
Abstract
Gestational diabetes mellitus (GDM) has a high heritability and frequently co-occurs with type 2 diabetes (T2D), indicating shared genetic mechanisms. Firstly, we employed RHOGE to ascertain the genetic correlation and putative causal directions between GDM and T2D. Subsequently, the Genotype-Tissue Expression Project v8 eQTls files and the FinnGen R11 dataset were employed to conduct cross-tissue transcriptome association studies, Functional Summary-based Imputation in single tissues, and Gene Analysis combined with Multimarker Analysis of Genomic Annotation for GDM and T2D, respectively. A total of 5 genes were identified as GDM susceptibility and 97 genes linked to T2D susceptibility. Of these, four genes (COBLL1, NRBP1, IFT172 and TRIM54) were identified as being shared. Mendelian randomization and colocalization analyses revealed the causal associations of them with GDM and T2D in distinct tissues. Subsequent analyses indicated COBLL1 may influence GDM and T2D risk by regulation of actin filament polymerization and interactions with chemical responses. NRBP1 may confer protective effects against diabetes through regulation of insulin secretion, while IFT172 and TRIM54 may play a role in energy balance signaling and metabolism of skeletal muscle, respectively. Our study provides insight into the shared genetic mechanism between GDM and T2D and identifies potential targets for pharmacological intervention.
Similar content being viewed by others


Introduction
Gestational diabetes mellitus (GDM) is a form of glucose intolerance that leads to hyperglycemia, either emerging or being first detected during pregnancy1. Over the past fifteen years, the prevalence of GDM has significantly risen2, currently affecting more than 16 million pregnancies globally each year3. GDM not only contributes to the cyclical nature of ‘diabetes begetting diabetes’ but also necessitates early detection and management to mitigate risks for adverse birth outcomes and reduce the future incidence of chronic metabolic diseases, such as type 2 diabetes (T2D)4,5. This condition notably increases the lifelong risk of developing T2D3,6. Despite its significant maternal and neonatal health implications, the genetic mechanisms underlying GDM remain poorly understood, with most research focusing on its genetic overlap with T2D. Previous studies have primarily examined loci associated with T2D7,8,9, and the three major genome-wide association studies (GWAS) of GDM10,11,12 have identified only five loci, limiting our ability to explore specific genetic variants or biological pathways unique to GDM. The most comprehensive GWAS of GDM identified 13 genome-wide significant loci13, most of which are located in non-coding regions, posing a significant challenge in understanding their functional roles14. Additionally, the complex patterns of linkage disequilibrium (LD) complicate the identification of causal variants15.
In recent years, transcriptomic-wide association studies (TWAS) have gained attention for their ability to predict gene-level associations using imputed gene expression from genetic data and their relevance to complex traits16. These advancements are feasible because some GWAS-identified risk variants overlap with expression quantitative trait loci (eQTLs), which regulate gene expression17. TWAS enhances statistical power by predicting gene expression from genetic data, reducing the confounding effects of environmental influences and mitigating reverse causation issues. This approach improves the identification of functional regions related to genetic associations18,19. Traditional TWAS analyses have been conducted separately in individual tissues, potentially overlooking shared regulatory mechanisms across tissues20. It has been demonstrated that eQTLs with significant effects are capable of regulating gene expression across a range of tissues21. Recent developments have introduced the unified test for molecular signatures (UTMOST), a novel cross-tissue TWAS methodology that conducts gene-level association analyses across multiple tissues. This enhancement refines the accuracy and broadens the scope of applicability of imputation models, which previously focused on single-tissue analyses22. The UTMOST approach employs a group-lasso penalty on the effect sizes of single nucleotide polymorphisms (SNPs) across diverse tissues, integrating eQTLs that exert multi-tissue effects while preserving the integrity of those that are tissue-specific yet significant. This cross-tissue strategy has been instrumental in identifying potential susceptibility genes for complex multisystem disorders, including migraine23, rheumatoid arthritis24, and autism spectrum disorder25.
The hypothesis that GDM and T2D share a common genetic foundation has recently been suggested3,6,26,27, but studies focusing on the genetic basis of GDM, especially concerning gene expression across different tissues, remain limited. This study initially explores the genetic correlations and potential causative links between GDM and T2D to confirm their interconnectedness. We conducted a cross-tissue TWAS using the UTMOST approach, integrating GWAS data for GDM and T2D from the FinnGen R11 study with eQTL data from the Genotype-Tissue Expression Project (GTEx) version 8. To identify associations within specific tissues, we applied the Functional Summary-based Imputation (FUSION) method28. FUSION is a toolset for performing transcriptome-wide and regulome-wide association studies, which predicts and tests the genetic components of molecular phenotypes for their association with disease using GWAS summary statistics. Additionally, we validated these associations through the application of the Multi-marker Analysis of Genomic Annotation (MAGMA)29, a tool for gene and gene-set analysis that aggregates genetic marker data at the gene level and tests the joint association of markers within the gene with the phenotype. The final set of candidate genes for both GDM and T2D was derived from the intersection of results from these three methods. Further analytical approaches, including Mendelian Randomization (MR) and colocalization analysis, Phenome-Wide Association Studies (PheWAS), gene network exploring using GeneMANIA, pathway enrichment analyses, and evaluations of tissue and cell-type specificity, as well as gene-chemical-disease interactions, were conducted to further investigate the biological characteristics and implications of these candidate genes.
Results
Genetic correlation and bidirectional causal relationships between GDM and T2D across 47 Tissues
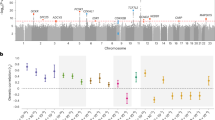
To explore the genetic interplay and bidirectional causality between GDM and T2D across various biological tissues, we employed a gene-expression-mediated approach to estimate genetic correlations between these two conditions. Unlike traditional genetic correlation methods that focus on SNPs, our approach emphasizes gene expression, providing insights into the genetic correlations that are influenced by gene expression levels. Utilizing TWAS data from 47 tissues, we identified significant expression-mediated genetic correlations between GDM and T2D in all tissues analyzed. Notably, the strongest genetic correlation was observed in the artery coronary (r2 = 0.92, p-value = 9.13E-62) as depicted in Fig. 1A. The genetic correlation of 0.92 in the artery coronary tissue is quite high, but it doesn’t necessarily mean this tissue is the most biologically relevant for GDM or T2D in a direct pathophysiological sense. It could reflect that the genetic loci influencing both GDM and T2D may have a pleiotropic effect, influencing coronary artery function or disease in a way that is not immediately apparent in terms of direct disease pathology but could still reflect shared genetic mechanisms. We additionally employed 1000 bootstrap analysis to test the robustness of the results. As shown in Supplementary Fig. 2, the genetic correlation between GDM and T2D in different tissues was robust.

A Genetic correlation between gestational diabetes mellitus and type 2 diabetes in 47 tissues. B A forest plot provides putative causal directions between gestational diabetes mellitus and type 2 diabetes in 47 tissues. RHOGE test is used for tests of individual coefficients. Each test is two-sided and the original P value is reported with no multiple comparisons. Horizontal lines indicate the 95% confidence intervals around the effect sizes. The GTEx’s final dataset (V8) contains DNA data from 838 postmortem donors and 17,382 RNA-seq across tissue sites and cell lines. The original datasets could be found in https://ftp.ebi.ac.uk/pub/databases/spot/eQTL/imported/GTEx_V8. ρGE the coefficient of genetic correlation between gestational diabetes mellitus and type 2 diabetes in distinct tissues, OR odds ratio.
We employed bi-variate linkage disequilibrium (LD) score regression to test the sample overlap between GDM and T2D. The regression intercept was 0.219 (se = 0.017), indicating a moderate degree of sample overlap. Although this overlap could slightly influence the genetic correlation results, its impact is minimal. As GDM and T2D are both metabolic disorders, and many tissues that influence metabolic pathways (such as adipose tissue, liver, and muscle) would be expected to have higher genetic correlations between these traits. However, tissues that are less directly involved in metabolism, like brain or skin, may not be as intuitively relevant. That said, there could still be indirect connections (e.g., brain tissues influencing appetite or lifestyle choices that contribute to metabolic diseases), but these tissues would not be the primary suspects for genetic correlations with GDM and T2D.
