
Abstract
Viral infection of living cells, exemplified by bacteriophage interaction with bacteria, is fundamental to biology and universal across living systems. Here, we establish an all-cell-free viral cycle where T7 phages infect synthetic cells, equipped with lipopolysaccharides on the outer leaflet of the lipid membrane, while encapsulating a cell-free gene expression system. We track each cycle step to demonstrate T7 phage-specific adsorption onto the liposomes, genome entry, replication, expression, and assembly of new infectious virions within the synthetic cells. We quantify key characteristics of the cycle, including the multiplicity of infection, replication efficiency, liposome size constraints, and phage rebinding dynamics. This work establishes a versatile, fully defined in vitro platform for reconstructing and investigating viral infections from individual molecular components.
Similar content being viewed by others

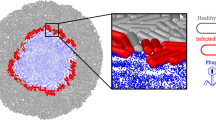
Introduction
Building biological systems from their molecular constituents enables isolating, quantifying, and characterizing biochemical mechanisms often entangled and thus difficult to probe in vivo. It is now possible to reconstitute complex biological processes in cell-free systems across multiple scales1,2,3. Cell-free gene expression (CFE) offers a testbed for validating and enriching our understanding of life’s fundamental principles and constructing synthetic life systems4,5. Among these challenges, establishing a phage cycle is particularly relevant due to the critical role of viral processes in biology. As Arthur Kornberg noted fifty years ago, “Despite the extraordinary complexity of the viral life cycle, its dissection and eventual reconstruction with chemically defined components in a cell-free system from start to finish remains an attractive prospect”6. Strikingly, despite our extensive knowledge of phage evolution7,8,9, ecology10,11,12, and molecular mechanisms13,14,15, a complete cell-free phage infection cycle based on a synthetic cell system has not been achieved, underscoring both conceptual and technical frontiers.
Previous studies have provided isolated elements toward this goal. First, phage φX174 genome ejection was achieved in vitro in the presence of a soluble receptor16. Later, the synthesis of infectious φX174 phages from purified components was demonstrated outside cells17, and its genome was synthetically assembled18. Further, phage T5 genome ejection into liposomes displaying the FhuA receptor19,20,21 showed that phages can be interfaced with single lipid bilayers via their receptor. Phage T7 infects Escherichia coli and is among the most characterized phages22,23. Every step of its infection cycle has been studied24, making it a relevant phage for assembling a synthetic, cell-free viral cycle. T7 is particularly adequate as a model system due to its obligate lytic cycle, minimal reliance on host factors25, and ease of genome engineering26,27. Previous studies have demonstrated the synthesis of infectious T7 phages in CFE systems28 and genome ejection in the presence of purified LPS (lipopolysaccharide)29. However, in contrast to phage T5, T7 genome translocation into liposomes via its LPS receptor has yet to be achieved. Furthermore, no study has integrated all the steps of a phage infection cycle into a single, self-contained framework.
Here, we set out to build a cell-free phage infection cycle (Fig. 1A) composed of the phage T7 and synthetic cells (SCs) (Fig. 1B). Despite the remarkable simplification compared to the in vivo framework, this synthetic platform reconstitutes all the steps of a phage cycle.

A The goal is to recapitulate key infection steps: phage adsorption, DNA entry, genome expression and replication, virion assembly, and SC lysis, releasing progeny phages into the external solution. B Schematic representation of RdLPS SC composition. The SCs consist of liposomes made from phospholipids (POPC and PE-PEG5000) and RdLPS. The RdLPS are exposed on the outer leaflet to enable phage T7 binding and subsequent infection. A cell-free expression (CFE, yellow) system devoid of DNA, messenger RNA, and phage DNA and proteins is encapsulated into the liposome to allow for phage genome expression upon DNA entry, initiating replication and virion production. The SCs are made in a buffered solution (blue).
Results
Phage T7 binds specifically to SCs displaying rough LPS
We hypothesized that cell-sized single-bilayer liposomes presenting rough LPS on the outer leaflet and encapsulating a CFE system would enable phage T7 binding, genome entry, genome expression, and subsequent synthesis of phage particles (Fig. 1A). This previously untested hypothesis assumes that rough LPS embedded within a single lipid membrane is a sufficient T7 receptor for genome entry into the SCs. However, creating CFE SCs incorporating rough LPS is challenging. LPS have high molecular weights and poor solubility compatibility with phospholipids, and they must remain stable in the membrane without compromising CFE inside the SCs30,31. Moreover, LPS must be incorporated into the outer leaflet while minimizing their presence in the inner leaflet to prevent unintended phage inactivation inside the SCs. Accordingly, we opted for RdLPS (LPS of the Rd chemotype containing only the inner core; Fig. 1A, a short rough LPS type (Supplementary Fig. 1). To enable interaction with this minimal receptor, we used a cognate T7 phage tail fiber mutant that can bind RdLPS in vitro, unlike wild-type T7 (T7 WT), which binds only to RaLPS (containing both inner and outer core; Supplementary Fig. 1). We assumed that our choice of RdLPS, one of the smallest LPS variants of E. coli, would enable stable integration into SCs, due to its greater hydrophobicity compared to larger LPS targets32,33.
Cell-sized liposomes loaded with a CFE reaction were prepared by emulsion transfer34,35 of a ternary lipid mixture comprised of phosphatidylcholine (POPC), phosphatidylethanolamine polyethylene glycol (PE-PEG), and RdLPS (Fig. 2A, Supplementary Fig. 2). The presence of PE-PEG was critical for preventing aggregation and facilitating the formation of stable liposomes. We explored the membrane composition while maintaining a total lipid concentration of 100 µM (Fig. 2B, Supplementary Fig. 3). A 55/15/30 mol% PC/PE-PEG/LPS mixture was chosen as it yielded a homogeneous and stable population of RdLPS-liposomes with diameters ranging from 1 µm to 50 µm. Notably, this ratio is consistent with reported LPS molar ratios in the E. coli outer membrane36.

A Size and volume distribution of RdLPS SCs generated in this study. The SCs exhibit polydispersity, with an average radius of 3.7 µm. Liposomes smaller than 20 µm in radius account for 65% of the total encapsulated CFE reactions. B Ternary phase diagram of SC lipid composition. The green region indicates the lipid mixtures that successfully form SCs at a total lipid concentration of 100 µM. Liposomes do not form in the absence of RdLPS and POPC, while liposomes lacking PE-PEG tend to aggregate. C Selective detection of RdLPS-containing liposomes. Two populations of liposomes, with and without RdLPS, were prepared separately and subsequently mixed in an outer solution containing GFP-TF*, a fluorescent RdLPS-binding probe derived from the tip of the tail fiber of a T7 phage mutant. Microscopy images confirm that GFP-TF* selectively binds to RdLPS liposomes. Scale bar: 20 µm. D A linear DNA circuit encoding a mcherry reporter gene under a T7 promoter and a transcriptional activation cascade (P70a-T7rnap) is mixed with the CFE reaction and encapsulated into RdLPS SCs. Microscopy images show red fluorescence inside the liposomes, confirming active CFE, while GFP-TF* fluorescence at the membrane confirms the presence of functional RdLPS on the SC surface. Scale bar: 20 µm. E Adsorption of fluorescent T7-Split-S* phages onto RdLPS SCs. Top: Schematic of the T7-Split-S* system, in which the gfp11 gene fragment is fused to the N-terminus of the T7 capsid gene (gene gp10B, supplementary text). The complementary gfp1-10 gene fragment is expressed downstream of gene 10B-gfp11 under a ribosome binding site, allowing fluorescence upon phage production. Bottom: Microscopy image of RdLPS SCs incubated with T7-Split-S* in the outer solution. Green fluorescence localized at the membrane indicates the specific binding of T7-Split-S* to RdLPS-rich SCs. Liposome image is split between fluorescence (left) and phase contrast (right). Scale bar: 20 µm. Experimental details are presented in methods and Supplementary Data 1. Source data are provided with this paper.
