
Abstract
RNA modifications (RMs) are critical for diverse biological processes, but the lack of accurate, quantitative detection methods has limited their study. A large-scale and high-quality training dataset is an essential component for accurate deep learning, but such dataset has been absent for RM detection, resulting in low accuracies. We developed DeepRM (Deep learning for RNA Modification), a sophisticated deep learning framework powered by Nanopore sequencing. DeepRM dataset is a massive-scale, three orders of magnitude larger than the comparable previous ones, and unprecedentedly high-quality dataset that closely mirrors endogenous transcript environments. Accordingly, DeepRM detects RM sites and measures their modification stoichiometries with a near-perfect accuracy. Using DeepRM, we constructed a comprehensive, human m6A atlas at single-molecule resolution that reveals a large number of previously underappreciated non-canonical m6A sites and differentially modified transcripts, highlighting the complexity and dynamic nature of the human epitranscriptome. DeepRM is freely available, providing a unique, powerful opportunity for understanding the biological functions of RMs. DeepRM can also be expanded to various other RMs and organisms, potentially becoming a future standard for investigating the epitranscriptome.
Similar content being viewed by others


Introduction
RNA modifications (RMs) are post-transcriptional modifications of ribonucleotides that regulate diverse biological processes, including gene expression1,2. Among these, N6-methyladenosine (m6A) is the most prevalent in eukaryotic mRNA and thus most extensively investigated2,3,4,5. Since m6A plays pivotal roles in RNA metabolism, controlling nuclear export6, processing7, splicing8, and stability9, dysregulation of m6A is associated with various pathological conditions, including neurodegenerative diseases10, immune disorders11, and cancers12. Understanding the precise roles of m6A requires accurate transcriptome-wide detection, a fundamental milestone that has remained elusive due to technical limitations in previous studies.
Previous m6A detection methods have suffered from low detection accuracy. For instance, antibody-based methods like MeRIP-seq and miCLIP are prone to high false positive rates due to the nonspecific binding of m6A-specific antibodies13,14,15,16. Chemical conversion-based methods, such as m6A-SEAL and m6A-SAC-seq, can produce false negatives due to incomplete chemical conversion17,18. Enzyme-based methods, including DART-seq and m6A-REF-seq, often rely on enzymes that act only at specific sequence motifs, resulting in low detection sensitivity19,20,21. The observation that the total number and positions of RM sites detected with previous methods do not agree well with each other clearly illustrates their limitations (Supplementary Fig. 1). Another critical constraint of the previous approaches is the lack of ability to determine the modification status of individual RNA molecules, that is crucial for studying the interplay between m6A and other biological processes such as mRNA splicing, translation, and other species of RM.
Recently, Nanopore direct RNA sequencing (DRS) has emerged as a promising technology for identifying RM at single-molecule resolution22. DRS determines the sequence of each RNA molecule based on the electric current generated by the RNA molecule when passing through a protein pore. Modified and unmodified RNA nucleotides can be distinguished by detecting subtle differences in electric current22,23. However, the previous approaches to detecting RM via DRS suffer from low detection accuracy caused by incomplete training datasets and ineffective modeling strategies24.
The whole transcriptome sequencing and in vitro transcription (IVT) have been widely used to generate training datasets for RM detection and yet both approaches have serious limitations that lower the accuracy of the models25,26,27,28,29,30,31. First, IVT and the whole transcriptome datasets encompass only a limited subset of local sequence contexts. For instance, the previously reported studies include a small subset (0.0011-1.2%) of possible local sequence contexts when considering flanking 11-mers (see Fig. 1a)25,28,31,32. Second, IVT replaces all adenosine (A) nucleotides with m6As. Such extensive modification does not occur in endogenous conditions, therefore resulting in an unrealistic bias when applied to train a deep learning model24,25,26,27,28,31,33. Third, the whole transcriptome datasets include a markedly larger number of sites with high m6A modification stoichiometry than those with low stoichiometry, because the high-stoichiometry sites can be more easily detected by the previous methods, causing a strong bias in the training dataset29,30. Here, the m6A modification stoichiometry refers to the number of m6As compared to the total number of As at a given site34. Fourth, many A and m6A sites are inaccurately labeled as m6A and A sites in the whole transcriptome datasets due to the low accuracy of the previous m6A detection methods (Supplementary Fig. 1)35,36.

a Overview of DeepRM (top) versus previous approaches (bottom). In the DeepRM dataset, signals of individual modifications are isolated (see panel b), while in most previous datasets, modification signals overlap because all adenosines are modified into m6A, reducing prediction accuracy. Since nanopore signals are largely affected by flanking sequences116, it is essential to include various local sequence contexts in the training dataset for sensitive RM detection. The DeepRM dataset encompasses all possible 11-mer sequence contexts (Fig. 2d), while the previous datasets contain limited and short sequence contexts (Fig. 2e). DeepRM uses raw electric currents as a feature, employing a large Transformer architecture to capture full information from Nanopore sequencing. In contrast, previous models use a few statistical features and shallow architectures. Consequently, DeepRM achieves unprecedentedly high accuracy in RM detection. The precision and recall values shown were calculated from the precision-recall curves of DeepRM (top) and m6Anet (bottom) in Fig. 4c, using the F1-maximizing threshold. b, c Effects of a single RM on surrounding nucleotides. Mean electric currents (b) and base quality (c) are plotted for 20 flanking random unmodified nucleotides centered on A or m6A (gray or red hexagons). The number of central 5-mer sequences was equally selected. n = 1,228,800 for both A and m6A. d Design of 87-nt building blocks (BBs) for large-scale dataset generation. Each BB contains three 21-nt local sequence context blocks (LCBs) in green and four 6-nt spacers in light blue. The base of interest is A (gray) and m6A (red). e Sequence of synthetic 49-nt RNA oligonucleotides and digested fragments for liquid chromatography tandem mass spectrometry (LC MS/MS). f MS chromatogram (left) and spectra (right) of a 13-nt fragment containing the designated site, showing different retention times between A (gray) and m6A oligonucleotides (red). Peak areas for each fragment were integrated to measure A or m6A purity. MS1 spectra of triply charged ions ([M-3H]−3) display measured and theoretical mass/charge ratio (m/z) values and mass difference (△m) between A and m6A oligonucleotides indicated.
Suboptimal computational designs of the previous methods have further lowered their detection accuracies. Most of the previous models relied on a small number of summary statistics such as base-calling errors and the mean and standard deviation of the electric current of DRS25,26,27,29,30,31,37,38,39,40. While these summary statistics contain partially useful information, a fraction of the richer information included in the raw electric current from DRS will be inevitably lost41. Especially, a base-calling error often coincides with the presence of RM, and thus it is one of the most frequently used statistics in the previous studies24. However, it is also affected by the sequencing quality of the reference sequence and often occurs irrelevant to RM, therefore it might not be a reliable feature for detecting RM25,31,39,40. Many of the previous methods identified an m6A site by analyzing 5-to-9-nt sequences flanking to the site, that is insufficient for capturing the full information embedded in the flanking sequences25,26,27,29,31,37,38,39.
To carefully address these limitations from the previous approaches, we developed DeepRM (Deep learning for RNA Modification), an RM detection framework based on an extensiveRM training dataset combined with a sophisticated deep learning model (Fig. 1a). First, we produced a massive-scale, high-quality training dataset, termed DeepRM dataset. Unlike IVT datasets, our DeepRM dataset closely mirrors endogenous transcripts where a single m6A nucleotide is flanked by unmodified nucleotides generated by employing chemical oligonucleotide synthesis42. Since individual entries included in the dataset are precisely labeled at single-molecule resolution, our DeepRM dataset enables the detection of RM in individual transcripts, also improving the quantification accuracy of modification stoichiometry. Second, the DeepRM dataset is free from the bias towards high-stoichiometry sites observed in the transcriptome datasets, because each entry is not a site represented by multiple aligned RNA molecules, but a single RNA molecule. Third, our deep learning model attempts to capture the full information embedded in the raw electric current generated during DRS, that most of the previous models have discarded. A state-of-the-art deep learning architecture, such as a Transformer combined with the raw electric current obtained from a wide range of 21 nucleotides, composed of an m6A or an A nucleotide and its flanking 20 unmodified nucleotides, will be able to overcome most of the previous limitations.
Accordingly, DeepRM shows near-perfect, unprecedentedly high accuracy in m6A detection and quantification, substantially outcompeting the previously published methods. Notably, DeepRM is the first model that accurately detects and quantifies m6As located on the previously determined consensus motif, DRACH (D = A/G/U, R = A/G, and H = A/C/U) as well as all of the non-DRACH motifs thanks to its diverse-context training dataset. DeepRM accurately quantifies m6A even in the low-depth regions, that has been a long-standing problem in epitranscriptomics43. Using DeepRM, we constructed a comprehensive map of the human m6A landscape in multiple cell lines. The human m6A map discovered a large number of non-DRACH m6A sites and differentially modified transcripts across the human transcriptome, illuminating the complexity and dynamic nature of the human epitranscriptome. DeepRM is a flexible framework that can be expanded to detect various species of RMs in diverse samples, organisms, and biological conditions, providing a unique and powerful opportunity to investigate the RM landscape. The DeepRM model, dataset, and m6A atlases are freely available for the research community to facilitate elucidation of the functions and regulatory mechanisms of RMs.
Results
Construction of a massive-scale training dataset for RNA modification detection representing diverse local sequence contexts
We generated a massive-scale training dataset for RNA modification (RM) detection, encompassing diverse local sequence contexts surrounding RM. Each entry in the training dataset is a 21-nt RNA sequenced via Nanopore direct RNA sequencing (DRS), termed local sequence context block (LCB). Each LCB consists of a single A or m6A flanked by 20 random unmodified nucleotides to represent diverse sequence contexts with minimal bias. We chose a large size of 21 nts for an LCB to ensure that most of the effect of local sequence contexts on the electric current can be captured. In the DRS data, modification-dependent variations in electric current and base quality were observed over a wide range of up to ±10 nts from m6A (Fig. 1b, c and Supplementary Fig. 2). We observed that a single m6A influences the DRS electric current beyond the range of 5–9 nts, which is the sequence context size that has been broadly used in the previous studies25,29,38. Hence, a larger sequence context size of 21 nts is beneficial for capturing the modification-induced variation. This result also indicates that electric current signatures from two m6As within a 20-nt window will overlap and therefore interfere with each other, that is one of the limitations of the previous IVT datasets where the distances between m6As are shorter than 20 nts24,25,26,27,28,31,33. On the other hand, our LCB size of 21 nts effectively minimizes such interference, allowing the trained model to correctly learn the electric current characteristics of individual m6As.
To maximize the number of LCBs sequenced in each DRS run, we decided to merge multiple LCBs into a single read. We synthesized 87-nt oligonucleotides containing three LCBs, termed the building blocks (BBs) (Fig. 1d). By employing chemical oligonucleotide synthesis, a single m6A and flanking unmodified nucleotides were incorporated together at designated positions in a single molecule, that is impossible via IVT42. We confirmed that m6A is accurately incorporated at the designated position with > 99.9% purity using liquid chromatography tandem mass spectrometry (LC MS/MS) (Fig. 1e, f and Supplementary Fig. 3). Optimized 5′ and 3′ end sequences of the BB were experimentally determined, so that multiple BB molecules are efficiently ligated together in a single reaction, producing long RNA reads that cannot be chemically synthesized (Fig. 2a and Supplementary Fig. 4a, b). Since RNAs shorter than 200 nts cannot be effectively sequenced by DRS44, we selected ligation products longer than 300 nts. The selected ligation products, each of which containing at least 12 LCBs, were sequenced via DRS.

a The experimental workflow for generating the DeepRM dataset. While some BBs are ligated intramolecularly to form circular RNAs, BBs ligated intermolecularly produce linear RNAs that can be further concatenated through iterated reactions (left). RNAs with lengths in multiples of 87 nts are confirmed via PAGE, and > 300-nt RNAs containing ≥ 12 LCBs are selected. Poly(A) tails are added, and these polyadenylated linear RNAs are isolated by poly(A) + selection with circular side products removed (middle). The isolated linear RNAs are sequenced via Nanopore direct RNA sequencing (right). b, c The amount of sequenced data for the DeepRM dataset. The number of reads obtained in each Nanopore direct RNA sequencing run (b) and the number of LCBs extracted from the sequenced reads (c) are displayed with A and m6A datasets in gray and red, respectively. d The depth distribution of individual 11-mer motifs in the DeepRM dataset, presented as an inverse survival function plot with 11-mer motifs harboring A (gray) or m6A (red) at the center (n = 1,048,576 for A and m6A each). The x-axis represents the cumulative fraction among all possible 11-mer motifs, and the y-axis represents log10(the number of LCBs containing each motif). The dotted line indicates the lowest depth observed in the DeepRM dataset. e The coverage comparison between the DeepRM dataset and the previous datasets (Curlcake25,26, ELIGOS31, IVET28, and mAFiA42). 11-mer coverage of each dataset, defined as the fraction of 11-mer motifs that have unmodified flanking nucleotides with depths of >10 for both A and m6A datasets among all possible 11-mers, is displayed as a box plot (left). For IVT datasets (Curlcake, ELIGOS, and IVET), 11-mers that contain A in the flanking region were excluded, since those As are modified into m6A. The number of 11-mers and the 11-mer coverage of each dataset are displayed (right) with the tables presenting consensus motifs (B = C/G/U, D = /G/U, H = A/C/U, N = A/C/G/U, and R = A/G) and examples of individual motifs included. Red hexagons represent m6A.
The size and sequence diversity of the DeepRM dataset largely surpass all previous datasets for RM detection. From DRS data composed of 33.9 million A and 30.6 million m6A reads (Fig. 2b and Supplementary Table 1), we obtained 181 million A and 116 million m6A LCBs (Fig. 2c), comprised of 6.24 billion nucleotides in total, surpassing the size of all previous m6A datasets with unmodified sequence contexts, including mAFiA by 2600 folds42. It covers all possible 1,048,576 11-mer motifs with a sequencing depth of >10 for both A and m6A (Fig. 2d). The mean depths of the 11-mer motifs are 172.7 for A and 110.7 for m6A. Indeed, the DeepRM dataset exceeds all previous datasets, showing an 85-to-87,000-fold increase in 11-mer motif coverage (Fig. 2e). Our DeepRM dataset that comprehensively represents the RM signal across a broad range of local sequence contexts, provides a pivotal foundation for training an accurate RM detection model.
RNA sequences and modification states are accurately annotated at single-molecule resolution in the DeepRM dataset
To train a deep learning model that can detect RM in individual transcripts, the sequence and modification states of each RNA molecule should be accurately annotated in the training dataset. While accurate single-molecule resolution annotation has been unavailable for the previous transcriptome datasets due to the absence of information on the modification states of individual transcripts, our DeepRM dataset allows precise single-molecule annotation by sequencing chemically synthesized LCBs that contain modification at predefined positions. The sequences and modification states can be accurately determined once LCBs are successfully reconstructed from the DRS data.
