
Abstract
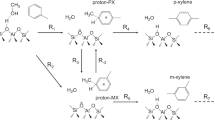
The selective production of high-value para-xylene (PX) through CO2 hydrogenation-coupled toluene methylation represents a promising strategy for sustainable carbon utilization. Herein, we report a ZnZrOx & HMCM-22 tandem catalyst that integrates CO2 hydrogenation with shape-selective toluene methylation. The optimized catalyst achieves 91% xylene selectivity with 70.4% PX dominance at 10.3% toluene conversion. Controlled tetraethyl orthosilicate (TEOS) deposition passivates Brønsted acid sites in HMCM-22 supercages, suppressing toluene disproportionation while preserving methylation activity in sinusoidal channels. Mechanistic studies reveal that formaldehyde species, generated on ZnZrOx and transferred to HMCM-22, act as kinetically favored methylating intermediates, enabling direct coupling between CO2 hydrogenation and toluene methylation. This pathway outperforms conventional methanol-mediated routes in both activity and selectivity. Our work establishes a dual-optimization strategy—zeolite microenvironment engineering and reactive intermediate control—offering mechanistic insights and design principles for high-performance tandem catalysis in carbon recycling aromatic synthesis.
Similar content being viewed by others

Introduction
Carbon dioxide, as an abundant C1 feedstock, presents a great opportunity for sustainable chemical synthesis when coupled with green hydrogen derived from renewable energy. Recent advances have demonstrated the feasibility of converting CO2/H2 mixtures into value-added products including methanol1,2, light olefins3,4,5,6, and aromatics7,8,9. Although direct CO2 hydrogenation to aromatics has been reported using tandem catalysis, the para-xylene (PX) selectivity within aromatic products remains suboptimal. This limitation contrasts sharply with the industrial demand for PX—a precursor for polyethylene terephthalate (PET) manufacturing—which currently relies predominantly on fossil-based routes such as naphtha cracking and methanol-mediated toluene alkylation. These considerations highlight the urgency to develop efficient strategies for synthesizing PX through CO2-derived methylating agents with toluene, thereby enabling PX production via a two-step CO2/H2 pathway.
Conventional toluene methylation processes employing methanol confront intrinsic thermodynamic limitations when implemented over H-ZSM-5 zeolites at 400–500 °C. Under these harsh thermal conditions, the concurrent activation of methanol-to-olefins (MTO) side reactions precipitates a cascade of operational challenges: competitive methanol consumption by olefin synthesis pathways leads to methylating agent depletion, while concomitant carbon deposition accelerates catalyst deactivation through pore blockage and active site coverage10,11. Furthermore, the kinetic rivalry between methylation and MTO pathways severely compromises reaction efficiency. This tripartite interplay of resource misallocation, catalyst instability, and suppressed target reaction kinetics collectively necessitates the development of catalytic architectures capable of circumventing methanol intermediation under thermodynamically favorable conditions.
Recent developments in tandem catalysis reveal promising alternatives through in situ-generated oxygenate intermediates. Oxide-zeolite composites (e.g., oxide: ZnZrOx, ZnGa2O4, In2O3-ZrO2, ZnCrOx; zeolites: SAPO-34, SSZ-13, H-ZSM-5)12,13,14,15,16 enable CO2 hydrogenation to lower olefins and aromatics via CHxO intermediates through formate-mediated pathways. Notably, these systems operate at significantly lower temperatures than conventional methanol-based processes, as CHxO intermediates exhibit lower activation barriers compared to free methanol. Our previous work further demonstrated that methanol synthesis in tandem systems is thermodynamically driven by downstream olefin formation—a critical coupling mechanism that facilitates intermediate-directed catalysis4. These findings suggest that judicious selection of reactive methylating intermediates could decouple methylation kinetics from MTO side reactions.
Emerging CO2 hydrogenation-coupled toluene methylation strategies underscore the pivotal influence of zeolite architectural design on product selectivity17,18,19,20. Whereas H-ZSM-5-based systems deliver moderate xylene yields, their inherent structural constraints—notably unidirectional channel geometry and pronounced Brønsted acidity—predispose the system toward competing toluene disproportionation and xylene isomerization pathways21,22. In contrast, MCM-22 zeolite demonstrates distinctive structural merits for shape-selective alkylation, characterized by sinusoidal 10-membered ring (10-MR) channels that impose steric restrictions on bulkier isomers, coupled with chemically tunable supercage configurations enabling precise acid site modulation. The spatial isolation of active sites within this architecture further suppresses disproportionation side reactions. These collective structural attributes establish MCM-22 as an exemplary scaffold for constructing tailored methylation microenvironments with enhanced selectivity23,24.
In this work, we develop a bifunctional ZnZrOx & HMCM-22 (ZZO & M-22) tandem catalyst that synergistically integrates CO2 hydrogenation and shape-selective methylation. Through strategic silica deposition in MCM-22 supercages, we selectively passivate Brønsted acid sites responsible for toluene disproportionation while preserving methylation-active sites in sinusoidal channels. Experimental results, advanced characterization via in situ synchrotron vacuum ultraviolet photoionization mass spectrometry (SVUV-PIMS) and density functional theory (DFT) calculations identify formaldehyde-like CH2O* as the dominant methylating intermediate, exhibiting a 0.86 eV lower activation barrier compared to methanol-mediated pathways. The optimized catalyst achieves 91% xylene selectivity with 70.4% PX dominance among isomers at 10.3% toluene conversion. This study establishes a dual-optimization strategy combining zeolite microenvironment engineering with reactive intermediate control, providing a blueprint for developing industrially viable CO2-to-aromatics systems.
