
Abstract
Human skeletal muscle comprises myofibers formed by fusion of thousands of myoblasts. This process depends on tightly regulated, muscle-specific fusogens, but its genetic control remains poorly understood. Here, we identify CHAMP1 (Chromosome Alignment Maintaining Phosphoprotein 1) as essential for human myoblast fusion in vitro and in vivo. Genomic and protein-interaction assays reveal a noncanonical role for CHAMP1 as a MyoD cofactor that directly activates expression of the key muscle fusogen Myomaker. As established in prior clinical reports, CHAMP1 mutations in patients cause developmental delay, hypotonia, and muscle weakness. Consistently, patient-derived cells show fusion defects that can be fully rescued by restoring Myomaker expression. Structure and function analyses identify C2H2-type zinc-finger motifs on CHAMP1 protein that are both necessary and sufficient for MyoD interaction and Myomaker expression. These findings highlight a cell-autonomous role for CHAMP1 in muscle development and disease and point to therapeutic avenues for treating CHAMP1-related muscle development defects.
Similar content being viewed by others


Introduction
A fundamental step in muscle development and regeneration is the fusion of muscle precursor cells or myoblasts to form multinucleated myofibers1,2,3,4,5,6,7. Vertebrate myoblast fusion is controlled by the functional cooperation between membrane fusion proteins Myomaker (MYMK) and Myomixer (MYMX, also known as Minion and Myomerger)8,9,10,11,12. Myomaker induces myoblast membrane fusion by forming a dimeric, GPCR-like structure with a conserved lipid-binding site that facilitates phospholipid insertion and enables transcellular interactions essential for fusogenic activity13. Myomixer, through interaction with Myomaker, likely facilitates this process through its conserved hydrophobic motif that disrupts the stability of the plasma membrane9,14. Importantly, mutations of MYMK or MYMX cause fusion myopathy in patients characterized by developmental delay, hypotonia, impaired muscle growth, and facial dysmorphism15,16,17.
The expression of muscle fusogens Myomaker and Myomixer is uniquely restricted to skeletal muscle cells, transiently present only during the brief developmental window of myoblast fusion8,9. Unlike most muscle proteins, their tightly controlled expression is essential because prolonged presence of these agents might disrupt sarcolemma stability and lead to muscle injury18. Although the expression and activity of these fusogens are known to be regulated by myogenic factors including MyoD and MyoG, as well as several key signaling pathways19,20,21,22,23,24,25 including Notch, ERK1/2, CaMKII, IRE1α, and TGFβ, the precise molecular mechanisms underlying the transcriptional activation of myoblast fusion program remain poorly understood.
We recently developed a CRISPR screening platform and identified a group of genes whose loss impairs human myoblast fusion ref. 26. CHAMP1 (Chromosome Alignment-Maintaining Phosphoprotein 1) emerged from this screen as a surprising hit, as its known role in chromosome alignment pertains to the mitotic phase of dividing cells, whereas myoblast fusion occurs in post-mitotic cells—a context in which CHAMP1 has not previously been studied. More importantly, mutations in CHAMP1 gene cause CHAMP1 syndrome27,28,29,30,31 with patients consistently exhibit developmental delay, muscle weakness and hypotonia. Despite the significance, little is known about the precise molecular function of CHAMP1 in muscle development and disease. This study aims to close the gap through detailed functional and mechanistic analyses of CHAMP1 in human muscle cells.
We demonstrate that CRISPR-mediated deletion of CHAMP1 in human myoblasts abolishes myoblast fusion both in vitro and in vivo after transplantation into immunodeficient mice. Notably, fibroblasts derived from CHAMP1 patients also exhibit severe fusion defect when reprogrammed into muscle cells. Transcriptome analysis revealed a marked reduction of MYMK expression in CHAMP1 deficient cells. Importantly, the fusion defect, in both CRISPR mutants and patient cells, can be fully rescued by compensated expression of Myomaker.
Mechanistically, chromatin binding and protein interaction assays uncovered a non-canonical function for CHAMP1 as a crucial cofactor of MyoD in directly activating MYMK expression. Structural and functional analyses identified key C2H2-type zinc finger motifs on CHAMP1 that are both necessary and sufficient for MyoD interaction, induction of MYMK and activation of human myoblast fusion. Together, these findings advance our understanding of the regulatory mechanisms governing human myoblast fusion and reveal an unexpected role for CHAMP1 in direct transcriptional regulation and human myogenesis, linking its activity to the muscle-autonomous component of a rare yet devastating congenital disorder caused by CHAMP1 mutations.
Results
The nuclear protein CHAMP1 is essential for human myoblast fusion in vitro and in vivo
As a first step to probe the molecular function of CHAMP1 protein in human muscle cells, we examined its expression and localization in myoblasts cultured in proliferation condition (growth medium, GM) or in myocytes at various time-points post myogenic induction (differentiation medium, DM). Immunostaining confirmed the robust inductions of myogenic differentiation makers including skeletal myosin proteins (detected by MF20 antibody) and myogenin (MyoG), and heavy multinucleations of cells after differentiation (Fig. 1a). In all these stages, CHAMP1 protein was consistently and exclusive detected inside the nucleus with exception of day three post differentiation when the CHAMP1 staining signal was observed additionally in the cytosol of multinucleated myotubes (Fig. 1a).

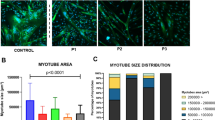
a Immunostaining result of CHAMP1 (top row), MyoG (middle row) and Myosin (MF20, gray signal in bottom row) for human myoblasts in growth medium or at various time points post myogenic induction. Scale bar: 100 μm. b Schematic of the human CHAMP1 protein and its domains, with the predicted CRISPR targeting sites for three selected gRNAs indicated by red arrows. The antibody recognition site at the C-terminus of the protein was highlighted. C2H2-ZNF: Cys2–His2 zinc-finger motif; WK, SPE, and FPE motifs are named after their consensus residues. c Representative immunostaining result. Numbers in the top panels denote CRISPR-induced indel sizes (bp) for the two CHAMP1 alleles (e.g., −14/ − 5 indicates 14-bp and 5-bp deletions on the two alleles). Scale bar, 10 μm for the top row and 100 μm for middle and bottom rows. Arrows point to multinucleated myotubes. GM: growth medium; DM: muscle differentiation medium. Quantification of myoblast differentiation (d) and fusion (e) at day 6 post myogenic differentiation from three independent experiments. The fusion index is normalized to the number of nuclei expressing myosin. WT: wildtype. f Design of human myoblast transplantation experiment. The hindlimb of the immunodeficient mice were pre-irradiated and pre-injured to eliminate endogenous muscle stem cells and promote human myoblast engraftment. g Representative immunostaining results of human myoblast transplantation experiments. LAMIN A/C labels human nucleus after transplantation; MYH3 marks regenerating muscle cells; laminin delineates the muscle cell periphery. Scale bar: 10 μm. h, Distribution of cross-sectional area of human myofibers from transplantation experiments. n = 4 for each genotype. P values were calculated using one-way ANOVA followed by Tukey’s multiple comparisons test (d, e) and two-sided unpaired Student’s t test (h). Data are presented as mean ± s.d. Source data are provided as a Source Data file.
Using CRISPR-Cas9 and gRNAs targeting different coding regions (Fig. 1b), we generated a panel of clonally derived human CHAMP1 deficient (CHAMP1∆) myoblasts. Genotyping and DNA sequencing confirmed the biallelic frame-shift mutations in all CHAMP1∆ clones (Supplementary Fig. 1). Immunostaining using an antibody raised against a C-terminus of CHAMP1 protein (amino acids 514–646, Fig. 1b) confirmed the complete depletion of CHAMP1 expression in CHAMP1∆ myoblasts (Fig. 1c). EdU incorporation assays showed similar proliferation rates between CHAMP1∆ and wildtype (WT) myoblasts, except for one CHAMP1∆ clone that exhibited a significant reduction in EdU incorporation (Supplementary Fig. 2a, b). Live/dead cell staining revealed comparable viability across all CHAMP1∆ and WT cells (Supplementary Fig. 2c, d). Karyotype analysis showed no gross chromosomal abnormalities for two CHAMP1∆ clones, while the third clone, which also exhibited reduced proliferation, showed minor regional chromosomal alterations (Supplementary Fig. 3). Importantly, since all CHAMP1∆ clones were derived from single cells following CRISPR-mediated gene inactivation and had undergone more than 20 divisions before karyotype analysis, these findings suggest that overall chromosome segregation during mitosis is largely preserved in CHAMP1∆ myoblasts.
Upon induction of myogenic differentiation, CHAMP1∆ myoblasts normally express myosin protein (Fig. 1c’) and exhibit a comparable differentiation index with that of WT cells (Fig. 1d). However, unlikely WT cells, which readily fuse into large and heavily multinucleated myotubes, the majority of CHAMP1∆ myoblasts fail to fuse after full-term induction of muscle differentiation (DM 6 days, Fig. 1c’, e). This defect persists after an extended period of culture in differentiation medium (DM 10 days, Fig. 1c”), suggesting an essential role of CHAMP1 for human myoblast fusion.