Additionally, we utilized bidirectional regression analyses to investigate the potential reciprocal associations between GDM and T2D, mediated by genetically regulated gene expressions across various tissues. These analyses revealed a significant causal effect of GDM on the development of T2D in all tissues studied, with the strongest evidence found in the artery aorta (odds ratio [OR] = 1.86, p-value = 2.14E-78). Conversely, the potential causal impact of T2D on GDM was also significant, particularly in the artery aorta (OR = 1.81, p-value = 3.94E-77). Figure 1B illustrates these significant bidirectional relationships across all 47 tissues, underscoring the complex interactions between GDM and T2D. We also observed bidirectional causal effects between GDM and T2D through the traditional bidirectional MR. For GDM to T2D, OR = 1.15, p-value = 8.17E-6; For T2D to GDM, OR = 1.58, p-value = 1.94E-77.
Identification of GDM and T2D candidate genes across multi-tissue and single-tissue TWAS analyses
Building upon the observed genetic correlations and causal relationships between GDM and T2D, we next aimed to identify specific genes associated with these conditions through comprehensive TWAS analyses.
In the cross-tissue TWAS analysis, 229 genes associated with GDM and 710 genes related to T2D were identified with p-values less than 0.05 (see Tables S1 and S2). Following adjustments for false discovery rates (FDR < 0.05), 11 genes linked to GDM and 319 genes related to T2D retained statistical significance, as depicted in Fig. 3A and B, respectively. Additionally, the single-tissue TWAS identified 193 genes linked to GDM and 4933 genes associated with T2D, with significant associations (FDR < 0.05) in at least one tissue (Tables S3 and S4). Notably, within these findings, 6 candidate genes for GDM (referenced in Fig. 3A and Table S5) and 168 candidate genes for T2D (illustrated in Fig. 2B and Table S6) met the rigorous selection criteria across both multi-tissue and single-tissue analyses.

A Upset plot illustrating the overlap of genes identified by various methods as being associated with gestational diabetes mellitus. B Upset plot presenting the overlap of genes identified by various approaches associated with type 2 diabetes. C Heatmap of fusion TWAS.P values for 5 identified gestational GDM in different tissues. D Heatmap of fusion TWAS.P values for 97 identified T2D in different tissues. E Venn diagram of GDM genes and T2D genes. GDM gestational diabetes mellitus, T2D type 2 diabetes, TWAS transcriptome-wide association studies, FUSION functional summary-based imputation.
Conditional and joint multiple-SNP analysis identifies independent and interacting genetic signals for GDM and T2D
To refine the identified candidate genes and minimize potential false positives arising from linkage disequilibrium (LD), we conducted conditional and joint multiple-SNP association (COJO) analyses. These analyses provided a deeper understanding of the independent and interactive effects of genes across tissues in GDM and T2D.
The 6 candidate genes for GDM and 168 for T2D, primarily located on chromosomes 2, 4, 18 and 22, were subjected to COJO analysis within their respective tissues to address potential LD-related false positives (Tables S7 and S8). To illustrate, we focused on the shared genes (COBLL1, NRBP1, IFT172 and TRIM54) between GDM and T2D. TWAS analysis conditioned on predicted expression revealed that COBLL1 in adipose visceral omentum (Fig. 3A), NRBP1 in the artery aorta (Fig. 3B), IFT172 in the brain cortex (Fig. 3C), and TRIM54 in the brain anterior cingulate cortex BA24 (Fig. 3D) did not significantly affect the TWAS signals for GDM, indicating their independence from LD. Conversely, conditioning on the expression of COBLL1 in the pancreas for T2D led to a marked decrease in the TWAS signal for SLC38A11 (Fig. 3E). Furthermore, the TWAS signals for ZNF513, SLC5A6 and ATRAID underwent a notable alteration when conditioned on the predicted expression of NRBP1 in the artery aorta for T2D (Fig. 3F). In the brain cerebellum, conditioning on IFT172 expression resulted in a substantial alteration in the TWAS signals for GPN1 in T2D (Fig. 3G). Notably, in the brain anterior cingulate cortex BA24 for T2D, the TWAS signals for ATRAID increased when conditioned on the expression of TRIM54 (Fig. 3H). Detailed effects of conditioning on other candidate genes across tissues are provided in Tables S7 and S8.

A Regional plot of COBLL1 gene in adipose visceral omentum for GDM. B Regional plot of COBLL1 gene in artery aorta for GDM. C Regional plot of IFT172 gene in brain cortex for GDM. D Regional plot of NRBP1 and TRIM54 genes in brain anterior cingulate cortex BA24 for GDM. E Regional plot of COBLL1 gene in pancreas for T2D. F Regional plot of NRBP1 gene in artery aorta for T2D. G Regional plot of IFT172 gene in brain cerebellum for T2D. H Regional plot of TRIM54 gene in brain anterior cingulate cortex BA24 for T2D. The top panel highlights all genes in the region. COJO analysis is used for tests of individual coefficients. Each test is two-sided and the P value is evaluated with false discovery rate. GDM gestational diabetes mellitus, T2D type 2 diabetes. The marginally associated TWAS genes are shown in blue, and the jointly significant genes are shown in green. The bottom panel shows a regional Manhattan plot of GWAS data before (grey) and after (blue) conditioning on the predicted expression of the green genes.
Gene association and cross-tissue integration identifies shared and distinct candidate genes for GDM and T2D
Building on the insights from previous analyses, we further refined the identification of candidate genes for GDM and T2D through MAGMA and cross-tissue integration. This approach enabled the discovery of both shared and distinct genetic signals underlying the two conditions.
MAGMA-based gene analysis revealed 155 genes significantly associated with GDM (FDR < 0.05), as illustrated in Fig. 2A and Table S9. For T2D, 3998 genes were identified as significantly associated, depicted in Fig. 2B and Table S10. To strengthen these findings, we integrated results from UTMOST cross-tissue analysis with significant genes identified in both FUSION and MAGMA analyses. This integration led to the identification of 5 promising candidate genes for GDM (depicted in Fig. 2A and E) and 97 for T2D (Fig. 2B and E). A heatmap displaying fusion TWAS p-values for these candidate genes is presented in Fig. 2C and D. Ultimately, four genes (COBLL1, NRBP1, IFT172 and TRIM54) were identified as shared by GDM and T2D through the combined analyses (Fig. 2E and Table S11).
Causal and colocalization analyses uncover tissue-specific associations for shared genes in GDM and T2D
To enhance our understanding of the causal relationships between the shared genes and the risk of GDM and T2D, we performed MR and colocalization analyses. These analyses provided robust evidence of tissue-specific gene associations and highlighted critical genetic loci involved in both conditions.
To investigate the causal roles of the four shared genes (COBLL1, NRBP1, IFT172 and TRIM54) in GDM and T2D, we employed MR and colocalization analyses. The COBLL1 gene is located on chromosome 2q24.3, and the results of FUSION demonstrate its significant expression in 2 tissues for GDM and 9 tissues for T2D (Table S11). The MR analysis underscored significant causal associations with GDM and T2D in four tissues (p < 0.05) (Fig. 4A and Table S12), with an OR (95%CI) of 0.85 (0.77, 0.93) for GDM and 0.82 (0.78, 0.86) for T2D in brain cortex. Colocalization analysis further supported these findings, with PP.H4 values of 0.81, 0.92 and 0.78 for Adipose_Visceral_Omentum, Artery_Aorta and Nerve_Tibial, respectively (Table S13). Notably, rs3769885 emerged as the most significant colocalization locus for GDM in Adipose_Visceral_Omentum (Fig. 4B) and Nerve_Tibial (Fig. 4C).