Results and discussion
Catalytic performance
The catalytic performance of tandem catalyst systems was systematically investigated in a fixed-bed reactor under controlled reaction conditions. Temperature-dependent studies show a monotonic increase in toluene conversion from 3% to 32% with simultaneous attenuation of PX selectivity with increasing reaction temperature from 280 to 400 °C (Fig. 1a, and Supplementary Fig. 1a). Thermodynamic redistribution of xylene isomers favored meta-xylene (MX) formation, accompanied by decreasing benzene selectivity and progressive accumulation of 1,2,4-trimethylbenzene (TMB) with increasing reaction temperature. This temperature-response profile suggests competing mechanisms: enhanced alkylation kinetics promoting active methyl species-toluene coupling at elevated temperatures, versus thermally activated processes including xylene isomerization to MX and over-methylation to TMB. Comparative evaluation of individual components in the ZZO & M-22 tandem system demonstrated distinct functionalities ZZO exclusively mediated CO2-to-methanol conversion while M-22 remained catalytically inert in isolation for CO2 hydrogenation (Fig. 1b). The combined system achieved integrated CO2 hydrogenation and toluene methylation without generating aromatization byproducts, with control experiments confirming toluene disproportionation as the dominant pathway on isolated M-22 with toluene as feedstocks.

a The effect of reaction temperature. b CO2 hydrogenation in the presence of toluene over ZZO & M-22; CO2 hydrogenation over ZZO, M-22, ZZO & M-22 and toluene disproportionation over M-22. c Influence of M-22-modified times by TEOS on the aromatics distribution and toluene conversion. d The Stability test. Standard reaction conditions: 3 MPa, 320 °C, GHSV of a mixture gas (CO2/H2/Ar = 24:72:4) = 12000 mL g−1 h−1, and WHSV of toluene = 1 h−1. When only toluene: 3.0 MPa, WHSV of toluene = 1 h−1, GHSV of mixture gas (CO2/H2/Ar = 24:72:4) = 12000 mL g−1 h−1. The color shadings represent the selectivity, and the red square represents the conversion.
Modification of M-22 via tetraethyl orthosilicate (TEOS) deposition induced three critical effects: pore architecture modulation favoring PX diffusion, passivation of external acidic sites, and suppression of disproportionation pathways. Progressive TEOS deposition cycles (1–4 cycles) enhanced PX selectivity from 43% to 64% while suppressing benzene selectivity from 10% to <1%, though with concomitant TMB accumulation (9%) due to restricted PX diffusion enabling secondary methylation (Fig. 1c). Five deposition cycles induced pore blockage, manifesting as activity loss with toluene conversion decreasing from 14% to 7%, establishing 4-cycle modification (ZZO & 4Si/M-22) as the optimal configuration. The ZZO & 4Si/M-22 catalyst achieves 91% xylene selectivity with 70.4% PX dominance among isomers at 10.3% toluene conversion. Compared to other catalyst systems, the ZZO & 4Si/M-22 can obtain a high xylene selectivity while achieving a high PX selectivity, and can completely inhibit the occurrence of toluene disproportionation side reactions (Supplementary Table 1). And the ZZO & 4Si/M-22 achieved a CO2 conversion of 4.2% with a CO selectivity of 22% at 320 °C (Supplementary Fig. 1b). Long-term stability tests confirmed sustained performance (>120 h) with undiminished PX selectivity (Fig. 1d).
Process parameter optimization revealed pressure-dependent behavior with elevated pressure from 1 to 6 MPa, which enhances toluene conversion from 5% to 12% but promotes TMB formation with selectivity increasing from 4% to 12% through PX retention-induced over-methylation (Supplementary Fig. 2a). Gas hourly space velocity (GHSV) studies identified 12,000 mL g−1 h−1 as optimal, balancing toluene conversion (10.3%) and selectivity (PX 64%, TMB 9%), with lower GHSV to 6,000 mL g−1 h−1 inducing disproportionation pathways (Supplementary Fig. 2b). Weight hourly space velocity (WHSV) adjustments demonstrated an intrinsic activity-selectivity trade-off: increasing WHSV from 1 to 3 h−1 improved PX selectivity from 64 to 80% but incurred 80% toluene conversion loss with decreasing from 10.3% to 2%, while simultaneously suppressing TMB formation (Supplementary Fig. 2c). These systematic investigations establish the ZZO & 4Si/M-22 system as an effective tandem catalyst for selective PX production, achieving precise control through synergistic component integration, strategic pore engineering, and optimized reaction parameters.
Structure characterization of the tandem catalysts
The ZZO solid solution oxide catalyst was synthesized through 20 mol% ZnO doping into ZrO2. X-ray diffraction analysis confirms that the resultant ZZO solid solution adopts a characteristic tetragonal crystal structure, as evidenced by the diffraction pattern in Fig. 2a. As shown in the transmission electron microscopy (TEM) image (Fig. 2b), the ZZO material exhibits a nanoparticle morphology with uniform dimensions. Elemental mapping analysis further demonstrates the homogeneous distribution of Zn atoms within the ZrO2 matrix (Fig. 2c), verifying the successful formation of a solid solution structure. The M-22 zeolite component displays well-defined nanosheet morphology, as observed in Fig. 2d. These crystalline sheets exhibit thicknesses of approximately 28 nm and lateral dimensions ranging from 300 to 500 nm. In the ZZO & M-22 tandem catalyst system, scanning electron microscopy (SEM) characterization reveals that ZZO nanoparticles are uniformly dispersed across the M-22 nanosheet surfaces (Fig. 2e). A critical observation from the high-resolution SEM image is the preservation of distinct structural features for both components, indicating that the interfacial interaction between ZZO and M-22 remains primarily physical rather than chemical. Additional structural validation is provided in Supplementary Fig. 3a, b, which present SEM images of the 4Si/M-22 modified sample. The morphology of M-22 nanosheets remains essentially unchanged after TEOS treatment, confirming that the structural integrity of the zeolite framework is maintained during the modification process. And the specific BET surface area, micropore area, external area and pore volume of M-22 all decreased, while the pore diameter remained almost unchanged. (Supplementary Table 2). This indicates that TEOS does not deposit extensively within the internal pores of the M-22, but predominantly at the entrances of all channels and on the external surface, thereby enhancing the shape selectivity of M-22.