We then tested the fusion capacity of human CHAMP1∆ myoblasts in vivo by transplanting the cells into pre-injured muscle tissue of immunodeficient mice (Fig. 1f). To promote engraftment of human myoblasts, we adopted a previously established irradiation protocol32 to treat the recipient mice that eliminates the regenerative function of endogenous satellite cells prior to muscle injury. After transplantation and tissue collection, human cells were identified by immunostaining for the expression of human-specific nuclear marker LAMIN A/C, while regenerating muscle cells were detected by immunostaining for embryonic myosin (MYH3). Notably, CHAMP1∆ myoblasts form markedly smaller myofibers compared to those formed by WT myoblasts (Fig. 1g, h). In both groups, no regenerating myonuclei of mouse origin (LAMIN A/C–/MYH3+/Hoechst+) were detected, confirming that regeneration by host satellite cells was effectively suppressed by the irradiation. Together, these results suggest that despite normal myogenic differentiation, the fusion program of human myoblasts is severely impaired in vivo upon genetic disruption of CHAMP1 function.
CHAMP1 controls human myoblast fusion by activating Myomaker expression
To elucidate the molecular mechanism underlying the fusion phenotype of human CHAMP1∆ myoblasts, we conducted a time-course transcriptome analysis by comparing CHAMP1∆ myoblasts with a rescue control group in which full-length CHAMP1 protein was reintroduced into CHAMP1∆ myoblasts through retrovirus infection. This allowed a direct comparison between CHAMP1∆ cells and their rescued counterparts in an isogenic background, enabling unambiguous examination of CHAMP1’s molecular function. For both groups, we collected cells under growth medium conditions (DM 0 day), at the two day of differentiation (DM 2 days), and at a later stage of differentiation (DM 6 days).
Consistent with our EdU incorporation and live/dead cell staining assays, transcriptome analysis showed comparable expression of proliferation markers (Supplementary Fig. 4a) and apoptosis markers (Supplementary Fig. 4b) between CHAMP1∆ cells and their isogenic rescued counterparts. Muscle differentiation also appeared largely normal in CHAMP1∆ myoblasts, as reflected by similar expression of key myogenic transcription factors (e.g., MYF5, MYOD, MYOG, MEF2C), fusion protein MYMX, and structural genes (e.g., ACTA1, CACNB1, CAV3, CAVIN4, MYBPH, MYH2, MYH3, MYH7, MYH8, MYOT, SYNPO2, TNNI1, TNNT2, TTN) relative to rescue controls (Fig. 2a). Comparison with WT myoblasts revealed a delayed expression of these muscle structural genes in CHAMP1∆ cells; however, most of these changes were not affected by CHAMP1 re-expression in CHAMP1∆ cells, suggesting that these difference likely reflects clonal variations of CHAMP1∆ lines rather than a specific molecular function for CHAMP1.

a Heatmap of RNA-seq results for selected muscle differentiation markers at various time points post-myogenic induction in wildtype, CHAMP1∆, and CHAMP1∆ isogenic rescue cells. Among them, several genes differentially expressed in CHAMP1∆ compared with the CHAMP1∆ isogenic rescue (fold change >2), including MYMK, are boxed. Muscle structural genes are listed in alphabetical order. Heatmap values are based on Z-scores. Diff: differentiation; Sig: significantly. Complete list of the differentially expressed genes is seen in Supplementary Data 1. b Myomaker immunostaining results. Human myoblasts were differentiated for 3 days. The cell boundary of a multinucleated myotube was outlined. Scale bar: 10 μm. c Western blot that confirmed the successful re-expression of CHAMP1 in the rescue experiment as shown in panels (d and e). Myosin immunostaining (d) and fusion quantification (e) results. The fusion index is measured from three biological replicates and normalized to the number of nuclei expressing myosin (instead of total number of nuclei). Cells were differentiated for 6 days. Scale bar: 100 μm in panel (d). The experiment was independently repeated two times for panel (b) and three times for panels (c and d) with similar results. Data are presented as mean ± s.d. P values were calculated using one-way ANOVA followed by Tukey’s multiple comparisons test (panel e). Source data are provided as a Source Data file.
Using a two-fold change threshold, no differentially expressed genes between CHAMP1∆ and the isogenic rescue control group were observed under the growth medium condition. In contrast, 52 genes were differentially expressed at day 2 of differentiation—all of which were downregulated—and 201 genes at day 6 of differentiation, with 176 of them downregulated in CHAMP1∆ cells compared to the rescue control (Supplementary Data 1). Among the genes commonly downregulated at both day 2 and day 6 of differentiation were CACNG1, CKM, MYMK, TNNC2, and TNNI2 (Fig. 2a).
Cross referencing of these differentially expressed genes with our previous myoblast fusion screen result revealed MYMK as the only gene standing out from both experiments. Notably, MYMK was the second most downregulated gene (by 7.1 folds) in CHAMP1∆ myoblasts at day 2 of differentiation, the peaking stage of myoblast fusion. Immunostaining confirmed the reduction of MYMK expression at protein level in CHAMP1∆ cells compared to WT cells (Fig. 2b). Notably, forced expression of Myomaker in CHAMP1∆ cells can rescue myoblast fusion, surpassing the fusion levels achieved by CHAMP1 rescue itself (Fig. 2c–e). By contrast, expressing another muscle fusogen Myomixer (MYMX), the function partner of Myomaker9, or MyoD, an upstream regulator of Myomaker24, did not improve fusion outcomes of CHAMP1∆ cells (Fig. 2d, e). These results highlight a crucial and specific role of Myomaker deficiency underlying the fusion defect of CHAMP1∆ cells.
CHAMP1 patient derived cells show reduced Myomaker expression and defective myoblast fusion which can be rescued by compensated Myomaker expression
De novo mutations of CHAMP1 gene cause CHAMP1 syndrome, a neurodevelopmental disorder showing muscle development delays, hypotonia, muscle weakness & facial dysmorphism27,30,31,33. These symptoms were also observed in Carey-Fineman-Ziter syndrome caused by hypomorphic MYMK mutations15. At present, the genetic connections between the two diseases remain unknown. A more critical question is whether the muscle defects in CHAMP1 disorders result from muscle-autonomous dysfunction.
We obtained skin primary fibroblasts from two CHAMP1 syndrome patients along with fibroblasts from their healthy family members as controls34. Genotyping and DNA sequencing confirmed distinct heterozygous CHAMP1 mutations in the patient cells (Fig. 3a). In one patient, a nonsense variant that replaces a cytosine with thymine at position 1489 of the CHAMP1 coding sequence was detected (c.1489 C > T), resulting in the substitution of arginine with a premature stop codon at amino acid position 497 (p.Arg497*); in the other patient, a frameshift variant caused by the deletion of two nucleotides (cytosine and thymine) at positions 542 and 543 was identified (c.542_543delCT), leading to a frameshift and a premature stop codon five residues downstream (p.Ser181Cysfs*5).

a Sanger sequencing results showing the CHAMP1 genotypes in primary skin fibroblasts isolated from healthy donors and CHAMP1 patients. p.: position; fs: frame shift; *, stop codon. b Myosin immunostaining of fibroblasts post-MyoD-induced trans-differentiation. Patient cells were also transduced with Myomaker expression vector to test its effect on fusion. Scale bar, 100 μm. c MYMK qPCR results in human fibroblasts with retroviral expression of empty vector (control) or MyoD protein. d Quantification results of cell fusion as in panel b. MYMK: Myomaker. The quantification data is measured from three biological replicates (c, d). The experiment was independently repeated three times with similar results. Data are presented as mean ± s.d. from three independent experiments. P values were calculated using one-way ANOVA followed by Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
EdU incorporation and live/dead staining assays revealed similar proliferation rate (Supplementary Fig. 5a, b) and viability (Supplementary Fig. 5c, d) between patient cells and healthy control cells. Because fibroblasts cannot be directly used as a model to study muscle development, we first converted them into myoblast-like cells by retroviral expression of MyoD, the master regulator of myogenesis35. Because retroviral transduction is limited to dividing cells and primary fibroblasts have relatively low mitotic activity, infection efficiency was modest (20%–25%, Supplementary Fig. 5e). Consequently, myogenic conversion was also limited (15%–30%, Supplementary Fig. 5f), as measured by the proportion of nuclei within MF20-positive cells relative to the total number of nuclei. To account for the myogenic conversion efficiency, we quantified cell fusion after normalizing to the muscle differentiation index.
In these assays, CHAMP1 patient-derived cells—unlike healthy controls—failed to fuse and did not form multinucleated myotubes (Fig. 3b). At the molecular level, the fusion defect of patient cells was accompanied by reduced MYMK expression for both patients (Fig. 3c) and can be rescued when the expression of Myomaker is ectopically compensated (Fig. 3b, d). These results suggest that CHAMP1 mutations may have cell-autonomously contributed to muscle development defects in CHAMP1 patients, and more importantly, compensated MYMK expression could be a therapeutic target to reverse such defects.
CHAMP1 controls Myomaker expression through interactions with MyoD
CHAMP1 protein is exclusively detected inside nucleus of differentiating myoblasts prior to myoblast fusion (Fig. 1a). Coincidently, all 52 genes including Myomaker, that were significantly changed in CHAMP1∆ myoblasts were in fact downregulated (Supplementary Data 1), suggesting that CHAMP1 might control human myogenesis by activating target gene expression.