Since our LCB sequences are designed to be filled with random nucleotides without their DNA templates, traditional alignment methods that align sequenced RNA reads to DNA templates cannot be applied to our DeepRM dataset (Fig. 1d). Besides, insertions and deletions introduced by DRS randomly shift LCBs within the reads, making reliably reconstructing LCBs from the DRS reads a significant challenge. To address this problem, we developed an algorithm that reconstructs LCBs by aligning the reads to 6-nt predefined sequences that are placed between the LCBs, termed spacers, and by finding the arrangement of aligned spacers and an A located in the center of an LCB that most closely matches our design (Figs. 1d, 3a and Supplementary Fig. 4c; see Methods for implementation details). The alignment result provides the sequences and positions of LCBs in each read, allowing accurate reconstruction of LCBs at single-molecule resolution.
We evaluated the accuracy of our template-free LCB reconstruction algorithm. For the evaluation purpose, a DNA template that exactly resembles an LCB ligation product with the random sequences replaced by arbitrarily chosen fixed sequences was prepared and regarded as the ground truth. We generated RNA sequences by in vitro-transcribing the DNA template, performed DRS with the transcribed RNA, and attempted to reconstruct LCBs only by analyzing the DRS reads without using the DNA template information. We compared the reconstructed LCBs with the ground truth DNA template by defining the precision as the fraction of correctly located LCBs among the reconstructed LCBs and the recall as the fraction of correctly located LCBs among the LCBs in the DNA template. Our LCB reconstruction algorithm achieved a precision of 0.999 with 0.515 and 0.445 of recall for A and m6A LCBs, respectively. We prioritized precision over recall to ensure that the entries in the DeepRM dataset are correctly annotated. Our algorithm outperformed the simpler position-based algorithms that showed much lower recall of 0.056-0.213 and 0.043-0.174 at the 0.999 precision level for A and m6A LCBs, respectively (Fig. 3b). We also verified an equivalent LCB reconstruction accuracy in another dataset whose sequence is highly different from our original one (Supplementary Fig. 4d), demonstrating that our LCB reconstruction algorithm accurately locates LCBs within the RNA reads without their DNA templates across diverse sequence contexts. Consistently, the reconstructed LCB sequences showed high percent identity to the DNA template across the entire 21-nt range, with a median of 99.5% (Fig. 3c), illustrating that the sequence and modification state of each 21-nt LCB in our DeepRM dataset are accurate and reliable.

a The reconstruction algorithm of local sequence context blocks (LCBs). Sequenced reads deviate from the design due to sequencing errors (top). To locate the LCBs (green) accurately, each read is aligned to the design using fixed-sequence spacers (blue; middle). LCBs are then located between the aligned spacers (bottom). b The precision-recall (PR) measurement of the LCB reconstruction alignment using an in vitro-transcribed dataset (see “Methods”). The PR curves of A and m6A LCB reconstruction are shown (right). The graph-based reconstruction algorithm (blue) was compared against simple algorithms using the distance from the first spacer from the 3′ end (orange), and using the distance from the 5′ end of the poly(A) tail (green). Dashed lines indicate extrapolated values between the maximum achievable recall and 1.0. The maximum achievable precision is indicated by red dots, and its corresponding recall is depicted by gray lines. c The accuracy of the reconstructed LCB sequences. The substitution (first from the top), insertion (second), and deletion (third) rates are calculated between the reconstructed LCBs and DNA template sequences. 1.0 minus the sum of the substitution, insertion, and deletion frequencies is shown (bottom). d The length of reads obtained from A (gray) and m6A (red) sequencing runs. Boxes, center lines, and whiskers represent the 25th-75th percentiles, the median, and ± 1.5 × interquartile range, respectively (n = 1000). e The correlation of electric currents for center A between replicates. Signal intensities at central A of an 11-mer motif at the center of LCB are compared between the two replicates. Each point represents an 11-mer motif with depth ≥50 in both replicates. Pearson’s R2 and Spearman’s ρ2 values are shown in each plot. The color scale indicates Gaussian kernel-estimated density. n = 618,976 (A rep. 1 vs. A rep. 2), 138,480 (A rep. 1 vs. m6A rep. 1), 505,623 (A rep. 1 vs. m6A rep. 2), 144,246 (A rep. 2 vs. m6A rep. 1), 661,463 (A rep. 2 vs. m6A rep. 2), and 144,081 (m6A rep. 1 vs. m6A rep. 2).
Beyond the high accuracy of LCB reconstruction, the similar length distribution between A and m6A reads indicates that the ligation step did not introduce a modification-dependent bias (Fig. 3d). The whole process of dataset construction was highly reproducible based on the strong correlation observed between the electric currents of independently synthesized and sequenced replicates (R2 ≥ 0.99 for all replicate pairs) (Fig. 3e). Taken together, these analyses suggest that our DeepRM dataset is a massive-scale, high-quality training dataset for m6A detection, facilitating the development of a highly accurate deep learning model.
DeepRM accurately detects m6A sites within diverse local sequence contexts
Based on the massive-scale DeepRM dataset, we developed a deep learning model (Fig. 4a, see Methods for the model architecture). The model detects the m6A signature from the DRS reads by distinguishing the raw electric currents generated by m6A and A, as well as their flanking 20-nt RNA sequence contexts (Fig. 4b). We adopted the Transformer architecture to capture the long-range interaction between the electric current and the sequence information embedded in the 21-nt LCBs. We compared the m6A detection accuracy of DeepRM with the previously reported Nanopore-based methods, m6Anet45, Dorado29, and SingleMod46. For the evaluation, consensus m6A sites based on four experimental m6A detection methods, GLORI47, m6A-SAC-seq18, m6ACE-seq48, and miCLIP249, none of which has been used to train DeepRM, were used as the ground truth.

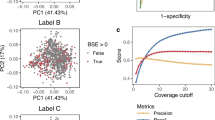
a The input features and architecture of DeepRM. Esig, i, Eseq, i, Epos, i, and Oi denote signal embedding, sequence embedding, positional encoding, and encoder output at position i. L, boi, and k denote the maximum token length, position of base of interest whose modification status is to be determined, and the number of tokens corresponding to the base of interest minus one. pmod is the modification probability and ⊙ is the element-wise product operator. b Tokenization of the input features. RNA sequence is tokenized with overlapping 5-mers. Base quality and dwell time are tokenized without transformation. Normalized electric current is tokenized with an overlapping sliding window. RNA sequence, base quality, and dwell time are repeated to align with the electric current. Token colors represent the corresponding nucleotides. c The precision-recall (PR, top) and receiver operating characteristic (ROC, bottom) curves of DeepRM, Dorado, m6Anet, and SingleMod. Parentheses show the area under the curve (AUC). Dotted lines indicate extrapolated values between the maximum achievable recall and 1.0. Sites with a sequencing depth of ≥ 20 were evaluated. d The PR (top) and ROC (bottom) curves at DRACH (left) and non-DRACH (middle) contexts. For the non-DRACH context, the results for the sites with a depth of ≥ 100 are also shown (right). Otherwise as in (c). e The PR (top) and ROC (bottom) curves at 15-mer motifs observed (left) and unobserved (right) in the training dataset. Otherwise, as in (c). f Ablation of the raw electric current feature using statistical features. The PR (top) and ROC (bottom) compare the original DeepRM with raw current (blue) to an ablated version using statistical features (orange). Otherwise as in (c). g The PR (top) and ROC (bottom) curves for identifying singletons (first column) and sparsely (second), moderately (third), and densely (fourth) clustered m6A sites. Sparsely, moderately, and densely clustered m6A sites are those within 15-21 nts (n = 754), 8-14 nts (n = 731), and 1-7 nts (n = 369) from another m6A site, respectively; singletons are non-clustered (n = 19,811). Otherwise as in (c).
DeepRM demonstrated high accuracy in detecting m6A sites within the human transcriptome (Fig. 4c). With an area under the precision-recall curve (AU-PR) of 0.943 and the receiver operating characteristic curve (AU-ROC) of 0.997. DeepRM outperformed m6Anet (AU-PR = 0.440 and AU-ROC = 0.738), Dorado (AU-PR = 0.559 and AU-ROC = 0.989) and SingleMod (AU-PR = 0.657 and AU-ROC = 0.899). Notably, DeepRM showed robust accuracy not only in DRACH (AU-PR = 0.958 and AU-ROC = 0.997) but also in non-DRACH (AU-PR = 0.715 and AU-ROC = 0.966) contexts (Fig. 4d). For the non-DRACH context, the accuracy was significantly increased when the sequencing depth was ≥100 (AU-PR = 0.833 and AU-ROC = 0.990). In contrast, m6Anet does not detect the non-DRACH sites, and Dorado and SingleMod showed a poor accuracy in non-DRACH contexts (AU-PR = 0.048 and AU-ROC = 0.966; AU-PR = 0.239 and AU-ROC = 0.821, respectively). Moreover, we verified that DeepRM outperformed the second-best performing tool, Dorado, regardless of its detection threshold choice (Supplementary Fig. 5a), and exhibited an equivalently robust performance even for 15-nt sequence contexts unobserved in the training dataset (AU-PR = 0.944 and AU-ROC = 0.999) (Fig. 4e). DeepRM’s unprecedentedly accurate detection of m6A sites across all contexts can be attributed to the DeepRM dataset that encompasses diverse local sequence contexts and the sophisticated deep learning model of DeepRM that uses the full electric current (Fig. 4f and Supplementary Fig. 6).
We examined whether DeepRM accurately detects clustered m6A sites47, even though it could be challenging given that the DeepRM training dataset only consisted of singleton m6As flanked by 20 unmodified nucleotides. For evaluation, we classified an m6A site >21 nts, 15–21 nts, 8–14 nts, or 1–7 nts apart from its nearest m6A site as a singleton, sparsely clustered, moderately clustered, or densely clustered site, respectively. To our surprise, DeepRM maintained a consistently high detection accuracy across the singletons (AU-PR = 0.940 and AU-ROC = 0.997), sparsely clustered (AU-PR = 0.959 and AU-ROC = 0.998), moderately clustered (AU-PR = 0.957 and AU-ROC = 0.999), and densely clustered (AU-PR = 0.903 and AU-ROC = 0.996) sites, outperforming Dorado and m6Anet in all four tested groups (Fig. 4g). To add another layer of validation, we sequenced a synthetic RNA produced via IVT and generated virtual m6A sites with designated sequencing depths and modification stoichiometries (Fig. 5a, see “Methods”). DeepRM showed the highest accuracy across a wide range of depths and stoichiometries, with the highest performance gain observed at low-stoichiometry sites (Fig. 5b). We reaffirmed the robust performance of DeepRM across both DRACH and non-DRACH contexts, and for clustered m6A sites (Fig. 5c, d). Moreover, we assessed the single-molecule resolution m6A detection accuracy of DeepRM using the synthetic evaluation dataset. DeepRM substantially outperformed all other tools in molecule-level m6A detection (Fig. 5e).

a Schematic of the IVT-based synthetic evaluation dataset. 5-mer motifs (red) centered with A are located across a 1000-nt RNA sequence, separated by randomized sequences of C, G, and U (black). The separation distances include all distances between 1 and 27 nts (green). b Site-level m6A detection performance tested on a synthetic evaluation dataset. 36 virtual mixes of the synthetic RNA reads, generated across six sequencing depths (x-axis) and six modification stoichiometries (y-axis) with 1000 resamplings, were used. PR curves of four m6A detection tools (DeepRM, Dorado, m6Anet, and SingleMod) are shown for combination. c m6A site detection accuracy for clustered m6A sites using the synthetic dataset. The sites were binned into six groups based on the distance to the nearest m6A site. AU-PR values of the four m6A detection tools are shown. d m6A site detection accuracy for DRACH and non-DRACH m6A sites using the synthetic dataset. AU-PR values of the models are plotted. n.d.: no m6A site detected by the corresponding model. e Molecule-level m6A detection performance tested on the synthetic dataset. The ROC and PR curves of the four m6A detection tools are shown with the AUC indicated in parentheses.
To experimentally evaluate the specificity of m6A detection by DeepRM, we produced a cDNA library by reverse-transcribing the HEK293T transcriptome and performed IVT of the cDNA library to generate the HEK293T transcriptome absent of any RM (Fig. 6a)50,51. Any m6A site detected in this IVT dataset would be a false positive, enabling reliable quantification of the false positive rate for DeepRM and the other methods. DeepRM detected only 90 A sites (0.00230%) in the IVT dataset as m6A, in contrast to identifying 111,527 m6A sites (0.948%) in the HEK293T whole transcriptome (Fig. 6b). The false positive rate of DeepRM was orders of magnitude lower than Dorado (0.331%), m6Anet (0.121%), and SingleMod (0.0354%), underscoring the remarkably high specificity of DeepRM. This was also 60-fold lower than that of GLORI, a chemical conversion-based m6A quantification method that has called 5006–5706 false positive m6A sites from their published HEK293T IVT dataset, suggesting that DeepRM has a substantially higher specificity than the leading non-Nanopore-based method as well47. To further verify the high specificity of DeepRM, we assessed whether m6A sites that disagree between DeepRM and Dorado in the HEK293T transcriptome are verified by GLORI. 74.9% of sites uniquely detected by DeepRM (n = 8672) were validated by GLORI, while only 1.20% of sites uniquely detected by Dorado (n = 7946) were validated, confirming the unparalleled specificity of DeepRM (Fig. 6c).

a A schematic illustration of the transcriptome-wide in vitro transcription (IVT) experiment to estimate the false positive rate. b The fraction of A sites identified as m6A sites in the HEK293T IVT transcriptome (left bars), that is the false positive rate, and the non-IVT whole transcriptome (right bars). c A Venn diagram of m6A sites detected by Dorado and DeepRM in the HEK293T whole transcriptome (left) and fraction of sites validated via GLORI (right). d SELECT result for sites detected by DeepRM (see “Methods”). Three sites (KDM4A A3515, NTN1 A5645, MKI67 A980) were identified by both DeepRM and Dorado, and the other three sites (KHDRBS1 A1533, KAT6A A777, MICU1 A1871) were identified by DeepRM but not Dorado. The mean and SEM of 2-ΔΔCt values of wild type (WT) and IVT samples are shown for each site, with the one-sided Student’s t test results (n = 3 for each sample). p-values: 2.75e-6 (KDM4A A3515), 8.30e-4 (NTN1 A5645), 4.41e-3 (MKI67 A980), 7.91e-5 (KHDRBS1 A1533), 2.06e-3 (KAT6A A777), and 2.20e-3 (MICU1 A1871). *: p < 0.05, **: p < 0.01, ***: p < 0.001, n.s.: not significant (p ≥ 0.05).
We additionally validated the non-DRACH m6A sites discovered by DeepRM via single-base elongation- and ligation-based qPCR amplification method (SELECT)52,53. Three sites identified by both DeepRM and Dorado (KDM4A A3515, NTN1 A5645, and MKI67 A980) along with three sites detected by DeepRM but not by Dorado (KHDRBS1 A1533, KAT6A A777, and MICU1 A1871) were tested. All of the six non-DRACH sites were validated as m6A sites by SELECT (Fig. 6d), confirming that DeepRM accurately detects m6A sites in non-consensus sequence contexts that many of the previously published methods are unable to detect25,26,29,30,42. Overall, these evaluation results clearly prove that DeepRM can accurately and reliably detect m6A sites within diverse local sequence contexts across the human transcriptome.
DeepRM precisely quantifies m6A modification stoichiometry
Accurate quantification of RM stoichiometry is a key to understanding the dynamic regulation of the epitranscriptome landscape20. Unlike the previous models trained at a site level29,30,37, DeepRM detects RM in individual transcripts at single-molecule resolution, enabling DeepRM to not only detect m6A but also quantify the modification stoichiometry of individual m6A sites. DeepRM computes the modification probability of the A base in each RNA read and then combines the single-molecule predictions for each site to calculate the modification stoichiometry with an information theory-based algorithm (see “Methods”).