a X-ray diffraction patterns of ZZO, M-22, and ZZO & 4Si/M-22. b The TEM and Aberration-corrected HRTEM images of ZZO. c Aberration-corrected high-angle annular dark-field scanning TEM images and elemental distribution of Zn and Zr for ZZO. d SEM image of M-22. e SEM image of ZZO & M-22.
Surface chemical properties of the tandem catalysts
CO2 temperature-programmed desorption (CO2-TPD) analysis reveals distinct adsorption behaviors between components (Supplementary Fig. 4a). The ZZO solid solution demonstrates sustained CO2 adsorption up to 500 °C, while the zeolite component exhibits negligible CO2 adsorption capacity. Quantitative evaluation of basic sites confirms that the physical integration of zeolite in the tandem catalyst exerts minimal influence on CO2 adsorption performance of ZZO (Supplementary Table 3). The superior PX selectivity observed in ZZO & 4Si/M-22 tandem catalysts highlights the critical role of TEOS modification. This performance enhancement correlates with the decisive influence of zeolite acid properties (both quantity and type) on PX selectivity. To elucidate this relationship, comprehensive acid characterization was conducted using NH3-TPD, pyridine Fourier-transform infrared (Py-FTIR) spectroscopy, 1H MAS NMR, and 27Al MAS NMR. NH3-TPD profiles (Supplementary Fig. 4b) exhibit two characteristic desorption regions: weak acid sites (50–250 °C) and strong acid sites (250–500 °C). As shown in Supplementary Table 4, the total acid content of the ZZO & M-22 tandem catalyst closely matches the arithmetic sum of individual components, confirming that physical mixing preserves the intrinsic acidity of both ZZO and M-22.
Systematic investigation of acid evolution for M-22 under successive TEOS modifications reveals progressive acid suppression (Fig. 3a). Notably, both weak and strong acid contents stabilize after four modification cycles, indicating saturation of surface passivation. Py-FTIR analysis identifies characteristic bands at 1540 cm−1 (Brønsted acid sites) and 1440 cm−1 (Lewis acid sites), with quantitative analysis following established protocols (Fig. 3b)25. Supplementary Table 5 demonstrates a marked reduction in both acid types with increasing modifications, particularly the near-complete elimination of Brønsted acidity after four cycles. This evidences TEOS-mediated encapsulation of acid sites within external surface pockets of M-22. 1H MAS NMR spectra (Fig. 3c) provide further validation of TEOS-induced acid modulation. The diminishing signal at 1.8 ppm (assigned to external silanol groups, Si-OH26) and attenuated resonance at 3.9 ppm (bridging hydroxyls, Si(OH)Al27,28) correlate with modification cycles. Concurrent chemical shift variations in the 3.9 ppm band reinforce the role of TEOS in external surface acid passivation, as quantified in Supplementary Table 6.

a NH3-TPD of M-22 with different Si modification times. b Pyridine-FTIR results for M-22 with different Si modification times. c 1H MAS NMR characterizations of M-22 with different Si modification time. d 27Al MAS NMR characterizations of M-22 with different Si modification times. Lewis Acid (LA) and Brønsted acid (BA).
Considering the intrinsic link between Brønsted acidity and framework aluminum (AlF) in zeolites (Supplementary Fig. 5), 27Al MAS NMR analysis was employed to probe Al speciation (Fig. 3d). Distinct resonances at 0 ppm (extra-framework AlEF, 6-coordinated) and 56 ppm (framework AlF, 4-coordinated) were identified29. Deconvolution of the 56 ppm band resolves three components: 50 ppm (T6 + T7 sites), 56 ppm (T1 + T3 + T4 + T5 + T8 sites), and 61 ppm (T2 sites)30. Structural analysis localizes T2 and T1 + T3 sites in supercages/surface pockets, T4 in inaccessible framework positions, T5 in sinusoidal channels with unstable and unfavorable for siting Al, and T8 in the sinusoidal channels connected to the bottom of the supercages, with thermodynamically stable bridging hydroxyls31,32,33, with T6 + T7 residing on external surfaces34. Progressive TEOS modification induces three critical changes: gradual attenuation of the AlEF signal at 0 ppm; systematic reduction of overall AlF intensity at 56 ppm; selective disappearance of the 61ppm component (T2 supercage sites) upon deconvolution (Supplementary Table 7, Supplementary Fig. 6a).
These observations suggest TEOS not only passivates external surface pocket acid sites but also modulates supercage acidity. This aligns with literature demonstrating toluene disproportionation (a benzene-forming side reaction) predominantly occurs in MWW-type supercages35,36. Consistent with Fig. 1c, reduced benzene selectivity parallels TEOS-induced supercage deactivation. Complementary 27Al MAS NMR analysis of precoked M-22 confirms analogous attenuation of 61 ppm signals (Supplementary Fig. 6b, c), validating efficacy of TEOS in suppressing toluene disproportionation through targeted acid site passivation.