Previously, we identified MyoD as a key factor for the transcriptional induction of MYMK expression and activation of human myoblast fusion24. Consistent with this notion, RNA-seq analysis of human MyoD∆ myoblasts, generated previously by CRISPR-induced mutagenesis24, revealed a complete loss of MYMK expression (Fig. 4a). As a confirmation of gene-specific CRISPR knockout effect, the MYMK expression can be robustly rescued upon re-expression of MyoD in MyoD∆ myoblasts (Fig. 4a). Likewise, expression of MYMK was greatly reduced in human CHAMP1∆ myoblasts, a change that was reversed upon re-expression of CHAMP1 in CHAMP1∆ myoblasts (Fig. 4a). Like Myomaker, majority of CHAMP1 target genes also showed consistent changes in MyoD∆ cells compared with the isogenic rescue group in which MyoD was restored in MyoD∆ cells (orange bars, Fig. 4b). Among these common targets for CHAMP1 and MyoD, many genes (e.g., MYMK) exhibited even greater reductions upon deletion of MyoD (Fig. 4b). These results suggest that CHAMP1 might regulate the expression of its target genes by working with MyoD.

a Human genome browser tracks showing the MYMK locus in human myoblasts at various time points post-myogenic induction. The enhancer region is outlined by a black box (a’). Diff.: Differentiation. ATAC-seq: assay for transposase-accessible chromatin sequencing. CUT&Tag: cleavage under targets and tagmentation. b Fold changes for 52 differentially expressed (DE) genes (padj <0.05) in CHAMP1∆ myoblast after normalization to isogenic rescue control group (as 1). The orange bars show the expression changes of the same set of 52 genes in MyoD∆ myoblasts normalized to its own isogenic rescue control group (as 1). Cells were differentiated for two days. p values were calculated using DESeq2 (version 1.40.2) in R (version 4.3.1) combined with Benjamini-Hochberg multiple testing correction. c–e Heatmaps depicting the CHAMP1 and MyoD co-occupied peaks in CUT&Tag data from human myotubes. Gene annotation results of the peaks are seen in Supplementary Data 2 and 3. Human myoblasts were differentiated for two days. f Highest ranking motifs for CHAMP1 and MyoD bound sites in human myotubes. g Representative immunostaining results of CHAMP1 and MyoD in human myoblasts of various genotypes. Scale bar: 2.5 μm. h Representative fluorescence images for proximity ligation assays (PLA) in human myoblasts of various genotypes. Cells were differentiated for two days. Scale bar: 15 μm. The experiment was independently repeated three times for panels (g and h) with similar results. Source data are provided as a Source Data file.
At present, little is known about the role and mechanism by which CHAMP1 works as a transcriptional regulator to control tissue development. To examine the chromatin binding of CHAMP1 and its functional relationship with MyoD, we conducted CUT&Tag experiments for this duo using WT human myoblasts at peak of myoblast fusion (DM 2 days). Intriguingly, these analyses identified 33,324 binding peaks for CHAMP1 protein (Supplementary Data 2). Moreover, 72.6% of CHAMP1 binding peaks were also bound by MyoD protein (Fig. 4c, d). Likewise, 42.4% of the 57,069 MyoD peaks (Supplementary Data 3) showed binding by CHAMP1 (Fig. 4d, e). The discovered motifs for CHAMP1 peaks closely resemble those for MyoD, with the highest ranking motif for both proteins being the CAGCTG-type E-box (Fig. 4f), a canonical MyoD binding motif36,37,38.
CHAMP1 and MyoD co-occupy a region in the intron 1 of MYMK that corresponds to an open chromatin region, as revealed by ATAC-seq (Fig. 4a’). This region is also enriched for the H3K27ac histone modification (Fig. 4a’), indicating it is an active enhancer. We also performed HiCAR (High-throughput Chromatin Accessibility and Regulatory interactions) to profile enhancer–promoter interactions in human myoblasts at day 2 post-induction. This analysis identified chromatin interactions at the MYMK locus (chr9:133,514,586–133,524,959), with the contact matrix revealing direct interactions between an intronic enhancer and the MYMK promoter within a single TAD (Supplementary Fig. 6), supporting their regulatory connection.
Importantly, the co-occupancy of CHAMP1 and MyoD on this enhancer appears to be dependent on each other because CUT&Tag signals for CHAMP1 binding were lost in MyoD∆ myoblasts; likewise, deletion of CHAMP1 also abolished MyoD binding to the same enhancer (Fig. 4a’). Such reciprocal effect is not simply due to a change in the expression or localization of one protein when the other factor is genetically deleted because the MyoD protein can be normally detected inside the nucleus of CHAMP1∆ myoblasts (Fig. 4g), and likewise, deletion of MyoD also did not affect the nuclear expression of CHAMP1 protein (Fig. 4g).
We then investigated whether CHAMP1 protein can directly interact with MyoD protein to control gene expression in human muscle cells. Indeed, super-resolution imaging revealed co-localization of this duo in differentiating WT myoblasts (Fig. 4g). Co-immunoprecipitation experiment and proximity ligation assay (PLA) confirmed the CHAMP1 and MyoD interaction in human myoblasts (Supplementary Fig. 7 and Fig. 4h). The protein-protein interaction signal was not detected when PLA was performed using either human CHAMP1∆ or MyoD∆ myoblasts, confirming the specificity of these interaction assays (Fig. 4h). Together, these results suggest that CHAMP1 protein functions as a key cofactor for MyoD in activating MYMK transcription and myoblast fusion.
C2H2-ZNF motifs, including those truncated from CHAMP1 patient mutations, are essential for CHAMP1 interaction with MyoD and activation of human myoblast fusion
We then performed the structure and function analysis of CHAMP1 protein for a deeper mechanistic understanding of its interaction with MyoD. CHAMP1 protein contains five C2H2-type zinc finger motifs39 (C2H2-ZNF): two located at N-terminus (amino acid positions: 16–37; 64–85) and three at C-terminus (amino acid positions: 711–732; 740–760; 767–786) (Fig. 5a). The central region of the protein includes several other domains, with the FPE domain critical for chromosome alignment as initially characterized in HeLa cells39. Patients with CHAMP1 syndromes frequently show truncations29 (e.g., at amino acid positions 181 or 497, Fig. 3a) that often delete a region that includes the C-terminal C2H2-ZNF motifs yet the precise role of these motifs remains largely unknown.

a Schematic representation of the human CHAMP1 protein and a series of truncation mutants. Numbers highlight the amino acid positions on human CHAMP1 protein. Two predicted nuclear location signals (NLS1: LFPSSKKLKKD; NLS2: MKRGKGK) are indicated with red arrows. C2H2-ZNF: Cys2–His2 zinc-finger motif; WK, SPE, and FPE motifs are named after their consensus residues. b Western blot confirming expression of Flag tagged CHAMP1 mutants depicted in panel (a). Representative myosin immunostaining images (c) and quantification results (d) of CHAMP1∆ myoblasts. Scale bar, 100 μm. The fusion index is normalized to the number of nuclei expressing myosin. e Western blot analysis of Flag tagged CHAMP1 point mutations that disrupt each individual C2H2-ZNF motifs. Numbers below the CHAMP1 blot showed the average band intensities from three independent Western blotting experiments. f Quantification results from proximity ligation assays (PLA) for the interaction between MyoD and CHAMP1 (WT or point mutations) after normalization to the protein expression level. g Quantification of myoblast fusion in CHAMP1∆ myoblasts after normalization to the protein expression level. The fusion index is normalized to the number of nuclei expressing myosin. h Representative FLAG immunostaining images of CHAMP1∆ myoblasts expressing the empty vector control, FLAG-tagged full-length CHAMP1 protein, or various truncation mutants. Scale bar: 10 μm. i Western blot confirming that the deletion of predicted NLS sites eliminates nuclear expression of CHAMP1. Nu.: nucleus; Cy.: cytosol. j Quantification of myoblast fusion in CHAMP1∆ myoblasts expressing empty vector control (EV), WT CHAMP1, or CHAMP1 mutants. Experiments were performed after 2 days of differentiation in panel f, and after 6 days in (c, d, g and j). Data are presented as mean ± s.d. from three independent experiments. P values were calculated using one-way ANOVA followed by Tukey’s multiple comparisons test (d, f, g, j). In panels d, f, and g, the p-values for CHAMP1-mutant groups are from comparisons with the WT group. Source data are provided as a Source Data file.
We generated a series of CHAMP1 truncation mutants fused with Flag tag and confirmed their expression in CHAMP1∆ cells by Western blotting (Fig. 5b). Interestingly, deletion of the C2H2-ZNF motifs, but not any other domains, of CHAMP1 abolished its activity in the myoblast fusion test using CHAMP1∆ cells (Fig. 5c, d). Moreover, CHAMP1 mutant containing only the C2H2-ZNF motifs (5x ZNF, 45% of protein sequence) showed comparable activity with that of the full-length CHAMP1 protein in CHAMP1∆ cells (Fig. 5c, d). Thus, the five C2H2-ZNF motifs are both necessary and sufficient to carry out CHAMP1’s function in human muscle cells.
The C2H2-ZNF motif, which is abundant in transcription factors, coordinates a zinc ion via two cysteine (C2) and two histidine (H2) residues and can mediate various functions, including protein-protein interactions40. To explore its role in CHAMP1, we then generated CHAMP1 mutants by changing the two histidine (H) residues in each C2H2-ZNF motif to alanine (A). These mutations are expected to cause major disruptions to the structure and function for each targeted C2H2-ZNF motif. Western blots confirmed the expression of these mutants after transduction into human CHAMP1∆ myoblasts (Fig. 5e). Interestingly, PLA tests showed that mutations of the 4th C2H2-ZNF motif (H756/760 A) drastically reduced the CHAMP1’s interaction with MyoD (Fig. 5f). Importantly, this mutation abolished CHAMP1 activity in the myoblast fusion test using CHAMP1∆ cells (Fig. 5g). Together, these results confirm the crucial role of MyoD–CHAMP1 interaction for human myoblast fusion and reveal the key role of the 4th C2H2-ZNF motif on CHAMP1 for mediating this interaction.
