
Abstract
With the rise in antimicrobial resistance, understanding the virulence factors utilized by pathogenic E. coli is essential for the development of alternative therapeutics. While previous work has shown that disruption of the E. coli rhomboid protease gene glpG leads to defects in bacterial colonization, here we provide mechanistic insight into the loss of fitness. We show GlpG is essential for the assembly of type 1 pili, a virulence factor required for the colonization of eukaryotic cells. Since pili are critical for biofilm formation and bacterial persistence, the absence of GlpG proteolytic activity reduces the production of biofilm. Working towards new potential antimicrobial targets for treating infections, we show that biofilm formation is hampered by GlpG inhibition. Our data demonstrates that GlpG plays a key role in protein quality control of type 1 pili and alters the paradigm for GlpG proteolysis, previously implicated in the cleavage of only membrane embedded substrates.
Similar content being viewed by others


Introduction
Escherichia coli is a diverse bacterial species with the ability to colonize and persist in various niches within animal hosts, often forming a symbiotic relationship by providing nutrients, key signals for homeostatic regulation, and protection against foreign pathogens1,2,3. Within the gastrointestinal tract, resident E. coli are typically commensal; however, these strains can take on a pathogenic course, causing disease outside the gut through combinations of acquired virulence factors such as adhesins, secreted toxins, or lipopolysaccharides4,5,6. Pathogenic strains are broadly categorized as either intestinal pathogenic E. coli, causing infection within the gastrointestinal tract, or extraintestinal pathogenic E. coli (ExPEC), as they have retained the ability to survive in the gut, but also wield virulence factors with the capacity to disseminate and colonize other sites, including the blood, brain, and urinary tract7,8,9. In addition, there are three unclassified pathogenic groups: adherent-invasive E. coli involved in Crohn’s disease, necrotoxic E. coli, which is associated with disease in humans and other mammals, and cell-detaching E. coli that involved in hemolysin production. Among ExPEC, the uropathogenic E. coli (UPEC) pathotype accounts for 80–90% of all urinary tract infections (UTI), one of the most common bacterial infections globally10,11,12. About 50% of women in the United States are expected to have at least one UTI in their lifetime with half experiencing a recurrent infection within 12 months of the initial episode13,14. The global death rate due to UTIs has increased by 140% from 1990 to 2019, largely attributed to the rise of antimicrobial resistance to classes of commonly prescribed antibiotics such as β-lactams, cephalosporins, and fluoroquinolones, ultimately making UPEC infections more challenging to treat15,16,17,18,19,20. Consequently, this places an urgent need for the development of alternate treatment strategies to combat these infections. However, in order to explore alternative therapeutics, a better understanding of bacterial virulence factors regulation is required.
Virulence factors play a crucial role in bacterial survival and persistence, largely through biofilm initiation and formation. Biofilms are surface-attached communities of bacteria embedded in a matrix of biopolymers such as proteins and polysaccharides, and are one of the critical properties responsible for most chronic infections by UPEC21. Pili and flagella, along with other adhesins and extracellular factors such as toxins, siderophores, enzymes, and polysaccharide coatings, are among the main virulence factors necessary for initial adhesion and colonization of host mucosal surfaces22,23,24. Once formed, biofilms are remarkably difficult to eradicate as these communities are more resistant to antimicrobial agents, surviving transient antibiotic treatment and regrowing when antibiotic is withdrawn, leading to infection persistence and recurrence25,26,27.
The expression, assembly, and turnover of virulence factors reflect a prokaryote’s ability to disseminate and infect a host, but how the proteins involved regulate these processes is not completely understood. The rhomboid protease superfamily are intramembrane proteases that are ubiquitously expressed across all kingdoms of life, with the eukaryotic members implicated in various functions such as mitophagy, growth factor signaling, amyloid precursor protein processing, and more28,29,30,31,32. Meanwhile, the roles of prokaryotic rhomboid proteases have been linked to functions such as quorum sensing with AarA in Providencia stuartii, biofilm formation with Rho1/2 in Haloferax volcanii, ion homeostasis with YqgP in Bacillus subtilis and protein quality control with GlpG in Shigella sonnei33,34,35,36. GlpG is a conserved and widely represented member of the rhomboid protease superfamily located in the plasma membrane of all known strains of E. coli as well as other prokaryotes37. Moreover, it has been shown that the glpG gene is essential for ExPEC persistence in the murine intestine and by knocking-out glpG, bacterial colonization is significantly reduced, however, by an unknown mechanism38. Additionally, while GlpG in S. sonnei targets and cleaves orphan proteins to prevent aggregation in the cell, a potential association to bacterial pathogenicity remains to be shown35.
Here, we provide a mechanistic insight into the link between bacterial colonization and proteostasis in pathogenic E. coli. Using proteomics, we sought to reveal physiological binding partners of GlpG and identified type 1 pili subunit, FimA, as well as periplasmic serine endoproteases DegQ and DegP39. We demonstrate that GlpG proteolytic activity is essential for type 1 pili formation in pathogenic pathotype UPEC, affecting the ability of E. coli to colonize and infect host cells. As adherence is essential for the initiation of biofilm formation, we show that the absence of GlpG catalysis, as well as GlpG inhibition, significantly reduces biofilm production. Our data demonstrates that GlpG regulates FimA protein quality control and turnover by cleaving FimA aggregates during type 1 pili formation. Moreover, proteomic analysis revealed GlpG association with housekeeping proteases DegP and DegQ. We demonstrated that these proteases are also involved in cleaving aggregated or misfolded FimA subunits, assisting in the clearance of aggregates accumulated in the periplasm. Thus, GlpG is part of an important quality control checkpoint of FimA subunits, ensuring assembly fidelity of type 1 pili. Overall, our study demonstrates that GlpG plays an essential role in UPEC virulence and may represent a viable drug target for the treatment of bacterial infections, such as UTIs, to combat the rise of antimicrobial resistant bacteria.
Results
Proteomic analyses of GlpG identified substrates associated with bacterial virulence
Rhomboid proteases were discovered in the early 2000’s, and E. coli rhomboid protease, GlpG, quickly became a key model for understanding the structure and mechanism of this family of intramembrane proteases40,41,42. Despite being the first family member to have the structure solved, the interacting partners, physiological substrate(s), and hence, the function of E. coli GlpG remained unknown. Our first focus was to identify binding partners/substrates of GlpG using a 3× FLAG tag genomically inserted downstream of the glpG gene to enable affinity pulldown and whole cell proteomics in the presence of non-denaturing detergent. This revealed that GlpG interacted with several proteins with roles in intracellular secretion and protein turnover when assigned to Clusters of Orthologous Groups (COGs) (Fig. 1a and Supplementary Data 1), in particular with Sec Y and G of the SecYEG translocon, its associated ATP-dependent motor protein SecA as well as its ancillary proteins YidC, SecD, and F. Together they drive Sec-mediated translocation of unfolded proteins across the membrane, a tightly regulated process that exports most secretory proteins in bacteria. GlpG may associate with the Sec translocon to readily cleave misfolded or aggregated proteins to prevent the toxic accumulation of aggregates, as part of a role in protein quality control, a function shared by other rhomboid proteases studied in the current literature. We also demonstrate GlpG association with DegQ and DegP (Fig. 1a), proteases with central roles in protein quality control that are closely involved in clearing unproductive or harmful protein intermediates, particularly under stress conditions. There is increasing evidence that both DegP and DegQ interact functionally with the SecYEG translocon43. DegP, in particular, has been shown to degrade preproteins that are stalled or improperly inserted into the membrane, suggesting a surveillance role at or near the translocon44.

a Cytoscape network of periplasmic and inner membrane proteins identified after affinity pulldown of whole cell extract with glpG-3XFLAG followed by Triton ×-100 solubilization and LC-MS/MS reveal an enrichment of proteins involved in proteostasis. n = 3. b Experimental procedure for LC-MS/MS proteomics of E. coli periplasmic proteins. Created with BioRender.com. https://BioRender.com/85mschr. c The volcano plot showing decreased (blue) and increased (orange) protein abundance levels in extracts after incubation with active relative to inactive GlpG. We quantified 895 proteins with high reproducibility (~95%) between biological replicates n = 4. Protein quantification was performed using Proteome Discoverer(v 2.4.1.15). Protein abundance changes between periplasmic proteins incubated with active and inactive GlpG with a significant p-value below 0.05 (−log10 p-value > 1.30) and a fold change (FC) greater than two (log2 FC > 1), are presented. Lines represent the boundaries for a twofold change and a significant p-value of <0.05. Proteins were identified with the UniProt ID. The number of proteins quantified in one sample but not the other is identified in curly brackets. d In-gel digestion and mass spectrometric characterization of periplasmic proteins of K12 substr. MG1655 ΔglpG separated by SDS-PAGE reveal accumulation of aggregated FimA (>135 kDa MW range) when incubated with inactive GlpG, but no accumulation when incubated with active GlpG, n = 2. e Western blot of K12 WT or mutant periplasmic proteins separated by SDS-PAGE, detecting FimA aggregates. Periplasmic proteins were isolated from K12 wild-type, K12 mutant strains harboring single gene deletions: ΔglpG, ΔdegQ, ΔdegP, and rescues of the mutant strains overexpressing GlpG. K12 ΔfimA was used as a negative control. Biological replicates for western blot samples were repeated n = 3 for confirmation.
The list of interacting partners from whole cell proteomics led us to take a narrower approach to analyze only the resident proteins in the periplasmic space. The active site of GlpG in E. coli faces the periplasm, allowing potential substrates to be identified when comparing isolated periplasmic proteins in the presence of active or inactive GlpG using LC-MS/MS (Fig. 1b). In addition, this minimizes cytosolic proteins that would otherwise be identified as non-physiological substrates45. Notably, the periplasmic fraction was isolated from a ΔglpG mutant of K12 substr. MG1655 E. coli to ensure any identifiable proteins were not prematurely cleaved by endogenous GlpG. We hypothesized that proteolysis by GlpG would lead to the generation of smaller protein fragments potentially leading to apparent decrease in protein abundance due to the fragment's degradation, precipitation in the lysate or running out of the gel. Therefore, proteins identified as significantly decreased in the presence of active GlpG, relative to inactive GlpG, would represent potential cleavage by GlpG (Fig. 1c). Several proteins were identified, including those involved in cell division, stress response, and chaperone mediated folding, such as FtsA, RcnB, and PpiD (Supplementary Fig. 1 and Supplementary Data 2). Importantly, virulence factor FimA, a subunit of type 1 pili, was identified among the proteins significantly decreased in the presence of active protease, drawing our attention in accordance with previous literature, describing colonization defects of pathogenic E. coli with a glpG knockout strain38.