Unraveling the key formaldehyde intermediates species and the reaction mechanism
The reaction mechanism was investigated through multiple complementary techniques. In situ DRIFTS analysis of ZZO during CO2 hydrogenation revealed formate (HCOO*) and methoxy (H3CO*) intermediates, with characteristic vibrations at 1595/1370 cm−1 (OCO stretching) and 2931/2824 cm−1 (C-H modes), respectively (Supplementary Fig. 7a). Isotopic substitution (CO2 + D2) caused rapid depletion of HCOO*/H3CO* signals within 5 min, replaced by transient DCOO* (2165 cm−1) and HD2CO* (2050 cm−1) species, confirming the formate pathway dominates CO2 hydrogenation over ZZO (Supplementary Fig. 7b)1. Isotope-switching (13CO2) experiments confirmed CO2 incorporation into products through 15 cm−1 redshift (2880 → 2865 cm−1) in methyl C-H vibrations and intensified aromatic signals (Fig. 4a)21,37,38. Compared to ZZO, ZZO & 4Si/M-22 showed selective disappearance of H3CO* species, while the HCOO* species still retain suggesting intermediate derived from formate species transfer to the zeolite component, thereby involving the formation of PX (Fig. 4b).

a In situ DRIFTS spectra over the ZZO & 4Si/M-22 catalyst exposed to the stream of C7H8 + CO2 + H2 and subsequently switched to C7H8 + 13CO2 + H2. b In situ DRIFTS of CO2 + H2 over ZZO and ZZO & 4Si/M-22. c Quasi in situ13C CP MAS NMR experiment for 13CO2 + H2 on ZZO and 13CO2 + H2 + C7H8 on ZZO & 4Si/M-22. d Quasi in situ 2D 13C-1H HETCOR MAS NMR experiment for 13CO2 + H2 on ZZO. e Quasi in situ 2D 13C-1H HETCOR MAS NMR experiment for 13CO2 + H2 + C7H8 on ZZO & 4Si/M-22. Reaction conditions: 320 °C, 3 MPa, 30 min tube-sealing test. f The GC-MS chromatograms of the others aromatics in the products from alkylation of toluene with different alkylation reagents (CO2 + H2, formaldehyde and methanol) over the ZZO & M-22 and M-22 catalyst. g The GC-MS chromatograms of the others aromatics in the products from alkylation of toluene with CO2 + H2 over the ZZO & M-22 and ZZO catalyst. h In situ SVUV-PIMS detection of gas-phase products and i photoionization efficiency spectra of the signals at m/z = 30 and 32 over ZZO catalyst during CO2 + H2. (300 °C, 0.8 MPa, 9,000 mL gcat−1 h−1 and H2/CO2 = 3/1). j The reaction pathway over the ZZO & M-22 catalyst.
Quasi in situ 13C NMR revealed surface species evolution (Fig. 4c): ZZO in 13CO2 + H2 showed formates (169 ppm), methoxy (52 ppm), and methanol (49 ppm)39,40 while ZZO & 4Si/M-22 in 13CO2 + H2 + C7H8 exhibited depleted CHxO signals alongside emerging xylene and trimethylbenzene signatures (20.9 ppm and 19.5 ppm)41,42. 1H-13C 2D heteronuclear correlation (HETCOR) NMR correlations confirmed these assignments formate (169/8.4 ppm), methoxy (52/3.7 ppm), and methanol (48/3.2 ppm) in ZZO (Fig. 4d), transitioning to PX and 1,2,4-trimethylbenzene signals (20.9/2.4 ppm and 19.5/1.84 ppm) in the tandem system (Fig. 4e). Collectively, these results demonstrate CHxO* identified as the key intermediate enabling selective PX formation through spatial proximity effects in the tandem catalyst system, and the depletion of CHxO species promotes formate conversion.
To further identify the CHxO intermediates and their migration behavior from ZZO to MCM-22, controlled experiments were performed (Fig. 4f, 4g). Significantly, side product—specifically 2-(m-methylbenzyl)toluene (2MMBT), 4-(p-methylbenzyl)toluene (4PMBT), and 2-(p-methylbenzyl)toluene (2PMBT)—were detected with selectivity of 3% during CO2 hydrogenation coupled with toluene over the ZZO & M-22 tandem catalyst (Fig. 4f, top panel; Supplementary Fig. 8a). This observation suggests that CH2O species, generated from CO2 hydrogenation, react with toluene, initially forming methylbenzyl alcohol intermediates. These intermediates then undergo further reactions with toluene to produce the observed side products. Crucially, when formaldehyde (CH2O) was used as a methylating agent, replacing CO2/H2, identical side products were formed. In contrast, methanol, as a methylating agent, did not lead to the formation of these compounds. Furthermore, formaldehyde exhibited more favorable reaction kinetics compared to methanol, facilitating PX formation at lower temperatures and achieving higher PX selectivity (Supplementary Fig. 8b). In situ SVUV-PIMS analysis enabled the identification of key intermediates through their characteristic ionization thresholds (Fig. 4h, i): m/z = 30 (H2CO, 10.88 eV) and m/z = 32 (CH3OH, 10.85 eV), which are consistent with values reported in the literature43,44. These findings confirm that CH2O* are generated over ZZO, and that formaldehyde—not methanol—acts as the alkylating agent formed from CO2 + H2 over the ZZO & M-22 tandem catalyst. Reaction kinetics further support the formation of CH2O* species formed over ZZO from CO2 hydrogenation, with methanol being produced over ZZO (Fig. 1b). To confirm that alkylation occurs exclusively over MCM-22 and not ZZO, the reaction of CO2 + H2 with toluene was carried out over isolated ZZO catalyst (Supplementary Fig. 9). This experiment unequivocally demonstrated that alkylation requires the presence of the MCM-22 component. These results indicate the CH2O* species, formed over ZZO, can migrate to M-22 site to contribute to the formation of PX.
