
Abstract
Ambient electrochemical CO2 capture powered by renewable energy offers a promising carbon removal route, exemplified by the emerging electrochemically mediated amine regeneration (EMAR) process demonstrated in lab-scale single cells and stacks. However, molecular-level insight into EMAR interfacial kinetics is still missing, particularly at the anode, where CO2 release involves a mechanistically non-trivial re-complexation process at the electrode–electrolyte interface, coupling heterogeneous metal-ion release with bulk complexation. Here, we report the time-resolved characterization of the interfacial molecular processes of the EMAR CO2 release process. Using in situ Fourier-transform infrared (FTIR) spectroscopy and ultraviolet-visible (UV-vis) spectroscopy, cyclic voltammetry, and real-time differential electrochemical mass spectrometry (DEMS), we examine how the nature of the electrolyte anion affects the CO2 release onset potentials. The time-resolved analyses reveal that Cl⁻ ions are more effective in releasing Cu ions and hence CO2 than nitrate or perchlorate. Molecular dynamics simulations show that strong surface Cu–Cl interactions likely facilitate favorable CO2 and carbamate adsorption kinetics. We expect that this study paves the way for broader use of interfacial in-situ analytics in electrified CO2 capture and release.
Similar content being viewed by others

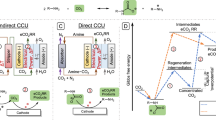

Introduction
Engineering carbon capture is poised to become a key technology to address global climate challenges, particularly targeting the capture and separation of CO2 from industrial point sources and off-gases1,2,3,4. Unlike its conventional temperature and pressure swing analogues, electrochemical CO2 capture, separation, and concentration strategies powered by clean electricity hold the promise to be a cheap, energy efficient, and scalable carbon capture technologies5,6,7. Among them, electrochemically mediated amine regeneration (EMAR) is emerging as an attractive electricity-based variant of more traditional thermal amine-based CO2 capture/release technologies8,9,10. The EMAR desorption process is the electrochemical analogue (electrochemical swing) of the circular thermal approaches (thermal swing), with the reduced-pressure high-temperature desorption stage replaced with a two-compartment electrochemical desorber cell that operates at the same low temperatures as the absorber11.
The circular electrochemical reactions of the EMAR CO2 capture and release cycle start with the adsorptive CO2 capture by an amine (Am) solutions yielding a carbamate (Am-CO2) solution
The KAm-CO2 value refers to the case of ethylenediamine (EDA). The carbamate solution exiting the absorber is fed to the anode compartment of a divided electrochemical EMAR cell for CO2 desorption and sorbent regeneration. At the anode, copper ions are electrochemically generated from a copper plate anode
to drive the fast competitive complexation of Am-CO2 carbamates into free CO2 and a colored Cu-Am complex
Combining the two latter reactions results in the effective anodic EMAR process in which metallic copper (Cu) undergoes a coupled redox/complexation reaction with Am-CO2 to form Cu(Am)n²⁺ complexes12,13,14. The difference in the two equilibrium (i.e., stability) constants, KAm -CO2 and KCu(Am)n, ensures fast and quantitative competitive release of CO2.
The CO2 is then separated from the liquid electrolyte, and the latter is fed to the cathode of the electrochemical EMAR cell where the amine sorbent is regenerated from the Cu(Am)n2+ complex by electrochemical Cu deposition
And the free amine fed back into the adsorber stage to close the EMAR cycle.
Generally, the electrochemical redox behavior of surface Cu species, such as metallic adatoms and Cu ions, during the interfacial CO2 release (Eqs. 2 and 3) will be strongly influenced not only by the applied overpotential and the nature of the Cu surface, but also by the composition and possible adsorption of electrolyte molecules and ions at the electrochemical interface. In other words, given a electrochemical driving force, the nature and concentration of carbamate, free amine, dissolved gases, anions, cations, and solvent molecules will influence the onset potential and kinetics of the local interfacial generation of Cu ions from Cu15,16,17,18,19. For instance, electrolyte ions may form a blocking adlayer on the electrode surface, hindering the Cu ion generating redox reaction20. Alternatively, ions at the electrode surface may enhance the generation of Cu ions through strong ion-ion interactions, thereby shifting the redox reaction and Cu ion onset potential21,22. Indeed, bulk electrolyte composition was reported to modulate the electric energy demand for CO2 release and amine regeneration in the EMAR process23. However, most prior work on EMAR processes has essentially focused on electrolyte and device engineering aspects at the full single EMAR cell level. By contrast, time-resolved exploration and understanding of the elementary interfacial EMAR process steps at the molecular scale, in particular using advanced in-situ and operando analytical techniques, have not been conducted to date. Hence, the detailed molecular reaction kinetics along with the nature and origin of kinetic barriers of individual EMAR steps are poorly understood to date. We intend to fill this knowledge gap, as the electrocatalytic science community is moving into the research area of electrified CO2 capture and concentration. A deeper understanding of the interfacial reaction kinetics will enable the design of structurally and energetically optimized electrochemical EMAR microenvironments that will bring to bear more efficient electrochemical CO2 capture and concentration technologies3,13,24.
Here, we report the discovery of a voltage-efficient Cl⁻-mediated interfacial EMAR CO2 release pathway that enables one of the lowest ever reported onset potentials of CO2 release (Fig. 1)23,25,26. Using time-resolved, online and in situ spectrometry and spectroscopy, we unravel relations between the kinetic onset potentials of interfacial CO2 release, closely linked to the generation of bulk Cu2+ species, and the nature of the electrolyte anions. Combining our experiments with computational modeling, we discuss the trends in CO2 release kinetics, with the interfacial adsorption of electrolyte species. Cu nanoparticles (NP) and smooth polycrystalline Cu surfaces in presence of supporting KCl electrolyte showed significantly less positive Cu redox onset potentials—as low as −0.36 VAg/AgCl – in 0.1 M CO2-saturated- EDA-KCl (KCl + EDA-CO2) bulk electrolytes compared to KNO3 and KClO4 electrolytes. Time-resolved Differential Electrochemical Mass Spectrometry (DEMS) confirmed this earlier onset to coincide with earlier CO2 release. Complementary in situ FTIR and in situ UV-vis were used, respectively, to track the formation of CO2 and the Cu-amine complex species at the interface. Moreover, the voltage and efficiency gains offered by the anion engineering in Cu-based systems were quantitatively evaluated, leading to significantly more energy savings. Molecular dynamics (MD) calculations provided a theoretical insights for our observations and suggested the anion-regulated molecular interactions at the Cu electrode. Our complementary experimental and computational methodological approaches highlight the importance of the local anion microenvironment for the design of EMAR systems, improving our understanding of the interfacial EMAR CO2 release process.