A The MR results confirmed the causal associations between three candidate genes and GDM and T2D. Wald ratio test is used for tests of individual coefficients. Each test is two-sided and the original P value is reported with no multiple comparisons. Horizontal lines indicate the 95% confidence intervals around the effect sizes. The GTEx’s final dataset (V8) contains DNA data from 838 postmortem donors and 17,382 RNA-seq across tissue sites and cell lines. B The results of colocalization analysis between COBLL1 and GDM in adipose visceral omentum. C The results of colocalization analysis between COBLL1 and GDM in nerve tibial. D The results of colocalization analysis between NRBP1 and GDM in esophagus gastroesophageal junction. E The results of colocalization analysis between NRBP1 and T2D in esophagus gastroesophageal junction. F The results of colocalization analysis between NRBP1 and T2D in small intestine terminal ileum. G The results of colocalization analysis between IFT172 and GDM in thyroid. H The results of colocalization analysis between IFT172 and T2D in thyroid. I The results of colocalization analysis between TRIM54 and GDM in brain anterior cingulate cortex BA24. J The results of colocalization analysis between TRIM54 and T2D in brain anterior cingulate cortex BA24. Colocalization analysis is used for tests of posterior probability (PP). A PP.H4 greater than 0.6 indicated the presence of shared causal variants. GDM gestational diabetes mellitus, T2D type 2 diabetes, OR odds ratio.
NRBP1, located on chromosome 2p23.3, was notably correlated with GDM in 23 tissues according to FUSION analysis (Table S11). MR analysis confirmed a causal relationship between NRBP1 and GDM in 16 tissues (p < 0.05) (Fig. 4A and Table S12), with the strongest association found in the skin sun exposed lower leg (OR = 0.77, p-value = 6.14E-09). Colocalization analysis supported this, with a PP.H4 value of 0.91 for Esophagus_Gastroesophageal_Junction (Table S13). rs1260326 emerged as the most significant colocalization locus for GDM in this tissue (Fig. 4D). Additionally, FUSION analysis revealed that the NRBP1 gene is a significant contributor to T2D in 22 tissues (Table S11). MR analyses confirmed a causal relationship in 16 tissues (p < 0.05) (Fig. 4A and Table S12), with the strongest association found in the artery aorta (OR = 0.80, p value = 2.67E-09). Colocalization analysis further supported this with PP.H4 values of 0.94 and 0.66 for Esophagus_Gastroesophageal_Junction and Small_Intestine_Terminal_Ileum, respectively (Table S14). rs1260326 was the most significant colocalization locus for T2D in Esophagus_Gastroesophageal_Junction (Fig. 4E), and rs2303370 was the most significant colocalization locus for T2D in Small_Intestine_Terminal_Ileum (Fig. 4F).
Similarly, MR analysis identified significant causal associations of IFT172 with both GDM and T2D in the spleen (p < 0.05) (Fig. 4A and Table S12). Notably, rs780096 emerged as the most significant colocalization locus for GDM and T2D in Thyroid (Fig. 4G and H) and rs780095 served as the most significant colocalization locus for GDM and T2D in Brain_Anterior_cingulate_cortex_BA24 (Fig. 4I and J).
PheWAS analysis reveals gene-phenotype associations for GDM and T2D shared genes
Building upon the previous MR and colocalization findings, a comprehensive PheWAS analysis was conducted to investigate the broader genetic associations of the four shared genes (COBLL1, NRBP1, IFT172, and TRIM54) with a wide array of diseases and traits, offering new insights into their potential clinical implications.
This study included a PheWAS (Phenome-Wide Association Study) analysis of the four genes shared between GDM and T2D (COBLL1, NRBP1, IFT172 and TRIM54), utilizing data from 17,361 binary phenotypes and 1419 quantitative phenotypes sourced from the AstraZeneca PheWAS portal database. The analysis revealed significant associations between genetic variants and specific diseases or traits. For COBLL1, IFT172 and TRIM54, the variants showed statistically significant correlations with binary phenotypes, particularly related to endocrine/metabolic diseases and musculoskeletal diseases (Fig. 5A, E, G). Moreover, these variants showed notable associations with quantitative phenotypes, including proteomics, NMR metabolomics, laboratory findings, and health services (Fig. 5B, F, H). These results suggest possible clinical implications, such as side effects and horizontal pleiotropy, warranting further investigation. In contrast, NRBP1 variants showed no significant associations with either binary or quantitative phenotypes (Fig. 5C and D).

A The Manhattan plot of phenome-wide association between COBLL1 and UKB binary traits. B The Manhattan plot of phenome-wide association between COBLL1 and UKB continuous traits. C The Manhattan plot of phenome-wide association between NRBP1 and UKB binary traits. D The Manhattan plot of phenome-wide association between NRBP1 and UKB continuous traits. E The Manhattan plot of phenome-wide association between IFT172 and UKB binary traits. F The Manhattan plot of phenome-wide association between IFT172 and UKB continuous traits. G The Manhattan plot of phenome-wide association between TRIM54 and UKB binary traits. H The Manhattan plot of phenome-wide association between TRIM54 and UKB continuous traits. Generalized linear model is used for phenome-wide association study. Each test is two-sided and the original P value is reported with no multiple comparisons. I GeneMania gene network of COBLL1 as the core. J GeneMania gene network of NRBP1 as the core. K GeneMania gene network of IFT172 as the core. L GeneMania gene network of TRIM54 as the core. UKB UK Biobank.
GeneMANIA network analysis reveals pathway enrichments for GDM and T2D-associated genes
Expanding on the phenotypic associations identified in the PheWAS analysis, a GeneMANIA network analysis was conducted to explore the interactive relationships and pathway enrichments of the genes COBLL1, NRBP1, IFT172, and TRIM54, providing further insights into their biological roles.
The GeneMANIA analysis revealed potential interactive gene networks, with COBLL1 positioned centrally, as shown in Fig. 5I. The associated networks with COBLL1 were primarily enriched in pathways related to actin dynamics, including actin nucleation, actin filament polymerization, and actin nucleation (Table S15). A second network, centered on NRBP1 (Fig. 5J), was predominantly enriched in pathways associated with nucleotide phosphorylation, such as DNA-templated transcription, elongation, phosphorus-oxygen lyase activity, and ribonucleoside diphosphate metabolism (Table S16). The network centered on IFT172 (Fig. 5K) showed significant enrichment in pathways related to protein transport along microtubule, microtubule-based protein transport, and intraciliary transport (Table S17). Finally, a network with TRIM54 at its core, presented in Fig. 5L, featured enrichment in pathways associated with ubiquitinyl hydrolase activity, omega peptidase activity, and protein modification by small protein removal (Table S18). This comprehensive genomic analysis underscores the complex interplay and functional diversity of these genes within various biological pathways, offering valuable insights into their potential involvement in the molecular mechanisms of GDM and T2D.
Pathway enrichment analysis uncovers biological functions of GDM and T2D-associated genes
Building on the interactive gene networks analyzed in the previous section, pathway enrichment analyses were conducted to explore the biological functions of the genes linked to GDM and T2D, revealing distinct pathway associations for each condition.
Following the identification of key genes linked to GDM and T2D, pathway enrichment analyses were performed to elucidate their potential biological functions. For the GDM-associated genes identified by UTMOST or FUSION, no significant enrichment was observed in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. However, Gene Ontology (GO) pathways revealed these genes predominantly participate in pathways such as iron-sulfur cluster binding, methylthiotransferase activity and protein metabolic process (Fig. 6A). Additional pathway analysis of the five GDM genes identified by UTMOST, FUSION, and MAGMA is shown in Supplementary Fig. 3. Conversely, the analysis of 97 genes linked to T2D revealed significant enrichment in GO pathways related to voltage-gated sodium channel activity, establishment of localization, microtubule-based processes, and components associated with the centrosome and microtubule organizing center (Fig. 6B). KEGG pathway analysis further highlighted the involvement of these genes in processes related to legionellosis (Fig. 6B), emphasizing the biological relevance of these findings in the context of T2D.

