
Abstract
We present a large-scale study of structural variation (SV) in the Qatari population, based on short-read whole-genome sequencing (WGS) of 6,141 individuals, identifying 153,946 variants across 5 classes reflecting the region’s diversity and evolutionary history. Leveraging consanguinity and biobank phenotypes, we identify >180 putative gene knockouts, and use proteomics to show functional consequences in homozygotes. Conversely, 52 genes show significant depletion of homozygous deletions, eight of which cause severe pediatric disease or murine embryonic lethality. Examining phenotypic extremes uncovers several non-exonic homozygous deletions with large effect, including in SPIRE2 (creatinine), MAGI2 (leanness) and a chr19 microRNA cluster (extreme obesity). Further, SV-GWAS reveals gene-trait associations independent of SNPs, including at ACY1 (acetylation), SLC2A9 (uric acid), UGT1A8 (bilirubin) and ZNF251 (alanine aminotransferase). Notably, 3.2% of Qataris carry findings in medically actionable genes, one-third attributable to SVs. Our findings offer a rich SV reference for a globally understudied population, and demonstrate the utility of consanguineous biobanks for studying SVs in health and disease. All common SVs and tag-SNPs are provided as imputation resource.
Similar content being viewed by others

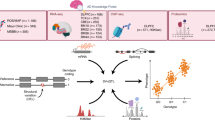

Introduction
The promise of precision population health can only be achieved once definitive, high-quality reference datasets are established and become openly accessible to scientific and medical communities worldwide. Yet, despite the rapid growth of public databases, populations of Arab ancestries remain under-represented1,2,3.
The Qatar Genome Programme (QGP) is the most extensively published to date, with a pilot phase dataset of 6,141 individuals for whom both genomic and phenotypic information from the Qatar Biobank (QBB) are made available4,5,6,7,8,9. However, most genetic studies on the Qatari population to date have focused on small variant classes, leaving the landscape and biomedical impact of structural variation (SVs) largely unexplored. Yet, this important class of variation, ranging in size from 50 bp up to 10 megabases, as longer SVs were removed3 is pervasive throughout genomic space, impacts an order of magnitude more bases than smaller variants do per individual, and contributes prominently to human health and disease7,8,10,11,12,13,14,15,16,17,18,19. In this study, we combine multiple approaches for SV inference into an ensemble pipeline and apply it to 6141 Qatari genomes. We report patterns and distribution of SVs across the five main Qatari subpopulations6, and assess their impact on both biobank quantitative traits and proteomics data available at the Qatar Biobank (QBB). Finally, we leverage high consanguinity rates in this population to identify genes depleted for homozygous deletions as well as predicted loss-of-function (pLoF) alleles appearing in homozygous state, and use proteomic data to validate the functional impact of the latter.
Results
Population-level SV characteristics
Using a pipeline with 10 tools (Supplementary Table 1) and stringent QC criteria (Fig. 1), a total of 153,946 high-confidence SVs were detected in 6141 individuals from the Qatari population (Supplementary Table 2), comprising 74,570 deletions, 17,147 duplications, 36,153 insertions, 20,628 translocations, and 5448 inversions (Fig. 2a; Supplementary Data 1), with a median size of 394 bp (Range: 50 bp – 9.94 Mb) (Fig. 2b; Supplementary Data 1). As expected, most SVs were rare (86.9 % with allele frequency (AF) < 1%), of which approximately one-third (34.4%) were singletons overall and nearly one-half (45.3%) were singletons once only unrelated individuals were considered. (Fig. 2c, Methods).

Our SV consensus calling pipeline begins by running the 10 SV inference tools, followed by quality control checks, consensus call generation for individual samples, re-genotyping of sample-level consensus call sets, population-level consensus call set creation, and extensive annotation.

a Distribution of SVs using the log scale, showing the number of deletions, duplications, insertions, translocations and inversions recovered by our SV consensus calling pipeline. b Size vs number of inferred SVs: most SVs were smaller than 100 kb. c, Most SVs were rare (allele frequency <1% in 86.8% of all SVs). d, e Principal component analysis (PCA) of common SV genotypes and SNP genotypes, shown as PC1 vs PC2. Points are colored by genetic ancestry and shaped by cohort (QGP, TGP). Figure e has been adapted from Razali et al. 20216. f pairwise FST values between populations, with colors ranging from blue (low differentiation, 0) to red (high differentiation, 0.06). g Waterfall plot depicting the SV counts per subpopulation. h, i Summary box plots showing per-sample numbers of homozygous (h) and heterozygous (i) deletions across QGP ancestries (n = 6141), with significant enrichment (determined using the two-sided Wilcoxon test) indicated relative to Peninsular Arabs (light blue) for homozygous deletions and relative to African Arabs (orange) for heterozygous deletions. Boxes show the median (centre line) and 25th–75th percentiles (bounds); whiskers extend to 1.5× IQR; points beyond whiskers are outliers. No error bars are plotted beyond box-plot elements. Pairwise p-values (2 h, vs PAR): PAR vs QGP_ADM, p < 2.2 × 10⁻¹⁶; PAR vs QGP_AFR, p < 2.2 × 10⁻¹⁶; PAR vs QGP_GAR, p < 2.2 × 10⁻¹⁶; PAR vs QGP_SAS, p = 7.71×10⁻¹²; PAR vs QGP_WEP, p < 2.2 × 10⁻¹⁶. Exact p-values (2i, vs AFR): AFR vs QGP_ADM, p < 2.2 × 10⁻¹⁶; AFR vs QGP_PAR, p < 2.2 × 10⁻¹⁶; AFR vs QGP_GAR, p < 2.2 × 10⁻¹⁶; AFR vs QGP_SAS, p < 2.2 × 10⁻¹⁶; AFR vs QGP_WEP, p < 2.2 × 10⁻¹⁶.
On average, each individual harbored 5026 SVs (SD = 493), including 2827 deletions (range: 1430–3546), 281 duplications (range: 58–556), 1588 insertions (range: 463–2693), 130 inversions (range: 55–302), and 198 translocations (range: 4–869) (Fig. 2g; Supplementary Data 2). These distributions are largely consistent with reports in other populations, except for duplications, which were lower in our cohort likely because of the highly-stringent filters used in our study (Methods)2,3.
Population genetic structure
Previously, this cohort was shown to consist of five major ancestries: peninsular Arabs (QGP_PAR, n = 1052), general Arabs (QGP_GAR, n = 2,312), West Eurasian and Persian Arabs (QGP_WEP, n = 1374), South Asian Arabs (QGP_SAS, n = 38), and African Arabs (QGP_AFR, n = 179), plus 1186 admixed individuals (QGP_ADM)6 (Supplementary Table 2). Principal component analysis (PCA) using SVs (AF > 0.01; Hardy–Weinberg equilibrium (HWE) P > 1 × 10−4) closely reproduced the PCA results from SNP data (Fig. 2e), showing similarly distinguishable clusters for these five major genetic ancestries (Fig. 2d). As expected fixation index (FST) analysis showed QGP_SAS and QGP_ AFR having the highest degree of differentiation (Fig. 2f, Supplementary Data 3). Moreover, despite a similar overall number of variants per individual, African (QGP_AFR) ancestry had a higher fraction of rare (AF < 1%) and unique SVs (Wilcoxon P < 2.2 × 10−16; Supplementary Fig. 1). Allele frequency concordance between common structural variants in the QGP dataset and gnomAD shows a strong correlation of r² ≈ 0.70. Subpopulation-specific comparisons reveal similar correlations: QGP African (AFR) to gnomAD African (r² = 0.707), QGP South Asian (SAS) to 1000 Genomes South Asian as a proxy (r² = 0.703), and QGP West Eurasian/Persian (WEP) to gnomAD European (r² = 0.705) allele frequencies (Methods, Supplementary Fig. 2). SVs were enriched around repetitive regions, including telomeres and centromeres, and the HLA region3,20 (Supplementary Fig. 3a–c). Of 20,628 translocations, 92.4% (n = 19,067) were located close to repetitive elements with relatively equal distribution across all chromosomes. This enrichment may reflect repeat expansions or transposon-mediated insertions rather than reciprocal chromosomal arm exchanges, consistent with prior observations21. Among classified translocation events, 48.20% were balanced-only, 16.08% unbalanced-only, and 35.72% both, indicating that most are balanced (Methods, Supplementary Data 1).