Illustration of the molecular interfacial processes during the electrochemical amine-mediated CO2 release, the anodic partial process of electrochemically mediated amine regeneration (EMAR) systems. Shown is absorption CO2 capture using ethylenediamine (EDA) to carbamate followed by the anodic electrochemical Cu2+-mediated competitive (indirect) CO2 release from the carbamates (EDA-carbamates) generating the strong [Cu(EDA)2]2+ complex. Color coding is Red (O), gray (C), yellow (Cu), blue (N) and white (H). The inset at right illustrates that electrolyte anion adsorption (blue negative ball) affects the CO2 capture and release kinetics by modulating the interacting of carbamate with the Cu electrode surface.
Results
Voltammetry of CO2 release on Cu in H-cell
To elucidate the role of electrolyte anions in the electrochemical CO2 release step of the EMAR process, we started by investigating the cyclic voltammetry (CV) over Cu foil and Cu nanoparticle (NP) electrodes in various 0.1 M KA (A = Cl⁻, NO3⁻, ClO4⁻, Br⁻, and I⁻) supporting electrolytes in a H-cell configuration in absence and presence of either 0.1 M ethylenediamine (EDA) or 0.1 M CO2-saturated EDA carbamate (EDA-CO2). Characteristic CV responses on the Cu electrodes for each anion and for all three electrolyte conditions were shown in Figs. S1 and S2. Anodic Cu/Cu+/2+ redox waves emerged in the order of EDA, EDA-CO2, and pure supporting electrolyte. EDA appeared to interact with the Cu surface very strongly and favored formation of Cu ions27. Fig. 2A and B evidenced that Cu foil and Cu NP in KCl + EDA-CO2 system owned apparently faster onset kinetics among all the supporting electrolytes. Notably, Cu foil and Cu NPs displayed Cu oxidation onset potentials of −0.36 VAg/AgCl and −0.21 VAg/AgCl respectively in 0.1 M KCl + EDA-CO2 electrolyte, which are significantly more cathodic compared to those obtained in KNO3 + EDA-CO2 and KClO4 + EDA-CO2 electrolyte. The faradaic efficiency (FE) for oxidation of the Cu foil to form [Cu(EDA)2]2+ (derived from quantification of the latter) was 90.2 ± 2.0% in 0.1 M KCl + EDA-CO2 electrolyte (Fig. S3, see Method for details). Notably, cyclic voltammetry in Fig. 2A, conducted in 0.1 M KCl with EDA–CO2 electrolyte, showed a distinct oxidation peak at ~0.08 V vs. Ag/AgCl followed by a consistent sharp current drop, indicating that the electrode surface formed via a dissolution–precipitation mechanism, with the diffusing species likely being the CuCl2⁻ complex28,29,30,31, whereas no obvious oxidative peak was observed in KNO3 + EDA-CO2 and KClO4 + EDA-CO2 electrolyte.

CO2 release-cyclic voltammetry for A Cu foil and B Cu nanoparticles in 0.1 M KA (A = Cl⁻, NO3⁻ and ClO4⁻) supporting electrolyte with 0.1 M EDA-CO2 carbamates in H cell. All the CVs were recorded at 5 mV s−1. The average resistance of Cu foil in 0.1 M EDA-CO2 electrolyte containing 0.1 M KCl, KNO3 and KClO4 at 25 °C was 5.4, 5.5 and 5.4 Ω, respectively. For Cu nps, the corresponding values were 6.6, 6.5, and 6.4 Ω. All CV curves were recorded without iR correction. Arrows indicated the onset potentials for Cu oxidation extracted from the voltammograms, and the numerical voltage labels denoted the corresponding onset values (vs. Ag/AgCl) for each supporting electrolyte. The arrow at 0.08 V (vs. Ag/AgCl) showed a distinct oxidation peak uniquely observed in the KCl + EDA–CO2 electrolyte.
The more cathodic Cu oxidation wave in chloride-containing EDA-CO2 suggests a much stronger interaction between Cl⁻ anions and Cu+/2+ 32,33. Indeed, chloride anions are known to readily undergo complexation with copper cations at or near the surface, specifically Cu ions. This Cu-Cl complexation likely acts as a thermodynamic driving force favoring Cu oxidation, thus shifting the Nernstian Cu redox potential, in turn shifting the apparent kinetic onset potential. Once interacting with Cl⁻ anions, Cu2+ ions were generated and subsequently complexed with EDA according to Eq. (3), inducing the release of free CO₂ into the solution. In addition, the anion concentration exhibited only a minimal impact on CO2 release behavior (Fig. S4). As shown in Fig. S5, similar Cl⁻-promoted Cu2+ release and CO2 desorption were also observed using N-methyldiethanolamine (MDEA), a tertiary amine with weaker Cu2+ binding affinity. This suggests that the anion effect could potentially extend to other classes of amines with varying coordination strengths and structures.