Incorporation of RdLPS into the outer leaflet of the lipid membrane and its expected function were assayed using a chimeric protein (GFP-TF*) consisting of superfolder GFP fused to the N-terminus of the C-terminal ‘tip’ domain of the T7 tail fiber mutant (TF*), which includes the S541R mutation responsible for RdLPS recognition (Supplementary Fig. 4)37. Two liposome populations (PC/PE-PEG and PC/PE-PEG/RdLPS) encapsulating CFE reactions were prepared separately. A red fluorescent dextran marker was added to the CFE reaction encapsulated within the RdLPS liposomes. Upon addition of GFP-TF* to a mixture of both populations, the fluorescence localized exclusively to RdLPS-containing membranes, confirming specific binding and stable LPS incorporation (Fig. 2C, Supplementary Figs. 5-8). We confirmed the asymmetrical localization of the RdLPS on the outer leaflet of the lipid membrane (Supplementary Fig.s 9-11, Supplementary Text). The presence of RdLPS in the membrane did not inhibit CFE gene expression activity inside the liposomes (Fig. 2D, Supplementary Figs. 12-13).
To further validate this system, the T7-Split-S* phage was engineered from T7 WT using PHEIGES27 to encode RdLPS-specific tail fibers and to tag capsid proteins fluorescently (Fig. 2E, Supplementary Fig. 14, Supplementary Text). A T7-Split-S* titer of 1011 PFU/mL added to the outer solution of the RdLPS SCs resulted in a clear fluorescent phage localization at the membrane. At an initial MOI (multiplicity of infection) of 1, where binding sites are in excess, over 90% of phages bind within the first 5 min, suggesting efficient initial adsorption (Supplementary Fig. 14)38. These findings confirmed RdLPS-specific phage adsorption onto RdLPS SCs.
T7 genome is internalized, expressed, and replicated into SCs
Bacteriophages eject their genomes into bacterial hosts through diverse mechanisms that have been explored theoretically and experimentally39,40,41. The genome translocation process, partially driven by internal capsid pressure, has been characterized in vivo for several bacteriophages42,43. For instance, phage T5 can eject its genome into single-bilayer liposomes displaying the FhuA protein receptor via the tip of the long non-contractile tail that crosses the lipid membrane19. In contrast, T7’s short tail prevents T7 from crossing the cell membranes. Inner core proteins are ejected and assemble a trans-membrane channel to translocate the genome into the cell44. Additionally, T7 requires its RNA polymerase to fully translocate its genome45. In a buffer solution, T7 spontaneously releases its genome upon incubation with purified rough LPS due to conformational changes in the binding region29. To track both genome internalization and subsequent gene expression inside the SCs, we engineered two T7 phage variants, either the WT or RdLPS-specific tail fibers (T7-mC-WT, T7-mC-S*), each carrying a mcherry reporter gene downstream of the major capsid gene under the control of a T7 promoter (Fig. 3A). We hypothesized that, as observed in bacterial infections, genome entry into SCs would lead to the expression of the T7 RNA polymerase, thereby initiating a cascade of mcherry gene expression. As designed, the addition of T7-mC-S* phages to the external solution of the RdLPS SCs, encapsulating a DNA-free CFE reaction, resulted in a red fluorescence signal accumulation inside the liposomes (Fig. 3B, Supplementary Figs. 15-16, Supplementary Movie 1), indicating successful genome translocation and the expression of T7 genes. The addition of GFP-TF* to the infected RdLPS SCs revealed that while RdLPS were homogeneously distributed across liposomes, not all liposomes were infected due to SCs leak, disruption or inactive CFE (Fig. 3C). Control experiments using T7-mC-WT, T7-WT, and T7-S* did not yield fluorescence above background levels, confirming that genome internalization was RdLPS-dependent (Fig. 3D, Supplementary Fig. 17).

A Schematic representation of the T7-mC-S* phage genome entry and subsequent gene expression within RdLPS SCs. The T7-mC-S* phage carries a mcherry reporter gene inserted downstream of the major capsid protein gene (gp10) under a T7 promoter. Following adsorption onto RdLPS SCs, the phage ejects its genome across the lipid bilayer into the inner CFE reaction, where transcription and translation occur. The encapsulated CFE reaction is initially devoid of phage DNA, RNA, or proteins. B Time-lapse fluorescence microscopy images of RdLPS SCs exposed to T7-mC-S* phages (MOI = 300) in the outer solution at 30 °C. The emergence of red fluorescence over time indicates successful phage genome entry, transcription, and translation of mCherry inside the SCs. Scale bar: 40 µm. C. 100x magnification fluorescence microscopy image of an RdLPS SC infected by T7-mC-S*. GFP-TF* (green) marks the RdLPS membrane, while mCherry (red) indicates phage genome expression. Scale bar: 10 µm. D Quantification of mCherry fluorescence intensity in RdLPS SCs incubated with different phage variants: T7-mC-WT, T7-mC-S*, T7 WT, and T7-WT-S*. T7-mC-WT and T7-WT cannot bind RdLPS, while T7-WT-S* lacks the mcherry reporter gene. The results confirm that T7-mC-S* specifically infects RdLPS SCs and drives mcherry expression. Data represent measurements from N = 30 individual liposomes per condition, derived from three independent biological replicate experiments. Data are presented as mean values ±SD. E Representative fluorescence microscopy images showing RdLPS SCs infection at different MOIs. Images were taken after 15 h of incubation at 30 °C. Dextran-FITC (green) was used as a volume marker to facilitate liposome quantification. Scale bar: 25 µm. F Proportion of infected SCs at different MOIs, calculated from four replicate experiments. Data are presented as mean values ±SD. G Scatter plot of mCherry concentration as a function of liposome radius. Red points represent liposomes infected by T7-mC-S* (N = 36 liposomes), while blue points correspond to liposomes expressing mcherry from an encapsulated genetic circuit (P70a-T7rnap and T7-mcherry at 0.1 nM and 1 nM, N = 62 liposomes). Linear fits indicate that mCherry concentration remains constant in SCs with pre-encapsulated circuits but decreases with increasing liposome radius upon T7-mC-S* infection. Data correspond to four independent biological replicates. H Detection of phage genome translocation using SYBR Gold, a DNA-intercalating dye. Left: Schematic of an RdLPS SC encapsulating CFE and SYBR Gold. Right: Microscopy images of SCs containing CFE and SYBR Gold incubated with (MOI 100) or without T7-S* phages. The green fluorescence of SYBR Gold indicates the presence of phage DNA inside SCs. Bottom: Time-lapse images of the same liposome at 4, 6.5, and 16 h after incubation with T7-mC-S*, attesting genome entry (green, SYBR Gold fluorescence) followed by mcherry expression (red). Scale bar: 20 µm. I Bar graph showing the evolution of green fluorescence intensity over time in SCs encapsulating SYBR Gold with (green) or without (gray) T7-mC-S* phages (MOI 100) in the outer solution. Data represent N = 23 individual liposomes per condition, obtained from three independent biological replicate experiments. Data are presented as mean values ±SD. J Detection of phage genome replication using FITC-dCTP, a fluorescent nucleotide incorporated into newly synthesized DNA. Left: Schematic of an RdLPS SC encapsulating CFE and FITC-dCTP. Right: Microscopy images of SCs containing CFE and FITC-dCTP incubated with (MOI 100) or without T7-S* phages. Green fluorescence indicates newly synthesized phage DNA. Bottom: Time-lapse images of the same liposome over 6, 10, and 16 h after incubation with T7-mC-S*, showing FITC-dCTP fluorescence (green) and subsequent mcherry expression (red). Scale bar: 20 µm. K Quantification of FITC-dCTP fluorescence clusters inside RdLPS SCs over time in four replicate experiments. Cluster counts represent the total number of observed replication foci per frame across replicates. Experimental details are presented in methods and Supplementary Data 1. Source data are provided with this paper.