Biological impact of SVs
Genic content impacted by SVs
Upon annotation, 12,058 (9%) SVs overlapped at least one exon across 17,510 genes (Supplementary Table 3). These included 5487 deletions, 4380 duplications, 1748 inversions, and 443 insertions, overlapping 6098, 10,738, 9103, and 501 genes, respectively (Supplementary Table 3). The higher genic content impacted by duplications and inversions is partly explained by the larger median size (3083 bp and 4914 bp) compared to deletions (875 bp). Notably, 4390 were predicted to cause loss of function of 4036 genes, including 3301 deletions, 84 duplications, 68 insertions and 937 inversions, with an average of 74.4 genes affected by pLoF SVs per genome (Supplementary Fig. 4; Supplementary Data 1). Pathway analysis of genes impacted by SVs revealed a significant enrichment for genes related to dilated cardiomyopathy (FDR 0.1%), insulin signaling (FDR 0.1%), and cancer (FDR 2%) (Supplementary Data 4). Notably, the Qatari population is reported to have a high burden of all three conditions21,22,23,24,25, with some sub-pathways such as those related to cardiomyopathy (arrhythmogenic right ventricular cardiomyopathy, dilated cardiomyopathy, and hypertrophic cardiomyopathy) appearing enriched in multiple subpopulations (QGP_PAR, QGP_GAR, QGP_WEP; FDR < 5%). Additionally, 3676 SVs overlapped topologically associating domains (TADs) defined by 32 distinct ENCODE or Roadmap chromatin-conformation experiments (954 deletions, 1705 duplications, and 1017 inversions) and 1427 SVs overlapped 1029 microRNA (miRNA) genes (445 deletions, 551 duplications, and 431 inversions) (Supplementary Data 1).
Actionable genes
We investigated the impact of pLoF SVs on 97 clinically actionable genes (ACMG v3.2 guidelines26). We identified 17 pLoF SVs affecting 12 ACMG genes in 58 individuals (1%) in our cohort, of which 13 were singletons. Notably, all of these were in heterozygous state, and 7 (41%) were novel across global SV repositories. Six genes related to cardiovascular health, including catecholaminergic polymorphic ventricular tachycardia, dilated cardiomyopathy, familial hypercholesterolemia, and long-QT syndrome types 1 and 14 (Supplementary Table 4).
Upon further manual curation, nine deletions met ACMG criteria for CNV reporting27. We investigated biobank phenotypic and questionnaire data28 on 25 carriers to assess functional impact (Supplementary Fig. 5) and found evidence in three individuals who share a 7.34 kb heterozygous exon 2 deletion in KCNQ1, a gene associated with Long QT syndrome 1 (MIM#192500, Mendelian Inheritance in Man index from the Online Mendelian Inheritance in Man database): one 57-year-old female carrier presented with consistent ECG findings – left atrial enlargement, a prolonged QT interval, and inferior myocardial infarction – while the two other carriers had abnormal ECG readings as well, including T-wave abnormalities, with lateral ischemia in one subject, and abnormal junctional rhythm in the other.
SV tagging by common SNPs
We investigated the extent to which common SVs in this cohort (8848 deletions, 1572 duplications, 552 inversions and 7843 insertions with AF > 0.01) were tagged by neighboring SNPs and what fraction of these were previously reported in the GWAS catalog29 (Supplementary Data 5). We identified 14,332 SVs tagged by 1,450,047 SNPs (r² > 0.1, distance <1 Mb), of which only 4652 (52.6 %) deletions, 198 (12.6 %) duplications, 161 (29.2 %) inversions and 581 (7.4 %) insertions were adequately tagged (r2 > 0.7, distance <1 Mb) by 176,613 unique SNPs in our population (Supplementary Fig. 6). Allele frequencies of these tagging SNPs showed high concordance with external datasets, with r² values exceeding 0.90 in all comparisons (r² = 0.92 vs. 1000 Genomes, r² = 0.94 vs. gnomAD, r² = 0.98 vs. GME, Supplementary Fig. 7). Notably, only 2857 of these overlap SNPs reported in the GWAS catalog (tagging 992 of our SVs, including 112 SVs novel in our dataset) (Supplementary Data 6).
A total of 1460 SNPs tag 42 SVs predicted to have a pLoF effect. These include 5 with previously reported GWAS associations (eg., deletions affecting LCE3B and LCE3C linked to psoriasis3,30) and 37 potentially functional SVs tagged by common SNPs in this population, which are absent from the GWAS database, including the 23 kb deletion in CD36, which is associated with hematological traits and discussed in detail in the next section.
Impact of consanguinity
Homozygous deletions and gene knockouts
Overall, 12% of the deletions found in QGP_PAR, QGP_GAR and QGP_WEP had distributions that deviated from HWE, reflecting the high levels of homozygosity in the population – a phenomenon previously observed for SNPs and indels in Qataris, driven by high ( > 50%) consanguinity rates6,31 (Supplementary Fig. 8). Inbreeding coefficients from SVs matched those derived from SNVs (median F = 0.11), with significantly higher levels in the peninsular Arab (QGP_PAR) group compared to the other subpopulations6 (Supplementary Fig. 9a, Wilcoxon P < 1.1 × 10−14), and lowest in QGP_AFR. Indeed, QGP_PAR had, on average, >60 more (41%) homozygous SVs per individual than any other subpopulation (Wilcoxon P < 2.2 × 10−16) (Fig. 2h).
This elevated consanguinity offers a unique opportunity to identify potential human knockouts. Of 74,570 deletions, 15,931 (21.3%) were present in a homozygous state in at least one individual, of which 428 overlapped at least one exon, and 175 were predicted to result in loss-of-function of 186 genes. Most exonic homozygous deletions (n = 265; 61%) were rare, and 32 were unique (Supplementary Data 1). Notably, 1279 were novel to all databases, and 5564 had MAF < 1% and were not previously observed in a homozygous state despite substantially larger cohort sizes.
Functional impact of homozygous pLoF deletions from proteomic data
We proceed to investigate the functional consequences of these homozygous deletions using plasma proteomics data generated on the SOMAscan v4 platform (1148 proteins) from 2,935 participants. Given the overlap between genes affected by homozygous deletions, assay proteins, and assayed carriers, we found five genes (CAPG, HGFAC, GHR, TPSAB1, and CD36) for which pLoF impact could be assessed (Supplementary Fig. 10, Supplementary Data 7). Four of these showed negligible protein levels in homozygous deletion carriers, while the deletion of exon 3 of the GHR gene did not appear to have a significant effect, consistent with previous reports32 (Fig. 3).

P-values were calculated using Kruskal–Wallis test. Boxplots show the inverse-normalized protein levels amongst three genotypes in samples from Phase 1 of the QGP, where proteomics data are available (Nc=c2935). The center line in the box indicates the median, the lower and upper hinges indicate the first and third interquartile range (IQR). Pairwise p-values (0/0 vs 0/1; 0/0 vs 1/1; 0/1 vs 1/1): GHR 2.6e-09/0.0022/0.051; TPSAB1 5.5e-06/0.087/0.26; CD36 < 2.22e-16/2.7e-05/0.00061; HGFAC < 2.22e-16/0.00054/0.00096; CAPG 8.1e-06/0.083/0.18.