Because CHAMP1 is a relatively large protein, it likely requires active nuclear import via nuclear localization signal (NLS) to access the nucleus and bind MyoD. Sequence analysis of CHAMP1 identified two NLS: NLS1 (residues 612–622, LFPSSKKLKKD) and NLS2 (residues 702–708, MKRGKGK) (Fig. 5a), which are retained in the 5× ZNF truncation mutant. Consistently, Flag immunostaining of CHAMP1 truncation mutants confirmed that the C-terminal 222 residues containing the two predicted NLS is required for the nuclear localization of CHAMP1 (Fig. 5h). Notably, deletion of NLS1 and NLS2, but not NLS1 alone, abolished CHAMP1’s nuclear localization, as shown by immunostaining (Fig. 5h) and nuclear fractionation followed by Western blotting analysis (Fig. 5i), this was accompanied by reduced CHAMP1 function in the myoblast fusion assay (Fig. 5j). Thus, CHAMP1 depends on NLS2 for its proper nuclear localization and function in human myoblast.
Discussion
De novo mutations in the CHAMP1 gene cause CHAMP1 syndrome, a neurodevelopmental disorder showing muscle development delays, hypotonia, muscle weakness & facial dysmorphism27,30,31,33,41. These symptoms appear at birth, with delayed motor milestones evident in the first year. Hypotonia and muscle weakness lead to motor delays and poor coordination, often requiring mobility aids and making daily tasks difficult. All affected individuals require 24/7 care. Despite the devastating outcome, the precise function of CHAMP1 in muscle cells remained unknown. In the present study, we identified a critical role for CHAMP1 in activating MYMK expression and promoting human myoblast fusion. Consistently, cells derived from CHAMP1 patients exhibited a marked reduction in MYMK expression and pronounced fusion defects, which were fully rescued by compensatory Myomaker expression. These findings suggest that the muscle symptoms observed in CHAMP1 syndrome may result from muscle-intrinsic dysfunction of CHAMP1. However, confirming fusion myopathy in patients requires future works of careful histopathological evaluation. Findings from individuals with MYMK mutations suggest that fusion defects are associated with pronounced imbalance in fiber size, with hypertrophied type II (fast twitch) fibers and atrophic type I (slow twitch) fibers, resembling congenital fiber type disproportion15.
At the molecular level, future studies are needed to elucidate the precise mechanism by which CHAMP1 patient mutations—predominantly heterozygous truncating variants—lead to muscle phenotypes, whether through haploinsufficiency, dominant negative effects, or gain-of-function mechanisms. Clinical studies show that individuals with CHAMP1 truncations exhibit more severe developmental impairments than those with full-gene deletions, suggesting a distinct pathogenic mechanism for each type of gene mutations29. In our functional analysis, re-expression of full-length CHAMP1 in CHAMP1∆ myoblasts (which retained CRISPR-induced truncated mutant expression when detected using an N-terminus targeting CHAMP1 antibody) partially rescued myoblast fusion but did not restore it to wild-type levels (Fig. 2d), supporting a dominant negative effect of the truncation mutants.
CHAMP1 was first discovered as a key factor for chromosome alignment, responsible for maintaining kinetochore-microtubule attachment in mitotic cells39,42. Recently, CHAMP1 was shown for heterochromatin assembly and can promote homologous recombination repair in cancer cells43,44,45. However, these function unlikely cause myoblast fusion defect for several reasons. First, although all CHAMP1∆ myoblasts show fusion defects, only one out of multiple clones exhibited regional chromosomal abnormalities in the karyotype analysis (Supplementary Fig. 3). Second, myoblast fusion only occurs when the cells withdraw from the cell cycle. In these post-mitotic cells, chromosome alignment unlikely plays direct role to control membrane fusion. Supporting this notion, none of CHAMP1’s known interactors, including CBX1/3/5, MAD2L2 and POGZ—proteins with well-established roles in chromosome alignment and DNA repair34—were identified in our initial fusion screen where CHAMP1 stood out as a highly significant hit. Last but most importantly, chromosomal alterations, by either misalignment or failure in the DNA repair, often occur randomly and are most likely irreversible; therefore, it is unexpected that the fusion phenotype of CHAMP1∆ myoblasts would be rescued by re-expressing CHAMP1 or Myomaker if global changes of chromosomal structure or random DNA mutations were the underlying cause.
However, since our experiments were conducted on immortalized human myoblasts stably expressing CDK4 and hTERT, it is possible that CHAMP1’s canonical functions in chromosome alignment and DNA repair were masked by the effects of cell immortalization. Thus, the muscle developmental defects observed in CHAMP1 patients may result from distinct yet non-exclusive mechanisms acting at different stages of human myogenesis—chromosomal alterations in proliferating muscle precursor cells, and defects in myoblast fusion after these cells exit the cell cycle and begin muscle differentiation. Although this study focused on myoblast fusion, our transcriptome analysis also revealed reduced expression of several muscle structural genes essential for locomotor function (CACNG1, MYL2, TNNC2, TNNI2, JUP, MYL5, ACTN3, STRIT1, MYL3) in CHAMP1∆ cells (Supplementary Data 1). This result may help explain the muscle weakness and hypotonia observed in CHAMP1 patients, even when a small portion of myoblasts managed to fuse.
Our previous study on human myoblast fusion established a crucial role for MyoD in activating the expression of MYMK24. In the present study, we showed that the deletion of CHAMP1 did not affect MyoD expression and its localization, and vice versa. Consistently, overexpression of MyoD also failed to rescue the fusion defect in CHAMP1-deficient myoblasts. Similarly, overexpression of CHAMP1 in 10T1/2 fibroblasts (Supplementary Fig. 8a), where MyoD expression is absent, failed to confer the fusion activity to fibroblast (Supplementary Fig. 8b). These results highlight an crucial role of both CHAMP1 and MyoD for the activation of MYMK expression and cell fusion.
Genome-wide profiling of MyoD binding in human myoblasts revealed ~57,000 peaks, closely matching the ~60,000 peaks reported in murine counterparts36,46. These binding sites were mapped close to 18,938 genes (Supplementary Data 3), yet only 1084 genes (5.7%) including MYMK exhibited significant expression changes upon genetic deletion of MyoD in human myoblasts. Thus, only small fraction of MyoD binding sites are associated with local transcription regulation, a notion that is also supported by previous studies using mouse C2C12 cells36,37,46. Similarly, CHAMP1 binding sites were detected near 14,148 genes in human myoblasts (Supplementary Data 2); however, only 52 genes and 201 genes showed significant expression changes ( > 2 folds) after 2 days and 6 days of differentiation, respectively. These findings underscore the functional significance of CHAMP1–MyoD co-occupancy yet also highlight the existence of additional mechanisms that dictate the gene specificity to MyoD or (and) CHAMP1 regulations during human myogenesis.
In line with this speculation, our structure and function analysis of CHAMP1 protein revealed four C2H2-ZNF motifs (#1, #2, #3, #5) that are required for CHAMP1’s function (Fig. 5g) but are dispensable for MyoD interaction (Fig. 5f). Thus, these motifs, including three C-terminal C2H2-ZNF motifs frequently missing from patient mutants, might be crucial for binding with other MyoD co-factors including p300, TCF3/12 that are essential for the transcription induction of MyoD target genes47,48,49,50. Thus, future studies aimed at identifying all components of this transactivation complex and determining its dynamic structures will be essential for a complete understanding of the target selection and CHAMP1’s regulation of MyoD during human myogenesis. Such knowledge will hold the key to understand the precise mechanism by which the CHAMP1 patient variants cause muscle development defect, especially when it incurs a dominant negative effect. Our study also encourages probing the transcriptional factor function of CHAMP1 in regulating the development and function of other cell types affected in CHAMP1 syndrome. Identifying both tissue-specific and shared mechanisms could inform the development of personalized therapies, tailored to individual patient mutations, with the ultimate goal of ameliorating or potentially curing CHAMP1-related disorders.
Methods
Human myoblast isolation, culture and karyotype analysis
Human myoblasts (AB1190) were isolated from paravertebral muscle of a healthy 16-year-old male donor51. Briefly, the muscle biopsies were finely minced and explants were plated onto noncoated Petri dishes in drops of fetal calf serum (Invitrogen, Carlsbad, CA). Primary cultures from muscle biopsies were co-transduced with two retroviral vectors expressing hTERT and CDK-4 cDNAs. Co-transduced cells were selected by neomycin and puromycin and then purified using magnetic beads coupled to antibodies directed against the myogenic marker CD56 (NCAM1). Individual clones with high myogenic and fusion capacity were selected. Human myoblasts are available upon request from Dr. Vincent Mouly at vincent.mouly@upmc.fr. Human MyoD∆ myoblasts were generated by CRISPR mutagenesis and authenticated previously24. Immortalized human myoblasts are maintained in skeletal muscle cell basal medium (PromoCell, C-23260) supplemented with 15% fetal bovine serum (FBS; Sigma-Aldrich, F2442), 5% growth medium supplement mix (PromoCell, C-39365), GlutaMAX, and 1% gentamicin sulfate. For differentiation, myoblasts were cultured in differentiation medium comprising DMEM supplemented with 2% horse serum, 10 μM DAPT (Cayman, 13197), and 1% penicillin/streptomycin. Karyotype analysis was performed by KaryoStat™ Assay using Genechip Probe Array containing 1.1 million probes across somatic and sex chromosomes.
