
Abstract
Highly pathogenic avian influenza H5N1 poses an increasing public health risk, particularly following its spillover into dairy cows and associated human infections in the U.S. since March 2024. Here, we systematically identified critical PB2 mutations emerged during avian-to-cattle transmission and subsequent adaptation in cattle, notably PB2 M631L, which conferred pathogenicity in mice comparable to the well-characterized PB2 E627K mutation. Retrospective analysis reveals that PB2 631L also circulated in avian and human H5N1 strains during the 2013–2014 outbreaks in Cambodia and Vietnam. Additional adaptive mutations include established markers (E627K, Q591R, D701N), and novel variants (I647V, G685R, K736R). These mutations enhance polymerase activity by improving the utilization of both bovine and human ANP32A proteins, thereby increasing viral fitness and pathogenicity in mammals. The convergence of these adaptations highlights the elevated zoonotic risk of cattle-adapted H5N1 viruses and underscores the urgent need for heightened surveillance across avian and mammalian hosts.
Similar content being viewed by others


Introduction
The potential for a pandemic triggered by the avian influenza H5N1 virus has been a major concern for decades1,2,3,4. Since its first detection in Hong Kong in 1997, H5N1 virus infections have been reported in over 954 individuals, with 464 fatalities, resulting in a mortality rate of 48.6%5. Since 2021, H5N1 clade 2.3.4.4b viruses have spread extensively among bird populations6. In March 2024, spillover events involving this clade virus were detected in dairy cows in the United States7,8,9. The virus subsequently spread to hundreds of farms, resulting in infections in various mammalian species10,11, including humans12,13,14, marking an unprecedented situation in the history of H5N1 outbreaks. Since the onset of the 2024 H5N1 outbreak, 70 cases of human infection have been reported in the United States15.
Infection by the avian influenza virus in mammalian cells is typically constrained by several factors16,17,18,19, including receptor preference20, virion stability21, and the activity of the viral polymerase22. The influenza polymerase, a heterotrimeric complex composed of PB2, PB1, and PA proteins, is responsible for viral RNA transcription and replication23. The restriction of avian influenza polymerase activity in mammalian cells can be significantly alleviated by adaptive mutations in the polymerase22. The PB2 E627K mutation is the most frequently identified adaptation site in viruses that have undergone interspecies transmission from avian to mammal hosts24,25. In human seasonal influenza viruses, PB2 residue 627 is predominantly a lysine, a hallmark of mammalian adaption. However, an exception was observed in 2009 pandemic H1N1 strain, which retains the avian-type glutamic acid at residue 627 but harbored a PB2 Q591R mutation, located spatially close to residue 627 within the polymerase structure26. Additional mammalian-adaptive mutations include PB2 D701N, D740N, T271A, and E627V27,28,29,30. In addition, it has also been reported that mutations in PB1 and PA proteins enhance the activity of avian influenza polymerase in mammalian cells22,31,32,33.
The host restriction of avian influenza virus polymerase activity is primarily linked to host species-specific differences in acidic nuclear phosphoprotein 32 protein A (ANP32A)34. Unlike mammalian ANP32A, avian ANP32A contains a 33-amino acid insertion that significantly enhances its support for polymerase activity from viral strains of various origins34,35. Functional and structural studies have shown that host ANP32 proteins play a critical role in forming viral RNA replication platform in conjunction with the viral polymerase36,37,38. Due to the inability of mammalian ANP32 proteins to effectively support avian influenza virus polymerase, adaptive mutations in the avian viral polymerase during cross-species transmissions have evolved to enhance its capacity to utilize mammalian ANP32 proteins for efficient viral RNA replication39,40.
In this study, we conducted a systematic investigation into the role of the H5N1 viral polymerase in facilitating cross-species transmissions. A series of critical mutations including PB2 M631L mutation within the PB2 protein were identified to enhance polymerase activity in mammalian cells while concurrently leading to increased pathogenicity in mice. We also found that the key mutation PB2 M631L had previously co-occurred in avian and human H5N1 infections during the 2013–2014 Cambodia/Vietnam H5N1 outbreaks. These findings highlight a persistent underestimation of H5N1 avian influenza virus adaptability and underscore the urgent need to strengthen surveillance and comprehensive pathogenicity assessments.
Results
Systematic identification of key mutations enhancing polymerase activity in mammalian cells during avian-to-cattle transmission of H5N1 virus in 2024
To investigate the contribution of the viral polymerase protein of the H5N1 clade 2.3.4.4b virus to cross-species transmission from birds to dairy cows, we selected a representative H5N1 virus strain, A/dairy cow/Ohio/078707-24-12/2024 (cowOH12-H5N1), isolated from a dairy cow (Fig. 1A). This strain was chosen because its PB2 protein sequence was the most prevalent among cattle isolates, with 417 identical sequences found among 1210 total sequences analyzed as of October 30, 2024 (Fig. S1A). For comparison, we selected an H5N1 clade 2.3.4.4b virus, A/chicken/Maine/24-007946-001/2024 (chMA01-H5N1), which was isolated from chicken in the United States in March 2024 and is significantly evolutionarily different from the cattle isolates (Figs. 1A and S1B).

A The chicken H5N1 virus (chMA01-H5N1) and cow H5N1 virus (cowOH12-H5N1) used in this study. Created in BioRender. lai, y. (2025) https://BioRender.com/x9td31y. B 293T cells were co-transfected with plasmids expressing chMA01-H5N1 or cowOH12-H5N1 PB2, PB1, PA, and NP proteins and a plasmid expressing green fluorescent protein (GFP) reporter vRNA. Cells were imaged 24 h post-transfection using a laser scanning fluorescence microscope. Magenta bars indicate viral segments from cowOH12-H5N1, while blue bars denote those from chMA01-H5N1. Images are representative of two independent experiments with consistent results. Scale bar, 100 μm. C 293T cells were co-transfected with plasmids expressing chMA01-H5N1 or cowOH12-H5N1 PB2, PB1, PA, and NP proteins and a plasmid expressing firefly luciferase reporter vRNA. At 24 h post-transfection, Firefly luciferase activity was measured and standardized to Renilla luciferase activity, presenting as relative polymerase activity. Data are presented as mean values ± SD from n = 3 independent biological replicate experiments. D 293T cells were co-transfected with plasmids expressing chMA01-H5N1 or cowOH12-H5N1 PB2, PB1, PA, and NP proteins and NA vRNA. At 12, 24, 36 and 48 h post-transfection, total RNAs were extracted and the steady-state RNA levels were determined by primer extension analysis. Images are representative of two independent experiments with consistent results. E Differential amino acid mutations between chMA01-H5N1 and cowOH12-H5N1 PB2 proteins are indicated. Firefly luciferase reporter assay assessed the effects of single (F) and combined (G) cow-specific PB2 mutations in the context of chMA01-H5N1 polymerase. Relative polymerase activity was normalized to the cowPB2 group. Magenta bars indicate the complete PB2 segments from cowOH12-H5N1, while blue bars denote that from chMA01-H5N1. Green bars indicate the notable substitutions. Data are presented as mean values ± SD from n = 4 independent biological replicate experiments. Statistical significance (C, F, G) was determined by one-way ANOVA with Dunnett’s post hoc test. Source Data are provided as a Source Data file.
To evaluate the activity of the viral polymerase in mammalian cells, we constructed viral mini-replicon systems for the both cow and chicken H5N1 viruses in human 293T cells. Green fluorescent protein (GFP) was used as a reporter (Fig. 1B), driven by an RNA polymerase I (Pol I) promoter that expressed a viral-like RNA encoding the GFP, flanked by the non-coding regions of the influenza A virus nucleoprotein (NP) segment. The results demonstrated that the H5N1 viral polymerase from cow isolate was highly active in mammalian cells, whereas the chicken H5N1 viral polymerase exhibited significantly restricted activity. To identify the contributions of the polymerase subunits and NP responsible for this difference, we performed one-to-one replacements of the polymerase subunits and NP. Reciprocal exchange of the PB2 segments between the two viruses resulted in a significant reversal of polymerase activity, while replacement of the PB1 segment has only a slight effect (Fig. 1B). Exchanges of the PA and NP segments has no measurable impact. Quantitative assays using firefly luciferase as a reporter confirmed that the PB2 protein from the cow H5N1 virus plays a predominant role in enhancing polymerase activity in mammalian cells (Fig. 1C).
