
Abstract
Mycorrhizal fungi serve as fundamental agents in forest establishment and progression, underpinning critical ecosystem functions through symbiotic root associations. Drylands, which cover nearly half of Earth’s land, have limited forest establishment, and factors influencing mycorrhization in these stressful environments remain unclear. Here, we integrate large-scale field surveys along aridity gradients with greenhouse experiments and over 33,000 microscopic mycorrhizal observations, revealing that aridity significantly enhances mycorrhization. Mycorrhizal fungi undergo niche modification, whereby facilitative microbial interactions promote mycorrhization under aridity stress. We identify a core synthetic microbial community linked to mycorrhization and provide mechanistic evidence that this community facilitates mycorrhization through physical attachment to fungal hyphae and by alleviating soil metabolite inhibition that otherwise suppresses mycorrhization under arid conditions. In this work, our findings highlight the role of microbial interkingdom interactions in driving tree mycorrhizal colonization in arid regions, offering critical insights for guiding tree planting and restoration efforts in drylands.
Similar content being viewed by others
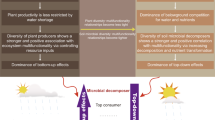

Introduction
Forest plantations provide significant ecological and economic benefits, including soil conservation, carbon sequestration, and timber production1,2,3. As a core strategy for landscape restoration, they are integral to the implementation of the United Nations Decade on Ecosystem Restoration (2021–2030)3. Similarly, forest plantations are fundamental for food production in agroecosystems. Unfortunately, the establishment and success of forest plantations is constrained by water scarcity across 41 percent of the global land area; i.e., in drylands4. This challenge is particularly significant in arid and hyperarid zones, where aridity thresholds critically limit forest ecosystem functions and have led to widespread plantations failures5. Global climate change further intensifies aridification processes, further threatening plantations viability4,6,7. Ectomycorrhizal (ECM) fungi are a key functional microbial community that enhances tree adaptability to aridity by optimizing root water absorption and physiological responses8,9. However, the mechanisms behind the ectomycorrhization of trees in drylands remain virtually unknown10,11,12,13, hampering our ability to design adaptive forest management strategies to mitigate aridity threats. Here, we considered two contrasting scenarios. (1) The prevalent harsh conditions in drylands, such as water scarcity, nutrient depletion, and carbon limitation in host trees, directly inhibit mycorrhizal symbiosis establishment14,15. (2) Tree establishment is likely more dependent on mycorrhizal fungi under highly arid and environmentally challenging conditions. In such settings, the soil microbiome can supportively maintain mycorrhizal colonization within these extreme ecosystems.
Advancing our understanding of how to support the establishment and success of forest plantations in drylands is urgently needed. The current lack of knowledge about the mechanisms behind the colonization of roots by mycorrhizal fungi in drylands exists for three main reasons. First, we do not know if mycorrhizal colonization follows an aridity-threshold driven distribution similar to that previously reported for biodiversity and soil function14. Revealing these aridity-shaped patterns and their mechanistic impacts requires comprehensive, cross-scale studies combining field surveys with microcosm experiments. Second, the contribution of the soil microbiome to support mycorrhizal establishment is poorly understood, yet soil microbes are known to undergo critical changes as aridity increases16,17. Mutualistic interactions within the microbiome represent a key ecological process mitigating aridity-induced suppression of mycorrhization18,19 (Fig. 1). Specifically, ECM fungal hyphae provide ecological niches for the soil microbiome, which in turn enhances ECMF hyphal growth and mycorrhization, thereby expanding tree resource acquisition capacity in drylands20,21. Nevertheless, the complexity of microbial interactions and their dynamic interplay with environmental factors constrain our mechanistic understanding of ectomycorrhization in drylands and pose significant challenges for constructing predictive models of mycorrhization22. Finally, we lack an ecological theory capable of explaining the role of the soil microbiome to support mycorrhizal establishment. Notably, the stress gradient hypothesis posits that facilitative microbial associations increase in frequency under stressful conditions23,24,25,26. We therefore hypothesize that aridity-shaped facilitative microbial interactions critically determine ectomycorrhization success.

Created in BioRender. Zi, H. (2025) https://BioRender.com/ehgollh.
In this work, we combined large-scale field surveys with greenhouse experiments and microscopic observations to evaluate mycorrhizal tree colonization across a wide aridity gradient. To evaluate the changes in ECM colonization with aridity, we conducted a regional survey across 11 poplar cultivation regions in China spanning 1400 km. This transect encompassed a natural aridity gradient (1-Aridity Index), ranging from 0.41 (dry subhumid) to 0.88 (arid; Supplementary Table 1, Fig. S1b). We performed over 33,000 microscopic examinations on 110 root samples, conducting at least 300 examinations per sample until no new ECM morphotypes were observed. We used Populus as our tree model. This tree species is fundamental in natural ecosystems, providing a niche for biodiversity while supporting multiple ecosystem services such as carbon sequestration. Populus is also one of the most commonly planted trees worldwide in restoration processes and in agrosystems to support timber production, playing a fundamental role in local and regional economies. Our sampling encompassed three planted Populus sections in China (Tacamahaca, Leuce, and Aigeiros) to evaluate species-independent aridity effect on ECM colonization. Subsequently, along the soil-ectomycorrhiza continuum, we profiled ECM fungal and bacterial community composition across the aridity gradient, revealing that cross-kingdom associations between ECM fungal and bacterial communities are primary drivers of ectomycorrhization along aridity gradients. We focused on three compartments: (i) bulk soil (reservoir of microbial diversity), (ii) ectomycorrhizosphere (hotspot for fungal-bacterial interactions), and (iii) ectomycorrhiza (functional interface for nutrient exchange), to dissect how aridity shapes microbial interaction driving ECM colonization. We further generated a synthetic microbial community (SynM) comprising nine key bacteria isolated from the ectomycorrhizosphere as promoters of ectomycorrhization. Finally, using integrated multi-omics and cultivation-dependent approaches27, we elucidated a dual mechanism for SynM-enhanced ectomycorrhization: (i) upregulation of genes involved in butyrate metabolism and DNA replication within ECM fungus, (ii) mitigation of the inhibitory effects of soil metabolites, specifically L-tyrosine and L-phenylalanine, on ECM fungus.
Results and discussion
Aridity enhances ectomycorrhizal fungal colonization and diversity
Our analyses revealed that aridity significantly increased ECM colonization. ECM colonization in arid zones (57.01%) was more than double those observed in dry subhumid zones (27.88%) (P < 0.05; Tukey-Kramer test; Fig. 2a). This strong aridity response supports the scenario that tree establishment in highly arid environments is more dependent on mycorrhizal fungi and underscores the importance of ECM symbiosis as a key functional trait for tree adaptation to water stress8. Further analysis indicated a nonlinear relationship between colonization and increasing aridity: colonization increased linearly across habitats with aridity values from 0.41 to 0.78 (P < 0.01) but plateaued under more severe aridity (P > 0.05; Fig. 2b). This aridity threshold (~0.78) aligns closely with previously reported thresholds (~0.8) associated with ecosystem breakdown and minimal plant photosynthesis14,28. Therefore, the stability of ECM colonization beyond this threshold suggests compensatory ecological processes (e.g., microbial interactions) may sustain mycorrhization within the observed aridity range.

a the ectomycorrhizal colonization rates of poplar in three aridity zones. b ectomycorrhizal colonization pattern of poplar along the aridity gradient. Black curve represents optimal cubic regression selected by lower Akaike information criterion (ΔAIC > 2), with error bands representing 95% confidence interval. Blue line represents segmented regression to identify aridity threshold (0.78). Statistical data are colored corresponding to their curves. Scatter colors match groupings in (a). c–f the Shannon diversity and community similarity of bacterial and ectomycorrhizal fungal communities along the aridity gradient. In all above panel: ECMF, ectomycorrhizal fungi; Arid (n = 38): aridity (1-aridity index) >0.8; Semi-arid (n = 52): 0.8 > aridity (1-aridity index) >0.55; Dry subhumid (n = 19): 0.55 > aridity (1-aridity index) >0.35; Soil: bulk soil; Rhiz: ectomycorrhizosphere; Root: ectomycorrhiza; Bar charts represent mean ± SD. Different lowercase letters indicate significant differences (one-way ANOVA followed by Tukey-Kramer test, P < 0.05); *P < 0.05; **P < 0.01; ***P < 0.001); nonsignificant differences presented as “ns”; The “Aridity” represents “1-aridity index”.