A notable example was the homozygous deletion (size = 23 kb; AF = 1.76%) overlapping exons 2 and 3 of CD36, a multi-ligand glycoprotein receptor that binds thrombospondin-1. This deletion was predicted pathogenic, and both heterozygous and homozygous deletion carriers had significantly lower CD36 protein levels (P < 2.22 × 10−16 and P = 2.7 × 10−5, respectively) compared to non-carriers (Fig. 3). Multiple phenotypes (MIM#610938, MIM#611162) have been reported for this gene, including susceptibility to macrothrombocytopenia (MIM#608404), and associations with hematologic traits, including white and red cell count, mean cell hemoglobin and total hemoglobin concentration31,33,34,35. Notably, none of the reported SNPs was in LD (r2 > 0.7) with this deletion in our population, suggesting an independent haplotype segregating in the Qatari population (Supplementary Data 6). Heterozygous and homozygous carriers had significantly higher red blood cell counts (Kruskal–Wallis P = 2.28 × 10−6) and significantly lower white blood cell counts (Kruskal–Wallis P = 1.82 × 10−11), mean cell hemoglobin concentrations (Kruskal–Wallis P = 1.02 × 10−7), and mean cell hemoglobin (Kruskal–Wallis P = 4.81 × 10−8; Fig. 4) compared to controls.

a A 23-kb deletion (AF 0.01759) identified on chromosome 7 and located in region between intron 1 and intron 3 of the CD36 gene showed decreased expression in heterozygous and homozygous carriers in proteomics data. Based on reported phenotypes in OMIM, we performed a quantitative analysis of the effect of this deletion on blood cell-related phenotypes. a white blood cells (N = 5995); b red blood cells (N = 5995); c mean cell hemoglobin concentration (N = 5995); d mean cell hemoglobin (N = 5995); e platelet counts (N = 5923); f mean platelet volume (N = 5994); g hemoglobin (N = 5994); h hematocrit (N = 5995); and i neutrophil hematocrit (N = 5990). P-values were calculated using Kruskal–Wallis test. Boxplots show the different values between three genotypes. The center line in the box indicates the median, the lower and upper hinges indicate the first and third interquartile range (IQR).
Impact of homozygous deletions on phenotypic extremes
Rare variants with large effects have been shown to drive phenotypic extremes across a range of human traits36,37,38. We hypothesized that individuals at the tails of distribution for the available phenotypes in the biobank might carry rare SVs with large effect28. We focused on homozygous genic deletions shared by at least two individuals, all of whom were within the top or bottom 5% of a given trait’s distribution.
We identified two such deletions, each in three individuals: a 3 kb deletion in intron 2 of MAGI2, previously associated by GWAS with BMI39,40, and a 2 kb deletion in intron 1 of SPIRE2, previously associated with creatinine levels41 (Supplementary Table 5, Supplementary Fig. 11). All three homozygous carriers of the MAGI2 deletion placed in the bottom 5% of the BMI distribution, and all three homozygous carriers of the SPIRE2 deletion were in the top 5% of creatinine levels. Neither of these deletions was tagged by common SNPs in our cohort or previously reported in association with these phenotypes (Supplementary Data 5).
We next investigated 55 homozygous exonic deletions in genes with known disease associations reported in OMIM, of which 11 (LPP, SYNE1, PMPCB, KIAA0556, ABCB7, AVPR2, DMD, TAF1, POF1B, CHM, and XPNPEP2) were novel (Supplementary Data 1) and 13 were predicted to result in loss of function, with no homozygous carriers reported previously (Supplementary Table 6). Screening the QBB database for clinically relevant phenotypes is challenging, given that the cohort is largely comprised of healthy adult volunteers. Yet, we found one 62-year-old male carrier of a hemizygous in-frame, partial deletion in the DMD gene who had normal serum creatine‑kinase levels (76 U/L; ref 39–232 U/L) and no skeletal symptoms reported, but an abnormal ECG and history of heart revascularization following myocardial infarction, consistent with previous reports of carriers of similar DMD mutations42,43,44,45 (Supplementary Fig. 12).
Depletion of homozygous deletions
The high levels of consanguinity could also provide a unique opportunity to explore the opposite end of the spectrum; specifically genomic intervals where homozygous deletions are absent despite high carrier frequency, potentially highlighting critical genes essential for fitness. We identified 19 loci that had a significant depletion of homozygous carriers (HWE P < 1.01 × 10−5). These overlapped 52 genes, of which 8 are listed in OMIM (Table 1; Supplementary Table 7). As expected, homozygous carriers of these deletions have never been reported. Two of these deletions overlapped genes known to cause severe pediatric phenotypes including epilepsy (GOSR2) and dystonia (PRKRA). Six genes (GOSR2, NSF, PDE4DIP, PRKRA, WNT3, and WNT9B) are known to cause lethality at different stages of embryonic development when knocked out in mouse46 (Table 1). The remaining 44 genes have no known disease associations thus far.
Genome-wide SV associations
SV associations with phenotypes
We tested 25,826 autosomal SVs (AF ≥ 0.1 %, ≤ 2 Mb) against 42 inverse‑normalised traits with an additive linear mixed model adjusting for age, sex and a SNP‑based kinship matrix. We identified 17 significant associations (P <4.6 × 10−8, 0.05/ [25,826 SVs x 42 clinical traits]) involving 15 unique SVs with nine clinical phenotypes (Table 2; Supplementary Fig. 13; Supplementary Table 8). Only two of these SVs are tagged by common SNPs in our population at r2 > 0.7. Among the most biologically plausible associations, we observed a 1.3-kb deletion affecting the last exon of the transferrin gene (TF) associated with increased total iron binding capacity (P = 4.85 × 10−112) and universal iron binding capacity (P = 2.94 × 10−59). This SV is in LD (r2 = 0.99) with rs1830084 located 3’ of the TF gene, previously associated with serum transferrin levels in Europeans47.
Most SVs were not tagged by SNPs including a novel 231 bp deletion in intron 5 of Solute Carrier Family 2 Member 9 (SLC2A9) associated with decreased uric acid levels (P = 3.77 × 10−18) and a common 1.6-kb deletion in intron 1 of UDP Glucuronosyltransferase Family 1 Member A8 (UGT1A8) associated with increased bilirubin levels (P = 3.65 × 10−10). The latter had a > 2.4-fold higher allele frequency in QGP_PAR (AF: 0.12) as compared with the second-highest subpopulation in our cohort, QGP_GAR (AF: 0.05). Another ethnic-specific association was found between a rare 411 bp deletion in intron 4 of Zinc Finger Protein 251 (ZNF251) and levels of the alanine aminotransferase (ALT) enzyme (P = 8.71 × 10−11), only observed in individuals of Western Eurasian/Persian ancestry (QGP_WEP AF: 0.012).
Association of rare SVs with protein levels
Rare SVs (AF < 1%) were tested for association with the inverse-normalized levels of 1305 proteins using a linear mixed model accounting for relatedness by generating a kinship matrix and adjusting it for sex, age, and three genetic principal components. We found 20 significant SV associations (P < 6.18 × 10−10, 0.05/[61,898 rare SVs × 1305 proteins]) with 14 unique proteins (Table 3; Supplementary Table 9). Eight of these associations were in cis at the following loci: ACY1, ASAH2, C9, CD177, ERBB3, IDUA, LTF, and MICB. Interestingly, a rare novel 414 bp deletion on Chr3 was associated in cis with ACY1 and LTF (P = 3.20 × 10−12, and P = 1.36 × 10−10, respectively). One carrier of this homozygous deletion had the lowest ACY1 levels48. ACY1 is required to remove the acetyl group from acetylated amino acids. Data from a metabolomics study previously performed on QBB showed extremely high levels of various acetylated amino acids in this homozygous carrier, which were previously unexplained49 (Supplementary Fig. 14). This finding suggests that ACY1 activity may be disrupted by this deletion. As expected, none of these rare SVs were in LD (r2 > 0.7) with common SNPs.