Based on the results presented above, the following reaction pathway is proposed, as illustrated in Fig. 4j. Initially, CO₂ and H₂ are adsorbed and activated on the ZZO component, leading to the formation of HCOO* species. These HCOO* species then undergo hydrogenation to produce CH2O* intermediates. The CH2O* intermediates subsequently migrate to the MCM-22 zeolite, where they react with toluene to form methylbenzyl alcohol, with para-methylbenzyl alcohol being the dominant product. The para-methylbenzyl alcohol then primarily undergoes dehydration, followed by hydrogenation, to yield the target product, PX. In parallel, some of the methylbenzyl alcohol can react with an additional toluene molecule, resulting in the formation of side products—2MMBT, 4PMBT, and 2PMBT.
DFT calculations on the reaction pathway
The experimental findings collectively suggest that CH2O* serves as the critical intermediate enabling kinetic coupling between the ZZO and M-22 components in the tandem catalytic system. To validate this hypothesis at the molecular level, density functional theory (DFT) calculations were conducted to compare three potential intermediates–CH2O*, CH3O*, and CH3OH in their ability to migrate from ZZO into sinusoidal channels of M-22. For the methanol-mediated pathway (Fig. 5a m1-m6), the mechanism initiates with methanol adsorption at the Brønsted acid site in the T8 position of sinusoidal channel in M-22 (Fig. 5a). Subsequent dehydration through transition state mTS2 generates a methyl group (CH3) and water, requiring a substantial activation barrier of 2.05 eV (Fig. 5b). Following water desorption (Fig. 5a m4), toluene enters the framework where the CH3 group attacks the para-carbon of the aromatic ring (Fig. 5a m5), ultimately yielding desorbed PX (Fig. 5a m6).

a Optimized geometries of all reaction intermediates and transition states. Reaction Gibbs free energy diagram: b Methanol reacting with toluene over M-22 zeolite, c CH2O* reacting with toluene on M-22 zeolite. d The initial adsorption energy of CH3O* relative to CH2O* and CH3OH in M-22.
In contrast, the CH2O*–mediated pathway demonstrates superior thermodynamic feasibility (Fig. 5a f1-f11). Initial adsorption of CH2O* at the T8 Brønsted site (Fig. 5a f1) is followed by protonation to form a hydroxymethyl cation (CH2OH*) (Fig. 5c f2). Concurrent toluene ingress enables para-carbon attack via transition state fTS1 (Fig. 5c), forming H--HO-CH2-Tol* (Fig. 5c f3-f4). The rate-determining step involves C-O bond cleavage through transition state fTS2 (1.19 eV barrier) (Fig. 5c f5), yielding CH2-Tol* and water (Fig. 5c f6). Subsequent hydrogen transfer from surface H* species (Fig. 5c f8) via transition state fTS3 (Fig. 5c f9) generates CH3-Tol*, culminating in PX desorption (Fig. 5c f11). Notably, CH3O* exhibits substantially higher adsorption energies compared to both CH2O* (+1.0 eV) and CH3OH (+3.0 eV) (Fig. 5d), rendering it thermodynamically unfavorable for alkylation within M-22. The maximum activation barrier of CH2O*-mediated pathway (1.19 eV) remains significantly lower than that of methanol barrier (2.05 eV), conclusively identifying CH2O* as the kinetically favored intermediate.
In summary, this work demonstrates a tandem catalytic strategy for direct PX synthesis via CO2 hydrogenation coupled with toluene methylation using a ZZO & M-22 system. Compartmentalized catalysis enables synergy: ZZO preferentially activates CO2/H2, while M-22 channels mediate C–C coupling via CH2O* intermediates. This approach overcomes thermodynamic constraints of conventional toluene methylation, achieving 91% xylene selectivity (70.4% PX/xylene) at 10.3% toluene conversion. TEOS modification of M-22 is critical, passivating supercage Brønsted acid sites (Si(OH)Al) via TEOS deposition to suppress disproportionation side reactions. Mechanistically, CH2O* intermediates kinetically couple CO2 activation and alkylation, bypassing methanol dehydration. Controlled diffusion of CH2O* from ZZO to M-22 optimizes thermodynamic-kinetic balance, facilitating confined electrophilic para-methylation. This establishes a paradigm for tandem catalyst design through zeolite architecture engineering, where precise intermediate dimension/configurational control governs selectivity. The strategy offers an efficient CO2 valorization route and a carbon recycling alternative to conventional PX production, underscoring structure-directed catalysis in bridging CO2 utilization with aromatics synthesis.
Methods
Materials
Zn(NO3)2 · 6H2O were purchased Sinopharm (China), (NH4)2CO3, m-xylene (99%), toluene (99.9%) and Zr(NO3)4 · 5H2O, tetraethylorthosilicate (TEOS, 99.98%) and n-hexane (99%) were obtained from Macklin, CO2/H2/Ar [24/72/4(v/v/v)], H2 (99.99%) and Ar (99.99%) were obtained from Lanzhou Yulong Gas Co., Ltd. HMCM-22 (Si/Al=15) were obtained from Nankai Catalysts.
Preparation of catalysts
The ZnZrO catalyst that doped 20% ZnO (Zn/ (Zn + Zr) molar ratio) was prepared as follows. 14.9 g of Zn (NO3)2 · 6H2O and 8.6 g of Zr (NO3)4 · 5H2O were dissolved in a flask by using 400 mL of deionized water, then the 250 mL aqueous solution of (NH4)2CO3 (43.2 g) as precipitant was dropwise added into the aforementioned solution under vigorous stirring at 70 °C to form a precipitate. The obtained suspension was continuously stirred for 4 h at 70 °C, followed by cooling down to room temperature and filtering. The precipitate was washed three times with deionized water, dried at 110 °C for 4 h, and calcined at 500 °C in static air for 3 h.