To further investigate the role of GlpG in proteostasis, an in-gel digestion was performed whereby periplasmic proteins isolated from the K12 ΔglpG mutant strain were incubated with active or inactive GlpG, separated by SDS-PAGE and followed by gel excision at different molecular weight ranges to be analyzed using LC-MS/MS. In line with the function of other rhomboid proteases that cleave aggregated substrates to prevent their accumulation34,35, we sought for a more detailed examination of which proteins may be forming aggregates in the absence of GlpG and could be cleaved by the re-addition of GlpG (Supplementary Data 3). When periplasmic proteins were incubated with inactive GlpG, FimA was found to be at a higher molecular weight (>135 kDa) instead of its monomeric size (18 kDa), whereas in the presence of active GlpG, this oligomeric form of FimA was not present (Fig. 1d and Supplementary Fig. 2). In wild-type (WT) E. coli FimA monomers were rarely detected, likely due to their incorporation into pili or rapid turnover (Fig. 1d, e).
In WT E. coli, homeostasis of FimA protein in periplasmic space is maintained, as evidenced by the lack of FimA aggregation when performing immunoblot detection by western blot. By contrast, FimA aggregation was detected in the periplasm of K12 MG1655 ΔglpG, even under SDS-denaturing conditions (Fig. 1e), suggesting that in the absence of GlpG catalytic activity, proteostasis of FimA is dysregulated, causing its accumulation and aggregation within the periplasm during its translocation. When K12 MG1655 ΔglpG was transformed with the plasmid bearing active GlpG, we observed notably decreased amount of aggregates, confirming GlpG’s role in protein quality control as seen in many other rhomboid proteases34,35. To test the hypothesis that periplasmic proteases DegQ and DegP found associated with GlpG (Fig. 1a), may also be involved in the biogenesis of type 1 pili, we detected FimA in the samples of periplasmic proteins isolated from single gene knockout of degQ and degP in K12 MG1655. Indeed, immunoblot analysis demonstrated aggregated FimA in the periplasm in the absence of either ΔdegQ and ΔdegP (Fig. 1e), identifying a link between DegQ and DegP proteases and quality control of type 1 pili for the first time. The more profound effect of ΔdegQ on FimA aggregation could be explained by the fact that ΔdegP protease is not constitutively expressed under normal growth conditions46, as opposed to ΔdegQ, which has relatively constant level regardless of specific environmental conditions47. Moreover, the K12 MG1655 ΔdegP and ΔdegQ mutants transformed with plasmid expressing active GlpG exhibited reduced amount of FimA aggregates. This suggests that there may be interplay between GlpG and DegQ/P that reflects a specialized proteostasis module that monitors and responds to the periplasmic aggregation of pilin subunits.
GlpG activity is essential for pili formation
FimA protein is the structural subunit of type 1 pili, comprising of up to 3000 subunits in a single pilus. It is an abundant and essential component for bacterial adherence to host cells, therefore maintaining its protein homeostasis is crucial in pathogenic E. coli. The main function of adhesion virulence factors, such as type 1 pili, is to facilitate attachment of the bacterium to the host cell, often as an initial step in invasion or uptake. To determine if the lack of GlpG catalysis affects type 1 pili formation, transmission electron microscopy (TEM) was used to visualize surface pili. UPEC strain NU149 was chosen to represent a clinically relevant isolate from a patient presenting cystitis48. We show that significantly more surface pili are present on NU149 wild-type relative to a genomically inactive glpG strain generated using CRISPR/Cas9, which harbors a Ser201Ala point mutation at the catalytic serine (Fig. 2a, b and Supplementary Fig. 3). A genomically inactive glpG mutant, hereby denoted as glpG-S201A, was used to gain a more physiological insight into the role of the protease compared to a gene knock-out. The growth curve and glpG mRNA expression of the resulting strain endogenously expressing catalytically inactive GlpG was unaffected compared to its wild-type counterpart (Supplementary Fig. 4). Importantly, it is known that mutation of the catalytic serine does not disrupt the folding of GlpG protease49. The significant impairment of pili formation observed in glpG-S201A mutant was rescued when the active GlpG protease was reintroduced to NU149 glpG-S201A using an inducible plasmid, with an apparent increase in not only pili but flagella, which was unexpected. The same trends were observed when fim protein chaperone, FimC, was overexpressed, assisting the folding, binding, and translocation of FimA (Fig. 2b and Supplementary Fig. 5) (discussed in detail below). To test if GlpG expression may be elevated in pathogenic E. coli relative to commensal E. coli, we quantified glpG mRNA transcript levels using RT-qPCR, and indeed detected higher expression in NU149 and J96 UPEC strains compared to K12 MG1655. This result was not affected by different nutrient environments such as LB, high nutrient terrific broth (TB), or M9 minimal media that could influence bacterial growth rate, biomass yield, gene expression, and protein production (Fig. 2c).

a Negatively stained electron micrographs of uropathogenic E. coli strain NU149 wild-type, the CRISPR/Cas9 glpG-S201A mutant, and glpG-S201A mutant transformed with inducible plasmid expressing active GlpG. Bar represents 0.5 µm. b Pili from micrographs were quantified by manual counting using n = 30 of E. coli cells per strain. Significant differences between strains were calculated using one-way ANOVA. c Quantification of glpG transcription using RT-qPCR analysis in commensal (K12) and uropathogenic (NU149 and J96) E. coli strains under different growth conditions. glpG mRNA is reported relative to housekeeping gene rrsB. Data are presented as mean values +/− SEM. Each strain was performed in biological triplicates n = 3 and technical triplicates n = 3. Significant differences were determined using one-way ANOVA. d Hemagglutination assay of NU149 E. coli strains confirms glpG mutant strains do not possess type 1 pili. The absence of type 1 pili on E. coli is confirmed by the binding to erythrocytes, forming a dense button of E. coli-bound erythrocytes at the bottom of the well. As a control, hemagglutination was inhibited by the presence of 2% of D-mannose prior to the addition of bacterial cultures. Assay performed in biological triplicate n = 3.
To confirm that the surface organelles detected using TEM were indeed type 1 pili, we performed a classical functional test for the presence of type 1 pili: a mannose-sensitive hemagglutination (MSHA) assay on guinea pig erythrocytes. Type 1 pili mediate the hemagglutination (HA) that occurs when piliated bacteria are mixed with erythrocytes of certain species. Since type 1 pili on UPEC bind to mannosylated proteins on host cells, particularly mannose found on uroplakins and integrins by FimH adhesion, the HA reaction can be inhibited by addition of 2% mannose50. The NU149 strains described above were grown under the same conditions as for TEM imaging and assessed for HA (Fig. 2d). NU149 wild-type and glpG-S201A strain overexpressing active GlpG protease were MSHA positive (type 1 piliated), whereas inactive glpG mutant as well as the cells transformed with an empty pBad vector did not exhibit HA, suggesting the absence of type 1 pili, in agreement with the TEM results. Overall, this data shows that the catalytic activity of GlpG influences type 1 pili, a critical virulence factor in E. coli, through the quality control of FimA.
GlpG activity influences pathogenic E. coli invasion
UPEC can infect both bladder and kidneys cells and account for the vast majority of all UTI infections12. To determine whether loss of pili observed in glpG-S201A strains results in reduced host colonization, both an in vitro adherence and invasion (gentamicin protection) assays were used to quantify bacterial adhesion and entry into epithelial cells (Fig. 3a). The glpG-S201A mutants of UPEC NU149 and commensal K12 substr. MG1655 showed significantly reduced invasion relative to their wild-type counterparts in both T24 human bladder (Fig. 3b) and mIMCD-3 mouse kidney cell lines (Fig. 3c). Notably, this reduction in invasion was rescued to wild-type levels when active GlpG was reintroduced using an inducible plasmid, confirming the loss of GlpG catalysis results in reduced host colonization in uropathogenic and commensal strains of E. coli. We also assessed invasion indices of UPEC strains against kidney and bladder host cells. The invasion index represents the ratio of invaded bacteria to adherent bacteria, quantifying invasion efficiency independent of adhesion. The invasion indices were not significantly different between NU149 WT and glpG-S201A mutant, therefore GlpG does not cause defect in initial attachment, but specifically impairs the ability to invade host cells (Supplementary Fig. 6). As observed by other groups, other adhesins besides pili, such as the Afa/Dr family of non fimbrial adhesions, or curli can also facilitate attachment playing roles in different host environments during different stages of infection51,52.

a Gentamicin protection assay used to quantify levels of invasion between different strains of E. coli. Created with BioRender.com. https://BioRender.com/bl2bx7c. b Quantification of infection between different strains of E. coli against mIMCD-3, mouse kidney cell line, and c T24, human bladder epithelial cell line reported as mean Colony Forming Units (CFU) per mL. Epithelial cells were infected using wild-type commensal K12 substr. MG1655 (green), or UPEC NU149 strains (blue) and their glpG mutants. Each strain was performed in biological and technical triplicates and with an n = 9. Data are presented as mean values +/− SEM. Significant differences between data sets were determined using one-way ANOVA.
Proximal and distal tubular epithelial cells of the kidney have the capability to phagocytose E. coli present in the tubular lumen, causing peritubular inflammation53. To determine if the GlpG protease impacts the E. coli infectivity for tubular epithelial cells, human kidney organoids differentiated to express segments of the proximal tubule, were infected with NU149 wild-type or NU149 glpG-S201A E. coli. Kidney organoids infected with wild-type NU149 had E. coli-FITC
stained structures present within LTL-Cy5 positive regions, which label proximal tubules (Fig. 4a, b and Supplementary Fig. 7). Meanwhile, a 3.5-fold reduction of detectable bacteria at 469 NB100-FITC per mm2 of LTL-Cy5 for NU149 glpG-S201A was observed compared to wild-type infected kidney organoids which had 1650 anti-E. coli-FITC per mm2 of LTL-Cy5 (Fig. 4c). Consistent with our results of the gentamicin protection assay, proximal tubular cell invasion for NU149 glpG-S201A was significantly lower compared to wild-type, further highlighting the importance of GlpG for bacterial pathogenicity.