Culture of patient and healthy control skin fibroblasts
Primary human skin fibroblasts—healthy control 1 (male, 32 years; GM27968), patient 1 (male, 23 months; GM27353), healthy control 2 (female, 30 years; GM27961), and patient 2 (female, 2 years; GM27963)—were obtained from the Coriell Institute and have been described previously34. No primary isolation was performed in this study. Cells are cultured according to the supplier’s recommended protocol. Cells were maintained in Eagle’s Minimum Essential Medium (EMEM) with Earle’s salts, non-essential amino acids, and 2 mM L-glutamine, supplemented with 15% non–heat-inactivated fetal bovine serum. All experiments were performed using cells between passages 3 and 5. For the sequencing validation of the CHAMP1 genotypes, genomic DNA was extracted and used for PCR amplification with the two pairs of primers. F1 (GCCCTCCTCTTCCTGAACAC) and R1 (GGATGGTCCCCAAGGTTCTG) for the authentications of GM27961 and GM27961 cells. F1&R1 primer pair amplifies 458 bp for WT; F2 (CCCACCACTATCCCCAGAGA) and R2 (GGAGTTCAGGGAAAAGGGCA) for the authentications of GM27353 and GM27961 cells. F2&R2 primer pair amplifies 422 bp for WT.
EdU incorporation assay
Cells were seeded at around 30% confluency one day before EdU incorporation assay. For the EdU assay, cells were cultured in growth medium containing 40 μM EdU for 3.5 h. EdU staining was performed by click chemistry following a published protocol52. Briefly, cells were fixed in PFA and were rinsed with PBS, permeabilized with 0.1% Triton X-100 for 10 min at room temperature, and then incubated for 30 min in the dark with a freshly prepared click reaction mixture containing 100 mM Tris, 1 mM CuSO₄, 1 μM Azide-Fluor 545, and 100 mM ascorbic acid. Nuclei were counterstained with Hoechst.
Cell viability assay
The viability of human myoblasts and primary fibroblasts was assessed by flow cytometry using the LIVE/DEAD™ Fixable Violet Stain Kit (ThermoFisher, L34963) according to the manufacturer’s protocol. Cells were detached with TrypLE (Gibco, 12605028), washed once with PBS, and resuspended at 1 × 10⁶ cells/ml in PBS containing 1 μl of dye per ml. Samples were incubated on ice for 20 min, washed once with PBS, and resuspended in 1% BSA/PBS prior to analysis on a CytoFLEX S Flow Cytometer (Beckman Coulter, Cat. No. B75408). Cells were gated by FSC-H and SSC-H to exclude debris and enrich for the main population. A dead cell control (freeze–thawed at –80 °C) was used to define LIVE/DEAD dye distribution; live cells were gated by excluding the ~10% tail overlapping with the dead cell population. Flow cytometry data were analyzed with FlowJo v10.8.1 to quantify the percentage of live and dead cells.
Fibroblast mixing and fusion experiment
C3H/10T1/2 fibroblasts (ATCC, CCL-226, Clone 8) were transduced with GFP-retrovirus together with either empty-retrovirus, myomaker-retrovirus, or CHAMP1-retrovirus. After infection, cells were thoroughly washed, detached with TrypLE, mixed with human WT myoblasts at a 1:1 ratio, and cultured in a mixed medium composed of equal volumes of 10% FBS–DMEM and human myoblast medium. Mixed cultures were allowed to attach and then switched to myogenic differentiation medium for three days, after which cells were subjected to myosin immunostaining and imaging.
Human myoblast transplantation experiment
All animal procedures were conducted with approval from the Institutional Animal Care and Use Committee (IACUC) at the University of Georgia. 2–3 months age NSG mice of both male and female (Jackson Laboratory, 005557) were anesthetized with isoflurane vapor during the entire procedure to perform targeted X-ray irradiation of the hind limbs (Rad320 X-ray, 1100 Rad)32. The mice’s main bodies were shielded with a lead covering, leaving only the hind limbs exposed to irradiation. One day after irradiation, a localized muscle injury was induced by intramuscular injection of 20 µL of 1.2% barium chloride into the periphery of the tibialis anterior (TA) muscle. On the third day post-injury, 0.5 × 10⁶ cultured human cells (WT or CHAMP1 knockout), suspended in 10 µl of sterile saline containing 0.5% BSA, were transplanted into the injured TA muscles of randomly allocated NSG mice using a Hamilton syringe. Prior to transplantation, human cells were confirmed negative for rodent pathogens (IMPACT Panel A). Mice remained anesthetized with isoflurane during the transplantation procedure. Thirty days after transplantation, TA muscles were harvested, sectioned, stained, and analyzed to evaluate muscle fiber contributions from the transplanted cells, following established protocols. Each experimental group consisted of four independent transplantations.
Gene knockout experiments in human myoblasts by lenti-CRISPR-Cas9
Three gRNAs were designed for CHAMP1: gRNA1 (GGAAGAGGTGTCTGTAGCTC), gRNA2 (TGCTGGACTACGGATCTCTG), and gRNA3 (GGGAAAAGGGCATGCTTCCG), and was individually cloned into plentiCRISPR v2-Blast gRNA vector (Addgene, 83480) in Golden Gate assembly. DNA sequencing validated the correct insertion of gRNA. For lentivirus production, Lenti-X 293 T cells (Clontech, 632180) were cultured in DMEM (containing 1% penicillin/streptomycin, 10% FBS). Transfection was performed using FuGENE6 (Promega, #E2692) with psPAX2 plasmid, pMD2.G plasmid. Two days later, lentivirus-containing supernatants were filtered and concentrated with Lenti-X Concentrator (Clontech, PT4421-2) following manufacturer’s protocol. psPAX2 vector was a gift from Didier Trono (Addgene plasmid # 12260). pMD2.G vector was a gift from Didier Trono (Addgene plasmid # 12259). Human myoblasts were infected with lentivirus in growth medium. Following blasticidin selection, single clones were isolated, expanded, and genotyped using PCR and subsequent Sanger sequencing for validation of mutations. CHAMP1 genotypes were determined by sequencing after PCR amplifications using the following primers. F1: GCCCTCCTCTTCCTGAACAC and R1: GGATGGTCCCCAAGGTTCTG; F1&R1 primer pair amplifies a 458 bp band in WT; F2: ATCAGTCTCACCCAGCTCCT and R2: GAGAACCACCACGGGAACTT; F2&R2 primer pair amplifies a 311 bp band in WT; F3: GCCTAGAAAACCTGCCCTGT and R3: CCCATGGGCTGCTACATGAT; F3&R3 primer pair amplifies a 712 bp band in WT;
Retroviral expression vector cloning and retrovirus preparation
The retroviral expression vector pMXs-Puro (Cell Biolabs, RTV-012) was used to construct gene expression vectors for this study. The CHAMP1 open reading frame and its mutants were codon-optimized and synthesized by Integrated DNA Technologies, with sequences verified through Sanger sequencing or whole plasmid sequencing. The expression vectors for Myomxier, Myomaker and MyoD were reported previously24. For rescue experiments, sgRNA-insensitive DNA cassettes with silent mutations destroying the protospacer or PAM sequence were designed and utilized. All retrovirus vectors used in this study have been sequenced to verify the correct insertions of DNA sequences. Retrovirus production was performed by transfecting human embryonic kidney 293 cells with the retroviral plasmid using FuGENE 6 (Promega, E2692). Viral supernatant was harvested two days post-transfection, filtered, and used to infect target cells in the presence of polybrene (Sigma-Aldrich, TR1003-G). Infected cells were switched to fresh culture medium 24 h post-infection.
RNA extraction and bulk RNA sequencing
Total RNA was extracted from human myoblast cells using TRIzol reagent (Thermo Fisher Scientific, 15-596-018) according to the manufacturer’s protocol. RNA quality and concentration were assessed using a TapeStation, and only samples with an RNA Integrity Number (RIN) above 9 were used for library preparation using the NEBNext Ultra II Directional RNA Library Prep Kit (E7760L) with poly(A) selection. Next-generation sequencing was performed on an Illumina platform by Admera, generating 150 bp paired-end reads with a depth of approximately 40 million reads per sample.
Bulk RNA sequencing data analysis
Raw sequencing data was processed with initial quality control (QC) using FastQC (version 0.11.9: http://www.bioinformatics.babraham.ac.uk/projects/fastqc) to assess read quality and MultiQC53 (version 1.14) was used to aggregate and visualize quality control metrics across all samples. After QC, raw sequencing reads were processed using fastp54 (version 0.23.2) for adaptor trimming and reads filtering with the following filtering parameters: quality score threshold: 20, maximum proportion of unknown bases (N): 10, maximum percentage of unqualified bases: 30%. A reference human hg38 genome index was constructed using HISAT2 (version 2.2.1) with the provided genome sequence55. Cleaned reads were aligned to the reference genome using HISAT2. Alignment outputs were converted to sorted BAM files using SAMtools56 (version 1.6), and BAM files were indexed for downstream analysis. Aligned reads were converted to normalized genome browser tracks (BigWig files) using deepTools57 (version 3.5.2) with 1 bp bin size for track visualization. Gene-level read counts were quantified using featureCounts58 (Subread version 2.0.3) and differential expression analysis was performed using DESeq259 (version 1.40.2) in R (version 4.3.1) combined with Benjamini-Hochberg multiple testing correction. Gene Ontology (GO) enrichment analysis was conducted using clusterProfiler60 (version 4.8.3) and the Homo sapiens annotation database (org.Hs.eg.db, version 3.17.0) in R. RNA-seq data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database under accession number: GSE307319.