Aridity also significantly elevated ECM fungal alpha diversity, measured both within the ectomycorrhizosphere and on the ECM root tips themselves (ectomycorrhiza) (P < 0.01; Fig. 2c). In the ectomycorrhizosphere, diversity increased by 242.12% in arid zones and 40.43% in semi-arid zones relative to dry subhumid zones (Tukey-Kramer, P < 0.05; Fig. S3a). A similar aridity-driven diversity increase was observed in the ectomycorrhiza (P < 0.01; Fig. S3a). These findings are consistent with our microscopic examinations, which found more ectomycorrhiza and greater diversity in arid zones. Furthermore, ECM fungal alpha diversity in bulk soil also increased significantly with aridity (P < 0.01), indicating their synergistic adaptation across the soil-ectomycorrhiza continuum to environmental stress gradients25. Bulk soil likely acts as a persistent reservoir of ECM fungal propagules (e.g., spores, hyphal fragments)29,30, potentially hosting a greater diversity of aridity-adaptive fungal strategies available for symbiosis.
Mycorrhizosphere microbiome determines mycorrhization under intensified aridity
We further demonstrated that the soil microbiome critically regulated mycorrhization in drylands. Our results showed that aridity significantly restructured ECM fungal and bacterial communities, though compartment-specific variation characterized these shifts (bulk soil, ectomycorrhizosphere, ectomycorrhiza; Fig. S4a, c). Notably, the bacterial community in the ectomycorrhizosphere displayed pronounced sensitivity to aridity, evidenced by its strongest correlation with aridity for alpha diversity (R² = 0.40, P < 0.01; Fig. 2c, d) and community similarity (Mantel r = −0.39, P < 0.01; Fig. 2e, f) relative to other compartments. This aridity-driven differentiation further amplified dissimilarity between ectomycorrhizosphere and bulk soil bacterial communities, peaking above an aridity value of 0.7 (Fig. S5). Collectively, our findings demonstrate progressive niche contraction for bacteria under increasing aridity, wherein the ectomycorrhizosphere constitutes a critical interface of available plant habitat, thereby fostering a bacterial community distinct from bulk soil31.
Next, to characterize the driving force of microbiome along the aridity gradient, we employed cohesion metrics to quantify potentially cooperative and competitive interactions between ECM fungi and bacteria. We found that in the ectomycorrhizosphere, aridity specifically enhanced cooperative interactions while suppressing competitive ones (Figs. 3a and S6). In contrast, in bulk soil and ectomycorrhiza, aridity exerted only minor effects on microbial interactions. Specifically, in the ectomycorrhizosphere, positive cohesion in arid zones was 1.37-fold and 2.42-fold higher than in semi-arid and dry subhumid zones, respectively (Fig. 3a). In this compartment, ECM fungal hyphal metabolites provide carbon sources supporting certain microorganisms, mitigating the lethal effect of water limitation under increasing aridity20,21. This aligns with the Stress Gradient Hypothesis, indicating that microbial cooperation is an important strategy for mycorrhizal adaptation to aridity24,32,33. Critically, the ectomycorrhizosphere exhibited the strongest association between positive cohesion and aridity (R² = 0.54, P < 0.001) compared with bulk soil (R² = 0.08, P < 0.01) and ectomycorrhiza (R² = 0.01, P > 0.05; Fig. 3a), confirming its status as the primary interface for aridity-driven microbial cooperation.

a the positive cohesion in soil, ectomycorrhizosphere and ectomycorrhiza, and their relationships with aridity. Soil: bulk soil; Rhiz: ectomycorrhizosphere; Root: ectomycorrhiza. Bar charts represent mean ± SD. Different lowercase letters indicate significant differences (two-way ANOVA followed by Tukey-Kramer test, P < 0.05); “ns” denotes nonsignificant differences. The “Aridity” represents “1-aridity index”. Arid (n = 38); Semi-arid (n = 52); Dry subhumid (n = 19). b the importance of community characteristics of ectomycorrhizal fungi (ECMF) and bacteria, and their cohesion for ectomycorrhizal colonization (random forests). %IncMSE refers to the Percentage Increase in Mean Squared Error. A thousand iterations of the permutation test were conducted, and Q-values were corrected using FDR methods (*P < 0.05). c bacterial ASVs positively associated with ECMF and ECMF colonization. The y-axis represents the mean of the significant positive connectedness between each bacterial ASV and the ECMF ASV; the x-axis indicates the correlation between bacterial ASVs and ECMF colonization. d analysis of key bacterial ASVs in extremely arid environments (aridity >0.7). e the Venn Diagram shows the number of ASVs shared between Method I and Method II. f the best matching strain for key ASVs. The relative abundance of corresponding ASVs in the ectomycorrhizosphere is included in the bar plots on the right.
We further evaluated the contributions of ECM fungal and bacterial community characteristics and their cross-kingdom interactions to ECM colonization using random forest (RFM) and generalized additive model (GAM). The bacterial community in the ectomycorrhizosphere dominated ECM colonization, with positive ECMF-bacterial cohesion emerging as the most influential factor (%IncMSE = 13.55, P < 0.05), followed by bacterial community composition (PCoA1) and alpha diversity (P < 0.05; Fig. 3b). Partial least squares regression confirmed a significant positive correlation between positive ECMF-bacterial cohesion and colonization in the ectomycorrhizosphere (R² = 0.22; Fig. S7a). This direct evidence supports our hypothesis that prolonged aridity drives ECM fungal niche optimization, promoting bacteria-ECMF cooperative assemblies to underpin mycorrhization. This represents a coevolutionary outcome of tripartite (bacteria-ECMF-host tree) systems responding to environmental stress. GAM analysis incorporating the six RFM-significant variables (total R2 = 0.65) strengthened these findings, with ectomycorrhizosphere bacteria explaining 34% of colonization variance (Fig. S7b). Although the identities of ECMF exhibit variable root colonization capacity34,35, RFM detected no direct contribution from ECM fungal community characteristics (Fig. 3b). This limited explanatory power of ECM fungi underscores the primacy of niche adjustment in driving ECM colonization under intensified aridity. Collectively, these findings suggest that under increasing aridity, tree mycorrhization depends on ectomycorrhizosphere microbiome, specifically on facilitative cross-kingdom interactions as pivotal drivers31. This underscores the necessity of prioritizing the dynamics of microbial interactions in predictive models of plant mycorrhization under conditions of global aridification.
The bacteriome of ectomycorrhizal colonization
We hypothesized that bacteria enriched specifically under high aridity contribute to ECM colonization. We then aimed to further understand the bacterial communities associated with mycorrhizal colonization. To such an end, we identified key ectomycorrhizosphere bacteria taxa for ECM colonization. We first detected 619 bacterial ASVs significantly enriched in the ectomycorrhizosphere compared to bulk soil (Fig. S8a). Subsequently, we employed two complementary analytical methods to identify key bacterial taxa. The first method utilized microbial interaction patterns. Given the importance of positive ECMF-bacterial interactions for ECM colonization (Fig. 3a), we calculated the positive connectedness (defined as the mean of significant positive correlations between each bacterial ASV and the ECMF ASV based on SparCC networks) and assessed the correlation of bacterial ASVs with ECM colonization. This approach identified 145 ASVs (Fig. 3c), indicating their potential to promote ectomycorrhization through positive interactions with ECM fungi. To further validate these candidates under high aridity, we quantified two key ecological metrics—occupancy and specificity (see “method section” for details)—in ectomycorrhizosphere samples with aridity >0.7. This additional screening step identified 36 candidate ASVs (Fig. 3d). Among these, 34 ASVs showed significant positive correlations with both ECMF ASVs and colonization (Fig. 3e), defining them as key bacteria that potentially facilitate ECM colonization. This key group represents four bacterial phyla and 19 genera, including Mesorhizobium (4 ASVs), Leifsonia (2 ASVs), and Flavobacterium (2 ASVs; Fig. S9a), demonstrating taxonomic diversity in bacteria supporting ECM colonization.