a Immunofluorescence images of kidney organoids (100 K) infected with wild-type NU149 or mutant strain NU149 glpG-S201A at a ratio of 1:100. E. coli were imaged with NC100-FITC antibody detecting O or K antigen serotypes in green, cell nuclei in blue, and proximal tubule visualized with LTL-Cy5 antibody. n = 5 images per strain. b Quantification of E. coli infection within tubule markers show significantly higher bacterial invasion in the wild-type NU149 sample. Each strain was performed in biological triplicates and with an n = 5. Data are presented as mean values +/− SEM. Significant differences between data sets were determined using one-way ANOVA.
GlpG influences biofilm formation of uropathogenic E. coli
Biofilms are a major cause in recurrent bacterial infections as it protects the pathogen from the host immune response as well as administered antibiotics, leading to persistence, chronic infection, and poorer patient outcomes54,55. Previous work showed that pathogenic E. coli can form biofilms on the surface of epithelial cells, which promote their persistence in vivo56. Importantly, surface pili were shown to play a key role in driving the formation of E. coli biofilms56,57,58. Given its role in the homeostasis of type 1 pili, we reasoned that GlpG is required for the assembly of biofilms by UPEC on biotic surfaces. To test this, we infected mIMCD-3 kidney cells with NU149 wild-type or glpG-S201A isogenic strains for 6 h to allow biofilm formation. To quantify these structures, we tallied the number of bacterial aggregates larger than 100 µm2, which was previously shown to correspond to the sizes of biofilms made by pathogenic E. coli56. Interestingly, we observed a significant reduction in biofilm formation (~25-fold) by the mutant relative to the wild-type (Fig. 5a, b). This was rescued by overexpressing GlpG in the mutant strain, confirming the role of this protease in facilitating biofilm assembly, likely through its role in FimA proteostasis.

a Immunofluorescence staining shows mIMCD-3 epithelial cells in red with DAPI stained nuclei in blue, infected with different UPEC stains in green. White arrows indicate examples of biofilms. Each panel is representative of n = 45 images per group obtained from biological triplicates. Scale bar is 100 µm. b Quantification of biofilm formation by the different UPEC stains relative to the wild-type. Data are presented as mean values +/− SD. Significance determined by Kruskal–Wallis test.
Another method to validate the effect GlpG has on the formation of bacterial biofilm is safranin staining, used to quantify biofilm mass produced in UPEC on an abiotic surface. Indeed, the NU149 glpG-S201A mutant, which displays impaired pili formation, showed significant reduction in biofilm production compared to the wild-type (Fig. 6a). Interestingly, the glpG-S201A strain overexpressing active GlpG protease showed a significant increase in biofilm biomass. These results demonstrate a correlative relationship between the catalytic activity of GlpG, pili formation, and biofilm forming abilities of E. coli. Indeed, negatively stained electron micrographs of NU149 demonstrated increased amount of GlpG protease in the cell significantly affected the amount of pili on the surface.

a Quantification of safranin-stained biofilms between different strains of NU149 with and without treatment with tosyl phenylalanyl chloromethylketone (TPCK), the same time as GlpG induction or 2 h after GlpG induction (b), measured by absorbance at 500 nm. The data represents biological and technical triplicates n = 3. Data are presented as mean values +/− SEM. Significant differences between data sets were determined using one-way ANOVA. c Electron micrographs of negatively stained NU149 glpG-S201A +GlpG cells without inhibitor and when TPCK was added the same time as GlpG induction (d) or 2 h after GlpG induction (e). Each strain was performed in biological and technical triplicates n = 3.
GlpG inhibition affects the expression of surface virulence factors
To investigate whether the expression of surface virulence factors can be blocked by inhibiting E. coli GlpG rhomboid protease, we used commercially available serine protease inhibitor, tosyl phenylalanyl chloromethylketone (TPCK). We determined the inhibitory concentration of TPCK for GlpG in vitro using a peptidic FRET substrate (Mca-FATA*AFGSP-Dpn) designed by our team based on the cleavage sequence of the transmembrane domain of TatA protein from P. stuartii—a known bacterial substrate of rhomboid protease AarA that E. coli GlpG is also able to cleave59. We measured the IC50 value of TPCK for GlpG to be 296 µM (Supplementary Fig. 8), consistent with previous observations60.
To explore the effect of TPCK on virulence factor expression, bacterial adherence, and formation of biofilm, biofilm inhibition and biofilm eradication assays were performed. In parallel, electron micrographs of E. coli cells treated with TPCK were taken to observe changes in surface organelles presentation. Biofilm inhibition was assessed by adding the inhibitor at the same time as the re-addition of active GlpG to the glpG-S201A strain (NU149 glpG-S201A + GlpG) and assessing the biofilm production. Overall, we observed a decrease in biofilm formation in the presence of TPCK for NU149 wild-type, but a more profound inhibitory effect was observed in NU149 glpG-S201A +GlpG, which we show produces more biofilm than the wild-type (Fig. 6a and d). We then explored whether inhibition of GlpG can aid in biofilm eradication, or the removal of pili that have already formed. To test this, biofilms were first pre-established with bacterial growth before the addition of TPCK inhibitor. When quantified, we did not observe a significant difference in adhered biomass for wild-type NU149 treated with TPCK; however, the NU149 glpG-S201A +GlpG strain, again, demonstrated a significant decrease in biofilm mass in the presence of TPCK (Fig. 6b and e). To ensure the reduction in biofilm was not due to toxic effects of TPCK, a metabolic activity assay was performed in parallel. Using tetrazolium salt dye, 2,3,5-triphenyl tetrazolium chloride (TTC), detecting cell redox activity and viability, we did not observe the decrease in metabolism for any of the strains used, both with and without inhibitor treatment (Fig. S9), indicating that biofilm eradication was not in fact due to cell death.
These observations were confirmed with negatively stained electron micrographs of NU149 glpG-S201A +GlpG. When TPCK was added along with the inducing agent for GlpG expression, the cells had a drastic decrease in pili (Fig. 6d), whereas when TPCK was added after surface elements were already formed, we observed a fragmented pili network (Fig. 6e). Pili are known to be fragile and constantly turned over and if assembly is faulty for reasons such as GlpG inhibition, the resulting pilus rods may have structural defects and reduced stability, making them more prone to dissociation. This indicates that GlpG is important for maintaining the integrity of the pili in pathogenic bacteria, a crucial feature for retaining the biofilm.
GlpG cleaves aggregated FimA to prevent accumulation in the periplasm
To confirm the mechanism of GlpG and its contribution to the quality control of FimA, we performed an in vitro endpoint activity assay capturing GlpG-mediated cleavage of FimA aggregates. Recombinantly expressed active GlpG, either dodecyl maltoside (DDM) solubilized or reconstituted into E. coli lipid proteoliposomes (PLS) to mimic the physiological membrane, was added to the purified periplasmic fraction of the K12 MG1655 ΔglpG mutant containing aggregated FimA. The time course cleavage was visualized by immunoblotting with FimA antibody (Fig. 7a, b). Indeed, upon incubation with active GlpG protease in DDM and in PLS, an observed decrease in FimA aggregates was evident over time confirmed by densitometry analysis of the corresponding bands (Fig. 7c, d and Supplementary Fig. 10), demonstrating GlpG can cleave FimA aggregates in the lipid bilayer.

Western blot analysis of FimA aggregates cleavage by a dodecyl maltoside (DDM)-solubilized GlpG or b proteoliposome (PLS)-reconstituted GlpG. FimA aggregates were isolated from K12 substr. MG1655 glpG-KO mutant and incubated with purified recombinant GlpG at 37 °C over 6 h. The sample of periplasmic proteins incubated without GlpG at 37 °C for 6 h was used as negative control, evidenced by the no observed self-disruption of aggregates. FimA antibody was used for visualization. Quantification by densitometry analysis of aggregated FimA by c DDM-solubilized GlpG or d PLS-reconstituted GlpG represented as % FimA cleaved relative to time = 0. e The structure of FimA (green) complexed with FimC chaperone (purple) (PDB code 4DWH; Crespo et al., 2013), showing the interaction of the C-terminal end of FimA with the FimC chaperone. The C-terminal peptide is shown in red with putative GlpG cleavage sites colored yellow. f Time course of GlpG-mediated cleavage of FRET-FimA peptide using DDM-solubilized, PLS reconstituted, or inactive GlpG was incubated with peptide at 37 °C with the reaction monitored by measuring fluorescence at 490 nm. Quantification of invasion by of NU149 glpG-S201A overexpressing FimC against g mIMCD-3 cells or h T24 cells compared to wild-type, NU149 glpG-S201A, and NU149 glpG-S201A overexpressing GlpG. Each strain was performed in biological and technical triplicates n = 3. Data are presented as mean values +/− SEM. Significant differences between data sets were determined using one-way ANOVA. i Immunofluorescence staining shows mIMCD-3 epithelial cells in red with DAPI stained nuclei in blue, infected with NU149 glpG-S201A + FimC. Panel is representative of n = 45 images per group obtained from biological triplicates. Scale bar is 100 µm. j Quantification of biofilm formation by the different UPEC stains relative to wild-type. Data are presented as mean values +/− SD. Significance determined by Kruskal–Wallis test.
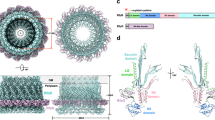
During FimA translocation through the Sec pathway, FimA has an exposed, hydrophobic C-terminal tail that is prone to aggregation in an aqueous periplasm. This C-terminal tail of FimA must immediately bind to FimC chaperone in the periplasm for its eventual transport to the outer membrane. Thus, if after SecYEG-export to periplasm, FimA is improperly folded, or when insufficient FimC chaperone molecules are available to bind and transport FimA through the periplasm, pilins will misfold and aggregate and are targeted by DegP and DegQ, the housekeeping periplasmic proteases. GlpG most likely influences this pathway by processing or regulating misfolded pilins, cleaving the C-terminus of FimA subunits and “untangling” big oligomers prior to its degradation upstream of DegP/Q-mediated degradation. To verify the cleavage site in the FimA molecule, we designed a FRET-FimA peptidic substrate (DABCYL-QARYFATGAATPGAANADATFKVQYQ-Glu(EDANS) based on the sequence of the FimA peptide identified in LC-MS/MS analysis of periplasmic proteins. The identified sequence is located on the C-terminus of FimA, residues 157 to 182, which forms the hydrophobic groove and binding site for FimC chaperone during translocation (Fig. 7e). Due to the hydrophobic nature of the region, the FRET-FimA peptide was prone to aggregation and had limited solubility, as expected. Cleavage of the peptide was demonstrated using purified GlpG in DDM and PLS with no cleavage by catalytically inactive GlpG as a control (Fig. 7f).