Discussion
The Qatari population shares significant genetic ancestry with neighboring populations in the Arabian Gulf, all of whom remain significantly underrepresented in global genomic databases. Here, we present a catalog of high-resolution SVs from 6,141 individuals from the Qatar Biobank – the largest study of genomic structural variation in Arab populations to date. We further leverage the availability of high-quality phenotypic data and high levels of consanguinity to explore the distribution and biological impact of SVs on health and disease in the Qatari population.
First, using a unique combination of 10 tools and extensive quality control, we present a detailed map of 153,946 high-confidence SVs. Our benchmarking revealed that concordance among different tools remains a challenge, highlighting the importance of using multiple algorithmic approaches to confidently detect SVs from short-read WGS data. Principal Component Analysis (PCA) analysis closely mirrored SNP-based PCA in separating the 5 main ancestral subpopulations present in Qatar, further validating the pipeline and rigorous quality filters used in this study.
Next, we explored the impact of SVs on genes and biological pathways. A total of 12,058 SVs (9%) affected exonic regions across 17,510 genes. Pathway analysis suggested significant enrichment for genes associated with cardiometabolic traits, consistent with elevated prevalence of metabolic and cardiovascular diseases in the Qatari populations24,25,50. Additionally, 4,036 genes appeared to contain variants predicted to cause loss of function (pLoF), 186 (4.6%) of which had at least one homozygote. Of these pLoF variants, 17 affected 12 ACMG genes in 1% of our cohort, which in addition to the 2.3% actionable findings picked up from small variants51 raises the total to 3.2% of the population, or 1 in 30, having a medically actionable genomic finding in the Qatar Biobank, a third of which come from SVs. Among these, we were able to confirm functional impact of an exon 1 deletion in KCNQ1 gene, for which biobank phenotypes revealed abnormal ECG readings and family history of heart disease.
A recurrent notable finding in this study was the impact of non-exonic SVs on biological phenotypes. For instance, we find a duplication on chromosome 19 affecting a miRNA cluster that included miR519D, which has been independently shown to be overexpressed in subcutaneous adipose tissue from obese subjects52. While the previous study did not reveal an underlying genetic driver for this association, it is possible that the overexpression was driven by a similar duplication which increased dosage of this locus. In our cohort, all four carriers of this 40-kb duplication had elevated BMIs (mean: 38.86; SD: 1.92), significantly higher (t-test, P = 2.13 × 10−3) than the average BMI of 29.4 (SD: 6.02) (Supplementary Fig. 15a, b). Another example is the common deletion in intron 5 of the SLC2A9 gene, that we observe associated with decreased uric acid levels. SLC2A9 encodes glucose transporter-9, recently found to transport uric acid, with genetic variants of the transporter accounting for 1.7%–5.3% of variance in serum uric acid concentrations53,54,. A decrease in uric acid levels in carriers in our cohort suggests a functional consequence of intron 5 deletion on biological activity in vivo. Yet another example is the deletion at the 3’ end of the transferrin (TF) gene which correlated with significantly increased total and universal iron binding capacity in carriers. Diseases associated with TF include atransferrinemia and iron deficiency anemia, while related pathways include insulin receptor recycling and clathrin-mediated endocytosis. Elevated TF levels in deletion carriers suggest the deletion potentially removes a negative regulator in the 3’ UTR, leading to increased expression and the phenotypic consequences observed. Finally, we identified a number of population-specific associations, such as a ZNF251 deletion strongly associated with alanine aminotransferase (ALT) levels in individuals of Persian ancestry only.
We further investigated the impact of rare SVs on protein levels, uncovering 20 significant pair-wise associations. In particular, a rare novel intronic deletion at chr3:53,690,870-53,691,283 located 1.6 Mb downstream of the ACY1 gene was associated with decreased protein levels, alongside elevated levels of various acetylated amino acids, thus highlighting a direct link between genetic variation, protein expression and metabolic endpoints (Supplementary Fig. 16).
A limitation of our association results is that missing genotypes were assigned as reference, which may lead to an underestimation of structural variant burden, particularly in regions with low genotyping confidence or high rates of missingness. While this strategy avoids false positives, it may bias results against detecting true variants in low-quality or complex regions.
One distinct feature of the Qatari population is the high ( >50%) degree of consanguinity23, with QGP_PAR individuals harboring significantly higher fraction (up to 41% more) of homozygous SVs compared to other Qatari subpopulations5,6. These present a unique opportunity to examine natural human knockouts and their impact on phenotypes. Human knockouts are crucial for understanding gene function, offering unique insights into mechanisms. Their identification requires genetic characterization of a very large number of individuals from outbred populations, or a modest number of individuals from highly consanguineous populations. Here, we discovered 186 genes with at least one individual harboring a homozygous exonic pLoF deletion. These included 21 genes with reported OMIM phenotypes, of which four had homozygous carriers in the gnomAD database and 10 showed a high global allele frequency (AF >0.01). Additionally, 13 genes had high haploinsufficiency scores (pLI >0.9), with two deletions showing at least one homozygous carrier in the gnomAD database, suggesting that these genes may be non-essential. We exploited available proteomics data to functionally validate pLOF knockouts. Five genes had overlapping data (CAPG, HGFAC, GHR, TPSAB1, and CD36) and four exhibited negligible protein expression in homozygous knockout carriers, with the exception of the well‑characterised exon‑3 deletion in GHR (GHRd3), which yields a functional receptor isoform as previously reported55. Interestingly, common variants at CD36 have been associated with hematologic traits, which we confirmed in our cohort by observing a significant reduction in white blood cell counts and an increase in red blood cell counts. This homozygous deletion at CD36 detected in our study in 13 subjects was absent in gnomAD SV, despite its larger sample size and an allele frequency of 0.017 in our cohort.
We next explored the association between homozygous deletions and extreme phenotypes, and identified a homozygous deletion affecting intron 2 in MAGI2, a gene previously associated by GWAS with BMI39,40, and another one affecting intron 1 at SPIRE2, previously associated with creatinine levels41. In examining these individuals’ quantitative data from the biobank, we found all MAGI2 deletion homozygotes were below the 5th percentile for BMI, while SPIRE2 homozygotes were in the top 95th percentile for creatinine levels, further supporting the significant impact of knockout variants. Given that these deletions were intronic, we examined the entire length of both genes in each set of individuals and ruled out the existence of putatively damaging small mutations (SNVs/indels) on the same haplotype driving these phenotypic extremes. Together with our previous observations above, these data reinforce the notion that mutations in non-exonic regions may play an under-appreciated role in missing heritability in complex traits.
Consanguineous populations offer another advantage at the opposite end of the genotype frequency spectrum – a significant absence of homozygous deletions despite high heterozygote carrier frequency may reveal loci crucial for survival and fitness. Despite the modest size of our cohort, we identified 19 such loci, overlapping 52 genes. Two of these deletions affected genes with high haploinsufficiency scores linked to severe pediatric conditions like epilepsy56,57 and dystonia58, while six affected genes known to cause embryonic lethality in mouse models (Supplementary Table 7).
Finally, it’s important to acknowledge certain limitations faced by this study. First, while our SV detection pipeline underwent extensive quality control and validation, the accuracy and concordance of SV callers from short-read WGS data remain a challenge, as different underlying algorithms yield varying results, leading to potential false positives or undetected variants. Second, the use of short-read sequencing, while effective for a broad survey, fails to detect more complex structural variations, such as large insertions, intricate rearrangements, and/or complex regions where multiple variations were introduced over the course of evolutionary history, creating a genomic landscape that is difficult to resolve with short-read methods alone. As orthogonal platforms such as long-read sequencing become mainstream, sensitivity for complex SVs and specificity in breakpoint identification will undoubtedly improve; notably, their enhanced read length and accuracy should also reduce the proportion of missing genotypes that arise when short reads cannot be confidently mapped, particularly in certain ancestries where many SVs appear novel and might get penalized due to high specificity filters. Additionally, the depth of phenotypic data in biobanks, while valuable for population-scale studies, can limit the ability to fully explore the consequences of novel variants, particularly rare variants that may drive unique phenotypes. Thus, future studies incorporating long-read technologies and more comprehensive phenotyping, potentially in a larger cohort and across diverse ancestries, will enhance the robustness of our understanding of the impact of SVs on health, making it important to generate long-read references for globally underrepresented populations59.