To obtain these key bacterial isolates, we first confirmed their predominant enrichment in the BJT, LZ, and GL regions through enrichment analysis (Fig. S8b). Subsequently, we isolated and purified bacteria from the ectomycorrhizosphere soil in these regions. A total of 417 bacterial strains were obtained, and after sequence alignment, 172 strains with sequence similarity of less than 98.65% (this measure is now widely used for species demarcation) were retained. Since promoting ECMF hyphae growth is a primary trait of ectomycorrhiza helper bacteria36, all 172 strains were co-cultured with the dominant ECM fungus Tuber from poplar plantations to assess hyphal growth promotion (Fig. S2c, S9a). Twelve bacterial strains (ASVs) that exhibited a negative association with ECM colonization significantly inhibited Tuber growth in vitro. Among these, seven strains were identified as Pseudomonas, and two strains were classified within the Bacillus (Fig. S10). Notably, these ASVs (e.g., Pseudomonas ASV9, B18) were found to be enriched in dry subhumid regions (Fig. S10). This finding corroborates our results (Fig. 3b), suggesting that competitive interactions inhibit ECM colonization in dry subhumid regions. Most importantly, nine key bacterial strains were identified based on sequence matching (≥98.65% similarity) to enriched ASVs and demonstrable growth-promoting abilities (Fig. 3f). Functionally, six of these nine strains significantly enhanced Tuber hyphal growth. Specifically, strain B1 (Flavobacterium, ASV196) showed the strongest promotion (12.34% increase versus control), while B188 (Sphingopyxis, ASV354) and B127 (Chryseobacterium, ASV727) ranked third (9.79%) and eighth (7.11%), respectively, among all isolates (Fig. S9a). These findings validate the rationality and scientific rigor of our methodology for selecting key bacterial taxa.
Experimental evidence on the contribution of bacteria to ECM colonization
We then seek to provide further experimental evidence that bacteria shape mycorrhizal tree colonization in drylands. To test whether key bacteria promote mycorrhization, we constructed a SynM comprising the nine key bacterial strains through bacterial co-culture. Subsequently, we established a pot experiment with four treatments: sterile control, SynM inoculation alone, Tuber inoculation alone (one of the dominant ECM fungi identified in field surveys), and SynM-Tuber co-inoculation. We observed that SynM significantly enhanced ECM colonization. Specifically, although Tuber inoculation alone achieved 18.83% ECM colonization, SynM-Tuber co-inoculation significantly increased colonization to 33.74% (Fig. 4a). This enhancement corresponded with 12.4% and 18.2% increases in poplar height and aboveground biomass, respectively, relative to the control (Fig. 4b–d). This result demonstrates that key bacterial taxa enriched in the ectomycorrhizosphere of trees under aridity conditions effectively promote mycorrhization. Furthermore, SynM inoculation alone showed no effect on tree growth, indicating that SynM facilitates primarily through direct interactions with ECM fungi rather than indirect tree-mediated pathways in this system.

a ectomycorrhizal fungi (ECMF) colonization. b plant height. c plant biomass based dry weight (dw). d shoot dry weight. e root dry weight. CK: sterile control; SynM: a synthetic microbial community comprising of the nine key bacterial strains) inoculation alone; ECMF: Tuber inoculation alone (one of the dominant ECM fungus identified in field surveys); SynM +ECMF: SynM-Tuber co-inoculation; ECMF + SE: individual inoculation with Tuber after adding soil extract to the substrate; SynM+ECMF + SE: co-inoculation with SynM and Tuber after adding soil extract to the substrate. Different lowercase letters indicate significant differences (one-way ANOVA followed by Tukey test, P < 0.05; n = 4 replicates with 3 poplars per replicate); Error bars indicate ± SD. Horizontal bars within boxes represent mean values. The tops and bottoms of boxes represent the 75th and 25th quartiles, respectively.
Previous studies have shown that soils contain ECMF-antagonistic metabolites (e.g., phenolics), which inhibit the growth and extension of ECMF hyphae37,38,39. Here, we tested whether arid soil extracts (SE) suppress mycorrhization and whether key bacteria mitigate this effect. We separately inoculated Tuber alone and co-inoculated SynM with Tuber in poplar pots amended with SE from arid regions (DX and LW, aridity: 0.74–0.87). Indeed, we found that arid SE inhibited ECM formation. SE addition reduced ECM colonization by 27% (P < 0.05; Fig. 4a), and decreased poplar height, shoot biomass, and total biomass by 5.44%, 9.78%, and 7.29% respectively, in Tuber-only treatments (Fig. 4b–d). Notably, SynM co-inoculation alleviated this suppression, maintaining ECM colonization and plant growth at non-extract level (P < 0.05; Fig. 4a). Several SynM constituents, such as Sphingomonas, Sphingopyxis, and Flavobacterium, are reported to degrade inhibitory organic acids and aromatic compounds40,41,42. Collectively, these results demonstrate that key bacteria promote ECM colonization and alleviate environmental suppression43.
Bacteria promote ECM growth via butanoate metabolism and DNA replication
We then aimed to understand the contribution of SynM to in vitro ECM fungal growth, and further investigate the mechanisms behind such relationship. To elucidate the mechanism by which key bacteria enhance ECM colonization, we assessed the effect of SynM on Tuber growth using three in vitro co-culture systems: contact coculture, bacterial supernatant, and non-contact coculture (methods detailed in “Methods”; Figs. 5a and S2c, S9a). These methods enable us to clarify the main interaction modes between bacteria and ECM fungi. We also constructed multiple SynMs in parallel as controls, which contained: SynM1 (three non-promoting strains from original SynM), SynM2 (six growth-promoting strains from original SynM), and SynM3 (nine randomly selected strains; Fig. S9a). Our in vitro co-culture systems revealed that direct physical contact (contact coculture) is important for SynM-mediated Tuber growth promotion. SynM showed the highest growth promotion (18.15%) when it contacts with Tuber hyphae (contact co-culture; Fig. 5b), surpassing other co-culture systems. Scanning electron microscopy confirmed bacterial colonization on Tuber hyphae (Fig. 5c), indicating a structural symbiosis. This contact-dependent interaction mode aligns with documented mechanisms for ECM fungal development facilitated by hyphal-colonizing bacteria27,36. Moreover, the rationally designed SynM outperformed all control communities (SynM1/2/3) and individual strains in Tuber growth promotion (Figs. 5a and S9b), confirming its rational design efficacy. Strikingly, removal of non-promoting strains from original SynM (SynM2) reduced the growth promotion by >50% relative to complete SynM (Fig. 5a), revealing functional synergy essential for SynM activity.

a the effects of synthetic microbiota on Tuber growth were tested by three methods. SynM, a synthetic microbial community comprising of the nine key bacterial strains. SynM1, three strains from the SynM with neutral effects on Tuber growth. SynM2, six strains from the SynM exhibiting Tuber growth promotion. SynM3, nine randomly selected strains corresponding to ectomycorrhizosphere-enriched ASVs. Bar charts represent mean ± SD; Different lowercase letters indicate significant differences (one-way ANOVA followed by Tukey test, P < 0.05; n = 4 biological replicates, in triplicates); b comparison of three methods for Tuber growth promotion by the rationally designed 11-strain SynM inoculation. Bar charts represent mean ± SD; Different lowercase letters indicate significant differences (one-way ANOVA followed by Tukey test, P < 0.05; n = 4 biological replicates, in triplicate). c a bacterial strain attached to Tuber hyphae. d KEGG pathway analysis for upregulated genes, ranked by rich factor (FDR < 0.05 cut-off). e Heat map and butanoate metabolism pathway for upregulated genes (KEGG). SynM (1–3) denotes three biological replicates of the SynM treatment group.
Tuber transcriptomics after contact coculture elucidated underlying mechanisms. SynM significantly altered Tuber gene expression, upregulating 937 genes compared to the control (Fig. S11a). KEGG enrichment analysis identified butanoate metabolism as the most enriched pathway, followed by DNA replication (Fig. 5d). The metabolic pathway and heatmap analysis revealed induced expression of genes encoding acetoacetyl-CoA synthetase (AACS), hydroxy methylglutaryl-CoA lyase (HMGCL), 4-aminobutyrate aminotransferase (puuE) and 3-hydroxybutyryl-CoA dehydrogenase (paaH) (Figs. 5e and S11c, d). These genes collectively enhance acetyl-CoA biosynthesis. AACS converts acetoacetate to acetoacetyl-CoA, HMGCL cleaves hydroxymethylglutaryl-CoA to generate acetoacetate, and paaH facilitates the conversion of 3-hydroxybutyryl-CoA to acetoacetyl-CoA. The accumulated acetoacetyl-CoA is further metabolized to acetyl-CoA, a central intermediate that enters the tricarboxylic acid cycle to produce ATP. This energy supply directly supports fungal hyphal growth and biomass accumulation44,45. Concomitantly, upregulation of DNA replication pathways indicates increased cellular proliferation, which is tightly coupled to enhanced energy availability from butanoate metabolism. Collectively, these results introduce a perspective on bacterial-ECM fungal interactions wherein SynM modulates butanoate metabolism and DNA replication pathways to stimulate ECM fungal growth46.