To confirm the role of GlpG in maintaining the fine balance between free FimA that is prone to aggregation, and FimA bound to FimC for pilin assembly, we investigated the effect of increasing the ratio of FimC relative to FimA in the NU149 glpG-S201A strain on cell invasion and biofilm formation. NU149 glpG-S201A, which we show has reduced pili and invasion of host cells, had pili (Fig. 2), as well as host invasion (Fig. 7g, h), return to wild-type levels when overexpressing FimC and. This is further supported by the partial rescue of biofilm formation upon overexpressing FimC in the glpG-S201A mutant where we observed ~75% restoration of biofilm biogenesis, suggesting that overexpression of this chaperone can prevent the aggregation of pilin subunits when GlpG is absent (Fig. 7i, j). This change in equilibrium, increasing available FimC to bind free FimA, rescues the pili-expressing, invasive phenotype. Since FimA requires FimC to reach the outer membrane, having excess chaperone available to bind and transport FimA during translocation limits the presence of unbound FimA and potential aggregation in the absence of GlpG proteostasis (Fig. 8). By contrast, when the cell has less FimC relative to FimA during type 1 pili formation, GlpG performs its role in protein homeostasis, cleaving FimA at the exposed, hydrophobic C-terminal tail. By identifying FimA as a physiological substrate of E. coli GlpG, our data supports the role of GlpG in maintaining type 1 pili quality control by cleaving FimA aggregates for subsequent degradation by other proteases such as DegP and DegQ that were identified as interactors of GlpG in whole cell proteomics. This prevents the accumulation of FimA in the periplasm and obstruction of the translocation pathway during type 1 pili assembly.

GlpG (blue) is closely associated with members of the SecYEG translocon (green) as well as other resident proteases in the periplasm, such as DegQ and DegP (grey), to readily prevent and clear FimA (yellow) aggregates. When FimC chaperone (purple) is not available to bind FimA, GlpG cleaves FimA at its hydrophobic C-terminal tail to facilitate its clearance and prevent the aggregation of FimA in the periplasm. The remainder of the FimA molecule is degraded by housekeeping proteases that cleave misfolded proteins in the periplasm, such as DegP and DegQ. Loss of GlpG catalytic activity causes the accumulation of FimA aggregates and impaired type 1 pili formation, which in turn affects host colonization of E. coli. Created with BioRender.com. https://BioRender.com/xstqntw.
Discussion
Our study provides mechanistic insight into a previously observed phenotype where deletion of the glpG gene significantly impaired ExPEC persistence in the murine intestine38. ExPEC use a variety of structural (pili, curli, and flagella) and secreted (toxins and iron-acquisition systems) virulence factors to disseminate and persist in sterile niches such as the urinary tract, bladder, and kidney. In type 1 pili, a single pilus contains thousands of repeating FimA monomers, which assemble non-covalently, extending outwards from the cell. Being a hydrophobic protein and physiologically high in abundance necessitates tightly regulated translocation and proteostasis mechanisms to ensure that any misfolded FimA is cleared from the cell.
Rhomboid proteases are known to cleave transmembrane substrates35,41. In this study, we identified FimA as the first physiological substrate for E. coli GlpG, shifting the paradigm for intramembrane proteases beyond sole recognition of transmembrane substrates and opening the door to explore more novel soluble substrates for this superfamily of proteases. Moreover, we identified several proteases associated with GlpG, including DegP and DegQ, which are also involved in protein quality control of the periplasmic space in E. coli by removing misfolded and aggregated proteins39, though more evidence is needed to confirm if DegQ or DegP physically interact with GlpG when cleaving FimA.
We showed that GlpG cleavage site is located on the C-terminus of the FimA molecule, a region critical for proteostasis of FimA subunits. As individual unfolded FimA is transported across the inner membrane via the SecYEG translocon, it is immediately bound by chaperone FimC. Upon binding, the chaperone donates its β-strand to interact with a hydrophobic groove located between N- and C-terminal β-strands within the FimA molecule. This event accelerates the folding of FimA and prevents premature assembly of the subunits in periplasm61. The same hydrophobic groove interacts with the incoming FimA subunit during pili assembly on the outer membrane, where the donor β-strand of FimC is replaced by β-strand of the next FimA molecule62. However, if FimA folding is unsuccessful upon FimA-FimC association, or if two orphan FimA molecules prematurely form a complex in periplasmic space, this can lead to accumulation of non-functional proteins. We propose that the cleavage of the C-terminal peptide in FimA molecule by GlpG disrupts the binding site for both FimC and the donor FimA, making FimA accessible for further degradation. Thus, GlpG cleaves membrane-proximal misfolded FimA clusters, reducing aggregation or converting them into smaller fragments. These fragments or unassembled pilins may then be then recognized and degraded by DegP/DegQ in the periplasm. Our also data supports physiological co-localization of GlpG, and these proteases required for protein trafficking. Together, they may form a proteostasis network that surveils and clears misassembled FimA, preventing toxic accumulation and allowing pilus biogenesis to proceed correctly. Indeed, this is in line with previous research on eukaryotic rhomboid protease Der1 and its role as a key regulator in endoplasmic reticulum (ER) associated protein degradation (ERAD) as a member of a ubiquitin ligase complex that degrades misfolded proteins in the ER upon its retrotranslocation into the cytosol63.
Interestingly, this is the first evidence of DegP and DegQ involvement in type 1 pili proteostasis. DegP has been identified as the protease that maintains the homeostasis of P pilus by cleaving pilin, PapA subunits, whereas degP mutant strains were found to accumulate higher levels of pilin subunits in the periplasm64,65.
Type 1 pili was observed to be affected by the absence of GlpG activity, resulting in reduced bacterial invasion and biofilm production. Type 1 pili is known to affect bacterial motility and locating surface contacts, a crucial step in the early stages of biofilm formation. Biofilms are major contributors to bacterial persistence as they provide bacterial communities protection from environmental stress and antibiotics, allowing increased potential for recurrence. Previous studies have also demonstrated that rhomboid proteases affect biofilm formation in Mycobacterium smegmatis and H. volcanii, showing less biofilm produced when one of the rhomboid genes was deleted36,66. Similarly, we show reduced biofilm formation on biotic and abiotic surfaces when glpG is inactivated in E. coli, in agreement with the role of GlpG in maintaining pilin homeostasis. Interestingly, biofilm formation was significantly increased when GlpG protease was expressed beyond wild-type levels. Importantly, overexpressing FimC rescued biofilm formation in the absence of a functional GlpG. This result further supports our model in which GlpG plays a key role in eliminating misfolded pilin subunits.
As the threat of antimicrobial resistant pathogens becomes increasingly more prevalent, surpassing multiple classes of antibiotics, new strategies that can help treat such bacterial infections are crucial. The gene encoding bacterial rhomboid protease, glpG, is evolutionarily widespread among prokaryotes and structurally conserved within the active site housing the serine-histidine catalytic dyad embedded in the plane of the membrane. Our results clearly show rhomboid protease activity is essential for generating virulence factor type 1 pili in UPEC and that the expression of type 1 pili can be blocked by inhibition of the GlpG protease. Currently, new antibacterials are in preclinical and clinical trials, many of which target bacterial proteins to disrupt the function of virulence factors. GlpG therefore represents a strong target in the development of antivirulents to combat AMR, given its critical role in type 1 pili.
-
Methods
-
LC-MS/MS proteomics on endogenously FLAG-tagged GlpG purified from whole cells
-
Periplasmic protein isolation
-
GlpG Purification
-
Reconstitution of GlpG in proteoliposomes
-
LC-MS/MS proteomics on periplasmic proteins
-
LC-MS/MS in-gel digestion proteomics on periplasmic proteins
-
FimA cleavage time point assay
-
Western blot
-
In vitro cleavage assay
-
Construction of mutant strains
-
Negatively stained transmission electron microscopy of E. coli
-
Hemagglutination assay
-
RT-qPCR of glpG
-
Mammalian cell culture conditions
-
Gentamicin protection assay
-
Infecting kidney organoids with E. coli
-
Immunofluorescence and confocal microscopy of kidney organoids
-
Quantification of bacteria infected proximal tubules
-
GlpG inhibition assay
-
Biofilm formation assay on biotic surfaces
-
Biofilm formation assay on abiotic surfaces
-
Biofilm inhibition assay
-
Biofilm eradication assay
-
Metabolic activity assay
-
Statistics & Reproducibility
-
Methods
LC-MS/MS proteomics on endogenously FLAG-tagged GlpG purified from whole cells
E. coli strain DY330 harboring a triple FLAG-tag (SPA-tag) inserted at the N-terminus of the genomic glpG gene was generated by targeted homologous recombination into the E. coli genome. For purification of endogenously expressed SPA-tagged GlpG, E. coli was grown in LB media with kanamycin and lysed in buffer containing 50 mM Tris, pH 8.0, 200 mM NaCl, 20% glycerol, and detergent, either 1% Triton ×-100, DDM or C12E8. No significant differences in pulldown data were observed therefore the three detergents samples were treated as replicates. Interactions in the presence of Triton ×-100 are presented for clarity. Cleared fractions containing solubilized GlpG bait and associated proteins were purified on anti-FLAG and calmodulin columns, following the SPA method67, except that after the first three washing steps, calmodulin wash buffer (30 mM Tris-HCl pH 7.9, 150 mM NaCl, 2 mM CaCl2, 0.1–0.5 mM Tris (2-carboxyethyl) phosphine (TCEP)) with 0.1% Triton was used, with the final two washes performed without detergent. Negative controls were performed in wild-type DY330 E. coli with anti-FLAG beads68. Purified protein preparation was resuspended in 25 μL of 8 M urea (1.5 M final), 20 mM HEPES (pH 8.0), reduced with 5 mM TCEP for 45 min at room temperature and alkylated with 15 mM iodoacetamide for 60 min in the dark. Modified trypsin (0.3 mg, Promega) was added, and digestion performed overnight at room temperature with gentle shaking. Peptide mixtures were acidified with formic acid and desalted using C18 TopTips (Glygen). Resulting peptides were loaded via autosampler onto a 75-μm inner diameter fused silica column packed with ~10 cm C18 resin (3 μm, 100 Å, Phenomenex) coupled to a Proxeon EASY nLC 1000 (Proxeon) and Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). Peptides were eluted by electrospray ionization with a 100 min water/acetonitrile gradient at ~0.30 µl min-1. Precursor ions [350–2000 m/z] underwent data-dependent collision-induced dissociation, with one full mass scan followed by 15 tandem mass scans of intense precursor ions, with dynamic exclusion.