A Results of the functional enrichment analysis for identified 198 GDM genes (UTMOST or FUSION). B Results of the functional enrichment analysis for identified 97 T2D genes. C Tissue-specific gene enrichment results of identified five GDM genes. D Tissue-specific gene enrichment results of identified 97 T2D genes. Bars with red color means significantly enriched DEG sets. E Cell-type-specific gene enrichment plot of identified 11 GDM genes by UTMOST. F Cell-type-specific gene enrichment plot of identified 97 T2D genes. Chemical interactions for COBLL1 gene (G), NRBP1 gene (H), IFT172 gene (I) and TRIM54 gene (J). The x-axis represents the number of interactions from the CTD (Comparative Toxicogenomics Database). The y-axis indicates the chemicals. The assessment of functional enrichment for the genes was assessed using the hypergeometric test. The g:SCS method was used for multiple testing correction with two-sided. GDM gestational diabetes mellitus, T2D type 2 diabetes, BP Biological process, GO Gene Ontology, MF Molecular function, CC Cellular Component, KEGG Kyoto Encyclopedia of Genes and Genomes, DEG Differentially Expressed Genes, UTMOST unified test for molecular signatures, FUSION functional summary-based imputation.
Tissue and cell-type specificity reveals key players in T2D and GDM
To further elucidate the tissue and cell-type specificity of the identified genes, an integrative analysis combining gene expression data with single-cell transcriptomics was performed, uncovering distinct tissue and cell-type enrichments for genes associated with T2D and GDM.
This analysis utilized gene expression data from the GTEx project30 combined with insights from 11 single-cell transcriptomics datasets31. It revealed significant enrichment of 97 T2D-associated genes in several tissues, particularly the Heart, Pancreas, Liver, Kidney, and Adrenal Gland (Fig. 6D). Specific cell types, including radial glial cells, type I pneumocytes, corneal epithelial cells, type II pneumocytes, and pancreatic ductal cells were identified as potential key players in organ homeostasis, functionality, and repair (Fig. 6F). For GDM, the five genes identified were notably enriched in the Adrenal Gland, Adipose Tissue, Nerve, Heart, and Muscle (Fig. 6C). Additionally, these genes were prominently enriched in diverse cell types including mast cells, corneal epithelial cells, acinar cells, decidual perivascular cells, and decidual macrophages (Fig. 6E). These findings illuminate the essential roles these genes may play in immune response regulation, tissue repair, and the maintenance of local homeostasis.
Chemical interactions and disease pathways of shared genes in GDM and T2D
Building on the tissue- and gene-specific findings, the next phase of the analysis explores the chemical interactions and disease pathways linked to genes commonly implicated in both GDM and T2D, underscoring their broader physiological relevance.
Utilizing the Comparative Toxicogenomics Database (CTD)32, this study predicted chemicals interactions for four genes shared between GDM and T2D (COBLL1, NRBP1, IFT172 and TRIM54). The analysis revealed a broad spectrum of chemical interactions for the COBLL1 gene, including exposure to Tetrachlorodibenzodioxin, bisphenol A, Valproic Acid, and dorsomorphin (Fig. 6G). Similarly, NRBP1 showed interactions with bisphenol A, Ethinyl Estradiol, Tetrachlorodibenzodioxin, and 1,2-Dimethylhydrazine (Fig. 6H). For IFT172, potential interactions with bisphenol A, Benzo(a)pyrene, Cisplatin, and Tretinoin (Fig. 6I). TRIM54 also showed potential for chemical interactions with Benzo(a)pyrene, Aflatoxin B1, bisphenol A, Tetrachlorodibenzodioxin, and Valproic Acid. Further analysis of Gene-Chemical-Disease pathways revealed that these genes are involved in a range of physiological disruptions including weight dynamics, liver and chemical diseases, weight loss, inflammation, fatty liver, kidney diseases, and various other endocrine and metabolic disorders (Tables S19–S22). Notably, COBLL1 was highlighted as a potential biomarker for Non-alcoholic Fatty Liver Disease and Myocardial Reperfusion Injury (Table S19), whereas the IFT172 gene was associated with potential markers for Gastrointestinal Stromal Tumors, Retinitis Pigmentosa 71, and Short Rib-Polydactyly Syndrome (Table S22).
Discussion
In this study, we utilized genetic data from GWAS and GTEx V8 eQTL datasets to comprehensively investigate the genetic links and potential bidirectional causal relationships between GDM and T2D. Our integrative approach assessed how genetically regulated gene expression influences disease risk for both conditions. Through cross-tissue TWAS, supported by single-tissue TWAS and MAGMA gene-based analyses, we identified five genes associated with GDM and 97 with T2D, among which four were shared. These shared genes were further validated through MR and colocalization analyses, providing robust evidence of their involvement. We also explored the potential biological functions of these susceptibility genes.
Our results demonstrated a significant genetic correlation and evidence for bidirectional causal effects between GDM and T2D, based on TWAS data from GTEx V8 and GWAS. Consistent with prior studies, most GDM-associated loci showed strong genetic overlap with T2D10,11,13,33,34,35, although a subset appeared specific to GDM. The largest GDM GWAS to date employed SCOUTJOY (Significant Cross-trait Outliers and Trends in Joint York regression), a novel method for evaluating whether the effect sizes of top loci follow a consistent trend across traits, indicating condition-specific associations while adjusting for sample overlap and trait-specific estimation errors13. This analysis uncovered substantial heterogeneity in effect sizes, highlighting both shared and unique genetic architectures between GDM and T2D. Furthermore, two large-scale GWAS studies reported a genetic correlation of approximately 0.70 between the two diseases, reinforcing their shared pathophysiology while suggesting the presence of distinct genetic contributors13,33. However, a key challenge remains that many risk loci identified by GWAS are located in non-coding regions14, with only a fraction functionally annotated36. This underscores the critical role of gene regulation, as reflected by the enrichment of eQTLs among disease-associated variants37. TWAS addresses this gap by linking genetically predicted gene expression with disease phenotypes16. Overall, our findings provide stronger evidence for both genetic correlation and causal interplay between GDM and T2D, while highlighting the utility of TWAS in uncovering the biological mechanisms driving diabetes susceptibility across multiple tissues.
Inflammation is a crucial component of the immune system’s defense, protecting the body from infections and aiding tissue repair. However, excessive or dysregulated immune responses can lead to harmful outcomes, such as septic shock, hypersensitivity reactions (including atopy, anaphylaxis, and contact hypersensitivity), or organ transplant rejection. Chronic or uncontrolled inflammation can contribute to the development of various diseases38. Recently, we identified metabolic loci and estimated the mediation effect of inflammatory biomarkers on chronic metabolic diseases39,40,41,42,43. The interplay between inflammatory processes and genetic factors is crucial in the onset and progression of GDM and T2D44,45.
Emerging research indicates that genetic variations affecting inflammatory pathways significantly influence GDM susceptibility46,47, with alterations in cytokine regulation, particularly TNF-alpha and IL-6, implicated in the inflammatory responses observed in pregnant women with GDM. These findings highlight a potential causative role of inflammation in GDM, pointing to novel therapeutic targets48. Pregnancy itself induces low-grade systemic inflammation, which may predispose individuals to metabolic complications49. Both GDM and T2D are characterized by chronic low-grade inflammation, affecting key tissues, including adipose tissue, liver, skeletal muscle, pancreatic islets, and the brain44,49,50. This persistent inflammatory state disrupts insulin signaling pathways, impairing glucose uptake and promoting unregulated lipolysis, leading to ectopic lipid accumulation and a cycle of insulin resistance. This accelerates the progression of diabetes and other metabolic disorders44. Given the complexity of these interactions, a comprehensive research approach is essential, integrating studies across multiple tissues to further understand the genetic and molecular determinants of metabolism in diabetes. Expanding research to encompass both tissue-specific and systemic inflammation could reveal novel genetic factors and pathways driving the onset and progression of these diseases, offering opportunities for more targeted therapeutic strategies44,49,50.