The efficacy of infection depended on the multiplicity of infection (MOI), defined as the ratio of plaque-forming units (PFU) to SCs (Fig. 3E, Supplementary Figs. 18-19, Supplementary Movie 2). At an MOI of approximately 3, fewer than 2% of SCs exhibited fluorescence. In contrast, nearly 70% of SCs were infected at an MOI of 300 (Fig. 3F). Quantitative analysis showed that, at constant phage concentrations in the outer solution, smaller SCs (<10 µm in diameter) exhibited greater mCherry concentration than larger ones (>15 µm in diameter) (Fig. 3G). We performed an analysis of mCherry concentration in SCs as a function of SCs’radii across four MOI values (3000, 300, 30, and 3). We observed the same trend, with smaller MOI favoring smaller liposomes while large MOI saturates the liposomes with phage genomes (Supplementary Fig. 20). This suggests that this effect is due to the phage concentration and not to noise associated with the cell-free reaction encapsulation method. This suggests also that the efficacy of genome translocation depends on the surface-to-volume ratio, where smaller SCs, despite fewer genome internalized, maintain a greater genomic concentration relative to their volume. We visualized genome entry into the SCs using SYBR Gold DNA intercalating fluorescent dye that does not inhibit CFE (Fig. 3H, Supplementary Figs. 21-24, Supplementary Movie 3). Upon the addition of T7-mC-S* (MOI ≈ 100) in the outer solution, an increase in fluorescence intensity inside the SCs was observed along with the formation of fluorescent clusters (Fig. 3I, Supplementary Fig. 24), indicating genome entry. Over time, these clusters dissipated as mCherry fluorescence increased, suggesting genome replication and subsequent packaging46,47. The observed cluster formation and disappearance likely reflect concatemer formation and processing during phage packaging. In addition, T7 genome replication was visualized with a fluorescent nucleotide encapsulated within the SCs to further understand the cluster dynamics. Consistent with the DNA intercalating agent results, green fluorescent clusters formed over time, followed by mcherry expression (Fig. 3J, Supplementary Figs. 25-28, Supplementary Movie 4). Cluster formation and subsequent dissipation were quantified, indicating genome replication and phage packaging, leading to the transition from heterogeneous to homogeneous fluorescence in SCs (Fig. 3K, Supplementary Figs. 27-29). We controlled for proper transcription in presence of the fluorescent nucleotide and discarded potential photobleaching (Supplementary Fig. 23).
Phage T7 infection and synthesis can be kinetically tracked in SCs
To estimate the genome entry time into the SCs, we used the engineered phages T7-mC-S* and T7-GFP-S*, in which the egfp gene replaces the mcherry gene at the same position in the genome (Supplementary Data 1). We compared the onset of fluorescence for both phages separately, when the genome was either encapsulated into the SCs or when phages were added in the outer solution. This assay provides two independent measures of the genome entry time, estimated to be 100 min and 105 min, respectively (Fig. 4A, Supplementary Fig. 30). This gives a genome entry time between one and two hours. To determine the genome packaging time and synthesis of infectious phage particles, we monitored the kinetics of PFUs per nanoliters at 30 °C in bulk and SCs. We chose nanoliter unit as it better reflects the small total volume of encapsulated CFE reaction into the SCs. The first synthesized infectious phages were observed after one hour. This experiment was also done at 24 °C and 37 °C (Supplementary Fig. 31). T7 packaging depends on temperature, the greater the temperature the faster the packaging. After one hour between 102 and 103 PFU/nL were obtained at 30 °C and 37 °C while no PFU were detected at 24 °C. Interestingly, similar titers are obtained between bulk and SCs packaging at 30 °C. After 5 h of incubation, we found titers in this range with 10 ± 2 × 104 PFU/nL, 7 ± 3 × 104 PFU/nL, 2 ± 1 × 104 PFU/nL in bulk CFE at 24 °C, 30 °C and 37 °C and 9 ± 3 × 104 PFU/nL in SCs at 30 °C. If all the genomes (0.1 nM) initially added to the CFE reaction become a PFU in absence of DNA replication, we expect 6 × 104 PFU/nL. We obtain slightly higher titer values than expected suggesting a DNA replication factor of at least 1.5 in SCs at 30 °C. It is a lower estimation considering that it is difficult to evaluate genome integrity while only viable phage particles are counted with the agar overlay method. Overall, we concluded that phage packaging time starts within the first hour. After one hour, 1–2% of the final titer is attained at 30 °C in bulk and in SCs and reaches >50% of final titer after two hours of incubation, while continuing to increase until five hours of incubation. We hence chose a characteristic packaging time of 1–2 h is our SC model at 30 °C.

A Gene expression times at 50% (t₅₀%) for encapsulated genomes and phage infection. Table showing the characteristic gene expression time (t₅₀%, min) for synthetic cells encapsulating T7-mC-S* or T7-GFP-S* genomes (Encapsulated DNA) compared to synthetic cells infected by the corresponding phages added externally (Phage outer feeding). The difference in t₅₀% reflects the additional delay associated with genome translocation during phage infection. Both reporters show a consistent ~1.5 h delay, while absolute values are shorter for GFP than for mCherry due to faster GFP maturation. B Phage production kinetics at 30 °C in bulk and synthetic cells. T7 phage titers (PFU/nL) were measured over time in bulk cell-free reactions (blue) and RdLPS liposomes (green). Both systems initiated phage production within 1 h and reached ~10⁵ PFU/nL after 5 h. Final titers slightly exceeded the ~6 × 10⁴ PFU/nL expected from input genomes alone, indicating a ~ 1.5-fold replication factor, estimated by comparing observed titers to the theoretical maximum in the absence of DNA replication. Data correspond to three independent biological replicates. Data are presented as mean values ±SD. C Bulk vs. liposome-encapsulated T7 phage assembly. T7 phage DNA is mixed into the CFE reaction and either incubated in bulk (test tube) or encapsulated within an RdLPS SCs. After incubation, the bulk reaction is diluted, and the liposomes are lysed. PFU quantification is performed using a plaque assay. D Comparison of T7 WT phage assembly efficiency in bulk vs. liposome-encapsulated CFE reactions. Phage assembly efficiency is evaluated at three different initial DNA concentrations. The total CFE volume in SCs is estimated from the liposome size distribution (Methods). Data were collected from four biological replicates for the liposome condition and three biological replicates for the bulk reactions. Data are presented as mean values ±SD. E Impact of RdLPS on T7 WT and T7-mC-S phage assembly. Genomes are mixed at 0.01 nM in the CFE reaction. PFU per liposome is estimated based on microscopy liposome counts and total PFU quantified via plating. Comparison is made between liposomes with and without RdLPS. Data correspond to three independent biological replicates per condition. Data are presented as mean values ±SD. F Time-lapse microscopy images of RdLPS SCs infected with T7-Split-S*. Green fluorescence indicates the production and localization of the fluorescent capsid protein (gp10b-GFP11 + GFP1-10). Fluorescent capsids initially cluster near the site of genome replication and later disperse throughout the SCs as packaging into new phage particles progresses. Scale: 25 µm; time in hours. G Productive phage replication requires active synthetic cells. RdLPS SCs encapsulating either active cell-free expression mix (‘active’ SCs, green) or CFE supplemented with RNase A (“dead” SCs, gray) were incubated with T7-mC-S* phages (MOI 100). After adsorption, washing, and further incubation, PFU were quantified following SC lysis. Active SCs produced ~81-fold more phages per vesicle compared to dead SCs, confirming that productive replication and assembly depend on active transcription–translation inside SCs. Data correspond to three independent biological replicates per condition. Data are presented as mean values ±SD. H Holin-induced membrane leakage in RdLPS versus LPS-less liposomes. RdLPS or LPS-less liposomes were loaded with red rhodamine–dextran dye (3 kDa) and a gene circuit expressing holin from different bacteriophages (T7, phi11, ICP3, and PRD1). Upon holin expression at 30 °C, pores formed in LPS-less membranes, resulting in dye release, whereas RdLPS membranes largely retained the dye, indicating protection against leakage. Leakage ratios were quantified in >50 liposomes per condition in biological duplicate, showing the distribution of individual vesicle responses. Box plots display the median (center line), the 25th and 75th percentiles (bounds of the box), and whiskers extending to the most extreme data points within 1.5 × the interquartile range from the box limits. Experimental details are presented in methods. Source data are provided with this paper.