We next monitored the kinetics of three viral RNA species accumulation for two viral polymerases using a primer extension assay. Overall, the levels of mRNA, cRNA, and vRNA synthesized by the cow H5N1 polymerase were significantly higher than those synthesized by the chicken H5N1 polymerase at all time points post-transfection (Fig. 1D). Consistent with above findings, the replacement of chicken H5N1 PB2 with cow H5N1 PB2 was sufficient to enable the chicken H5N1 polymerase to efficiently accumulate all three viral RNA species in mammalian cells (Fig. 1D). These results indicate that the PB2 protein is the primary determinant of the enhanced activity of the cow H5N1 polymerase in mammalian cells.
To further elucidate the differences between the two polymerases responsible for enhanced activity in mammalian cells, we compared the amino acid sequences of the PB2 proteins from the cow and chicken H5N1 viruses. Our analysis revealed a total of 13 distinct amino acids differences between the two PB2 proteins (Figs. 1E and S1C). We then individually introduced these cow-specific PB2 amino acids into the chicken PB2 protein and assessed the resulting changes in polymerase activity. As shown in Fig. 1F, the PB2 M631L substitution significantly enhanced the chicken H5N1 polymerase activity by 23.3-fold. Additionally, substitutions such as PB2 T58A, A129T, V495I and V649I also increased polymerase activity by 1.8- to 9.0-fold (Fig. 1F). While individual mutations improved polymerase activity, none achieved the activity levels of the cow H5N1 polymerase. Therefore, we tested combination of these amino acid substitutions. Notably, the combination of PB2 A129T, V495I, or V649I with M631L was sufficient to elevate the chicken H5N1 polymerase activity to match that of the cow H5N1 polymerase (Fig. 1G). It is important to note that the PB2 segment of the cattle H5N1 virus was recently identified as being derived from reassortment with North American low pathogenic avian influenza viruses1,9. Here, our functional data further demonstrate that the specific amino acids acquired through this event are the primary drivers of increased polymerase activity observed in the avian-to-cattle cross-species transmission of the H5N1 virus, with the PB2 M631L mutation playing a major role.
The PB2 M631L mutation previously co-occurred in both avian and human H5N1 virus isolates during outbreaks in Cambodia and Vietnam between 2013 to 2014
Given the significant role of the PB2 M631L mutation in cross-species transmission, we analyzed its occurrence across all H5N1 viruses available on GISAID database. Surprisingly, in addition to the recent detection of PB2 631L in human infections linked to cattle in the United States in 2024, this variant was also present in human H5N1 cases in Cambodia and Vietnam during 2013–2014 (Figs. 2A and S2A). Further genomic surveillance revealed that PB2 631L was concurrently circulating in both avian and human H5N1 viruses from those regions with highest frequency observed between 2013 and 2014 (Figs. 2A, and S2A, B). Notably, the PB2 M631L mutation first emerged in an avian influenza virus as early as 2005 in Thailand (Figs. 2A and S2B). These findings suggest that PB2 M631L emerged earlier than previously recognized and may have contributed to past, underappreciated zoonotic events. This underscores the importance of retrospective analysis of archived viral genomes to better understand the early emergence of mammalian-adaptative mutations in H5N1 viruses.

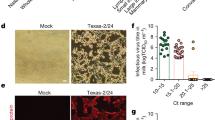
A Statistics on the number of human (top panel) and avian (bottom panel) H5N1 viruses carrying the PB2 631L. Different countries are represented by distinct colors as shown in figure. B Statistics on the number of human (top panel) and avian (bottom panel) H5N1 viruses with PB2 E627K or M631L mutations in Cambodia and Vietnam. The schematic diagram of human and avian (A, B) were created in BioRender. lai, y. (2025) https://BioRender.com/x9td31y. Six-week-old female BALB/c mice were intranasally inoculated with 2000 PFU of wild-type (WT) or indicated PB2 mutant recombinant H5N1 viruses. Mouse body weight curves (C) and survival curves (D) were monitored (n = 3). Body weight data are presented as mean values ± SD from n = 3 mice. E Viral titers in the lungs of mice euthanized on day 5 post-infection were determined by plaque assay. Data are presented as mean values ± SD from n = 3 mice. Statistical significance was determined by one-way ANOVA with Dunnett’s post hoc test. F Hematoxylin-eosin (H&E) and immunohistochemistry (IHC) staining of the lungs of mice infected with WT or indicated PB2 mutant recombinant H5N1 viruses on day 5 post-infection. Scale bar, 100 μm. Source Data are provided as a Source Data file.
According to records from the World Health Organization (WHO), Cambodia and Vietnam have reported 74 and 130 cases of human H5N1 infections, respectively, since 2003 (Fig. S2C). Specifically, Cambodia documented 26 and 9 human infections in 2013 and 2014, respectively, accounting for 47% of its total cases5. To investigate potential viral factors underlying this spike, we collected and analyzed all available PB2 sequences from H5N1 viruses isolated from both humans and avian sources in Cambodia and Vietnam. Our analysis revealed that all human H5N1 viruses isolated during 2013–2014 harbored the PB2 M631L mutation (Fig. 2B). In contrast, the well-characterized PB2 E627K mutation, though present in roughly half of the other human H5N1 infections reported in the region over the years, was absent in the 2013–2014 human cases that harbored PB2 631L (Fig. 2B). Among H5N1 viruses from avian sources, primarily chickens and ducks, PB2 E627K was detected at low frequencies, while PB2 M631L was presence in 20–50% of local avian isolates during 2013–2015 (Fig. 2B). These findings suggest a strong temporal and geographic association between the increased prevalence of the PB2 M631L variant in poultry and the high incidence of human infections during this period. This supports the hypothesis that PB2 M631L contributed to enhanced zoonotic potential and highlights the importance of ongoing molecular surveillance in animal reservoirs.
We further conducted a phylogenetic analysis of PB2 amino acid sequences from H5N1 viruses isolated in Cambodia and Vietnam during 2013–2014 (Fig. S2D). The analysis indicated that all viruses carrying the PB2 631L clustered within a single monophyletic clade, which was clearly distinct from PB2 631M-containing viruses, located at the base of the phylogenetic tree. The tree topology showed that human isolates were embedded within avian viruses carrying the PB2 631L mutation, supporting the hypothesis of poultry-to-human interspecies transmission during this period. Notably, H5N1 viruses isolated from a civet cat in Cambodia and a tiger in Vietnam also clustered within the PB2 631L clade, suggesting that this variant was capable of infecting a broader range of mammalian hosts. These findings indicate that the PB2 M631L mutation may have facilitated expanded host tropism and contributed to multiple interspecies transmission events, including spillover into both humans and nonhuman mammals.
To investigate whether additional potential adaptive mutations emerged following H5N1 virus infection in humans, we analyzed variations in the PB2 protein from viral sequences obtained from human cases in Cambodia and Vietnam during the 2013–2014 outbreak (Fig. S2E). Despite the limited dataset of only 24 PB2 sequences, we observed a relatively high degree of amino acid variation. For example, the PB2 T521A mutation was present in 10 sequences, alongside other substitutions such as V356I, N456D, T524A, and D740N. Importantly, PB2 D740N has been previously characterized as a mammalian-adaptive mutation29, indicating that these variations may have contributed to enhanced polymerase activity and replication efficiency of avian H5N1 viruses in human cells. These mutations were rarely observed in contemporaneous avian H5N1 viruses, confirming their role in host adaptation (Fig. S2F). Although direct correlations between individual clinical outcomes and specific mutations cannot be established with current data, WHO reports indicated that the 2013–2014 outbreak resulted in 21 deaths among 39 confirmed human infections in Cambodia and Vietnam, corresponding to a mortality rate exceeding 50%5. These findings suggest the high pathogenic potential of H5N1 viruses harboring PB2 631L, particularly when accompanied by additional adaptive mutations that may arise during human infections, and reinforce the importance of genomic surveillance during zoonotic outbreaks.