To assess DeepRM’s accuracy of modification stoichiometry measurement, we obtained the datasets of GLORI and eTAM-seq, a chemical-based and an enzyme-based experimental method, respectively, for m6A quantification47,54, and compared DeepRM with other Nanopore-based methods. When evaluated with the GLORI dataset in the HEK293T transcriptome, DeepRM exhibited a remarkably stronger correlation of m6A modification stoichiometry with GLORI (R² = 0.912), outperforming Dorado (R² = 0.551), m6Anet (R² = 0.572), and SingleMod (R² = 0.756) (Fig. 7a). Similarly, the eTAM-seq dataset in the HeLa transcriptome confirmed that DeepRM achieved a significantly higher correlation (R² = 0.850) than Dorado (R² = 0.383), m6Anet (R² = 0.491), and SingleMod (R² = 0.710) (Fig. 7b). For additional validation, we evaluated the modification stoichiometry accuracy using commonly detected m6A sites between DeepRM and each of the other tools, confirming that DeepRM has the highest accuracy in both DRACH and non-DRACH contexts (Supplementary Figs. 7, 8).

a The scatter plots of m6A modification stoichiometry in the HEK293T transcriptome quantified by GLORI (x-axis) and four Nanopore-based methods (DeepRM, Dorado, m6Anet, and SingleMod; y-axis). m6A modification stoichiometry of those sites with sequencing depths of > 40, 31–40, 21−30, 11–20, and 5–10 are shown (top to bottom). For depths of ≤ 20, the m6Anet result is not shown since m6Anet requires a minimum sequencing depth of 20. The color scale indicates Gaussian kernel-estimated density. Pearson’s R2, Spearman’s ρ2, and the number of samples are shown. b The scatter plots of m6A modification stoichiometry in the HeLa transcriptome quantified by eTAM-seq (x-axis) and the four Nanopore-based models (y-axis). Otherwise, as in (a).
As a single-molecule resolution model, DeepRM can quantify even the low-depth m6A sites that were not readily quantifiable with the previous methods29,37. To evaluate DeepRM’s ability to quantify m6A in the low-depth regions, we evaluated its performance in HEK293T and HeLa transcriptomes at various sequencing depths. For moderate sequencing depths between 31 and 40, DeepRM exhibited strong correlations (R² = 0.834 in HEK293T and R² = 0.755 in HeLa), notably outperforming Dorado (R² = 0.575 in HEK293T and R² = 0.454 in HeLa), m6Anet (R² = 0.545 in HEK293T and R² = 0.430 in HeLa), and SingleMod (R² = 0.588 in HEK293T and R² = 0.571 in HeLa). Similarly, for lower sequencing depths between 21 and 30, DeepRM maintained strong correlations (R² = 0.796 in HEK293T and R² = 0.718 in HeLa), again surpassing Dorado (R² = 0.559 in HEK293T and R² = 0.457 in HeLa), m6Anet (R² = 0.546 in HEK293T and R² = 0.418 in HeLa), and SingleMod (R² = 0.546 in HEK293T and R² = 0.516 in HeLa).
Next, we assessed DeepRM’s performance in low-depth regions of ≤ 20 where the prediction results of m6Anet were unavailable, and thus it was excluded from the comparison. For sequencing depths between 11 and 20, DeepRM showed a strong correlation (R2 = 0.690 in HEK293T and R2 = 0.598 in HeLa), outperforming Dorado (R2 = 0.482 in HEK293T and R2 = 0.386 in HeLa) and SingleMod (R2 = 0.448 in HEK293T and R2 = 0.401 in HeLa). Even for lower depths between 5 and 10, DeepRM exhibited a much more robust correlation (R2 = 0.477 in HEK293T and R2 = 0.348 in HeLa) than Dorado (R2 = 0.314 in HEK293T and R2 = 0.221 in HeLa) and SingleMod (R2 = 0.281 in HEK293T and R2 = 0.225 in HeLa). Also, we used the IVT-based synthetic dataset to evaluate the modification stoichiometry accuracy of DeepRM across diverse depths and stoichiometries, showing that DeepRM is the most accurate tool across all of the evaluated depth and stoichiometry ranges (Supplementary Fig. 9). In conclusion, these results demonstrate that DeepRM accurately quantifies m6A across a broad range of sequencing depths, maintaining its high accuracy even in regions with low sequencing depths.
DeepRM discovers a large body of non-DRACH m6A sites in the human transcriptome
With DeepRM, we detected 134,706 and 115,494 m6A sites in the transcriptome of HEK293T and HeLa cell lines, respectively, with a false discovery rate of 0.005 (Supplementary Data 1 and 2). Our estimated number of m6A sites in the HEK293 transcriptome places between the numbers previously reported by miCLIP2 (n = 36,556), m6ACE-seq (n = 33,163), m6A-SAC-seq (n = 12,234), and GLORI (n = 176,642)18,47,48,49. Similarly, our estimated number of m6A sites in the HeLa transcriptome is in line with that reported by eTAM-seq (n = 80,941)54.
We discovered that 10.1% (n = 13,650 in HEK293T) to 10.2% (n = 11,760 in HeLa) of the m6A sites are located within non-DRACH motifs (Fig. 8a), also consistent with the previous reports, miCLIP2 (6.33%) and GLORI (14.7%). Almost all detected non-DRACH sites (96.1% in HEK293T and 95.9% in HeLa) had a single nucleotide deviation from the DRACH motif, with GGACG (31.1% in HEK293T and 29.7% in HeLa) and GGAUU (17.5% in HEK293T and 17.7% in HeLa) being the most frequent motifs (Fig. 8b). We also noticed that a small fraction of non-DRACH m6A sites is located in the contexts that diverge far from the DRACH motif (0.0934% in HEK293T and 0.167% in HeLa), including CCAUG, CCAGG, and CUAUG motifs. Intriguingly, the non-DRACH m6A sites displayed quite distinct characteristics compared to the DRACH sites. The m6A modification stoichiometry distribution of the non-DRACH sites was unimodal and centered at low modification stoichiometry, in contrast to the clear bimodal distribution observed for the DRACH sites (Fig. 8c). The non-DRACH m6A sites were more concentrated on the CDS compared to the DRACH m6A sites, while the background distribution of the non-DRACH and DRACH motifs exhibited the opposite pattern (Fig. 8d). The non-DRACH m6A sites detected in both cell lines exhibited significantly elevated sequence conservation than their unmodified controls and a similarly increased sequence conservation was observed for the DRACH m6A sites (Fig. 8e), suggesting important biological functions of both DRACH and non-DRACH m6A sites. The biological significance of the differences between the DRACH and non-DRACH sites remains elusive, making it a compelling focus for future research, that an accurate and versatile method, such as DeepRM, will be able to facilitate.

a The composition of m6A sites detected in the HEK293T (n = 134,706, left) and HeLa (n = 115,494, right) transcriptomes. The number and relative fraction of DRACH mRNA, DRACH ncRNA, non-DRACH mRNA, and non-DRACH ncRNA sites are shown. b The 20 most common 5-mer contexts with their numbers of sites and the 11-mer consensus sequence logo of the m6A sites detected in the HEK293T (left) and HeLa (right) transcriptomes. The x- and y- axes of the sequence logo indicate the relative position from m6A and information content117, respectively. c The distribution of the m6A modification stoichiometry detected in the HEK293T and HeLa transcriptomes, shown as a kernel density estimate (KDE)118,119, at all (left), DRACH (top right), and non-DRACH (bottom right) contexts. d The metagene profiles of DRACH and non-DRACH motifs. The positional distributions of DRACH (blue) and non-DRACH (orange) motifs are shown for the m6A sites (top) and all A sites (bottom) detected in the HEK293T (left) and HeLa (right) transcriptomes, shown as a KDE. Sites with a sequencing depth of ≥ 20 were used. The plotted lengths of the 5′ UTR, CDS, and 3′ UTR on the x-axis are scaled by their average lengths of human mRNAs. e, The phyloP conservation scores of A and m6A sites within DRACH and non-DRACH contexts in HEK293T (DRACH A and m6A (n = 18,495 each) and non-DRACH A and m6A (n = 2215 each)) and HeLa (DRACH A and m6A (n = 15,105 each) and non-DRACH A and m6A (n = 1837 each)) transcriptomes. The boxes, center lines, and whiskers represent the 25th-75th percentiles, the median, and ± 1.5 × interquartile range, respectively. The two-sample one-sided Kolmogorov–Smirnov test results are shown. *: p < 0.001.
m6A modification is a global and dynamic process across the whole transcriptome
To gain a global insight into the impact of m6A on the human epitranscriptome, the m6A sites detected in the transcriptome of HEK293T and HeLa cell lines were analyzed for individual genes. The vast majority of the expressed genes (87.2% in HeLa and 87.4% in HEK293T, see Methods) harbored at least one m6A site (Fig. 9a). The median number of m6A sites in each gene was 6 in both HEK293T and HeLa. Thousands of genes were heavily m6A modified with each of 1648 (13.3%) and 2051 (14.8%) of the expressed genes in HeLa and HEK293T, respectively, accommodating >20 m6A sites. The most heavily modified genes, including EXD3, ADAT3, and MFSD5, had >10% of their A sites modified into m6A (Fig. 9b), demonstrating that m6A modification is a widespread and frequently occurring process across the human transcriptome.

a The histogram (top) and cumulative distribution function (bottom) of the number of m6A-modified sites within a gene detected in the HEK293T (blue) and HeLa (orange) transcriptomes. Dashed lines and the corresponding numbers indicate 50th and 90th percentiles for each cell line, and gray lines indicate 50th and 90th percentiles of the expressed genes. b Top 10 genes harboring the largest number of m6A sites and their respective number of m6A sites detected in the HEK293T (left) and HeLa (right) transcriptomes. c Examples of the differentially modified genes between the HEK293T and HeLa transcriptomes. Modification stoichiometries of detected m6A sites are shown as bars (left y-axis), and log10(sequencing depth) is shown as area plots (right y-axis). Those sites with a sequencing depth of < 20 in either transcriptome were not included in the plots. d The relative fraction of DRACH and non-DRACH contexts among the static and differential m6A sites. Static and differential sites are those sites modified in both and only in one of the transcriptomes, respectively. The relative fraction of the non-DRACH sites is indicated (left). Examples of genes containing differentially modified non-DRACH sites are shown (right). Otherwise, as in (c). e The relative fraction of static and differential m6A sites among the m6A sites detected within weakly (with < 5 m6A sites), moderately (with 5–10 m6A sites), and heavily (with > 10 m6A sites) modified genes. The relative fractions of the static and differential m6A sites are indicated (middle). Examples of genes only containing the static sites (left) and containing the differential sites (right) are shown. Otherwise, as in (c).
By comparing the epitranscriptome of HeLa and HEK293T, we discovered that most of the commonly expressed genes (66.4%, n = 7604) were differentially modified between the two cell lines (Fig. 9c). These differentially modified genes include SNX25 (TGF-β signaling regulator)55, HTT (autophagy regulator)56, NCOA2 (cell proliferation regulator)57, and MAP2K7 (stress response and apoptosis regulator)58. Particularly, APOBEC3B, a DNA cytosine deaminase that drives tumorigenesis59, had two m6A sites in HEK293T but none in HeLa. In contrast, VASP, associated with tumor cell proliferation and migration60, was exclusively methylated in HeLa. Differentially modified sites, defined herein as sites exclusively modified in one of the cell lines, are enriched with non-DRACH m6A sites, consistent with a previous report37. 8.7% of static and 19.3% of differential m6A sites were placed within non-DRACH m6A sites, resulting in 2.2 times enrichment (Fig. 9 d).
We further found that the weakly modified genes with <5 m6A sites per gene were enriched with differentially modified m6A sites (Fig. 9e). Specifically, 29.8% of the m6A sites located on the genes with <5 m6A sites were differentially modified, including DCAF11 (E3 ubiquitin ligase substrate adapter) and BCAM (cell adhesion molecule). In contrast, only 18.9% of the m6A sites located on the genes with >10 m6A sites were differentially modified, including NHSL3 (cell migration regulator) and STAU2 (mRNA trafficking regulator) (Fig. 9e). These results on differentially modified non-DRACH m6A sites reveal the dynamic nature of m6A sites on the human transcriptome, again highlighting the importance of accurate detection of m6A across diverse sequence contexts.
Single-molecule resolution detection of m6A reveals co-occurring m6A sites and those sites associated with alternative mRNA splicing
One of the key advantages of DeepRM over the previous RM detection methods13,14,15,16,17,18,19,20,21 is its single-molecule resolution. By accurately detecting m6A within individual reads, DeepRM enables the single-molecule analysis of the epitranscriptome, such as studying the co-occurrence between RM sites. Using DeepRM, we discovered 4819 pairs of significantly co-occurring m6A sites across the HEK293T transcriptome (Fig. 10a). One of the most striking examples was discovered in SHQ1 (Fig. 10b), where the m6A co-occurred in 40.0% of the reads, about twofold higher than the expected level of 21.8% (q = 3.0 × 10−10). This result suggests a possibility of molecule-level coordination between m6A sites.

a Schematic of a co-occurring pair of m6A sites. The reads mapped to a pair of m6A sites were classified into four groups (modified at both, only at the first, only at the second, and in neither of the sites), and the co-occurrence was examined by Fisher’s exact test. b An example of a co-occurring pair of m6A sites in SHQ1. The positions of the significantly co-occurring m6A sites are shown in red (top left) with m6A modification scores of individual reads, categorized into four groups as in a (bottom left). The fraction of reads belonging to each group and the fraction of modified reads at each site are shown (bottom left). The numbers of reads classified based on modification status at the two co-occurring sites are shown (right) with the q-value from Fisher’s exact test with Benjamini-Hochberg correction. c Schematic of an exon skipping-associated m6A site. Each read was classified into either exon-included (yellow) or exon-skipped (green) group, and subsequently classified into either modified or unmodified group. The association between m6A and exon skipping was examined by Fisher’s exact test. d An example of an exon skipping-associated m6A site in TPT1. The read depth of the two types of isoforms generated by exon skipping is shown with the positions of m6A sites in red (top). The transcript structures are shown in the middle genomic track with the per-read m6A modification scores of individual reads, categorized as in (c) (bottom). 50 reads were randomly subsampled from each group, either exon-included (yellow) or exon-skipped (green), for visualization. The fractions of modified and unmodified reads in each type of isoforms are shown with the q-value from Fisher’s exact test with Benjamini-Hochberg correction.
Another example where the single-molecule resolution of DeepRM would be particularly useful is alternative mRNA splicing. The association between m6A and RNA splicing has been known8,16,61, but systematic analysis has been limited by the lack of an accurate single-molecule RM detection technique62,63. We searched for m6A sites associated with exon skipping, the most prevalent type of alternative mRNA splicing, and identified dozens of sites across the human transcriptome (Fig. 10c). One example was located within TPT1 (Fig. 10d), where the exon-skipped isoform had a higher fraction of modified reads (29.8%) compared to the exon-retained isoform (0.6%; q = 2.8 × 10−26). This result demonstrates that the unique single-molecule RM detection capability of DeepRM can help illuminate the relationship between RM and alternative mRNA processing through comprehensive, transcriptome-wide analysis (manuscript in preparation).