Real-time kinetic study of interfacial CO2 release on Cu
To evaluate the precise kinetic onset potential of the EMAR CO2 release, time-resolved detection of evolved product gases at the interface as a function of applied potential was conducted using DEMS, which allowed the simultaneous recording of voltametric responses and product-specific ion current signals34. DEMS measurements were carried out in saturated EDA-CO2 carbamate electrolyte in a custom-made capillary flow cell in presence of three different KA electrolytes (A= Cl⁻, NO3⁻ and ClO4⁻), and the applied electrode potential on a Cu disc electrode was cycled ~ 10 times until a stable CV had been reached. The capillary flow cell showed in Fig. S6 combines a continuous convective electrolyte flow toward the electrified interface with a rapid continuous capillary sampling of products toward the liquid–vacuum interface. This ensured plug-flow conditions and sub-second response times for all volatile electrochemical products. During the anodic sweep, the CO2 evolution onset potential was defined as the potential at which the mass spectrometer ion current signal for the m/z = 44 species (CO2) reached 2% of the peak value observed during CV cycling, a reasonably low detection threshold to consider as the CO2 release onset. The DEMS results (Fig. 3) revealed the detailed potential dependence of CO2 evolution, with interfacial molecular CO2 intensity increasing monotonically during anodic potential sweeps, consistent with anodic CO2 release at the Cu surface according to Eqs. (2) and (3). No competing O2 evolution (Fig. S7) was detected during anodic polarization, confirming a high faradaic efficiency of the CO2 release process. The limited faradaic losses in Fig. S3 may be related to competing processes near the double layer, which likely include the formation of free Cu2+ and Cu–Cl species. While presence of free EDA in the feed would also result in inefficiencies due to [Cu(EDA)2]2+ complex formation, this inefficiency route is likely not dominant under our CO2 saturated carbamate feed conditions. Experimental kinetic onset potential values of CO2 release showed a reproducible trend with the nature of the anion, in particular in the order Cl⁻ < NO3⁻ < ClO4⁻. More specifically, Cu disc electrodes displayed a kinetic CO2 onset potential of −0.23 VAg/AgCl in KCl (Fig. 3D and E), −0.12 VAg/AgCl in KNO3, and −0.10 VAg/AgCl in KClO4. We conclude that our DEMS investigations firstly provided accurate onset potentials for EMAR-based CO2 release. Different supporting electrolytes revealed a significant anion impact at the Cu-EMAR interface, and thus suggesting the presence of Cl⁻ induces earlier CO2 release.

Real-time DEMS analysis links the applied Cu electrode voltametric potential sweeps to the m/z = 44 mass current signal revealing kinetic onsets of interfacial CO2 release in 0.1 M EDA-CO2 (carbamate) electrolyte by continuous cyclic voltammetric scan at 5 mV s−1 in presence of 0.1 M A KCl, B KNO3 and C KClO4. The bottom plot shows the concurrent cyclic voltametric sweep over time. D Voltammetric potential sweeps with the m/z = 44 mass current signal corresponding to the cycle outlined by dashed lines in (A–C). Arrows and numerical labels marked the CO2 release onset potentials versus Ag/AgCl, defined as the anodic potential at which the m/z = 44 ion current reaches 2% of its peak value. The numerical voltages indicated the corresponding onset potential for each supporting electrolyte. E Comparison of the CO2 evolution onset potentials for each cycle in various supporting electrolytes, as detected by an operando DEMS capillary flow cell system. The kinetic onset potential was defined as the potential at which the mass spectrum ion current signal reached 2% of peak maximum for the m/z = 44 species. CVs were recorded by applying potential cycles within a specific range. The potential windows for each electrolyte were selected relative to their respective open-circuit potentials (OCP). The scan begins at the open-circuit potential, where no CO₂ release occurs. Electrolyte flow is 2 µL s−1. F Photo of custom-made capillary flow cell.
Since the applied overpotential for an electrochemical reaction is related to the amount of excess energy needed to drive the reaction, we sought to estimate and compare values for this “excess energy consumption” of the EMAR anode process operating in the different electrolytes. The calculation, using our measured values of CO2 onset potential (Fig. 3D and Table S1) related to the measured open circuit potential (Fig. S8), and the measured faradaic efficiency (Fig. S3), is explained in Supplementary Note 2. By this approach, we estimate that initiating CO2 release in the KCl + EDA-CO2 system required an excess energy consumption of 0.64 kJe/molCO2 to overcome overpotential barriers, which is significantly lower than the energy requirements for the KNO3 + EDA-CO2 (6.4 kJe/molCO2) and KClO4 + EDA-CO2 (10.2 kJe/molCO2) systems, corresponding to energy savings of 5.76 kJe/molCO2 compared to the KNO3 + EDA-CO2 system and 9.56 kJe/molCO2 compared to KClO4 + EDA-CO2 system. These results highlight the importance of optimizing the electrolyte composition to minimize energy consumption in electrochemical CO2 capture and release processes. Comparison of our system with the state-of-the-art in regeneration energy consumption is inherently challenging due to the significant variations in assumptions across the studies reviewed. This complexity is exemplified by a statement regarding the relatively mature Carbon Engineering Direct Air Capture (DAC) and Negative Emissions Technologies (NETs) process: “CE has spent several tens of millions of dollars developing DAC technology, yet our performance and cost estimates still carry substantial uncertainty.”35 Thus, to ensure a focused comparison on the energetics of a specific half-cell electrode process, we consider only the core components of the electrochemical system related to overpotential.
Real-time tracking of interfacial Cu-amine complexation
In situ UV-vis absorption spectroscopy experiments were conducted to track the evolution of electrochemically generated Cu complex species at the surface diffusion layer according to Eq. (3), wherein the generation of colored Cu(Am)n2+ can be detected by absorption spectroscopy (Fig. S9)36. The UV-vis spectroelectrochemical cell as shown in Fig. S10 includes a patterned “honeycomb” electrode mounted in a thin-layer quartz cuvette, with a secure cap ensuring precise alignment of the electrode and reference. UV-vis spectra were collected in 0.1 M KCl + EDA-CO2 electrolyte under potential sweeps from −0.24 to +0.36 VAg/AgCl, which corresponds to 0–0.6 VOCP. At open circuit potentials (OCP) of ~ −0.24 VAg/AgCl, there was no observable absorption in the 400–800 nm wavelength range, confirming no Cu dissolution before EMAR CO2 release reaction. With increasing applied anodic potential from −0.24 to +0.36 VAg/AgCl, absorption at 550 nm gradually increased (Fig. 4A and B), which was attributed to the formation of the colored [Cu(EDA)2]2+ complex by the re-complexation reaction of Eq. (3). As the electrode potential decreased, the 550 nm peak intensity slowly faded due to diffusion (Fig. 4B). To compare anion effects, the measurements were also carried out in KNO3 and KClO4 electrolyte. They displayed similar spectroscopic behavior (Fig. S11). It is noted that after three cycles of EMAR CO2 release reaction, only the voltametric sweep in KCl showed sustained [Cu(EDA)]2+absorbance Fig. S12, given Fig. S11 as a reference, confirming a fast formation of [Cu(EDA)2]2+, and thus CO2 released faster in presence of KCl than in other KA electrolytes.