To further investigate phage assembly within the SCs, we first encapsulated the T7 WT genome at various concentrations into RdLPS-coated SCs and compared, after incubation and lysis by osmotic shock, the phage titers to those obtained from batch 10-µL CFE reactions (Fig. 4C, D, Supplementary Figs. 32-33). Infectious phages were detected in both batch and SCs across all tested genome concentrations. At the lowest DNA concentration (10 pM), phage synthesis was more prevalent inside the SCs than in batch mode, likely due to favorable colocalization of gene expression machinery and phage components under confinement as compared to batch reactions. However, at higher concentrations, phage titers were comparable between the two systems, with slightly lower titers observed in SCs (Fig. 4D). Next, to assess the effect of RdLPS incorporation on phage titer, we encapsulated T7-WT or T7-mC-S* genomes (10 pM) inside SCs with or without RdLPS into the membrane (the RdLPS facing toward the outer solution). After incubation and lysis by osmotic shock, the PFU titers per SC were estimated. While similar PFU per SC were obtained for liposomes lacking RdLPS, a 94 ± 4% reduction of infectious RdLPS-susceptible T7-mC-S* phages was observed in RdLPS-containing SCs (Fig. 4E). These findings confirm that infectious phage particles are assembled inside LPS SCs. However, phage titers are reduced for RdLPS-susceptible phages, likely due to inactivation by irreversible phage binding to RdLPS.
To track phage assembly inside RdLPS SCs, we incubated SCs with T7-Split-S* at an MOI of 100. At this MOI, no detectable fluorescence was observed at the membrane during adsorption. However, once the T7-Split-S* genome entered the SCs, the capsid protein fused to GFP11, and the complementary GFP1-10 were concurrently expressed to form fluorescent capsid proteins as a result of split-GFP complementation. We observed large fluorescent clusters, consistent with capsids self-assembly and binding to the genomic concatemer replicons to package the genomes. Further, this approach enabled visualizing genome concatemer packaging maturation into fluorescent phage capsids. Initially, green fluorescence clusters appeared inside the liposomes suggesting DNA entry and replication (Supplementary Movie 5), as fluorescent capsids localized near DNA concatemers (Fig. 4F, Supplementary Figs. 34-35). Over time, the clusters dispersed into smaller fluorescence foci, suggesting concatemer processing into progeny phage particles.
Phage Release via Osmotic Shock
At the final step of the lytic cycle, phages induce lysis of the host once progeny phages are assembled. In bacterial hosts, lysis is first mediated by a lysis cassette, which is typically expressed late in infection and consists of endolysins (which degrade the cell wall), holins (which form pores into the inner membrane), and spanins (which fuse the inner and outer membranes)48,49. This process results in spheroplast formation that spontaneously lyse due to osmotic shock50. Unlike bacteria, which experience lysis in a hypotonic environment, our SCs are incubated in isotonic conditions, making the phage lysis cassette insufficient, by construction, to induce liposome rupture. Indeed, no significant lysis was observed when phage genomes were expressed inside the RdLPS SCs. To determine the concentration of phages synthesized into the SCs when phages are added to the outer solution, we applied an osmotic shock to an active and a dead SC populations. The dead SC population was created by adding Rnase A into the encapsulated CFE reaction, which ensures that no phage is synthesized. We observed 80 times more phages per liposome in the active compared to dead SCs.
We investigated LPS-carrying SCs as a model for the bacterial outer membrane, and SCs devoid of LPS as a model for the bacterial inner membrane. CFE of the T7 holin into PC/PE-PEG SCs led to significant membrane disruption, as evidenced by leakage of a co-encapsulated dextran dye (Fig. 4H). However, in PC/PE-PEG/RdLPS SCs, no dye leakage was observed, indicating that RdLPS inhibits holin-mediated membrane disruption (Fig. 4H, Supplementary Movie 6). Similar results were obtained with holins from other bacteriophages, supporting that LPS prevents holin activity in this system, which is consistent with its natural function in forming pores in the LPS-less inner membrane of bacteria50. To mimic natural phage release conditions, a simple osmotic shock can induce liposome disruption and phage release (Fig. 4C, D, G), replicating the final step of the lytic cycle.
Discussion
For half a century, achieving a cell-free SC phage infection cycle seemed an ambitious undertaking, considering the complexity of the infection mechanism and molecular architecture. Yet, using a systematic and quantitative approach, we demonstrated that phage T7 can effectively infect CFE-loaded single-bilayer SCs and produce progeny. Most tailed phages initiate infection by reversibly binding to a primary receptor, often a surface glycan like LPS, followed by irreversible attachment to a secondary receptor, typically a core LPS sugar or an outer membrane protein, which stimulates genome entry. This study demonstrates that RdLPS alone is a sufficient membrane component for T7-S* genome internalization into SCs, providing insights into the minimal requirements for phage infection. A distinction between T7 and T5 lies in their structural differences and mechanisms of genome translocation. T5 utilizes FhuA as a docking site but does not use it as a direct genome channel. Instead, its long non-contractile tail crosses the bilayer at the periphery of FhuA, facilitating DNA internalization. In contrast, T7 possesses a short non-contractile tail, preventing it from penetrating the bacterial membrane. Unlike previous liposome studies with T5, which lacked cytoplasmic CFE activity19, our system incorporates a CFE reaction inside the liposomes, thereby enabling all the steps of a phage infection process in an SC system. While our current system is optimized for T7, which uses only RdLPS as its receptor, the strategy could be extended to phages like T4 that require both LPS and specific outer membrane protein receptors. To accommodate such phages, the protein receptor could be incorporated into the outer membrane either by direct CFE within the liposomes, allowing spontaneous membrane insertion, or via reported secretion systems capable of inserting membrane proteins51. The T7-based platform reported here is a foundational step toward a more generalizable in vitro system for studying diverse phage infections.
Achieving a self-amplifying SC phage cycle with a greater burst size would require optimization of several critical steps, including DNA translocation efficacy, genome replication, minimization of phage rebinding and engineering of the phage genome to express an enzyme capable of lyzing the LPS membrane of the SCs. With such improvements, this system could be expanded to achieve multiple infection cycles and evolutionary experiments. While external intervention for phage release may limit selection on the lysis cassette, potentially leading to its deletion, such a cycle would offer a unique advantage by allowing experimental control over phage population dynamics, including the suppression of fast replicating ‘cheaters.’ This setup could enable targeted selection for traits at the membrane such as tail fiber mutations, or in the cytoplasm such as defense systems, facilitating directed evolution of host specificity.
This work demonstrates the extent to which biological systems can function far from their natural conditions and in far simpler molecular settings. Unlike bacteria, the LPS-coated SCs lack a second lipid bilayer and a cell wall, which in natural conditions provides mechanical resistance to osmotic stress. E. coli maintains an internal osmolarity of approximately 300 mOsm greater than its surrounding broth, allowing the difference in osmotic pressure to burst the cells and release phage progeny. In contrast, in the cell-free phage cycle presented in this work, T7 infection proceeds but bursting does not occur spontaneously because of the isotonic conditions (around 600 mOsm inside and outside the SCs). Lysis is achieved by transferring the SCs from isosmotic to hypoosmotic milieus (Fig. 4C, D, G). We found that while LPS enables T7 DNA internalization, it inhibits holin pore formation preventing lysis and release. The efficacy of the phage replication cycle inside SCs is limited by several factors. The large multiplicity of infection (MOI around 100) constrains the number of productive infections, while DNA replication in CFE achieves 1–2-fold amplification28 compared to the 50–300-fold observed in bacterial hosts due to finite resources in CFE. Newly synthesized phages often rebind to inactive SCs via persistent LPS receptors, reducing the effective phage titer and hindering further propagation.
This work offers a tractable bottom-up approach to studying viral infection in a controlled environment, decoupling phage biology from the complexities of living bacteria. This approach allows the screening of individual infection variables, free from the complex effects of bacterial metabolism, growth, and division. It also provides a modular platform for investigating intracellular phage-host interactions, including phage defense mechanisms. As a proof of concept, we showed that T7 infection can be aborted by encapsulating CRISPR-Cas and guide RNAs targeting the phage genome within SCs (Supplementary Fig. 36), illustrating the potential of this system for studying antiviral strategies.
Large integrative projects meant for building complex biological systems from scratch are taking shape around the globe. Building SCs from the ground up has become major research in the USA, in Europe and in Asia5,52,53,54. The bottom-up construction of synthetic viral cycles has been recently discussed. The phage cycle built in this work captures these two emerging trends observed in cell-free constructive biology. As bioengineering capabilities are rapidly developing, safety concerns arise too. Biocontainment solutions must be anticipated and imagined, considering that bottom-up SCs are currently not designed to be infectable by phages. In that respect, the cell-free phage infection mechanism devised in this work delivers fundamental information for developing one type of biocontainment solution to control SC systems.