This study builds on previous work by employing the UTMOST framework for cross-tissue TWAS, which offers a significant improvement over traditional single-tissue analyses. By integrating gene expression data from multiple tissues, the UTMOST framework provides a more comprehensive view of gene-trait associations, enhancing statistical power and improving the detection of associations that may be overlooked in isolated tissue studies22. Using this robust approach and subsequent validation, we identified five genes associated with GDM susceptibility and 97 genes linked to T2D. Notably, four of these genes are shared between GDM and T2D, representing novel findings not previously reported. Further bioinformatics analyses revealed that these candidate genes are primarily involved in processes such as ovarian steroidogenesis, insulin secretion, and legionellosis. They also show significant enrichment in various metabolic tissues and cell types. Overall, our integrative approach underscores the genetic overlap between GDM and T2D, highlighting four shared genes. This framework not only deepens our understanding of the genetic mechanisms underlying these complex diseases but also provides a foundation for developing targeted therapeutic strategies.
The COBLL1 gene encodes cordon-bleu protein-like 1, a relatively understudied gene linked to carbohydrate-lipid metabolism and metabolic diseases51,52. It has been suggested as a risk factor for T2D, influencing lipid and carbohydrate metabolism, body fat percentage, and obesity, which are associated with metabolic and cardiovascular complications51. COBLL1 may also contribute to insulin resistance and inflammation53,54. Although studies on COBLL1 polymorphisms in GDM are limited, with no significant associations found in Indian or Polish populations52,55, our study employed MR analysis to confirm a causal link between COBLL1 expression and GDM/T2D risk, supported by bioinformatics analyses of actin dynamics. Nuclear receptor binding protein 1 (NRBP1), a pseudokinase conserved across species, is crucial for cellular homeostasis and plays roles in various cancers56. Its tumorigenic effects are context-dependent, showing both tumor-suppressive and promoting functions57,58. While NRBP1’s effects on the central nervous system and vascular cells are poorly understood, it may influence neurodegenerative processes via amyloid-beta regulation59. Emerging evidence also links NRBP1 to apo CIII sialylation and hypertriglyceridemia60,61, impacting insulin resistance and cardiovascular disease62. This study used MR analysis to identify a causal link between NRBP1 expression and decreased GDM/T2D risk, highlighting its potential as a therapeutic target.
The IFT172 gene encodes a peripheral component of the intraflagellar transport subcomplex-B (IFT-B), which is crucial for primary cilia function63. Mutations in IFT172 are associated with ciliopathies, but no direct link to glycosylation has been established63. Primary cilia play a key role in metabolic processes, including obesity, insulin signaling, and T2D, suggesting an indirect involvement in lipoprotein metabolism64. Ciliopathies such as Bardet-Biedl and Alström syndromes exhibit both obesity and T2D phenotypes64. Ciliary genes regulate energy homeostasis through leptin and insulin signaling64,65, implicating IFT172 in GDM and T2D by modulating ciliary functions and energy pathways. TRIM54 (MURF), a RING-type E3 ubiquitin ligase, regulates skeletal myogenesis and myotube fusion by mediating ubiquitin-proteasomal protein turnover66,67,68,69. It also serves as a diagnostic biomarker for acute myocardial infarction, and heterozygous variants cause cardiac/skeletal myopathies70,71. The role of TRIM54 in skeletal muscle metabolism suggests its involvement in glucose regulation and potential contributions to GDM/T2D pathogenesis.
This study successfully identified four previously uncharacterized genes associated with susceptibility to both GDM and T2D. Based on these findings, we proposed potential roles for these genes in the pathophysiology of these metabolic disorders. By uncovering common genetic risk factors, this research improves our understanding of the mechanisms connecting GDM and T2D. Clinically, this knowledge could lead to more precise risk stratification, enabling earlier identification of individuals at higher risk for T2D following GDM. Additionally, genetic markers could guide personalized treatment approaches, improving the management of both conditions. From a public health perspective, incorporating genetic insights into screening protocols could facilitate earlier identification of high-risk populations, promoting timely interventions to prevent the progression from GDM to T2D. Ultimately, understanding the genetic overlap could inform the development of novel prevention strategies, including gene-based therapies, and improve overall health outcomes for affected individuals. This research lays the groundwork for future clinical and public health practices aimed at reducing the burden of T2D, particularly in populations with a history of GDM. However, the study does have some limitations that warrant attention. One limitation is potential bias due to the age and gender distribution in the GDM and T2D populations. GDM primarily affects younger women, while T2D is more prevalent in older adults of both sexes, potentially introducing age- and gender-related confounding factors. Additionally, collider bias could affect the results if the populations were not properly matched, leading to false associations. Future research address these limitations through stratification, age-adjustment, and techniques like propensity score matching to ensure more accurate and generalizable findings. Another limitation is the exclusive focus on a European ancestry cohort, which limits the generalizability of the results to other populations. Furthermore, while efforts were made to minimize false-positive associations, the absence of an independent replication dataset restricts the ability to validate the robustness of the identified genetic signals. To overcome these challenges, future studies should prioritize the inclusion of ancestrally diverse populations, enabling a more comprehensive understanding of the genetic architecture underlying GDM and T2D. Further investigation through rigorous biological experiments is also necessary to validate these findings and elucidate the precise molecular and pathophysiological mechanisms involved.
In summary, our multi-tissue TWAS identified four novel genes whose expression is associated with susceptibility to both GDM and T2D. These findings provide valuable new insights into the genetic mechanisms underlying these conditions. Despite certain limitations, this research lays a solid foundation for future studies aimed at elucidating the biological significance and therapeutic potential of these genetic signals. A deeper understanding of these mechanisms could ultimately enhance the diagnosis, treatment, and prevention of GDM and T2D.
Methods
Supplementary Fig. 1 illustrates the analytical framework employed in this study. We mainly employed three approaches (UTMOST, FUSION and MAGMA) to ascertain the candidate genes for GDM and T2D. We conducted these three approaches independently for selecting the candidate genes, and then we took the intersection of the candidate genes screened by these three methods for the final identified genes for GDM and T2D, respectively. In the process of screening candidate genes using each method independently, associations achieving an FDR below 0.05 after applying the false discovery rate (FDR) correction for each method were selected. The purpose of simultaneously conducting the intersection is to ensure a reduced FDR and to corroborate the association with GDM or T2D from multiple perspectives. All analyses in this study were conducted using publicly available data. Summary-level statistics, gene expression data, and other datasets were approved by their respective institutional review boards, as indicated in the ‘Data Availability’ section of the website. Ethical guidelines were adhered to in accordance with the standards set by each institutional review board.