A In situ UV-vis revealing CO2 release on Cu nanoparticles in 0.1 M KCl scanned from −0.24 to +0.36 VAg/AgCl, B normalized peak area of Cu-amine complex signals obtained from in situ UV-vis in (A). The peak centered at 550 nm can be attributed to [Cu(EDA)2]2+. The bottom plot shows the concurrent cyclic voltammetric sweep over time. Spectra were collected for wavelengths between 400 and 800 nm with a time resolution of 400 ms at a scan rate of 5 mV s-1. C In situ FTIR revealing CO2 release on Cu nanoparticles as a function of applied potential in 0.1 M KCl, D normalized peak area of CO2 release signals on Cu nanoparticles obtained from in situ FTIR in (C). The peak at 2340 cm−1 could be assigned to released CO2. The bottom plot shows the concurrent cyclic voltammetric sweep over time. All the FTIR spectra were obtained in the range of 2500 and 2100 cm−1 with a spectral resolution of 4 cm−1. Each spectrum was collected from an average of 64 scans. The potential was scanned at a scan rate of 5 mV s-1. The background spectrum was taken before applying any potential. Error bars represent the ± s. d. between triplicate measurements.
We further study the EMAR CO2 release kinetics of Eq. (3) using in-situ Fourier Transform Infrared Spectroscopy (FT-IR) in an ATR configuration. Thereby we probed surface reaction intermediates and product over Cu NP electrodes37,38. No free CO2 absorption could be observed at open circuit potential of −0.24 VAg/AgCl. FTIR spectra recorded during anodic voltammetric scans from OCP (−0.24 V to +0.26 VAg/AgCl, corresponding to 0–0.5 VOCP) revealed a distinct vibrational feature at 2340 cm−1 (Fig. 4C, D, and S13), attributable to linear free CO₂, confirming its evolution according to Eq. (3). On the cathodic scan (+0.26 VAg/AgCl to OCP), the intensity of ~2340 cm−1 gradually decreased and eventually disappeared, in accordance with the decreasing reaction rate.
Our experimental in situ studies on the EMAR CO2 release process of Eq. (3) revealed consistency between in situ FTIR, in-situ UV-vis and real-time DEMS data. The above in situ spectroelectrochemical results presented clarified that Cl⁻ effect plays a crucial role in interfacial interactions, influencing the formation of Cu-amine complexation step and CO2 release step25.
Interfacial molecular dynamics simulations
To elucidate the role of anions in modulating the onset potential kinetics of CO2 release, molecular dynamics (MD) simulations on the Cu/electrolytes interfaces were performed for the investigated anions of chloride, nitrate and perchlorate. Our computations could help assess how variations in electrolyte chemistry could affect EMAR CO2 release performance, primarily through modifications in the electrochemical double-layer structure39,40. The systems were constructed following established methodologies in the literature, where the liquid electrolytes involving the associated ions and molecules were placed between two Cu electrodes39,40. Assuming that the local interfacial accumulation of carbamates and CO2 molecules around the active electrode will promote the subsequent CO2 release, we investigated the anion effect through MD simulations, specially qualifying and comparing the distribution behavior of anions and these two molecules. As shown in Fig. 5A–C, at the constant potential difference of 0 V (no potential difference), a considerable accumulation tendency of Cl⁻ ions was observed around the bottom Cu electrode (anode), while contrastingly, nitrate and perchlorate anions distributed more uniformly throughout the electrolyte. This preferential local enrichment of Cl⁻ at the Cu electrode is evident from the density peak (Fig. 5D), suggesting the distinct electrostatic adsorption between anions and the Cu surface. When analyzing the origin of the anion interaction discrepancy, the intrinsic features of anions such as size configurations, ionic conductivity and mobility, the diffusion coefficient may collectively make contributions, leading to the distinct ion distribution40,41. Variations in the specific anion adsorption control the local electric fields near the Cu electrode surface (extent of diffuse double layer for instance), thereby mediating the distribution of the molecular compounds39,40. As evidenced in Fig. 5A–C, the near-surface density of EDA-CO2 carbamate and CO2 molecules in the case of chloride anions was also higher than that in nitrate and perchlorate anions, for which the neutral molecules appeared more dispersed in the electrolyte. This enhanced accumulation of molecules could be intuitively observed from the density peak near the Cu surface, demonstrating the role of Cl⁻ anions in promoting molecular adsorption (Fig. 5E and F). As a result, Cu2+ will be generated more readily, potentially promoting an earlier release of molecular CO2 from carbamate.

Molecular dynamics predictions of atomic configurations for the A Cu-carbamates-Cl interface, B Cu-carbamates-nitrate interface and C Cu-carbamates-perchlorate interface at the fixed potential difference (ΔU) of 0 V. Comparison of density distribution of D anions, E carbamates and F CO2 molecules versus distance from the Cu electrode. ΔU, voltage difference between the bottom anode and top cathode of the Cu(111) surfaces, ΔU = 0 V indicates no voltage difference. The concentrations of molecules and anions were set to approximately 0.1 M to align with the experimental conditions. Color coding is Red (O), gray (C), yellow (Cu), green (Cl), purple (K), blue (N) and white (H).
In addition, the application of an external cell voltage appears to further stimulate the inhomogeneous distribution of ions and molecules at the Cu electrode surface, accelerating the anion-induced Cu ion formation and subsequent CO2 release process. As reflected in Fig. S14, at a constant cell voltage of 0.1 V (the potential of bottom anode is 0.1 V higher than that of the top cathode), more Cl⁻ ions were found to aggregate near the anodic Cu surface. While the near surface concentrations of ClO4⁻ and NO3⁻ rose somewhat, their distribution remained spatially more uniform compared to Cl⁻. The local accumulation of molecular compounds (CO2 and Carbamate) near the Cu anode increased, as well, in line with variations of the anionic near-surface charge and the corresponding local electric field at the Cu surface under non-equilibrium conditions. Strong Cu-Cl interaction with chloride anions could facilitate the oxidation of Cu to Cu2+ ion, and thus enhance the CO2 release dynamics (Fig. S15)28,29,32,33,42. Thus, the choice of the anions at the interfacial microenvironment offers an effective strategy to modulate the CO2 release kinetics.