Bacteria facilitate mycorrhization by eliminating soil metabolic constraints
To better understand the mechanism by which key bacteria facilitate ECM colonization through the elimination of soil constraints, we first assessed the impact of SE from arid zones on Tuber growth. SE significantly inhibited Tuber growth, reducing colony diameter by 23.73% versus the control (Fig. 6a). Crucially, pre-cultivating the SE with SynM (SE+SynM) eliminated this suppression, demonstrating bacteria-mediated microenvironmental improvement supporting ECM fungi (Fig. 6a). Metabolic profiling revealed that SynM specifically reduced two aromatic amino acids in SE: L-tyrosine (20.05-fold reduction) and L-phenylalanine (14.01-fold reduction) (Fig. 6b–f). These metabolites involved in various metabolic pathways (Fig. 6d, e), and their accumulation disrupts metabolic homeostasis of fungi43. We propose that SynM members, notably Sphingomonas and Sphingopyxis, likely eliminate the inhibitory by utilizing these aromatic amino acids as carbon and nitrogen sources, thereby optimizing the microenvironment for ECM fungal growth41,47.

a effect of soil extract on Tuber growth. Control, 1/5 MMN medium; Soil extract, 1/5 MMN medium with soil extract; Soil extract + SynM supernatant, 1/5 MMN medium with soil extract and synthetic community supernatant; Soil extract + SynM, 1/5 MMN medium with soil extract after SynM cultivation. Bar charts represent mean ± SD; Different lowercase letters indicate significant differences (one-way ANOVA followed by Tukey test, P < 0.05; n = 4 biological replicates, in triplicates). b supervised Partial Least Squares Discriminant Analysis. c compared with the original soil extract, the downregulated metabolites in soil extract after inoculation of SynM (DESeq2, |log2 FC| > 1 and P < 0.05, two-sided Wald test with Benjamini-Hochberg adjustment). d KEGG pathway enrichment analysis of downregulated metabolites shows the significantly enriched pathways (Q value < 0.05). Q value was calculated using a two-tailed Chi-squared test with Bonferroni correction. e metabolites involved in KEGG pathway. f differential analysis of L-tyrosine and L-phenylalanine between treatments. Soil extract, 1/5 MMN medium with soil extract; Soil extract + SynM, 1/5 MMN medium with soil extract after SynM cultivation. Bar charts represent mean ± SD; * P < 0.05 (two-sided Wilcoxon rank sum, n = 3 biological replicates, in triplicates). g downregulated genes in soil extract treatment and upregulated genes in soil extract after inoculation of SynM. h KEGG pathway enrichment analysis of differential genes. Significant pathways are bold (Q value < 0.05, marked with *). Q value was calculated using two-tailed Chi-squared test with Bonferroni correction.
Transcriptomic analysis of Tuber in control, SE, and SE+SynM offered further mechanistic insight. 507 genes exhibiting sequential downregulation in SE and recovery in SE+SynM (Fig. 6g). KEGG enrichment of these genes showed their significant roles for glutathione metabolism and arachidonic acid metabolism pathways (Figs. 6h and S12). This transcriptional response indicates that soil extract compromises antioxidant defenses and disrupts membrane integrity through dysregulated arachidonic acid metabolism48,49. SynM effectively counteracted these detrimental effects at the transcriptional level. Collectively, these findings establish that SynM facilitates ECMF colonization by eliminating inhibitory soil metabolites, specifically removing inhibitory aromatic amino acids, thereby alleviating metabolic stress and restoring essential cellular functions for oxidative defense and membrane integrity in the ECM fungus. This bacteria-mediated soil metabolic change establishes a conducive microenvironment for successful mycorrhization.
Taken together, ECM colonization represents a key adaptive strategy of tree plantation for addressing the intensified aridity stress, characterized by a distinct threshold effect. ECM fungi undergo niche adjustment as aridity intensifies, wherein facilitation of the microbiome supports ECM colonization under aridity stress. Given the ECM fungal diversity, this ecological regulatory strategy exhibits broader applicability for mitigating the negative impacts of global aridity on mycorrhizal systems. Through the construction of a targeted SynM, we mechanistically demonstrated that bacteria facilitate ECM colonization by direct physical attachment to fungal hyphae and by eliminating soil inhibitory metabolites. Our findings elucidate an ecological mechanism sustaining mycorrhization under severe aridity and provide a microbiome-based strategy to enhance plantation resilience against climate change stressors.
Methods
Site description and sampling
To investigate how ECM colonization in poplar roots responds to an aridity gradient, we conducted a regional-scale survey spanning 11 poplar cultivation regions (Table S1). These regions were located across a climate gradient from west to east in China, spanning dry sub-humid to arid zones, and were distributed along gradients of climatic aridity (ranging from 0.41 [dry subhumid] to 0.88 [arid]; Fig. S1a). Regions were selected based on the five poplar cultivation areas in China, with the westernmost and easternmost sites separated by 1400 km. Geographic coordinates were recorded in the field using handheld GPS receivers (Fig. S1a). Corresponding mean annual temperature (MAT) and mean annual precipitation values (MAP) for each region were obtained from the WorldClim2 Global Climate Data at a spatial resolution of 30 arc-seconds based on the sampling coordinates. Aridity (1-aridity index) was estimated using the Global Aridity Index dataset (http://www.cgiar-csi.org/data/global-aridity-and-pet-database), higher values denote drier conditions. Further details regarding the 11 regions are provided in Supplementary Table S1.
Sampling was conducted in September 2021 across 11 regions. In each region, 7–13 poplar plantations with minimal recent disturbance were selected as sampling sites, spaced at least 100 m apart to prevent cross-interference between poplar root systems (Table S1). At each sampling site, the sample comprised two to three individual poplar trees, which were pooled to form a single composite sample per site. From each poplar tree, we collected the following compartments in the field: roots and bulk soil. Roots were collected and gently shaken to remove loosely adhering soil. In total, 110 root samples and 110 bulk soil samples were collected from 11 regions. All these samples were placed on ice for transport and processed within 48 h. The 110 bulk soil samples were divided into two subsamples: one was air-dried at room temperature for the determination of soil physicochemical properties; the other was stored at −80 °C for subsequent DNA extraction.
Morphotyping and colonization rate measurement
Fine roots (<2 mm diameter) were cut to approximately 2 cm segments (Fig. S2). After carefully removing adhering soil particles that obscured observation using sterile tweezers (flame-sterilized with an alcohol lamp), root tips were examined under a dissecting microscope (10–50× magnification; Nikon SMZ18, Kanagawa, Japan) for the presence of a mantle. Root tips with a mantle were identified as ECM roots. Subsequently, based on morphological distinctions in branching pattern, tip morphology, emanating hyphae, ECM root tips were classified into distinct morphotypes50,51. Per sample, at least ten root tips per ECM morphotype were preserved for molecular analysis; all morphotypes from the same sample were pooled into a single composite sample52, yielding a total of 110 ECM root samples. For rare morphotypes with low abundance, a minimum of two root tips per morphotype was collected in per sample53. No minimum number of root tips per sample was predefined: observations continued until no new morphotypes were detected. Approximately 300 ~ 400 root tips were examined per sample, totaling over 33,000 root tips. The number of ECM root tips and total root tips observed per sample was recorded. ECM colonization rate was calculated as: (Number of ECM root tips/Total root tips observed) × 100%. For clarity, the root samples referenced in this manuscript are understood to pertain to ECM samples, unless stated otherwise.
Collection of ectomycorrhizosphere soil samples
Mycorrhizosphere soil was defined as the soil layer adhering to ECM roots with an approximate thickness of ~1 mm54. For its collection, the protocol of He et al55. was followed: ECM roots were transferred to sterile 50-mL centrifuge tubes containing 20 mL of sterile 10 mM phosphate-buffered saline (PBS) and shaken at 120 rpm for 20 min at 4 °C56. During shaking, mantle integrity was monitored in real-time, with parameters adjusted if necessary to prevent dislodgement. Subsequently, using sterile tweezers, the ECM root tips were picked out and taken as ectomycorrhiza samples. The remaining suspension was centrifuged (6000 × g, 4 °C, 20 min) to pellet ectomycorrhizosphere soil. These steps were repeated as needed until sufficient samples were obtained. 110 ectomycorrhizosphere soil samples and 110 ectomycorrhiza samples were collected and stored at −80 °C for DNA extraction.
