fig. 8f, g). Upon successful reprogramming, the Best1 promoter was expected to drive EGFP expression specifically in induced cells. We added the 15 selected chemical compounds to the reprogramming medium to initiate HEFs reprogramming. During the early induction phase, HEFs exhibited rapid proliferation; however, by day 12, only a few clones emerged. To support clone growth, we removed surrounding cells, but their proliferation remained slow. By day 24, only a limited number of cells were BEST1-EGFP positive. Continued induction showed minimal change, with cell numbers declining. We then introduced three compounds (NIC, RA, and Activin A, M3) to enhance RPE differentiation and maturation, but the proportion of BEST1-EGFP-positive cells remained low.
Due to the suboptimal efficiency and potential cytotoxicity of excessive compounds, further optimization of the induction system was required. Starting with the initial 15 small molecules and 3 differentiation and maturation compounds, we explored various combinations and culture conditions. We found that using 9 small molecules (M9: CHIR-99021, Hh-Ag1.5, LDN193189, RG108, BMS-345541, R-268712, BIX-01294, VPA, SB-431542) in the initial phase and 2 compounds (M2: NIC, and Activin A) during the differentiation maturation phase resulted in 6.74% BEST1-EGFP-positive (hciRPE) cells by day 38 (Fig. 6b, c), whereas other combinations yielded lower proportions of positive cells.
We further characterized the molecular and biological features of hciRPE cells. After purification and expansion, induced hciRPE cells exhibited characteristic hexagonal morphology and notable melanin granules (Fig. 6d), while maintaining a normal karyotype (Supplementary Fig. 8h). qRT-PCR analysis and immunofluorescence collectively confirmed the expression of key RPE markers, including MITF, CRALBP, BEST1, and RPE65, with EGFP specifically co-expressed in induced hciRPE cells (Fig. 6e, f). Transcriptomic analysis indicated that the expression profile of hciRPE cells closely resembled that of primary human RPE (hRPE) cells used as a positive control (Fig. 6g, h). In both hciRPE cells and intermediate reprogramming states, fibroblast-specific genes (e.g., FN1 and RUNX2) were downregulated, while RPE-specific genes (e.g., RPE65, TYR, LHX2, and CRALBP) were upregulated. Notably, TFs such as RORB, PAX3, DES, and TFAP2A were upregulated during the intermediate stages, suggesting a possible activation of neuroectodermal transcriptional programs and a potential transition toward RPE lineage commitment. By day 24, markers associated with OV and retinal progenitor cells (PAX6, SIX3, VSX2, MITF and BEST1) were activated (Supplementary Fig. 8i), indicating the successful transition into OV-like cells, a critical intermediate state for RPE lineage commitment. With continued differentiation, these cells ultimately resembled hRPE cells (Fig. 6g, h). Further analysis showed that hciRPE cells secreted growth factors such as VEGF and PEDF (Fig. 6i), and TEER analysis confirmed the presence of tight junctions and barrier function (Fig. 6j). Moreover, hciRPE cells maintained typical RPE morphology, molecular identity, and stable proliferative capacity over multiple passages (Supplementary Fig. 8j-l). These findings demonstrate that our optimized two-stage chemical reprogramming system effectively converts HEFs into functional hciRPE cells, providing a safe and non-integrative approach for studying RPE biology and developing cell-based therapies.
ciRPE cell transplantation restores retinal function in RCS rat
Leveraging the functional and safety advantages of ciRPE cells, we initiated in vivo transplantation studies to evaluate their therapeutic potential for RD diseases. The Royal College of Surgeons (RCS) rat, characterized by a mutation in the Mertk gene that impairs RPE phagocytosis, serves as a well-established RD model18,38. FACS-purified tdTomato+ ciRPE cells were transplanted into the subretinal space of 3-week-old RCS rats (Fig. 7a and Supplementary Fig. 9a, b), with PBS, mESCs, MEFs, or pRPE cells transplanted as controls. Postoperative Optical Coherence Tomography (OCT) revealed transient subretinal bulges that resolved within three weeks, suggesting initial engraftment and tissue adaptation (Fig. 7b). During a 4-month follow-up, no tumor formation was observed in the ciRPE group, whereas 13 out of 15 rats receiving tdTomato-labeled mESCs developed visible intraocular tumors (Fig. 7c), consistent with results from subcutaneous teratoma assays (Supplementary Fig. 9c), supporting the safety profile of ciRPE cells.

a Schematic diagram of subretinal ciRPE cell transplantation in RCS rats. b Representative OCT images showing the subretinal transplantation site of ciRPE cells in RCS rats at 0, 1, 2 and 3 weeks post-transplantation. Scale bar, 600 μm. c Representative photographs from in vivo imaging showing tumor formation in nude mice following subretinal transplantation of tdTomato-labeled mESCs (via lentivirus) and tdTomato-ciRPE cells. Quantitative analysis data are displayed in the right panel (n = 10 independent mice per group). d Representative images of bright-field (top) and immunofluorescence (bottom) from eye tissue sections 8 weeks after subretinal transplantation of tdTomato-ciRPE into RCS rats. The dashed box indicates the area of cell transplantation. The nuclei were counterstained with DAPI (blue). Scale bars, 200 μm. e Immunofluorescence analysis of whole retinal sections at 12 weeks post-transplantation showed clusters of tdTomato+ ciRPE cells in the transplanted area, forming a monolayer structure. Magnified views of the regions delineated by the dashed-line boxes show: (i) a non-transplanted region and (ii) transplanted areas. Quantitative analysis data are displayed in the right panel (n = 10 independent rats per group). Scale bars, 500 μm. f–h Representative immunostaining showing tdTomato+ ciRPE cells coexpressing Mitf (F), Best1 (G), and Pax6 (H). Scale bars, 50 μm. i Representative TUNEL-stained micrographs of retinal cryosections from RCS rats 12 weeks after ciRPE cell transplantation (left), with statistical results shown on the right, using the sham-transplanted group as a control (n = 10 independent rats per group). Scale bars, 50 μm. j Representative immunostaining showing transplanted tdTomato+ ciRPE cells coexpressing Rhodopsin in the retina of RCS rats 12 weeks after transplantation. Scale bars, 50 μm. k Representative b-wave responses of the ciRPEs-transplanted and sham-transplanted groups were assessed through fERG with an intensity of 0.48 log cd*s/m² (dark 3.0) at 4w, 8w, 12w and 16w post transplantation (left). Statistical analysis of b-wave amplitudes in the ciRPEs-transplanted and sham-transplanted groups (right) (n = 6 independent rats per group). l Schematic illustration of the qOMR test setup (left). Quantitative evaluation of visual acuity in the ciRPEs-transplanted and sham-transplanted groups, obtained through the qOMR test at 4w, 8w, 12w and 16w post transplantation (right) (n = 10 independent rats per group). Data are mean ± SD. Unpaired, two-tailed Student’s t-test was used to assess statistical significance. Three independent experiments were performed with similar results and representative results are shown. Source data are provided as a Source Data file.
Histological and immunostaining analyses revealed that tdTomato+ ciRPE cells formed clusters in the subretinal space at 4 weeks and organized into a structured monolayer by 12 weeks (Fig. 7d, e), with expression of mature RPE markers such as Mitf, Cralbp, Pax6, and Rpe65 (Fig. 7f–h). Typically, RCS rats exhibit severe retinal dysfunction between 2 to 3 months of age, characterized by substantial photoreceptor loss and thinning of the outer nuclear layer (ONL)38. At 12 weeks post-transplantation, the ONL in the ciRPE-transplanted eyes was thicker compared to both the sham and MEF groups, and moderately greater than in the pRPE group (Fig. 7e and Supplementary Fig. 9d–f), indicating that ciRPE transplantation partially protected photoreceptors and slowed their degeneration, thereby contributing to the preservation of retinal structure and function. Consistently, TUNEL staining showed fewer apoptotic cells in the ONL of ciRPE-transplanted eyes compared to the Sham, MEF, and pRPE groups (Fig. 7i and Supplementary Fig. 9g, h), further supporting the protective effect of ciRPE cells against photoreceptor degeneration. The loss of RPE cell phagocytic function due to the Mertk mutation in RCS rats results in impaired clearance of POSs53,54. To assess whether ciRPE cells could restore this function in vivo, we evaluated their uptake of Rhodopsin, a major component of photoreceptor outer segments. After 12 weeks post-transplantation, tdTomato and Rhodopsin co-localization was clearly observed within the transplanted ciRPE cells (Fig. 7j), while such signal was not detected in the MEF group and was barely detectable in the pRPE group (Supplementary Fig. 9i), indicating that ciRPE cells more effectively integrated into the host RPE layer and regained phagocytic activity.
To comprehensively evaluate the impact of ciRPE cell transplantation on retinal function, we performed dark-adapted flash electroretinography (fERG) and quantitative optomotor response (qOMR) tests. fERG recordings from 4 to 16 weeks post-transplantation showed that ciRPE-transplanted eyes exhibited higher b-wave amplitudes than those in the sham, MEF, and pRPE groups, particularly at earlier time points (Fig. 7k and Supplementary Fig. 9j), suggesting that ciRPE cells contributed to photoreceptor preservation and partial functional recovery. Consistent with this, qOMR tests revealed improved visual behavior in the ciRPE group, including increased sensitivity to spatial frequency and motion direction, as reflected by elevated qOMR scores (Fig. 7l and Supplementary Fig. 9k). In contrast, the MEF group showed no improvement relative to the sham group, while the pRPE group exhibited a degree of functional rescue that was slightly weaker than that observed in the ciRPE group (Supplementary Fig. 9k). These results support the survival, safety, and functional integration of ciRPE cells over the examined period in the RCS model, and suggest their potential to partially restore retinal function in degenerative conditions.