In conclusion, we present a rich dataset of SVs that enhances the understanding of the genetic landscape of this understudied population and emphasizes the critical role of genomic-scale variants in driving genetic diversity and disease susceptibility. Our findings reveal novel associations between SVs and clinically relevant phenotypes, the majority of which are not captured by common SNPs, and position the Middle East as a prime region for further study, leveraging consanguinity and detailed phenotypic/multi-omic traits across large cohorts. Further, the discovery of several novel variants with large effects in non-coding regions underscores the critical role of structural variants outside of exons in genomic studies. Finally, we found that 29.7% of common SV’s (AF >0.01) and only a minor fraction (3.6%) of all SVs are adequately tagged by SNPs (r2 >0.7), and provide imputation information for the most common GWAS platforms as a valuable resource to centers conducting research on Arab populations worldwide.
Methods
Study Cohort
The cohort of 6,218 high-quality genomes was provided by the Qatar Genome Programme, of which 77 were excluded due to quality control (QC) issues such as gender mismatch, excess heterozygosity, or a call rate below 95%. Metrics used to evaluate the quality of 6,141 genomes appear in refs. 4,10,60 and (https://www.qphi.org.qa/genomicdata). A wide range of physical traits, biochemical measurements, and lifestyle information was collected via questionnaires from adult Qatari nationals and long-term residents28,61. Participants in the QBB undergo whole-genome sequencing (WGS). All subjects provided written informed consent. The Qatar Biobank (QBB) study was approved by the Qatar Biobank Institutional Review Board (IRB) Committee (IRB protocol E/2017/RES-ACC-0032/0002).
Whole-genome sequencing
Details regarding sample preparation, library preparation, clustering, and sequencing have been reported elsewhere5. Briefly, DNA extracted from blood samples was sequenced on an Illumina HiSeqX sequencer using a 150-base paired-end single-index-read format, achieving an average coverage of 30×. The sequencing reads were mapped to GRCh37 using BWAkit (version 0.7.1229)62
Determination of subject ethnicity
Subject ethnicity was determined as reported previously, and analysis was performed using ADMIXTURE, PCA, FST, and F3 methods6. Analysis was performed on autosomal single-nucleotide variants6.
SV-calling and discovery
We selected 10 SV-calling algorithms (Supplementary Table 1), aiming to cover all the algorithmic approaches commonly used for SV detection. To identify a comprehensive set of high-confidence SVs, we developed an ensemble pipeline combining 10 SV callers representing four orthogonal algorithmic approaches – read-depth, split-read, paired-end, and combinatorial approaches63,64,65,66,67,68,69,70,71,72(see Methods, Supplementary Table 1and Fig. 1). CNVnator66, ERDS68, and Genomestrip69 identify SVs by looking for changes in read depth (RD); Breakseq265 and Breakdancer64 use clusters of discordantly aligned paired-end reads (PE); SvABA71 is based on de novo genome assembly (AS); DELLY67, LUMPY (as part of the SpeedSeq SV discovery pipeline)73, WHAM72, and Manta70 combine PE with the identification of split reads (SR). The insertion and translocation datasets were generated using SV calls from Manta70. We used Delly to classify translocation calls, defining those with Cn > 2.5 or Cn < 1.5 as unbalanced. The SV-calling algorithms were run independently for each sample using default parameters. Due to differences in output content and format among tools, a set of tool-specific curation steps was required to standardize the outputs. Breakdancer and CNVnator text outputs were converted into standard variant call format (VCF) using their respective scripts. The Genomestrip multi-sample VCF file was converted to a single-sample VCF using bcftools/1.1174. In addition, SvABA reports SVs as breakends, and we developed a Java tool (https://github.com/idraktt/qgp_sv_paper/tree/main/htsSidra) to convert these SVs to deletions (DEL), duplications (DUP), translocations (TRA), and inversions (INV).
Identification of tool-specific SV quality thresholds
Despite several advancements in the field, the reliable detection of SVs remains challenging, with SV-calling algorithms often producing a high number of false positives63,75. To mitigate, we developed tool-specific quality thresholds as follows: we used twenty randomly selected trios belonging to an independent dataset sequenced on the same platform11. To determine tool-specific quality thresholds, we then merged the SV calls for the trios using SURVIVOR (version 1.0.7) and, for each trio, calculated the percentage of “putative de novo” SVs (i.e., SVs present in the child, but not overlapping with any SVs identified in the parents) at different thresholds for the quality parameters (e.g., number of reads, quality scores; Supplementary Data 8). Next, we binned the support filters into ten non-overlapping windows for each tool and determined the first window at which putative de novo variants for each child were at most 5%. These thresholds were then selected as the cutoffs for the primary analysis to call SVs across the 6,141 individuals (Supplementary Fig. 16, Supplementary Data 1).
Per-sample consensus call set
Per-sample consensus calls were generated using SURVIVOR v.1.0.776, and we retained only SVs reported by at least two tools. The sizes ranged from 50 bp to 10 Mb, concordant with the previous study3, and were considered overlapping SVs if they were of the same type (e.g., deletion) and their start and end coordinates were within 500 bp.
Regenotyping of the per-sample consensus call set
Combining per-sample consensus call sets using SURVIVOR v.1.0.776 from different SV callers introduces challenges due to discrepancies in genotyping values and variations in ‘FORMAT’ column data generated by each caller. To tackle these challenges, we conducted re-genotyping, whereby we removed the additional columns and performed breakpoint genotyping of SVs using SVTyper (version 0.7.0) with the default parameters. To add depth of sequence to support and filter SVs, we used Duphold (version 0.2.3)77 where for deletions we used DHFFC <0.7 and for duplications DHFBC >1.3 which is fold-change for the variant relative to the rest of the chromosome and fold-change for the variant depth relative to bins in the genome, respectively. Upset plots were used to examine concordance of SV callers for deletions (Supplementary Fig. 17), duplications (Supplementary Fig. 18), and inversions (Supplementary Fig. 19), and illustrate the importance of using a multi-algorithm calling approach, as different combinations can identify unique SVs that may be missed by other combinations. For example, the top two most effective combinations for identifying duplications were SpeedSeq + Manta (combining SR- and PE-based approaches) and ERDS + CNVnator (RD-based approaches), which identified 618,513 and 202,544 non-unique SVs in the 6,141 Qatari genomes, representing the total SVs detected by each combination in all samples rather than the consensus SVs within the population.
Population-level consensus call set
Population-level consensus calls were generated using SURVIVOR v.1.0.776. We used all SVs belonging to the per-sample consensus call set; as before, SVs were considered overlapping if they were of the same type and their start and end coordinates were within 500 bp, with a minimum SV size of 50 bp. All missing genotypes were converted to references using bcftools v1.9 plugin+missing2ref74. AF values were calculated using the allele_freq.py module of SVTyper (version 0.7.0)73. The final VCF file contained SV calls from 6141 samples.
Filtering out SVs overlapping with segmental duplications and ENCODE blacklist regions
Segmental duplication bed files (downloaded from hg19 tracks) and the ENCODE blacklist regions file78 were used to filter problematic regions of SVs called from our population-level consensus call set. We used bedtools intersect (v.2.26)79 to filter regions showing 90% minimum reciprocal overlap between our calls and the segmental duplications and ENCODE blacklist region files; SVs overlapping >90% were discarded from our analysis.
Unrelated allele‑frequency calculation
Pairwise kinship coefficients were computed with KING v2.3.880. For every pair showing second‑degree‑or‑closer relatedness, one sample was removed, reducing the dataset from 6141 to 3754 individuals. Allele‑frequency estimates were then recalculated on this subset; these values are included in the master structural‑variant call‑set (Supplementary Data 1) released with this study.