Soil properties
Sieved (<2 mm) soil samples were analyzed for soil properties. Soil pH was measured potentiometrically in a 1:2.5 (w/v) soil-water suspension. Total carbon (TC) and total nitrogen (TN) were quantified by combustion using a vario MICRO cube elemental analyzer (Elementar, Germany). According to China’s Soil Standard protocols for forest regions (LY/T 1232-2015, LY/T 1234-2015, in Chinese), total phosphorus (TP) was determined via alkali fusion-Mo-Sb anti spectrophotometric method, and Available phosphorus (AP) was extracted with 0.05 mol·L–1 HCl − 0.025 mol·L–1 1/2 H2SO4; Total potassium (K) was determined by flame photometry on a NaOH melt, and available K was measured using 1 mol·L–1 ammonium acetate extraction flame photometry. Nitrate nitrogen (NO₃⁻-N) and ammonium nitrogen (NH₄⁺-N) were measured following the procedures outlined by DeForest (Ohio University, Athens 2009), specific protocol for each assay can be found at https://www.ohio.edu/cas/plant-biology/research/facilities-laboratories/forest-soil-ecology-lab/optimized-lab-protocols. The soil properties for each region are presented in Table S2 (Supplementary material).
DNA extraction, PCR amplification and sequencing
DNA from bulk soil, rhizosphere and ECM root samples was isolated using the Fast DNA spin kit for soil (MP Biomedicals, Santa Ana, CA, USA) according to the manufacturer’s instructions. Quality and quantity of the DNA were checked by NanoDrop5000 spectrophotometer (Thermo Scientific, Wilmington, USA) and gel electrophoresis (BG-gdsAUTO 130). The extracted DNA was amplified using primers targeted to the V5-V7 region of the bacterial 16S rRNA gene (799F-AACMGGATTAGATACCCKG57, 1193R-ACGTCATCCCCACCTTCC58). The fungal internal transcribed spacer region 1 (ITS1 region) of ribosomal RNA was amplified using ITS1F (CTTGGTCATTTAGAGGAAGTAA)59 and ITS2 (GCTGCGTTCTTCATCGATGC) primers60. All PCR reactions were carried out in 25 µL reactions: 5 × reaction buffer (5 µL), 5 × GC buffer (5 µL), dNTP (2.5 mM, 2 µL), Forward primer (10 μM, 1 µL), Reverse primer (10 μM, 1 μL), DNA Template (2 µL), ddH2O (8.75 µL), DNA Polymerase (Q5, 0.25 μL). The PCR amplification program was as follows: 94 °C for 2 min; 30 cycles of 98 °C for 15 s, 55 °C for 30 s, 72 °C for 30 s; and 72 °C for 5 min. Following purification with Vazyme VAHTSTM DNA clean beads (Vazyme, Nanjing, China), the PCR products were sequenced on an Illumina MiSeq platform at Shanghai Personal Biotechnology Co., Ltd (Shanghai, China).
Bioinformatic analysis
After sequencing, QIIME2 (open-source, http://qiime2.org) was used to trim (16S rRNA sequences were trimmed to 300 bp length (240 bp for fungi)) and denoise raw data into amplicon sequencing variants (ASVs) via the DADA2 plugin (q2-dada2). ITSx 1.0.11 was used to remove the flanking small ribosomal subunit (SSU) and 5.8 S genes, leaving the ITS1 region for further analyses. Taxonomy was assigned using the SILVA database (v138.1) for 16S rRNA gene amplicons and the UNITE database (v9-16.10.2022 release) for ITS amplicons. To reduce bias from variation in sample read numbers, samples were rarefied (the " rrarefy function in the R “vegan” package v2.6-4) to the sample with the lowest read count (15,637 counts for the 16S rRNA gene dataset and 30,712 counts for the ITS dataset). Then, in order to reduce the influence of sparse features and eliminate possible error ASVs, ASVs that exist in only a single sample and have fewer than 10 sequences have been removed.
FungalTraits database61, which includes ECM fungal groups identified later than the publication of FUNGuild, was used to extract the ECM fungal ASVs (EASVs) from fungal ASVs according to “primary lifestyle”, based on genus-level identification, with > 90% sequence similarity. Subsequent analyses of alpha and beta diversities of ECM fungal community were based on the EASVs.
Isolation and identification of ectomycorrhizosphere bacteria
From the ectomycorrhizosphere soil samples (Baijitan, Lanzhou, and Gulang), 417 isolates were successfully cultured. Briefly, 1 g of each fresh ectomycorrhizosphere soil was suspended in 9 mL sterile saline solution (0.9% [w/v] NaCl in ddH2O), and vortexed vigorously for 3 min followed by shaking at 200 rpm for 30 min at 30 °C. Three gradient dilutions (10–3, 10–4, 10–5) of soil suspensions were plated on Luria-Bertani agar (LB), Nutrient agar (NA), Tryptic soy agar (TSA) and Reasoner’s 2 A agar (R2A) media, then incubated at 30 °C in the dark for 2–7 days. Colony development was monitored daily. Distinct colonies were selected based on morphological characteristics and purified through three successive subcultures on NA. All isolates were identified by Sanger sequencing of 16S rRNA genes amplified with the 27 F (5’-AGAGTTTGATCCTGGCTCAG-3’) and 1492 R (5’-GGTTACCTTGTTACGACTT-3’) primers. The 16S sequences were submitted to NCBI databases for taxonomic identification and homologous sequence similarity. Taxonomic identification employed a 98.65% similarity threshold with > 95% coverage for homologous sequences62. Among all isolates, 172 exhibited sequence similarity below this threshold and were retained for subsequent experiments. Pure cultures were preserved in 30% (v/v) glycerol at −80 °C.
Effect of bacteria on ECM fungal growth in vitro
To assess bacterial effects on ECM fungal growth in vitro, 172 isolated strains were co-cultured with Tuber, one of dominant genus identified in the field investigation, according to the methods of Deveau et al63. and Zhao et al64. Bacterial cultures were grown in nutrient broth at 30 °C with 180 rpm shaking for 48 h, harvested by centrifugation (5000 × g, 5 min), washed thrice with sterile water, and resuspended to OD600 = 1. Tuber cultures were pre-grown on modified Melin-Norkrans agar (MMN, 1:5 dilution) for 72 h. Mycelial plugs (4 × 4 mm) were transferred to fresh 1/5 MMN plates and inoculated with 25 μL bacterial suspension (treatment) or sterile water (control). Then, parafilm-sealed plates were incubated at 25 °C in darkness. Fungal radial growth was measured daily in two perpendicular directions for 4 days. Four biological replicates were performed. To test the significance of growth promotion or inhibition, fungal radial growth between treatment and control groups was determined by exact Wilcoxon rank sum tests (two-sided, P < 0.05). Growth-promoting and growth-inhibiting strains were defined as those showing significant stimulation or suppression relative to controls. Percentage growth change was calculated as [(Rtreatment - Rcontrol)/Rcontrol] × 100%, where R represents mean radial growth65.
The construction of synthetic microbial communities
To obtain bacterial isolates corresponding to key ectomycorrhizosphere taxa, 16S rRNA gene sequences of 172 isolates were aligned against ASVs identified as the key bacterial isolates using a 97% similarity threshold. Nine strains that best matched nine bacterial ASVs were selected to construct a SynM. We also constructed multiple SynMs in parallel as controls, which contained: SynM1 (Three strains from the original nine with neutral effects on Tuber growth), SynM2 (Six strains from the original nine exhibiting Tuber growth promotion), and SynM3 (Nine randomly selected strains corresponding to ectomycorrhizosphere-enriched ASVs; Fig. S9a). Selected strains were activated on NA for 48 h followed by subculture in liquid NA with a total culture period of 72 h at 30 °C. Bacterial suspensions were prepared by centrifuging cultures at 5000 × g for 5 min and resuspending pellets in sterile water to OD600 = 1. We mixed equal volumes of suspensions of each single strain to construct a total of above four different SynMs.
Effect of synthetic microbial community on ECM colonization in a pot experiment
To assess SynM effects on ECM formation, a pot experiment was conducted using hybrid poplar (Populus alba × P. glandulosa ‘84 K’). Five treatments were applied: T1) Tuber inoculation alone; T2) SynM inoculation alone; T3) co-inoculation with SynM and Tuber; T4) individual inoculation with Tuber after adding soil extract to the substrate; T5) co-inoculation with SynM and Tuber after adding soil extract to the substrate. Each treatment comprised four replicates with three poplars per replicate. The steps are as follows: a tissue-cultured poplar tree was performed in Murashige and Skoog (MS) medium as described in our previous work66, then transplanted into a plastic pot containing autoclaved substrate (profile porous ceramic: vermiculite: peat = 3:1:1 v/v). In treatments T4 and T5, substrates were given 30 mL of pre-prepared soil extract. After five days of acclimation, microbial inoculations were performed. SynM suspensions (10 mL, OD600 = 1) were injected into pots. For Tuber inoculants, we first cultured them in 1/5 MMN liquid medium for 72 h (25 °C, 180 r/min) and their hyphae were chopped and blended using a sterile stirrer in a new conical flask. 10 mL of Tuber inoculant was inoculated into the pot using a sterile syringe. Pots were randomized in a greenhouse under 16-h light/8-h dark cycles at 25/20 °C day/night temperatures, with sterile water replenishment every 72 h.