Discussion
The RPE is crucial for maintaining retinal structure and supporting photoreceptor function, with its dysfunction directly contributing to various RD diseases. While RPE cell replacement therapy holds significant potential for treating these conditions, current methods face challenges in safety, efficiency, and scalability for RPE cell production55,56,57. Our study demonstrates that chemical reprogramming efficiently generates functional RPE cells without genetic manipulation, offering a safer and more clinically promising approach for cell replacement therapies. By leveraging a predictive platform informed by single-cell transcriptomic data, we systematically identified small-molecule combinations that guide fibroblasts toward an EF/OV-like state, forming the basis of our two-stage reprogramming protocol. Using a two-stage reprogramming protocol, fibroblasts were directed into an intermediate state with RPE precursor characteristics and subsequently matured into functional RPE cells. Mechanistic studies identified key small molecules and TFs involved in this process. Transplantation experiments demonstrated the therapeutic potential of ciRPE cells, which integrated into the host RPE layer, reduced apoptosis, and improved visual function in an RD model.
Compared to traditional TF-based reprogramming strategies that rely on genomic integration, chemical modulation of TF activity offers a safer and more clinically viable approach. However, identifying optimal small molecules through conventional large-scale chemical library screening remains laborious and time-consuming. Zheng et al. developed the SiPer platform, which uses single-cell data to predict compounds that target specific TFs for cellular conversion19. While effective, its reliance on static network models and pre-existing datasets limits its ability to capture dynamic signaling interactions. In contrast, our scRCF platform integrates scRNA-seq data, a broadened chemical perturbation database, and dynamic gene co-expression networks to enable pathway-specific compound selection via effect score calculations using a random walk-based module partitioning algorithm, scRCF identifies key signaling proteins essential for cell fate transformation. This pipeline, further validated by DRUG-seq2, enhances predictive accuracy and reprogramming efficiency. Compared to SiPer, scRCF offers superior capabilities in dynamically integrating signaling networks, refining small molecule prioritization, and enabling experimental feedback, thereby improving both precision and stability.
Based on our predictive platform, we established a two-stage chemical reprogramming strategy for converting both mouse and human fibroblasts into RPE cells. In the mouse system, the initial stage involved LDN193189, A 83-01, CKI-7, Hh-Ag1.5, CHIR-99021, BMS-345541, and RG108, which facilitated neuroectodermal induction and EF/OV-like transitions. This was followed by a shared maturation stage (M3) using NIC, RA, and Activin A to drive RPE differentiation. For human fibroblasts, an extended cocktail was required by adding R-268712, BIX-01294, VPA, and SB-431542 to overcome chromatin-related reprogramming barriers and facilitate lineage progression.
The key small molecules contributed to the regulation of transcription, signaling pathways, and epigenetic remodeling. Hh-Ag1.5 activated the SHH pathway and induces TFs such as Bmi1, Sox2, and N-Myc29,58,59,60,61; LDN193189, a BMP inhibitor, promoted, and neuroectodermal induction29,62; RG108, a DNA methyltransferase inhibitor, relieved DNA methylation-mediated silencing and improved transcriptional accessibility63. BMS-345541 inhibited NF-κB signaling to reduce apoptosis and support reprogramming64,65. and CHIR-99021, a GSK-3β inhibitor, activated the Wnt/β-catenin signaling to enhance proliferation and differentiation66,67,68. In human cells, epigenetic regulators such as VPA, BIX-01294, and SB-431542 further increased chromatin accessibility and promoted lineage conversion63,69,70,71. The maturation stage compounds—NIC, RA, and Activin A—were selected based on established protocols and previous organoid studies to promote the final transition from EF/OV-like cells to mature, functional RPE. The use of certain small molecules in our study—such as Hh-Ag1.5, LDN193189, and CHIR-99021—overlaps with compounds previously applied in neuroectodermal induction or reprogramming. This convergence likely reflects the shared developmental origin of RPE and neural lineages. Crucially, these molecules were not selected arbitrarily but systematically prioritized and validated by our prediction platform from a large candidate pool, underscoring the accuracy and biological relevance of our strategy. Moreover, we identified molecules such as BMS-345541 and CKI-7 as novel functional contributors in RPE reprogramming, offering new potential targets for future mechanistic exploration.
Although we achieved high-efficiency ciRPE induction in the mouse system and demonstrated preliminary therapeutic effects in RD model—including host integration and partial restoration of visual function—these effects remain limited to a specific preclinical context. The long-term safety, immune compatibility, and functional stability of transplanted cells require further systematic evaluation through extended in vivo studies to support future clinical translation.
In contrast, the efficiency of ciRPE induction from human fibroblasts remains relatively low, which limits its immediate scalability for clinical-grade cell production. Several factors may underlie this challenge. One major consideration is the lack of high-quality single-cell RNA-seq datasets from early EF stages, which constrained our predictive model to rely on OV data as a surrogate reference. While organoid-derived OV datasets expanded the framework, their relatively late developmental timing may have reduced prediction resolution and reprogramming efficiency. In addition, primary HEFs are inherently heterogeneous in epigenetic state, transcriptional networks, and lineage potential, which likely contributes to variable chemical responses and divergent reprogramming trajectories. Moreover, although several small molecules effective in the mouse system were incorporated into the human cocktail, the overall efficiency remained suboptimal, pointing to species-specific differences in signaling dependencies and reprogramming routes.
These observations highlight both the opportunities and the challenges of extending chemical reprogramming to human fibroblasts. Overcoming these barriers will require more refined characterization of human fibroblast subpopulations, deeper understanding of their lineage plasticity, and optimization of small-molecule combinations tailored to human-specific regulatory landscapes. Advancing along these directions may ultimately enable scalable and clinically relevant generation of RPE and other retinal lineages from adult autologous cells for personalized regenerative therapies. In conclusion, this study outlines an innovative, gene-editing-free, and efficient two-stage approach using a small-molecule cocktail to convert fibroblasts into functional RPE cells. Our single-cell transcriptomics-based platform enabled the precise identification of optimal small molecules for enhancing reprogramming. Functional and mechanistic analyses revealed core TFs driving RPE conversion, and successful ciRPE cell transplantation in RD models demonstrated their therapeutic potential. These findings advance our understanding of cellular reprogramming mechanisms and open new avenues for developing safe, scalable, and effective regenerative therapies targeting RD diseases.
Methods
Human tissues
The primary HEFs and primary hRPE cells used in this study were isolated from donated tissues of 16- and 18-week embryos, with the research approved by the Ethics Committee of the Eye Hospital of Wenzhou Medical University (Research License 2021-238-k-208-01). Informed consent forms were prepared in compliance with ISSCR guidelines for fetal tissue donation. All donors provided written informed consent and willingly contributed fetal tissue samples after deciding to undergo legal pregnancy termination. All experimental procedures adhered to the Regulations on the Management of Human Genetic Resources of the People’s Republic of China.
Animals
The Best1-Cre [C57BL/6-Tg (BEST1-cre)1Jdun/J], Fsp1-cre [BALB/c-Tg (S100a4-cre)1Egn/YunkJ], and ROSA26-tdTomato [B6. Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J] mice were obtained from the Jackson laboratory. C57BL/6J mice were obtained from Jiangsu GemPharmatech, while 129S4/SvJae and BALB/c- Nude mice were sourced from Shanghai Vital River Laboratory. RCS rats, a well-recognized and classical animal model of RD, were kindly provided by Professor Guoping Fan. Animals were randomly assigned to experimental groups regardless of sex, as no sex-specific differences were anticipated in the context of this study. All experimental animal strains underwent genotype verification prior to breeding and were housed under specific pathogen-free conditions with a 12-h light/dark cycle, ambient temperature of 22 ± 2 °C, and relative humidity of 50–60%. Efforts were made to minimize animal suffering and to reduce the number of animals used. All procedures conducted on these animals were in compliance with ethical animal license protocols and received approval from the Laboratory Animal Ethics Committee of Wenzhou Medical University (Research License wydw2024-0200).
Predicting and screening small molecules using scRCF
This scRCF pipeline consists of three parts: 1. Screen signal proteins and network based on SiPer; 2. Two-step optimization for small molecule prioritization; 3. Refinement and validation of predicted small molecules.
Screen signal proteins and network based on SiPer
The scRCF pipeline requires single-cell data from the initial and target cell types. Differentially expressed transcription factors (DETFs) are identified using Seurat (v4.3.0 or v5.1.0)72. The processing steps include creating a Seurat object, integrating scRNA-seq data, normalizing the data, identifying differentially expressed genes, and selecting TFs with an absolute avg_log2FC > 1 and an adjusted p value < 0.05. The list of TFs, including TFs and co-factors, is obtained from the AnimalTFDB (3.0) database73. The expression of DETFs is booleanized based on their regulation status relative to the initial cell type, with up-regulation assigned as 1 and down-regulation as −1.
Signal networks and candidate signaling proteins were identified using SiPer19, with binarized differentially expressed TFs as input. SiPer incorporates three built-in prior knowledge databases: a small molecule perturbation database, a small-molecule target database, and a signaling network. The small molecule perturbation database was constructed from the LINCS L1000 database (level 5)74, retaining signatures within six hours post-perturbation in normal or primary cell lines19,74. Only TFs were included, with regulation statuses assigned as +1 for up-regulation and −1 for down-regulation based on z-scores. This database contains 5570 entries of small molecule treatment information. The small-molecule target database integrates data from the Drug Repurposing Hub75, STITCH v5.076, and MedChemExpress (www.medchemexpress.com). For STITCH, only protein targets with a confidence score > 0.4 were retained. Interaction values between small molecules and targets were assigned as follows: 1 for activation, −1 for inhibition, and 2 for unknown. The database contains 4389 small molecules and 46,493 interactions. The signaling network is based on the prior knowledge network described by Zheng et al.19, including signal proteins, TFs, and hierarchical network information. Using SiPer, the input TFs are integrated into the signaling network, producing outputs that include signal proteins, TFs, and signaling proteins (candidate SPs) identified as critical drivers of chemical reprogramming.
Two-step optimization for small molecule prioritization
Community partitioning using short random walks. Community partitioning identifies groups of functionally similar proteins, allowing the targeting of only a subset of these proteins to achieve desired effects77. A random walk method is used to detect communities within the signaling network. Initially, each node is treated as an independent community. A fixed-length random walk is then simulated, where the walker transitions from the current node to a randomly selected neighboring node. The transition probability is defined as the likelihood of signal transmission. The similarity between node pairs is assessed by calculating the transition probability during the random walk. The distance measure \({{{\rm{r}}}}_{{{\rm{ij}}}}\) is defined as:
where \({P}_{{ik}}^{t}\) represents the probability of transitioning from node \(i\) to node k in a t-step random walk. Nodes or communities with high similarity are merged iteratively until a stopping condition is met, set here as four merges. The post-merge community distance is defined as:
where \({P}_{C1k}^{t}\) is the probability of transitioning from community C1 to node k in a t-step random walk, averaged over all nodes in the community. Gain the community partitioning results of the graph. This process produces a partitioned graph, with signal proteins assigned to specific communities.