The Orbitrap Elite mass spectrometer was operated with Thermo XCalibur software. All RAW files were converted to mzXML using ReAdW-4.3.1 and submitted for database searching using SEQUEST -PVM v.27 (rev. 9)69 under standard workflow conditions with a non-redundant E. coli proteome sequence FASTA file from Ecocyc. Search parameters were set to allow for dynamic modification of methionine oxidation and fixed modification of cysteine carbamidomethylation using precursor ion tolerance of 20 ppm. A stringent false discovery rate of 1% (P < 0.01) was used to filter candidate peptide and protein identifications. Precursor mass tolerance was set to 3 Da (daughter mass ion tolerance set to the default of 0), while enabling partial tryptic enzyme and single site missed cleavages. The STATQUEST filtering algorithm70 was then applied to all putative SEQUEST search results to assign statistical confidence. High confidence (>90% likelihood) spectral counts from SEQUEST/STATQUEST were used to calculate Normalized Spectral Abundance Factor (NSAF) score71. To be more conservative, the proteins were then filtered on median NSAF score or STATQUEST probability of 99%. Proteins localized in the periplasm and inner membrane, belonging to the COG categories O and U72 were visualized as Cytoscape network73. All raw data is deposited and publicly available in the MassIVE repository (MSV000097249) and Supplementary Data. 1.
Periplasmic protein isolation
Periplasmic proteins were isolated using cold osmotic chock and a Tris-Sucrose -EDTA buffer45. K12 substr. MG1655 ΔglpG mutant was grown to OD600 1.0 and centrifuged at 5000 × g for 10 min at 4 °C. Cells were resuspended in a Tris-Sucrose-EDTA buffer (200 mM Tris pH 8.0, 500 mM sucrose, 1 mM EDTA, 1 mM PMSF) with a cOmplete EDTA-free protease inhibitor cocktail tablet (Roche, Cat. # 11836170001) and incubated on ice for 30 min. The sample was centrifuged 20,000 × g for 30 min at 4 °C where the final supernatant contains the periplasmic proteins.
GlpG purification
Recombinantly expressed E. coli GlpG and inactive mutant GlpG Ser201Ala were purified following standard membrane protein purification protocols74. Briefly, E. coli GlpG-TEV-6 × His or GlpG-S201A-TEV-6 × His in a pBAD vector was expressed in TOP10 cells with 0.002% arabinose at OD600 0.8 and collected by centrifugation at 5000 × g for 15 min at 4 °C. The pellet was resuspended in lysis buffer containing 50 mM Tris, pH 8.0, 200 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktail tablet (Roche Cat. #), and lysed using high-pressure with an Emulsiflex. The proteins were separated from cellular debris at 13,000 × g for 30 min at 4 °C and the resulting supernatant was centrifuged at 40,000 × g for 2 h at 4 °C to isolate the membrane fraction. Membranes were resolubilized in a lysis buffer containing 1% Dodecyl-β-D-maltoside, incubated with Ni-NTA resin and eluted with increasing concentrations of imidazole in the lysis buffer. Fractions containing GlpG-TEV-6 × His were further incubated with TEV protease and incubated in Ni-NTA resin again. The flow-through contains full-length GlpG while TEV-6 × His remains bound to the resin. The resulting protein fraction was concentrated with a 30 MWCO Amicon spin concentrator (Millipore, Cat. #UFC9030) and injected into a HiLoad 16/600 Superdex 200 pg column (Cytiva, Cat. #28-9893-33).
Reconstitution of GlpG into proteoliposomes
400 μg E. coli polar lipids (Avanti) suspended in chloroform were dried under nitrogen stream in a glass tube, followed by overnight incubation in a desiccator to yield a thin film of lipid. 50 μl of water and DDM detergent was added to the lipid film for resuspension at room temperature for 10 min, followed by the addition of purified GlpG protease (400 μg) in 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 20% glycerol, 0.1% DDM, to yield the final weight ratio of 1:1:2, GlpG:lipid:detergent. The detergent was slowly removed by the addition of SM2 Biobeads (Bio-Rad) while stirring on ice for 6 h to allow for the generation of proteoliposomes (PL); the process was controlled by the regimen of Biobead addition. 50%:20% sucrose density gradient ultracentrifugation was used to purify PL. The concentration of reconstituted GlpG was assessed by BCA assay (Pierce BCA Protein Assay Kit, Thermo Fisher Scientific) and the purity was confirmed with Coomassie staining after SDS-PAGE.
LC-MS/MS proteomics on periplasmic proteins
Periplasmic proteins isolated from K12 substr. MG1655 ΔglpG mutant were incubated with 5 µM of active or inactive GlpG and separated based on molecular weight using 4–20% Mini-PROTEAN TGX Precast Gels (Bio-Rad, Cat. # 4561094) at either (1) 100 V for 5 min, or a migration of 1.5 cm or (2) 150 V for 30 min where proteins were analyzed separately based on molecular weight range. Gels were fixed for 20 min (50% ethanol, 2% phosphoric acid), washed for 20 min in MilliQ H2O and stained overnight with a blue silver Coomasie stain (10% phosphoric acid, 20% ethanol, 750 mM ammonium sulfate, 13 mM Coomassie blue G-250) and washed for 20 min in MilliQ H2O.
The band or bands were excised from the gel and further cut into 1 mm pieces. The gel pieces were transferred to a round bottom 96-well plate for destaining (50 mM ammonium bicarbonate, 50% acetonitrile). The plate was incubated for 10 min at 37 °C. The solution was removed from the wells and the destaining procedure was repeated 4 times. The solution was removed and replaced by acetonitrile for dehydration. The samples were incubated again at 37 °C for 10 min. Dehydration was repeated until the gel pieces became white (3 times). The samples were dried at 37 °C for 10 min, rehydrated in a reducing solution (100 mM ammonium bicarbonate and 10 mM β-mercaptoethanol), and incubated for 30 min at 37 °C. The reducing solution was removed. The samples were alkylated with iodoacetamide (10 mg/mL of iodoacetamide and 100 mM ammonium bicarbonate) and incubated for 30 min at 37 °C. The gel bands were subsequently washed with 100 mM ammonium bicarbonate and incubated for 10 min at 37 °C (repeated twice). The samples were then dehydrated in acetonitrile for 10 min at 37 °C (repeated twice). Acetonitrile was removed and the samples were dried at 37 °C for 10 min. The gel bands were then trypsinized (0.3 μg of mass-spec grade trypsin per well, Promega) overnight at room temperature. The solutions containing tryptic peptides were transferred to a V-bottom 96-well plate. Tryptic peptides were further extracted from the gel with 2% acetonitrile and 1% formic acid followed by incubation at 37 °C for 1 h. The extraction was repeated using 50% acetonitrile and 0.5% formic acid followed by incubation at 37 °C for 1 h. The samples were dried with vacuum and resuspended in 20 mL of 0.1% formic acid for analysis on liquid chromatography tandem mass spectrometry (LC-MS/MS). Peptides were separated using a nanoflow-HPLC (Thermo Fisher Scientific EASY-nLC 1200 or 1000 System) coupled to a Orbitrap Fusion Lumos or QExactive mass spectrometer (Thermo Fisher Scientific).
Raw data were analyzed using Proteome Discoverer (v2.4.1.15, Lumos) or Protein Prospector (v5.22.1, QExactive) software against the E. coli proteome (https://www.uniprot.org). Search parameters include a precursor and a fragment mass tolerance of 15 ppm and 0.8 Da (Lumos) or 10 ppm and 0.01 Da (QExactive), respectively. Searches included up to two missed trypsin cleavages, and charge states of 2, 3, and 4. Static modification was set to carbamidomethylation of cysteine and dynamic modifications include oxidation of methonines, deamidated glutamine and asparagine, and acetylated protein N-termini. 895 proteins were quantified with high reproducibility (~95%) between biological replicates n = 2. Protein abundance changes between periplasmic proteins incubated with active and inactive GlpG with a significant p-value below 0.05 (−log10 p-value > 1.30) and a fold change (FC) greater than two (log2 FC > 1), are presented. Identified peptides were searched against a random decoy protein database to evaluate the false-positive rate (set to 1%). All raw data is deposited and publicly available in the MassIVE repository (MSV000097236) and Supplementary Data 2 and 3. Gene ontology was performed using https://metascape.org against the E. coli proteome using: GO biological processes, KEGG pathway, Reactome Gene Sets, CORUM, TRRUST, PaGenBase, and Wiki Pathways.
Hemagglutination assay
E. coli strains were grown statically at 37 °C until OD600 = 1.0; for strains harboring an inducible plasmid, cells were grown till OD600 = 0.6, the gene was induced and grown for another 2 h. The OD600 then was adjusted to 1.0 for comparison between different strains. Guinea pig erythrocytes (Innovative Research, Inc., USA) were washed with PBS and diluted to the final concentration of 3% (v/v). In a 96-well round bottom plate, 50 µL of bacterial suspension was mixed with 50 µL of 3% red blood cell (RBC) suspension. The plate was incubated at room temperature for 60 min without agitation. Absence of type 1 pili is indicated by a “button” formed by the settled RBCs at the round bottom well, whereas presence of type 1 pili binds the RBCs forming a diffuse mixture. For MSHA, 2% mannose was added to bacterial suspension to selected wells before adding RBC and incubated for 15 min to inhibit type 1 pili-mediated hemagglutination.