After six months, the plant height of each poplar was recorded, and the root and aboveground parts of each poplar were collected for physiological index determination. Firstly, the ECMF colonization rate of the poplar was observed according to our previous method66. In short, the fine roots of poplar trees were carefully washed with sterile water, cut into 2 cm segments, and examined under a microscope. ECM root tips displaying fungal mantles with emanating hyphae were counted across ≥100 segments per plant. ECM colonization rates were calculated as described previously. Roots and shoots were oven-dried (80 °C, 72 h) for biomass determination.
Effect of synthetic microbial community on ECM fungal growth in vitro
We then tested the effect of synthetic bacteria on the growth of Tuber by three coculture systems: contact coculture (physical contact-dependent interactions), bacterial supernatant (metabolite-mediated interactions), and noncontact coculture (volatile organic compound-mediated interactions; Fig. S2c). These methods enable us to clarify the main interaction modes between bacteria and ECM fungi27. In the contact coculture system, 25 μL SynM suspension was injected concentrically around a 4-mm Tuber mycelial plug on 1/5 MMN agar. In the bacterial supernatant system, sterile supernatant (0.22-μm filtered after centrifugation at 5000 × g for 5 min) from 72-h SynM cultures was applied similarly. In the noncontact coculture system: SynM spread on NA (200 μL, 72 h, 25 °C); Tuber transferred to fresh 1/5 MMN agar; fungal plate inverted over bacterial plate and sealed with Parafilm67. In this way, Tuber and SynM grow separately in the same space without contact, allowing us to evaluate the impact of volatile organic compounds from SynM on fungal growth. Control groups received equivalent volumes of sterile water to replace SynM (or supernatant). Radial growth of Tuber was measured after incubation for 72 h at 25 °C. Experiments were done in triplicate.
Transcriptome sequencing of ECM fungi
To elucidate the molecular mechanisms underlying SynM-mediated promotion of Tuber growth, transcriptome analysis was performed on the control group and the SynM-treated group from the contact coculture system. Three biological replicates were selected per treatment group, yielding a total of six samples. First, Tuber hyphae corresponding to each treatment were collected from the culture medium for RNA extraction. Total RNA was extracted using the Trizol method following grinding in liquid nitrogen. After digestion with DNase I to remove genomic DNA contamination, RNA integrity and purity were assessed by agarose gel electrophoresis and Nanodrop spectrophotometry.
Libraries were constructed using the Illumina Truseq™ RNA Sample Prep Kit. Eukaryotic mRNA containing polyA tails was enriched using magnetic beads coated with oligo(dT). The enriched mRNA was then fragmented by ultrasonication. Using the fragmented mRNA as a template and random oligonucleotides as primers, the first strand of cDNA was synthesized in an M-MuLV Reverse Transcriptase system. Subsequently, the RNA strand was degraded with RNase H, and the second strand of cDNA was synthesized using DNA polymerase I with dNTPs. The purified double-stranded cDNA underwent end repair, A-tailing, and adapter ligation. cDNA fragments approximately 200 bp in size were selected using AMPure XP beads, amplified by PCR, and the PCR products were purified again using AMPure XP beads to obtain the final libraries.
Sequencing was performed on the Illumina platform using the TruSeq SBS Kit (300 cycles). Raw reads were processed with Trimmomatic (http://www.usadellab.org/cms/index.php?page= trimmomatic) to remove low-quality sequences: reads containing >10% ambiguous bases (N), bases at the ends with quality scores <20, adapter sequences, and reads shorter than 75 bp after trimming. Following quality control, high-quality reads were mapped to the previously assembled transcriptome using Trinity (http://trinityrnaseq.sourceforge.net/), which de novo assembled all sequencing fragments into contigs and singletons. The assembled sequences were functionally annotated by aligning them against the NR, Swiss-Prot, eggNOG, KEGG, GO, and CAZy databases. Differentially expressed genes (DEGs) were identified based on the criteria of |log2(fold change)| ≥1 and false discovery rate (FDR) < 0.05. Functional enrichment analysis of DEGs was conducted using the GO and KEGG databases.
In vitro assessment of SynM-mediated soil metabolite modification on ECM fungal growth
To assess the effect of soil metabolites on Tuber growth, soil samples from Dingxi and Lingwu (aridity: 0.74–0.87) were selected for extract preparation. Briefly, 30 g of soil was combined with 90 mL of 80% methanol and agitated at 180 rpm for 3 h. The supernatant was then collected and subjected to rotary evaporation to remove methanol. The precipitate was re-dissolved with 1 mL of methanol under ultrasonic conditions. A 10 mL soil extract solution was prepared by diluting this concentrate with 9 mL sterile water, followed by filtration through a 0.2 μm membrane to remove microorganisms. This procedure was repeated in subsequent experiments. This procedure was repeated for subsequent experiments.
Next, we established three treatments to investigate how SynM modifies soil metabolic components to facilitate the ECMF growth. (1) Soil extract control: 5 mL of soil extract was combined with 15 mL of 1/5 MMN and inoculated with Tuber. (2) soil extract + SynM supernatant: Soil extract was mixed with an equal volume of sterile SynM supernatant. Subsequently, 5 mL of this mixture was combined with 15 mL of 1/5 MMN and inoculated with Tuber. (3) soil extract + SynM: The SynM was inoculated into 20 mL soil extract and incubated at 28 °C for 72 h. Following incubation, the culture was centrifuged, and the supernatant was passed through a 0.2 μm filter to remove the bacteria. Then, 5 mL of this treated SE was combined with 15 mL of 1/5 MMN and inoculated with Tuber. In the control group, an equal volume of sterile water was substituted for the soil extract. Radial growth of Tuber was measured after 72 h of incubation in darkness at 25 °C.
Transcriptome sequencing of Tuber from Control, Soil extract control, and Soil extract + SynM treatments provided molecular-level insights, following the sampling and sequencing protocols described above in the section titled “Transcriptome sequencing of ECM fungi”.
LC-MS metabolomic analysis of modified soil extracts
To characterize changes in soil metabolite composition induced by the SynM, LC-MS was used to profile metabolites in both original SE and SynM-treated SE. For metabolite extraction, 100 μL of each sample was vortex-mixed with 400 μL of 80% methanol-water solution, incubated on ice for 5 min, and centrifuged at 15,000 × g for 20 min at 4 °C. A defined volume of the supernatant was diluted to 53% methanol using spectrometry-grade water. This dilution was transferred to a new centrifuge tube and re-centrifuged at 15000 × g for 20 min at 4 °C. The final supernatant was loaded onto the LC-MS system for analysis (Vanquish UHPLC and Orbitrap Q ExactiveTM HF; Thermo Fisher Scientific, Germany). Chromatographic separation was achieved using a Hypersil Gold column (100 × 2.1 mm, 1.9 μM) with a flow rate of 0.2 mL/min and a 17-min linear gradient. For the positive ionization mode, mobile phase A consisted of water containing 0.1% formic acid (FA), and mobile phase B was methanol. For negative ionization mode, mobile phase A was 5 mM ammonium acetate (pH 9.0), and mobile phase B was methanol. The solvent gradient program was set as follows: 2% B, 1.5 min; 2-100% B, 12.0 min; 100% B, 14.0 min; 100–2% B, 14.1 min; 2% B, 17 min. The Q ExactiveTM HF mass spectrometer was operated in both positive and negative ionization modes with spray voltage of 3.2 kV, capillary temperature of 320 °C, sheath gas flow rate of 40 arbitrary units (arb) and auxiliary gas flow rate of 10 arb.
Statistical analyses
Statistical analyses and data visualization were performed conducted in Origin Pro 2023, R software (v4.0.2; https://www.R-project.org/). Residual normality and variance homogeneity were verified using the Shapiro-Wilk and Levene tests, respectively. Differences in ECM colonization rates were assessed by one-way ANOVA followed by Tukey-Kramer post hoc tests. Alpha diversity indices and beta diversity (weighted UniFrac-based) for bacterial and ECMF communities were calculated with the “vegan” R package (v2.5-2; https://cran.rproject.org/ package=vegan). Two-way ANOVA with Tukey-Kramer post hoc testing evaluated alpha diversity variations across compartments and aridity zones. Beta diversity patterns were visualized via principal coordinate analysis (PCoA) and statistically tested using permutational multivariate analysis of variance (PERMANOVA) with the adonis function (vegan). Linear and quadratic relationships of ECMF colonization and microbial diversity along aridity gradients were examined. Model fit was evaluated via Akaike Information Criterion (AIC; lower values indicating better fit68; Table S3). Thresholds were detected when the quadratic regressions fit the data better. We fit segmented regressions using the “segmented” package of R to identify the tipping point where the values abruptly changed along the aridity gradient69, with the tipping point detected as the aridity threshold, as many previous studies28,70. Geographic distances (Km) between sampling sites were calculated from coordinates using the “geosphere” package. Distance-decay relationships for community similarity (1-weighted UniFrac distance) were fitted with ordinary least squares regression, with correlations assessed via Mantel tests.