Data sources for GDM GWAS and T2D GWAS
We obtained data for GDM and T2D from the FinnGen Release 11 cohort (https://www.finngen.fi/en/access_results). This cohort comprises data from 16,802 individuals with GDM and 237,816 control subjects, as well as 71,728 individuals with T2D and 369,007 control subjects, all of whom are of European ancestry. We began with data from pregnancies involving genotyped FinnGen participants, defining a “pregnancy window” as the period from 40 weeks before delivery to 5 weeks postpartum. A pregnancy was classified as having gestational diabetes if it met any of the following criteria: (I1) the presence of gestational diabetes diagnoses (ICD codes) within the pregnancy window, (I2) any diabetes-related codes (e.g., ICD8) within the same window, or (I3) abnormal blood glucose test results recorded in the pregnancy registry. Exclusion criteria were applied to pregnancies based on the following conditions: (E1) a diabetes diagnosis outside the pregnancy window, (E2) any prior history of significant pancreatic diseases such as chronic pancreatitis, pancreatic necrosis, pancreatic cancer, or cystic fibrosis, or (E3) prior diagnoses of type 1 (T1D) or type 2 diabetes (T2D). Pregnancies that did not meet any inclusion criteria for gestational diabetes but passed the exclusion criteria were categorized as ‘unaffected’. To define the study cohort, cases were selected from the genotyped women with pregnancy history, identifying those with at least one pregnancy that met the inclusion criteria for gestational diabetes and passed the exclusion criteria. Controls were defined as women with only ‘unaffected’ pregnancies, meaning no history of diabetes or significant pancreatic diseases before or during pregnancy, and no abnormal blood glucose results in the pregnancy registry. Detailed information could be found in Supporting Information.
eQTL data acquisition
Expression data were retrieved from the Genotype-Tissue Expression (GTEx) Project, Version 8 dataset30, which provides gene expression profiles across 47 diverse tissues (excluding testis and prostate) from 838 deceased donors (https://ftp.ebi.ac.uk/pub/databases/spot/eQTL/imported/GTEx_V8/ge/). The number of samples per tissue varies considerably, with the lowest number being 73 in the renal cortex and the highest number reaching 706 in skeletal muscle.
Genetic correlations and putative causal directions between GDM and T2D
RHOGE19 is an R package that assesses the local genetic correlation between gene expression and complex traits and to compute the genetic correlations derived from predicted expression between trait pairs. This approach is extended to a bi-directional regression that provides putative causal directions between traits. In this study, RHOGE was further utilized to assess the influence of predicted gene expression on mediating these correlations and to estimate the correlation of effect sizes between GDM and T2D. The method incorporates a bidirectional regression framework to infer potential causal directions among traits exhibiting non-zero correlations19. Utilizing this technique, we conducted TWAS using GTEx tissue data and GWAS results from the FinnGen R11 dataset to explore potential expression-mediated causal relationships between GDM and T2D based on single-tissue analyses (see below). Furthermore, 1000 bootstrap analysis was employed to evaluate the robustness of the genetic correlation between GDM and T2D in different tissues.
Cross-tissue analyses for GDM and T2D
We employed the UTMOST method (https://github.com/Joker-Jerome/UTMOST) for cross-tissue analysis to estimate gene-trait associations at an organismal level22. This method enabled the identification of a multitude of genes within tissues that exhibited augmented trait heritability and enhanced imputation accuracy22,72. We integrated these gene-trait associations using the generalized Berk-Jones (GBJ) test, which leverages covariance derived from single-tissue statistics22,73. Associations achieving an FDR below 0.05 after applying the false discovery rate (FDR) correction were considered statistically significant.
Single tissue analyses for GDM and T2D
The FUSION tool (http://gusevlab.org/projects/fusion/) was deployed to perform TWAS that amalgamate GDM and T2D GWAS data with eQTL data across 47 tissues from the GTEx V8 dataset18. Initially, LD assessment between the predictive model and SNPs at each GWAS locus was conducted using 1000 Genomes Project samples of European ancestry. Subsequently, FUSION implemented a series of predictive models, encompassing LASSO, Elastic Net, BSLMM, BLUP, and the top 1 model, to determine the most accurate predictive model based on their impact on gene expression weights. The model exhibiting the highest predictive accuracy was selected to determine the gene weights. The selected model was then used to integrate these weights with genetic effects ascertained from the Z-scores of GDM and T2D GWAS, to facilitate TWAS for both conditions. Genes that met the criterion of an FDR < 0.05 in both cross-tissue and single-tissue analyses for each condition were identified as candidate genes.
Conditional and joint analysis
Within the same FUSION framework, a conditional and joint analysis module (COJO) was utilized to discern conditionally independent genetic variants within each locus18. This post-processing step was pivotal in refining our understanding of the genetic underpinnings that influence trait variability, by accounting for LD among markers74. In this study, the genes identified by both UTMOST and FUSION (6 significant genes for GDM and 168 significant genes for T2D) were selected for COJO analysis. COJO was conducted separately for the tissues that associated with above significant genes identified by FUSION, genes identified through this analysis as independently associated were classified as jointly significant, whereas those that did not maintain significance were categorized as marginally significant. FDR correction was applied for multiple testing in this stage.
Gene analysis by MAGMA
Gene-level investigations were conducted employing MAGMA software (version 1.10), which leveraged data from Phase 3 of the 1000 Genomes Project (Build 37, European data only) to amalgamate SNP-level association statistics into comprehensive gene scores. The objective of this process was to quantify the strength of the association between each gene and the phenotype under study29,75. For a comprehensive understanding of the parameters and methodological approaches employed, the original MAGMA documentation should be consulted29.
Mendelian randomization and colocalization analysis
The application of MR was executed via the “TwoSampleMR” package in R76. This analysis utilized cis-eQTL SNPs as instrumental variables (IVs), with gene expression delineated as the exposure, and GWAS datasets on GDM and T2D as the outcomes. The preliminary phase comprised the identification of genome-wide significant SNPs (p < 5E-08), followed by LD clumping to segregate independent SNPs (r2 < 0.001)77. Before MR analysis, we removed the pleiotropic eQTLs as many eQTLs may be associated with multiple tissues. The MR effect estimation was facilitated using the Wald ratio method, with statistical significance established at p < 0.05 for IFT172 (1 tissue), p < 0.0125 for COBLL1 (4 tissue), and p < 3.12E-3 for NRBP1 (16 tissues) considering multiple testing.
Subsequently, a Bayesian colocalization analysis was deployed using the “coloc” package in R78. This method aimed to determine the concurrence of causal loci between GWAS and eQTL signals, with a particular focus on the posterior probability (PP) of five hypothetical relationships78. A PP.H4 greater than 0.6 indicated the presence of shared causal variants within the examined datasets.
PheWAS analysis of shared genes between GDM and T2D
A phenome-wide association study (PheWAS) was conducted utilizing exome sequencing and phenotype data accessible through the AstraZeneca PheWAS Portal (https://azphewas.com/)79. This portal enabled an extensive investigation into the correlations between gene variants and a wide range of phenotypes. The initial study examined the association of shared genes with a range of binary and quantitative phenotypes, derived from the medical records of approximately 500,000 UK Biobank participants79. This analysis included an exploration of horizontal pleiotropy and potential adverse effects related to four genes shared between GDM and T2D (COBLL1, NRBP1, IFT172 and TRIM54) within the context of the UK Biobank.
GeneMANIA analysis
The GeneMANIA platform (https://genemania.org/) was employed to facilitate access to a plethora of datasets, including genetic interactions, pathways, and co-expression data pertinent to the target genes80. This platform was instrumental in elucidating potential biological functions associated with these genes81, thereby enhancing the understanding of their roles within the cellular and molecular frameworks of the studied traits.
Pathway enrichment analysis
Enrichment analyses were conducted utilizing the g:Profiler tool, employing the Gene Ontology and KEGG databases for the selection of gene sets82. To address the issue of multiple testing, the g:SCS (Set Counts and Sizes) methodology was utilized for correction.
GTEx Tissue Specific Expression Analysis (TSEA)
Tissue-specific expression analysis (TSEA) was performed leveraging RNA-seq data from the GTEx project, which provides expression profiles from a variety of tissues sourced from post-mortem subjects83. The analyses aimed to determine whether genes associated with GDM and T2D exhibited enriched expression in particular tissues, applying hypergeometric tests. Corrections for multiple comparisons were made using the Benjamini–Hochberg method, aiming to maintain a FDR below 0.05.