Discussion
In this study, we introduced DeepRM, a framework that detects m6A modification with an unprecedentedly high accuracy. DeepRM is based on the largest m6A modification dataset to date, encompassing all possible 11-mer sequence contexts. DeepRM provides a comprehensive view of the m6A landscape across the human transcriptome, identifying a large number of non-DRACH as well as DRACH m6A sites. DeepRM successfully quantified m6As even in the low-depth regions where the previous methods often failed to quantify, underscoring its high sensitivity and robustness. The comprehensive and quantitative human m6A atlas generated by DeepRM highlights the prevalent and dynamic nature of m6A modification in the human transcriptome.
The DeepRM dataset presents several significant improvements over previous datasets. We constructed the DeepRM dataset by designing RNA oligonucleotides that represent RM within diverse local sequence contexts to closely mimic the endogenous transcripts, unlike the IVT datasets. In the dataset, each RM nucleotide is surrounded by an unmodified 20-nt sequence context, ensuring that the effect of flanking sequences on the electric current can be sufficiently captured. The DeepRM dataset is designed at single-nucleotide resolution such that RMs are embedded exclusively at predefined positions via chemical oligonucleotide synthesis. This allows the trained model to pinpoint the m6A site in individual transcripts, substantially elevating the accuracy of modification stoichiometry measurements. Thanks to these improvements, the information trained from the DeepRM dataset can be readily applied to m6A sites located on the endogenous transcripts in the transcriptome.
We released DeepRM as a free, publicly available software to facilitate its application in diverse biological and medical research. DeepRM achieves a competitive running time compared to the Nanopore manufacturer’s official tool, Dorado, while delivering much higher accuracy (Supplementary Table 2). With DeepRM, researchers can perform end-to-end RM detection in only two simple steps (Supplementary Fig. 10). We anticipate that DeepRM, powered by its unprecedented performance and usability, will serve as a future standard for epitranscriptome research.
While DeepRM enables remarkably accurate detection of m6A, several unexplored avenues could potentially enhance its performance. First, A nucleotides in the real transcriptome are modified into various other species such as N6,2′-O-dimethyladenosine (m6Am), N1-methyladenosine (m1A), and 2′-O-methyladenosine (Am), yet our dataset only includes A and m6A, potentially leading to misclassification of other A modifications as m6A. Incorporating other A modifications could further improve the model’s precision. Second, while DeepRM accurately detects clustered m6A sites, a slight accuracy drop was observed for densely clustered sites (Fig. 4g). We postulate that adding local sequence context blocks (LCBs) that contain multiple m6A sites on a single LCB to the training dataset would alleviate this limitation. Third, we observed that the effect of m6A modification extends beyond the 21-nt span of an LCB (Fig. 1b, c). Although the current size of an LCB perhaps captures most of the impact of a single m6A on flanking sequences, extending it by several nucleotides might allow DeepRM to fully capture the impact of a single m6A, leading to a more accurate detection. Fourth, DeepRM, like other Nanopore-based RM detection tools, may exhibit lower accuracy for m6A sites adjacent to some other RM species. For instance, m6A sites in the human rRNAs, that are adjacent to N4-acetylcytidine (ac4C) and pseudouridine (ψ), were not reliably detected by the RM detection tools (Supplementary Fig. 11 and Supplementary Tables 3, 4)64. Nevertheless, this result only shows a limited picture because there are only two m6A sites in the human rRNA. For a more comprehensive assessment, we evaluated DeepRM for m6A sites near 5-methylcytosine (m5C), another widespread RM in the human transcriptome, and confirmed that DeepRM maintains its robust performance near m5C (Supplementary Fig. 5b)65,66. In our future work, we will improve the performance in these circumstances by adding various other RNA modification species into the DeepRM dataset, as described below.
DeepRM can be used to accurately detect and quantify RMs in not only the human transcriptome but also various other organisms, clinical and environmental samples, and biological contexts, providing a powerful and unique opportunity to explore diverse epitranscriptomic landscapes. The flexible design of our DeepRM library also enables DeepRM to be extended for detecting diverse species of other RMs. By introducing different types of RMs into LCBs, we successfully produced massive-scale datasets for various RMs such as ψ, m5C, and 2′-O-methylguanosine (Gm) (manuscript in preparation). Training on these RM datasets together with the m6A dataset, DeepRM will be able to simultaneously detect various species of RMs with a single Nanopore sequencing experiment. We anticipate that the DeepRM framework proposed in this study will pave the way for a universal platform for detecting various RMs, accelerating future investigations that reveal unique insights into their functional roles.
Methods
Selection of 5′ and 3′ end sequences for building blocks
The DeepRM (Deep learning for RNA Modification) dataset was constructed from chemically synthesized 87-nt oligonucleotides termed building blocks (BBs). The BBs were ligated with each other and sequenced via DRS. The 5′ and 3′ end sequences of the BB were designed to optimize ligation efficiency. To select these sequences, we measured the sequence preference of RNA ligase via an assay modified from AQ-seq (Supplementary Fig. 4b). In this assay, random RNA sequences are ligated, and the ligation efficiency of each random sequence is measured by its enrichment in the sequenced reads.
5′ adapters with four random nucleotides at their 3′ ends, 3′ adapters with four random nucleotides at their 5′ ends, and 59-nt oligonucleotides only consisting of random nucleotides, named central random oligonucleotides, were purchased from Integrated DNA Technologies (IDT) (Supplementary Data 3). Pre-adenylated 3′ adapter was ligated to the central random oligonucleotides with T4 RNA ligase 2, truncated KQ (NEB, M0373L), followed by the ligation of 5′ adapter with T4 RNA ligase 1 (NEB, M0204L). The ligated products were reverse-transcribed and amplified following the TruSeq® small RNA library preparation guide (Illumina). The cDNA was sequenced with HiSeq 2500 (single-end, 101 bp, Illumina).
From the sequencing result, enrichment values of all 8-mer ligation motifs were calculated. The 8-mer motifs of the junctions between the 5′ adapter and the central random oligonucleotide were counted. The junctions between the 3′ adapter and the central random oligonucleotide were excluded from analysis, because T4 RNA ligase 2, truncated KQ, used for ligating the 3′ adapter was not used for producing the DeepRM dataset. Assuming a uniform prior, a one-sided p-value was calculated for each 8-mer motif via the χ2 test and corrected for multiple testing via the Benjamini-Hochberg correction67. Fold enrichment was calculated as the number of reads for each motif divided by \(n\times {4}^{-8}\). 8-mer motif with the highest fold enrichment was selected, split in half, and its 5′ and 3′ portions were used as the 3′ and 5′ end sequences, respectively (Supplementary Fig. 4b). The selected end sequences were 5′-CGAC and AGUC-3′.
Selection of spacer sequences for building blocks
In the BB, spacers (four predefined 6-nt sequences) were inserted between the local sequence context blocks (LCBs) to enable the reconstruction of LCBs from sequenced reads. We developed a systematic method for generating optimal RNA spacer sequences. The spacer sequences were selected to maximize LCB reconstruction and sequencing efficiencies by optimizing their nucleotide composition, sequence diversity, sequencing error, dimerization, and folding tendency.
First, all possible ordered sets of four 6-nt RNA sequences that meet the following constraints, hereby referred to as candidates, were generated. The 5′ and 3′ sequences of the first and the last spacer are CGAC and AGUC, which are the optimized 5′ and 3′ end sequences (see Selection of spacer sequences for building blocks). Each of the four nucleotides should occur six times in total and should occur once or twice in each spacer.
The candidates were filtered to minimize sequence similarity between spacers, calculated by Levenshtein distance, to ensure the diversity of spacer sequences. The sequence diversity enables the reconstruction of LCBs within the sequenced reads (see Reconstruction algorithm of local sequence context blocks). Specifically, candidates including a spacer pair with a Levenshtein distance below 3 were discarded.
Candidates that can form complementary base pairs between spacers were excluded to prevent the formation of undesired secondary structures and dimers. Next, the candidates that minimize the Nanopore sequencing error were selected. Specifically, error rate, defined as the sum of substitution, insertion, and deletion rates, was calculated for all possible 5-mer sequences from the HEK293T sequencing data. Candidates with mean 5-mer error rates below the 5th percentile were selected.
Finally, the thermodynamic properties of the remaining candidates were evaluated to minimize folding and dimer formation. For each candidate, 1000 randomized RNA sequences were filled in between spacers to simulate the folding of the synthesized 87-nt BB, and the results were averaged. The mean minimum free energy (MFE) of the BBs was calculated for each candidate, and candidates with a mean MFE above the 95th percentile were selected. Subsequently, the mean MFE of the BB dimers was calculated, and candidates with mean dimer MFE above the 95th percentile were selected. Among the remaining candidates, the sequence with the lowest error rate was selected (Supplementary Fig. 4c). The MFE values were calculated with ViennaRNA (ver. 2.6.4), and the Levenshtein distances were calculated with polyleven (ver. 0.8)68,69.
Building block synthesis, ligation, and size selection
BBs, that are chemically synthesized RNA oligonucleotides, were purchased from IDT. The quality of the BBs was checked by loading 1/10x aliquots of BBs on 12% TBE-Urea gel system (National Diagnostics, SequaGel UreaGel System) with 20/100 single-stranded oligo length standards (IDT). Sequences of the purchased BBs are listed in Supplementary Data 3.
We generated longer RNA molecules for sequencing by ligating the BBs iteratively. 640 pmol of 87-nt BBs were ligated by incubating with 19% PEG8000, 1 mM ATP, 10% DMSO, 1 x T4 RNA ligase buffer (NEB, B0216S), and 50 U T4 RNA ligase 1 at 33 °C for 16 hrs. For every 100 μl solution, ligated RNA was isolated using guanidium thioisocyanate-phenol-chloroform (Trizol) extraction using 300 μl of QIAzol® Lysis Reagent (Qiagen) and 120 μl chloroform (Sigma-Aldrich), and purified with RNA Clean & Concentrator-5 (Zymo Research, R1013) according to the manufacturer’s protocol.
We selected RNA molecules longer than 300 nts for sequencing. The purified BB ligates were separated using an 8% TBE-Urea gel system (National Diagnostics) (Supplementary Fig. 4a). Gel slices containing RNAs longer than 300 nts were obtained and eluted with 3x volume of 300 mM NaCl buffer overnight at 4 °C. To remove the gel debris, the eluted samples were purified with 0.22 μm Spin-X Centrifuge Tube Filter (Corning). RNAs were recovered by Oligo Clean & Concentrator (Zymo Research, D4061) following the manufacturer’s instructions.
Poly(A) tailing of ligation products and selection of poly(A)+ products
The ligated BBs were polyadenylated to allow sequencing via DRS. E-PAP Poly(A) Tailing Kit (Thermo Fisher Scientific, AM1350) was used for polyadenylation. We modified the manufacturer’s protocol to achieve the optimal poly(A) tail length of 30 nts for DRS (see below, Supplementary Fig. 4e). 30 pmol of ligated RNA was incubated with 1 x E-PAP buffer, 2.5 mM MnCl2, 10 μM ATP, 100 U RNaseOUT recombinant ribonuclease inhibitor (Invitrogen, 10777019), and 8 U E-PAP in 50 μl solution at 37 °C for 45 min. The product was purified with Oligo Clean & Concentrator (Zymo Research, D4061). To remove circular RNAs and select poly(A) + products, the purified RNAs were treated with Dynabeads Oligo(dT)25 (Invitrogen, 61002) following the manufacturer’s protocol.
We identified the optimal poly(A) tail length by sequencing in vitro-transcribed (IVT) RNAs with various poly(A) tail lengths (Supplementary Fig. 4e). 274-nt RNAs were in vitro-transcribed from DNA templates that include 6-nt index sequences. IVT RNAs including indices 1,2, and 3 were ligated to 20-nt, 30-nt, and 40-nt RNA oligonucleotides consisting of only A nucleotides, respectively. IVT RNAs including indices 4 and 5 were polyadenylated with E-PAP Poly(A) Tailing Kit, to include ~ 100-nt and ~ 300-nt poly(A) tails. The polyadenylated RNAs were mixed at equimolar concentration and prepared for Nanopore sequencing (RNA002) according to the manufacturer’s protocol. The sequenced reads were aligned to the DNA templates and classified into the five poly(A) length groups by their indices. The optimal poly(A) length was selected by analyzing the sequencing yield. The sequencing yield was measured by the number of reads uniquely mapped to each DNA template. 30 nts was selected as the optimal poly(A) tail length for the DeepRM dataset construction.
Validation of the quality of chemically synthesized RNA oligonucleotides
Unambiguous oligonucleotides were used to validate the m6A modification status, as orthogonal validation methods such as SELECT52 and GLORI47 require an unambiguous reference sequence and thus cannot be applied to random sequences. A 49-nt RNA derived from the MALAT1 lncRNA was synthesized, containing either a single A or m6A at the 28th residue (Supplementary Data 3). For partial fragmentation, 300 pmol of the synthesized products were denatured in 3 M urea at 90 °C for 5 min and digested with 1 x NEBufferTM r1.1 and 150 U RNase 4 (NEB, M1284) at 37 °C for 1 hr. The resulting 13-nt fragment containing the designated residue was analyzed for the accuracy of m6A modification status and position using liquid chromatography tandem mass spectrometry (LC MS/MS) in technical triplicate (Fig. 1e).
Samples were centrifuged at 20,000 × g for 20 min at 4 °C, and the supernatants were subjected to LC MS/MS analysis using an ACQUITY UPLC I-Class system (Waters) coupled to a Tribrid Lumos or Ascend mass spectrometer (Thermo Fisher Scientific) equipped with a HESI source. An aliquot of the sample was directly injected onto a BEH C18 analytical column (2.1 × 100 mm, 1.7 µm, 130 Å; Waters). Mobile phase A consisted of 10 mM TEAA (triethylammonium acetate) in 5% ACN (acetonitrile), and mobile phase B consisted of 10 mM TEAA in 15% ACN. Separation was performed at a flow rate of 250 µL/min using the following gradient: 0% B (0 min), 15% B (17 min), 95% B (20 min), followed by column washing and re-equilibration at 0% B for 8 min. Electrospray ionization was operated in negative mode with a spray voltage of 2.8 kV, a sheath gas flow rate of 45, an auxiliary gas flow rate of 10, and an ion transfer tube temperature of 325 °C. Full MS scans were acquired in the range of m/z 400–3500 at a resolution of 60 K, with an RF lens setting of 30%, AGC target of 125%, and a maximum injection time of 50 milliseconds (ms). Data-dependent MS/MS (DDA) acquisition was performed using HCD with stepped normalized collision energy (NCE). Fragment ions were detected at a resolution of 30,000 with an AGC target of 300% and a maximum injection time of 86 ms.
For assessment of A and m6A purity, we quantified the ratio of A and m6A nucleotides by integrating the peak areas of signal intensity detected in the MS chromatogram (Fig. 1f). LC MS/MS was then performed on 13-nt fragments from A or m6A oligonucleotides to verify the exact position of m6A. All MS/MS spectra for each target sequence were manually annotated, with the fragment m/z values calculated using the Ariadne web tool70. Using MS/MS spectra with full sequence coverage, the pairs of the obtained fragment ions containing each A residue were compared to distinguish m6A position with a + 14 Da mass increase in m6A oligonucleotides, that corresponds to the addition of a single methyl group (-CH3) (Supplementary Fig. 3a–c). To reaffirm the modification site, LC MS/MS of the non-cleaved, intact 49-nt RNA was conducted by Bioneer. In total, six MS/MS runs of A-oligonucleotides and four runs of m6A-oligonucleotides were analyzed (Supplementary Data 4), comparing the pairs of representative fragment ions containing each A residue (Supplementary Fig. 3d, e).