PB2 M631L mutation confers high virulence in mice, comparable to the canonical mammalian adaptive PB2 E627K mutation
Previous studies have shown that the PB2 M631L mutation enhances polymerase activity in mammalian cells, suggesting a potential role in mammalian adaptation similar to that of the well-characterized PB2 E627K mutation41,42,43,44. However, the in vivo pathogenic effects of PB2 M631L remain poorly characterized. To address this, we employed reverse genetics to generate a 2 + 6 reassortant virus by combining the hemagglutinin (HA) and neuraminidase (NA) segments from the laboratory-adapted A/WSN/1933 (H1N1) virus with the remaining six segments from A/dairy cow/Ohio/078707-24-12/2024 (H5N1). This strategy enabled safe handling of the virus under biosafety level 2 conditions and allowed us to evaluate the impact of polymerase mutations on viral infectivity. We generated all possible recombinant viruses carrying single or combined mutations at PB2 positions 627 and 631. Groups of female BALB/c mice were intranasally inoculated with 2000 PFU of each virus, and body weight and survival were monitored daily over a 14-day period. On day 5 post-infection, lung tissues were harvested for viral titration, histopathological examination, and immunohistochemistry (IHC) analysis.
The results demonstrated that the wild-type PB2 of the cattle-derived H5N1 virus (PB2 627E + 631L) enabled efficient infection in mice, leading to mortality between days 7 and 10 post-infection (Fig. 2C, D). In contrast, introducing a reversion mutation at position 631 (PB2 627E + 631M) markedly attenuated viral pathogenicity, as evidenced by minimal weight loss and 100% survival. Strikingly, the addition of the E627K mutation on the L631M background (PB2 627K + 631M) restored high pathogenicity, causing rapid mortality within 6–7 days. The combination of E627K and M631L (PB2 627K + 631L) further augmented virulence, with mice succumbing as early as days 4–6. Consistently, viral titer assays revealed that both PB2 631L and E627K mutations independently conferred elevated viral loads in lung tissues, and their combination produced the highest titers (Fig. 2E). In contrast, the L631M mutation significantly diminished viral replication. Histopathological and immunohistochemical (IHC) analyses of lung tissues corroborated these findings: extensive tissue damage and high viral antigen expression were observed in mice infected with viruses harboring 627K and/or 631L, whereas limited pathology and viral antigen were detected in lungs infected by the PB2 627E + 631M variant (Fig. 2F). The WSN virus served as a positive control and confirmed successful infection in all experimental settings. Together, these results provide compelling evidence that PB2 M631L, akin to E627K, significantly enhances mammalian adaptation and contributes to increased pathogenicity of the H5N1 virus in a mammalian host.
Ongoing H5N1 outbreaks in cattle and other mammals have facilitated the emergence of additional mammalian-adapted mutations in PB2
Between March 2024 and June 2025, more than 1073 cattle herds in the United States were confirmed to be infected with H5N1 virus15. This unprecedented scale of cross-species transmission over a relatively short timeframe raises serious concerns about the virus acquiring adaptive mutations that enhance its replication in mammals. To assess this, we analyzed a total of 3028 PB2 sequences from cattle-derived H5N1 viruses available as of March 31, 2025. This analysis identified 172 amino acid substitutions, with several positions showing notably high variation frequencies (Figs. 3A and S3).

A Variations of cow H5N1 PB2 amino acids. The PB2 sequences of H5N1 derived from dairy cows from 2024 to March 31, 2025 (n = 3,028) were aligned and compared for mutations. The horizontal axis represents amino acid positions on PB2, and the vertical axis shows the base-10 logarithm of mutation frequency. The notable mutations are marked. Four well-known mammal-adaptive mutations are highlighted in magenta. B Accumulation of representative H5N1 PB2 mutations derived from dairy cows. In the left panel, red denotes H5N1 genotype B3.13 (all bearing PB2 631L), while blue denotes H5N1 genotype D1.1 (all bearing PB2 631M). The right panel details the accumulation of key mutations in H5N1 B3.13. PB2 sequences are sorted by virus isolation date, with each column representing an individual viral isolate. Substitutions are highlighted in pink. Firefly luciferase reporter assay assessed the effects of the cow-related (C), nonhuman mammal-related (D) and human-related (E) mutations on cow H5N1 polymerase activity. 293T cells were co-transfected with plasmids expressing cowOH12-H5N1 PB2 (WT or mutant), PB1, PA, and NP proteins and a plasmid expressing firefly luciferase reporter vRNA. The relative polymerase activity was normalized to the wild-type (WT) cowPB2 group. Mutations that significantly enhanced polymerase activity are shown in red. Data are presented as mean values ± SD from n = 3 independent biological replicate experiments. Statistical significance was determined by one-way ANOVA with Dunnett’s post hoc test. Source Data are provided as a Source Data file.
Of particular concern, mammalian-adaptive mutations such as E627K and Q591R were found to emerge on the genetic background of the PB2 631L variant, with E627K showing a marked increase in prevalence during 2025 (Figs. 3B and S3). The well-characterized PB2 D701N mutation was also detected, predominately in a distinct genotype D1.1 virus associated with recent H5N1 2.3.4.4b clade infections in dairy cows45. Notably, the PB2 D740N mutation, previously reported in human H5N1 infections in Cambodia and Vietnam (2013–2014), was identified in multiple cattle-derived viral sequences (Figs. 3B and S2E). In addition, PB2 A58T and K670R have shown significant accumulation since September 2024 (Figs. 3B and S3). Collectively, these findings suggest that PB2 mutations emerging in H5N1 viruses circulating among cattle may enhance viral fitness and adaptation in mammalian hosts.
To assess the functional consequences of newly emerged PB2 mutations, 28 amino acid substitutions with either high variation frequencies or potential mammalian-adaptive significance were introduced individually into the PB2 gene of the cowOH12-H5N1 strain and performed by viral mini-replicon system (Fig. 3C). The results showed that multiple PB2 mutations significantly enhanced polymerase activity. As expected, the PB2 E627K mutation led to the most substantial increase, a 19.2-fold enhancement, consistent with its previously observed role in promoting virulence and pathogenicity in in mammalian models (Fig. 2C–F). Several other mutations also conferred moderate but notable increases in polymerase activity, such as PB2 R589K, Q591R, I647V, and D701N elevated polymerase activity by 2.2 to 4.4-fold, while PB2 D740N, previously associated with human infections in Cambodia and Vietnam, increased activity by 2.9-fold (Fig. 3C). Taken together, these findings indicate that the widespread and sustained transmission of H5N1 viruses in cattle populations is fostering the occurrence of mammalian-adaptive mutations in PB2. These mutations significantly enhance polymerase activity in mammalian cells, potentially contributing to increased viral infectivity and pathogenicity in mammalian hosts.
In addition to widespread infections in cattle, H5N1 infections have also been reported in other mammals, including humans, cats, red foxes and raccoons, as documented in recent surveillance data5,15. To further investigate potential mammalian-adaptive mutations, we obtained and analyzed PB2 sequences from viruses isolated from nonhuman mammals, particularly those closely related to the cattle-derived H5N1 lineage (Fig. S4). Among these, we identified that the PB2 K736R mutation in viruses isolated from infected cats enhanced polymerase activity by 2.8-fold (Fig. 3D), indicating that this mutation may contribute to mammalian adaptation of the virus. We also observed that the PB2 V596A mutation led to a significant reduction in polymerase activity, although the reason for this effect is currently unknown.