Duo-link proximity ligation assay (PLA)
Proximity Ligation Assay (PLA) experiments were conducted using Duo-link kits (Sigma-Aldrich, DUO92013, DUO92001, DUO92005, DUO82049, DUO82040, DUO92202). Myoblasts with the indicated genotypes were cultured on chamber slides and washed with 1× PBS before fixation in 4% paraformaldehyde (PFA) for 10 min at room temperature (RT). Fixed cells were permeabilized with 0.5% Triton X-100 in PBS for 10 min at RT and then washed with 1× PBS. Duolink II Blocking Solution (1×) was added to each well, and slides were incubated in a pre-heated humidity chamber at 37 °C for 1 h. Primary antibodies used for PLA experiments are anti-MyoD (Novus Biologicals, NBP1-54153, NB100-56511, 1.0 mg/ml, dilution 1:500), anti-CHAMP1 (Thermofisher Scientific, HPA008900, 0.1 mg/ml, dilution 1:200) and anti-FLAG (Sigma-Aldrich, F3165, 3.8-4.2 mg/ml, dilution 1:500). Primary antibodies were diluted in Duolink II Antibody Diluent (1×), and 40 μl of the antibody solution was applied per well, followed by overnight incubation at 4 °C. The next day, PLA probe incubation, oligonucleotide ligation, and amplification steps were performed following the manufacturer’s instructions. Finally, ~100 μl of Duolink II Mounting Medium with DAPI was added, and coverslips were applied. Images were captured using a Zeiss LSM 980 confocal microscope with Airyscan2.
Assay for transposase-accessible chromatin sequencing (ATAC-seq)
ATAC-seq experiments were conducted following a published protocol61, with minor modifications. Specifically, Adaptor A and Adaptor B were prepared at a concentration of 50 µM by mixing 100 µM AdaptorA-ME oligo (5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3’) or AdaptorB-ME oligo (5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3’) with 100 µM pMENTs oligo (5’- [Phos]CTGTCTCTTATACACATCT-3’) in a 1:1 ratio, followed by annealing in a thermocycler (95 °C for 5 min, then ramping down at −0.1 °C/s to 20 °C). Annealed adaptors were combined with recombinant Tn5 protein (0.5 mg/mL) in a 1:6 ratio, incubated at room temperature for 30 min, and then placed on ice for 10 min. Tn5-Adaptor A and Tn5-Adaptor B were then mixed in a 1:1 ratio and stored at −20 °C for long-term use. For ATAC-seq, cells were permeabilized with ice-cold Nuclei Isolation Buffer (5% BSA, 0.2% IGEPAL CA-630, 1x cOmplete protease inhibitor cocktail in PBS) for 5 min. A total of 50,000 cells were collected in a PCR tube by centrifugation at 500 x g, 4 °C for 5 min, then resuspended in 20 µL transposition mixture (10 µL 2x TD buffer, 6.6 µL PBS, 2 µL H2O, 0.2 µL 10% Tween-20, 0.2 µL 1% digitonin, 1 µL Tn5-AdaptorA/B mix). The transposition reaction was carried out in a ThermoMixer at 37 °C, 550 rpm for 30 min. Tagmented DNA was purified using a Qiagen MinElute kit following the manufacturer’s protocol and eluted in 20 µL Qiagen Buffer EB. Subsequently, 25 µL NEBNext Ultra II Q5 Master Mix, 2.5 µL 10 µM Nextera i5 indexing primer, and 2.5 µL 10 µM Nextera i7 indexing primer were added to the eluted DNA, followed by PCR amplification using the following conditions: 72 °C for 5 min; 98 °C for 30 s; 9 cycles of 98 °C for 10 s, 63 °C for 30 s, and 72 °C for 1 min; final extension at 72 °C for 2 min; hold at 4 °C. The PCR-amplified product was purified using the Qiagen MinElute kit and eluted in 30 µL Qiagen Buffer EB. The quality of ATAC-seq libraries was assessed using the Agilent Tapestation 4200 system, quantified by qPCR, and sequenced on an Illumina NextSeq 2000 using the following parameters: Read 1–51 cycles, Index 1–8 cycles, Index 2–8 cycles, Read 2–51 cycles.
Cleavage under targets and tagmentation (CUT&Tag)
CUT&Tag experiments were performed as previously described62, with minor modifications. Specifically, annealed adaptors (as described above for ATAC-seq) were loaded onto recombinant Protein A/G-Tn5 (0.7 mg/mL) using the same procedure. Myoblasts (differentiated for two days)and Drosophila S2 cells were subjected to light crosslinking with 0.2% formaldehyde in PBS on ice for 5 min, followed by quenching with 0.125 M glycine. Crosslinked cells were permeabilized with ice-cold Nuclei Isolation Buffer for 5 min. A total of 300,000 human myoblasts and 30,000 Drosophila S2 cells (spike-in control) were collected by centrifugation at 1000 x g, 4 °C for 5 min and resuspended in 75 µL MED1 buffer (300 mM NaCl, 2% BSA, 20 mM HEPES, 2 mM EDTA, 1 mM DTT, 1x cOmplete protease inhibitor cocktail, 0.5 mM spermidine, 0.01% digitonin, 0.01% IGEPAL CA-630 in nuclease-free H2O). The following antibodies were used for overnight incubation at 4 °C (1 µg per reaction): anti-Drosophila histone H2Av (Active Motif #61751), anti-CHAMP1 (Atlas Antibodies #HPA008900), anti-MyoD (Novus #NBP1-54153), or anti-H3K27ac (Abcam #ab4729). After primary antibody incubation, the nuclei were washed once with 75 µL MED1 buffer, resuspended in 75 µL MED1 buffer with 2 µg secondary antibody (Rabbit anti-Mouse IgG, Invitrogen #A27022; or Guinea Pig anti-Rabbit IgG, Novusbio #NBP1-72763, depending on the host species of the primary antibody), and incubated on a rotator at room temperature for 45 min. For H3K27ac CUT&Tag, secondary antibody incubation step was skipped. The nuclei were then washed once with 75 µL MED1 buffer, resuspended in 75 µL MED1 buffer with 2 uL loaded Protein A/G-Tn5, and further incubated on a rotator at room temperature for 45 min. After Protein A/G-Tn5 incubation, the nuclei were washed 3 times with 75 µL MED2 buffer (300 mM NaCl, 2% BSA, 20 mM HEPES, 1 mM DTT, 1x cOmplete protease inhibitor cocktail, 0.5 mM spermidine, 0.01% digitonin, 0.01% IGEPAL CA-630 in nuclease-free H2O) and resuspended in 75 µL MED2 buffer. The tagmentation reaction was initiated by adding 3 µL 250 mM MgCl2 and incubating at 37 °C, 550 rpm for 1 h. Tagmented DNA was purified using a Qiagen MinElute kit and eluted in 20 µL Qiagen Buffer EB. Library preparation was performed as described for ATAC-seq, using PCR amplification with 15 cycles for transcription factor CUT&Tag and 10 cycles for H3K27ac CUT&Tag. Libraries were purified, quality-controlled, and sequenced using the same procedures and parameters as above described for ATAC-seq.
HiCAR library preparation
HiCAR was performed as previously described63,64. Briefly, 100,000 crosslinked cells were lysed in 400 μl NPB buffer (PBS with 5% BSA, 1 mM DTT, 0.2% IGEPAL, Roche Complete Protease Inhibitor, 12.5 μl RNaseOUT) at 4 °C for 15 min to isolate nuclei. Nuclei were resuspended in 350 μl 2X TB buffer (66 mM Tris-acetate pH 7.8, 132 mM potassium acetate, 20 mM magnesium acetate, 32% DMF), 335 μl water, and 15 μl assembled Tn5 transposome and rotated at 37 °C for 1.5 h. Reactions were quenched with 350 μl 40 mM EDTA, nuclei washed with 0.075% BSA, and treated with 32.5 μl water, 5 μl 10X NEBuffer 3.1 (NEB, B7203S), and 12.5 μl 2% SDS at 62 °C for 10 min; supernatants containing nuclear RNA were saved. Nuclei were resuspended in 100 μl H2O, 14 μl 10X NEBuffer 3.1, and 25 μl 10% Triton X-100, and incubated at 37 °C for 15 min. For CviQI digestion and ligation, nuclei were washed with 1 ml 1.1X NEBuffer 3.1, incubated with 90 μl 1.1X NEBuffer 3.1 containing 100 U CviQI (NEB, R0639L) and 1 μl 20 μM TruseqR1 oligo at room temperature for 2 h, followed by ligation with 48 μl 10X T4 ligation buffer, 6 μl T4 DNA ligase (400 U/μl, NEB, M0202S), 2.4 μl 20 mg/ml BSA (NEB, B9000S), 40 μl 10% Triton X-100, and 283.6 μl H2O for 4 h at room temperature. After centrifugation, nuclei were resuspended in 200 μl 2X RCB (100 mM Tris-HCl pH 8.0, 100 mM NaCl, 0.4% SDS) and incubated at 68 °C for ≥1.5 h to reverse crosslink, and DNA was recovered by ethanol precipitation, washed with 80% ethanol, and dissolved in 21 μl 10 mM Tris-HCl (pH 8.0). DNA gap repair was performed with 4 μl 10 mM dNTPs, 5 μl 10X CutSmart buffer, 1.5 μl T4 DNA polymerase (NEB, M0203L), and 20.5 μl H2O at room temperature for 30 min, followed by heat inactivation at 75 °C for 20 min. Samples were digested with 1 μl NlaIII (NEB, R0125L) at 37 °C for 1 h, purified with 0.9X SPRI beads (Beckman, B23319), and dissolved in 80 μl 10 mM Tris-HCl. DNA was diluted to 1 ng/μl and circularized in T4 ligation buffer with T4 DNA ligase (400 U/μl, NEB, M0202S) for ≥2 h at room temperature, then purified with DNA Clean & Concentrator kit (Zymo, D4013) and eluted in 16 μl H2O. For PmeI digestion and PCR, 14 μl DNA was incubated with 1.7 μl 10X CutSmart buffer and 1.3 μl PmeI at 37 °C for 1 h, followed by PCR with 16 μl 5X Q5 buffer, 1.6 μl 10 mM dNTPs, 1.6 μl Nextera-pcr-i7–10-L primer, 1.6 μl NEB primer i501 (both 10 μM), 0.8 μl Q5 polymerase (NEB, M0491L), and 58.4 μl H2O using the following program: 72 °C for 5 min, 98 °C for 30 s; then 11 cycles of 98 °C for 10 s, 59 °C for 30 s, 72 °C for 45 s; followed by 72 °C for 5 min and hold at 4 °C. Amplified DNA (300–750 bp) was gel-extracted with DNA Recovery Kit (Zymo, D4002) for deep sequencing.