Si/M-22 zeolite was prepared by tetraethoxysilane (TEOS) modification. Typically, the steps to modify the M-22 once were to immerse 1 g M-22 into the mix solution of 1 g TEOS and 20 ml of n-hexane for 4 h; after that, organic solvent was removed by evaporation at 80 °C and the powder was dried overnight at 110 °C, followed by calcination at 550 °C for 4 h in air. While the above steps were repeated, a new symbol of nSi/M-22 (n means the number of repetitions) was used. After repeating the procedure above for four times, the 4Si/M-22 was obtained. The tandem catalyst was typically prepared by physical mixing. The weight ratio of oxides and zeolites was 1:1. For preparation of tandem catalyst ZZO & M-22, ZZO and M-22 were grinded in an agate mortar for 5 min, pressed under 10 MPa and granulated into the required size in the range of 40-80 mesh.
M-22 with the pre-coked supercages, denoted as pre-coked M-22 was prepared following the procedure: M-22 was pretreated at 500 °C for 2 h and then cooled down to 350 °C under Ar flow (100 mL min−1). Subsequently, the transformation of m-xylene (99%) was carried out at 350 °C, atmospheric pressure and 8 h−1 of m-xylene WHSV with Ar flow (40 mL min−1) for 12 h, to form pre-coked M-22.
Catalyst evaluation
Catalytic performance was evaluated on a high-pressure fixed-bed reactor equipped with a quartz sample tube. Typically, 0.52 g catalyst (40-80 mesh) were loaded into the quartz sample tube without mixed quartz sand. Before evaluating the catalytical performances, raised to the specified temperature under the protection of argon. For the investigation of toluene methylated with CO2 and H2, a mixture gas of CO2/H2/Ar [24/72/4(v/v/v)] was induced into a quartz sample tube, and then toluene was carried into reaction tube (90 °C) at a constant flow rate by using a piston pump. The reaction was conducted at 320 °C and 3.0 MPa, with a GHSV of 12,000 mL g−1 h−1 and a toluene weight hourly space velocity (WHSV) of 1 h−1, and the data were collected at the reaction time of 12 h. For the contrast experiment of toluene methylated with methanol/Formaldehyde (s-Trioxane), a mixed solution of C7H8/CH3OH or HCHO [5/1 (mol/mol)] was pumped into reaction tube with a toluene WHSV of 1 h−1 at 320 °C /260 320 °C and 3.0 MPa, while 12,000 mL g−1 h−1, H2/Ar was used as a carry gas simultaneously. In the quantitative analysis, we use diphenyl as an internal standard to determine the C balance, and the C balance was above 96%. The liquid products were analyzed by using the gas chromatography equipped with CBX-WAX columns to analyze aromatics, the conversion of toluene and production selectivity was calculated as follows:
Where ni is the molar amount at the outlet of the individual aromatic product i.
Catalyst characterization
The XRD analysis was performed using a Philips PW1050/81 diffractometer equipped with a CuKα radiation source (λ = 1.5418 Å). The instrument was operated in Bragg-Brentano geometry at 40 kV and 30 mA.
The specific surface area was calculated using the Brunauer-Emmett-Teller (BET) method. The textural properties of the samples, including specific surface area and pore size distribution, were characterized by N2 physisorption using a MicrotracBEL BELSORP-mini II analyzer. Prior to analysis, approximately 0.2 g of each catalyst was subjected to degassing at 300 °C for 2 h.
The microstructural and elemental composition of the catalysts were characterized by aberration-corrected HAADF-STEM and STEM-EDX mapping, respectively, using an FEI Cubed Titan G2 60-300 microscope at an accelerating voltage of 300 kV. Morphological analysis was performed by scanning electron microscopy (SEM) on a Thermo Fisher Scientific Apreo S instrument. The SEM specifications were as follows: electron gun: field emission (FEG); accelerating voltage: 30–200 kV.
CO2 and NH3 temperature-programmed desorption (TPD) experiments were conducted using a Micromeritics ASAP 2920 system. Approximately 0.1 g of catalyst, loaded in a quartz reactor, was first pretreated at 500 °C under Ar flow for 3 h and subsequently cooled to 50 °C. The sample was then saturated with a flow of either 10 vol% CO2/He or 10 vol% NH3/He (50 mL min−1) for 1 h at this temperature. Subsequently, the flow was switched to pure He for 1 h at 50 °C to purge physically adsorbed molecules. After stabilizing the baseline, the temperature was ramped to 700 °C at a rate of 10 °C min−1 under He flow, and the desorption signal was monitored by a thermal conductivity detector (TCD).
In-situ DRIFT spectra of surface species formed from the CO2/13CO2 + H2/D2 reaction. Prior to the experiment, the samples were heated to 450 °C for 2 h under an Ar atmosphere to remove any water contained in the cell and on the catalyst surface. In situ DRIFT spectra of surface species over ZZO from CO2 + H2 and subsequently switched to D2. Reaction conditions: ZZO, 0.1 MPa, 320 °C, 30 mL min−1 CO2 + H2, D2 10 mL min−1. In situ DRIFT spectra of surface species over ZZO & 4Si/M-22, 30 ml/min CO2 + 10 mL min−1 H2 +toluene, subsequently switched to 13CO2 was injected into the cell, and infrared spectrum information of the catalyst surface and species was collected.