We further analyzed PB2 protein variations in H5N1 viruses associated with human infections, focusing on strains belonging to the B3.13 genotype that originated from dairy cow transmission. Notably, the first human H5N1 isolate (huTX37-H5N1), obtained in late March 202414, contained the mammalian-adaptive PB2 E627K mutation, along with avian-like signatures 631M and 362E41 (Fig. S5). In addition to huTX37-H5N1, all other human H5N1 PB2 sequences harbored the PB2 631L substitution, while none contained E627K substitution (Fig. S5). Functional analysis showed that E627K significantly enhanced polymerase activity by 19.2-fold in the cowOH12-H5N1 mini-replicon system (Fig. 3C), and G362E increased activity by 4.1-fold (Fig. 3E). In contrast, L631M reduced polymerase activity, but the combination of E627K and L631M retained high activity (Fig. 3E). Among additional human PB2 sequences analyzed, all retained the 631L mutation, yet none carried the E627K mutation, reinforcing prior evidence that PB2 M631L plays a significant role in mammalian adaptation. Furthermore, four other mutations (A58T, E249G, I562V, K670R) detected in human isolates were also found in cattle-derived viruses, but did not significantly alter polymerase activity in our assays (Fig. 3E). The expression levels of all PB2 mutant proteins were confirmed to be unaffected by the mutations (Fig. S6A). Overall, these findings suggest that the cow H5N1 virus possesses an inherently robust polymerase function in human cells.
The adaptive mutations in PB2 arising from H5N1 virus transmission in cattle and other mammals facilitate the virus polymerase to utilize both cow and human ANP32 proteins
The host ANP32 protein family is essential for influenza virus RNA synthesis34,46. Specifically, chicken ANP32A (chANP32A) supports the polymerase activity of both avian and mammal-adapted influenza viruses, while human ANP32A (huANP32A) and ANP32B (huANP32B) primarily support only mammal-adapted strains39,40. In contrast, avian ANP32B, ANP32E and human ANP32E do not support influenza virus polymerase activity due to inherent functional limitations47. Phylogenetic analysis indicated that cow ANP32A and ANP32B are closely related to their human counterparts, implying a comparable role in supporting influenza virus replication (Fig. 4A). To test this, we constructed mini-replicons in 293T ANP32A, ANP32B, and ANP32E triple-knockout cells (293T TKO48) complemented with ANP32 proteins from different species (Figs. 4B and S6B). The results indicated that chANP32A effectively supported cow H5N1 polymerase activity, while cow and human ANP32A and ANP32B supported replication to a significantly lesser extent. Among cow ANP32 proteins, ANP32A provided stronger support than ANP32B, mirroring the pattern observed with human ANP32 proteins. Increasing ANP32 expression levels enhanced polymerase activity in all cases (Fig. 4B).

A Phylogenetic analysis of ANP32 proteins derived from different hosts. B Dose-dependent effects of different ANP32 proteins on the cowOH12-H5N1 polymerase activity in the firefly luciferase reporter assay in 293T ANP32A, ANP32B, and ANP32E triple-knockout cells (293T TKO). Data are presented as mean values ± SD from n = 3 independent biological replicate experiments. Statistical significance was determined by two-way ANOVA with Dunnett’s multiple comparisons test. C Structure of the influenza viral polymerase complexed with the ANP32 protein (PDB: 8R1J). Mutations which affected polymerase activities are highlighted in red. D Electrostatic isosurface visualization of the viral polymerase showing the electrical changes (red negative, blue positive) due to cow-adaptive mutations and mammal-adaptive mutations, with a red dashed line indicating the putative path of the ANP32 LCAR domain. Firefly luciferase reporter assay was implemented to compare the effects of key PB2 substitutions on the cowOH12-H5N1 polymerase activity in 293T TKO cells complemented with chicken ANP32A (E), cow ANP32A (F), and human ANP32A (G). The relative polymerase activity was normalized to the wild-type (WT) cowPB2 group. Data are presented as mean values ± SD from n = 3 independent biological replicate experiments. Statistical significance was determined by one-way ANOVA with Dunnett’s post hoc test. Source Data are provided as a Source Data file.
Structural analysis of the complex of influenza virus polymerase and ANP32 protein revealed that most of these adaptive mutations are located at key functional interfaces, including the region adjacent to the ANP32 binding site and the dimerization interface of the viral polymerase complex (Fig. 4C). Consistent with previous studies, these mutations enhance the positive charge in the PB2 627 domain and nuclear localization signal (NLS) domain, thereby strengthening electrostatic interactions with the negatively charged mammalian ANP32 proteins (Fig. 4D)36,37,38. To assess the functional impact of these mutations, we tested polymerase activity in 293T TKO cells complemented with either avian or mammalian ANP32 proteins (Fig. 4E–G). The avian-specific PB2 L631M mutation only modestly reduced activity with chicken ANP32A but severely impaired polymerase activity with cow and human ANP32A, confirming its role as a mammalian-restrictive mutation (Fig. 4E–G). In contrast, other adaptive mutations had minimal effects on polymerase activity with chicken ANP32A, but significantly enhanced activity when cow or human ANP32A was present (Fig. 4E–G). Taken together, these findings demonstrate that H5N1 viruses circulating in cattle have acquired adaptive PB2 mutations that enhance the polymerase’s ability to utilize mammalian ANP32 proteins, thereby facilitating efficient replication in bovine hosts and increasing the risk of zoonotic transmission to humans.
Mammalian-adaptive PB2 mutations increase the pathogenicity of cattle-derived H5N1 virus in mice
Given the emergence of multiple PB2 mutations in cattle-derived H5N1 viruses that enhance polymerase activity in mammalian cells, these genetic adaptations may also elevate viral virulence, posing a growing public health concern. To systematically evaluate their impact on pathogenicity, we used reverse genetics to generate recombinant viruses containing the HA and NA segments from A/WSN/33 (H1N1) and the six internal gene segments from the cowOH12-H5N1 virus, each engineered with specific PB2 mutations of interest. Female BALB/c mice were intranasally inoculated with these reassortant viruses, and pathogenicity was assessed by monitoring body weight loss, viral titers in lung tissue, and histopathological changes, following the same protocols as in earlier experiment.
Consistent with previous findings, the PB2 L631M mutation significantly attenuated viral pathogenicity in mice, while the PB2 E627K mutation markedly enhanced virulence (Fig. 5A). Several established mammalian-adaptive mutations, including Q591R, D701N, and D740N, led to more rapid weight loss and earlier mortality compared to the wild-type virus, confirming their role in increased pathogenicity (Fig. 5A). In addition, newly identified mutations from this study, such as I647V, G685R, and K736R, also substantially elevated pathogenicity. Notably, I647V induced rapid weight loss and resulted in complete mortality by day 5 post-infection, closely resembling the impact of the E627K mutation. The G362E mutation caused moderately accelerated weight loss, while K670R showed a pathogenic profile similar to the wild-type virus (Fig. 5A). Histopathological analyses revealed extensive lung damage in mice infected with all PB2 mutants, except for L631M, indicating severe pulmonary involvement (Fig. 5B). Moreover, viral titers in lung tissues were elevated in all mutants except G362E and K670R, further supporting their enhanced replication capacity in vivo (Fig. 5C). Taken together, these results demonstrate that both known and newly emerging PB2 mutations in cattle-derived H5N1 viruses contribute to increased viral replication and pathogenicity in mammals, thereby heightening the risk of zoonotic transmission and severe disease.

A Six-week-old female BALB/c mice were intranasally inoculated with 2000 PFU of wild-type (WT) or indicated PB2 mutant recombinant H5N1 viruses. Mouse body weight curves (left) and survival curves (right) were monitored (n = 4). Body weight data are presented as mean values ± SD from n = 4 mice. B Hematoxylin-eosin (H&E) staining of the lungs of mice infected with WT or PB2 mutant recombinant H5N1 viruses on day 5 post-infection. Scale bar, 100 μm. C Viral titers in the lungs of mice euthanized on day 5 post-infection were determined by plaque assay. Data are presented as mean ± SD from n = 3 mice. Source Data are provided as a Source Data file.