ATAC-seq, CUT&Tag and HiCAR data analysis
Quality control of the raw sequencing reads were performed using MultiQC (v1.14)53. Reads were quality trimmed using trim-galore (v0.6.7)65. Sequencing reads were mapped to GRCh38 (hg38) reference genome using Bowtie2 (v2.4.4)66. Duplicated sequencing reads were removed using picard (v2.26.11, https://broadinstitute.github.io/picard/). Peak calling was performed for deduplicated sequencing reads using MACS2 (v2.2.7.1)67. For ATAC-seq and H3K27ac CUT&Tag, genome alignment was normalized by RPKM using deeptools (v3.5.3)68, and visualized using IGV (v2.15.4)69. For transcription factor CUT&Tag, genome alignment was normalized using deeptools bamCoverage68, with a scaling factor calculated as [Control Drosophila reads]/[Sample Drosophila reads], and visualized using IGV. Motif analysis was performed using HOMER (v4.11.1)70. HiCAR sequencing data were processed using the nf-core/hicar pipeline under default instructions71. Raw reads were subjected to quality control, adapter trimming, alignment to hg38, and extraction of valid paired-end interactions. Downstream analysis including ATAC-seq peak calling, chromatin loops identification, and genomic interaction annotation was performed using default setting. Processed outputs were converted into multiple visualization formats, and final contact matrices and ATAC-seq tracks were visualized with pyGenomeTracks72.
Immunostaining and microscopy
Cells were fixed in 4% paraformaldehyde (PFA) in PBS for 10 min at room temperature, permeabilized with 0.5% Triton X-100 in PBS, and blocked with 3% bovine serum albumin (BSA) in PBS for 1 h at room temperature. Primary antibodies (1: 500 dilution) used for immunostaining experiments are anti-MyoD (Novus Biologicals, NBP1-54153, NB100-56511, 1.0 mg/ml), anti-MYOG (DSHB, F5D), anti-myosin (DSHB, MF20), MYH3 (DSHB, F1.652), anti-laminin (Novus Biologicals, NB300-144, 1.0 mg/ml), anti-Lamin A/C (Thermofisher Scientific, MA3-1000, 1.0 mg/ml), anti-CHAMP1 (Thermofisher Scientific, HPA008900, 0.1 mg/ml), anti-Myomaker (mouse monoclonal antibody) and anti-FLAG (Sigma-Aldrich, F3165, 3.8-4.2 mg/ml). Following blocking, cells were incubated with the primary antibody overnight at 4 °C, then with an Alexa Fluor–conjugated secondary antibody (1:1,000 dilution, Thermofisher Scientific, A21127, A21242, A27034, A27036, A28177). Nuclei were counterstained with Hoechst dye. Phase-contrast images were captured using a BioTek Lionheart FX automated microscope, and fluorescence images were acquired using either the BioTek system or an Olympus FLUOVIEW FV1200 confocal laser scanning microscope. Super resolution imaging was conducted with a Zeiss LSM 980 confocal microscope with Airyscan 2.
Differentiation index and fusion index measurements
The differentiation index was calculated as the proportion of nuclei within MF20-positive cells relative to the total number of nuclei. The fusion index was determined as the proportion of nuclei within myotubes (cells containing ≥3 nuclei) relative to the total number of myosin expressing nuclei. Both indices were derived from manual cell counts, with treatment information blinded to maintain objectivity.
Western blotting analysis
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich, R0278) supplemented with cOmplete protease inhibitor (Sigma-Aldrich, 04693159001) and incubated on ice for 15 min. The lysates were centrifuged at 15,000 × g for 10 min at 4 °C, and the supernatant was collected and mixed with 4× Laemmli sample buffer (Bio-Rad, 161-0747). A total of 30 µg of protein was resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane (Sigma-Aldrich, ISEQ00010). The membrane was blocked with 5% non-fat milk for 1 h at room temperature and incubated overnight at 4 °C with primary antibodies diluted 1 in 1000 in 5% milk: GAPDH (Santa Cruz Biotechnology, sc-32233), FLAG (Sigma-Aldrich, F3165), and CHAMP1 (Sigma-Aldrich, HPA008900), anti-Lamin A/C (Thermofisher Scientific, MA3-1000). After washing with Tris-buffered saline containing 0.1% Tween-20 (TBST), the membrane was incubated with secondary antibodies (1 in 2500 dilution)—goat anti-mouse IgG–HRP (Invitrogen, A28177) and goat anti-rabbit IgG–HRP (Invitrogen, A27036)—in blocking buffer for 1 h at room temperature. Proteins were detected using Western Blotting Luminol Reagent (Thermofisher Scientific, 34075). Band intensities from the Western blot images were quantified using ImageJ (version 1.44p). Uncropped, unprocessed scans of all blots are provided in Supplementary Fig. 9.
Statistics & reproducibility
All mice and human cells were randomly allocated to the experimental group. All measurements were taken from distinct samples. For RNA-seq analysis, the differentially expressed genes were identified with a cutoff of fold change greater than 2 and p value < 0.05. Sample sizes are provided in the figure legends. Data were processed using GraphPad Prism 9 software. Error bars in all bar graphs represent standard deviation. Experiments were not randomized, and investigators were not blinded to sample allocation or outcome assessment. No statistical method was used to predetermine sample size. No data were excluded from the analyses.
Ethical statement
All experiments involving recombinant DNA, human cells, third-generation lentiviral and retroviral vectors were conducted in compliance with institutional biosafety regulations and under the approval of the University of Georgia Institutional Biosafety Committee (IBC protocol no. 2023-0076). Human myoblasts and skin fibroblast samples were generated previously34,51 and were obtained as de-identified materials. All animal experiments followed the ARRIVE guidelines and were approved by the University of Georgia Institutional Animal Care and Use Committee (IACUC; Animal Use Protocol A2025 01-008-Y1-A0). Mice were fed PicoLab Rodent Diet 20, irradiated (5053) with ad libitum access to chow and fresh water under standard vivarium conditions. No dietary restrictions were applied during experiments. Mice were housed in a UGA facility on a 12:12 h light/dark cycle, at 20–22 °C, with 40–60% relative humidity, in accordance with UGA IACUC and the Guide for the Care and Use of Laboratory Animals.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All differentially expressed genes from the CHAMP1 RNA-seq analysis was provided in Supplementary Data 1. All annotated CUT&Tag peaks for CHAMP1 and MyoD were provided in Supplementary Data 2 and 3. All RNA-sequencing data generated in this study is available in Gene Expression Omnibus (GEO) repository under the accession number GSE307319 and are publicly available. All other data are available in the article and its Supplementary files or from the corresponding author upon request. Source data are provided with this paper.
Code availability
Code used for the DNA sequencing data analysis performed in this study is fully available on GitHub: https://github.com/ZhengZhang991012/CHAMP1_NatCom.
References
Bentzinger, C. F., Wang, Y. X. & Rudnicki, M. A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 4, a008342 (2012).
von Maltzahn, J., Jones, A. E., Parks, R. J. & Rudnicki, M. A. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc. Natl. Acad. Sci. USA 110, 16474–16479 (2013).
Rudnicki, M. A. et al. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 75, 1351–1359 (1993).
Hasty, P. et al. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364, 501–506 (1993).
Black, B. L. & Olson, E. N. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev. Cell Dev. Biol. 14, 167–196 (1998).
Fung, C. W. et al. Cell fate determining molecular switches and signaling pathways in Pax7-expressing somitic mesoderm. Cell Discov. 8, 61 (2022).
Kim, J. H. & Chen, E. H. The fusogenic synapse at a glance. J. Cell Sci. 132, jcs213124 (2019).
Millay, D. P. et al. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature 499, 301–305 (2013).
Bi, P. et al. Control of muscle formation by the fusogenic micropeptide myomixer. Science 356, 323–327 (2017).
Zhang, Q. et al. The microprotein Minion controls cell fusion and muscle formation. Nat. Commun. 8, 15664 (2017).
Quinn, M. E. et al. Myomerger induces fusion of non-fusogenic cells and is required for skeletal muscle development. Nat. Commun 8, 15665 (2017).
Zhang, H. et al. Evolution of a chordate-specific mechanism for myoblast fusion. Sci. Adv. 8, eadd2696 (2022).