Nanopore direct RNA sequencing
For Oxford Nanopore Technologies (ONT) direct RNA sequencing (DRS), libraries were prepared with the SQK-RNA004 sequencing kit (ONT). 500 ng of poly(A)-tailed RNA was ligated with RTA adapter, reverse-transcribed with SuperscriptTM III reverse transcriptase (Invitrogen, 18080085), and purified with 1.8x RNAClean XP beads (Beckman Colter). Then, the sample was ligated with RLA adapter, purified with 1 x XP beads, and eluted with 33 μl REB elution buffer. The sample was mixed with SB buffer and LIS solution, and loaded onto a primed PromethION RNA flow cell (ONT, FLO-PRO004RA). The sequencing was performed using a PromethION 2 Solo device (ONT).
Reconstruction algorithm of local sequence context blocks
The LCBs were reconstructed from the reads produced by sequencing the LCB ligation products via DRS. To accurately locate the LCBs without their DNA templates while tolerating the high error rate of Nanopore DRS, we devised an LCB reconstruction algorithm based on the weighted directed acyclic graph (DAG) (Fig. 3a). The algorithm can be conceptually understood as an alignment algorithm, but instead of aligning a query to DNA templates, it aligns a query to the oligonucleotide design that is mostly random nucleotides. In the algorithm, each read is represented as a DAG. A vertex of the DAG corresponds to a spacer, and an edge corresponds to a translocation between spacers. Edges are scored based on their conformity to the BB design, and each edge is classified as an LCB or non-LCB. By searching for the highest-scoring path on the DAG, the most probable alignment between the BB design and the sequenced read is discovered. Finally, LCB edges in the highest scoring path, which are the most probable LCBs from the read, were extracted.
The algorithm is similar to the Hidden Markov Model (HMM): it emits a sequence of read positions and penalizes or favors certain transitions between the read positions. The DAG-based formulation was adopted to enforce the same constraints as HMM while avoiding parameter learning. To learn the parameters that perform well across diverse sequence contexts, Nanopore sequencing data with diverse motifs and ground truth of m6A positions was needed, but such data was not available. Therefore, the DAG-based formulation provides a robust, training-free solution. The detailed implementation of the algorithm is demonstrated below.
The algorithm takes the following parameters. \({l}_{{{{\rm{spacer}}}}}=6\) is the length of the spacers, \({n}_{{{{\rm{anc}}}}}=4\) is the number of spacers in each BB. \({l}_{{{{\rm{LCB}}}}}=21\) and \({l}_{{{{\rm{BB}}}}}=87\) are the length of the designed LCB and BB, respectively. The algorithm uses four tolerance parameters, \(({t}_{{{{\rm{spacer}}}}},{t}_{{{{\rm{disp}}}}},{t}_{{{{\rm{indel}}}}},{t}_{{{{\rm{ligation}}}}})=({{\mathrm{3,4,3,1}}})\), and three weight parameters, \(({w}_{{{{\rm{spacer}}}}},{w}_{{{{\rm{indel}}}}},{w}_{{{{\rm{skip}}}}})=({{\mathrm{2,3,6}}})\). The functions and optimization method of these tolerance and weight parameters are detailed below.
To create the vertices of the DAG, the spacer sequences were searched in each sequenced read, allowing for errors. In the BB design, there are ordered spacer sequences \({A}=\{{a}_{1},\ldots,{a}_{{n}_{{{{\rm{spacer}}}}}}\}\). For each position \(i\) of the \({{{\rm{read}}}}\), substring \({{{\rm{read}}}}[i:i+{l}_{{{{\rm{spacer}}}}}]\) (\({{{\rm{read}}}}[i:j]\) denotes a substring of \({{{\rm{read}}}}\) from position \(i\) to \(j-1\)) was compared to each designed spacer sequences \({a}_{k}\in A\) by calculating the Levenshtein distance71. For each position \(i\), we found the index of the best-matching spacer sequence, \(m(i)\), that has the minimum Levenshtein distance, \(d\left(i\right)\). \(m(i)\) and \(d\left(i\right)\) are defined as per Eq. (1). For each \(i\), if and only if \(d\left(i\right)\le {t}_{{{{\rm{spacer}}}}}\) (spacer error tolerance factor), \({{{\rm{read}}}}[i:i+{l}_{{{{\rm{spacer}}}}}]\) was considered as a spacer, and thus \(i\) was used as a vertex of the DAG. Formally, \(V=\{i|d (i) \le {{t}_{{{{\rm{spacer}}}}}}\}\) is a set of vertices of the DAG of a read.
DAG \(G=(V,E)\) was built by connecting the spacers found in the read in a 5′-to-3′ direction. \(E=\){\(\left\langle i,j\right\rangle |{i},j\in V\,{{{\rm{and}}}}\,i < j\)} is the set of edges in the DAG. Then, the DAG was pruned by only keeping the edges satisfying the following two conditions. First, the sum of the vertex Levenshtein distances, \(d\left(i\right)+d\left(j\right)\le {t}_{{{{\rm{spacer}}}}}\). Second, the displacement, \(j-i\), is close to the designed displacement \({D}_{{{{\rm{design}}}}}\left(i,j\right)\). \({D}_{{{{\rm{design}}}}}\left(i,j\right)\) is the most probable displacement between \(i\) and \(j\) if the BB was sequenced without error, calculated as per Eq. (2). The tolerance for the error between (\(j-i\)) and \({D}_{{{{\rm{design}}}}}\left(i,j\right)\) is a product of displacement error tolerance factor \({t}_{{{{\rm{disp}}}}}\) and \({N}_{{{{\rm{LCB}}}}}\left(i,j\right)\). \({N}_{{{{\rm{LCB}}}}}\left(i,j\right)\) is the most probable number of LCBs between positions \(i\) and \(j\), estimated as per Eq. (3). Formally, the set of vertices in the pruned DAG, \(E^{\prime}\), is defined as per Eq. (4). Herein we denote \(\left\langle i,j\right\rangle \in E^{\prime}\) simply as \({{{{\bf{e}}}}}_{{{{\bf{i}}}},{{{\bf{j}}}}}\). This pruning strategy allowed us to process >107 reads per sequencing experiment in a reasonably short amount of time.
-
\({D}_{\min }\left(x,y\right)\) is the minimum designed displacement between \(x\)-th and \(y\)-th spacers.
-
\({N}_{{{{\rm{BB}}}}}\left(i,j\right)\) is the most probable number of BBs between positions \(i\) and \(j\).
After pruning, we checked if each edge represents an LCB. For \({{{{\bf{e}}}}}_{{{{\bf{i}}}},{{{\bf{j}}}}}\) to represent an LCB, it should satisfy two conditions. First, \({N}_{{{{\rm{LCB}}}}}\left(i,j\right)=1\). Second, it should be possible to modify the sequence \({{{\rm{read}}}}[i+{l}_{{{{\rm{spacer}}}}}:j]\), with at most \({t}_{{{{\rm{indel}}}}}\) insertions and deletions, to a sequence of length \({l}_{{{{\rm{LCB}}}}}\) with “A” at the center (\({l}_{{{{\rm{LCB}}}}}\) is odd). \(M(i,j)\) is defined as a set of insertion and deletion operations satisfying the aforementioned condition for positions \(i\) and \(j\), as per Eq. (5). \(M(i,j)\) should not be empty for \({{{{\bf{e}}}}}_{{{{\bf{i}}}},{{{\bf{j}}}}}\) to represent an LCB. Formally, \(C\), a set of edges that represent an LCB, is defined as per Eq. (6).
-
\({n}_{{{{\rm{ins}}}}}\) and \({n}_{{{{\rm{del}}}}}\) are numbers of insertions and deletions, respectively.
-
\(\delta\) is the change in the number of nucleotides between the 5′ spacer and the central A.
-
\({{{\rm{read}}}}\left[i\right]\) denotes the \(i\)-th nucleotide of \({\mbox{read}}\).
We checked if each non-LCB edge represents a ligation between two BBs. Formally, \(L\), a set of edges that represent a ligation, is defined as per Eq. (7).
We quantified the conformity of each edge to the BB design with three penalty terms. \({P}_{{{{\rm{spacer}}}}}\left(i,j\right)\) is a spacer penalty that measures the conformity of the spacers to the design, as per Eq. (8). \({P}_{{{{\rm{indel}}}}}\left(i,j\right)\) is an indel penalty that measures the conformity of the sequence between the spacers to the design, as per Eq. (9). \({P}_{{{{\rm{skip}}}}}\left(i,j\right)\) is a skip penalty for skipping an LCB between the spacers, as per Eq. (10).
The length of each edge, \(|{{{{\bf{e}}}}}_{{{{\bf{i}}}},{{{\bf{j}}}}}|\), was calculated from the weighted sum of \({P}_{{{{\rm{spacer}}}}}\left(i,j\right)\), \({P}_{{{{\rm{indel}}}}}\left(i,j\right),\) and \({P}_{{{{\rm{skip}}}}}\left(i,j\right)\), as per Eq. (11). Finally, the longest path in the DAG was searched for each weighted DAG via dynamic programming implemented in NetworkX (ver. 3.1)72. Among the edges of the longest path, edges that represent LCBs, formally \({{{{\bf{e}}}}}_{{{{\bf{i}}}},{{{\bf{j}}}}}\in C\), were extracted and stored.
The tunable tolerance and weight parameters used in this algorithm were optimized via a Markov chain Monte Carlo method. A discrete-time Markov chain that transforms an input RNA sequence into a DRS-sequenced read was constructed using an alignment error profile of DRS calculated from the HEK293T transcriptome sequencing data. In a Monte Carlo simulation, a pool of LCB ligation products was randomly generated and transformed into DRS-sequenced reads using the Markov chain. Then, LCBs were reconstructed from the reads using our algorithm, and the accuracy of the LCB location was measured by the maximum F1 score. Each simulation was conducted with a random combination of the tolerance and weight parameters, and the combination with the best LCB reconstruction accuracy was selected.
Nanopore data processing and dataset construction
The DRS sequencing data was basecalled with Dorado (ver. 0.7.3) with rna004_130bps_sup@v5.0.0 basecalling model and options “--chunksize 12000 --min-qscore 0 --emit-moves --estimate-poly-a”45. The resulting BAM files were sorted and indexed with SAMTools (ver. 1.16.1), then parsed with pysam (ver. 0.22.0)73. LCBs were reconstructed from the basecalled reads (see Reconstruction algorithm of local sequence context blocks). Electric current was extracted from the POD5 files based on the indices of the reconstructed LCBs and the move table produced by Dorado. The currents were scaled according to the range between the 20th and 80th percentiles and were shifted according to the average of the 20th and 80th percentiles. Each file in the DeepRM dataset consisted of normalized electric current, nucleotide sequence, base quality, dwell time, and modification label.
For the whole poly(A)+ transcriptome and in vitro-transcribed transcriptome samples, the reads were aligned to the human reference transcriptome (see below) during basecalling using Minimap2 and Dorado45,74. Only the primary alignments were selected, and all transcriptomic positions with adenosine (A) as the reference base were extracted from the alignment. The normalized electric current, nucleotide sequence, base quality, dwell time, and reference position were stored for each extracted A nucleotide. The human reference transcriptome was obtained by merging NCBI RefSeq (GCF_000001405.40-RS_2024_08) and ENSEMBL (GCA_000001405.29-Release 113) annotations with AGAT (ver. 1.4.1)75,76,77, including only the transcripts to the 24 standard chromosome assemblies. For the total RNA sample, reads were aligned to the human rRNA sequences from NCBI RefSeq, composed of NR_003285.3, NR_003286.4, and NR_003287.4. For the synthetic test dataset, reads were aligned to the IVT template sequence (Supplementary Data 3).
Comparative analysis of dataset sizes and motif coverages
For Curlcake dataset25, motif coverage and dataset size were obtained from the supplementary files of GSM3528749, GSM3528750, GSM3528751, and GSM3528752. For the mAFiA dataset42, motif coverage and dataset size were obtained from the original publication. For the ELIGOS dataset31, sequencing results were downloaded from SRR11550246 and SRR11550255, and processed as described in the original publication via Minimap274. For the IVET dataset28, sequencing results were downloaded from SRR23804248 and SRR23804250, and processed as described in the original publication via Tombo. Dataset size was defined as the number of nucleotides used for training, equivalent to the number of entries multiplied by 5 for Curlcake, mAFiA, ELIGOS, and IVET; 21 for DeepRM. Motif coverage was defined as the fraction of 11-mer motifs sequenced for >10 times for both A and m6A samples.
Accuracy evaluation of reconstruction algorithms of local sequence context blocks
A DNA template containing 15 LCBs, with all random nucleotides substituted by fixed ones, was in vitro-transcribed (IVT) and sequenced via DRS using the method described above. Sequenced reads were aligned to the DNA template sequence, and mapped reads whose length is within ±30 nts of the template length were selected. To match the base quality distribution of the IVT reads to those of A and m6A LCB reads in the DeepRM dataset, 100,000 reads were randomly sampled for A and m6A each. The sampling weight was given by the base quality distribution of A and m6A LCB reads divided by the distribution of the IVT reads.
The graph-based LCB reconstruction algorithm and two distance-based baseline algorithms were used to reconstruct LCBs from the sampled reads. The first distance-based algorithm searches for LCBs using distance from the 3′-most spacer, and the second algorithm uses distance from the 5′ end of the poly(A) tail. Both distance-based algorithms score each LCB based on the Levenshtein distance of the spacer sequences. Each reconstructed LCBs were classified as correct or incorrect based on the alignment to the DNA template. Precision was defined as the fraction of correct LCBs among the reconstructed LCBs, and recall was defined as the fraction of correctly reconstructed LCBs among all 1,500,000 LCBs. Substitution, insertion, and mismatch rates were calculated for each of the 21 individual positions in the LCBs based on the alignment to the DNA template.
Training of the DeepRM model and detection of m6A sites
The DeepRM model was trained using the knowledge distillation framework, where the knowledge learned by a larger teacher model is transferred to a smaller student model, improving the computational efficiency78. For DeepRM, a teacher model with 15 million parameters was initially trained and distilled to a student model with 4 million parameters. This method allowed DeepRM to achieve a competitive running time compared to Dorado, while delivering much higher accuracy (Supplementary Table 2).
The teacher model was trained using AdamW optimizer with binary cross-entropy (BCE) loss79. A constant global dropout rate of 0.1 and a weight decay factor of 0.1 were used for regularization, and the gradient norm was clipped at 1.080. The learning rate was scheduled using the cosine annealing method81 with an initial learning rate of 0.0003, and the model was trained for 8 epochs. For each training batch of size 12,800, A and m6A samples were independently and randomly drawn from the respective training data pool. The ratio of A and m6A in each batch was randomly determined following a binomial distribution centered at the ratio of A and m6A data pool sizes. 5% of the training data was randomly sampled for validation, and the checkpoint with the lowest validation loss was selected for evaluation. Distributed data parallel strategy was used for multi-GPU training.