Discussion
The emergence and sustained spread of H5N1 2.3.4.4b clade virus currently represent a substantial pandemic threat, as evidenced by its continued transmission following the infection of dairy cows in the U.S. in March 2024. In this study, we systematically identified key mammalian adaptative mutations within the PB2 subunit of the H5N1 viral polymerase that emerged during the recent unprecedented avian-to-cattle cross-species transmission and rapid spreading of H5N1 clade 2.3.4.4b viruses in dairy cows and its spillover to other mammals. Among these, consistent with recent reports, the PB2 M631L mutation emerged as a major contributor to enhanced polymerase activity41,42,43,44. Using a mouse model, we further demonstrate that PB2 M631L increases viral pathogenicity to a degree comparable to the well-characterized PB2 E627K mutation. In addition to M631L, we identified several mutations, including the canonical E627K, Q591R, and D701N, as well as newly discovered I647V, G685R, and K736R, that further enhance polymerase activity in mammalian cells and significantly increase virulence in mice. Interestingly, our retrospective sequence analysis revealed that PB2 631L also appeared in human H5N1 outbreaks in Cambodia and Vietnam during 2013–2014 and co-circulated in avian strains. These findings underscore the escalating risk posed by emerging H5N1 strains and highlight the urgent need for enhanced surveillance and proactive intervention strategies.
Interestingly, our retrospective analysis uncovered a long-overlooked observation: the PB2 631L variant emerged in human H5N1 infections during the 2013–2014 outbreaks in Cambodia and Vietnam. According to WHO records, Cambodia reported 35 human H5N1 infections in 2013–2014, representing 47% of the country’s total 74 confirmed cases from 2005 to 20255 (Fig. S2C). The case fatality rate during this period was notably high, reaching 50%5. In this study, we directly compared the pathogenicity of the PB2 M631L mutation with that of the well-characterized E627K mutation using a mouse model (Fig. 2C–F). Our results demonstrated that PB2 M631L significantly increased viral pathogenicity, comparable to the effect of E627K. More importantly, the combination of both mutations (631L + 627K), which has been detected in cattle H5N1 viruses in 2025, further enhanced pathogenicity in mice. It should be noted, however, that the presence of PB2 631L does not fully explain the pathogenicity of H5N1 viruses in humans, as most recently reported human H5N1 cases in the United States in 2024 carrying this mutation presented with mild symptoms12. This discrepancy in clinical severity is likely multifactorial, including the specific viral genotype, the primary route of infection (e.g., conjunctival exposure versus respiratory inhalation), and early antiviral intervention1,49. Specifically, a large proportion of recent U.S. cases presented with mild conjunctivitis and received timely oseltamivir treatment, which may have contributed to the less severe outcomes12. Despite these complexities, the repeated emergence of PB2 M631L mutation across independent H5N1 outbreaks supports its potential role as a mammalian-adaptation marker worthy of continued surveillance.
An intriguing observation from our retrospective analysis is the concurrent prevalence of the PB2 M631L mutation in both avian and human H5N1 infections during the 2013–2014 outbreaks, a pattern that has re-emerged during the ongoing 2024 H5N1 outbreaks (Fig. 2A, B). This raises a key question: in which host did the PB2 M631L mutation initially arise? Recent studies suggest that this mutation may have emerged following the spillover of H5N1 viruses into cattle during the 2024 outbreak7,41. Supporting this hypothesis, comparison of early limited B3.13 genotype of avian viruses (isolated between November 2023 and February 2024) with cattle-derived viruses reveals the emergence of PB2 M631L mutation, along with accompanying E362G and D441N mutations in cattle, implying that these changes may have arisen post-spillover7. However, the alternative possibility, that PB2 631L was already present in avian viruses prior to cattle infection, cannot be ruled out. Global sequence analysis indicated that the PB2 M631L mutation first appeared as early as 2005 in an avian influenza virus, and has since been sporadically detected in several Southeast Asian countries (Figs. 2A and S2B). More recently, PB2 631L-bearing avian viruses have been detected in Nigeria (2021), Canada (2022), and the United States (2023–2024), suggesting this variant has been circulating at low frequencies in avian populations over an extended period. Notably, between 2013 and 2015, PB2 631L viruses represented a significant proportion of avian virus populations in Cambodia and Vietnam, which may have contributed to the elevated number of human infections observed during that time (Fig. 2B). Consistently, all available H5N1 human isolates from Cambodia and Vietnam in 2013–2014, as well as 2024 H5N1 cattle isolates, carried the PB2 631L mutation (Figs. 2B, and 3A, B). Furthermore, among the 13 available PB2 sequences from 2024 human H5N1 genotype B3.13 isolates, 12 harbored the 631L variant (Fig. S5). The only exception, huTX37-H5N1, which possessed the PB2 E627K mutation, is believed to have arisen independently via direct avian-to-human transmission41. Given the strong adaptability conferred by PB2 631L, it is plausible that the PB2 M631L mutation initially occurred in avian viruses and subsequently facilitated cross-species transmission to mammals, including cattle and humans, thereby contributing to recent H5N1 outbreaks. However, due to the lack of genomic data from early cattle infections, likely occurring between January and February 20247,9, the precise timeline of the emergence and interspecies spread of this mutation remains unresolved. Nevertheless, ongoing genomic surveillance of PB2 631L in both avian and mammalian hosts is critical to elucidate its role in cross-species transmission and to better understand the evolutionary dynamics underlying H5N1 outbreaks.
In addition to the PB2 M631L mutation, we systematically identified additional mammalian-adaptive mutations in the PB2 subunit of the viral polymerase that arose during cross-species transmission of H5N1 clade 2.3.4.4b viruses from avian hosts to dairy cows, as well as during subsequent viral adaptation within cattle. During avian-to-cattle transmission, we identified three mutations A129T, V495I and V649I that significantly enhanced polymerase activity (Fig. 1F). When combined with the M631L mutation, the polymerase activity of the avian-origin virus increased to levels comparable to that of the cow-adapted virus (Fig. 1G). As H5N1 clade 2.3.4.4b viruses continued to spread among cattle and other mammals, we detected the emergence of well-known mammalian-adaptive mutations E627K, Q591R and D701N, as well as several previously unrecognized substitutions including I647V, G685R, and K736R (Fig. 3A), all of which were shown to enhance viral polymerase activity and increase pathogenicity in mice (Fig. 5A–C). Furthermore, a major outbreak of H5N1 clade 2.3.4.4b with high mortality in marine mammals in South America during 2022–2023 was reported to carry PB2 Q591K and D701N mutations, both associated with mammalian adaptation and heightened virulence50,51,52. Given the growing diversity of PB2 mutations associated with mammalian adaptation, and the ongoing, sustained transmission of H5N1 among dairy cow populations, the virus is progressively acquiring enhanced capacity to infect and replicate in mammals, thereby increasing the risk of human infection.
It is well-documented that the interaction between host-specific ANP32 proteins and the influenza virus polymerase is a key determinant of polymerase activity34,36,37,38. By integrating structural analyses of the ANP32–polymerase complex with our functional studies, we characterized mammalian-adaptive mutations found in currently circulating H5N1 viruses. Interestingly, most of these mutations are clustered near the polymerase interface adjacent to ANP32, enhancing the virus’s ability to utilize mammalian ANP32 proteins and thereby facilitating cross-species adaptation (Fig. 4C–G). These findings support the critical role of the polymerase–ANP32 interaction in host adaptation and highlight the need for ongoing surveillance of these and other mutations at this functional interface.
In summary, our data demonstrate that the H5N1 clade 2.3.4.4b viruses are rapidly acquiring mammalian-adaptive mutations in the PB2 subunit that enhance polymerase activity, thereby increasing virulence and zoonotic potential (Fig. 6). In line with the framework of adaptive evolution for zoonotic influenza A viruses18, polymerase mutations often arise early during cross-species infection, lowering the initial barrier to mammalian adaptation. The persistent circulation of these polymerase-adapted viruses in mammals provides an evolutionary platform that may facilitate the accumulation of further critical changes, such as alterations in HA receptor binding preference53 or escape from host restriction factors16, thereby increasing the likelihood of the virus achieving pandemic potential18. Therefore, targeted global surveillance of key adaptive mutations, coupled with functional characterization of emerging variants and the implementation of proactive containment strategies, is urgently needed to mitigate the threat of zoonotic spillover.