Methods
Synthetic cell experiments
Lipopolysaccharides from Escherichia coli F583 (Rd mutant) (L6893-5MG, Sigma), 16:0-18:1 PC (POPC) (850457, Avanti Polar Lipids), and 16:0 PEG5000 PE (880200, Avanti Polar Lipids) were used. Additional materials included 4 mL glass vials with Teflon-lined caps (600460, Avanti), Posi-Click tubes from Thomas Scientific (1138W14), liquid paraffin (128-04375, Wako), Optical-Quality Imaging Plates (384-well, black-walled, clear bottom, A58941, Thermo Scientific), dNTP set (100 mM, 10297018, Thermo Fisher), SYBR Gold Nucleic Acid Gel Stain (S11494, Thermo Fisher), Fluorescein-12-dCTP (NU- 809-FAMX-S, Jena Bioscience), Tetramethylrhodamine-Dextran (3000 MW, Anionic, Lysine Fixable, Thermo Fisher), and Fluorescein isothiocyanate-dextran (46945-100MG-F, Sigma).
Phage spotting and lysate purification
0.22 μm centrifuge filter tubes were from (8160, Costar), v-bottom 96-well plates (701201, NEST), and square petri dishes (688102, Greiner Bio-One). Difco agar (214010, BD), and LB broth (BP1426-500, Fisher). 100 kDa membrane Amicon Ultra-15 centrifugal filter units (UFC910024, Millipore). Centrifuge tubes, culture tubes, and 25 mL reservoirs were generic.
Phage DNA extraction
DNase I recombinant RNAse-free (M0303S, NEB), RNase A (T3018L, NEB), Proteinase K (P8111S, NEB). SDS (20% wt/vol, AM9820, Sigma), Phenol:Chloroform Alcohol (25:24:1, 15593049, Sigma), 3 M sodium acetate solution (pH 5.2, AM9740, Sigma). Absolute ethanol and chloroform were generic, as available.
Assembly and PCR
The T7 genome was obtained from Boca Scientific (310025). KOD1 polymerase was sourced from Diagnocin (TYB-KMM-101), and a PCR clean-up kit from Invitrogen (K310001). PCR tubes (8 strips) were generic, as available. 10× TAE buffer was purchased from Invitrogen (AM9869), and a 1 kb+ DNA ladder from Invitrogen (10-787- 018) and Greenglo (NC1620342, Thomas Scientific). For assembly, exonuclease III (M0206S, NEB), PEG8000 from Sigma (P2139), and Tris-HCl 1 M (15568-025, Invitrogen). All buffers were prepared freshly, pH adjusted, autoclaved and filter sterilized. Large bore low binding tips were used when handling phage genomic DNA and liposomes.
Engineering and phenotyping of phages T7-mC-S* and T7-Split-S* (Supplementary Fig. 37)
Genome assembly was performed following the PHEIGES workflow method1. Preparation of the 2X assembly buffer should be done in advance, aliquoted into 50 μL tubes, and stored at −20 °C. 2X Assembly Buffer: 20 mM Tris-HCl (pH 7.9), 100 mM NaCl, 20 mM MgCl2, 10% (w/v) PEG 8000, 2 mM Dithiothreitol (DTT) and 1 U/μL Exonuclease III. Genome Assembly: 2X assembly buffer, DNA fragments, and CFE reaction are thawed on ice. Equal volumes of DNA fragments (1–5 nM) are mixed to create an equimolar DNA mix. T7-mC-S* and T7-Split-S* DNA mixes were prepared following assembly maps in Supplementary Data 1 at 1 nM for each fragment. 2.5 μL of DNA mix is mixed to 2.5 μL of 2X assembly buffer in a 1.5 mL tube on ice. The mixture is homogenized, spin down and incubated 1 min at room temperature. The tube is then transferred on a heat block prewarmed at 75 °C for 1 min, then at 25 °C for 5 min. 7.5 μL of 75% CFE reaction is added to 2.5 μL of the assembly mix. The CFE reaction is gently mix, spined down and incubate at 29 °C for at least 4 h. CFE reactions containing the synthetized T7-mC-S* or T7-Split-S* were then spotted on an E. coli rfaC lawn (RdLPS sensitive tail fiber mutants). Isolated plaques are picked and mixed to a liquid culture of E. coli rfaC at OD = 0.05. 200 µL of this culture containing the phage is incubated at 37 C in a shaking plate reader (Agilent Biotek H1m or Neo2) to follow optical density, GFP and mCherry fluorescence over time. The obtained signals from the plate reader (Agilent Biotek H1m or Neo2) were acquired with the Gen5 software. The data presented shows OD, red and green fluorescence from four wells containing a T7-mC-S* isolated plaque, a T7-Split-S* isolated plaque, only the LB medium and the E. coli rfaC culture without phage. The plate reader (Agilent Biotek H1m or Neo2) was incubated at 37 C, with orbital shaking and signals were recorded every 5 minutes for 6 hours.
Phage stock preparation for SCs experiments
Engineered phage lysates are amplified in 2–5 liter of E. coli rfaC as the host until lysis. Phage lysates are treated with 0.5% chloroform to clear remaining bacteria. The lysates are then filter-sterilized through 0.22 μm filters. Lysates are then washed and purified using 100 kDa ultrafiltration columns in phage buffer (10–mM K-glutamate, 1 mM Mg-glutamate, 10 mM Tris-HCl). Phage lysates are washed until no more fluorescence is detected in the pass-through solution (generally 3–5 10-mL wash). Phage lysates are recovered in 0.5 mL−1 mL, phage titers are recorded (typically reaching 1010−1011 PFU/mL) and the phage stock stored at 4 °C (phages stocks are used for SCs experiments within 2–3 weeks).
Phage DNA purification and sequencing
Phage DNA Purification T7 phage genomic DNA was extracted using the phenol-chloroform method2. Briefly, 10 mL of pre-treated lysate (1011 PFU/mL) and 10 mL of phenol-chloroform-isoamyl alcohol (PCI) to a separatory funnel. Invert gently, release pressure via the stopcock, and allow layers to separate (20–30 min). Remove the phenol phase (bottom layer) and interphase. Repeat PCI extraction twice more, then perform a final wash with pure chloroform. Recover the aqueous phase, add 1 mL sodium acetate, and 20 mL pre-chilled ethanol. Incubate at −80 °C for 1 h, centrifuge at 4000 x g for 20 min, and wash the DNA pellet with 70% ethanol. Resuspend the DNA pellet in 100 μL deionized water (typically 20 nM of T7 phage is obtained). Measure DNA concentration by Nanodrop. The tree phage genomes generated in this study, sequenced using long-read nanopore sequencing, have been deposited in the European Nucleotide Archive (ENA) under the BioProject PRJEB104191. The genome assemblies correspond to the three engineered variants T7-mC-S*, T7-GFP-S*, and T7-Split-S*, and are available under the accession codes OZ370878, OZ370879, and OZ370880, respectively.
Emulsion transfer method for Synthesis of RdLPS SCs (Supplementary Fig. 2)
The lipid composition to incorporate RdLPS in liposomes is composed of POPC (760.076 g/mol), PEG-PE 5000 (5745.03 g/mol) and RdLPS (considered here 2700 g/mol55). The final concentration in the oil should be in the range: POPC: 25 µM–125 µM, PEG-PE: 5 µM–35 µM RdLPS: 20 µM – 80 µM. In this work, the composition used was POPC/RdLPS/PEGPE 55/25/20 (molar ratio) at a total lipid concentration of 100 µM. This corresponds to 55 µM of POPC (42 µg/mL), 15 µM PEG-PE (85 µg/mL) and 30 µM of RdLPS (80 µg/mL).
Lipid stock preparation
Lipids (POPC, PEGPE, RdLPS) were dissolved in chloroform at appropriate concentrations (5 mg/mL for POPC and RdLPS; 10 mg/mL for PEGPE) and stored in airtight glass vials with Teflon-lined caps at −20 °C. Lipid stocks are stable for up to one month at −20 °C in the dark.