Discussion
We have investigated the interfacial CO2 release step of the EMAR process using in situ spectroscopy (FTIR, UV-vis) and real time DEMS. The CO2 release step is a complex reaction step coupling interfacial anodic Cu2+ ion release with re-complexation yielding free CO2 molecules (Eqs. 1–3). In our investigations, scientific focus was placed on the impact of the nature of electrolyte anions. We have demonstrated a Cl⁻-mediated electrochemical anodic CO2 release with a significantly lower (less positive) and hence energetically more favorable onset potential compared to other electrolyte anions. The Cl⁻ ions enabled onset potentials as low as −0.36 VAg/AgCl on Cu electrodes. Trends in onset potentials were confirmed for Cu nanoparticles and foils. Poor activity was observed in KI and KBr electrolytes due to the formation of CuA precipitates. In situ FTIR and in situ UV-vis analyses confirmed the formation of CO2 and [Cu(EDA)2]2+, with their concentrations increasing and decreasing in response to the applied potential. UV-vis data enabled the estimation of faradaic efficiencies of over 90% for the EMAR CO2 release process for each anion. Parasitic residual amine Cu corrosion and formation of Cu+ surface species can account for the imperfect faradic efficiencies. Finally, MD calculations suggested anion-regulated preferentially distribution of neutral CO2 and carbamates molecules near the Cu interface in the Cu-Cl system favoring the ready formation of Cu2+ ions associated with lower CO2 onset potentials.
To our knowledge, this is the first in-situ, time-resolved study of the anodic interfacial molecular CO2 release in the EMAR desorption and regeneration process. We expect that this report will spark wider use of interfacial in-situ analytics to provide insight into the kinetic barriers and mechanisms of molecular interfacial processes on solid electrodes during the electrified CO2 capture, release, and concentration. Besides, within the KA + EDA-CO2 system, our findings reveal that initiating CO2 release in the KCl + EDA- CO2 configuration requires an excess energy consumption of 0.64 kJe/molCO2—substantially lower than the 6.4 kJe/molCO2 and 10.2 kJe/molCO2 required by the KNO3 + EDA-CO2 and KClO4 + EDA-CO2 systems, respectively—thereby achieving energy savings of 5.76 kJₑ/molCO2 and 9.56 kJₑ/molCO2.
MD calculations further provided theoretical insights into the anion-regulated preferential distribution of neutral CO2 and carbamates molecules at the electrode interface within the Cu-Cl system, thereby favoring the formation of Cu2+ ions associated with lower CO2 onset potentials. Cl⁻ ions tended to accumulate around the Cu electrode, leading to a preferential distribution of neutral CO2 and carbamates molecules. In contrast, NO3⁻ and ClO4⁻ show inferior affinity to the Cu electrode, inducing the suboptimal interfacial accumulation of neutral molecules. This heterogeneous molecular distribution implies the anion-modulated molecular interactions at the Cu surface, wherein specific ionic solvation dynamically controls the local electrical microenvironment, thereby dictating the spatial arrangement of adsorbates. Our findings on the anion effect offer a facile strategy to lower regeneration energy consumption in NETs and DAC, while our insights into electrolyte compatibility and interfacial engineering enable localized, controllable CO2 release, providing a foundation for integration with downstream CO2 conversion.
Methods
Materials
Potassium chloride (KCl, ≥99%) and potassium nitrate (KNO3, ≥99%) were purchased from VWR. Potassium perchlorate (KClO4, ≥99%) was purchased from Thermoscientific. Copper nanoparticles (nanopowder, 25 nm particle size) and ethylenediamine (≥99%) were purchased from Sigma Co., Ltd.
Preparation of Cu nanoparticle electrode
50 μL of commercial Cu nanoparticle ink (0.125 mg cm-2 loading) was drop-cast onto the polished glassy carbon working electrode. The ink was prepared by dispersing 20 mg of powder in 8 mL of water, 1.5 mL of isopropanol, and 50 μL of a 5 wt% Nafion solution, followed by sonication.
Preparation of Cu foil electrode
Electrodes polishing
Before use, copper foil (99.99%, 1 mm thickness, 1 cm × 2.5 cm) was polished via electropolishing in 85% phosphoric acid by applying a potential of + 1 V for 1 min. Both sides of copper foil were exposed to solution. Copper electrode, Pt mesh and Ag/AgCl was used as the working electrode, counter electrode and reference electrode, respectively.
Characterizations
Electrochemical measurements
The electrochemical tests were carried out in H cell, separated by a Nafion 117 membrane (Ion Power, GmbH). Prior to use, the membrane was sequentially treated with H2O2, H2SO4 and deionized water at 80 °C. The experiments were conducted in freshly prepared aqueous solutions containing 0.1 M ethylenediamine with different supporting electrolytes (KCl, KNO3, KClO4, KI, and KBr). Cu electrodes, platinum mesh, and a leak-free Ag/AgCl electrode were used as working electrodes, counter electrode, and reference electrode, respectively, measured with a Biologic SP 300 potentiostat. The leak-free Ag/AgCl electrode was calibrated versus a Pt/H2 electrode. Before the electrochemical reaction, electrolyte in the working compartment was sparged continuously with CO2 overnight at a flow rate of 10 sccm (standard cubic centimeters per minute) from the bottom of the cell, triggering the formation of Am-CO2 (Eq. 1). The pH of CO2-saturated 0.1 M EDA–CO2 electrolyte containing 0.1 M KCl, KNO3 and KClO4 at 25 °C (7.2 ± 0.02, 7.1 ± 0.02, and 7.2 ± 0.02, respectively) was measured using a calibrated pH meter. All CV curves recorded in the H-cell were presented without iR correction. The resistance was measured using the electrochemical workstation prior to the measurements. For each measurement, fresh electrolyte was used.