FimA cleavage time point assay
Periplasmic proteins were dialyzed in GlpG activity buffer containing 50 mM MES, pH 6.0, 150 mM NaCl, 20% glycerol, 0.1% DDM, as determined by our group75. The periplasmic proteins were incubated with 5 µM of E. coli GlpG or the catalytically inactive mutant (Ser201Ala) at 37 °C for 6 h, taking fractions at various time points for western blot analysis.
Western blot
To measure the level of FimA over time, fractions of FimA incubated with GlpG were analyzed by standard western blot methods and probed with rabbit anti-FimA antibody (dilution = 1:2000; Biomatik, CAC11912) and HRP conjugated goat anti-rabbit antibody (dilution = 1:5000; Thermo Fisher Scientific, Cat. #31460). Amersham Western Blot Detection Reagent (Cytiva, Cat. #45-002-401) was used and the blot was imaged on the Li-COR using the ImageJ program.
In vitro cleavage assay
FimA FRET peptide, DABCYL-QARYFATGAATPGAANADATFKVQYQ-Glu(EDANS) (BioMatik) was incubated either GlpG in DDM or in PLS. For GlpG in DDM, activity buffer consisting of 50 mM MES, pH 6.0, 150 mM NaCl, 20% glycerol, 0.1% DDM. The reaction was started with 0.5 µM of purified GlpG solubilized in DDM and was monitored for 3 h at 37 °C. The activity assay with GlpG reconstituted into PLS was performed the same way as for the protease in DDM with the only difference being the activity buffer, where DDM was omitted (50 mM MES pH 6.0, 150 mM NaCl, 20% glycerol). Prism 9.0, GraphPad Software, was used to determine cleavage of the peptide.
Construction of mutant strains
Ser201Ala point mutations in the glpG gene were generated in E. coli K12 substr. MG1655 and NU149, denoted as glpG-S201A, by method of CRISPR/Cas9 gene-editing76. Briefly, electrocompetent cells harboring pCas (Addgene: #62225) were co-transformed with pTargetGlpG and 500 ng of donor template DNA containing the glpG gene with a S201A mutation. Cells were recovered at 30 °C for 2 h and spread on LB agar containing kanamycin (50 µg/mL) and spectinomycin (50 µg/mL) and incubated overnight at 30 °C. Transformants were identified by colony PCR and Sanger sequencing.
Negatively stained transmission electron microscopy of E. coli
Different strains of E. coli were grown statically to preserve pili integrity in LB for 48 h at 37 °C. Strains harboring a GlpG plasmid were induced after the first 8 hof growth. 7 µL of cell culture were gently added to glow-discharged carbon-coated copper grids (Electron Microscopy Sciences, Cat. # CF200-Cu-50) and left at room temperature for 5 min. Excess sample was wicked away using Wattman paper, washed with nuclease-free water and stained with 2% (w/v) uranyl acetate. Grids were imaged on a Hitachi H-7650 Transmission Electron Microscope.
RT-qPCR of glpG
E. coli strains were grown in LB media, TB media, or M9 minimal media to an OD600 of 1.0 and centrifuged at 5000 × g. RNA was extracted using TRIzol™ Max Bacterial RNA Isolation kit (Invitrogen, Cat. #16096020). cDNA was amplified using iScript Reverse Transcription Supermix (Bio-RAD, Cat. #1708840) and RT-qPCR was performed with PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific, Cat. #A25741) and analyzed for mRNA gene expression using the QuantStudio5 system. Both glpG and housekeeping gene rrsB were measured with glpG mRNA expression was measured as ΔΔCt.
Mammalian cell culture conditions
Mouse inner medullary collecting duct (mIMCD-3; ATCC, Cat. # CRL-2123) cells were grown in DMEM:F12 medium (Gibco, #Cat. #11320033) containing 10% fetal bovine serum (Sigma-Aldrich, Cat. #12103 C) at 37 °C, 5% CO2. Human bladder (T24; ATCC, Cat. # HTB-4) cells were grown in McCoy’s medium (Sigma-Aldrich, Cat. #30-2007) containing 10% fetal bovine serum at 37 °C, 5% CO2.
Gentamicin protection assay
Various strains of E. coli were grown to an OD600 of 1.0 and added to a confluent layer of mouse inner medullary collecting duct (mIMCD-3; ATCC, Cat. # CRL-2123) or human bladder (T24; ATCC, Cat. # HTB-4) cell line to facilitate infection for 4 h at 37 °C. Cells were washed with sterile PBS and grown in medium containing 100 μg/mL of gentamicin for 2 h to kill all remaining extracellular bacteria, leaving invasive bacteria unaffected. Invasive bacteria were collected by lysing mammalian cells with 2% (v:v) TritonX-100. Serial dilutions of lysate were plated on LB agar plates and grown for 18 h at 37 °C to quantify bacterial invasion by manual counting of colony-forming units.
Generation of kidney organoids
Kidney organoids were differentiated using the Takasato protocol with minor modifications as previously described by the Chun group77,78. Briefly, induced pluripotent stem cells were grown in 6 well plates coated with Matrigel matrix (Corning, Cat. #354277). Cells were treated with 8 μM CHIR99021 (R&D Systems, Cat. #4423) for 4 days followed by recombinant human FGF-9 (200 ng/mL; R&D Systems, Cat. #273-F9-025) and heparin (1 μg/mL, Millipore Sigma, Cat. #H4784) for an additional 3 days in APEL2 medium (Stem Cell Technologies, Cat. #05275) supplemented with 5% PFHM-II (Thermo Fisher, Cat. #12040077). On day 7, cells were dissociated using trypsin-EDTA (0.05%) for 2 min and 100,000 cells were transferred onto a six-well transwell plate (Corning, Cat. # 07-200-170) with nine pellets per well without an aggregation step. Cell clusters were incubated with a pulse of 5 μM CHIR99021 in APEL2 + PFHM-II medium for 1 h at 37 °C. After 1 h, the medium was changed to APEL2 + PFHM supplemented with 200 ng/mL FGF-9 and 1 μg/mL heparin for an additional 5 days and then were maintained in APEL2 + PFHM-II medium for 14 days with medium change every other day.
Infecting kidney organoids with E. coli
UPEC E. coli NU149 wild-type and NU149 glpG-S201A were grown overnight in 5 ml LB at 37 °C, 225 RPM. 300 μl of overnight culture for each strain were transferred to 2.7 ml of LB and grown for 4 h at 37 °C, 225 RPM. Cultures were centrifuged at 2000 × g for 2 min with the supernatant aspirated and discarded. Each pellet of bacteria was resuspended in 1 mL APEL2. On day 26 of differentiation, kidney organoids were infected in triplicates with 1:10 and 1:100 concentrations of E. coli strain or vehicle control and infected at 37 °C, 5% CO2 for 6 h with agitation every 1 h. After 6 h, supernatant was discarded, and kidney organoids were washed 2 times with PBS for 5 min with rocking for each wash. To inactivate E. coli, kidney organoids were treated with an APEL2 solution containing 2× Pen/Strep (200 units/mL penicillin and 200 μg/mL streptomycin) (Gibco; Cat. #15140122) at 37 °C, 5% CO2 for 2 h. The organoids were then washed 2 times with sterile PBS for 5 min and fixed with 2% paraformaldehyde for 20 min at 4 °C. After two washes with PBS, kidney organoids were stored at 4 °C until processed for immunofluorescence and imaging.
Immunofluorescence and confocal microscopy of kidney organoids
Kidney organoids were processed prior to immunofluorescence microscopy78. In brief, kidney organoids were blocked in a blocking buffer (10% donkey serum and 0.05% Triton-X in PBS) for 2 h at room temperature. Primary antibodies to E. coli (anti-E. coli-FITC; Novus Cat. #NB100 64448, 1:25), anti-NPHS1 (R&D Systems, Cat. #AF4269, 1:200), Lotus tetragonolobus lectin-Cy5 (GlycoMatrix Cat. # 21761117-1; 1:200) were incubated at 4 °C overnight. Following the overnight incubation, organoids were washed 3 times with wash buffer (0.3% Triton ×-100 in PBS) for 10 min each wash followed by incubation in wash buffer with fluorescent conjugated secondary antibodies that include Alexa Fluor donkey anti-sheep 594 (Thermo Fisher Scientific, 1:250) for 2 h at room temperature. After 3 washes with PBS and 10 min labeling with 5 μg/ml Hoescht 33342 (Thermo Fisher Scientific, Cat. #H3570), confocal images were acquired with a Zeiss LSM 880 confocal microscope.
Quantification of bacteria infected proximal tubules
LTL-Cy5 labeled proximal tubules were identified and isolated for quantification of E. coli infected proximal tubules of the kidney organoids. Confocal images of E. coli-FITC positive structures within the LTL-Cy5 positive regions were analyzed using ImageJ as were described previously for SARS-CoV-2 infected kidney organoids79.
GlpG inhibition assay
Inhibition was calculated by using 0.2 µM of purified GlpG that was incubated with tosyl phenylalanyl chloromethylketone (TPCK) in a concentration range of 0.1 µM to 5 mM at 37 °C for 60 min in activity buffer consisting of 50 mM MES, pH 6.0, 150 mM NaCl, 20% glycerol, 0.1% DDM. The reaction was started with 15 µM of modified TatA-FRET substrate (KM for modified TatA-FRET is 10 µM; Mca-FATA*AFGSP-Dpn, Biomatik, 98% purity) and was monitored for 3 h at 37 °C. Prism 9.0, GraphPad Software, was used to determine the IC50 value.