The schema highlights two significant H5N1 outbreaks associated with the PB2 M631L substitution: the 2013–2014 Cambodia and Vietnam outbreak and the ongoing 2024–2025 North American outbreak. During the North American H5N1 outbreak, transmission within cattle herds resulted additional adaptive substitutions (Q591R, I647V, G685R, D701N, D740N), together with the PB2 631L. Additionally, the K736R substitution was identified in H5N1 viruses infecting cats. The co-occurrence of 631L and E627K (M631L + E627K) has also emerged in cattle herds in 2025. The mutations E627K, Q591R, and D701N are well-known mammalian adaptive mutations, while I647V, G685R, and K736R represent newly discovered mammalian adaptive mutations in this study. These mutations further enhance viral polymerase activity in mammalian cells and increase viral pathogenicity in mice, posing a greater risk to mammals. Created in BioRender. lai, y. (2025) https://BioRender.com/x9td31y.
Methods
Ethics statement and biosafety
All animal experiments were performed according to the protocols approved by the Animal Use and Care Committee of the Institute of Biophysics, Chinese Academy of Sciences (ABSL-2-2025068). Before experimentation, animals were housed in individually ventilated cages under specific pathogen-free conditions, with a room temperature maintained between 20 °C and 24 °C, a 12 h/12 h light/dark cycle, and 40-70% humidity. Food and water were provided ad libitum. Humane endpoints for euthanasia included a body weight loss of 25% or more with no possibility of recovery. The rescue and infection of the recombinant viruses were performed under Biosafety Level 2 (BSL-2) containment at the Institute of Microbiology, Chinese Academy of Sciences. All experiments were approved by the Institutional Biosafety Committee of the Institute of Microbiology, Chinese Academy of Sciences.
Cells
Human embryonic kidney HEK 293T (ATCC, CRL-3216) cells and Madin-Darby canine kidney (MDCK) (ATCC, CCL-34) cells were purchased from the American Type Culture Collection and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, C11995500CP) supplemented with 10% fetal bovine serum (FBS; Gibco, 10437–028), and 1% penicillin-streptomycin (Pen-Strep; Gibco, 15140-122). Human embryonic kidney 293T ANP32A, ANP32B, and ANP32E triple-knockout cells (293T TKO48) were kindly provided by Prof. Xiaojun Wang (State Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, The Chinese Academy of Agricultural Sciences, Harbin, China) and were cultured under the same conditions as those described above for 293T cells. Cells were cultured in humidified incubators at 37 °C with 5% CO2.
Mice experiments
Six-week-old female BALB/c mice were purchased from HuaFuKang Bioscience Co., Ltd (Beijing, China) and were kept in individually ventilated cages under specific pathogen-free conditions, with room temperature maintained between 20 °C and 24 °C on a 12 h dark/12 h light cycle and 40–70% humidity. After acclimating for at least 3 days, mice were randomly allocated into different groups. For infection, mice were randomly allocated into different groups. Mice were anesthetized via intraperitoneal injection of 200 μL of 1.25% tribromoethanol and subsequently inoculated intranasally with 2000 PFU of each virus in 30 μL PBS. Mice were monitored for 2 weeks post-infection, with daily assessment of weight loss and other signs of disease. Any mouse losing ≥25% of its initial body weight was euthanized. To determined viral titers of lungs, three mice per group were euthanized on day 5 post-infection, and viral titers in lung tissues were determined by plaque assays. For histology, tissues were fixed in 4% buffered formalin, embedded in paraffin, sectioned and stained with hematoxylin and eosin (H&E). Tissue sections were also immunostained for viral nucleoprotein (NP) with a monoclonal primary antibody (Millipore, MAB8251).
Plasmids
The RNP reconstitution system plasmids (pcDNA-PB2, pcDNA-PB1, pcDNA-PA, pcDNA-NP) of influenza A/chicken/Maine/24-007946-001/2024 (chMA01-H5N1) and A/dairy cow/Ohio/078707-24-12/2024 (cowOH12-H5N1) viruses and the pCAGGS plasmids expressing cowANP32A (GenPept accession no. NP_001181948.1), cowANP32B (GenPept accession no. NP_001030246.1) were generated by gene synthesis (Azenta Life Sciences, China). pPOLI-NP/NCR-GFP was derived from the pPOLI-WSN/NP plasmid by subcloning the enhanced green fluorescent protein (eGFP) gene in place of the original NP ORF. pPOLI-Firefly was generated by replacing the firefly luciferase gene into the ORF region of the pPOLI-WSN/NP plasmid. The reverse genetics vectors of cowOH12-H5N1 were in vitro subcloned to pHW2000 (pHW2000-cowOH12-H5N1 PB2, PB1, PA, NP, M and NS). pHW2000-WSN/HA and pHW2000-WSN/NA were stored in our laboratory54. All mutant plasmids were generated by site-directed PCR mutagenesis (NEB, E0554S) and were identified by DNA sequencing.
Generation of recombinant viruses
Recombinant H5N1 viruses (the wild-type and PB2 mutants) were generated using a previously described eight-plasmid rescue system55. Briefly, 293T cells were co-transfected with pHW2000-H5N1-PB2 (or PB2 mutant), PB1, PA, NP, M, NS, and pHW2000-WSN-HA, NA plasmids. At 48 h post-transfection, the supernatant was harvested and the rescued viruses were plaque purified. The purified viruses were subsequently passaged for two generations on MDCK cells, and viral genomes were sequenced. Viral titers were determined by a standard plaque assay in MDCK cells.
Plaque assay
Confluent monolayers of MDCK cells (2.3 × 105 cells/well, 12-well plate) were seeded the day prior to infection. For each virus assayed, 10-fold serial dilutions (10-1 to 10-6) were prepared. For infection, cells were washed twice with PBS and 300 μL of each viral dilution were added. Viral absorption was allowed to occur for 1 h at room temperature and gently shaken every 15 min. After viral absorption, cells were overlaid with media with a final concentration of 1% low-melting agarose (Sigma-Aldrich, A9414). Infection was allowed to proceed for 3 days at 37 °C with 5% CO2. Subsequently, cells were fixed with 4% paraformaldehyde at 4 °C overnight. The supernatant was then removed and cells stained for 10 min with 1% crystal violet in PBS. Crystal violet was removed and cells carefully washed with water. Plaques were counted and PFU/ml were calculated.
Western blot
Cell pellets were lysed with RIPA Lysis Buffer (Beyotime, P0013B) for 25 min at room temperature, and then the debris were removed by centrifugation at 13,800 g for 15 min. The lysates were denatured with reducing SDS loading buffer and boiled at 95 °C for 5 min. Protein samples fractionated by SDS-PAGE were then transferred onto nitrocellulose membranes. The membranes were blocked with 5% skim milk in TBST and incubated with primary antibodies, followed by secondary antibodies. Protein expression levels were visualized using an enhanced chemiluminescence detection kit (NCM Biotech). Mouse monoclonal anti-β-actin (TransGene, HC201-01) and mouse monoclonal anti-Flag (TransGene, HT201-01) were purchased from TransGen Biotech Co., LTD (Beijing). Rabbit polyclonal anti-PB2 was stored in our laboratory56. Goat anti-rabbit IgG (H&L)-HRP conjugated (Easybio, BE0101-100) and goat anti-mouse IgG (H&L)-HRP conjugated (Easybio, BE0102-100) were purchased from Boao Yijie Technology Co., LTD (Beijing).
Mini-replicon GFP reporter assay
HEK 293T cells in 24-well plates were transfected with a mixture of plasmids pcDNA-PB2 (50 ng), pcDNA-PB1 (50 ng), pcDNA-PA (25 ng), and pcDNA-NP (100 ng) together with pPOLI-NP/NCR-GFP (25 ng) using Lipofectamine 2000 (Invitrogen, 11668019) according to the manufacturer’s instructions. The plasmids of chicken H5N1 and cow H5N1 polymerase subunits (PB2, PB1, PA) and NP were reciprocally exchanged on a one-to-one basis. After transfection for 24 h, the expression level of GFP was detected by fluorescence microscopy.