In situ FTIR spectroscopy was carried out in a modified attenuated total reflection (ATR)-FTIR setup. The Cu nanoparticles were dispersed in water, and the solution of samples was deposited directly onto the ATR prism, avoiding intensity loss due to the lack of a sputtered layer of a conducting material such as Au. The electrochemical connection was made via a carbon cloth material that provides a reservoir of electrolyte over the samples, minimizing mass transport limitations and polarization effects as shown in Fig. S16. The spectra were collected in a custom-made glass cell with a Bruker Vertex 70 v FTIR spectrometer equipped with a Mercury-Cadmium-Telluride detector cooled with liquid nitrogen. A platinum mesh and a leak-free Ag/AgCl were used as counter electrode and reference electrode, respectively. All in situ electrochemical measurements were controlled using a Metrohm Autolab PGSTAT204 potentiostat. An unpolarized beam was focused with a Pike Veemax II onto the sample spot of the cell. The spectral resolution was set to 4 cm−1 and 100 to 107 interferograms were collected and averaged for each presented spectrum. Here, the reference spectrum was collected in the same electrolyte (0.1 M CO2 saturated EDA with various supporting electrolytes) immediately before the investigated potential scan at the respective start potential. A Si hemisphere was used as the ATR crystal and sample was deposited on the prism on the IR beam ATR focus spot and contacted with Toray Paper 030 carbon cloth and a Pine glassy carbon rod fixating the carbon cloth. The complete beam pathway was under vacuum more than 24 h prior to each measurement. The positions of the bands were determined by taking the band maximum, or, for bipolar bands, by taking the mean value of the maximum and the minimum. The potential was scanned over the specified range at a scan rate of 5 mV s−1. The background spectrum was taken before applying any potential.
In situ UV–vis spectra were collected using a UV/VIS/NIR spectrometer from Avantes (AvaLight-DH-S-BAL) equipped with a deuterium and halogen lamp and a light guide. The spectrometer was connected via fiber optic cables to the electrochemical cell. Transmittance spectra were collected between 400 and 800 nm with a time resolution of 400 ms. The electrochemical cell constituted a 1 cm quartz cuvette, Ag/AgCl reference electrode with a Ø = 1 mm shaft (Pine ltd.). The potential was scanned over the specified range at a scan rate of 5 mV s-1, measured with a Biologic SP 300 potentiostat. The background spectrum was taken before applying any potential.
DEMS was measured in a dual thin layer electrochemical flow cell from Liquidloop ltd., “PoGASi DEMS 3”. DEMS was recorded in 0.1 M CO2 saturated EDA with various supporting electrolytes. The electrochemistry was controlled using a BioLogic potentiostat, a leak-free Ag/AgCl as a reference electrode (Warner Instruments), and a Pt mesh as a counter electrode. The DEMS cell was connected to a mass spectrometer (QMS 200, Pfeiffer Vacuum) via a 50 μm thick microporous PTFE membrane with 20 nm pore size (Cobetter®, Cat. No. PF-002HS). Two turbomolecular pumps (HiPace 80) were operating at 10–6 mbar. Electropolished Cu disc electrodes were used as working electrode, CVs were recorded by potential cycling at a specific range, measured by a Biologic SP 300 potentiostat. The scan limits varied slightly for the different electrolyte pHs to account for differences in resistance and activity. The samples were first preconditioned at a scan rate of 5 mV s-1 for ~10 cycles.
Classical molecular dynamics (MD) simulations
To investigate the distribution of CO2 and CO2-EDA around Cu electrode surface in the presence of different anions (chloride, nitrate, and perchlorate), molecular dynamics (MD) simulations were performed to examine the confined behavior of anions, CO2, and CO2-EDA in aqueous electrolyte system between two Cu electrodes. The simulation system was constructed using PACKMOL43, with a box size of 62.6 Å × 41.0 Å × 70 Å, the atomic positions of the Cu(111) electrodes were frozen during the entire simulation. Periodic boundary conditions (PBC) were applied in the x and y directions, while the z direction was fixed. The system contained 4560 water molecules, 9 EDA-CO2 carbamate and CO2 molecules, 9 anions. K⁺ ions were introduced to ensure charge neutrality, corresponding to an anion concentration of approximately 0.1 M. Short-range interactions between molecules were described by Lennard-Jones (LJ) potentials, while long-range Coulombic interactions were calculated using the particle-particle particle-mesh (PPPM) method, with a precision of 10⁻⁵ and a cutoff radius of 12 Å.
The force field parameters for the system were obtained from multiple references: Cu atoms from ref. 44, Cl⁻ from ref. 41, NO3⁻ from ref. 42, ClO4⁻ from ref. 45, K⁺ from ref. 46, and CO2 from ref. 47. The parameters for CO2-EDA were based on the second-generation general Amber force field (GAFF2)48 and generated using the AuToFF49 tool. Cross-interaction parameters between atoms were calculated using the Lorentz-Berthelot mixing rules50. To eliminate initial energy hotspots, energy minimization was performed before each simulation, followed by a 2 ns preliminary equilibration simulation in the NVE ensemble. This ensured the establishment of a thermodynamically stable interfacial structure for subsequent simulations. In the production simulation phase, the equilibrated system was transferred to a 2D-PBC setting, and electrode potentials were adjusted using the USER-CONP2 package51, which implements the finite field method. Simulations were performed at a fixed potential difference of ΔU = 0.0 V and 0.1 V in the NVT ensemble at 298.15 K for at least 10 ns. Temperature control was achieved using a Nosé-Hoover thermostat, with Hamiltonian dynamics regulated by global velocity rescaling52. The final 1 ns of trajectory data was analyzed to ensure statistical reliability. Data visualization was performed using OVITO software to reveal key features of the interfacial microstructure and molecule affinity behavior53.