Lipid-in-Oil Mixture Preparation
In a 7 mL glass vial, lipids are mixed in 200 μL hexadecane. Typically, 10 μL of POPC, 20 μL RdLPS and 12 μL PEG-PE were added and vortexed for 15 s. 1 mL long-chain mineral oil (Wako liquid paraffin) is added, and the mixture is heated for 1–2 h at 70 °C, vortexed briefly to ensure homogeneity, and used directly.
CFE reaction Emulsification
In a microcentrifuge tube, 200 μL of the lipid-in-oil mixture was combined with 10 μL of the CFE reaction containing the desired components. The solution was emulsified by vortexing or rubbing on a rack, producing CFE droplets encapsulated in lipid micelles. The emulsion was transferred onto a new tube containing 250 μL of cationic solution and centrifuged at 5000 x g for 10 min to form liposomes.
RdLPS SCs Transfer and Phage Infection
Transfer of LPS liposomes: After centrifugation, the oil phase is removed. 10 µL of liposome pellet is transferred in a new 1.5 ml tube containing 90 µL of outer solution. Phage Addition and Imaging: Prepare 100 μL outer solution to which phage is added. Typically, 107 −108 PFU/mL were added. In a 384-well plate, 90 μL of outer solution with phages was combined with 10 μL of liposomes. The plate was centrifuged at 600 x g for 5 min, followed by fluorescence microscopy imaging. Acquisition was done with an inverted microscope IX81 (Olympus) equipped with a camera and proper sets of filters. A 20x air objective or 40x and 100x oil objectives were used for observation. Image analysis was conducted with ImageJ and custom-made programs on Python.
Buffer compositions
Cationic solution: 270 mM K-glutamate, 25.5 mM Mg-glutamate, 45 mM Tris-HCl, autoclaved and adjusted to pH 8 and filter sterilized. Outer solution: 30 mM Maltodextrin, 90 mM K-glutamate, 4 mM Mg-glutamate, 1.5% PEG 8000, 4 mM amino acids, and an energy mix identical to the CFE reaction. The buffer is adjusted to pH 8 and filter sterilized.
CFE reaction and buffers
The CFE reactions used in this work were based on the CFE toolbox 2.056. The E. coli strain BL21-∆recBCD Rosetta2, with a recBCD knockout to prevent the degradation of linear DNA57, was used for lysate preparation. Briefly, Cells were grown in 2xYT medium supplemented with phosphates, pelleted, washed, and lysed using a cell press. The lysate was centrifuged, and the supernatant was incubated at 37 °C for 80 minutes. After a second centrifugation, the supernatant was dialyzed at 4 °C for 3 h. Following a final centrifugation step, the lysate was aliquoted and stored at −80 °C. The volume of the CFE reactions consisted of 33% cell extract, and 67% of the other components (energy mix, and amino acids). The reaction buffer included 50 mM Hepes (pH 8), 1.5 mM ATP and GTP, 0.9 mM CTP and UTP, 0.26 mM coenzyme A, 0.33 mM NAD, 0.75 mM cAMP, 0.068 mM folinic acid, 1 mM spermidine, 30 mM 3-phosphoglyceric acid (3-PGA), 1 mM dithiothreitol (DTT), 1.5% PEG8000, and 20-40 mM maltodextrin. Amino acids were added at concentrations between 1.5 mM and 6 mM for each of the 20 amino acids (Supplementary Data 1). Magnesium (2–5 mM) and potassium (50-100 mM) concentrations were calibrated for deGFP synthesis and T7 phage production to obtain reproducible batches. Reactions were incubated at 30 °C in either 1.5 mL tubes, 96-well plates, or encapsulated within liposomes in 384 well-plates. Fluorescence data from CFE reactions (Agilent Biotek H1m or Neo2) were treated with Microsoft PowerPoint or Kaleidagrah.
Determination of PFU per nL (PFU/nL)
For bulk cell-free reactions, PFU/nL titers were determined from 10 µL reactions. For liposome experiments, individual liposomes’ radii were measured under the microscope, and assuming spherical geometry, the volume of each liposome was calculated. The total encapsulated CFE reaction volume was then calculated by summing the volumes of all liposomes in the well. PFU counts were divided by this total volume, yielding PFU per nanoliter.
Microscopy data analysis
Microscopy images were acquired using an Olympus IX81 inverted microscope controlled by Metamorph software. Liposomes were imaged in 384-well plates, mounted on a custom-made microplate stage. Timelapse acquisitions across multiple wells were performed simultaneously. Temperature was maintained at 30 °C by adjusting the microscope room temperature. Bright-field, green, and red fluorescence channels were recorded using phase contrast, GFP, and Texas Red filter sets. Image processing including segmentation and particle analysis was performed with ImageJ. exported data were plotted using Python scripts.
Figure 2A: Fluorescent images of RdLPS liposome populations encapsulating a dye were acquired at 20x magnification. Liposome segmentation and particle analysis were performed with ImageJ. Radius and volume measurements were used to plot size and volume distributions. The dataset includes 37,663 liposomes analyzed from three replicates. The mean liposome radius was 3.7 μm, with liposomes smaller than 20 μm in radius accounting for over 65% of the total encapsulated CFE reaction volume.
Figure 3D: RdLPS synthetic cells were infected by T7-mC-WT, T7-WT, T7-S* and T7-mC-S* phages, and their red fluorescence intensity was recorded after 13 hours of incubation with a microscope at a 20x magnification. For each condition and each replicate, the maximum fluorescence intensity inside each liposome characterized is measured. For the four conditions, a total of N = 28, 27, 28 and 31 liposomes respectively were characterized from three replicate.
Figure 3F: RdLPS synthetic cells encapsulating FITC-dextran were incubated with a serial dilution of T7-mC-S*. Liposome count was estimated by segmenting the green fluorescence channel using consistent thresholding across all datasets. Infected liposome count was determined by segmenting the red fluorescence channel after 15 h of incubation, using the same calibrated thresholding from a negative control (no phage). The multiplicity of infection (MOI) was calculated as the ratio of PFU added per well to the total liposome count. The infected liposome ratio was determined by dividing the red-channel liposome count by the total green-channel liposome count. Ratios were calculated from four replicate.
Figure 3G: RdLPS SCs either infected by T7-mC-S* phages or encapsulating mcherry expression gene cascade were incubated over 15 hours. Red fluorescence intensity was recorded with a microscope at 20x magnification. Using the procedure described in Supplementary text, the radius and concentration of in-situ synthesized mCherry were measured for N = 62 (mcherry gene encapsulation) and N = 36 (T7-mC-S* infection) liposomes. Liposomes were randomly selected from three replicate experiments in each condition. The linear fit slopes, intercept and R2 are (-0.76, 21.3, 0.35) for T7-mC-S* infection and (0.23, 22.1, 0.02) for mcherry gene encapsulation.
Figure 3I: RdLPS SCs either infected by T7-mC-S* phages or left uninfected had their green fluorescence intensity recorded for 15 hours every 15 minutes with a microscope at a 20x magnification. For each condition and each replicate, the timelapse images were cropped around isolated liposomes so that only one liposome would be present in each cropping series. The maximum intensity of each cropped image was then taken as the measurement of the green fluorescence intensity inside the liposome. For the two conditions, a total of N = 27 and 17 liposomes from three replicates respectively were characterized.
Figure 3K: Cluster count over time was analyzed using the green fluorescence channel. To isolate clusters, images from the timelapse green channel were processed by subtracting duplicates blurred time series (with different Gaussian sigma factors), effectively removing non-cluster fluorescence. The resulting cluster images were converted into masks, and clusters were counted over time using particle analysis. Identical parameters were applied to all conditions. Data represent clusters from 4 replicates. No clusters were detected in the absence of phage. Data were exported as CSV files and plotted using Python.
Figure 4A: To determine the typical time scale of DNA entry in liposomes, we recorded fluorescence intensity of SCs either encapsulating phage DNA or in presence of phage in the outer solution. For each of these two conditions, we considered two replicates observed simultaneously and obtained from the same initial liposome creation batches. Images were acquired every 15 minutes. For each replicate, the average fluorescence intensity of the 0.1% brightest pixels of each image was recorded and combined into the average over both replicates (master curve), then plotted as a function of time. A sigmoidal fit was then used to evaluate the initial and final plateau values of the master curve. The typical times of gene expression were evaluated from the time point where the master curve reaches halfway between the initial and final plateaus.