The student model was trained using the AdamW optimizer with a sum of BCE loss and cosine embedding loss as the loss function. The cosine embedding loss is one minus the cosine similarity between the output embeddings of the Transformer encoder of the teacher and the student model. The learning rate was scheduled using the cosine annealing method with an initial learning rate of 0.0005, and the model was trained for 16 epochs. Otherwise, the same settings were used as the teacher model.
For the detection of RM sites using DeepRM, inference data was prepared by running Dorado basecaller and DeepRM preprocess simultaneously using command “dorado basecaller --reference <reference-path > --min-qscore 0 --emit-moves <dorado-model-path > <pod5-path > --chunksize 12000 | tee <bam-path> | deeprm call prep -c 160 -p <pod5-path > -b - -o <deeprm-preprocessing-path > ”. This simultaneous execution of Dorado and DeepRM reduces the total running time of DeepRM. Then, the prepared inference data was used to detect RM using command “deeprm call run -d <deeprm-preprocessing-path > -o <deeprm-output-path > -m <deeprm-model-path > -b <bam-path > ”.
The training and inference programs were implemented with PyTorch (ver. 2.7.0), CUDA (ver. 12.8), TorchMetrics (ver. 1.3.0), and NumPy (ver. 2.1.2)82,83,84. Dell PowerEdge XE9680 machine with 2 Intel Xeon Platinum 8480+ CPUs (total 112 cores, 3.8 GHz), 2 TB memory (DDR5), and 8 NVIDIA H100 GPUs (80 GB HBM3) were used for both training and inference.
Architecture design of the DeepRM model
There are two types of input tokens in the DeepRM model. The first token is the sequence embedding, where each token represents an RNA pentamer85. The input RNA sequence of 21 nucleotides is split into 17 overlapping pentamers with a stride of 1. Then, the sequence is warped along the time dimension to align with the normalized electric current based on the move table, as in Remora86. The sequence embedding is produced via a lookup table where 1,024 5-mer nucleotides are represented as 512-dimensional vectors87.
The second token is the signal embedding, which is a linear projection of the electric current, base quality, and dwell time. The electric current is processed as follows. First, the electric current of each read is normalized using robust scaling, adapted from Dorado45. Second, for each A site, the electric current corresponding to the 17-nt region centered at the site is sliced. Third, the sliced electric current is tokenized using an overlapping sliding window with a size of 30 and a stride of 688,89, where the \(N\)-th token contains \(6N\) to \((6N+29)\)-th data points of the sliced electric current. Base quality and dwell times are aligned to the electric current as described above for nucleotide sequence. For each nucleotide, dwell time is calculated at + 0 and + 10-nt positions, which corresponds to the time taken for passing through the pore constriction and motor proteins, respectively90,91. The electric current, base quality, and dwell time are stacked and linearly projected to a 512-dimensional embedding space. Both the sequence and signal tokenizers are trained end-to-end. The two input tokens and sinusoidal positional encoding are added together and passed to the Transformer encoder block of the DeepRM model.
We trained two versions of the DeepRM model: a larger teacher model and a smaller student model (see Training of the DeepRM model and detection of m6A sites). The DeepRM teacher model is composed of a Transformer encoder and a fully connected regression head92,93. The Transformer encoder block consists of 6 standard Transformer encoder layers with a dimension of 512 and 8 self-attention heads. Gaussian error linear unit (GeLU) was used as an activation function. FlashAttention-v2 was used to improve computational performance94. The output of the Transformer encoder is masked so that only the tokens corresponding to the base of interest are passed to the regression head. The regression head consists of 6 fully connected layers of dimension 512, 1 layer of dimension 128, and a final layer of dimension 1. The output of the regression head was transformed with a sigmoid function. The DeepRM teacher model consists of 14,802,641 parameters in total.
The DeepRM student model is composed of a convolutional neural network (CNN), a Transformer encoder, and a fully connected regression head92,93. The CNN consists of four pre-activation ResNet blocks95, with kernel sizes of 5, 15, 5, and 5 from upstream to downstream. All blocks used a stride of 1 and grouped convolution with 8 groups. GeLU was used as an activation function. The output of the CNN was fed into the Transformer encoder, comprising 3 standard Transformer encoder layers with a dimension of 256 and 8 self-attention heads. The regression head consists of 6 fully connected layers of dimension 256, 1 layer of dimension 128, and a final layer of dimension 1. For knowledge distillation training, a single fully connected layer was used to upscale the 256-dimensional embedding of the student model to 512 dimensions. Otherwise, the same architecture was used as the teacher model. In total, the DeepRM student model consists of 3,914,833 parameters. The knowledge distillation framework and the CNN-Transformer hybrid architecture were both needed for the smaller DeepRM model, which uses half the encoder layers and hidden dimension of the teacher model, to match the performance of the teacher model (Supplementary Fig. 6c and Supplementary Table 5).
Ablation studies were performed to validate the advantage of DeepRM’s architecture. To verify the importance of the overlapping sliding window tokenization of the electric current, an ablation model was constructed by replacing the overlapping sliding window with a non-overlapping sliding window, where the \(N\)-th token contains \(6N\) to \((6N+5)\)-th data points of the sliced electric current. To confirm the necessity of the electric current feature, an ablation model was constructed by replacing the raw electric current feature with the statistical features used in m6Anet45: the mean, standard deviation, and length of the electric current corresponding to each nucleotide. To assess the advantages of the Transformer architecture, an ablation model was constructed by replacing the Transformer encoder layers with the long short-term memory (LSTM) layers, using the identical number of encoder layers and hidden dimensions. The ablation models used the same setting as the DeepRM teacher model unless otherwise specified, and their performances were compared against the DeepRM teacher model.
Algorithms for modification stoichiometry estimation and modification score calculation
During inference, a modification probability for each site for each read is calculated. These molecule-level modification probabilities are piled up to estimate the modification stoichiometry of each site, that is the number of modified bases compared to the total number of bases at a given site. To estimate the modification stoichiometry accurately, we used Kullback-Leibler (KL) divergence instead of calculating the ratio of reads exceeding a certain threshold96. This allowed DeepRM to effectively utilize all molecule-level predictions without discarding any information. Here, the modification probability of read \(i\) estimated by DeepRM is noted as \({p}_{i}\). First, the divergence between each observed read and a theoretical uninformative read was calculated. The divergence of read \(i\), \({D}_{{{{\rm{KL}}}}}({p}_{i}{||}0.5)\), is the KL divergence between \({{{\rm{Bernoulli}}}}\left({p}_{i}\right)\) and a non-informative distribution \({{{\rm{Bernoulli}}}}\left(0.5\right)\). For each site, total divergence is defined as the sum of the divergence of all mapped read, and positive divergence as the sum of the divergence of reads with \({p}_{i}\ge 0.5\). Finally, the modification stoichiometry (\(s\)) was estimated by dividing the positive divergence by total divergence of each site, as per Eq. (12). We confirmed that this information theory-based estimation of modification stoichiometry is more accurate than simply using the fraction of modified reads (Supplementary Fig. 12).
-
Each parameter \(k\) of \({D}_{{{{\rm{KL}}}}}\) indicates \({{{\rm{Bernoulli}}}}\left(k\right)\).
We developed a modification score to accurately represent the probability of modification of each site. Each m6A site in the transcriptome has a different modification stoichiometry. Since the modification stoichiometry of each site was estimated as above, we used the information to calculate the modification probability by weighing the modification evidence from each read based on the modification stoichiometry of the site.
We defined the modification score of each modified read (\({p}_{i}\) ≥ \(t=0.98\)) as a linear combination of Shannon information content and KL divergence. The Shannon information content \(I\left({\neg M|}{p}_{i}\right)\) quantifies the information gained by observing the unmodified site \(({\neg M})\) given the estimated modification probability \({p}_{i}\), which is higher when \({p}_{i}\) is high97. The KL divergence \({D}_{{{{\rm{KL}}}}}\left({s||}{p}_{i}\right)\) of read \(i\) is the divergence between \({{{\rm{Bernoulli}}}}\left(s\right)\) and \({{{\rm{Bernoulli}}}}\left({p}_{i}\right)\), quantifies the difference between the modification evidence observed in the read and the estimated modification stoichiometry \((s)\) of the site. Finally, the site modification score \((r)\) was defined as the sum of modification scores of the modified reads normalized by the total number of reads mapped to the site \((n)\), as per Eq. (13).
-
Parameter \(Q\) of \(I({P|Q})\) indicates \({{{\rm{Bernoulli}}}}\left(Q\right)\).
For evaluating the accuracy of modification stoichiometry quantification, sites containing more information than a minimal information given by the estimated modification stoichiometry were used, as per Eq. (14).
Comparative analysis of modification detection and stoichiometries
The modification detection and stoichiometry measurement accuracies of DeepRM were evaluated against Dorado (ver. 0.7.3), the official basecalling and modification detection model developed by Oxford Nanopore Technologies (unpublished), m6Anet (ver. 2.1.0), and SingleMod (ver. 1.0)29,45,46. For m6Anet, the sequencing data was preprocessed with blue-crab (ver. 0.1.0), slow5tools (ver. 1.3.0), and f5c (ver. 1.4), as described by the authors98,99. m6Anet was run with the option “--pretrained_model HEK293T_RNA004”. Dorado was run with rna004_130bps_sup@v5.0.0 basecalling model, rna004_130bps_sup@v5.0.0_m6A@v1 RM calling model, and options “--chunksize 12000 --min-qscore 0 --emit-moves --estimate-poly-a --modified-bases m6A”. The modification calls were extracted by the pileup function of modkit with default options (ver. 0.2.6)100. For SingleMod, the sequencing data was preprocessed with Picard (ver. 2.18.29) and f5c (ver. 1.4), as described by the authors98,101. SingleMod was run with the options “-v 004 -g 0 -b 30000”.
To evaluate the modification detection results, we compared the m6A sites detected by the three models against the ground truth of m6A sites in the human transcriptome (see Generation of the ground truth dataset). For all-context and DRACH site evaluations, All A sites with sequencing depth ≥ 20 were evaluated. For the non-DRACH site evaluation, All A sites with depth ≥ 100 were evaluated. Sites without prediction output were considered as zero-predicted for each model. The receiver operating characteristic (ROC) curve, precision-recall (PR) curve, their area under the curve (AUC), and maximum F1 score were computed with Scikit-learn (ver. 1.3.2)102.
To evaluate the modification stoichiometry measurement results, we compared the modification stoichiometry of m6A sites measured by each model against the modification stoichiometry label (see Generation of the ground truth dataset)47,54. Sites identified as m6A-modified by each model were used to evaluate the respective model. For DeepRM, m6A sites were defined as per Eq. (14). For Dorado, sites with predicted modification stoichiometry above zero were used, since the model does not provide a probabilistic score. For m6Anet, sites with modification probability above zero were used. SciPy (ver. 1.10.1) and pandas (ver. 2.1.4) were used to compute Pearson’s R, Spearman’s ρ, and Gaussian KDE103,104. Matplotlib (ver. 3.7.1) and Seaborn (ver. 0.12.2) were used to generate the plots105,106. Matplotlib-venn (ver. 1.1.1) was used to plot the Venn diagram of Fig. 6c107.
Generation of the ground truth dataset
For generating the ground truth of m6A sites in the human transcriptome, we processed the published RNA-seq raw data of GLORI47, m6A-SAC-seq18, m6ACE-seq48, and miCLIP249 experiments in HEK293T cell line, and eTAM-seq54 experiment in HeLa cell line. All raw RNA-seq reads were trimmed for the adapters, and low-quality bases with cutadapt (ver. 4.6), and barcodes were extracted and removed with umi_tools (ver. 1.1.6). The reads were aligned to the human reference genome (GCF_000001405.40) with STAR (ver. 2.7.10b).
Based on the reference human transcriptome annotation described in a previous section (see Nanopore data processing and dataset construction), we selected exonic sites whose reference sequences are As. Then, we defined the m6A signature for each site. Truncation at the A site was defined as the m6A signature for m6ACE-seq, and A-to-N mutation for m6A-SAC-seq. For miCLIP2, we selected C sites located 1 nt downstream from the A sites and defined C-to-T mutation and truncation as the m6A signature. For eTAM-seq and GLORI, where unmodified A is converted to guanine, unconverted A was defined as the m6A signature. Signatures derived from 5-nt ends of each read or within 10 nts of any insertion or deletion were excluded. Mutation-based signatures from known single-nucleotide polymorphism sites were also excluded.
After collecting the m6A signatures for each site, we constructed a contingency table of the signature, using 100 neighboring sites as a control group, and performed Fisher’s exact test on these tables. Sites with false discovery rate (FDR) < 10−7 in the whole transcriptome sample and FDR ≥ 0.1 in the m6A-free control sample were determined as m6A sites. Sites with FDR ≥ 0.1 in the whole transcriptome sample were determined as unmodified A sites. For miCLIP2, the m6A sites were determined using only the whole transcriptome sample because the published data lacks a m6A-free control.
For evaluation of modification detection, m6A sites called in at least three of the four methods (miCLIP2, m6ACE-seq, m6A-SAC-seq, and GLORI) were labeled as actual positives. Sites within the same transcripts with the actual positives and called as unmodified As in all four experiments were labeled as actual negatives. For the evaluation of modification stoichiometry measurement, all m6A sites called in GLORI and eTAM-seq were used as labels for HEK293T and HeLa cell lines, respectively.
Preparation of human transcriptome sequencing library
Human transcriptome data were generated from HeLa (CCL-2) and HEK293T (CRL-3216) cell lines purchased from ATCC company (Supplementary Table 6). Both cell lines were cultured in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin under 5% CO2. Total RNA was extracted with QIAzol (Qiagen), precipitated with isopropanol, and washed with ethanol. After DNase I treatment, RNA was purified with RNA Clean & Concentration kit-25 (Zymo Research, R1017). From 20 μg of total RNA, poly(A) + RNA was selected with Dynabeads Oligo(dT)25 according to the manufacturer’s instructions. The DRS sequencing library was prepared from 500 ng of poly(A) + RNA.
Generation of the transcriptome-wide in vitro transcription dataset and estimation of false positive rates
To generate the transcriptome-wide in vitro transcription (IVT) dataset, we reverse-transcribed the HEK293T transcriptome and in vitro-transcribed the HEK293T cDNA, following published protocols50,51. 100 ng of HEK293T poly(A) + RNA was combined with the VN primer and the Strand-switching primer. The primers had sequences identical to the cDNA-PCR Sequencing Kit (ONT, SQK-PCS109) and were manufactured by Bionics. For reverse transcription, the poly(A) + RNA and primer mix was incubated with Maxima H Minus Reverse Transcriptase (Thermo Scientific, EP0751) at 42 °C for 90 min, 85 °C for 5 min, and held at 4 °C. The first-stranded cDNA was amplified using LongAmp Taq 2X Master Mix (NEB, M0287S) and IVT_T7_Forward and Reverse primers as follows: initial denaturation step at 95 °C for 30 s (1 cycle); denaturation at 95 °C for 15 s, annealing at 62 °C for 15 s, and extension at 65 °C for 15 min (10 cycles); final extension at 65 °C for 15 min (1 cycle); and hold at 4 °C. To eliminate any remaining single-stranded DNA, the amplified cDNA was treated with Exonuclease I (NEB, M0293S) and purified with AMPure XP beads (Beckman Colter) according to the manufacturer’s guidelines.