Quantitative analysis by UV-vis absorption spectroscopy of [Cu(EDA)2]2+ complex
The [Cu(EDA)2]2+ complex was analyzed by UV-vis spectroscopy using a UV/VIS/NIR spectrometer from Avantes (AvaLight-DH-S-BAL) equipped with a deuterium and halogen lamp and a light guide. The Cu-EDA complex exhibits an absorbance at 550 nm, so the selected wavelength range was 400–800 nm. A blank solution containing 0.1 M EDA and KCl was used for baseline correction by filling a cuvette, inserting it into the spectrophotometer, and setting the absorbance to zero. A calibration curve was prepared using standard solutions with known concentrations (1.64, 1.75, 1.85, 1.93 and 2.00 mM), measuring their absorbance at 550 nm, and plotting absorbance against concentration. The sample absorbance was measured at 550 nm, with samples collected at various potentials after 30 min of reaction. The sample concentration was determined using the calibration curve with the formula: c = A − yintercept/slope. The calibration curve for UV-vis spectrophotometric determination of [Cu(EDA)2]2+ complex was shown in Fig. S17.
Faradaic efficiency, FE, was expressed as the ratio of the actual charge involving Cu redox reaction to its theoretical charge, is calculated by dividing the observed concentration of Cu-EDA complex.
Where nCu are the moles of Cu, determined by measuring the concentration of the Cu-amine complex using UV-vis spectroscopy.
Data availability
The MD simulation structures are provided at https://doi.org/10.5281/zenodo.17501792. Source data are provided with this paper.
References
Sullivan, I. et al. Coupling eectrochemical CO2 conversion with CO2 capture. Nat. Catal. 4, 952–958 (2021).
Eltayeb, A. O., Stern, M. C., Herzog, H. & Hatton, T. A. Energetics of electrochemically-mediated amine regeneration. Enrgy Proced. 63, 595–604 (2014).
Massen-Hane, M., Diederichsen, K. M. & Hatton, T. A. Engineering redox-active electrochemically mediated carbon dioxide capture systems. Nat. Chem. Eng. 1, 35–44 (2024).
Zito, A. M. et al. Electrochemical carbon dioxide capture and concentration. Chem. Rev. 123, 8069–8098 (2023).
Shen, K. Z. et al. On the origin of carbon sources in the electrochemical upgrade of CO2 from carbon capture solutions. Joule 7, 1260–1276 (2023).
Halliday, C. & Hatton, T. A. Sorbents for the capture of CO2 and other acid gases: a review. Ind. Eng. Chem. Res 60, 9313–9346 (2021).
Leung, D. Y. C., Caramanna, G. & Maroto-Valer, M. M. An overview of current status of carbon dioxide capture and storage technologies. Renew. Sustain. Energy Rev. 39, 426–443 (2014).
Stern, M. C., Simeon, F., Herzog, H. & Hatton, T. A. Post-combustion carbon dioxide capture using electrochemically mediated amine regeneration. Energ. Environ. Sci. 6, 2505–2517 (2013).
Kang, J.S., Kim, S. & Hatton, T. A. Redox-responsive sorbents and mediators for electrochemically based CO2 capture. Curr. Opin. Green Sustain. 31, 100504 (2021).
Digdaya, I. A. et al. A direct coupled electrochemical system for capture and conversion of CO2 from oceanwater. Nat. Commun. 11, 4412 (2020).
Wang, M. et al. Flue gas CO2 capture via electrochemically mediated amine regeneration: system design and performance. Appl. Energ 255, 10120–10129 (2019).
Stern, M. C. & Hatton, T. A. Bench-scale demonstration of CO2 capture with electrochemically-mediated amine regeneration. Rsc Adv. 4, 5906–5914 (2014).
Voskian, S. & Hatton, T. A. Faradaic electro-swing reactive adsorption for CO2 capture. Energ. Environ. Sci. 12, 3530–3547 (2019).
Wang, M., Herzog, H. J. & Hatton, T. A. CO2 capture using electrochemically mediated amine regeneration. Ind. Eng. Chem. Res. 59, 7087–7096 (2020).
Aleta, P., Refaie, A., Afshari, M., Hassan, A. & Rahimi, M. Direct ocean capture: the emergence of electrochemical processes for oceanic carbon removal. Energ. Environ. Sci. 16, 4944–4967 (2023).
Hassan, A. et al. Reviving the absorbent chemistry of electrochemically mediated amine regeneration for improved point source carbon capture. Chem. Eng. J. 484, 149566 (2024).
Rahimi, M. et al. Carbon dioxide capture using an electrochemically driven proton concentration process. Cell Rep. Phys. Sci. 1, 100033 (2020).
Rahimi, M., Catalini, G., Puccini, M. & Hatton, T. A. Bench-scale demonstration of CO2 capture with an electrochemically driven proton concentration process. Rsc Adv. 10, 16832–16843 (2020).
Bruggeman, D. F., Rothenberg, G. & Garcia, A. C. Investigating proton shuttling and electrochemical mechanisms of amines in integrated CO2 capture and utilization. Nat. Commun. 15, 9207 (2024).
Angelucci, C. A., Souza-Garcia, J. & Feliu, J. M. The role of adsorbates in electrocatalytic systems: an analysis of model systems with single crystals. Curr. Opin. Electrochem. 26, 100666 (2021).
Wu, F. et al. Identification of K+-determined reaction pathway for facilitated kinetics of CO2 electroreduction. Nat. Commun. 15, 6972 (2024).
Zhan, C. et al. Key intermediates and Cu active sites for CO2 electroreduction to ethylene and ethanol. Nat. Energy 9, 1485–1496 (2024).
Wang, M., Hariharan, S., Shaw, R. A. & Hatton, T. A. Energetics of electrochemically mediated amine regeneration process for flue gas CO2 capture. Int J. Greenh. Gas. Control 82, 48–58 (2019).
Renfrew, S. E., Starr, D. E. & Strasser, P. Electrochemical approaches toward CO2 capture and concentration. Acs Catal. 10, 13058–13074 (2020).
Rahimi, M. et al. An electrochemically mediated amine regeneration process with a mixed absorbent for postcombustion CO2 capture. Environ. Sci. Technol. 54, 8999–9007 (2020).
Hassan, A., Afshari, M. & Rahimi, M. A membraneless electrochemically mediated amine regeneration for carbon capture. Nat. Commun. 16, 6333 (2025).
Guan, Y. et al. Origin of copper dissolution under electrocatalytic reduction conditions involving amines. Chem. Sci. 15, 14485–14496 (2024).