SV annotation
SVs identified at the population level were annotated using AnnotSV (version 2.2)81. We used VCF files without columns containing sample-level genotyping information as input. We set the SVinputInfo parameter to 1 to precisely extract the ALT and INFO fields from the structural variation data. This data was then included in the output file. A structural variant was considered novel when it was absent from four large population‑scale resources bundled with AnnotSV v2.2-(i) Database of Genomic Variants (DGV, 2020‑02‑25); (ii) 1000 Genomes Project SV map (Sudmant et al., 2015); (iii) gnomAD‑SV v2.1 (Collins et al., 2020); and (iv) the Hall‑lab meta‑catalogue of 17,795 genomes (Abel et al., 2018)-using AnnotSV’s default threshold of ≥ 70 % reciprocal overlap and identical SV type. For all other settings, we used the default values. All available annotations produced by AnnotSV 2.2 can be found at https://lbgi.fr/AnnotSV/annotations.
Structural‑variant allele‑frequency benchmarking
The QGP SV call‑set was re‑annotated using SVAFotate v0.0.182 and the reference file SVAFotate_core_SV_popAFs.GRCh37.bed.gz (downloaded from https://github.com/fakedrtom/SVAFotate). This composite BED merges population allele‑frequency annotations from the CCDG SV catalogue, gnomAD‑SV v4, and the high‑coverage 1000 Genomes Project SV call‑set (Supplementary Data 9).
Gene enrichment analysis
Genes affected by SVs in three major subpopulations (QGP_GAR, QGP_PAR, QGP_WEP) were evaluated against the Kyoto Encyclopedia of Genes and Genomes (KEGG) (version KEGG FTP Release 2024-01-22) using gprofiler283,84.
Evaluation of pLoF effect of SVs
To assess the pLoF impact of SVs, we annotated our SVs using the SVAnnotate module from GATK package version 4.4.0.085. The input parameter for this annotation process was based on encode gene annotations, version 4486.
Proteomics data
Based on aptamer binding technology, the SOMAscan platform (SomaLogic, Boulder, CO, USA)87 was used to quantify protein levels for 1305 blood-circulating proteins in 2935 samples from Phase 1 of the QGP88. Protein levels (processed relative fluorescence unit values) were log-scaled and adjusted for age, sex, the first ten genetic principal components, and HSP90 levels (SeqId 2625-53_4) using linear regression. Residuals from the regression models were further inverse-normalized to ensure normality before being associated with SVs.
Principal‑component analysis (PCA) of the normalised protein matrix shows no separation by ancestry; instead, the top PCs capture subtle technical factors such as low‑grade haemolysis, consistent with recent multi‑cohort reports that technical variation outweighs genetic structure in large plasma‑proteome datasets which can be seen below89 (Supplementary Fig. 20).
Selection of homozygous deletions for proteomics analysis
To select homozygous deletions for proteomics analysis, we filtered homozygous deletions evaluated as predicted loss-of-function (pLoF) events in the gene and had at least one individual carrying the variant in a homozygous state.
Depletion of homozygous deletions
We evaluated genotype distributions per SV under the null expectations set by the Hardy–Weinberg equilibrium (HWE; 1 = p2 + 2pq + q2). Here, we tabulated genotype distributions per population for each biallelic, autosomal SV, and computed a chi-squared test P-value using the “Hardy-Weinberg” package in R90. To identify SVs that violated HWE, we filtered our calls, retaining only the deletions that disrupted exonic regions, had zero homozygous carriers, and calls that significantly departed from HWE equilibrium (P < 0.05 / [4970 number of autosomal deletions that overlap exonic regions of genes and doesn’t have individuals with homozygous genotype] = 1.00 × 10−5).
Genome-wide SV associations
SV–phenotype association analysis with allele frequency AF > 0.1% and with size ≤ 2 megabases, was performed for the SV calls and 42 clinically relevant quantitative phenotypic traits collected at the QBB during volunteer admission and described elsewhere (Supplementary Table 8)60. Rank-based inverse normal transformation was applied to the traits prior to the association analysis. The association analysis was performed using the q.emmax test in the EPACTS v3.4.291 software pipeline using default parameters, with AF cutoff criteria of 0.1%. SNP files for the same individuals were used to generate the kinship matrix for 6141 individuals using the EPACTS v3.4.291 make-kin function, which was used as an input file along with the SV files for the q.emmax function. We used age and sex as covariates.
Association between proteins levels and rare SVs
Rare SV (AF < 1%) association with protein levels was performed using RVtests v2.1.092. To include related individuals, we first created a kinship matrix for the 2935 QBB participants using the vcf2kinship function of RVTests. We ran a linear mixed model adjusted for rare variants using a function, namely FastLMM from RVTests software, to generate summary statistics for the association of 61,898 rare SVs with the inverse normal distribution of 1,305 proteins. We adjusted for sex, age, and the first three genetic principal components. We used a genome-wide significance threshold of P< 0.05 / 61,898 rare SVs × 1305 proteins = 6.18 × 10^-10 to correct for multiple testing.
SNP tagging
To conduct SNP tagging, we generated artificial SNPs’ specific positions: one base pair before the start and one base pair after the end of each structural variation. For each of the 8,848 non-redundant deletions, 1572 duplications, and 552 inversions, and for 7843 insertions, we created a new VCF file containing these representative SNPs of SVs, incorporating the same genotype values as those reported for the SV genotypes. In other words, if an individual was heterozygous for a given SV, that individual’s two pseudo-SNPs would also be marked as heterozygous in the new VCF file. This approach allows each SV to be encoded as a pair of pseudo-SNPs, enabling downstream SNP-based tools, such as PLINK, to compute LD between these SVs (in SNP-like format) and nearby SNPs present in the WGS dataset. We concatenated dummy SNP and SV VCF files with the QGP cohort SNP VCF file using bcftools/1.1174 and ran PLINK (1.90b6.18)93 on the merged VCF file with --r2 and 2 Mbp window size options, before filtering for SNPs with r2 > 0.7. Then, SNPs with the highest r2 values were used to tag SNPs. When two or more tagging SNPs had the same r2 value, the closest SNP to the SV was selected. To identify SNPs previously reported in the GWAS catalog, we queried the GWAS catalog using the timesaveR::GWAS.catalog.(accessed on 27th September 2023) SNP function (https://github.com/alesssia/timesaveR) providing a list of SNPs tagged by SVs. Entries with a trait value not equal to 0 and entries with a P-value < 5 × 10−8 were retained for further analysis. The Infinium Global Diversity Array v1.0 was downloaded from (https://webdata.illumina.com/downloads/productfiles/global-diversity-array/infinium-global-diversity-array-8-v1-0-D1-manifest-file-csv.zip) and merged with a list of SNPs tagging SVs (r² > 0.1, distance <1 Mb), available on GitHub with a list of all SNPs tagged by SVs (https://github.com/idraktt/qgp_sv_paper/blob/main/data/all_taggins_snps_r0.1_snp_array.zip).
Coordinate lift‑over to GRCh38
To support future analyses and facilitate comparisons with studies that use more recent references, we include additional columns in our results that map variants to GRCh38 coordinates. Structural‑variant coordinates in the QGP callset were converted from GRCh37 to GRCh38 using CrossMap v0.6.194. The bed module was run with the chain file grch37_to_grch38.over.chain.gz (http://ftp.ensembl.org/pub/assembly_mapping/homo_sapiens/GRCh37_to_GRCh38.chain.gz). The resulting GRCh38 positions are provided in Supplementary Data 10.
Data visualization
A manual review of variants was performed using Integrative Genomics Viewer (IGV) (v2.4.0)95 and samplot (Version 1.0.17)96. Results were plotted using ggplot2 (version 3.3.5)97, upsetr library98 and scripts from the gnomad-sv pipeline3 within the R framework (v 3.4)99.