Biofilm formation assay on biotic surface
mIMCD-3 cells were grown in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12 1:1, Gibco). Cells were seeded on coverslips pre-coated with poly-L-lysine and grown to 90% confluency. UPEC NU149 strains were grown overnight at 37 °C in lysogeny broth (LB). The following day, cells were subcultured in LB at 37 °C until the OD600 reached 0.2. Protein expression was induced by adding 0.002% arabinose or 1 mM IPTG for 2 h. mIMCD-3 cells were infected with NU149 strains at a multiplicity of infection of 1:25 for 1.5 h. Non-adherent bacteria were washed off with 1× PBS twice, then cells were incubated with fresh media for an additional 4 h. At the end of the infection period, cells were washed twice with 1× PBS, then fixed with 3% paraformaldehyde (Sigma-Aldrich) for 30 min at room temperature. Coverslips were blocked overnight with 3% goat serum (Invitrogen) and 1% bovine serum albumin in PBS (BioShop) at 4 °C. To stain the cells and bacteria, samples were incubated with rabbit anti-O-Ag conjugated to FITC (1:50 dilution, Novus Biologicals) and phalloidin Alexa Fluor 568 (1:40 dilution, Invitrogen, Cat. #A12380) for 1 h at room temperature. Coverslips were washed three times in 1× PBST, then stained with 300 nM DAPI (Invitrogen, Cat. #D1306) for 5 min at room temperature and washed three times with 1× PBST prior to mounting on glass slides. All images were captured with BioTek Cytation 5 Imager using a 20× objective. Samples were imaged blindly and processed with BioTek Gen5 software for biofilm quantification. Acquired images were preprocessed then analyzed to quantify aggregates with surface area of 100 µm2 or more, shown previously to represent biofilms formed by pathogenic E. coli56. Images were obtained from three independent experiments.
Biofilm formation assay on abiotic surface
E. coli NU149 wild-type, E. coli NU149 glpG-S201A strain and E. coli NU149 glpG-S201A + GlpG, the inactive glpG strain transformed with pBad plasmid bearing active GlpG protease, were streaked on LB agar with the appropriate antibiotic. Bacteria cultures were grown in LB medium from colonies and grown for 16–18 h at 37 °C with shaking. Overnight cultures were diluted to OD600 of 0.01 where 200 µl of bacterial sample was added per well on a 96-well plate. LB media alone was always included as a negative control of contamination. Bacterial cultures were grown at 37 °C under static conditions. After 3 h, arabinose was added to the wells with NU149 glpG-S201A + GlpG strain to the final concentration of 0.02%. After 24 h, the optical density at 600 nm was recorded on a BioTeK Cytation5 microplate reader to quantify the overall bacterial growth in each well. Media with planktonic cells was discarded and wells were rinsed three times with PBS. Once excess PBS was removed, adhered biomass was stained by adding 230 µl of 0.1% Safranin solution to each well and placing the plate on an orbital shaker for 30 min at room temperature (Ommen et al., 2017). The stain was discarded, the wells were rinsed with PBS three times again and the excess of the buffer was removed. Safranin dye bound to the adhered biofilm was solubilized in 230 µl of 30% acetic acid per well. The absorbance at 595 nm was recorded to evaluate the amount of biomass adhered.
Biofilm inhibition assay
TPCK inhibitor was added at the same time as a freshly prepared bacterial suspension with OD600 of 0.01 at the final concentration of 400 µM and then incubating for a certain time followed by quantification of biofilm biomass by Safranin the same as described above.
Biofilm eradication assay
A requirement is pre-establishing consistent biofilms in the well prior to inhibition; thus biofilm was allowed to be formed as described above. After incubation for 24 h the adhered biomass was rinsed with fresh LB media, treated with TPCK (400 µM final concentration), incubated for another 24 h and quantified with Safranin dye.
Metabolic activity assay
The 96-well plate for metabolic activity assay was set up the same way as the biofilm eradication assay plate above. After pre-establishing biofilms, washed with fresh LB media and treated with TPCK inhibitor, TTC dye (TTC, 0.05% final concentration) was added to the wells and the plate was incubated for 24 h under static conditions at 37 °C. The wells were rinsed three times with PBS, and 230 µl of methanol was added to each well followed by quantification by reading the absorbance at 500 nm.
Statistics and reproducibility
All statistical analyses for in vitro experiments were performed using the one-way ANOVA test except for the analysis biotic biofilm formation, which was performed with the Kruskal–Wallis test, using GraphPad v10.6.1 for Windows. Figures 1b, 3a, and 8 were created using BioRender.com. No statistical method was used to predetermine sample size.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The glpG-FLAG pulldown proteomic data generated in this study are provided in the Supplementary Data 1 and deposited in the MassIVE database under accession code MSV000097249 [https://doi.org/10.25345/C54B2XH58]. The proteomic data on in-gel digestion of periplasmic proteins in this study are provided in Supplementary Data 2 and 3 as well as deposited in the MassIVE database under accession code MSV000097236 [https://doi.org/10.25345/C5T14V221]. Source data are provided with this paper.
References
Nataro, J. P. & Kaper, J. B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11, 142–201 (1998).
Tenaillon, O., Skurnik, D., Picard, B. & Denamur, E. The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 8, 207–217 (2010).
van Elsas, J. D., Semenov, A. V., Costa, R. & Trevors, J. T. Survival of Escherichia coli in the environment: fundamental and public health aspects. ISME J. 5, 173–183 (2010).
Croxen, M. A. et al. Recent advances in understanding enteric pathogenic Escherichia coli. Clin. Microbiol. Rev. 26, 822–880 (2013).
Kaper, J. B., Nataro, J. P. & Mobley, H. L. T. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140 (2004).
Vila, J. et al. Escherichia coli: an old friend with new tidings. FEMS Microbiol. Rev. 40, 437–463 (2016).
Croxen, M. A. & Finlay, B. B. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 8, 26–38 (2010).
Pakbin, B., Brück, W. M. & Rossen, J. W. A. Virulence factors of enteric pathogenic Escherichia coli: a review. Int. J. Mol. Sci. 22, 9922 (2021).
Sarowska, J. et al. Virulence factors, prevalence and potential transmission of extraintestinal pathogenic Escherichia coli isolated from different sources: recent reports. Gut Pathog. 11, 10 (2019).
Marrs, C. F., Zhang, L. & Foxman, B. Escherichia coli mediated urinary tract infections: are there distinct uropathogenic E. coli (UPEC) pathotypes?. FEMS Microbiol. Lett. 252, 183–190 (2005).
Subashchandrabose, S. & Mobley, H. L. T. Virulence and fitness determinants of uropathogenic Escherichia coli. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.uti-0015-2012 (2015).
Terlizzi, M. E., Gribaudo, G. & Maffei, M. E. UroPathogenic Escherichia coli (UPEC) infections: virulence factors, bladder responses, antibiotic, and non-antibiotic antimicrobial strategies. Front. Microbiol. 8, 1566 (2017).
Rozwadowski, M. & Gawel, D. Molecular factors and mechanisms driving multidrug resistance in uropathogenic Escherichia coli—An update. Genes 13, 1397 (2022).
Salvatore, S. et al. Urinary tract infections in women. Eur. J. Obstet. Gynecol. Reprod. Biol. 156, 131–136 (2011).
Yang, B. & Larson, T. J. Multiple promoters are responsible for transcription of the glpEGR operon of Escherichia coli K-12. Biochim. Biophys. Acta (BBA) - Gene Struct. Expr. 1396, 114–126 (1998).
Darby, E. M. et al. Molecular mechanisms of antibiotic resistance revisited. Nat. Rev. Microbiol. 21, 280–295 (2023).
Flores-Mireles, A. L., Walker, J. N., Caparon, M. & Hultgren, S. J. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 13, 269–284 (2015).
Gupta, K. et al. International clinical practice guidelines for the treatment of acute uncomplicated cystitis and pyelonephritis in women: a 2010 update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin. Infect. Dis. 52, 103–120 (2011).
Mazzariol, A., Bazaj, A. & Cornaglia, G. Multi-drug-resistant gram-negative bacteria causing urinary tract infections: a review. J. Chemother. 29, 2–9 (2017).
Murray, C. J. L. et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
Costerton, J. W., Stewart, P. S. & Greenberg, E. P. Bacterial biofilms: a common cause of persistent infections. Science 284, 1318–1322 (1999).
Belas, R. Biofilms, flagella, and mechanosensing of surfaces by bacteria. Trends Microbiol. 22, 517–527 (2014).
Pratt, L. A. & Kolter, R. Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemotaxis and type I pili. Mol. Microbiol. 30, 285–293 (1998).
Tabibian, J. H. et al. Uropathogens and host characteristics. J. Clin. Microbiol. 46, 3980–3986 (2008).
Ito, A., Taniuchi, A., May, T., Kawata, K. & Okabe, S. Increased antibiotic resistance of Escherichia coli in mature biofilms. Appl. Environ. Microbiol. 75, 4093–4100 (2009).
Mah, T.-F. Biofilm-specific antibiotic resistance. Future Microbiol. 7, 1061–1072 (2012).
Sharma, G. et al. Escherichia coli biofilm: development and therapeutic strategies. J. Appl. Microbiol. 121, 309–319 (2016).
Düsterhöft, S., Künzel, U. & Freeman, M. Rhomboid proteases in human disease: mechanisms and future prospects. Biochim. Biophys. Acta (BBA) - Mol. Cell Res. 1864, 2200–2209 (2017).
Freeman, M. Rhomboid proteases and their biological functions. Annu. Rev. Genet. 42, 191–210 (2008).
Lysyk, L., Brassard, R., Touret, N. & Lemieux, M. J. PARL protease: a glimpse at intramembrane proteolysis in the inner mitochondrial membrane. J. Mol. Biol. 432, 5052–5062 (2020).
Paschkowsky, S., Hamzé, M., Oestereich, F. & Munter, L. M. Alternative processing of the amyloid precursor protein family by rhomboid protease RHBDL4. J. Biol. Chem. 291, 21903–21912 (2016).
Wunderle, L. et al. Rhomboid intramembrane protease RHBDL4 triggers ER-export and non-canonical secretion of membrane-anchored TGFα. Sci. Rep. 6, 27342 (2016).
Stevenson, L. G. et al. Rhomboid protease AarA mediates quorum-sensing in Providencia stuartii by activating TatA of the twin-arginine translocase. Proc. Natl. Acad. Sci. USA 104, 1003–1008 (2007).
Began, J. et al. Rhomboid intramembrane protease YqgP licenses bacterial membrane protein quality control as adaptor of FtsH AAA protease. EMBO J. 39, e102935 (2020).
Liu, G. et al. Bacterial rhomboid proteases mediate quality control of orphan membrane proteins. EMBO J. 39, e102922 (2020).
Costa, M. L. et al. Rhomboid proteases: key players at the cell surface within haloarchaea. Front. Microbiol. 16, 1547649 (2025).
Koonin, E. V. et al. The rhomboids: a nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol. 4, R19 (2003).