Rana, M. S., Rahman, M. A. & Alam, A. M. S. A CV study of copper complexation with guanine using glassy carbon electrode in aqueous medium. Int. Sch. Res. Not. 2014, 308382 (2014).
Butcher, D. P. & Gewirth, A. A. Nitrate reduction pathways on Cu single crystal surfaces: effect of oxide and Cl−. Nano Energy 29, 457–465 (2016).
Tilak, B. V., Perkins, R. S., Kozlowska, H. A. & Conway, B. E. Impedance and formation characteristics of electrolytically generated silver oxides—I formation and reduction of surface oxides and the role of dissolution processes. Electrochim. Acta 17, 1447–1469 (1972).
Crousier, J., Pardessus, L. & Crousier, J. P. Voltammetry study of copper in chloride solution. Electrochim. Acta 33, 1039–1042 (1988).
Fan, H. et al. Performance comparation of MEA and EDA in electrochemically-mediated amine regeneration for CO2 capture. Sep. Purif. Technol. 311, 123282 (2023).
Bartonícek, R. & Lukasovská, M. A potential pH diagram for the system Cu-NH3-Cl−-H2O. Corros. Sci. 9, 35–42 (1969).
Wang, X. et al. Mechanistic reaction pathways of enhanced ethylene yields during electroreduction of CO2–CO co-feeds on Cu and Cu-tandem electrocatalysts. Nat. Nanotechnol. 14, 1063–1070 (2019).
Keith, D. W., Holmes, G., Angelo, D. S. & Heidel, K. A process for capturing CO2 from the atmosphere. Joule 2, 1573–1594 (2018).
Zhang, D., Wang, R., Wang, X. & Gogotsi, Y. In situ monitoring redox processes in energy storage using UV–Vis spectroscopy. Nat. Energy 8, 567–576 (2023).
Schott, J. A. et al. FTIR investigation of the interfacial properties and mechanisms of CO2 sorption in porous ionic liquids. Green. Chem. Eng. 2, 392–401 (2021).
Liang, L. et al. Electroreduction of CO2 on Au(310)@Cu high-index facets. Angew. Chem. Int Ed. 62, e202218039 (2023).
Fan, M. et al. Cationic-group-functionalized electrocatalysts enable stable acidic CO2 electrolysis. Nat. Catal. 6, 763–772 (2023).
Yoo, J. M., Ingenmey, J., Salanne, M. & Lukatskaya, M. R. Anion effect in electrochemical CO2 reduction: from spectators to orchestrators. J. Am. Chem. Soc. 146, 31768–31777 (2024).
Zhong, C. et al. A Review Of Electrolyte Materials And Compositions For Electrochemical Supercapacitors. Chem. Soc. Rev. 44, 7484–7539 (2015).
Anagnostopoulos, A., Alexiadis, A. & Ding, Y. Molecular dynamics simulation of solar salt (NaNO3-KNO3) Mixtures. Sol. Energy Mater. Sol. Cells 200, 109897 (2019).
Martínez, L., Andrade, R., Birgin, E. G. & Martínez, J. M. PACKMOL: a package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157–2164 (2009).
Heinz, H., Vaia, R. A., Farmer, B. L. & Naik, R. R. Accurate simulation of surfaces and interfaces of face-centered cubic metals using 12−6 and 9−6 lennard-jones potentials. J. Phys. Chem. C. 112, 17281–17290 (2008).
Doherty, B., Zhong, X., Gathiaka, S., Li, B. & Acevedo, O. Revisiting OPLS force field parameters for ionic liquid simulations. J. Chem. Theory Comput. 13, 6131–6145 (2017).
Jensen, K. P. & Jorgensen, W. L. Halide, ammonium, and alkali metal ion parameters for modeling aqueous solutions. J. Chem. Theory Comput. 2, 1499–1509 (2006).
Sun, C., Wen, B. & Bai, B. Application of nanoporous graphene membranes in natural gas processing: molecular simulations of CH4/CO2, CH4/H2S and CH4/N2 separation. Chem. Eng. Sci. 138, 616–621 (2015).
He, X., Man, V. H., Yang, W., Lee, T. S. & Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 153, 114502 (2020).
Wang, C., Liao, K., Wang, Z., Wang, Y. & Gong, K. AuToFF Program. Version 1.0. (Hzwtech, Shanghai, China, 2023).
Lennard-Jones, J. E. Cohesion. Proc. Phys. Soc. 43, 461 (1931).
Tee, S. R. & Searles, D. J. Fully periodic, computationally efficient constant potential molecular dynamics simulations of ionic liquid supercapacitors. J. Chem. Phys. 156, 184101 (2022).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the open visualization tool. Model. Simul. Mater. Sci. Eng. 18, 015012 (2010).
Acknowledgements
This work was financially supported by the Sol2H2 project of European Union’s Horizon 2020 research and innovation program under grant No. 101006701, EcoFuel, the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, grant No. STR 596/18-1 and LI 4184/2-1), Natural Science Foundation of Anhui Province (2308085MB47), Anhui Provincial Department of Education university natural science research project (2024AH050070), and the Initiative and Networking Fund of the Helmholtz Association (grant agreement No. KA2-HSC-12, ‘A Comprehensive Approach to Harnessing the Innovation Potential of Direct Air Capture and Storage for Reaching CO2-Neutrality’, DACStorE). The authors thank Hefei Advanced Computing Center and the High-Performance Computing Platform of Anhui University for computational support.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
P.S. and L.L. conceived the idea and co-wrote the paper; L.L. carried out the electrode preparation, characterization, and electrochemical evaluation; F.F. conducted the DEMS measurements and analyzed data; L.Y. and J.W. carried out the MD simulation and analyzed the results; L.Y. supervised the MD simulation; X.W. and W.J. participated in the discussion of the electrochemical results; M.M. participated in the discussion of reaction mechanism; all authors contributed to the discussions and assisted during manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Fengwang Li and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liang, L., Firschke, F., Wang, J. et al. Anion effects govern efficiency of electrochemical amine-mediated CO2 capture/release. Nat Commun 17, 489 (2026). https://doi.org/10.1038/s41467-025-67177-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67177-7