Prioritizing candidate small molecules. Based on the community partitioning results, an effect score is calculated for each small molecule in the small-molecule target database. The effect score is defined as:
where C represents the communities affected by the small molecule, and the indicator function is defined as:
Here, D denotes the mode of action of the small molecule on a protein in the database, and P denotes the influence mode of the signaling protein. Small molecules with an effect score of 0 are then removed, yielding a refined list of candidate small molecules.
Selection of signal pathways. Small molecules achieve cell identity transitions by influencing signal transduction and epigenetic signaling pathways78. To refine signal pathway classifications, we referred to the KEGG and Reactome79 databases, which provide detailed categorizations of signal transduction pathways. Additionally, we incorporated classification information from the compound supplier (MedChemExpress) to standardize pathway classifications across these databases. Based on this integration, we identified the 10 most relevant signal pathways for reprogramming. Signal pathways were further subdivided based on specific molecular mechanisms and reaction steps (Supplementary Table 3), creating correspondences between pathways and their sub-pathways. Information on small molecule drugs, along with their associated signaling and sub-pathway classifications, was compiled using data from compound suppliers (Supplementary Data 9). Then, the effect score is normalized:
where ES denotes the effect score of each drug, and \(\max ({{ES}}_{{pathway}})\) represents the highest ES value in the pathway corresponding to the small molecule compound. Finally, retain the small molecule with the highest effect score within the sub-pathways of each signaling pathway classification. Output the list of candidate small molecules.
Refinement and validation of predicted small molecules
(Proceed with DRUG-seq2 and analysis.).
Isolation and culture of primary HEFs
HEFs were isolated as previously described80,81. Briefly, fetal skin tissue (2–4 cm²) was washed twice with Dulbecco’s Phosphate-Buffered Saline (DPBS, Gibco, c14190500cp) containing 2% Penicillin-Streptomycin (P/S, 10,000 U/mL, Gibco,15140122), then cut into 1–2 mm² pieces and digested with 2 mg/mL Collagenase IV (Gibco, 17104019) at 37 °C for 1 h. Digestion was then terminated by adding 15% fetal bovine serum (FBS, Biological Industries, 04-001−1ACS) and 2% P/S to high-glucose Dulbecco’s Modified Eagle Medium (DMEM, Gibco, C11995500bt). The suspension was collected into a 50 mL tube, allowed to stand for 10–15 min, and the supernatant containing single-cell suspensions was transferred to a cell culture dish for cultivation. After the cells adhered the next day, the medium was replaced with fresh growth medium. HEFs were then cultured in DMEM supplemented with 15% FBS, 1% P/S, 1% MEM Non-Essential Amino Acids (NEAA, Sigma-Aldrich, M7145-100ML), 1% GlutaMAX (Gibco, 35050061), and 0.055 mM 2-Mercaptoethanol (Gibco, 21985023).
Isolation and culture of primary human RPE cells
For isolation of primary hRPE cells, the dissected eye tissue was washed twice in PBS containing 2% P/S, and the anterior segments and surrounding connective tissue were removed, leaving the eyecup. Using fine tweezers, the RPE layer was separated from the neural retina and incubated in Dispase solution (0.5–1.0 mg/mL, CORNING, 534253) containing 50 μg/mL DNase Ⅰ (Roche, 11284932001) at 37 °C for 45 min. After incubation, the eyecup was gently shaken to release RPE cells, and further digestion with 0.25% trypsin (Thermo Fisher Scientific, 25200056) was performed if necessary. The cell suspension was collected, centrifuged, and resuspended in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12/GlutaMAX (DMEM/F-12/GlutaMAX, Thermo Fisher Scientific, 10565042) supplemented with 20% FBS and 1% P/S. Cells were then plated onto Matrigel-coated (CORNING, 354234) culture plates and incubated at 37 °C with 5% CO₂. The medium was changed every 2–3 days until the cells reached confluence.
Cell isolation and culture from mice
Primary MEFs were derived from E13.5 mouse embryos with genetic backgrounds including C57BL/6J, 129S4/SvJae, Best1-Cre/ROSA26tdTomato (generated by crossing Best1-Cre mice with ROSA26 tdTomato mice), and Fsp1-Cre/ROSA26tdTomato (generated by crossing Fsp1-Cre mice with ROSA26 tdTomato mice), following standardized protocols16. Specifically, neural tissues (including the brain, spinal cord, and tail), limbs, gonads, and visceral organs were carefully removed and discarded prior to the isolation of MEFs. The remaining tissues were then finely minced, enzymatically dissociated using trypsin, and plated onto 10-cm culture dishes in DMEM supplemented with 10% FBS and 1% MEM NEAA.
The extraction of Tail-Tip Fibroblasts (TTFs) was carried out following established protocols52. Tail tips (approximately 0.3 mm) from 3-week-old mice were collected, minced, enzymatically digested with trypsin, and subsequently cultured in MEF medium to isolate TTFs.
The protocol for isolating and culturing primary mouse RPE cells was optimized based on previous research82,83. Briefly, eyes from 8–12-week-old C57BL/6J mice were dissected under a microscope. The eyes were then transferred to a dish containing DPBS supplemented with 1% P/S. The surrounding connective tissue and anterior segment were carefully removed, and the retina was delicately peeled from its internal attachment to the optic nerve using fine tweezers, ensuring the RPE layer remained intact within the eyecup. The eyecup was subsequently placed into a 24-well plate and incubated with dispase I (Sigma-Aldrich, D4818) for 45 min. The plate was gently agitated to facilitate the detachment of RPE cells from the eyecup. Detached RPE cells were then transferred to a Matrigel-coated culture plate and resuspended in complete medium (DMEM + 15% FBS + 2% P/S).
Generation of ciRPE cells from MEFs using a two-stage chemically defined reprogramming protocol
M7 reprogramming medium (RM) preparation
Equal volumes of Neurobasal (Thermo Fisher Scientific, 21103049) and DMEM/F12/GlutaMAX supplemented with 1% N2 (Thermo Fisher Scientific, 17502048), 1% B27 supplement without vitamin A (Thermo Fisher Scientific, 12587010), 7.5% BSA (Solarbio, H1130), 1% NEAA, 1% P/S, bFGF (10 ng/ml, PeproTech, 100-18B) and the small molecules CHIR-99021 (3 μM, MCE, HY-10182), LDN193189 (0.1 mM, Selleckchem, S2618), A 83-01 (0.5 mM, Selleckchem, S7692), Hh-Ag1.5 (0.5 mM, Xcess Biosciences, M60004-10S), RG108 (10 μM, Selleckchem, S2821), BMS-345541 (0.2 mM, MCE, HY-10519) and CKI-7 (5 μM, Sigma-Aldrich, C0742).
M3 differentiation and maturation medium (DM) preparation
DMEM/F12/GlutaMAX supplemented with 10% KSR (Gibco, 10828010), 1% NEAA, 1% P/S, 0.055 mM 2-mercaptoethanol, Y27632 (10 μM, Selleckchem, S6390) and Nicotinamide (NIC, 10 mM, Sigma-Aldrich, 47865-U), Retinoic acid (RA, 1 μM, Sigma-Aldrich, R2625), Activin A (0.2 mM, MCE, HY-P70311).
ciRPE maintenance medium preparation
ciRPE maintenance medium includes proliferation medium and function-maintaining medium. The proliferation medium consisted of DMEM/F12/GlutaMAX supplemented with 1% N2, 2% B27 without vitamin A, 1% NEAA, 1% P/S, 0.1 mM 2-mercaptoethanol, 10 ng/mL bFGF, 20 ng/mL EGF (Gibco, PMG8045), 10 μM Y27632, and 0.5 μM A 83-01. The function-maintaining medium consisted of DMEM/F12/GlutaMAX supplemented with 1% N2, 2% B27 with vitamin A, 1% NEAA, 1% P/S, 0.1 mM 2-mercaptoethanol, 0.2 μM Activin A, 0.5 μM RA, 1 μM BMP4, and 10 mM NIC.
All small molecules were dissolved and diluted in DMSO (Sigma-Aldrich, D4540) following the manufacturer’s instructions and are detailed in Supplementary Data 10.
Stage I: Chemical conversion of eye-field cells from MEFs
-
(1)
Four days before the chemical conversion, Matrigel was thawed overnight at 4 °C. The following day, 6-well plates were pre-cooled at 4 °C for a minimum of 1 h. Matrigel was diluted 1:40 in DMEM/F12/GlutaMAX and 1 mL of this diluted Matrigel solution was added to each pre-cooled well of the 6-well plate, which was then incubated at 4 °C overnight.
Note: To ensure optimal performance, Matrigel must be thawed at 4 °C and should not be subjected to repeated freeze-thaw cycles.
-
(2)
Two days before chemical induction, thaw the cryopreserved primary MEFs in a 37 °C water bath and centrifuge to remove the cryoprotectant. Resuspend the cells in MEF medium and seed them into 6-well plates at a density of 20 × 104 cells per well. Prior to seeding the MEFs, pre-warm the Matrigel-coated 6-well plates at 37 °C for at least 30 min.
Note: The density of MEFs is crucial for the success of chemical induction. Ensure that cells are evenly distributed; too few cells can lead to poor proliferation and cell health, while overly dense cultures may affect the success rate of induction. Regularly monitor the health and growth of MEFs after seeding to ensure they adhere well and proliferate as expected. Proper pre-warming of Matrigel-coated plates is essential for optimal cell attachment and performance.
-
(3)
One day before chemical induction, replace the culture medium of MEFs with fresh medium and allow the cells to proliferate for one day. Regularly check the growth and condition of the MEFs to ensure that the cell health and density are optimal.
Note: The health of MEFs is crucial for successful conversion. Long-term storage of MEFs in liquid nitrogen may also reduce conversion efficiency. This study used freshly prepared MEFs or those stored in liquid nitrogen for no more than 6 months.