Sigmoid: \(f:x\rightarrow\frac{{I}_{i}-{I}_{f}}{1+{{{\rm{e}}}}^{k(t-{t}{*})}}+{I}_{f}\) where \({I}_{i}\) and \({I}_{f}\) are respectively the initial and final plateau values of the master curve. We considered experimental points and not the sigmoid of best fit to evaluate \({t}_{50\%}\), which was found close to \({t}{*}\).
Figures 4B, D, E, G: In these experiments, liposomes encapsulated rhodamine-dextran (red fluorescence dye) and the T7 genome. Liposome count and radius were obtained through red-channel image segmentation, and liposome volume was calculated assuming spherical geometry. The total encapsulated CFE volume was estimated as the sum of all liposome volumes in a well. PFU production in liposomes was determined by liposome lysis and plating on an E. coli B lawn. PFU per nanoliter was estimated by dividing the recovered PFU by the total estimated liposome volume. Data represents triplicates for 4B, E, G and 4 replicates for 4D.
Figure 4H: A red fluorescent dextran dye and a phage holin gene circuit were co-encapsulated in liposomes with or without RdLPS. Red fluorescence intensity was recorded with a microscope at 20x magnification every 5 minutes for three hours. Leak ratios were obtained by superposing initial (average of the first 10 images colorized in red) and final (average of the last ten images artificially colorized green) images. Using the superpositions the liposomes were sorted in two groups red liposomes (leaked liposomes) and orange liposomes (no leak). After subtracting the fluorescence intensity of the background, the complement of the ratio of those differences was used as the leak ratio of each liposome. A leak ratio of \(1\) indicates that the entirety of the red fluorescent dextran dye leaked out of the liposome, while a leak ratio of \(0\) indicates that no dye has leaked from the liposome.
\({{\rm{Leak\; ratio}}}=1-\frac{{I}_{f}-{{BG}}_{f}}{{I}_{i}-{{BG}}_{i}}\) where \({I}_{i}\) and \({I}_{f}\) are the maximum fluorescence intensities inside the liposomes measured respectively right after creation and at the end of the incubation, while \({{BG}}_{i}\) and \({{BG}}_{f}\) and the corresponding background fluorescence intensities. Over 50 liposomes were individually assessed from two replicate experiments.
Supplementary Fig. 35: Kinetic plots were generated in Python. mCherry fluorescence (T7-mC-S* infection) and Sybr Gold fluorescence data (from Fig. 3I and Supplementary Fig. 24) were analyzed, along with cluster formation data from Fig. 3K. Sigmoidal fits were applied to normalized mean Sybr Gold and mCherry fluorescence, while a Gaussian fit was used for normalized dCTP-FITC cluster counts. Min-max normalization was applied, and individual data points were included in Supplementary Fig. 35.
Statistics and reproducibility
For each experiment at least three independent replicates were made, except for experiments 4 A and 4H (duplicate liposome batches). Plaque uncertainties were estimated at the time of counting (duplication or plaque belonging to the same spot). The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment. For liposome analysis, liposomes were selected to be entirely contained within the imaging frame and have a well-defined outline that does not overlap with other liposomes. Liposomes were selected on a channel containing the volume marker and not the signal of interest. The micrographs shown in Figs. 2, 3 and 4 are representative images from one experiment. Each experiment was independently repeated three times with similar results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data (graphs, microscopy, movies, DNA sequences) generated and used in this study are available at the University of Minnesota Digital Conservancy (https://conservancy.umn.edu/) with the permanent URL: https://doi.org/10.13020/hmz3-yf4358, and under public available license, and in the supplementary file Supplementary Data 1. Source data are provided with this paper. The tree phage genomes generated in this study have been deposited in the European Nucleotide Archive (ENA) under the BioProject PRJEB104191. The genome of the three engineered phages T7-mC-S*, T7-GFP-S*, and T7-Split-S* are available under the accession codes OZ370878, OZ370879, OZ370880 respectively. Associated BioSample accessions are available at: T7-mC-S* (ERS27427136 [https://www.ebi.ac.uk/ena/browser/view/ERS27427136?dataType=SAMPLE]), T7-GFP-S* (ERS27427138 [https://www.ebi.ac.uk/ena/browser/view/ERS27427138?dataType=SAMPLE], and T7-Split-S* (ERS27427137 [https://www.ebi.ac.uk/ena/browser/view/ERS27427137?dataType=SAMPLE]). Source data are provided with this paper.
Code availability
The code and plotting scripts used in this study are available in the GitHub repository Noireauxlab-TXTL/A-synthetic-phage-cycle, archived on Zenodo under the DOI 10.5281/zenodo.1759335159.
References
Garenne, D. & Noireaux, V. Cell-free transcription-translation: engineering biology from the nanometer to the millimeter scale. Curr. Opin. Biotechnol. 58, 19–27 (2019).
Silverman, A. D., Karim, A. S. & Jewett, M. C. Cell-free gene expression: an expanded repertoire of applications. Nat. Rev. Genet 21, 151–170 (2020).
Garenne, D. et al. Cell-free gene expression. Nat. Rev. Methods Prim. 1, 1–18 (2021).
Abil, Z. & Danelon, C. Roadmap to building a cell: an evolutionary approach. Front Bioeng. Biotechnol. 8, 927 (2020).
Kriebisch, C. M. E. et al. A roadmap toward the synthesis of life. Chemistry 11, (2025).
Kornberg, A. DNA Replication. (San Francisco: W. H. Freeman, (1974).
Bull, J. J. & Lauring, A. S. Theory and empiricism in virulence evolution. PLoS Pathog. 10, e1004387 (2014).
Stern, A. & Sorek, R. The phage-host arms race: shaping the evolution of microbes. Bioessays 33, 43–51 (2011).
Salmond, G. P. C. & Fineran, P. C. A century of the phage: past, present and future. Nat. Rev. Microbiol 13, 777–786 (2015).
Abedon, S. T. Phage evolution and ecology. Adv. Appl. Microbiol. 67, 1–45 (2009).
Blazanin, M. & Turner, P. E. Community context matters for bacteria-phage ecology and evolution. ISME J. 15, 3119–3128 (2021).
Chevallereau, A., Pons, B. J., van Houte, S. & Westra, E. R. Interactions between bacterial and phage communities in natural environments. Nat. Rev. Microbiol. 20, 49–62 (2022).
Maffei, E. et al. Systematic exploration of Escherichia coli phage-host interactions with the BASEL phage collection. PLoS Biol. 19, e3001424 (2021).
Maxwell, K. L. The Anti-CRISPR Story: A Battle for Survival. Mol. Cell 68, 8–14 (2017).
Häuser, R. et al. Bacteriophage protein-protein interactions. Adv. Virus Res. 83, 219–298 (2012).
Incardona, N. L. & Selvidge, L. Mechanism of adsorption and eclipse of bacteriophage phi X174. II. Attachment and eclipse with isolated Escherichia coli cell wall lipopolysaccharide. J. Virol. 11, 775–782 (1973).
Aoyama, A., Hamatake, R. K. & Hayashi, M. In vitro synthesis of bacteriophage phi X174 by purified components. Proc. Natl. Acad. Sci. USA 80, 4195–4199 (1983).
Jaschke, P. R., Lieberman, E. K., Rodriguez, J., Sierra, A. & Endy, D. A fully decompressed synthetic bacteriophage øX174 genome assembled and archived in yeast. Virology 434, 278–284 (2012).
Lambert, O., Plançon, L., Rigaud, J. L. & Letellier, L. Protein-mediated DNA transfer into liposomes. Mol. Microbiol. 30, 761–765 (1998).
Böhm, J. et al. FhuA-mediated phage genome transfer into liposomes: a cryo-electron tomography study. Curr. Biol. 11, 1168–1175 (2001).
Mangenot, S., Hochrein, M., Rädler, J. & Letellier, L. Real-time imaging of DNA ejection from single phage particles. Curr. Biol. 15, 430–435 (2005).
Studier, F. W. Bacteriophage T7. Science 176, 367–376 (1972).
Dunn, J. J. & Studier, F. W. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol. 166, 477–535 (1983).