Cell-type-specific enrichment analysis (CSEA)
Cell-type-specific enrichment analysis (CSEA) was executed using WebCSEA to compare gene sets against tissue-cell-type (TCs) expression signatures, derived from 11 single-cell expression datasets31. Dai et al.31 consolidated expression data from over 5.5 million cells across 111 tissues and 1355 TCs, eliminating low-expression genes and utilizing the “deTS” t-statistic-based method to identify signature genes for each cell type. Genes that ranked in the top 5% of t-statistic scores for specific cell types were categorized as cell type-specific. Fisher’s exact test was employed to investigate if these genes were overrepresented among the shared genes for each disease trait.
Gene-chemical-disease analysis for shared genes between GDM and T2D
Investigation into chemical agents associated with the shared genes between GDM and T2D was facilitated through searches in the Comparative Toxicogenomics Database (CTD) database (https://ctdbase.org/)32. This database, a comprehensive public repository, aggregates extensive information on chemical-gene/protein interactions, as well as chemical-disease and gene-disease relationships, thus supporting predictions regarding the environmental impacts on disease via the curation of gene-chemical-disease interaction data.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The obesity and metabolic disorder GWAS data were obtained from the FinnGen R11 dataset (https://www.finngen.fi/en/access_results). Gene expression and eQTL data are freely available at https://ftp.ebi.ac.uk/pub/databases/spot/eQTL/imported/GTEx_V8. GTEx multi-tissue gene expression weight for FUSION analysis (http://gusevlab.org/projects/fusion/#gtex-v8-multi-tissue-expression). LD reference data of 1000 Genomes for FUSION analysis (https://data.broadinstitute.org/alkesgroup/FUSION/LDREF.tar.bz2). Data for UTMOST analysis files (https://zhaocenter.org/UTMOST). Data for MAGMA analysis files (https://fuma.ctglab.nl/downloadPage).
Code availability
Publicly available code and software were used to perform the analyses. Analysis code and software include RHOGE, UTMOST, FUSION, MAGMA, TwoSampleMR (https://mrcieu.github.io/TwoSampleMR), PheWAS (https://azphewas.com/), GeneMANIA (https://genemania.org/), g:Profiler (https://biit.cs.ut.ee/gprofiler/gost), TSEA, WebCSEA (https://bioinfo.uth.edu/webcsea) and CTD, corrplot package (https://github.com/taiyun/corrplot), forestplot package (https://cran.r-project.org/web/packages/forestplot/vignettes/forestplot.html), UpSetR package (https://github.com/hms-dbmi/UpSetR), pheatmap package (https://github.com/raivokolde/pheatmap), and ggvenn package (https://github.com/NicolasH2/ggvenn).
References
Saravanan, P. Gestational diabetes: opportunities for improving maternal and child health. Lancet Diab. Endocrinol. 8, 793–800 (2020).
Shah, N. S. et al. Trends in gestational diabetes at first live birth by race and ethnicity in the US, 2011-2019. JAMA 326, 660–669 (2021).
Dennison, R. A. et al. The absolute and relative risk of type 2 diabetes after gestational diabetes: a systematic review and meta-analysis of 129 studies. Diab. Res. Clin. Pr. 171, 108625 (2021).
Vounzoulaki, E. et al. Progression to type 2 diabetes in women with a known history of gestational diabetes: systematic review and meta-analysis. BMJ 369, m1361 (2020).
Zhang, C. & Catalano, P. Screening for gestational diabetes. JAMA 326, 487–489 (2021).
Auvinen, A.-M. et al. Type 1 and type 2 diabetes after gestational diabetes: a 23 year cohort study. Diabetologia 63, 2123–2128 (2020).
Kawai, V. K. et al. A genetic risk score that includes common type 2 diabetes risk variants is associated with gestational diabetes. Clin. Endocrinol. (Oxf.) 87, 149–155 (2017).
Ding, M. et al. Genetic variants of gestational diabetes mellitus: a study of 112 SNPs among 8722 women in two independent populations. Diabetologia 61, 1758–1768 (2018).
Powe, C. E. & Kwak, S. H. Genetic studies of gestational diabetes and glucose metabolism in pregnancy. Curr. Diab Rep. 20, 69 (2020).
Kwak, S. H. et al. A genome-wide association study of gestational diabetes mellitus in Korean women. Diabetes 61, 531–541 (2012).
Wu, N. N. et al. A genome-wide association study of gestational diabetes mellitus in Chinese women. J. Matern Fetal Neonatal Med 34, 1557–1564 (2021).
Pervjakova, N. et al. Multi-ancestry genome-wide association study of gestational diabetes mellitus highlights genetic links with type 2 diabetes. Hum. Mol. Genet 31, 3377–3391 (2022).
Elliott, A. et al. Distinct and shared genetic architectures of gestational diabetes mellitus and type 2 diabetes. Nat. Genet. 56, 377–382 (2024).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet 20, 467–484 (2019).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet 47, 1091–1098 (2015).
Hormozdiari, F. et al. Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet 99, 1245–1260 (2016).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet 48, 245–252 (2016).
Mancuso, N. et al. Integrating gene expression with summary association statistics to identify genes associated with 30 complex traits. Am. J. Hum. Genet 100, 473–487 (2017).
Liu, X. et al. Functional architectures of local and distal regulation of gene expression in multiple human tissues. Am. J. Hum. Genet. 100, 605–616 (2017).
Battle, A., Brown, C. D., Engelhardt, B. E. & Montgomery, S. B. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Hu, Y. et al. A statistical framework for cross-tissue transcriptome-wide association analysis. Nat. Genet. 51, 568–576 (2019).
Gui, J. et al. A cross-tissue transcriptome-wide association study reveals novel susceptibility genes for migraine. J. Headache Pain. 25, 94 (2024).
Ni, J. et al. Novel insight into the aetiology of rheumatoid arthritis gained by a cross-tissue transcriptome-wide association study. RMD Open 8, e002529 (2022).
Rodriguez-Fontenla, C. & Carracedo, A. UTMOST, a single and cross-tissue TWAS (Transcriptome Wide Association Study), reveals new ASD (Autism Spectrum Disorder) associated genes. Transl. Psychiatry 11, 256 (2021).
Robitaille, J. & Grant, A. M. The genetics of gestational diabetes mellitus: evidence for relationship with type 2 diabetes mellitus. Genet. Med. 10, 240–250 (2008).
McIntyre, H. D. et al. Gestational diabetes mellitus. Nat. Rev. Dis. Prim. 5, 47 (2019).
Liufu, C. et al. Integration of multi-omics summary data reveals the role of N6-methyladenosine in neuropsychiatric disorders. Mol. Psychiatry 29, 3141–3150 (2024).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Lonsdale, J. et al. The genotype-tissue expression (GTEx) project. Nat. Genet 45, 580–585 (2013).
Dai, Y. et al. WebCSEA: web-based cell-type-specific enrichment analysis of genes. Nucleic Acids Res. 50, W782–W790 (2022).
Davis, A. P. et al. Comparative toxicogenomics database’s 20th anniversary: update 2025. Nucleic Acids Res. 53, 1328–1334 (2024).
Schmidt, M. I. et al. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet 353, 1649–1652 (1999).
Yue, S. et al. Genome-wide analysis study of gestational diabetes mellitus and related pathogenic factors in a Chinese Han population. BMC Pregnancy Childbirth 23, 856 (2023).
Zhen, J. et al. Genome-wide association and Mendelian randomisation analysis among 30,699 Chinese pregnant women identifies novel genetic and molecular risk factors for gestational diabetes and glycaemic traits. Diabetologia 67, 703–713 (2024).
Price, A. L., Spencer, C. C. & Donnelly, P. Progress and promise in understanding the genetic basis of common diseases. Proc. Biol. Sci. 282, 20151684 (2015).
Nicolae, D. L. et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 6, e1000888 (2010).
Lawrence, T. & Gilroy, D. W. Chronic inflammation: a failure of resolution? Int J. Exp. Pathol. 88, 85–94 (2007).
Fu, L. et al. Mediating role of inflammatory biomarkers in the causal effect of body composition on glycaemic traits and type 2 diabetes. Diab. Obes. Metab. 26, 5444–5454 (2024).