-
(4)
On the day of chemical induction, prepare the fresh RM of Stage I, working in the dark to ensure complete dissolution of all components, particularly the chemical agents. After incubating MEFs overnight in MEF medium, wash the cells twice with 1× PBS and replace the medium with the freshly prepared RM. Refresh the RM the next day.
-
(5)
During Stage I induction, cells will rapidly proliferate within the first 1–4 days, with their morphology transitioning from elongated spindle-shaped to small oval-shaped. After one week, proliferation of the clones will slow. At this point, mechanically remove surrounding non-clonal cells using a yellow pipette tip to facilitate the continued expansion of the clones. About one week later, cells will begin to adopt a cobblestone-like morphology. Thereafter, refresh the medium daily until EF-like cells are formed.
Stage II: Generation of ciRPE cells from eye-field cells
For Stage II induction, the cells exhibit initial RPE cell morphology but still lack maturity and pigmentation. At this stage, replace the medium with DM of Stage II to induce pigmentation and promote further maturation of ciRPE cells. The medium is changed daily. After two weeks of treatment, the cells reach near-full maturity and show significant pigmentation. Subsequently, ciRPE cells are passaged and expanded in maintenance medium.
Dynamic alternating culture for ciRPE cells
To balance proliferation and RPE-specific functionality, ciRPE cells derived from MEFs were cultured using a dynamic alternating medium protocol.
After the first passage, during the initial expansion phase (day 0-7), the cells were cultured in proliferation medium to maximize growth. Following this, the cells were briefly exposed to function-maintaining medium for 1–2 days every 3–5 days. This alternating approach effectively supported both cell expansion and the preservation of early RPE-like characteristics.
Flow cytometry
Cells were washed twice with DPBS to eliminate residual culture medium. Adherent cells were then dissociated into single-cell suspensions by treatment with 0.25% trypsin at 37 °C for 5 min. The enzymatic activity was neutralized by adding an equal volume of culture medium supplemented with 10% FBS. Following centrifugation at 300 × g for 5 min, the resulting cell pellet was resuspended in precooled staining buffer (DPBS, 1.5% FBS, and 0.5% BSA) to reduce non-specific binding and prevent aggregation. The suspension was passed through a 25 μm cell strainer to ensure a uniform single-cell preparation. For fluorescence staining and sorting, cells requiring antibody labeling were incubated with primary antibodies at the recommended dilution for 30 min on ice, protected from light to preserve fluorescence integrity. After incubation, cells were washed twice with precooled staining buffer to remove unbound antibodies. When secondary antibody labeling was necessary, fluorophore-conjugated secondary antibodies were applied under the same conditions for 30 min on ice in the dark, followed by additional washing steps.
Prior to flow cytometry and sorting, a viability dye was added to the staining buffer to exclude dead cells. The prepared cell suspension was analyzed and sorted using a BD FACSAria™ cell sorter. Gating strategies were carefully designed based on fluorescence markers and viability staining to ensure the precise isolation of the target cell population. Compensation controls were applied to correct for fluorescence spillover and enhance gating accuracy. Sorted cells were collected directly into tubes containing culture medium supplemented with 10% FBS to maintain viability. Post-sort purity was assessed to verify sorting accuracy. Data acquisition and analysis, including both flow cytometry and sorting results, were performed using FlowJo software (FlowJo V10).
Quantitative RT-PCR
RNA was extracted using the FastPure Cell/Tissue Total RNA Isolation Kit V2 (Vazyme, RC112) following the manufacturer’s instructions. The RNA was reverse-transcribed using the HiScript III 1st Strand cDNA Synthesis Kit ( + gDNA wiper) (Vazyme, R312) to synthesize cDNA. qRT-PCR was performed on a CFX Manager Real-Time PCR system (Bio-Rad) using specific primers and Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Q712). Analyses were conducted using the ΔΔCt method with GAPDH normalization. The primer sequences are listed in Supplementary Table 4.
Immunofluorescence
Immunofluorescence for Cells: Cells were washed with DPBS twice and then fixed with 4% paraformaldehyde (4%PFA, Beyotime, P0099) for 15–20 min at room temperature. Afterward, the cells were washed with 1×PBS twice and then permeabilized with 0.3% Triton X-100 (Solarbio, T8200) for 10 min, blocked with 1% bovine serum albumin (BSA, Sigma-Aldrich, V900933) in 1×PBS for 1 h at 37 °C. The cells were then incubated with primary antibodies overnight at 4 °C. The next day, they were incubated with secondary antibodies for 2 h at room temperature in the dark. The cells were then stained with 4′, 6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific, D1306) to indicate the nucleus for 5–10 min. Labeled cells were imaged with confocal laser scanning microscopy (LSM900 with Airyscan, ZEISS, Germany). Antibodies are described in Supplementary Table 5.
Immunofluorescence for Tissue Sections: Surgically removed eyeballs had their lenses peeled away, and the residual eye cups were fixed overnight in 4% PFA solution. The fixed tissues were subsequently dehydrated and embedded in OCT cryo-embedding medium (SAKURA, 4583) for rapid freezing. These tissues were sectioned into 15 μm thick slices using a Thermo Scientific Micron HH 560 cryomicrotome, mounted onto glass slides, and stored at –80 °C. When immunostaining was required, the tissue frozen sections were air-dried at room temperature and blocked with 1% BSA. The subsequent steps were identical to those used for cell immunofluorescence staining.
Transmission electron microscopy
Cells were fixed in 2.5% glutaraldehyde buffer (Electron Microscopy Sciences, 16000) at room temperature for 2 h and then washed three times with PBS buffer. The cells were subsequently treated with 1% cold osmium tetroxide for 1 h. After fixation, the samples were washed with PBS buffer, dehydrated through a graded series of ethanol solutions (50%, 70%, 85%, and 100%), and embedded in epoxy resin. The samples were then sectioned into thin slices using an LKB ultramicrotome and stained with 1% uranyl acetate and lead citrate. Micrographs were obtained using a transmission electron microscope (Hitachi HT7800, Japan).
Phagocytosis assays
Cells were incubated with 1 μm yellow-green fluorescent latex beads (Sigma-Aldrich, L4655) at 37 °C for 12 h, or with POSs isolated from fresh pig eyes and labeled with fluorescein isothiocyanate (FITC, Sigma, F7250) for 6 h38,84,85. The cells were then washed rigorously with PBS to remove unphagocytosed POSs, fixed with 4% PFA, permeabilized with 1% Triton X-100, and blocked with 1% BSA in 1×PBS for 1 h. ZO-1 (Invitrogen, 61-7300) immunostaining was used to show the boundary of cells. DAPI was used to label nuclei. If required, FITC fluorescence was quenched with 0.4% trypan blue (Gibco, 15250061) for 10 min before fixation. Finally, the samples were imaged using a fluorescence microscope. Z-stack images were obtained using a Nikon confocal microscope (LSM900 with Airyscan, ZEISS, Germany).
Transepithelial electrical resistance (TEER) assays
Seed cells at a density of 1.0 × 104/cm² into the upper chambers of 24-well Transwell membranes (0.4 μm, CORNING). Fill the inner and outer chambers with 0.2 mL and 0.7 mL of culture medium, respectively, and cultured in an incubator at 37 °C with 5% CO2, replacing the culture medium every 3 days. Measure the TEER every 5 days using a Millicell-ERS ohmmeter (ERS-2, Millipore, USA). A cell-free chamber group was set up as a blank control. To measure TEER, remove the 24-well Transwell plate from the incubator, allow it to equilibrate at room temperature for 30 min, then insert the electrodes on either side of the filter and record the TEER readings. The TEER was calculated through the formula: TEER (Ω cm2) = (treatment resistance (Ω)-background resistance (Ω))×membrane area (cm2).
Enzyme-linked immunosorbent assay (ELISA)
Mouse and human ciRPE cells were plated at a density of 2.5 × 105/cm² and cultured on Transwell membranes coated with Matrigel. Cell culture supernatants from the apical and basal sides of the ciRPE cells, corresponding to the upper and lower compartments of the Transwell, respectively, were collected after one week of cell culture. Standard ELISA was performed using antibody-coated plates, biotin-labeled antibodies, and streptavidin-HRP for detection. Detailed procedures were followed according to the instructions of the PEDF ELISA kit (CUSABIO, CSB-E08820m and CSB-E08818h) and the VEGF ELISA kit (CUSABIO, CSB-E04756m and CSB-E11718h).
Cell proliferation assay
The proliferation rate of ciRPE and pRPE cells was determined using the Cell-Light EdU Apollo488 In Vitro Kit (RiboBio, C10310-3) according to the manufacturer’s instructions, as previously described86. Briefly, cells were incubated with 50 μM EdU for 2 h. After incubation, the cells were fixed with 4% PFA for 15 min and permeabilized with 0.5% Triton X-100 for 10 min. The cells were then incubated with the Apollo reaction cocktail for 15 min in the dark. After staining, the cells were washed, resuspended in PBS, and analyzed using flow cytometry to quantify EdU incorporation.
Cell cycle analysis
Cells were carefully dissociated into single-cell suspensions by trypsin, washed twice with DPBS, and then fixed overnight with cold 70% ethanol. Fixed cells were washed twice with PBS, followed by RNase (100 µg/ml, Thermo Fisher Scientific, EN0531) treatment and PI (50 µg/mL, Sigma-Aldrich, 537060) staining for 30 min at 37 °C. Approximately 2 × 104 cells were analyzed using FACSCanto II (Becton Dickinson) to determine the cell cycle distribution pattern. The percentages of cells in the G1, S, and G2/M phases of the cell cycle were analyzed using Flowjo V10.
Karyotype analysis
To conduct karyotype analysis, cells were initially treated with colchicine (0.1 µg/mL, Sigma-Aldrich, C-207) at 37 °C for 2 h to synchronize them in metaphase. After colchicine treatment, the cells were trypsinized, resuspended, and subjected to hypotonic treatment using 0.075 M potassium chloride for 15 min at 37 °C to facilitate cell swelling and chromosome spread. Following hypotonic treatment, the cells were fixed with a 3:1 methanol acid solution. The fixed cell suspension was then applied to slides to achieve chromosome spread. Finally, chromosomes were visualized by staining with Giemsa (Solarbio, G1015).