FTIR spectra of pyridine was performed for both M-22 and nSi/M-22. The sample (about 30 mg) was pressed into a wafer, and then treated under vacuum at 400 °C for 1 h. After cooling to 50 °C, pyridine was introduced to the transmission cell until saturation. Then, the signal vs. the reference signal was recorded at 150 °C until the signal unchanged.
Magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra of 1H and 27Al were collected on a Bruker Avance III 600 MHz Wide Bore spectrometer operating at a magnetic field of 14.1 T, 4 mm dural-channel probe; the rotate speed of samples is 10.0 kHz; The chemical shifts for 27Al MAS NMR spectra were referenced to the aqueous solution of Al(NO3)3 (0 ppm), respectively. The chemical shifts for 1H MAS NMR spectra were referenced to tetramethylsilane (TMS). Quantitative 1H MAS NMR measurement was performed by comparing the signal area of the zeolite sample under study with that of adamantine as an external standard.
Quasi in situ13C CP MAS NMR experiment and Quasi in situ 2D 13C-1H HETCOR MAS NMR experiment are carried out as follows: the basic experimental device is a home-built high-pressure fixed-bed catalytic reactor. Standard reaction conditions: 3 MPa, 320 °C, 10 mL min−1 H2 + 60 mL min−1 13CO2. The reactor was quickly immersed into liquid nitrogen to quench the reaction, so that highly reactive intermediates on the catalysts can be capture. Subsequently, the reactor was sealed, after waiting for recovery to room temperature, and further transferred into 4 mm rotor for further NMR experiment.
In situ SVUV-PIMS experiments were carried out beamline BL03U at the National Synchrotron Radiation Laboratory, Hefei, P.R. China. This technique is employed for the detection of oxygen-containing intermediates during the CO₂ hydrogenation process. The photoionization efficiency spectra were collected for 300 s in the energy range of 10.5-11.3 eV with a step of 0.05 eV. Reaction conditions: the ZnZrO catalyst (0.5 g); reaction gas (H2/CO2 = 3/1), at 300 °C, 0.8 MPa, GHSV = 12,000 mL gcat−1 h−1.
Data availability
All data needed to support the findings of this study are included in the main text or in the Supplementary Information. Data are available from the corresponding authors upon request.
References
Wang, J. et al. A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol. Sci. Adv. 3, e1701290 (2017).
Wang, J. et al. High-Performance MaZrOx (Ma= Cd, Ga) Solid-Solution Catalysts for CO2 Hydrogenation to Methanol. ACS Catal 9, 10253–10259 (2019).
Ma, Z. Q. et al. Development of Tandem Catalysts for CO2 Hydrogenation to Olefins. ACS Catal 9, 2639–2656 (2019).
Li, Z. L. et al. Highly Selective Conversion of Carbon Dioxide to Lower Olefins. ACS Catal 7, 8544–8548 (2017).
Xu, Y. et al. Highly Selective Olefin Production from CO2 Hydrogenation on Iron Catalysts: A Subtle Synergy between Manganese and Sodium Additives. Angew. Chem. Int. Ed. 59, 21736–21744 (2020).
Yang, H. Y. et al. Selective synthesis of olefins via CO2 hydrogenation over transition-metal-doped iron-based catalysts. Appl. Catal. B Environ. Energy 321, 122050 (2023).
Ni, Y. M. et al. Selective conversion of CO2 and H2 into aromatics. Nat. Commun. 9, 3457 (2018).
Wang, Y. et al. Rationally Designing Bifunctional Catalysts as an Efficient Strategy to Boost CO2 Hydrogenation Producing Value-Added Aromatics. ACS Catal 9, 895–901 (2019).
Li, Z. L. et al. Highly Selective Conversion of Carbon Dioxide to Aromatics over Tandem Catalysts. Joule 3, 570–583 (2019).
Ahn, J. H. et al. Methanol Usage in Toluene Methylation with Medium and Large Pore Zeolites. ACS Catal 3, 817–825 (2013).
Bi, Y. et al. Improved Selectivity toward Light Olefins in the Reaction of Toluene with Methanol Over the Modified HZSM-5 Catalyst. ChemCatChem 6, 713–718 (2014).
Liu, X. L. et al. Selective transformation of carbon dioxide into lower olefins with a bifunctional catalyst composed of ZnGa2O4 and SAPO-34. Chem. Commun. 54, 140–143 (2018).
Gao, P. et al. Direct Production of Lower Olefins from CO2 Conversion via Bifunctional Catalysis. ACS Catal 8, 571–578 (2018).
Chen, S. Y. et al. Hydrogenation of CO2 to Light Olefins over ZnZrOx/SSZ-13. Angew. Chem. Int. Ed. 63, e202316874 (2024).
Zhou, C. et al. Highly Active ZnO-ZrO2 Aerogels Integrated with H-ZSM-5 for Aromatics Synthesis from Carbon Dioxide. ACS Catal 10, 302–310 (2020).
Zhang, J. F. et al. Hydrogenation of CO2 into aromatics over a ZnCrOx-zeolite composite catalyst. Chem. Commun. 55, 973–976 (2019).
Miao, D. et al. Selective synthesis of para-xylene and light olefins from CO2/H2 in the presence of toluene. Catal. Sci. Technol. 11, 4521–4528 (2021).
Shang, X. et al. Regulating anisotropic diffusion in zeolite for reinforced para-xylene synthesis via CO2 hydrogenation in the presence of toluene. Appl. Catal. B Environ. Energy 359, 124523 (2024).
Hong, B. et al. Low-pressure CO2 hydrogenation coupled with toluene methylation to para-xylene using atomic Pd-doped ZnZrOx-HZSM-5. Appl. Catal. B Environ. Energy 361, 124606 (2025).