The purified HEK293T cDNA was in vitro-transcribed using HiScribe T7 High Yield RNA Synthesis Kit (NEB, E2040S) and purified with RNA Clean & Concentrator Kit-25 (Zymo Research, R1017). The IVT product was polyadenylated with E-PAP Poly(A) Tailing Kit (Thermo Fisher Scientific, AM1350) according to the manufacturer’s protocol and cleaned using RNA Clean & Concentrator Kit-5 (Zymo Research, R1016). Finally, the DRS library was prepared from 500 ng of the RNA. Primers are listed in Supplementary Data 3.
The false positive rate and false discovery rate of DeepRM, Dorado, and m6Anet were estimated from the transcriptome-wide IVT dataset that is completely absent of RNA modification, as a reliable ground truth. The false positive rate was estimated by dividing the number of m6A sites identified in the transcriptome-wide IVT dataset by the total number of evaluated A sites. The false discovery rate was estimated by dividing the fraction of m6A sites in the IVT dataset by the fraction of m6A sites in the HEK293T whole transcriptome dataset. The m6A detection threshold of each model was selected to maximize the F1 score of m6A site detection in the HEK293T transcriptome (see Comparative analysis of modification detection and stoichiometries). The values of these F1-optimal thresholds are 0.842 (DeepRM), 0.500 (Dorado), 0.570 (m6Anet), and 0.251 (SingleMod). These thresholds were consistently used for all comparative evaluations of RM detection tools.
Experimental validation of m6A sites by single-base elongation- and ligation-based qPCR amplification method (SELECT)
SELECT was employed to validate non-DRACH (D = A/G/U, R = A/G, and H = A/C/U) m6A sites identified by DeepRM. We selected six sites for validation based on the following criteria. Transcripts with transcript per million reads (TPM) below the 90th percentile were filtered out, and transcripts with a fraction of m6A among all A above 0.005 were also filtered out. TPM was estimated by NanoCount (ver. 1.1.0)108. Among the sites within the remaining transcripts, non-DRACH sites identified as m6A by DeepRM with FDR < 0.0005 were selected. Sites that had surrounding (±50 nts) GC content above 60% or below 40%, sites with the minimum free energy (MFE) of the surrounding (±50 nts) region below -30 kcal/mol, and sites with neighboring (±30 nts) single-base repeat with at least 5 nts, were excluded. MFE was calculated by ViennaRNA (ver. 2.6.4), and GC contents were calculated by Biopython (ver. 1.79)109. Among the remaining sites, three sites identified as m6A by both DeepRM and Dorado, with another three sites identified as m6A by DeepRM but not by Dorado, were chosen for validation.
The SELECT protocol was modified from the original publications52,53. 3 μg of total RNA from HEK293T and 50 ng of the transcriptome-wide IVT dataset, absent of RNA modification, were mixed with 40 nM Up probe, 40 nM Down probe, and 10X rCutsmart buffer (NEB, B6004S). To anneal the probes to the samples, the mixture was incubated as follows: 90 °C for 1 min, 85 °C for 1 min, 80 °C for 1 min, 75 °C for 1 min, 70 °C for 1 min, 65 °C for 1 min, 60 °C for 1 min, 55 °C for 1 min, 50 °C for 1 min, 45 °C for 1 min, and 40 °C for 6 min, 30 °C for 1 min, 20 °C for 1 min, 10 °C for 1 min, and hold at 4 °C. Annealed samples were incubated with 3 μl of 0.0267 U/μl Bst 2.0 DNA polymerase (NEB, M0537S), 0.5 mM dTTP, and 25 mM ATP at 40 °C for 15 min. Subsequently, 1 μl of 1.25 μ/μl SplintR ligase (NEB, M0375S) was added to the sample. The sample was incubated at 37 °C for 20 min, denatured at 80 °C for 20 min, and kept at 4 °C. The final SELECT product was quantified via qPCR. The reaction mixture (10 μl) for qPCR consisted of 1 μl of the final reaction sample, 5 μl of PowerUp™ SYBR™ Green Master Mix for qPCR (ThermoFisher, A25742), and 200 nM SELECT universal primers. qPCR was performed as follows: 95 °C for 5 min; 95 °C for 10 s and 60 °C for 30 s (40 cycles); 95 °C for 15 s; 60 °C for 1 min; 95 °C for 15 s (collect fluorescence at a ramping rate of 0.05 °C/s); and hold at 4 °C. The SELECT products of each target site were normalized to the non-m6A sites located in the same gene. Probes and primers are listed in Supplementary Data 3.
Preparation of total RNA sequencing library
For total RNA sequencing library preparation, DNase I-treated total RNA from HEK293T cell lines was in vitro polyadenylated using E-PAP Poly(A) Tailing Kit (Thermo Fisher Scientific, AM1350). The polyadenylation was optimized from the manufacturer’s protocol with minor changes. 2 μg of total RNA was incubated with 1x E-PAP buffer, 2.5 mM MnCl2, 0.1 mM ATP, 10 U RNaseOUT recombinant ribonuclease inhibitor (Invitrogen, 10777019), and 1 U E-PAP in 10 μl solution at 37 °C for 10 min, and purified with Oligo Clean & Concentrator (Zymo Research, D4061). The DRS sequencing library was prepared from 500 ng of poly(A)-tailed total RNA using the SQK-RNA004 sequencing kit (ONT), following the manufacturer’s protocol.
Production of a synthetic evaluation dataset
To generate an independent set of synthetic evaluation datasets separated from the DeepRM dataset or human transcriptome dataset, we designed sequences including all 18 DRACH motifs and 68 non-DRACH motifs. A ~ 1 kb sequence with T7 polymerase promoter was synthesized and cloned into the pBHA vector using blunt-end EcoRI by Bioneer. In vitro transcription was conducted using HiScribe® T7 High Yield RNA Synthesis Kit (NEB, E2040S), using 500 ng of EcoRI-treated, linearized DNA, following the manufacturer’s protocol. The control synthetic RNA was generated with rNTPs, and the m6A-modified RNA was produced by substituting rATP with N6-methyladenosine-5′-triphosphate (m6ATP) (Trilink, N-1013-1). Following DNase I treatment, IVT RNA was purified using a 5% TBE-Urea gel (National Diagnostics), eluted with 3 x volume of 300 mM NaCl buffer overnight at 4 °C, and recovered with the Oligo Clean & Concentrator (Zymo Research, D4061). Polyadenylation was performed using E-PAP Poly(A) Tailing Kit (Thermo Fisher Scientific, AM1350), following the optimized protocol (see Preparation of total RNA sequencing library). Poly(A)-tailed RNAs were purified with Dynabeads Oligo(dT)25 (Invitrogen, 61002) following the manufacturer’s protocol. The DRS sequencing library was prepared from 500 ng of poly(A)-tailed RNA using the SQK-RNA004 sequencing kit (ONT), following the manufacturer’s protocol.
To generate virtual m6A sites with designated modification stoichiometry and sequencing depths, reads were randomly sampled from the sequencing data of unmodified and m6A-modified synthetic test RNA. Six stoichiometries (0.1, 0.2, 0.3, 0.4, 0.5, and 1.0) and six depths (10, 20, 30, 40, 50, and 100) were used, resulting in a total of 36 combinations. For each combination of stoichiometry \({{{\rm{s}}}}\) and depth \(d\), \(1000\cdot d\cdot (1-s)\) and \(1000\cdot d\cdot s\) reads were randomly sampled from the unmodified and modified sequencing data, respectively, producing \(1000\cdot n\) virtual m6A sites, where \(n=108\) is the number of A sites in the IVT template sequence, excluding sites within 10 nt from both ends of the template sequence. For each combination, \(1000\cdot d\) reads were additionally sampled from unmodified sequencing data, producing \(1000\cdot n\) virtual A sites. Binary classification accuracies were measured using these 108,000 A and 108,000 m6A sites, independently for each combination. For the single-molecule evaluation, 100,000 reads were sampled from modified and unmodified sequencing data, respectively, and binary classification accuracies were measured for A sites in the IVT template sequence, excluding those within 10 nt from both ends of the template sequence.
Detection of m6A sites in the whole transcriptome
We obtained the m6A modification landscape across the HEK293T and HeLa transcriptomes with DeepRM. A modification score threshold value corresponding to FDR < 0.005 was used to detect m6A. Genomic positions within the annotated exons that have adenosine as a reference sequence and sequencing depth ≥ 20 were selected for analyses, resulting in 11,763,619 and 10,051,097 positions in HEK293T and HeLa cell lines, respectively. Genes that contain any of the filtered genomic positions were used for analyses, resulting in 13,905 and 12,416 genes in HEK293T and HeLa cell lines, respectively.
Genomic positions that correspond to at least one DRACH site within the sequenced transcript were classified as DRACH sites, and the other sites were classified as non-DRACH sites. Genomic positions that correspond to at least one sequenced RefSeq or ENSMBL protein-coding transcript were considered mRNA sites, and the other sites were classified as non-coding RNA (ncRNA) sites. Pre-computed phyloP score for multiple sequence alignment of 100 vertebrate genomes was downloaded from the UCSC Genome Browser for conservation analysis in Fig. 8b110. For the conservation analysis, m6A and A sites were selected in pairs to match the combination of 5-mer motif, gene, and log10 sequencing depth distributions between the m6A and A groups. Logomaker (ver. 0.8) was used to plot the consensus sequence logo in Fig. 8b111.
Detection of differentially methylated sites
Differentially methylated sites were defined as sites identified as m6A in one transcriptome and identified as A in another transcriptome at FDR < 0.005. Only the sites sequenced in both transcriptomes with sequencing depth ≥20 were considered. Differentially modified genes were defined as genes containing at least one differentially methylated site. Differentially modified genes plotted in Fig. 9c, e are genes whose cosine similarity values of the m6A vector of the two transcriptomes are below 0.8. The m6A vector is the array of modification stoichiometry of all A sites in a gene, with unmodified A sites marked as 0. Differentially modified genes plotted in Fig. 9d are genes that have at least five differentially methylated non-DRACH sites.
Identification of co-occurring and exon skipping-associated m6A sites
Co-occurring pairs of m⁶A sites were identified using the m6A detection result across the HEK293T transcriptome by DeepRM (see Detection of m6A sites in the whole transcriptome). For every pair of m⁶A sites in the same reference transcript, a 2 × 2 contingency table was constructed for co-occurrence testing using the following categories: (1) reads modified at both sites, (2) reads modified at site 1 but unmodified at site 2, (3) reads unmodified at site 1 but modified at site 2, and (4) reads unmodified at both sites. A site was considered modified when the molecule-level modification score predicted by DeepRM was ≥ 0.5. Reads with mismatches or deletions at either site or lacking coverage across both sites were excluded. Two-sided Fisher’s exact tests were conducted using SciPy104 (ver. 1.10.1), and resulting p-values were adjusted for multiple testing with the Benjamini–Hochberg procedure. Pairs of m⁶A sites with q < 0.05 and odds ratio > 1 were classified as significantly co-occurring.
To identify exon-skipping-associated m⁶A sites, we first identified unannotated isoforms and quantified isoform expression, modifying the method suggested by Glinos et al.112. Raw reads were aligned to the GRCh38 reference genome using Minimap2 (ver. 2.30-r1287) with the options “-ax splice -uf --secondary = no“73. Alignments with a mapping quality of < 10 and with secondary or supplementary alignments were discarded. Isoform discovery was performed using FLAIR (ver. 2.2.0)113. Misaligned splice sites were corrected with command “flair correct -w 20”. Isoforms were determined using command “flair collapse -s 10 --quality 10 --stringent --check_splice -n longest”, and isoform expression was quantified with command “flair quantify --quality 10 --stringent --check_splice”. For the isoform determination and the expression quantification steps, we used full-length reads determined as the reads with their 5′ ends mapped within 10 nt upstream to 50 nt downstream of the annotated transcription start sites. Isoforms supported by ≥ 20 full-length reads were retained for downstream analysis. Exon skipping events were identified using SUPPA (ver. 2.3)114. For each exon skipping event, all A sites shared between exon-included and exon-skipped isoforms were extracted. For each A site, a 2 × 2 contingency table was generated using the following categories: (1) modified A in exon-skipped isoforms, (2) unmodified A in exon-skipped isoforms, (3) modified A in exon-included isoforms, and (4) unmodified A in exon-included isoforms. Reads not covering the site or containing mismatches/deletions were excluded. The Fisher’s exact test was performed as described above, and those sites with q < 0.05 were determined as significantly associated sites with exon skipping.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing data generated in this study has been deposited in the Sequence Read Archive (SRA) under accession code PRJNA1222552. The raw Nanopore electric current data is available upon request to D.B. for non-commercial academic purposes, as the data cannot be deposited to the SRA. The SELECT data and LC MS/MS data generated in this study are provided in the Source Data file and in Mass Spectrometry Interactive Virtual Environment (MasssIVE) under accession code MSV000098979 [https://doi.org/10.25345/C5862BQ87], respectively. Source data are provided in this paper.
Code availability
DeepRM model and code are available on GitHub (https://github.com/vadanamu/DeepRM) for non-commercial academic purposes115.
References
Wang, Y. et al. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 16, 191–198 (2014).
Roundtree, I. A., Evans, M. E., Pan, T. & He, C. Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017).
Desrosiers, R., Friderici, K. & Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 71, 3971–3975 (1974).
Fu, Y., Dominissini, D., Rechavi, G. & He, C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 15, 293–306 (2014).
Jia, G. F. et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011).
Roundtree, I. A. et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. ELife 6, e31311 (2017).
Alarcón, C. R., Lee, H., Goodarzi, H., Halberg, N. & Tavazoie, S. F. N6-methyladenosine marks primary microRNAs for processing. Nature 519, 482–485 (2015).
Xiao, W. et al. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol. Cell 61, 507–519 (2016).
Wang, X. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014).
Shafik, A. M. et al. N6-methyladenosine dynamics in neurodevelopment and aging, and its potential role in Alzheimer’s disease. Genome Biol. 22, 17 (2021).
Paramasivam, A., Priyadharsini, J. V. & Raghunandhakumar, S. Implications of m6A modification in autoimmune disorders. Cell. Mol. Immunol. 17, 550–551 (2020).
Barbieri, I. & Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 20, 303–322 (2020).
Meyer, K. D. et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646 (2012).
Linder, B. et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772 (2015).
McIntyre, A. B. R. et al. Limits in the detection of m6A changes using MeRIP/m6A-seq. Sci. Rep. 10, 6590 (2020).
Dominissini, D. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012).
Wang, Y., Xiao, Y., Dong, S., Yu, Q. & Jia, G. Antibody-free enzyme-assisted chemical approach for detection of N6-methyladenosine. Nat. Chem. Biol. 16, 896–903 (2020).
Hu, L. L. et al. m6A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat. Biotechnol. 40, 1210–1219 (2022).
Meyer, K. D. DART-seq: an antibody-free method for global m6A detection. Nat. Methods 16, 1275–1280 (2019).
Garcia-Campos, M. A. et al. Deciphering the “m6A code” via antibody-independent quantitative profiling. Cell 178, 731–747 (2019).
Zhang, Z. et al. Single-base mapping of m6A by an antibody-independent method. Sci. Adv. 5, eaax0250 (2019).
Garalde, D. R. et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 15, 201–206 (2018).
Workman, R. E. et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods 16, 1297–1305 (2019).