Lentiviral package and cell transfection
Lentiviral particles were produced by transient transfection of HEK293T cells using a second-generation packaging system. HEK293T cells were seeded in 10 cm dishes at a density of 3 × 106 cells per dish and cultured overnight in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. The next day, when the cells reached 70–80% confluence, the constructed vectors (pLKO.1-shAscl1-1, pLKO.1-shAscl1-2, pLKO.1-shOlig2-1, pLKO.1-shOlig2-2 and pLKO.1-shControl) were co-transfected with the lentiviral packaging vectors pMD2.G (Addgene, 12259) and psPAX2 (Addgene, 12260) into HEK293 T cells using Lipofectamine 3000 (Invitrogen, L3000015) transfection reagent, following the manufacturer’s instructions. Viral supernatants were collected at 48- and 72-h post-transfection, filtered through a 0.45 µm syringe filter to remove cellular debris, and concentrated overnight using the Universal Virus Concentration Kit (Beyotime, C2901S). The concentrated viral particles were used within 2 days for cell infection without undergoing a freeze-thaw cycle.
For cell infection, cells were exposed to lentiviral particles at a multiplicity of infection (MOI) of 10 in the presence of 8 µg/mL polybrene (Sigma-Aldrich, TR-1003) to enhance transduction efficiency. After 24 h, the infection medium was replaced with fresh complete medium, and the cells were incubated for an additional 48–72 h. Subsequently, 2 µg/mL puromycin (Yeasen, ISY1130) was used to select successfully transduced cells.
Subretinal transplantation
The transplantation therapy was performed on RCS rats, a recognized model of RD. All experiments conducted in this study followed the guidelines and permissions provided by the Institutional Committee of Laboratory Animal Ethics. For subretinal injection, tdTomato+ ciRPE cells (harvested either immediately after the 32-day induction or after an additional short-term maturation culture of up to one week) were sorted by FACS and prepared into a 2 μL cell suspension with a concentration of 5×104 cells/μL. For subretinal injection, RCS rats (P21) were anesthetized with pentobarbital sodium (40 mg/kg, Sigma-Aldrich, P3761) after dilating the pupils with 1% tropicamide (Kangye Pharmaceutical, H20044926) and applying the topical anesthetic 0.5% proparacaine hydrochloride (ALCON-COUVREUR, H20103352). Under a surgical microscope, a channel was created by inserting a 31-gauge needle into the subretinal space. Subsequently, the injection was performed using a Hamilton syringe fitted with a 31-gauge needle. After the injection, the needle was left in place for a few seconds to reduce reflux and enable maximal cell release before being slowly withdrawn. The eyelid was then returned to its original position, and a drop of Ofloxacin Ointment (Sinqi Pharmaceutical, H10940177) was applied. The rats were kept on a 37 °C warming bed until fully awake. Postoperatively, ofloxacin ointment was applied topically for 3 days to prevent dryness and infection. All rats, including sham-transplanted ones, were administered dissolved Cyclosporine A (250 mg/L, Sangon, A600352) from 48 h before transplantation until the end of the experiment to mitigate immune rejection. The effectiveness of the transplantation treatment was then evaluated at designated time points.
In vivo imaging of transplanted cells
After cell transplantation, RCS rats were anesthetized and their pupils were dilated as described above. Fundus photographs of the RCS rat eyes were obtained using an Eyemera FUNDUS retinal imaging microscope (IIscience, China) to immediately assess the success of the cell transplantation. Bright field images were captured using the bright channel. Successful transplantation was indicated by the presence of bright bubbles in the subretinal space visible in the fundus images.
Optical coherence tomography analysis
Optical Coherence Tomography (OCT; Heidelberg Engineering, Germany) was employed to document and monitor the development of the transplant in the host retina from 0 to 3 weeks post-cell transplantation. OCT imaging of the retina in RCS rats was performed using a Bioptigen Envisu R2200 Spectral Domain Ophthalmic Imaging System (Bioptigen, Research Triangle Park, NC) following anesthesia with pentobarbital sodium and pupil dilation with 1% tropicamide.
Teratoma formation
To develop teratoma in vivo, 1 × 107 ciRPE cells were injected subcutaneously into BALB/c Nude mice. mESCs cells were used as a control. Teratoma formation was seen 6–8 weeks after cell transplantation. Then, a cellphone imaging system was used to capture images of the teratoma formation in the mice. All animal experiments were approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University. The maximal tumor size permitted by the ethics committee was 1.5 cm in diameter or 1500 mm³ in volume, and this limit was not exceeded in any of the experimental animals.
Electroretinogram
Corneal scotopic flash electroretinogram recordings were conducted on the eyes of RCS rats during weeks 2–16 post-transplantation. ERG data was recorded with a RETI-scan system (Roland Consult, RETI-scan, Germany). Rats were dark-adapted overnight before testing, then anesthetized with pentobarbital sodium. The pupils of the rats were dilated using 1% tropicamide. Proparacaine hydrochloride eye drops were applied for ocular surface anesthesia, and ofloxacin eye ointment was used to prevent dry eyes and bacterial infection. For flash ERG recordings, two gold wire loop corneal electrodes were attached to the eyes. Additionally, two reference electrodes were attached to the forehead, and a ground electrode was attached to the tail. The ERG parameters for photopic responses were set as follows: stimulation intensities at 0.48 log candela (cd)•s/m² (light 3.0). The ERG parameters for scotopic responses were configured as follows: stimulation intensities at –2.02 log cd•s/m² (dark 0.01), 0.48 log cd•s/m² (dark 3.0), and 0.98 log cd•s/m² (dark 10.0). The rats were placed on a thermostat platform at 37 °C to keep them warm throughout the ERG test. All procedures for the dark/scotopic-adapted ERG tests were performed in dim red light. Analysis of b-wave amplitudes was performed using ERG data analysis software.
Optomotor response-based visual function assessment
qOMR was recorded from 2 to 16 weeks post-transplantation to assess the visual function of RCS rats. The visual acuity of RCS rats was measured by recording videos of optomotor responses to a virtual cylinder with alternating black and white qOMR system (PhenoSys, Berlin, Germany). Rats were placed on the white platform of the qOMR system. The system tracks the movement of the animal’s head in real-time relative to the presented visual stimulus and objectively quantifies the OMR results. The stimulation protocols were as follows: the spatial frequency of the pattern was set at full check with several different spatial frequencies (0.05, 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.375, 0.4, 0.425, 0.45, and 0.5 cycles/degree), with movement at 12°/s for 60 s. The qOMR score is calculated as the ratio of concordant-to-discordant body/head movements in response to the moving gratings displayed on the screens surrounding the animal. A qOMR score of 1.0 served as a threshold for sensory perception: animals with qOMR scores below 1.0 were considered unable to perceive the specific spatial frequency being investigated.
Hematoxylin-eosin (HE) staining
For HE staining, the frozen sections were air-dried at room temperature and stained with the Modified Hematoxylin-Eosin Stain Kit (Solarbio, G1121). The staining process included 8 min of hematoxylin staining, 60 s in bluing solution, and 5 s of eosin staining. After staining, the sections were washed with PBS, coverslipped, and imaged using a DM4b microimaging system (Leica, USA). Images were acquired with LAS X software.
Small living animal imaging
Small animal imaging was conducted using the IVIS Spectrum system (PerkinElmer, USA), equipped with fluorescence and bioluminescence detection capabilities. Animals housed under SPF conditions were anesthetized with isoflurane and positioned on a pre-warmed imaging platform to ensure stability and minimize stress. To reduce background fluorescence, areas such as the mouth, nose, paws, and urination sites were gently cleaned using warm water-moistened gauze prior to imaging. The mice were then placed on the imaging platform within a dark box, and the software-controlled platform elevation was adjusted to optimize the field of view. An initial background image was captured under bright-field illumination, followed by fluorescence imaging in dark-field mode to eliminate external light sources and record emitted fluorescence. Superimposing bright- and dark-field images allowed precise localization and quantification of photons. A fluorescence imaging module (dsRed/RFP) was used for these measurements. The small animal in vivo imaging software provided photon intensity-scaled images and facilitated the analysis of luminescence area, total photon count, and photon intensity.
TUNEL assay for apoptosis detection
Apoptotic cells in frozen tissue sections were detected using a TUNEL assay kit (Alexa Fluor 488, Yeasen, 40307ES60) following the manufacturer’s instructions. Briefly, the eye sections were fixed with freshly prepared 4% PFA at room temperature for 20 min, followed by three washes with PBS. Permeabilization was performed using a 20 μg/mL Proteinase K solution for 10 min at room temperature. After rinsing with 1×PBS, the TUNEL reaction mixture was freshly prepared and applied to the sections. The slides were incubated in a humidified chamber at 37 °C in the dark for 1 h. Following the incubation, the sections were washed three times with PBS to remove excess reagents. Nuclei were counterstained with DAPI (1 µg/mL) for 10 min, followed by a final PBS wash. The slides were then mounted with antifade mounting medium and imaged using a fluorescence or confocal microscope. Apoptotic cells labeled with Alexa Fluor 488 fluoresced green, while nuclei stained with DAPI fluoresced blue. The proportion of apoptotic cells was determined by calculating the ratio of TUNEL-positive cells to the total number of nuclei in randomly selected fields.
Generation of the Best1 Pr-EGFP reporter HEF cell Line
The sgRNA plasmids were constructed by cloning protospacers downstream of the U6 promoter into the px330 backbone (Addgene, 68807) using the BbsI restriction site, following a standard protocol designed for AAVS1 targeting. For ligation, single-stranded DNAs were annealed to form double-stranded DNAs with 4-bp overhangs on both ends, which served as substrates for T4 DNA ligase. The cloning backbone was digested with BbsI-HF (NEB, R3539). Ligation was carried out by adding 1 µL of T4 DNA ligase (5 U/µL, Thermo Fisher Scientific, EL0014) to achieve a total reaction volume of 10 µL.
The BEST1 Pr-EGFP cell line was generated by inserting stop-BEST1 Pr-EGFP-WPRE-PA-LoxP-PuroR-LoxP at the AAVS1 locus. The homology arm DNA sequences were obtained from HEF cell genomic DNA by PCR and purified using the FastPure Gel DNA Extraction Mini Kit (Vazyme, DC301-01). Seamless cloning of the HDR vector (constructed in our lab) with BEST1 Pr-EGFP was performed using the Uniclone One Step Seamless Cloning Kit (Genesand, SC613), following digestion with AsisI (NEB, R0630) and SwaI (NEB, R0604S).