Mini-replicon luciferase reporter assay
To examine the effect of the PB2 mutations on viral polymerase activity, HEK 293T cells in 24-well plates were co-transfected with plasmids pcDNA-PB2 (50 ng), pcDNA-PB1 (50 ng), pcDNA-PA (25 ng), pcDNA-NP (100 ng), pPOLI-Firefly (50 ng), and a Renilla luciferase expression plasmid (5 ng) as an internal control for the dual-luciferase assay. To determine the effect of ANP32 proteins on viral polymerase activity, 293T TKO cells were co-transfected with the viral RNP reconstitution system plasmids together with a pCAGGS-ANP32 (10 ng) expression plasmid using Lipofectamine 2000 according to the manufacturer’s instructions. After transfection for 24 h, cells were lysed, and luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega, E1910) and a GLOMAX 96 Microplate luminometer (Promega). The relative polymerase activity was calculated by dividing the Firefly luciferase signal by the Renilla luciferase signal, followed by normalization. All experiments were independently replicated at least three times.
Primer extension analysis
Approximately 106 293T cells were co-transfected with 0.5 μg of protein expression plasmids encoding pcDNA-PB2, pcDNA-PB1, pcDNA-PA, pcDNA-NP and 0.5 μg of the vRNA expression plasmids pPOLI-WSN/NA using Lipofectamine 2000 and Opti-MEM (Gibco, 31985070) according to the manufacturer’s instructions. At 12, 24, 36, and 48 h post-transfection, total RNA was extracted using TRIzol reagent. The steady-state levels of mRNA, cRNA and vRNA were analyzed by primer extension with 32P-labeled specific primers for negative- or positive-sense RNA of NA. The primers used for the detection of the NA-specific RNA species and internal control 5S rRNA were described previously57. The primer extension products for 5S rRNA, vRNA, cRNA, and mRNA were approximately 100 nt, 130 nt, 160 nt, and 170-175 nt in length, respectively, consistent with previous reports58,59. These extension products were analyzed by 6% PAGE gel containing 7 M urea in Tris-borate-EDTA (TBE) buffer and detected by autoradiography.
Bioinformatics analysis
Sequences from H5N1 clade 2.3.4.4b viruses were retrieved from the GISAID database (http://gisaid.org/) for avian, dairy cows, humans or other mammals. Sequences were aligned using MAFFT (v7.526). Specific residues of interest were summarized along with sequence metadata using a custom Python script and plotted using the R program (v4.4.1). The custom code has been deposited on GitHub (https://github.com/laiyuerong/H5N1).
Phylogenetic analysis
PB2 amino acid sequences of H5N1 viruses isolated from human, mammalian, and avian infections in Cambodia and Vietnam during 2013 and 2014 were obtained from the GISAID database. The dataset included 138 sequences derived from avian viruses (of which 92 strains carried the 631M residue and 44 strains carried 631L), 24 from human viruses (all of which possessed the 631L), one from tiger virus (carried the 631L), and one from civet cat virus (carried the 631L). The sequences were aligned using MAFFT (v7.526) and a maximum likelihood phylogenetic tree was constructed using MEGA (v7.0.20) software with 1000 bootstrap replications. The analysis revealed that all sequences with the M631L mutation clustered together, while the 92 sequences carrying 631M were located at the base of the phylogenetic tree. For clearer visualization, 15 representative sequences with 631M were selected and analyzed together with all 631L mutant sequences to generate a final maximum likelihood tree using MEGA (v7.0.20).
Statistics
GraphPad Prism software (v8) was used for statistical analysis. One-way analysis of variance (ANOVA) with Dunnett’s correction and two-way ANOVA with Dunnett’s multiple comparisons test were used for comparisons.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated in this study are provided in the main text, Supplementary Information, and Source Data file. The virus sequence data analyzed were obtained from the GISAID EpiFlu Database (accession numbers are available in Supplementary Data 1). Source data are provided with this paper.
Code availability
The code used to perform the analyses in this study is publicly available and has been deposited in a GitHub repository (available at https://github.com/laiyuerong/H5N1).
References
Peacock, T. et al. The global H5N1 influenza panzootic in mammals. Nature 637, 304–313 (2024).
Imai, M. et al. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 486, 420–428 (2012).
Wang, D., Zhu, W., Yang, L. & Shu, Y. The epidemiology, virology, and pathogenicity of human infections with avian influenza viruses. Cold Spring Harb. Perspect. Med. 11, a038620 (2021).
Liu, J., Gao, G. F., Yuen, K.-Y., Leo Poon, L. M. & Song, N. Time is now: Preparing for the next pandemic. hLife 3, 113–117 (2025).
World Health Organization. Cumulative Number of Confirmed Human Cases for Avian Influenza A(H5N1) Reported to WHO, 2003-2024, 12 December 2024, <https://www.who.int/publications/m/item/cumulative-number-of-confirmed-human-cases-for-avian-influenza-a(h5n1)-reported-to-who–2003-2024–20-december-2024> (2024).
Xie, R. et al. The episodic resurgence of highly pathogenic avian influenza H5 virus. Nature 622, 810–817 (2023).
Caserta, L. C. et al. Spillover of highly pathogenic avian influenza H5N1 virus to dairy cattle. Nature 634, 669–676 (2024).
Tarbuck, N. et al. Detection of A(H5N1) influenza virus nucleic acid in retail pasteurized milk. Res. Square https://doi.org/10.21203/rs.3.rs-4572362/v1 (2024).
Nguyen, T. Q. et al. Emergence and interstate spread of highly pathogenic avian influenza A(H5N1) in dairy cattle in the United States. Science 388, eadq0900 (2025).
Burrough, E. R. et al. Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4b Virus Infection in Domestic Dairy Cattle and Cats, United States, 2024. Emerg. Infect. Dis. 30, 1335–1343 (2024).
Chothe, S. K. et al. Marked Neurotropism and Potential Adaptation of H5N1 Clade 2.3.4.4.b Virus in Naturally Infected Domestic Cats. Emerg. Microbes Infect. 14, 2440498 (2024).
Garg, S. et al. Highly Pathogenic Avian Influenza A(H5N1) Virus Infections in Humans. N. Engl. J. Med 392, 843–854 (2024).
Morse, J. et al. Influenza A(H5N1) Virus Infection in Two Dairy Farm Workers in Michigan. N. Engl. J. Med 391, 963–964 (2024).
Uyeki, T. M. et al. Highly Pathogenic Avian Influenza A(H5N1) Virus Infection in a Dairy Farm Worker. N. Engl. J. Med 390, 2028–2029 (2024).
CDC. H5 Bird Flu: Current Situation, <https://www.cdc.gov/bird-flu/situation-summary/index.html> (2025).
Pinto, R. M. et al. BTN3A3 evasion promotes the zoonotic potential of influenza A viruses. Nature 619, 338–347 (2023).
Mehle, A., Petric, P. P., Schwemmle, M. & Graf, L. Anti-influenza A virus restriction factors that shape the human species barrier and virus evolution. PLOS Pathog. 19, e1011450 (2023).
Long, J. S., Mistry, B., Haslam, S. M. & Barclay, W. S. Host and viral determinants of influenza A virus species specificity. Nat. Rev. Microbiol 17, 67–81 (2019).
Kupferschmidt, K. Why hasn’t the bird flu pandemic started?. Science 386, 1205–1206 (2024).
Shi, Y., Wu, Y., Zhang, W., Qi, J. & Gao, G. F. Enabling the ‘host jump’: structural determinants of receptor-binding specificity in influenza A viruses. Nat. Rev. Microbiol 12, 822–831 (2014).
Mair, C. M., Ludwig, K., Herrmann, A. & Sieben, C. Receptor binding and pH stability - How influenza A virus hemagglutinin affects host-specific virus infection. Biochim Biophys. Acta 1838, 1153–1168 (2014).
Gilbertson, B., Duncan, M. & Subbarao, K. Role of the viral polymerase during adaptation of influenza A viruses to new hosts. Curr. Opin. Virol. 62, 101363 (2023).
Te Velthuis, A. J. W. & Fodor, E. Influenza virus RNA polymerase: insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol 14, 479–493 (2016).
Subbarao, E. K., London, W. & Murphy, B. R. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J. Virol. 67, 1761–1764 (1993).