Long, T. et al. Cryo-EM structures of Myomaker reveal a molecular basis for myoblast fusion. Nat. Struct. Mol. Biol. 30, 1746–1754 (2023).
Gamage, D. G. et al. Phosphatidylserine orchestrates Myomerger membrane insertions to drive myoblast fusion. Proc. Natl. Acad. Sci. USA 119, e2202490119 (2022).
Di Gioia, S. A. et al. A defect in myoblast fusion underlies Carey-Fineman-Ziter syndrome. Nat. Commun. 8, 16077 (2017).
Ramirez-Martinez, A. et al. Impaired activity of the fusogenic micropeptide Myomixer causes myopathy resembling Carey-Fineman-Ziter syndrome. J. Clin. Invest. 132, e159002 (2022).
Ganassi, M., Muntoni, F. & Zammit, P. S. Defining and identifying satellite cell-opathies within muscular dystrophies and myopathies. Exp. Cell Res 411, 112906 (2022).
Petrany, M. J., Song, T., Sadayappan, S. & Millay, D. P. Myocyte-derived Myomaker expression is required for regenerative fusion but exacerbates membrane instability in dystrophic myofibers. JCI Insight 5, e136095 (2020).
Eigler, T. et al. ERK1/2 inhibition promotes robust myotube growth via CaMKII activation resulting in myoblast-to-myotube fusion. Dev. Cell 56, 3349–3363 e3346 (2021).
Bjornson, C. R. et al. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 30, 232–242 (2012).
Gioftsidi, S., Relaix, F. & Mourikis, P. The Notch signaling network in muscle stem cells during development, homeostasis, and disease. Skelet. Muscle 12, 9 (2022).
Girardi, F. et al. TGFbeta signaling curbs cell fusion and muscle regeneration. Nat. Commun. 12, 750 (2021).
Melendez, J. et al. TGFbeta signalling acts as a molecular brake of myoblast fusion. Nat. Commun. 12, 749 (2021).
Zhang, H. et al. Human myotube formation is determined by MyoD-Myomixer/Myomaker axis. Sci. Adv. 6, eabc4062 (2020).
Joshi, A. S. et al. The IRE1alpha/XBP1 signaling axis drives myoblast fusion in adult skeletal muscle. EMBO Rep. 25, 3627–3650 (2024).
Haifeng, Z. et al. Development of a split-toxin CRISPR screening platform to systematically identify regulators of human myoblast fusion. Nat. Commun. https://doi.org/10.1038/s41467-025-67583-x (2025).
Hempel, M. et al. De Novo Mutations in CHAMP1 Cause Intellectual Disability with Severe Speech Impairment. Am. J. Hum. Genet 97, 493–500 (2015).
Deciphering Developmental Disorders, S Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228 (2015).
Levy, T. et al. Prospective phenotyping of CHAMP1 disorder indicates that coding mutations may not act through haploinsufficiency. Hum. Genet 142, 1385–1394 (2023).
Isidor, B. et al. De novo truncating mutations in the kinetochore-microtubules attachment gene CHAMP1 cause syndromic intellectual disability. Hum. Mutat. 37, 354–358 (2016).
Tanaka, A. J. et al. De novo pathogenic variants in CHAMP1 are associated with global developmental delay, intellectual disability, and dysmorphic facial features. Cold Spring Harb. Mol. Case Stud. 2, a000661 (2016).
Arpke, R. W. et al. A new immuno-, dystrophin-deficient model, the NSG-mdx(4Cv) mouse, provides evidence for functional improvement following allogeneic satellite cell transplantation. Stem Cells 31, 1611–1620 (2013).
Levy, T. et al. CHAMP1 disorder is associated with a complex neurobehavioral phenotype including autism, ADHD, repetitive behaviors and sensory symptoms. Hum. Mol. Genet 31, 2582–2594 (2022).
Yoshizaki, Y. et al. CHAMP1 premature termination codon mutations found in individuals with intellectual disability cause a homologous recombination defect through haploinsufficiency. Sci. Rep. 14, 31904 (2024).
Davis, R. L., Weintraub, H. & Lassar, A. B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51, 987–1000 (1987).
Cao, Y. et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev. Cell 18, 662–674 (2010).
Dall’Agnese, A. et al. Transcription Factor-Directed Re-wiring of Chromatin Architecture for Somatic Cell Nuclear Reprogramming toward trans-Differentiation. Mol. Cell 76, 453–472 e458 (2019).
Blum, R., Vethantham, V., Bowman, C., Rudnicki, M. & Dynlacht, B. D. Genome-wide identification of enhancers in skeletal muscle: the role of MyoD1. Genes Dev. 26, 2763–2779 (2012).
Itoh, G. et al. CAMP (C13orf8, ZNF828) is a novel regulator of kinetochore-microtubule attachment. EMBO J. 30, 130–144 (2011).
Fedotova, A. A., Bonchuk, A. N., Mogila, V. A. & Georgiev, P. G. C2H2 Zinc Finger Proteins: The Largest but Poorly Explored Family of Higher Eukaryotic Transcription Factors. Acta Nat. 9, 47–58 (2017).
Abi Raad, S., Yazbeck Karam, V., Chouery, E., Mehawej, C. & Megarbane, A. CHAMP1-related disorder: Sharing 20 years of thorough clinical follow-up and review of the literature. Genes (Basel) 14, 546 (2023).
Vermeulen, M. et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142, 967–980 (2010).
Li, F. et al. CHAMP1 binds to REV7/FANCV and promotes homologous recombination repair. Cell Rep. 40, 111297 (2022).
Li, F. et al. CHAMP1 complex directs heterochromatin assembly and promotes homology-directed DNA repair. Nat. Commun. 16, 1714 (2025).
Fujita, H. et al. CHAMP1-POGZ counteracts the inhibitory effect of 53BP1 on homologous recombination and affects PARP inhibitor resistance. Oncogene 41, 2706–2718 (2022).
Fong, A. P. et al. Genetic and epigenetic determinants of neurogenesis and myogenesis. Dev. Cell 22, 721–735 (2012).
Sartorelli, V., Huang, J., Hamamori, Y. & Kedes, L. Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol. Cell Biol. 17, 1010–1026 (1997).
Wang, S. et al. Tcf12 is required to sustain myogenic genes synergism with MyoD by remodelling the chromatin landscape. Commun. Biol. 5, 1201 (2022).
Puri, P. L. et al. Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol. Cell 1, 35–45 (1997).
Dilworth, F. J., Seaver, K. J., Fishburn, A. L., Htet, S. L. & Tapscott, S. J. In vitro transcription system delineates the distinct roles of the coactivators pCAF and p300 during MyoD/E47-dependent transactivation. Proc. Natl. Acad. Sci. USA 101, 11593–11598 (2004).
Mamchaoui, K. et al. Immortalized pathological human myoblasts: towards a universal tool for the study of neuromuscular disorders. Skelet. Muscle 1, 34 (2011).
Salic, A. & Mitchison, T. J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 105, 2415–2420 (2008).
Ewels, P., Magnusson, M., Lundin, S. & Kaller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–W191 (2014).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Wei, X. et al. HiCAR is a robust and sensitive method to analyze open-chromatin-associated genome organization. Mol. Cell 82, 1225–1238 e1226 (2022).
Wei, X., Tran, D. & Diao, Y. HiCAR: Analysis of Open Chromatin Associated Long-range Chromatin Interaction Using Low-Input Materials. Curr. Protoc. 3, e899 (2023).
Krueger, F. et al. FelixKrueger/TrimGalore: v0.6.10 – add default decompression path. Zenodo https://doi.org/10.5281/zenodo.7598955 (2023).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Robinson, J. T., Thorvaldsdottir, H., Turner, D. & Mesirov, J. P. igv.js: an embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV). Bioinformatics 39, btac830 (2023).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Ewels, P. A. et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 38, 276–278 (2020).
Lopez-Delisle, L. et al. pyGenomeTracks: reproducible plots for multivariate genomic datasets. Bioinformatics 37, 422–423 (2021).
Acknowledgements
We extend our heartfelt gratitude to Jeff D’Angelo at CHAMP1 foundation, Bing Ren at UCSD, Kaixiong Ye at UGA for their invaluable support of our research, thank trainees Stephanie Campanano, Moira Alejandra La Fuente, Lindsey Erin Hossfeld, Emilia Keys and Kaitlyn Burtt in Bi laboratory for technical assistance. A. Bigot and V. Mouly from the Myoline platform of the Myology Institute for immortalized human cell lines. M. Kandasamy from Biomedical Microscopy Core at UGA for imaging assistance. Funding: This work was supported by the NIH R35 award GM147209 and R21 award AR080330 to B.P.; the DoD CDMRP grant HT94252410734 and the NIH grants R01CA257851& R01CA247769 to J.X. Z.W. is a DDBrown Awardee of the Life Sciences Research Foundation.
Author information
Authors and Affiliations
Contributions
H.Z., and P.B. designed research; H.Z., M.Z., Z.Z., Z.W., R. Shi., Y.W., X.W., R. Shang., J.L., C.H., Y.D., and P.B. performed research; H.Z., M.Z., Z.Z., Z.W., R. Shi., Y.W., R. Shang., C.H,. and P.B. analyzed data; J.X. and Y.D. for Resources. P.B. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
P.B. is a holder of the Canada patent CA3052705A1. The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, H., Zhou, M., Zhang, Z. et al. CHAMP1 is an essential regulator for human myoblast fusion and muscle development. Nat Commun 17, 546 (2026). https://doi.org/10.1038/s41467-025-67584-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67584-w