For plasmid electroporation, HEF cells were dissociated into single cells using trypsin. Approximately 1 × 106 cells were resuspended in a nucleofection solution prepared according to the manufacturer’s instructions, consisting of 82 μL P3 primary cell solution and 18 μL Supplement 1 (Lonza). To this mixture, 5 μg of plasmid DNA, including 2.5 μg of pX330-VSX2 plasmid and 2.5 μg of the VSX2-tdTomato targeting vector, was added. The cell-plasmid mixture was then transferred to nucleofection cuvettes (Lonza). Nucleofection was performed using the CA137 program on the Nucleofector 4D (Lonza). After nucleofection, cells were gently transferred to Matrigel-coated plates with HEFs media and incubated at 37 °C with 5% CO2. Following electroporation, cells were exposed to 2 μg/mL puromycin for approximately 7 days. Puromycin-resistant clones were then selected and expanded for genotyping. If necessary, the Puro resistance gene (PuroR) located between the two LoxP sites can be excised using Cre recombinase. The oligonucleotide sequences and vector sequences used for plasmid construction in this study are listed in Supplementary Table 4.
Generation of hciRPE cells from HEFs using a two-stage chemically defined reprogramming protocol
M9 RM preparation
Equal volumes of Neurobasal and DMEM/F12/GlutaMAX supplemented with 1% N2, 1% B27 supplement without vitamin A, 7.5% BSA, 1% NEAA, 1% P/S, bFGF and the small molecules CHIR-99021 (10 μM), LDN193189 (0.5 mM), Hh-Ag1.5 (0.5 mM), RG108 (10 μM), BMS-345541 (0.2 mM), R-268712 (10 μM, MCE, HY-12953), BIX-01294 (1 μM, MCE, HY-10587), Valproic acid (VPA, 0.2 mM, MCE, HY-10585) and SB-431542 (10 μM, Sigma-Aldrich, S4317).
M2 DM preparation
DMEM/F12/GlutaMax supplemented with 10% KSR, 1% NEAA, 1% P/S, 0.055 mM 2-mercaptoethanol, NIC (10 mM), Activin A (0.2 mM).
hciRPE maintenance medium preparation
The ciRPE maintenance medium is designed to support the long-term culture and functionality of mature hciRPE cells. It consists of DMEM/F12/GlutaMAX as the base, supplemented with 1% N2, 2% B27 with vitamin A, 1% NEAA, 1% P/S, and 0.1 mM 2-Mercaptoethanol. To promote cell health and expansion, 10 ng/mL bFGF and 20 ng/mL EGF (Gibco, PHG0311) are included. Additionally, low concentrations of Activin A (0.1 μM), RA (0.5 μM), and BMP4 (0.5 μM, R&D, 314-BP) are added to maintain RPE-specific gene expression, pigmentation, and structural integrity. The medium is freshly prepared, and all components are dissolved thoroughly before use.
Stage I: Chemical conversion of OV cells from HEFs
-
(1)
Matrigel was thawed overnight at 4 °C. The following day, 6-well plates were pre-cooled at 4 °C for at least 1 h. Matrigel was diluted 1:40 in DMEM/F-12/GlutaMAX, and 1 mL of this diluted Matrigel solution was added to each pre-cooled well of the 6-well plate. The plates were then incubated at 4 °C overnight.
-
(2)
The next day, thaw the cryopreserved primary HEFs in a 37 °C water bath, and centrifuge to remove the cryoprotectant. Resuspend the cells in 15% FBS-DMEM medium and seed them into 6-well plates at a density of 15 × 104 cells per well. Prior to seeding the HEFs, pre-warm the Matrigel-coated plates at 37 °C for at least 30 min.
Note: This study utilized either freshly prepared HEFs or those stored in liquid nitrogen for no longer than 2 months.
-
(3)
When the density of HEFs reaches 60–70%, chemical induction can be initiated. For Stage I induction, prepare the fresh RM in the dark to ensure the complete dissolution of all components, especially the chemical agents. Wash the HEFs twice with 1×PBS and replace the medium with the freshly prepared RM. Refresh the RM every two days.
-
(4)
During Stage I induction, the cells will rapidly proliferate within the first week, transitioning in morphology from elongated spindle-shaped to small oval-shaped. By approximately day 12, a few stable clones of uniform size and morphology will have formed. At this stage, mechanically remove surrounding non-clonal cells using a yellow pipette tip to facilitate the continued expansion of the clones. Thereafter, refresh the medium daily until OV-like cells are formed.
Stage II: Generation of hciRPE cells from OV cells
For Stage II induction, replace the medium with DM to induce pigmentation and promote the further maturation of hciRPE cells. The medium is refreshed daily. After two weeks of treatment with the maturation medium, the cells exhibit RPE-like morphology and display some pigmentation. Subsequently, the hciRPE cells are passaged and expanded in maintenance medium.
Maintenance culture of hciRPE cells
For long-term culture, hciRPE cells are passaged using mechanical dissociation methods and seeded onto Matrigel-coated plates, maintaining a confluency of 60–80%. The maintenance medium is refreshed every 2–3 days to ensure optimal cell health. Cells are regularly monitored for RPE-specific characteristics, including morphology, pigmentation, and the expression of key markers.
DRUG-Seq2
After treating cells with different small molecules in a 96-well plate for a specified period, cells were collected from the wells and RNA was extracted. RNA-seq libraries were prepared according to the manufacturer’s instructions using the VAHTS® Universal Pro DNA Library Prep Kit for Illumina, and sequencing was performed on an Illumina NovaSeq 600024,25. For DRUG-seq2 data processing, the gene expression matrix was generated according to the Drop-seq analysis protocol (https://github.com/broadinstitute/Drop-seq). Briefly, reads from each well were demultiplexed using 10-base well and 10-base plate barcodes, then aligned to the reference genome. Unique alignments were further processed by demultiplexing based on the transcript UMI for each gene.
DRUG-Seq2 analysis
We employed the DRUG-Seq2 method to evaluate the effects of various drugs on a specific gene set. Gene expression data were first collected for each drug, with multiple biological replicates included for each treatment. The mean expression value for each gene across all replicates was calculated, and a targeted gene set related to the EF and neuroectoderm was selected for analysis. To standardize the expression data and correct for technical variability, we calculated Z-scores for each gene. Specifically, for each gene in the selected set, the Z-score for its expression in each drug was calculated by subtracting the mean expression of that gene across all drugs (μ) from the drug-specific expression value (X), and dividing the result by the standard deviation of the gene’s expression across all drugs (σ). The formula used is as follows:
where X is the expression value of the gene in a specific drug, μ is the mean expression value of the gene across all drugs, and σ is the standard deviation of the gene’s expression across all drugs. Finally, for each drug, the average Z-score across the selected gene set was computed to provide an overall measure of the drug’s effect on the gene set. High average Z-scores indicate significant upregulation of the gene set, while low average Z-scores suggest significant downregulation.
RNA-seq and analysis
For RNA-seq sample collection, we scraped cells at each time point that exhibited a relatively consistent and successful induction phenotype, minimizing the inclusion of cells with failed induction. For each sample, total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) and subsequently purified using the FastPure Cell/Tissue Total RNA Isolation Kit V2 (Vazyme), following the manufacturer’s instructions. RNA-seq libraries were constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB England BioLabs). Paired-end sequencing was performed using Illumina NovaSeq 6000 with 150 base pairs (bp) read length. For RNA-seq data analysis, the clean reads were aligned to the mouse genome (mm10) using STAR87 (v2.7.11a) with the default parameter settings. The read counts of each gene were calculated using program featureCounts88 of Subread package. The gene counts were normalized using R package edgeR (4.0.16)88. The differential expression analysis was performed by R package limma (3.58.1), and the thresholds of significantly DEGs were |log2 fold change| > 1.5 and adjusted p value < 0.01. Perform unsupervised clustering analysis on the obtained differentially expressed genes using Mfuzz (2.26.0)89. GO analysis was performed on the genes in each cluster using the R package clusterProfiler90.
scRNA-seq and analysis
For scRNA-seq sample collection, we scraped all cells with different induction phenotypes at each time point, aiming to analyze all cell types involved in the induction process. Cells were washed in ice-cold PBS and dissociated using SeekMate Tissue Dissociation Kits (SeekGene). DNase I treatment was applied if needed. Cell viability was assessed using a Fluorescence Cell Analyzer (Countstar® Rigel, S2), after removing erythrocytes and filtering out debris and dead cells. Fresh cells were resuspended in RPMI1640 (Gibco) with 2% FBS at 1 × 106 cells/mL. Single-cell RNA-Seq libraries were prepared using the SeekOne® Digital Droplet Single Cell 3’ Kit (SeekGene). Cells were mixed with reverse transcription reagents and loaded into SeekOne® chip S3 for droplet generation. Reverse transcription was performed at 42 °C for 90 min, followed by inactivation at 85 °C for 5 min. cDNA was purified, amplified, fragmented, end-repaired, and ligated to sequencing adaptors. Indexed PCR was performed to amplify the 3’ polyA regions, including Cell Barcodes and Unique Molecular Indices (UMIs). Libraries were cleaned, quantified, and sequenced on an Illumina NovaSeq 6000 or DNBSEQ-T7 platform.
For scRNA-seq data analysis, the demultiplexing, barcoded processing, gene counting and aggregation were performed using the SeekSoulTools (v1.2.2) software with the default parameters. All scRNA-seq reads were aligned to the mouse reference genome (mm10). The scRNA-seq expression profiles were analyzed using R package Seurat (v4.1.0)72. The cells within a minimum of 200 genes expressed and the percentage of mitochondrial gene counts<10% were kept for subsequent analysis. We followed the standard Seurat pipeline by running function FindVariableFeatures, ScaleData, RunPCA, FindNeighbors, and FindClusters in order. We performed single-cell regulatory network inference and clustering (SCENIC) analysis using python package pySCENIC (0.12.1)91 to identify activated TFs and subnetwork of each TF (regulons) for each cell type. To calculate RNA velocity, both velocyto (v0.17)91 and scVelo (0.2.5)92 were used to analyze reads that passed the quality control after clustering as instructed. First, the standard velocyto pipeline was run to count spliced and unspliced reads for each sample based on the filtered SeekSoulTools-generated bam files. Then the output loom file was used as input for scVelo based on the dynamic model to estimate velocity embedding.