Microbial cohesion between ECM fungi and bacteria was quantified per sample based on SparCC-constructed interkingdom networks (SparCC |r| > 0.4, P < 0.05)71,72. Positive cohesion (reflecting the strength of positive interkingdom connections) and negative cohesion (indicating negative connectivity) were derived72. Compartmental differences in cohesion were tested with two-way ANOVA and Tukey-Kramer tests. We then employed a linear mixed model (LMM) to assess ECMF-bacterial interaction changes along aridity gradients. RFM identified key predictors of ECMF colonization, with model significance evaluated via “RfUtilities” R package and predictor importance via “RfPermute” R package. A GAM incorporated the six RFM-selected predictors, and compartmental contributions to ECM colonization were quantified by decomposing GAM-adjusted R².
To identify key ectomycorrhizosphere bacterial taxa influencing ECM colonization, bacterial ASVs significantly enriched in the ectomycorrhizosphere (relative to bulk soil) were first detected using DESeq2 (“DESeq2” R package). Subsequently, two complementary approaches identified key taxa. The first method utilized microbial interaction patterns. Positive connectedness (defined as the mean value of significant SparCC-derived positive correlations between bacterial ASVs and ECMF ASVs) was calculated, and correlations between bacterial ASVs and ECM colonization were assessed. Another strategy targeting environmental filtering utilized the finding that bacterial community divergence between the ectomycorrhizosphere and bulk soil peaked at an aridity level >0.7. Ectomycorrhizosphere bacteria specifically enriched under high aridity were hypothesized as ECM colonization contributors. These taxa were characterized by occupancy (the relative frequency of occurrence of species in the samples of habitat) and specificity (the mean abundance of species in the samples of habitat) with both metrics exceeding 0.7 a threshold referenced from methods for identifying specialist species in environments73,74.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw plant RNA-seq data, amplicon sequencing data generated in this study have been deposited in the Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra). Raw amplicon sequences derived from the field survey are publicly available under NCBI BioProject number PRJNA1311795 (16S rRNA gene) and PRJNA1311818 (ITS). All 16S rRNA sequence data of bacterial strains are publicly available under NCBI BioProject number PRJNA1312253. The RNA-seq data for Tuber are publicly available under NCBI BioProject number PRJNA1312988. Source data are available in the Figshare database (https://doi.org/10.6084/m9.figshare.30775883). Source data are provided with this paper.
Code availability
Codes used in this study are available in the Figshare database (https://doi.org/10.6084/m9.figshare.30685235).
References
Ren, S. et al. Storage potential of soil functional carbon fractions in the World’s largest plantations. Adv. Sci. 12, e04995 (2025).
Wang, Y. J. et al. Land availability and policy commitments limit global climate mitigation from forestation. Science 389, 931–934 (2025).
Garrett, L., Lévite, H., Besacier, C., Alekseeva, N. & Duchelle, M. The key role of forest and landscape restoration in climate action. (FAO, 2022).
Reynolds, J. F. et al. Global desertification: building a science for dryland development. Science 316, 847–851 (2007).
Ma, L. L. et al. Planted forests in China have higher drought risk than natural forests. Glob. Change Biol. 31, e70055 (2025).
Koppa, A. et al. Dryland self-expansion enabled by land-atmosphere feedbacks. Science 385, 967–972 (2024).
Gu, L., Schumacher, D. L., Wang, H. M., Yin, J. & Fischer, E. M. Land-atmosphere feedbacks drive dryland drought and expansion under climate warming. Innovation 6, 100863 (2025).
Cosme, M. Mycorrhizas drive the evolution of plant adaptation to drought. Commun. Biol. 6, 346 (2023).
Sachsenmaier, L. et al. Forest growth resistance and resilience to the 2018–2020 drought depend on tree diversity and mycorrhizal type. J. Ecol. 112, 1787–1803 (2024).
Poudel, M. et al. The role of plant-associated bacteria, fungi, and viruses in drought stress mitigation. Front. Microbiol. 12, 743512 (2021).
Hu, M. Y. et al. Plant functional traits affect biomass responses to global change: a meta-analysis. J. Ecol. 113, 2046–2065 (2025).
Van Nuland, M. E. et al. Global hotspots of mycorrhizal fungal richness are poorly protected. Nature 645, 414–422 (2025).
Jiang, F. et al. Mycorrhizal symbioses and tree diversity in global forest communities. Sci. Adv. 11, eadt5743 (2025).
Berdugo, M. et al. Global ecosystem thresholds driven by aridity. Science 367, 787–790 (2020).
Stuart, E. K. et al. Acquisition of host-derived carbon in biomass of the ectomycorrhizal fungus Pisolithus microcarpus is correlated to fungal carbon demand and plant defences. FEMS Microbiol. Ecol. 99, fiad037 (2023).
Canarini, A. et al. Soil fungi remain active and invest in storage compounds during drought, independent of future climate conditions. Nat. Commun. 15, 10410 (2024).
Xie, L. L. et al. Variations in ectomycorrhizal exploration types parallel seedling fine root traits of two temperate tree species under extreme drought and contrasting solar radiation treatments. Plant Cell Environ. 47, 5053–5066 (2024).
Guarnizo, A. L., Navarro-Ródenas, A., Calvo-Polanco, M., Marqués-Gálvez, J. E. & Morte, A. A mycorrhizal helper bacterium alleviates drought stress in mycorrhizal Helianthemum almeriense plants by regulating water relations and plant hormones. Environ. Exp. Bot. 207, 105228 (2023).
Berrios, L. et al. Positive interactions between mycorrhizal fungi and bacteria are widespread and benefit plant growth. Curr. Biol. 33, 2878–2887.e2874 (2023).
Navarro-Rodenas, A., Berna, L. M., Lozano-Carrillo, C., Andrino, A. & Morte, A. Beneficial native bacteria improve survival and mycorrhization of desert truffle mycorrhizal plants in nursery conditions. Mycorrhiza 26, 769–779 (2016).
Jorgensen, K., Clemmensen, K. E., Wallander, H. & Lindahl, B. D. Do ectomycorrhizal exploration types reflect mycelial foraging strategies? New Phytol. 237, 576–584 (2023).
Durán, P. et al. Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 175, 973–983.e914 (2018).
Bertness, M. D. & Callaway, R. Positive interactions in communities. Trends Ecol. Evol. 9, 191–193 (1994).
Hammarlund, S. P. & Harcombe, W. R. Refining the stress gradient hypothesis in a microbial community. Proc. Natl. Acad. Sci. USA 116, 15760–15762 (2019).
Hernandez, D. J., David, A. S., Menges, E. S., Searcy, C. A. & Afkhami, M. E. Environmental stress destabilizes microbial networks. ISME J. 15, 1722–1734 (2021).
Gao, C. et al. Co-occurrence networks reveal more complexity than community composition in resistance and resilience of microbial communities. Nat. Commun. 13, 3867 (2022).
Berrios, L., Ansell, T. B., Dahlberg, P. D. & Peay, K. G. Standardizing experimental approaches to investigate interactions between bacteria and ectomycorrhizal fungi. FEMS Microbiol. Rev. 49, fuae035 (2025).
Hao, Z. G. et al. Thresholds in aridity and soil carbon-to-nitrogen ratio govern the accumulation of soil microbial residues. Commun. Earth Environ. 2, 236 (2021).
Miyamoto, Y., Maximov, T. C., Kononov, A. & Sugimoto, A. Soil propagule banks of ectomycorrhizal fungi associated with Larix cajanderi above the treeline in the Siberian Arctic. Mycoscience 63, 142–148 (2022).
Shemesh, H., Bruns, T. D., Peay, K. G., Kennedy, P. G. & Nguyen, N. H. Changing balance between dormancy and mortality determines the trajectory of ectomycorrhizal fungal spore longevity over a 15-yr burial experiment. New Phytol. 238, 11–15 (2023).