Fu, L. et al. Effect of life course body composition on lipids and coronary atherosclerosis mediated by inflammatory biomarkers. Free Radic. Biol. Med. 227, 157–165 (2025).
Fu, L., Han, X., Wang, Y. & Hu, Y. Q. Identifying genetic overlaps in obesity and metabolic disorders unlocking unique and shared mechanistic insights. Free Radic. Biol. Med. 231, 80–93 (2025).
Fu, L. et al. Role of inflammatory biomarkers in mediating causal effect of life course body composition on hypertension. Hypertension 82, e57–e69 (2025).
Fu, L. et al. Insights into causal effects of genetically proxied lipids and lipid-modifying drug targets on cardiometabolic diseases. J. Am. Heart Assoc. 14, e038857 (2025).
Rohm, T. V., Meier, D. T., Olefsky, J. M. & Donath, M. Y. Inflammation in obesity, diabetes, and related disorders. Immunity 55, 31–55 (2022).
Ray, G. W. et al. Genetic and inflammatory factors underlying gestational diabetes mellitus: a review. Front. Endocrinol. 15, 1399694 (2024).
Kuzmicki, M. et al. High resistin and interleukin-6 levels are associated with gestational diabetes mellitus. Gynecol. Endocrinol. 25, 258–263 (2009).
Volkan, G., Emin, Ş, Gözde, A., Hamit, U. & Mustafa, M. Association of gestational diabetes and proinflammatory cytokines (IL-6, TNF-α and IL-1β). J. Embryol. 1, 6–11 (2017).
Pan, X., Jin, X., Wang, J., Hu, Q. & Dai, B. Placenta inflammation is closely associated with gestational diabetes mellitus. Am. J. Transl. Res 13, 4068–4079 (2021).
Mor, G., Cardenas, I., Abrahams, V. & Guller, S. Inflammation and pregnancy: the role of the immune system at the implantation site. Ann. N. Y Acad. Sci. 1221, 80–87 (2011).
Tsalamandris, S. et al. The role of inflammation in diabetes: current concepts and future perspectives. Eur. Cardiol. 14, 50–59 (2019).
Lu, Y. et al. New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat. Commun. 7, 10495 (2016).
Ustianowski, P. et al. COBLL1 and IRS1 gene polymorphisms and placental expression in women with gestational diabetes. Biomedicines 10, 1933 (2022).
Kraja, A. T. et al. Pleiotropic genes for metabolic syndrome and inflammation. Mol. Genet. Metab. 112, 317–338 (2014).
Chen, Z. et al. Functional screening of candidate causal genes for insulin resistance in human preadipocytes and adipocytes. Circ. Res. 126, 330–346 (2020).
Arora, G. P. et al. Association between genetic risk variants and glucose intolerance during pregnancy in north Indian women. BMC Med. Genomics 11, 64 (2018).
Kerr, J. S. & Wilson, C. H. Nuclear receptor-binding protein 1: a novel tumour suppressor and pseudokinase. Biochem Soc. Trans. 41, 1055–1060 (2013).
Ruiz, C. et al. High NRBP1 expression in prostate cancer is linked with poor clinical outcomes and increased cancer cell growth. Prostate 72, 1678–1687 (2012).
Wilson, C. H. et al. Nuclear receptor binding protein 1 regulates intestinal progenitor cell homeostasis and tumour formation. EMBO J. 31, 2486–2497 (2012).
Yasukawa, T. et al. NRBP1-containing CRL2/CRL4A regulates amyloid β production by targeting BRI2 and BRI3 for degradation. Cell Rep. 30, 3478–3491.e6 (2020).
Sengupta, U., Nilson, A. N. & Kayed, R. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 6, 42–49 (2016).
Li, H. et al. Based on systematic druggable genome-wide Mendelian randomization identifies therapeutic targets for diabetes. Front. Endocrinol. 15, 1366290 (2024).
Atkinson, M. A., Eisenbarth, G. S. & Michels, A. W. Type 1 diabetes. Lancet 383, 69–82 (2014).
Taschner, M., Bhogaraju, S. & Lorentzen, E. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation 83, S12–S22 (2012).
Lee, H., Song, J., Jung, J. H. & Ko, H. W. Primary cilia in energy balance signaling and metabolic disorder. BMB Rep. 48, 647–654 (2015).
Zick, Y. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci. STKE 2005, pe4 (2005).
Spencer, J. A., Eliazer, S., Ilaria, R. L. Jr., Richardson, J. A. & Olson, E. N. Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING-finger protein. J. Cell Biol. 150, 771–784 (2000).
Pizon, V. et al. Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MURF2 RING-finger protein. J. Cell Sci. 115, 4469–4482 (2002).
Perera, S., Holt, M. R., Mankoo, B. S. & Gautel, M. Developmental regulation of MURF ubiquitin ligases and autophagy proteins nbr1, p62/SQSTM1 and LC3 during cardiac myofibril assembly and turnover. Dev. Biol. 351, 46–61 (2011).
Perera, S., Mankoo, B. & Gautel, M. Developmental regulation of MURF E3 ubiquitin ligases in skeletal muscle. J. Muscle Res. Cell Motil. 33, 107–122 (2012).
Han, Q. Y. et al. Circulating E3 ligases are novel and sensitive biomarkers for diagnosis of acute myocardial infarction. Clin. Sci. 128, 751–760 (2015).
Olivé, M. et al. New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum. Mol. Genet. 24, 3638–3650 (2015).
Lo Faro, V. et al. Novel ancestry-specific primary open-angle glaucoma loci and shared biology with vascular mechanisms and cell proliferation. Cell Rep. Med. 5, 101430 (2024).
Sun, R., Hui, S., Bader, G. D., Lin, X. & Kraft, P. Powerful gene set analysis in GWAS with the generalized Berk-Jones statistic. PLoS Genet. 15, e1007530 (2019).
Liao, C. et al. Transcriptome-wide association study of attention deficit hyperactivity disorder identifies associated genes and phenotypes. Nat. Commun. 10, 4450 (2019).
de Leeuw, C. A., Stringer, S., Dekkers, I. A., Heskes, T. & Posthuma, D. Conditional and interaction gene-set analysis reveals novel functional pathways for blood pressure. Nat. Commun. 9, 3768 (2018).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. ELife 7, e34408 (2018).
Yuan, S. et al. Plasma proteins and onset of type 2 diabetes and diabetic complications: proteome-wide Mendelian randomization and colocalization analyses. Cell Rep. Med 4, 101174 (2023).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Wang, Q. et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature 597, 527–532 (2021).
Mostafavi, S., Ray, D., Warde-Farley, D., Grouios, C. & Morris, Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 9, S4 (2008).
Warde-Farley, D. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38, W214–W220 (2010).
Raudvere, U. et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Dougherty, J. D., Schmidt, E. F., Nakajima, M. & Heintz, N. Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res. 38, 4218–4230 (2010).
Acknowledgements
This study was supported by grants to F.L.W from the Capital’s Funds for Health Improvement and Research (grant no. Capital’s Funds for Health Improvement and Research 2024-4-20911), National Natural Science Foundation of China (grant no. 82204063), and H.Y.Q from the National Key R&D Program of China (grant no. 2023YFF1205101).
Author information
Authors and Affiliations
Contributions
Study concept and design: L.F. Acquisition of data: L.F. and X.H. Analysis and interpretation of data: L.F. and X.H. Drafting of the manuscript: L.F. and X.H. Critical revision of the manuscript for important intellectual content: L.F., X.H., Y.W., and Y.Q.H. Funding recipients: L.F. and Y.Q.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fu, L., Han, X., Wang, Y. et al. Genetic insights and mechanistic parallels in gestational diabetes mellitus and type 2 diabetes. Nat Commun 17, 660 (2026). https://doi.org/10.1038/s41467-025-67385-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67385-1