Cleavage under targets and tagmentation (CUT&Tag) and analysis
Approximately 1 × 105 cells from each sample were processed using the Hyperactive Universal CUT&Tag Assay Kit for Illumina (Vazyme, TD903-02) following the manufacturer’s protocol. Briefly, cells were harvested, and 1 × 105 cells from each sample were counted. The cells were then incubated with Concanavalin A beads at room temperature for 10 min. After removing the liquid, primary antibody was added in ice-cold Antibody Buffer, and the mixture was incubated overnight at 4 °C. The following day, the liquid was discarded, and a secondary antibody in Dig-Wash Buffer was added, followed by a 1-h incubation at room temperature. After sufficient washing, CUT&Tag pA/G-Tn5 Transposomes were added and incubated at room temperature for 1 h. Following a final wash, tagmentation was carried out at 37 °C for 1 h. DNA was then extracted using DNA extraction beads. PCR amplification was performed using i7 and i5 Indexed Primers, followed by library cleanup, quantification, and Illumina sequencing. All antibody information is summarized in Supplementary Table 5 are included in CUT&Tag Kit.
For data analysis, we performed quality control to raw data using FastQC (v0.12.1). Then the reads were trimmed using TrimGalore (v0.6.10) to remove detected adapters automatically. The clean reads were mapped to mouse reference genome mm10 using Bowtie2 (v2.3.4.2)93. After converting and sorting the output SAM files to BAM files using Samtools (v1.9), we removed the duplicated reads and reads with low mapping quality using Picard and Samtools. Next, we downsample the deduplicated BAM files to the same number of bases and perform quantile normalization using DANPOS94.
Key TF mining
Seurat (v4.1.0)19 was used to obtain TFs with significant changes (logFC>1, adjust. p < 0.05) from the SCENIC’s AUC matrix which estimate the regulon activity of the TFs across cell types. TFs passing the significance threshold were queried in the STRING database95, with a confidence score threshold of 0.4 to obtain the protein-protein interaction (PPI) data. The resulting PPI network, characterized by 125 nodes and 1041 edges, was imported into Cytoscape for visualization and further analysis. To identify key transcriptional regulators, the CytoHubba plugin96 was used to calculate the MCC score for each node, highlighting nodes with potential functional importance. MCC was chosen due to its ability to prioritize hubs that may participate in tightly connected clusters, potentially representing critical regulatory modules.
Statistical and reproducibility
All experiments were conducted independently and repeated at least three times. Results are presented as mean ± SD. To compare two groups, an unpaired two-tailed Student’s t-test was used. For comparisons involving multiple groups, one-way ANOVA followed by Tukey’s test or Dunnett’s multiple comparisons test was applied. Statistical analyses were carried out using SPSS Statistics 19.0 software, with significance set at p values less than 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The main data supporting the results of this study are available within the paper and its Supplementary Information. The RNA-Seq, scRNA-seq and CUT&Tag data generated in this study have been deposited in the Gene Expression Omnibus repository (GEO, NCBI) under accession number GSE287516, GSE287517 and GSE287785. The publicly available scRNA-seq dataset used in this study can be accessed via the GEO under the accession numbers GSE22859020, GSE15732921, GSE16905526, GSE18145127, GSE18883428, GSE18357297 and GSE18846198. All data generated in this study are provided in the Source Data file. Source data are provided with this paper.
Code availability
Source code for the sc-RCF method is publicly available in the GitHub repository: https://github.com/ZhengBiaoZhu/sc-RCF.
References
Jin, Z. B. et al. Stemming retinal regeneration with pluripotent stem cells. Prog. Retin Eye Res. 69, 38–56 (2019).
Tong, Y. et al. Comparative mechanistic study of RPE cell death induced by different oxidative stresses. Redox Biol. 65, 102840 (2023).
Algvere, P. V., Berglin, L., Gouras, P., Sheng, Y. & Kopp, E. D. Transplantation of RPE in age-related macular degeneration: Observations in disciform lesions and dry RPE atrophy. Graefes Arch. Clin. Exp. Ophthalmol. 235, 149–158 (1997).
Weisz, J. M. et al. Allogenic fetal retinal pigment epithelial cell transplant in a patient with geographic atrophy. Retina 19, 540–545 (1999).
Del Priore, L. V. et al. Retinal pigment epithelial cell transplantation after subfoveal membranectomy in age-related macular degeneration: clinicopathologic correlation. Am. J. Ophthalmol. 131, 472–480 (2001).
Mandai, M., Kurimoto, Y. & Takahashi, M. Autologous induced stem-cell-derived retinal cells for macular degeneration. N. Engl. J. Med 377, 792–793 (2017).
Sharma, R., Bose, D., Maminishkis, A. & Bharti, K. Retinal pigment epithelium replacement therapy for age-related macular degeneration: Are we there yet? Annu Rev. Pharm. Toxicol. 60, 553–572 (2020).
Leach, L. L. et al. Induced pluripotent stem cell-derived retinal pigmented epithelium: A comparative study between cell lines and differentiation methods. J. Ocul. Pharm. Ther. 32, 317–330 (2016).
Araki, R. et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature 494, 100–104 (2013).
Zhang, K. et al. Direct conversion of human fibroblasts into retinal pigment epithelium-like cells by defined factors. Protein Cell 5, 48–58 (2014).
Woogeng, I. N. et al. Inducing human retinal pigment epithelium-like cells from somatic tissue. Stem Cell Rep. 17, 289–306 (2022).
Li, X. et al. Small-molecule-driven direct reprogramming of mouse fibroblasts into functional neurons. Cell Stem Cell 17, 195–203 (2015).
Hu, W. et al. Direct conversion of normal and Alzheimer’s disease human fibroblasts into neuronal cells by small molecules. Cell Stem Cell 17, 204–212 (2015).
Tian, E. et al. Small-molecule-based lineage reprogramming creates functional astrocytes. Cell Rep. 16, 781–792 (2016).
Bai, Y. et al. Direct chemical induction of hepatocyte-like cells with capacity for liver repopulation. Hepatology 77, 1550–1565 (2023).
Pan, S. H., Zhao, N., Feng, X., Jie, Y. & Jin, Z. B. Conversion of mouse embryonic fibroblasts into neural crest cells and functional corneal endothelia by defined small molecules. Sci. Adv. 7, eabg5749 (2021).
Federation, A. J., Bradner, J. E. & Meissner, A. The use of small molecules in somatic-cell reprogramming. Trends Cell Biol. 24, 179–187 (2014).
Farjood, F. et al. Identifying biomarkers of heterogeneity and transplantation efficacy in retinal pigment epithelial cells. J. Exp. Med. 220, e20230913 (2023).
Zheng, M. et al. A single cell-based computational platform to identify chemical compounds targeting desired sets of transcription factors for cellular conversion. Stem Cell Rep. 18, 131–144 (2023).
Qiu, C. et al. A single-cell time-lapse of mouse prenatal development from gastrula to birth. Nature 626, 1084–1093 (2024).
Xu, Y. et al. A single-cell transcriptome atlas profiles early organogenesis in human embryos. Nat. Cell Biol. 25, 604–615 (2023).
Gabriel, E. et al. Human brain organoids assemble functionally integrated bilateral optic vesicles. Cell Stem Cell 28, 1740–1757 e1748 (2021).
Surendran, H. et al. Transplantation of retinal pigment epithelium and photoreceptors generated concomitantly via small molecule-mediated differentiation rescues visual function in rodent models of retinal degeneration. Stem Cell Res Ther. 12, 70 (2021).
Pan, Z. et al. Generation of iPSC-derived human venous endothelial cells for the modeling of vascular malformations and drug discovery. Cell Stem Cell 32, 227–245.e9 (2024).
Li, J. et al. Transcriptome-based chemical screens identify CDK8 as a common barrier in multiple cell reprogramming systems. Cell Rep. 42, 112566 (2023).
Kim, I. et al. Integrative molecular roadmap for direct conversion of fibroblasts into myocytes and myogenic progenitor cells. Sci. Adv. 8, eabj4928 (2022).
Huyghe, A. et al. Comparative roadmaps of reprogramming and oncogenic transformation identify Bcl11b and Atoh8 as broad regulators of cellular plasticity. Nat. Cell Biol. 24, 1350–1363 (2022).
Chen, X. et al. A fast chemical reprogramming system promotes cell identity transition through a diapause-like state. Nat. Cell Biol. 25, 1146–1156 (2023).
Zhang, M. et al. Pharmacological reprogramming of fibroblasts into neural stem cells by signaling-directed transcriptional activation. Cell Stem Cell 18, 653–667 (2016).
Pham, T. X. A. et al. Modeling human extraembryonic mesoderm cells using naive pluripotent stem cells. Cell Stem Cell 29, 1346–1365.e1310 (2022).
Castel, G. et al. Induction of human trophoblast stem cells from somatic cells and pluripotent stem cells. Cell Rep. 33, 108419 (2020).
Amirpour, N. et al. Differentiation of eye field neuroectoderm from human adipose-derived stem cells by using small-molecules and hADSC-conditioned medium. Ann. Anat. 221, 17–26 (2019).
Salehi, H. et al. Application of hanging drop culture for retinal precursor-like cells differentiation of human adipose-derived stem cells using small molecules. J. Mol. Neurosci. 69, 597–607 (2019).
Maruotti, J. et al. Small-molecule-directed, efficient generation of retinal pigment epithelium from human pluripotent stem cells. Proc. Natl. Acad. Sci. USA 112, 10950–10955 (2015).
Hazim, R. A. et al. Differentiation of RPE cells from integration-free iPS cells and their cell biological characterization. Stem Cell Res. Ther. 8, 217 (2017).
Buchholz, D. E. et al. Rapid and efficient directed differentiation of human pluripotent stem cells into retinal pigmented epithelium. Stem Cells Transl. Med 2, 384–393 (2013).