Molineux, I. J. The T7 Group. in The Bacteriophages (ed. Calendar, R.) 0 (Oxford University Press, https://doi.org/10.1093/oso/9780195168778.003.0020 (2005).
Kulczyk, A. W. & Richardson, C. C. The replication system of bacteriophage T7. Enzymes 39, 89–136 (2016).
Pires, D. P., Cleto, S., Sillankorva, S., Azeredo, J. & Lu, T. K. Genetically engineered phages: a review of advances over the last decade. Microbiol Mol. Biol. Rev. 80, 523–543 (2016).
Levrier, A. et al. PHEIGES: all-cell-free phage synthesis and selection from engineered genomes. Nat. Commun. 15, 2223 (2024).
Shin, J., Jardine, P. & Noireaux, V. Genome replication, synthesis, and assembly of the bacteriophage T7 in a single cell-free reaction. ACS Synth. Biol. 1, 408–413 (2012).
González-García, V. A. et al. Conformational changes leading to T7 DNA delivery upon interaction with the bacterial receptor. J. Biol. Chem. 290, 10038–10044 (2015).
Nomura, K. et al. Interaction of lipopolysaccharide and phospholipid in mixed membranes: solid-state 31P-NMR spectroscopic and microscopic investigations. Biophys. J. 95, 1226–1238 (2008).
Stephan, M. S. et al. Biomimetic asymmetric bacterial membranes incorporating lipopolysaccharides. Biophys. J. 122, 2147–2161 (2023).
Raetz, C. R. H. & Whitfield, C. Lipopolysaccharide endotoxins. Annu Rev. Biochem 71, 635–700 (2002).
Kubiak, J., Brewer, J., Hansen, S. & Bagatolli, L. A. Lipid lateral organization on giant unilamellar vesicles containing lipopolysaccharides. Biophys. J. 100, 978–986 (2011).
Noireaux, V. & Libchaber, A. A vesicle bioreactor as a step toward an artificial cell assembly. Proc. Natl. Acad. Sci. USA 101, 17669–17674 (2004).
Garamella, J., Garenne, D. & Noireaux, V. TXTL-based approach to synthetic cells. Methods Enzymol. 617, 217–239 (2019).
Smit, J., Kamio, Y. & Nikaido, H. Outer membrane of Salmonella typhimurium: chemical analysis and freeze-fracture studies with lipopolysaccharide mutants. J. Bacteriol. 124, 942–958 (1975).
Garcia-Doval, C. & van Raaij, M. J. Structure of the receptor-binding carboxy-terminal domain of bacteriophage T7 tail fibers. Proc. Natl. Acad. Sci. USA 109, 9390–9395 (2012).
De Paepe, M. & Taddei, F. Viruses’ life history: towards a mechanistic basis of a trade-off between survival and reproduction among phages. PLoS Biol. 4, e193 (2006).
Letellier, L., Boulanger, P., de Frutos, M. & Jacquot, P. Channeling phage DNA through membranes: from in vivo to in vitro. Res Microbiol 154, 283–287 (2003).
Van Valen, D. et al. A single-molecule Hershey-Chase experiment. Curr. Biol. 22, 1339–1343 (2012).
Iglesias, S. M., Li, F., Briani, F. & Cingolani, G. Viral genome delivery across bacterial cell surfaces. Annu Rev. Microbiol 78, 125–145 (2024).
Molineux, I. J. & Panja, D. Popping the cork: mechanisms of phage genome ejection. Nat. Rev. Microbiol 11, 194–204 (2013).
Ponchon, L., Mangenot, S., Boulanger, P. & Letellier, L. Encapsidation and transfer of phage DNA into host cells: from in vivo to single particles studies. Biochim Biophys. Acta 1724, 255–261 (2005).
Molineux, I. J. No syringes please, ejection of phage T7 DNA from the virion is enzyme driven. Mol. Microbiol. 40, 1–8 (2001).
Kemp, P., Gupta, M. & Molineux, I. J. Bacteriophage T7 DNA ejection into cells is initiated by an enzyme-like mechanism. Mol. Microbiol 53, 1251–1265 (2004).
Serwer, P., Greenhaw, G. A. & Allen, J. L. Concatemers in a rapidly sedimenting, replicating bacteriophage T7 DNA. Virology 123, 474–479 (1982).
White, J. H. & Richardson, C. C. Processing of concatemers of bacteriophage T7 DNA in vitro. J. Biol. Chem. 262, 8851–8860 (1987).
Wang, I. N., Smith, D. L. & Young, R. Holins: the protein clocks of bacteriophage infections. Annu Rev. Microbiol. 54, 799–825 (2000).
Cahill, J. & Young, R. Phage lysis: multiple genes for multiple barriers. Adv. Virus Res. 103, 33–70 (2019).
Young, R. Phage lysis: three steps, three choices, one outcome. J. Microbiol. 52, 243–258 (2014).
Matsubayashi, H., Kuruma, Y. & Ueda, T. In vitro synthesis of the E. coli Sec translocon from DNA. Angew. Chem. 126, 7665–7668 (2014).
Adamala, K. P. et al. Present and future of synthetic cell development. Nat. Rev. Mol. Cell Biol. 25, 162–167 (2024).
Staufer, O. et al. Building a community to engineer synthetic cells and organelles from the bottom-up. Elife 10, e73556 (2021).
Frischmon, C., Sorenson, C., Winikoff, M. & Adamala, K. P. Build-a-Cell: Engineering a Synthetic Cell Community. Life (Basel) 11, 1176 (2021).
Schmidt, G., Jann, B. & Jann, K. Immunochemistry of R lipopolysaccharides of Escherichia coli. Studies on R mutants with an incomplete core, derived from E. coli O8:K27. Eur. J. Biochem. 16, 382–392 (1970).
Garamella, J., Marshall, R., Rustad, M. & Noireaux, V. The All E. coli TX-TL Toolbox 2.0: a platform for cell-free synthetic biology. ACS Synth. Biol. 5, 344–355 (2016).
Batista, A. C. et al. Differentially optimized cell-free buffer enables robust expression from unprotected linear DNA in exonuclease-deficient extracts. ACS Synth. Biol. 11, 732–746 (2022).
Levrier, A. et al. Data supporting A synthetic cell phage cycle. Data Repository for the University of Minnesota (DRUM). https://doi.org/10.13020/hmz3-yf43 (2025).
Levrier, A. et al. A synthetic cell phage cycle. Noireauxlab-TXTL/A-synthetic-phage-cycle (Zenodo), https://doi.org/10.5281/zenodo.17593351 (2025).
Acknowledgements
The authors would like to thank Aset Khakimzhan and Seth Thompson for their help in the preparation of the CFE system used in this work, Lucienne Letellier, Jerome Bonnet, Peter Voyvodic, Petra Schwille, Damien Baigl and David Bikard for the insightful scientific discussions. This work and the materials are based on funding provided by the National Science Foundation (CBET FMRG 2228971) (AL, PS, DG, SB, VN), the École Doctorale Frontières de l’Innovation en Recherche et Éducation—Programme Bettencourt (AL, ABL), the Bettencourt Schueller Foundation support to Engaged Life Science Department of the Learning Planet Institute (ABL), the Human Frontier Science Program (LT0007/2024-L) (PS), and Fulbright France (AL).
Author information
Authors and Affiliations
Contributions
Conceptualization: A.L., A.B.L., V.N. Methodology: A.L., D.G., P.S. Investigation: A.L., P.S., D.G. Visualization: A.L, Z.I. Funding acquisition: A.L, A.B.L, V.N, P.S, S.B. Project administration: A.L, A.B.L, V.N. Supervision: V.N, A.B.L. Writing – original draft: A.L, A.B.L, V.N. Writing – review & editing: A.L, A.B.L, V.N, S.B.
Corresponding authors
Ethics declarations
Competing interests
Antoine Levrier, Ariel B. Lindner and Vincent Noireaux have submitted a patent application to the European Patent Office pertaining to the reconstitution of a complete cell-free synthetic cell-based phage cycle of this work (application number EP25305837, pending). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kevin Yehl, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Levrier, A., Soudier, P., Garenne, D. et al. A synthetic cell phage cycle. Nat Commun 17, 557 (2026). https://doi.org/10.1038/s41467-025-67249-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67249-8