Russell, C. W., Richards, A. C., Chang, A. S. & Mulvey, M. A. The rhomboid protease GlpG promotes the persistence of extraintestinal pathogenic Escherichia coli within the gut. Infect. Immun. https://doi.org/10.1128/iai.00866-00816 (2017).
Kolmar, H., Waller, P. R. & Sauer, R. T. The DegP and DegQ periplasmic endoproteases of Escherichia coli: specificity for cleavage sites and substrate conformation. J. Bacteriol. 178, 5925–5929 (1996).
Wang, Y. & Ha, Y. Open-cap conformation of intramembrane protease GlpG. Proc. Natl. Acad. Sci. USA 104, 2098–2102 (2007).
Maegawa, S., Koide, K., Ito, K. & Akiyama, Y. The intramembrane active site of GlpG, an E. coli rhomboid protease, is accessible to water and hydrolyses an extramembrane peptide bond of substrates. Mol. Microbiol. 64, 435–447 (2007).
Ben-Shem, A., Fass, D. & Bibi, E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc. Natl. Acad. Sci. USA 104, 462–466 (2007).
Merdanovic, M., Clausen, T., Kaiser, M., Huber, R. & Ehrmann, M. Protein quality control in the bacterial periplasm. Annu. Rev. Microbiol. 65, 149–168 (2011).
Braselmann, E., Chaney, J. L., Champion, M. M. & Clark, P. L. DegP chaperone suppresses toxic inner membrane translocation intermediates. PLoS ONE 11, e0162922 (2016).
Quan, S., Hiniker, A., Collet, J.-F. & Bardwell, J. C. A. Isolation of bacteria envelope proteins. In Bacterial Cell Surfaces: Methods and Protocols (ed. Delcour A. H.). (Humana Press, 2013).
Kim, S. & Sauer, R. T. Distinct regulatory mechanisms balance DegP proteolysis to maintain cellular fitness during heat stress. Genes Dev. 28, 902–911 (2014).
Farn, J. & Roberts, M. Effect of inactivation of the HtrA-like serine protease DegQ on the virulence of Salmonella enterica serovar typhimurium in mice. Infect. Immun. 72, 7357–7359 (2004).
Schaeffer, A. J., Schwan, W. R., Hultgren, S. J. & Duncan, J. L. Relationship of type 1 pilus expression in Escherichia coli to ascending urinary tract infections in mice. Infect. Immun. 55, 373–380 (1987).
Vinothkumar, K. R. Structure of rhomboid protease in a lipid environment. J. Mol. Biol. 407, 232–247 (2011).
Evans, D. J., Evans, D. G. & DuPont, H. L. Hemagglutination patterns of enterotoxigenic and enteropathogenic Escherichia coli determined with human, bovine, chicken, and guinea pig erythrocytes in the presence and absence of mannose. Infect. Immun. 23, 336–346 (1979).
Martinez, J. J., Mulvey, M. A., Schilling, J. D., Pinkner, J. S. & Hultgren, S. J. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 19, 2803–2812 (2000).
Whelan, S., Lucey, B. & Finn, K. Uropathogenic Escherichia coli (UPEC)-associated urinary tract infections: the molecular basis for challenges to effective treatment. Microorganisms 11, 2169 (2023).
Maesaka, J. K. & Shimamura, T. Phagocytosis of E. coli by renal tubular epithelia. Yale J. Biol. Med. 57, 817–824 (1984).
Sahoo, K. & Meshram, S. Biofilm formation in chronic infections: a comprehensive review of pathogenesis, clinical implications, and novel therapeutic approaches. Cureus 16, e70629 (2024).
Flemming, H.-C. et al. Biofilms: an emergent form of bacterial life. Nat. Rev. Microbiol. 14, 563–575 (2016).
Elhenawy, W. et al. High-throughput fitness screening and transcriptomics identify a role for a type IV secretion system in the pathogenesis of Crohn’s disease-associated Escherichia coli. Nat. Commun. 12, 2032 (2021).
Patkowski, J. B. et al. The F-pilus biomechanical adaptability accelerates conjugative dissemination of antimicrobial resistance and biofilm formation. Nat. Commun. 14, 1879 (2023).
Ghigo, J.-M. Natural conjugative plasmids induce bacterial biofilm development. Nature 412, 442–445 (2001).
Strisovsky, K., Sharpe, H. J. & Freeman, M. Sequence-specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol. Cell 36, 1048–1059 (2009).
Zoll, S. et al. Substrate binding and specificity of rhomboid intramembrane protease revealed by substrate–peptide complex structures. EMBO J. 33, 2408–2421 (2014).
Vetsch, M. et al. Mechanism of fibre assembly through the chaperone–usher pathway. EMBO Rep. 7, 734–738 (2006).
Puorger, C., Vetsch, M., Wider, G. & Glockshuber, R. Structure, folding and stability of FimA, the main structural subunit of type 1 pili from uropathogenic Escherichia coli strains. J. Mol. Biol. 412, 520–535 (2011).
Wu, X. et al. Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science 368, eaaz2449 (2020).
Hal Jones, C., Danese, P. N., Pinkner, J. S., Silhavy, T. J. & Hultgren, S. J. The chaperone-assisted membrane release and folding pathway is sensed by two signal transduction systems. EMBO J. 16, 6394–6406 (1997).
Jones, C. H. et al. Escherichia coli DegP protease cleaves between paired hydrophobic residues in a natural substrate: the PapA pilin. J. Bacteriol. 184, 5762–5771 (2002).
Kateete, D. P. et al. Rhomboids of mycobacteria: characterization using an aarA mutant of Providencia stuartii and gene deletion in Mycobacterium smegmatis. PLoS ONE 7, e45741 (2012).
Babu, M. et al. Global landscape of cell envelope protein complexes in Escherichia coli. Nat. Biotechnol. 36, 103–112 (2018).
Babu, M., Kagan, O., Guo, H., Greenblatt, J. & Emili, A. Identification of protein complexes in Escherichia coli using sequential peptide affinity purification in combination with tandem mass spectrometry. JoVE 69, e4057 (2012).
Eng, J. K., McCormack, A. L. & Yates, J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 (1994).
Kislinger, T. et al. PRISM, a generic large scale proteomic investigation strategy for mammals*, S. Mol. Cell. Proteom. 2, 96–106 (2003).
Florens, L. et al. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods 40, 303–311 (2006).
Tatusov, R. L., Koonin, E. V. & Lipman, D. J. A genomic perspective on protein families. Science 278, 631–637 (1997).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Arutyunova, E., Panigrahi, R., Strisovsky, K. & Lemieux, M. J. Chapter ten - production of recombinant rhomboid proteases. In Methods in Enzymology (ed. Gelb, M. H.). (Academic Press, 2017).
Arutyunova, E., Strisovsky, K. & Lemieux, M. J. Chapter fifteen - activity assays for rhomboid proteases. In Methods in Enzymology (ed. Gelb, M. H.). (Academic Press, 2017).
Jiang, Y. et al. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 System. Appl. Environ. Microbiol. 81, 2506–2514 (2015).
Chun, J. et al. DGAT2 inhibition potentiates lipid droplet formation to reduce cytotoxicity in APOL1 kidney risk variants. J. Am. Soc. Nephrol. 33, 889–907 (2022).
Rahmani, W. et al. Attenuation of SARS-CoV-2 infection by losartan in human kidney organoids. iScience 25, 103818 (2022).
Chung, H., Bui-Marinos, M. P., Rahmani, W., Corcoran, J. A. & Chun, J. Infecting kidney organoids with a cDNA reporter clone of SARS-CoV-2. STAR Protoc. 3, 101617 (2022).
Acknowledgements
We thank Jack Moore and the Alberta Proteomics and Mass Spectrometry Facility for technical support. We also thank Sara Amidian, Joesph Primeau for microscopy support at the University of Alberta Imaging Core, and the SPP-ARC Cryo-EM Facility. We thank Dr. Tracy Raivio for supplying pathogenic E. coli and K12 single gene knockouts. We received funding from the Canadian Foundation for Innovation (37833 and 39051) and Natural Sciences, Engineering Research Council of Canada (DGECR-2018-00142 discovery grant to O.J.) and Alberta Graduate Excellence Scholarship (B.H). This work was supported by the Women’s and Children’s Health Research Institute at the University of Alberta (M.J.L.), the Natural Science and Engineering Research Council of Canada (RGPIN-2016-06478 to M.J.L.; RGPIN-2019-20234 to M.B.) and RGPIN-2023-04396 (M.J.L.), Striving for Pandemic Preparedness—The Alberta Research Consortium, and Canada Foundation for Innovation (CFI), Canada Research Chair CRC-2021-00172 (HKA), Canadian Institutes of Health Research (PJT-180387 to H.S.Y.; PJT-426160 to E.W.). For experiments performed at the University of Alberta Faculty of Medicine & Dentistry Cell Imaging Core, RRID:SCR_019200, which receives financial support from the Faculty of Medicine & Dentistry, the University Hospital Foundation, Striving for Pandemic Preparedness—The Alberta Research Consortium, and Canada Foundation for Innovation (CFI) awards to contributing investigators.
Author information
Authors and Affiliations
Contributions
Conceptualization, J.L., E.A., and M.J.L.; Affinity-purification-mass spectrometry, S.P. and M.B., In-gel digestion proteomics, B.H. and O.J.; Generation of mutant E. coli, protein purification, immunoblotting, sample preparation for electron microscopy, RT-qPCR, J.L., Gentamicin protection assay, J.L., L.B., H.A., and E.W., Abiotic biofilm assays, inhibition assays, metabolic assays, enzyme kinetics, E.A., Kidney organoid generation and infection, microscopy of organoids, H.J.C. and J.C., Biotic biofilm assays, J.W. and W.E., Writing–original draft, J.L. and E.A.; Writing–review & editing, J.L., E.A., and M.J.L.; Supervision, M.J.L., H.S.Y., O.J., J. C., W.E and M.B.; Funding acquisition, M.J.L., H.S.Y., O.J., J. C., W.E., and M.B.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, J., Arutyunova, E., Hartley, B. et al. Rhomboid protease GlpG regulates type 1 pili quality control and virulence in pathogenic E. coli. Nat Commun 17, 968 (2026). https://doi.org/10.1038/s41467-025-67697-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67697-2