Duester, G. Towards a Better Vision of Retinoic Acid Signaling during Eye Development. Cells 11, 322 (2022).
Idelson, M. et al. Directed differentiation of human embryonic stem cells into functional retinal pigment epithelium cells. Cell Stem Cell 5, 396–408 (2009).
Lepletier, A. et al. Interplay between follistatin, activin A, and BMP4 signaling regulates postnatal thymic epithelial progenitor cell differentiation during aging. Cell Rep. 27, 3887–3901 e3884 (2019).
Lu, S. et al. Generation of a BEST1 Pr-EGFP reporter human embryonic stem cell line via CRISPR/Cas9 editing. Stem Cell Res 82, 103625 (2025).
Wang, S. et al. The retinal pigment epithelium: Functions and roles in ocular diseases. Fundam. Res 4, 1710–1718 (2024).
Krohne, T. U. et al. Generation of retinal pigment epithelial cells from small molecules and OCT4 reprogrammed human induced pluripotent stem cells. Stem Cells Transl. Med. 1, 96–109 (2012).
Ford, K. M., Saint-Geniez, M., Walshe, T., Zahr, A. & D’Amore, P. A. Expression and role of VEGF in the adult retinal pigment epithelium. Invest Ophthalmol. Vis. Sci. 52, 9478–9487 (2011).
Shang, P., Stepicheva, N. A., Hose, S., Zigler, J. S., Jr & Sinha, D. Primary cell cultures from the mouse retinal pigment epithelium. J. Vis. Exp. 133, 56997 (2018).
Wang, C. et al. Reprogramming of H3K9me3-dependent heterochromatin during mammalian embryo development. Nat. Cell Biol. 20, 620–631 (2018).
Wang, H., Yang, Y., Liu, J. & Qian, L. Direct cell reprogramming: Approaches, mechanisms and progress. Nat. Rev. Mol. Cell Biol. 22, 410–424 (2021).
Xu, R. et al. Stage-specific H3K9me3 occupancy ensures retrotransposon silencing in human pre-implantation embryos. Cell Stem Cell 29, 1051–1066 e1058 (2022).
Qin, H., Zhao, A. & Fu, X. Small molecules for reprogramming and transdifferentiation. Cell Mol. Life Sci. 74, 3553–3575 (2017).
Suva, M. L. et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 157, 580–594 (2014).
Chanoumidou, K. et al. One-step Reprogramming of Human Fibroblasts into Oligodendrocyte-like Cells by SOX10, OLIG2, and NKX6.2. Stem Cell Rep. 16, 771–783 (2021).
Imayoshi, I. et al. Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science 342, 1203–1208 (2013).
Vierbuchen, T. et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041 (2010).
Gal, A. et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat. Genet 26, 270–271 (2000).
Martinez-Vacas, A. et al. Systemic taurine treatment affords functional and morphological neuroprotection of photoreceptors and restores retinal pigment epithelium function in RCS rats. Redox Biol. 57, 102506 (2022).
Moy, A. B., Kamath, A., Ternes, S. & Kamath, J. The challenges to advancing induced pluripotent stem cell-dependent cell replacement therapy. Med. Res. Arch. 11, 4784 (2023).
Binder, S., Stanzel, B. V., Krebs, I. & Glittenberg, C. Transplantation of the RPE in AMD. Prog. Retin Eye Res. 26, 516–554 (2007).
Kurzawa-Akanbi, M. et al. Pluripotent stem cell-derived models of retinal disease: Elucidating pathogenesis, evaluating novel treatments, and estimating toxicity. Prog. Retin Eye Res 100, 101248 (2024).
Copp, A. J., Greene, N. D. & Murdoch, J. N. The genetic basis of mammalian neurulation. Nat. Rev. Genet 4, 784–793 (2003).
Hatton, B. A. et al. N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. 66, 8655–8661 (2006).
Leung, C. et al. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 428, 337–341 (2004).
Moon, J. H. et al. Induction of neural stem cell-like cells (NSCLCs) from mouse astrocytes by Bmi1. Biochem Biophys. Res Commun. 371, 267–272 (2008).
Lee, J. H. et al. Single transcription factor conversion of human blood fate to NPCs with CNS and PNS developmental capacity. Cell Rep. 11, 1367–1376 (2015).
Qin, J. et al. Direct chemical reprogramming of human cord blood erythroblasts to induced megakaryocytes that produce platelets. Cell Stem Cell 29, 1229–1245.e1227 (2022).
Cuenca-Zamora, E. J. et al. miR-146a(-/-) mice model reveals that NF-kappaB inhibition reverts inflammation-driven myelofibrosis-like phenotype. Am. J. Hematol. 99, 1326–1337 (2024).
Hahn, A. et al. Serum amyloid A1 mediates myotube atrophy via Toll-like receptors. J. Cachexia Sarcopenia Muscle 11, 103–119 (2020).
Jin, C. et al. The combination of bFGF and CHIR99021 maintains stable self-renewal of mouse adult retinal progenitor cells. Stem Cell Res. Ther. 9, 346 (2018).
Elling, U. et al. Derivation and maintenance of mouse haploid embryonic stem cells. Nat. Protoc. 14, 1991–2014 (2019).
Xu, Y. et al. Derivation of totipotent-like stem cells with blastocyst-like structure forming potential. Cell Res 32, 513–529 (2022).
Han, Y. C. et al. Direct reprogramming of mouse fibroblasts to neural stem cells by small molecules. Stem Cells Int 2016, 4304916 (2016).
Mohamed, T. M. et al. Chemical enhancement of in vitro and in vivo direct cardiac reprogramming. Circulation 135, 978–995 (2017).
Li, W. et al. Generation of rat and human induced pluripotent stem cells by combining genetic reprogramming and chemical inhibitors. Cell Stem Cell 4, 16–19 (2009).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
Hu, H. et al. AnimalTFDB 3.0: A comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 47, D33–D38 (2019).
Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000. Profiles Cell 171, 1437–1452.e1417 (2017).
Corsello, S. M. et al. The drug repurposing hub: A next-generation drug library and information resource. Nat. Med 23, 405–408 (2017).
Szklarczyk, D. et al. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res 44, D380–D384 (2016).
Zhang, Y. Y., Liang, D. M. & Du, P. F. iEssLnc: quantitative estimation of lncRNA gene essentialities with meta-path-guided random walks on the lncRNA-protein interaction network. Brief Bioinform 24, bbad097 (2023).
Wang, J., Sun, S. & Deng, H. Chemical reprogramming for cell fate manipulation: Methods, applications, and perspectives. Cell Stem Cell 30, 1130–1147 (2023).
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 48, D498–D503 (2020).
Du, Y. et al. Human hepatocytes with drug metabolic function induced from fibroblasts by lineage reprogramming. Cell Stem Cell 14, 394–403 (2014).
Xie, B. et al. A two-step lineage reprogramming strategy to generate functionally competent human hepatocytes from fibroblasts. Cell Res 29, 696–710 (2019).
Fernandez-Godino, R., Garland, D. L. & Pierce, E. A. Isolation, culture and characterization of primary mouse RPE cells. Nat. Protoc. 11, 1206–1218 (2016).
Shen, J., He, J. & Wang, F. Isolation and culture of primary mouse retinal pigment epithelial (RPE) Cells with Rho-Kinase and TGFbetaR-1/ALK5 inhibitor. Med Sci. Monit. 23, 6132–6136 (2017).
Tian, H. et al. A cell culture condition that induces the mesenchymal-epithelial transition of dedifferentiated porcine retinal pigment epithelial cells. Exp. Eye Res. 177, 160–172 (2018).
Zhu, X. et al. Direct conversion of human umbilical cord mesenchymal stem cells into retinal pigment epithelial cells for treatment of retinal degeneration. Cell Death Dis. 13, 785 (2022).
Li, S. et al. ESRG is critical to maintain the cell survival and self-renewal/pluripotency of hPSCs by collaborating with MCM2 to suppress p53 pathway. Int J. Biol. Sci. 19, 916–935 (2023).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Kumar, L. & E Futschik, M. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2, 5–7 (2007).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Aibar, S. et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Chen, K. et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat. Genet 47, 1149–1157 (2015).
McGuffin, L. J. et al. Prediction of protein structures, functions and interactions using the IntFOLD7, MultiFOLD and ModFOLDdock servers. Nucleic Acids Res 51, W274–W280 (2023).
Chin, C. H. et al. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8, S11 (2014).
Lee, H. et al. Single-cell transcriptome of the mouse retinal pigment epithelium in response to a low-dose of doxorubicin. Commun. Biol. 5, 722 (2022).
Guan, J. et al. Chemical reprogramming of human somatic cells to pluripotent stem cells. Nature 605, 325–331 (2022).
Acknowledgements
We thank Dr. Shukuan Ling and Dr. Qingran Kong for technical assistance. We also thank Dr. Guoping Fan for kindly providing the RCS rats. This study was funded by the National Natural Science Foundation of China (T2525030 to J.S., 32488101 and 92168205 to S.G., 82101143 to H.L.), Zhejiang Provincial Natural Science Foundation of China (LZ24H120003 to H.L., LQ24C120003 to S.L., LY23H120003 to S.P.), “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2025C02153 to J.S.), the Summit Advancement Disciplines of Zhejiang Province (Wenzhou Medical University-Pharmaceutics) (to J.S.), Space Application System of China Manned Space Program(KJZ-YY-NSM0608 to J.S.) and Postdoctoral Fellowship Program of CPSF (GZC20231952 to S.L.).
Author information
Authors and Affiliations
Contributions
J.S., S.G., J.Q. and S.P. designed and supervised whole study; S.L., H.L., S.Z., J.L., N.G., X.L., M.C., H.H., W.P., Q.Z. and Y.C. performed the experiments; Z.Z., Y.Y., X.W. and J.S. performed computational analysis; J.S., S.G., H.L. and S.L. provided materials and funding support; S.L., H.L., S.Z., J.S., S.G., and S.P. wrote the manuscript; All authors revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Gilbert Bernier, Julia Joung and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, S., Liu, H., Pan, SH. et al. Chemical reprogramming of fibroblasts into retinal pigment epithelium cells for vision restoration. Nat Commun 17, 409 (2026). https://doi.org/10.1038/s41467-025-67104-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67104-w