Williams, A., Sinanaj, B. & Hoysted, G. A. Plant-microbe interactions through a lens: tales from the mycorrhizosphere. Ann. Bot. 133, 399–412 (2024).
Hoek, T. A. et al. Resource availability modulates the cooperative and competitive nature of a microbial cross-feeding mutualism. PLoS Biol. 14, e1002540 (2016).
Piccardi, P., Vessman, B. & Mitri, S. Toxicity drives facilitation between 4 bacterial species. Proc. Natl. Acad. Sci. USA 116, 15979–15984 (2019).
Lofgren, L. et al. Suillus: an emerging model for the study of ectomycorrhizal ecology and evolution. New Phytol. 242, 1448–1475 (2024).
Voller, F., Ardanuy, A., Taylor, A. F. S. & Johnson, D. Maintenance of host specialisation gradients in ectomycorrhizal symbionts. New Phytol. 242, 1426–1435 (2024).
Frey-Klett, P., Garbaye, J. & Tarkka, M. The mycorrhiza helper bacteria revisited. New Phytol. 176, 22–36 (2007).
Tedersoo, L., Bahram, M. & Zobel, M. How mycorrhizal associations drive plant population and community biology. Science 367, eaba1223 (2020).
Peng, L. et al. A facultative ectomycorrhizal association is triggered by organic nitrogen. Curr. Biol. 32, 5235–5249.e5237 (2022).
Rasheed, Z. et al. Production of some secondary metabolites of antibiotic nature from mycorrhizal helper bacteria (MHB) associated with conifers. Pol. J. Environ. Stud. 33, 4325–4334 (2024).
Singh, B. K. & Walker, A. Microbial degradation of organophosphorus compounds. FEMS Microbiol. Rev. 30, 428–471 (2006).
Yang, F. et al. Genome-wide analysis reveals genetic potential for aromatic compounds biodegradation of Sphingopyxis. Biomed. Res. Int. 2020, 5849123 (2020).
Zhao, Q. et al. Comparative genomic analysis of 26 Sphingomonas and Sphingobium strains: dissemination of bioremediation capabilities, biodegradation potential and horizontal gene transfer. Sci. Total Environ. 609, 1238–1247 (2017).
Tong, S. et al. Characterization of a fungal competition factor: production of a conidial cell-wall-associated antifungal peptide. PLoS Pathog. 16, e1008518 (2020).
Zhong, Z. X. et al. Differential gene expression profiling analysis in Pleurotus ostreatus during interspecific antagonistic interactions with Dichomitus squalens and Trametes versicolor. Fungal Biol. 121, 1025–1036 (2017).
Mochizuki, K. et al. The ASCT/SCS cycle fuels mitochondrial ATP and acetate production in Trypanosoma brucei. Biochim. Biophys. Acta Bioenerg. 1861, 148283 (2020).
Labbe, J. L., Weston, D. J., Dunkirk, N., Pelletier, D. A. & Tuskan, G. A. Newly identified helper bacteria stimulate ectomycorrhizal formation in Populus. Front. Plant Sci. 5, 579 (2014).
Al Farraj, D. A., Alkufeidy, R. M., Alkubaisi, N. A. & Alshammari, M. K. Polynuclear aromatic anthracene biodegradation by psychrophilic Sphingomonas sp., cultivated with Tween-80. Chemosphere 263, 128115 (2021).
Park, J. et al. Sulfur metabolism-mediated fungal glutathione biosynthesis is essential for oxidative stress resistance and pathogenicity in the plant pathogenic fungus Fusarium graminearum. mBio 15, e0240123 (2024).
Hanna, V. S., Abd El-Ghany, M. N., Ibrahim, M. I. M., Abdel-Rahman, T. M. & Tallima, H. Novel approaches to Mortierella alpina identification and arachidonic acid production optimization. ACS Omega 9, 34456–34463 (2024).
Agerer, R. Exploration types of ectomycorrhizae: a proposal to classify ectomycorrhizal mycelial systems according to their patterns of differentiation and putative ecological importance. Mycorrhiza 11, 107–114 (2001).
van der Linde, S. et al. Environment and host as large-scale controls of ectomycorrhizal fungi. Nature 558, 243–248 (2018).
Põlme, S. et al. Biogeography of ectomycorrhizal fungi associated with alders (Alnus spp.) in relation to biotic and abiotic variables at the global scale. New Phytol. 198, 1239–1249 (2013).
Reis, F. et al. Ectomycorrhizal fungal diversity and community structure associated with cork oak in different landscapes. Mycorrhiza 28, 357–368 (2018).
Edwards, J. et al. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 112, E911–E920 (2015).
He, X. H., Yang, Y. Z. & Yuang, Z. L. Protocol for sampling of root and rhizosphere soils from trees in natural fields. Bio 101, e2003655 (2021).
Beckers, B., Op De Beeck, M., Weyens, N., Boerjan, W. & Vangronsveld, J. Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. Microbiome 5, 25 (2017).
Chelius, M. K. & Triplett, E. W. The diversity of archaea and bacteria in association with the roots of Zea mays L. Microb. Ecol. 41, 252–263 (2001).
Bodenhausen, N., Horton, M. W. & Bergelson, J. Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. PloS ONE 8, e56329 (2013).
Gardes, M. & Bruns, T. D. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 2, 113–118 (1993).
White, T. J., Bruns, T., Lee, S. & Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols. https://doi.org/10.1016/b978-0-12-372180-8.50042-1 (Academic Press, 1990).
Põlme, S. et al. FungalTraits: a user-friendly traits database of fungi and fungus-like stramenopiles. Fungal Divers. 105, 1–16 (2021).
Kim, M., Oh, H. S., Park, S. C. & Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351 (2014).
Deveau, A. et al. The mycorrhiza helper Pseudomonas fluorescens BBc6R8 has a specific priming effect on the growth, morphology and gene expression of the ectomycorrhizal fungus Laccaria bicolor S238N. New Phytol. 175, 743–755 (2007).
Zhao, L., Wu, X. Q., Ye, J. R., Li, H. & Li, G. E. Isolation and characterization of a mycorrhiza helper bacterium from rhizosphere soils of poplar stands. Biol. Fertil. Soils 50, 593–601 (2014).
Founoune, H. et al. Interactions between ectomycorrhizal symbiosis and Fluorescent pseudomonads on Acacia holosericea: isolation of mycorrhiza helper bacteria (MHB) from a Soudano-Sahelian soil. FEMS Microbiol. Ecol. 41, 37–46 (2002).
Zi, H. Y. et al. Co-inoculation with rhizobacterial community and an ectomycorrhizal fungus promotes poplar ectomycorrhization. Appl. Soil Ecol. 206, 105833 (2025).
Yu, L. et al. Rhizosphere microbiome of forest trees is connected to their resistance to soil-borne pathogens. Plant Soil 479, 143–158 (2022).
Cavanaugh, J. E. & Neath, A. A. The Akaike information criterion: background, derivation, properties, application, interpretation, and refinements. Wiley Interdiscip. Rev. Comput. Stat. 11, e1460 (2019).
Muggeo, V. Segmented: an R package to fit regression models with broken-line relationships. R New. 8, 20–25 (2008).
Hu, W. G. et al. Aridity-driven shift in biodiversity-soil multifunctionality relationships. Nat. Commun. 12, 5350 (2021).
Friedman, J. & Alm, E. J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8, e1002687 (2012).
Herren, C. M. & McMahon, K. D. Cohesion: a method for quantifying the connectivity of microbial communities. ISME J. 11, 2426–2438 (2017).
Dufrêne, M. & Legendre, P. Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol. Monogr. 67, 345–366 (1997).
Gweon, H. S. et al. Contrasting community assembly processes structure lotic bacteria metacommunities along the river continuum. Environ. Microbiol. 23, 484–498 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China 32430069, W2412011 (to X.L.); The Jiangsu Special Fund on Technology Innovation of Carbon Dioxide Peaking and Carbon Neutrality BE2022420 (to X.L.). We appreciate Professor Nan Yang of Nanjing Forestry University for providing the Tuber strains.
Author information
Authors and Affiliations
Contributions
X.L. conceived the project, designed the experiments. H.Z. and Y.W. conducted data curation, methodology, and writing of the original draft. M.D.-B. contributed to data interpretation, writing – review & editing. Z.H., Y.L., S.B., F.C., and M.D.-B. worked on the manuscript. All authors have discussed the results, read and approved the contents of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zi, H., Hua, Z., Wang, Y. et al. Mycorrhizal colonization of dryland tree establishment depends on soil microbial cooperation. Nat Commun 17, 1041 (2026). https://doi.org/10.1038/s41467-025-67797-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67797-z