Data availability
The data collected or generated by QBB are made available to bona fide researchers employed within or otherwise contractually bound to public and private institutions that conduct scientific research and that meet the requirements detailed in the QBB Research Access policy. The raw, unfiltered individual-level calls have been deposited with the Qatar Biobank (QBB), now part of the Qatar Precision Health Institute (QPHI). Genome sequencing data and population-level multi-sample SV VCF generated in this study can be accessed through the project Accession ID: QF-QGP-RES-ACC-0032 upon submission and approval of a data access request. Data concerning phenotypic and genotypic characteristics of study participants, including proteomics data used in this study, cannot be made publicly available in accordance with their informed consent and national data-protection regulations. Researchers may apply for access via the Qatar Precision Health Institute (QPHI) portal at https://www.qphi.org.qa/research/how-to-apply. Applications are reviewed by the QPHI Access Committee and require approval by the QBB Institutional Review Board. Data access requests are typically processed within 12–20 days, depending on the completeness of the application and the required compliance checks. Access is governed by a Data Use Agreement that limits use of the data to the approved study scope, permits provision of de-identified data only with no attempts to re-identify individuals and strict confidentiality in line with Qatari law, and recognizes that QPHI remains the data owner. The whole-genome sequencing dataset of 20 trios used in this study was generated with written informed consent under the institutional review board of Sidra Medicine (IRB No. 1500767). Whole-genome sequencing data for these families have been submitted to the MSSNG project and are included in the DB7 release; controlled access can be requested by completing a data access agreement at https://research.mss.ng. For researchers affiliated with recognized institutions, MSSNG applications are typically reviewed by the independent Data Access Compliance Office within approximately 10 business days. Summary statistics from the genome-wide SV–trait association analyses and rare SV–protein level association analyses are available via Zenodo (DOI: 10.5281/zenodo.17602656) under a Creative Commons Attribution 4.0 International license. All other data supporting the findings of this study are available either within the article, the supplementary information and supplementary data files.
Code availability
The study utilized previously published analysis tools and custom tools described in the Methods section. Custom tools and scripts developed for this study are archived at Zenodo (https://doi.org/10.5281/zenodo.17604635) and are available in a public GitHub repository at https://github.com/idraktt/qgp_sv_paper.
References
Narasimhan, V. M. et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science 352, 474–477 (2016).
Abel, H. J. et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature 583, 83–89 (2020).
Collins, R. L. et al. A structural variation reference for medical and population genetics. Nature 581, 444–451 (2020).
Fakhro, K. A. et al. The qatar genome: a population-specific tool for precision medicine in the middle east. Hum. Gen. Variation 3, 16016 (2016).
Mbarek, H. et al. Qatar genome: Insights on genomics from the middle east. Hum. Mutat. 43, 499–510 (2022).
Razali, R. M. et al. Thousands of qatari genomes inform human migration history and improve imputation of arab haplotypes. Nat. Commun. 12, 5929 (2021).
Fakhro, K. A. et al. Copy number variations in the genome of the qatari population. BMC Genomics 16, 834 (2015).
Rossi, N. et al. Ethnic-specific association of amylase gene copy number with adiposity traits in a large Middle Eastern biobank. NPJ Genom. Med. 6, 8 (2021).
Rodriguez-Flores, J. L. et al. Indigenous arabs are descendants of the earliest split from ancient eurasian populations. Genome Res 26, 151–162 (2016).
Aamer, W. et al. Burden of mendelian disorders in a large middle eastern biobank. Genome Med 16, 46 (2024).
Abdi, M. et al. Genomic architecture of autism spectrum disorder in qatar: The baraka-qatar study. Genome Med 15, 81 (2023).
Sanchis-Juan, A. et al. Complex structural variants in mendelian disorders: identification and breakpoint resolution using short- and long-read genome sequencing. Genome Med 10, 95 (2018).
Stankiewicz, P. & Lupski, J. R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 61, 437–455 (2010).
Zhang, F., Gu, W., Hurles, M. E. & Lupski, J. R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 10, 451–481 (2009).
Weischenfeldt, J., Symmons, O., Spitz, F. & Korbel, J. O. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 14, 125–138 (2013).
Mills, R. E. et al. Mapping copy number variation by population-scale genome sequencing. Nature 470, 59–65 (2011).
Conrad, D. F. et al. Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712 (2010).
Gandhi, G. D. et al. Assessing the genetic burden of familial hypercholesterolemia in a large middle eastern biobank. J. Transl. Med. 20, 502 (2022).
Gandhi, D. et al. Mapping the genetic landscape of treatable inherited metabolic disorders in a large Middle Eastern biobank. Genet. Med. 26, 101268 (2024).
Audano, P. A. et al. Characterizing the major structural variant alleles of the human genome. Cell 176, 663–675.e19 (2019).
Sherif, S. et al. The immune landscape of solid pediatric tumors. J. Exp. Clin. Cancer Res. 41, 199 (2022).
Yousri, N. A. et al. Metabolic and metabo-clinical signatures of type 2 diabetes, obesity, retinopathy, and dyslipidemia. Diabetes 71, 184–205 (2022).
Bener, A. & Alali, K. A. Consanguineous marriage in a newly developed country: the Qatari population. J. Biosoc. Sci. 38, 239–246 (2006).
Al-Shafai, K. N. et al. Genetic evaluation of cardiomyopathies in qatar identifies enrichment of pathogenic sarcomere gene variants and possible founder disease mutations in the arabs. Mol. Genet. Genomic Med. 9, e1709 (2021).
Aburawi, E. H., Aburawi, H. E., Bagnall, K. M. & Bhuiyan, Z. A. Molecular insight into heart development and congenital heart disease: An update review from the Arab countries. Trends Cardiovasc. Med. 25, 291–301 (2015).
Miller, D. T. et al. ACMG SF v3.2 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the american college of medical genetics and genomics (ACMG). Genet. Med. 25, 100866 (2023).
Aarabi, M. et al. Copy number variants in the ACMG secondary finding genes: A reporting framework for clinical cytogeneticists. Genetics in medicine open 101839 https://doi.org/10.1016/j.gimo.2024.101839 (2024).
Al Kuwari, H. et al. The qatar biobank: Background and methods. BMC Public Health 15, 1208 (2015).
Sollis, E. et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res 51, D977–D985 (2023).
de Cid, R. et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat. Genet. 41, 211–215 (2009).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
Bougnères, P. The exon-3 deletion of the growth hormone receptor (GHR) gene still has a limited impact in clinical endocrinology. J. Clin. Endocrinol. Metab. 95, 56–59 (2010).
Kanai, M. et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 50, 390–400 (2018).
Vuckovic, D. et al. The polygenic and monogenic basis of blood traits and diseases. Cell 182, 1214–1231.e11 (2020).
Chen, M.-H. et al. Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell 182, 1198–1213.e14 (2020).
Cohen, J. et al. Low LDL cholesterol in individuals of african descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 37, 161–165 (2005).
Li, D., Lewinger, J. P., Gauderman, W. J., Murcray, C. E. & Conti, D. Using extreme phenotype sampling to identify the rare causal variants of quantitative traits in association studies. Genet. Epidemiol. 35, 790–799 (2011).
Peloso, G. M. et al. Phenotypic extremes in rare variant study designs. Eur. J. Hum. Genet. 24, 924–930 (2016).
Akiyama, M. et al. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat. Genet. 49, 1458–1467 (2017).
Hoffmann, T. J. et al. A large multiethnic genome-wide association study of adult body mass index identifies novel loci. Genetics 210, 499–515 (2018).
Sinnott-Armstrong, N. et al. Genetics of 35 blood and urine biomarkers in the UK biobank. Nat. Genet. 53, 185–194 (2021).
Kamdar, F. & Garry, D. J. Dystrophin-deficient cardiomyopathy. J. Am. Coll. Cardiol. 67, 2533–2546 (2016).