Hatta, M., Gao, P., Halfmann, P. & Kawaoka, Y. Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 293, 1840–1842 (2001).
Mehle, A. & Doudna, J. A. Adaptive strategies of the influenza virus polymerase for replication in humans. Proc. Natl. Acad. Sci. USA 106, 21312–21316 (2009).
Bussey, K. A., Bousse, T. L., Desmet, E. A., Kim, B. & Takimoto, T. PB2 residue 271 plays a key role in enhanced polymerase activity of influenza A viruses in mammalian host cells. J. Virol. 84, 4395–4406 (2010).
Li, Z. et al. Molecular basis of replication of duck H5N1 influenza viruses in a mammalian mouse model. J. Virol. 79, 12058–12064 (2005).
Peacock, T. P. et al. Mammalian ANP32A and ANP32B Proteins Drive Differential Polymerase Adaptations in Avian Influenza Virus. J. Virol. 97, e0021323 (2023).
Kirui, J., Bucci, M. D., Poole, D. S. & Mehle, A. Conserved features of the PB2 627 domain impact influenza virus polymerase function and replication. J. Virol. 88, 5977–5986 (2014).
Arai, Y. et al. PA Mutations Inherited during Viral Evolution Act Cooperatively To Increase Replication of Contemporary H5N1 Influenza Virus with an Expanded Host Range. J. Virol. 95, e01582–20 (2020).
Lutz, M. M., Dunagan, M. M., Kurebayashi, Y. & Takimoto, T. Key Role of the Influenza A Virus PA Gene Segment in the Emergence of Pandemic Viruses. Viruses-Basel 12, 365 (2020).
Bussey, K. A. et al. PA residues in the 2009 H1N1 pandemic influenza virus enhance avian influenza virus polymerase activity in mammalian cells. J. Virol. 85, 7020–7028 (2011).
Long, J. S. et al. Species difference in ANP32A underlies influenza A virus polymerase host restriction. Nature 529, 101–104 (2016).
Domingues, P. et al. Profiling host ANP32A splicing landscapes to predict influenza A virus polymerase adaptation. Nat. Commun. 10, 3396 (2019).
Carrique, L. et al. Host ANP32A mediates the assembly of the influenza virus replicase. Nature 587, 638–643 (2020).
Staller, E. et al. Structures of H5N1 influenza polymerase with ANP32B reveal mechanisms of genome replication and host adaptation. Nat. Commun. 15, 4123 (2024).
Arragain, B. et al. Structures of influenza A and B replication complexes give insight into avian to human host adaptation and reveal a role of ANP32 as an electrostatic chaperone for the apo-polymerase. Nat. Commun. 15, 6910 (2024).
Zhang, H. L. et al. Fundamental contribution and host range determination of ANP32A and ANP32B in influenza A virus polymerase activity. J. Virol. 93, e00174–19 (2019).
Staller, E. et al. ANP32 proteins are essential for influenza virus replication in human cells. J. Virol. 93, e00217–e00219 (2019).
Gu, C. et al. A human isolate of bovine H5N1 is transmissible and lethal in animal models. Nature 636, 711–718 (2024).
Bayoumi, M. et al. Identification of amino acid residues in polymerase PB2 responsible for differential replication and pathogenicity of avian influenza virus H5N1 isolated from human and cattle in Texas, US. Emerg. Microbes Infect. 14, 2542247 (2025).
Idoko-Akoh, A. et al. Creating resistance to avian influenza infection through genome editing of the ANP32 gene family. Nat. Commun. 14, 6136 (2023).
Zhang, X. et al. Enhanced pathogenicity and neurotropism of mouse-adapted H10N7 influenza virus are mediated by novel PB2 and NA mutations. J. Gen. Virol. 98, 1185–1195 (2017).
USDA. APHIS Confirms D1.1 Genotype in Dairy Cattle in Nevada, <https://www.aphis.usda.gov/news/program-update/aphis-confirms-d11-genotype-dairy-cattle-nevada-0> (2025).
Sugiyama, K., Kawaguchi, A., Okuwaki, M. & Nagata, K. pp32 and APRIL are host cell-derived regulators of influenza virus RNA synthesis from cRNA. eLife 4, e08939 (2015).
Long, J. S. et al. Species specific differences in use of ANP32 proteins by influenza A virus. eLife 8, e45066 (2019).
Zhang, Z. et al. Selective usage of ANP32 proteins by influenza B virus polymerase: Implications in determination of host range. PLoS Pathog. 16, e1008989 (2020).
Kupferschmidt, K. How did cow flu start? Scientists still don’t know. Science 387, 1342–1342 (2025).
Leguia, M. et al. Highly pathogenic avian influenza A (H5N1) in marine mammals and seabirds in Peru. Nat. Commun. 14, 5489 (2023).
Pardo-Roa, C. et al. Cross-species and mammal-to-mammal transmission of clade 2.3.4.4b highly pathogenic avian influenza A/H5N1 with PB2 adaptations. Nat. Commun. 16, 2232 (2025).
Uhart, M. M. et al. Epidemiological data of an influenza A/H5N1 outbreak in elephant seals in Argentina indicates mammal-to-mammal transmission. Nat. Commun. 15, 9516 (2024).
Jassem, A. N. Critical Illness in an Adolescent with Influenza A(H5N1) Virus Infection. N. Engl. J. Med 392, 927–929 (2024).
Zhang, L. et al. Fine regulation of influenza virus RNA transcription and replication by stoichiometric changes in viral NS1 and NS2 proteins. J. Virol. 97, e0033723 (2023).
Hoffmann, E., Neumann, G., Kawaoka, Y., Hobom, G. & Webster, R. G. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. USA 97, 6108–6113 (2000).
Zhang, L. et al. Twenty natural amino acid substitution screening at the last residue 121 of influenza A virus NS2 protein reveals the critical role of NS2 in promoting virus genome replication by coordinating with viral polymerase. J. Virol. 98, e0116623 (2024).
Robb, N. C., Smith, M., Vreede, F. T. & Fodor, E. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J. Genl Virol. 90, 1398–1407 (2009).
Zhang, L. et al. Influenza A virus NS2 protein acts on vRNA-resident polymerase to drive the transcription to replication switch. Nucleic Acids Res 53, gkaf027 (2025).
te Velthuis, A. J. W., Robb, N. C., Kapanidis, A. N. & Fodor, E. The role of the priming loop in influenza A virus RNA synthesis. Nat. Microbiol. 1, 16029 (2016).
Acknowledgements
This work was supported by grants from the National Key R&D Program of China (2022YFF1203200 to T.D.), the National Natural Science Foundation of China (32400123 to L.Z. and 82472248 to T.D.), Beijing Natural Science Foundation (L242116 to T.D.), Shenzhen Science and Technology Program (JCYJ20240813112504007 and JCYJ20250604180053068 to L.Z.), and Autonomous Deployment Project of State Key Laboratory of Biopharmaceutical Preparation and Delivery (2024-FX-A-06 to Y.C.). We thank Prof. Xiaojun Wang of the Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences, for providing the 293T TKO cells. We thank Xinyi Wu (IBP, CAS), Longlong Chen (IBP, CAS), Xin Wen (IBP, CAS) for breeding and management of laboratory animals, excellent animal technical support and experimental data collection services. We thank the data contributors the EpiFlu Database of the Global Initiative on Sharing All Influenza Data (GISAID).
Author information
Authors and Affiliations
Contributions
T.D., G.F.G., and L.Z. conceived the study. L.Z., Y.L., and L.W. performed bioinformatic analyses. L.Z., Y.L., Y.C., Q.Y., Y.S., S.D., and H.W. conducted biologic experiments. L.Z. and Y.L. analyzed the structures. T.D., G.F.G., L.Z. and Y.L. wrote the manuscript. All authors participated in the discussion and manuscript editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kevin Ciminski and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, L., Lai, Y., Cui, Y. et al. Emergence of mammalian-adaptive PB2 mutations enhances polymerase activity and pathogenicity of cattle-derived H5N1 influenza A virus. Nat Commun 17, 1011 (2026). https://doi.org/10.1038/s41467-025-67753-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67753-x