Zhong, Z.-D. et al. Systematic comparison of tools used for m6A mapping from nanopore direct RNA sequencing. Nat. Commun. 14, 1906 (2023).
Liu, H. et al. Accurate detection of m6A RNA modifications in native RNA sequences. Nat. Commun. 10, 4079 (2019).
Gao, Y. et al. Quantitative profiling of N6-methyladenosine at single-base resolution in stem-differentiating xylem of Populus trichocarpa using Nanopore direct RNA sequencing. Genome Biol. 22, 1–17 (2021).
Acera Mateos, P. et al. Prediction of m6A and m5C at single-molecule resolution reveals a transcriptome-wide co-occurrence of RNA modifications. Nat. Commun. 15, 3899 (2024).
Wu, Y. et al. Transfer learning enables identification of multiple types of RNA modifications using nanopore direct RNA sequencing. Nat. Commun. 15, 4049 (2024).
Hendra, C. et al. Detection of m6A from direct RNA sequencing using a multiple instance learning framework. Nat. Methods 19, 1590–1598 (2022).
Lorenz, D. A., Sathe, S., Einstein, J. M. & Yeo, G. W. Direct RNA sequencing enables m6A detection in endogenous transcript isoforms at base-specific resolution. RNA 26, 19–28 (2020).
Jenjaroenpun, P. et al. Decoding the epitranscriptional landscape from native RNA sequences. Nucleic Acids Res. 49, e7 (2021).
Yu, B. et al. m6ATM: a deep learning framework for demystifying the m6A epitranscriptome with Nanopore long-read RNA-seq data. Brief. Bioinform. 25, https://doi.org/10.1093/bib/bbae529 (2024).
Teng, H., Stoiber, M., Bar-Joseph, Z. & Kingsford, C. Detecting m6A RNA modification from nanopore sequencing using a semi-supervised learning framework. Genome Res. 34, 1987–1999 (2024).
Lewis, C. J. T., Pan, T. & Kalsotra, A. RNA modifications and structures cooperate to guide RNA–protein interactions. Nat. Rev. Mol. Cell Biol. 18, 202–210 (2017).
Cruciani, S. et al. De novo basecalling of m6A modifications at single molecule and single nucleotide resolution. Genome Res. 34, 1987–1999 (2024).
Ge, R. et al. m6A-SAC-seq for quantitative whole transcriptome m6A profiling. Nat. Protocols 18, 626–657 (2023).
Pratanwanich, P. N. et al. Identification of differential RNA modifications from nanopore direct RNA sequencing with xPore. Nat. Biotechnol. 39, 1394–1402 (2021).
Leger, A. et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat. Commun. 12, 7198 (2021).
Parker, M. T. et al. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. ELife 9, e49658 (2020).
Abebe, J. S. et al. DRUMMER-rapid detection of RNA modifications through comparative nanopore sequencing. Bioinformatics 38, 3113–3115 (2022).
Ni, P., Xu, J. R., Zhong, Z. Y., Luo, F. & Wang, J. X. RNA m6A detection using raw current signals and basecalling errors from Nanopore direct RNA sequencing reads. Bioinformatics 40, https://doi.org/10.1093/bioinformatics/btae375 (2024).
Chan, A. et al. Detecting m6A at single-molecular resolution via direct RNA sequencing and realistic training data. Nat. Commun. 15, 3323 (2024).
Oberdoerffer, S. & Gilbert, W. V. All the sites we cannot see: Sources and mitigation of false negatives in RNA modification studies. Nat. Rev. Mol. Cell Biol. 26, 237–248 (2024).
Maguire, S. & Guan, S. Rolling circle reverse transcription enables high fidelity nanopore sequencing of small RNA. PLoS ONE 17, e0275471 (2022).
Oxford Nanopore Technologies. Dorado. GitHub https://github.com/nanoporetech/dorado (2024).
Xie, Y. Y. et al. Single-molecule direct RNA sequencing reveals the shaping of epitranscriptome across multiple species. Nat. Commun. 16, 5119 (2025).
Liu, C. et al. Absolute quantification of single-base m6A methylation in the mammalian transcriptome using GLORI. Nat. Biotechnol. 41, 355–366 (2023).
Koh, C. W. Q., Goh, Y. T. & Goh, W. S. S. Atlas of quantitative single-base-resolution N6-methyl-adenine methylomes. Nat. Commun. 10, 5636 (2019).
Körtel, N. et al. Deep and accurate detection of m6A RNA modifications using miCLIP2 and m6Aboost machine learning. Nucleic Acids Res. 49, e92 (2021).
Tavakoli, S. et al. Semi-quantitative detection of pseudouridine modifications and type I/II hypermodifications in human mRNAs using direct long-read sequencing. Nat. Commun. 14, 334 (2023).
Mulroney, L. et al. A comprehensive survey of RNA modifications in a human transcriptome. Preprint at https://doi.org/10.1101/2024.10.22.619587 (2024).
Xiao, Y. et al. An elongation- and ligation-based qPCR amplification method for the radiolabeling-free detection of locus-specific N(6)-methyladenosine modification. Angew. Chem. Int. Ed. 57, 15995–16000 (2018).
Wang, Y. et al. LEAD-m(6)A-seq for Locus-specific detection of N(6) -methyladenosine and quantification of differential methylation. Angew. Chem. Int. Ed. 60, 873–880 (2021).
Xiao, Y. L. et al. Transcriptome-wide profiling and quantification of N6-methyladenosine by enzyme-assisted adenosine deamination. Nat. Biotechnol. 41, 993–1003 (2023).
Hao, X. et al. SNX25 regulates TGF-beta signaling by enhancing the receptor degradation. Cell. Signal. 23, 935–946 (2011).
Rui, Y. N. et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 17, 262–275 (2015).
Yu, J. et al. Disruption of NCOA2 by recurrent fusion with LACTB2 in colorectal cancer. Oncogene 35, 187–195 (2016).
Takahashi, S. et al. RASSF7 negatively regulates pro-apoptotic JNK signaling by inhibiting the activity of phosphorylated-MKK7. Cell Death Differ. 18, 645–655 (2011).
Burns, M. B. et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370 (2013).
Li, K. et al. The Wnt/β-catenin/VASP positive feedback loop drives cell proliferation and migration in breast cancer. Oncogene 39, 2258–2274 (2020).
Haussmann, I. U. et al. m6A potentiates alternative pre-mRNA splicing for robust sex determination. Nature 540, 301–304 (2016).
Choquet, K., Patop, I. L. & Churchman, L. S. The regulation and function of post-transcriptional RNA splicing. Nat. Rev. Genet. 26, 378–394 (2025).
Gleeson, J. et al. Isoform-level profiling of m(6)A epitranscriptomic signatures in human brain. Sci. Adv. 11, eadp0783 (2025).
Squires, J. E. et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 40, 5023–5033 (2012).
Taoka, M. et al. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 46, 9289–9298 (2018).
Boileau, E. et al. Sci-ModoM: a quantitative database of transcriptome-wide high-throughput RNA modification sites. Nucleic Acids Res. 53, D310–D317 (2024).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Lorenz, R. et al. ViennaRNA Package 2.0. Algorithms Mol. Biol. 6, https://doi.org/10.1186/1748-7188-6-26 (2011).
Seiji, F. polyleven. GitHub https://github.com/fujimotos/polyleven (2024).
Nakayama, H. et al. Ariadne: a database search engine for identification and chemical analysis of RNA using tandem mass spectrometry data. Nucleic Acids Res. 37, e47 (2009).
Levenshtein, V. Binary codes capable of correcting deletions, insertions, and reversals. Dokl. Akad. Nauk SSSR 163, 845–848 (1966).
Hagberg, A., Swart, P. J. & Schult, D. A. Exploring network structure, dynamics, and function using NetworkX. in Proc. of the 7th Python in Science Conference (SciPy2008) (ed. Varoquaux, G., Vaught, T. & Millman, J.) 11–15 (Pasadena, USA, 2008).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094 (2018).
Daniat, J. AGAT: Another Gff Analysis Toolkit to handle annotations in any GTF/GFF format. (Version v1.4.1). Zenodo https://doi.org/10.5281/zenodo.3552717.
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Harrison, P. W. et al. Ensembl 2024. Nucleic Acids Res. 52, D891–D899 (2023).
Hinton, G., Vinyals, O. & Dean, J. Distilling the knowledge in a neural network. Preprint at https://doi.org/10.48550/arXiv.1503.02531 (2015).
Loshchilov, I. Decoupled weight decay regularization. In Proc. of The Seventh International Conference on Learning Representations (2019).
Srivastava, N., Hinton, G., Krizhevsky, A., Sutskever, I. & Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 15, 1929–1958 (2014).
Loshchilov, I. & Hutter, F. SGDR: Stochastic gradient descent with warm restarts. In Proc. of The Fifth International Conference on Learning Representations (2017).
Paszke, A. et al. PyTorch: An imperative style, high-performance deep learning library. In Proc. Advances in Neural Information Processing Systems m32 (Nips 2019) (2019).
Detlefsen, N. S. et al. TorchMetrics - measuring reproducibility in PyTorch. J. Open Source Softw. 7, 4101 (2022).
Harris, C. R. et al. Array programming with NumPy. Nature 585, 357–362 (2020).
Ji, Y. R., Zhou, Z. H., Liu, H. & Davuluri, R. DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics 37, 2112–2120 (2021).
Oxford Nanopore Technologies. Remora. GitHub https://github.com/nanoporetech/remora (2024).
Bengio, Y., Ducharme, R., Vincent, P. & Jauvin, C. A neural probabilistic language model. J. Mach. Learn. Res. 3, 1137–1155 (2003).
Gong, Y., Chung, Y. A. & Glass, J. AST: Audio spectrogram transformer. In Proc. Interspeech 2021 (2021).
Dosovitskiy, A. et al. An image is worth 16x16 words: Transformers for image recognition at scale. In Proc. of The Ninth International Conference on Learning Representations (2021).
Fleming, A. M., Mathewson, N. J., Manage, S. A. H. & Burrows, C. J. Nanopore Dwell time analysis permits sequencing and conformational assignment of pseudouridine in SARS-CoV-2. ACS Central Sci. 7, 1707–1717 (2021).
Stephenson, W. et al. Direct detection of RNA modifications and structure using single-molecule nanopore sequencing. Cell Genom. 2, 100097 (2022).
Vaswani, A. et al. Attention Is All You Need. In Proc. of the Advances in Neural Information Processing Systems 30 (2017).
Devlin, J., Chang, M. W., Lee, K. & Toutanova, K. BERT: Pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies (2019).
Dao, T. FlashAttention-2: Faster attention with better parallelism and work partitioning. In Proc. of The Twelfth International Conference on Learning Representations (2024).
He, K., Zhang, X., Ren, S. & Sun, J. Identity mappings in deep residual networks. In Proc. European Conference on Computer Vision, 630-645 (2016).
Kullback, S. & Leibler, R. A. On information and sufficiency. Annal. Math. Stat. 22, 142–143 (1951).
Shannon, C. E. A mathematical theory of communication. Bell Syst. Tech. J. 27, 623–656 (1948).
Gamaarachchi, H. et al. GPU accelerated adaptive banded event alignment for rapid comparative nanopore signal analysis. BMC Bioinform. 21, 343 (2020).
Gamaarachchi, H. et al. Fast nanopore sequencing data analysis with SLOW5. Nat. Biotechnol. 40, 1026–1029 (2022).
Oxford Nanopore Technologies. Modkit. Github https://github.com/nanoporetech/modkit (2024).
Broad Institute. Picard toolkit. GitHub https://broadinstitute.github.io/picard/ (2025).
Pedregosa, F. et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python (vol 33, pg 219, 2020). Nat. Methods 17, 352–352 (2020).
McKinney, W. Data structures for statistical computing in Python. SciPy 445, 51–56 (2010).
Waskom, M. L. Seaborn: statistical data visualization. J. Open Source Softw. 6, 3021 (2021).
Hunter, J. D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007).
Tretyakov, K. matplotlib-venn. GitHub https://github.com/konstantint/matplotlib-venn (2024).
Gleeson, J. et al. Accurate expression quantification from nanopore direct RNA sequencing with NanoCount. Nucleic Acids Res. 50, https://doi.org/10.1093/nar/gkab1129 (2022).
Cock, P. J. A. et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423 (2009).
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R. & Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121 (2010).
Tareen, A. & Kinney, J. B. Logomaker: beautiful sequence logos in Python. Bioinformatics 36, 2272–2274 (2020).
Glinos, D. A. et al. Transcriptome variation in human tissues revealed by long-read sequencing. Nature 608, 353–359 (2022).
Tang, A. D. et al. Full-length transcript characterization of mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 11, https://doi.org/10.1038/s41467-020-15171-6 (2020).
Trincado, J. L. et al. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 19, 40 (2018).
Kang, G. et al. Comprehensive discovery of m6A sites in the human transcriptome at single-molecule resolution. DeepRM. https://doi.org/10.5281/zenodo.17636697 (2025).
Boža, V., Brejová, B. & Vinař, T. DeepNano: deep recurrent neural networks for base calling in MinION nanopore reads. PLoS ONE 12, e0178751 (2017).
Schneider, T. D., Stormo, G. D., Gold, L. & Ehrenfeucht, A. Information-content of binding-sites on nucleotide-sequences. J. Mol. Biol. 188, 415–431 (1986).
Rosenblatt, M. Remarks on some nonparametric estimates of a density-function. Annal. Math. Stat. 27, 832–837 (1956).
Parzen, E. On estimation of a probability density function and mode. Annal. Math. Stat. 33, 1065–1076 (1962).
Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT, Republic of Korea (MSIT; RS-2020-NR049538, RS-2022-NR067483, and RS-2025-02273009 awarded to D.B.), by a grant of Korean ARPA-H Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (MOHW; RS-2025-25422732 awarded to D.B.), by the National Institute of Health (NIH) research project (2022-ER1605-00 awarded to D.B.), by the Starting Growth Technological R&D Program funded by the Ministry of SMEs and Startups, Republic of Korea (MSS; RS-2023-00258711 awarded to D.B.), by the Technological Innovation R&D Program funded by MSS (RS-2025-25463995 awarded to S.P.), and by a grant of National Bio Bigdata Project funded by four ministries (MOHW, MSIT, Ministry of Trade, Industry and Energy, and Korea Disease Control and Prevention Agency) of Korea (RS-2024-00438566 awarded to D.B.). This study was also supported by Artificial Intelligence Industrial Convergence Cluster Development Project funded by MSIT and Gwangju Metropolitan City, by National IT Industry Promotion Agency (NIPA) funded by MSIT, and by Korea Research Environment Open Network (KREONET) managed and operated by Korea Institute of Science and Technology Information (KISTI).
Author information
Authors and Affiliations
Contributions
Conceptualization and supervision by D.B.; experiment by G.K., H.C., H.R.C., J.P., N.S., E.J., J.J., J.K., and Y.K.K.; bioinformatics analysis by H.H., H.J., N.Y., J.L., J.Y., W.C., and S.P.; writing by G.K., H.H., H.J., H.C., H.R.C., N.Y., and D.B.; resources and funding acquisition by S.P. and D.B.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Christoph Dieterich, Guan-Zheng Luo and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kang, G., Hwang, H., Jeon, H. et al. Comprehensive discovery of m6A sites in the human transcriptome at single-molecule resolution. Nat Commun 17, 664 (2026). https://doi.org/10.1038/s41467-025-67417-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67417-w