Nakamura, A. et al. Deletion of exons 3-9 encompassing a mutational hot spot in the DMD gene presents an asymptomatic phenotype, indicating a target region for multiexon skipping therapy. J. Hum. Genet. 61, 663–667 (2016).
Fortunato, F., Tonelli, L., Farnè, M., Selvatici, R. & Ferlini, A. DMD deletions underlining mild dystrophinopathies: literature review highlights phenotype-related mutation clusters and provides insights about genetic mechanisms and prognosis. Front. Neurol. 14, 1288721 (2023).
Lester, G., Femia, G., Ayer, J. & Puranik, R. A case report: X-linked dystrophin gene mutation causing severe isolated dilated cardiomyopathy. Eur. Heart J. Case Rep. 3, (2019).
Baldarelli, R. M. et al. The mouse gene expression database (GXD): 2021 update. Nucleic Acids Res 49, D924–D931 (2021).
Benyamin, B. et al. Common variants in TMPRSS6 are associated with iron status and erythrocyte volume. Nat. Genet. 41, 1173–1175 (2009).
Thareja, G. et al. Differences and commonalities in the genetic architecture of protein quantitative trait loci in European and Arab populations. Hum. Mol. Genet. 32, 907–916 (2023).
Belkadi, A. et al. Identification of PCSK9-like human gene knockouts using metabolomics, proteomics, and whole-genome sequencing in a consanguineous population. Cell Genomics 3, 100218 (2023).
Haris, B. et al. Epidemiology, genetic landscape and classification of childhood diabetes mellitus in the State of Qatar. J. Diabetes Investig. 12, 2141–2148 (2021).
Elfatih, A. et al. Actionable genomic variants in 6045 participants from the Qatar Genome Program. Hum. Mutat. https://doi.org/10.1002/humu.24278 (2021).
Martinelli, R. et al. miR-519d overexpression is associated with human obesity. Obesity (Silver Spring) 18, 2170–2176 (2010).
Vitart, V. et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 40, 437–442 (2008).
Döring, A. et al. SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat. Genet. 40, 430–436 (2008).
Urbanek, M., Russell, J. E., Cooke, N. E. & Liebhaber, S. A. Functional characterization of the alternatively spliced, placental human growth hormone receptor. J. Biol. Chem. 268, 19025–19032 (1993).
Dibbens, L. M. & Rubboli, G. GOSR2: a progressive myoclonus epilepsy gene. Epileptic Disord 18, 111–114 (2016).
Corbett, M. A. et al. A mutation in the Golgi Qb-SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am. J. Hum. Genet. 88, 657–663 (2011).
Dos Santos, C. O. et al. The prevalence of PRKRA mutations in idiopathic dystonia. Parkinsonism Relat. Disord. 48, 93–96 (2018).
Ghorbani, M. et al. Near-complete middle eastern genomes refine autozygosity and enhance disease-causing and population-specific variant discovery. Nat. Genet. https://doi.org/10.1038/s41588-025-02173-7 (2025).
Thareja, G. et al. Whole genome sequencing in the middle eastern qatari population identifies genetic associations with 45 clinically relevant traits. Nat. Commun. 12, 1250 (2021).
Al Thani, A. et al. Qatar biobank cohort study: study design and first results. Am. J. Epidemiol. 188, 1420–1433 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Mahmoud, M. et al. Structural variant calling: the long and the short of it. Genome Biol 20, 246 (2019).
Chen, K. et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6, 677–681 (2009).
Abyzov, A. et al. Analysis of deletion breakpoints from 1,092 humans reveals details of mutation mechanisms. Nat. Commun. 6, 7256 (2015).
Abyzov, A., Urban, A. E., Snyder, M. & Gerstein, M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21, 974–984 (2011).
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012).
Zhu, M. et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am. J. Hum. Genet. 91, 408–421 (2012).
Handsaker, R. E. et al. Large multiallelic copy number variations in humans. Nat. Genet. 47, 296–303 (2015).
Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016).
Wala, J. A. et al. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res 28, 581–591 (2018).
Kronenberg, Z. N. et al. Wham: identifying structural variants of biological consequence. PLoS Comput. Biol. 11, e1004572 (2015).
Chiang, C. et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat. Methods 12, 966–968 (2015).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Liu, Z. et al. Towards accurate and reliable resolution of structural variants for clinical diagnosis. Genome Biol 23, 68 (2022).
Jeffares, D. C. et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 8, 14061 (2017).
Pedersen, B. S. & Quinlan, A. R. Duphold: scalable, depth-based annotation and curation of high-confidence structural variant calls. Gigascience 8, giz040 (2019).
Amemiya, H. M., Kundaje, A. & Boyle, A. P. The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep. 9, 9354 (2019).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Geoffroy, V. et al. AnnotSV and knotAnnotSV: a web server for human structural variations annotations, ranking and analysis. Nucleic Acids Res 49, W21–W28 (2021).
Nicholas, T. J., Cormier, M. J. & Quinlan, A. R. Annotation of structural variants with reported allele frequencies and related metrics from multiple datasets using SVAFotate. BMC Bioinformatics 23, 490 (2022).
Raudvere, U. et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 47, W191–W198 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics 11, 11.10.1–11.10.33 (2013).
Frankish, A. et al. GENCODE 2021. Nucleic Acids Res 49, D916–D923 (2021).
Gold, L. et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 5, e15004 (2010).
Suhre, K. et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun. 8, 14357 (2017).
Searfoss, R. et al. Impact of hemolysis on multi-OMIC pancreatic biomarker discovery to derisk biomarker development in precision medicine studies. Sci. Rep. 12, 1186 (2022).
Puig, X., Ginebra, J. & Graffelman, J. A bayesian test for hardy-weinberg equilibrium of biallelic X-chromosomal markers. Heredity 119, 226–236 (2017).
Kang, H. M. EPACTS (efficient and parallelizable association container toolbox). EPACTS: efficient and parallelizable association container toolbox (2014).
Zhan, X., Hu, Y., Li, B., Abecasis, G. R. & Liu, D. J. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 32, 1423–1426 (2016).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Zhao, H. et al. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007 (2014).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Belyeu, J. R. et al. Samplot: a platform for structural variant visual validation and automated filtering. Genome Biol 22, 161 (2021).
Villanueva, R. A. M. & Chen, Z. J. ggplot2: Elegant graphics for data analysis (2nd ed.). Measurement: Interdisciplinary Research and Perspectives 17, 160–167 (2019).
Conway, J. R., Lex, A. & Gehlenborg, N. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940 (2017).
R Core Team, R. R: A language and environment for statistical computing. (2013).
Acknowledgements
We would like to thank the QBB and the QGP for providing access to the phenotyping and genomic datasets used in this study. This study was partly funded by Qatar National Research Fund’s Path Towards Precision Medicine award (PPM1-1229-15002) and supported by (PPM1-1122-150008). We are grateful to all the participants.
Author information
Authors and Affiliations
Contributions
The overall study was initiated and designed by M.F. and K.F. Data analysis and results generation was performed by E.A. with assistance from N.S., A.V., T.A., A.B., M.G., H.N., G.T., R.R., and Y.M. Manuscript writing and figure preparation was performed by E.A., M.F., K.F. with contributions from N.S., A.V., A.B., M.G., G.G., A.A., F.A. and Y.M. Advice on data interpretation was provided by A.M., W.A., R.S., N.R. and A.I. Input on scientific discussions and feedback on manuscript was provided by Y.M., F.S., K.S. and A.A.
Corresponding author
Ethics declarations
Competing interests
F.S. receives research support from Illumina, PacBio, and Oxford Nanopore. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ryan Collins, Wan-Ping Lee, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Aliyev, E., Syed, N., Visconti, A. et al. The biomedical landscape of genomic structural variation in the qatari population. Nat Commun 17, 1019 (2026). https://doi.org/10.1038/s41467-025-67763-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67763-9