Tuo, J. et al. Shape-selective synthesis of para-xylene through tandem CO2 hydrogenation and toluene methylation over ZnCeZrOx/MCM-22 catalyst. Chin. J. Catal. 73, 174–185 (2025).
Zuo, J. C. et al. Selective methylation of toluene using CO2 and H2 to p-xylene. Sci. Adv. 6, eaba5433 (2020).
Xiao, Z. K. et al. Designing a bifunctional ZrCuOx/HZSM-5 catalyst for selective methylation of toluene with carbon dioxide to para-xylene. Fuel 319, 123848 (2022).
Zhu, Z. R. et al. Catalytic performance of MCM-22 zeolite for alkylation of toluene with methanol. Catal. Today 93, 321–325 (2004).
Parmar, D. et al. Spatiotemporal Coke Coupling Enhances para-Xylene Selectivity in Highly Stable MCM-22 Catalysts. J. Am. Chem. Soc. 144, 7861–7870 (2022).
Madeira, F. F. et al. Ethanol transformation into hydrocarbons on ZSM-5 zeolites: Influence of Si/Al ratio on catalytic performances and deactivation rate. Study of the radical species role. Appl. Catal. A Gen. 443, 171–180 (2012).
Hunger, M. et al. Multinuclear solid-state nmr investigation of zeolite MCM-22. Zeolites 15, 188–192 (1995).
Ma, D. et al. An investigation of the roles of surface aluminum and acid sites in the zeolite MCM-22. Chem. Eur. J. 8, 162–170 (2002).
Ma, D. et al. MAS MMR studies on the dealumination of Zeolite MCM-22. J. Phys. Chem. B 105, 1770–1779 (2001).
Corma, A. et al. Infrared spectroscopy, thermosprogrammed desorption, and nuclear magnetic resonance study of the acidity, structure, and stability of zeolite MCM-22. Zeolites 15, 576–582 (1995).
Kennedy, G. J. et al. Multinuclear MAS NMR studies of zeolites MCM-22 and MCM-49. Catal. Today 49, 385–399 (1999).
Zhou, D. H. et al. DFT studies on the location and acid strength of Bronsted acid sites in MCM-22 zeolite. J. Mol. Catal. A Chem. 244, 11–19 (2006).
Sastre, G. et al. Preferential siting of bridging hydroxyls and their different acid strengths in the two-channel system of MCM-22 zeolite. J. Phys. Chem. B 104, 4349–4435 (2000).
Li, Y. et al. A DFT study on the distributions of Al and Bronsted acid sites in zeolite MCM-22. J. Mol. Catal. A Chem. 338, 24–32 (2011).
Mériaudeau, P. et al. On the localization of tetrahedral aluminum in MCM-22 zeolite. Catal. Lett. 61, 89–92 (1999).
Wu, P. et al. Selective formation of p-xylene with disproportionation of toluene over MCM-22 catalysts. Micropor. Mesopor. Mat. 22, 343–356 (1998).
Laforge, S. et al. m-Xylene transformation over H-MCM-22 zeolite 1: Mechanisms and location of the reactions. J. Catal. 220, 92–103 (2003).
Muller, G. et al. Infrared microscopic study of sorption and diffusion of toluene in ZSM-5. J. Phys. Chem. 98, 7436–7439 (1994).
El-Sabbagh, O. I. et al. Synthesis and antiviral activity of new pyrazole and thiazole derivatives. Eur. J. Med. Chem. 44, 3746–3753 (2009).
Larmier, K. et al. CO2-to-Methanol Hydrogenation on Zirconia-Supported Copper Nanoparticles: Reaction Intermediates and the Role of the Metal-Support Interface. Angew. Chem. Int. Ed. 56, 2318–2323 (2017).
Ji, Y. et al. Oxygenate-based routes regulate syngas conversion over oxide-zeolite bifunctional catalysts. Nat. Catal. 5, 594–604 (2022).
Wang, C. et al. Unraveling Spatially Dependent Hydrophilicity and Reactivity of Confined Carbocation Intermediates during Methanol Conversion over ZSM-5 Zeolite. J. Am. Chem. Soc. 146, 8688–8696 (2024).
Shang, X. et al. Xylene Synthesis Through Tandem CO2 hydrogenation and toluene methylation over a composite ZnZrO Zeolite Catalyst. Angew. Chem. Int. Ed. 62, e202309377 (2023).
Yang, H. Y. et al. Tuning the selectivity of CO2 hydrogenation to alcohols by crystal structure engineering. Chem 10, 2245–2265 (2024).
Hu, J. T. et al. Sulfur vacancy-rich MoS2 as a catalyst for the hydrogenation of CO2 to methanol. Nat. Catal. 4, 242–250 (2021).
Acknowledgements
This work thank beamline BL04B at the National Synchrotron Radiation Laboratory, Hefei, P.R. China, for the beam time and assistance with experiments. This work was supported by the National Key R & D Program of China (2023YFB4104501) and the National Natural Science Foundation of China (grant nos. 223B2204 and U24A20493).
Author information
Authors and Affiliations
Contributions
J.T. and Y.X. contributed equally. J.T. conceived and conducted the experiments, and analyzed data; Y.X. conducted the DFT calculations; J.C., J.Z., H.W., F.R., J.Y., C.H., and H.Y. offered the help in the experiments and data analysis; C.L. conducted the in situ SVUV-PIMS experiments. The manuscript was written by Z.L.; Z.L. and C.L. proposed the project, analyzed data and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tian, J., Xi, Y., Cheng, J. et al. CO2 hydrogenation toward toluene methylation for selective para-xylene synthesis. Nat Commun 17, 967 (2026). https://doi.org/10.1038/s41467-025-67695-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67695-4
